

Abstract
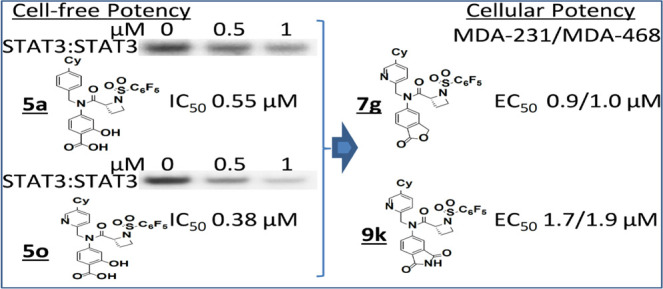
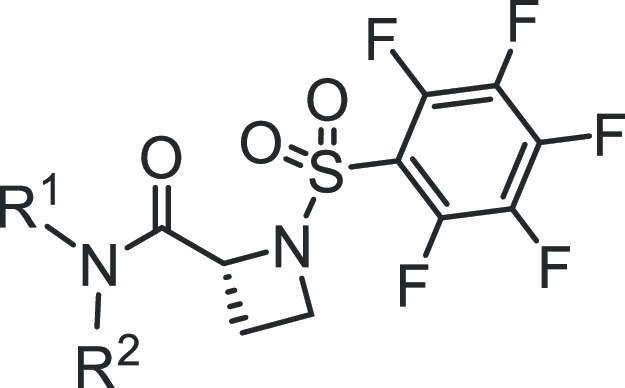
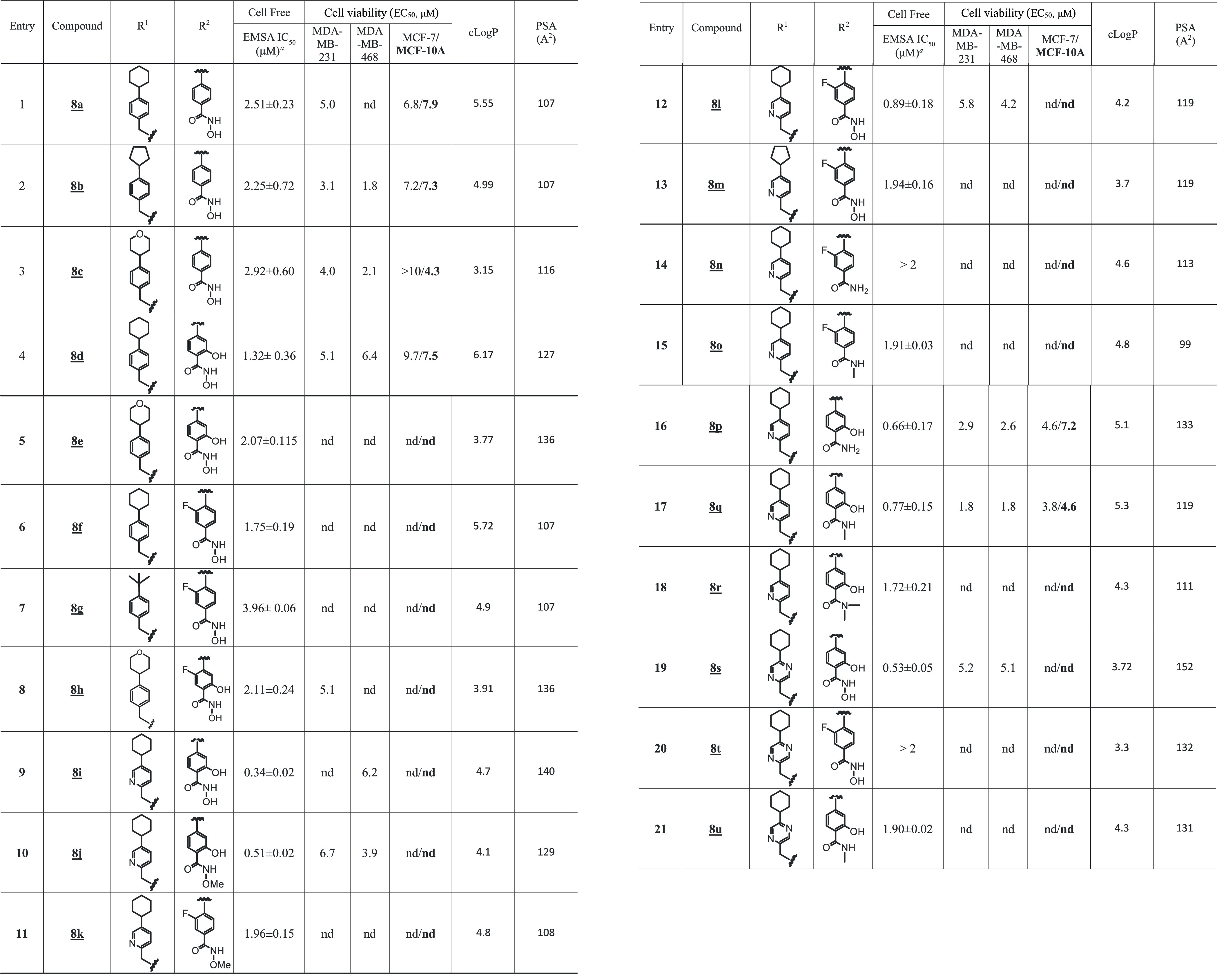
We optimized our previously reported proline-based STAT3 inhibitors into an exciting new series of (R)-azetidine-2-carboxamide analogues that have sub-micromolar potencies. 5a, 5o, and 8i have STAT3-inhibitory potencies (IC50) of 0.55, 0.38, and 0.34 μM, respectively, compared to potencies greater than 18 μM against STAT1 or STAT5 activity. Further modifications derived analogues, including 7e, 7f, 7g, and 9k, that addressed cell membrane permeability and other physicochemical issues. Isothermal titration calorimetry analysis confirmed high-affinity binding to STAT3, with KD of 880 nM (7g) and 960 nM (9k). 7g and 9k inhibited constitutive STAT3 phosphorylation and DNA-binding activity in human breast cancer, MDA-MB-231 or MDA-MB-468 cells. Furthermore, treatment of breast cancer cells with 7e, 7f, 7g, or 9k inhibited viable cells, with an EC50 of 0.9–1.9 μM, cell growth, and colony survival, and induced apoptosis while having relatively weaker effects on normal breast epithelial, MCF-10A or breast cancer, MCF-7 cells that do not harbor constitutively active STAT3.
Introduction
The signal transducer and activator of transcription (STAT) family of cytoplasmic transcription factors mediate the cellular responses to cytokine and growth factors, including cell growth and differentiation, inflammation, and immune responses.1−3 STAT activation is initiated upon receptor–ligand binding that induces STAT recruitment to the receptor and STAT phosphorylation by Janus kinases (JAKs) and Src family kinases. Two phospho-STAT monomer proteins form STAT:STAT dimers through a reciprocal pTyr-Src homology (SH)2 domain interactions, translocate to the nucleus, and bind to specific DNA-response elements in target gene promoters to induce gene transcription.2−5
While normal STAT activation is rapid and transient, aberrant activation of one family member, STAT3, is prevalent and has a causal role in many human cancers.6,7 STAT3 is therefore a valid and an attractive target for the development of novel anticancer therapeutics.7−10 One of the main approaches to develop inhibitors of STAT3 is focused on targeting the key pTyr:SH2 domain interaction and the STAT3:STAT3 dimerization event.8,9 Several small-molecule inhibitors have been developed that target the STAT3 SH2 domain and disrupt the STAT3:STAT3 dimerization.11−26 However, there has been limited success in the advancement of these STAT3 inhibitors into clinical application due to low potency and pharmacokinetic (PK) limitations and toxicity.27 More recently, a new class of inhibitors of the STAT3 signaling pathway has emerged that focuses on protein degradation. The PROTAC-STAT3 degraders, represented by SD-36, promoted STAT3 degradation with nanomolar potency, and SD-36 induced a complete tumor growth inhibition in vivo in multiple tumor models.28,29
We have previously provided proof-of-concept for the in vivo antitumor efficacy of the micromolar potent lead inhibitors, BP-1-102, SH5-07, and SH4-54, which are based on the N-methylglycinamide scaffold, with its two amine moieties condensed with three different functionalities.24,26 We took steps to address the challenges with potency and PK properties and hence advance the development of these amino acid amide-based inhibitors and recently published an extensive study on the structure–activity relationship (SAR) analysis using an iterative medicinal chemistry approach in which the amino acid linker was varied along with the simultaneous optimization of the three functionalities to improve potency and physicochemical properties, and this led to new proline-based analogues.30 With the focus on potency, we further extended the optimization of the proline-based analogues into other cyclic amino acids and have now derived more exciting new series of (R)-azetidine-2-carboxamide analogues of BP-1-102, including 5a, 5o, and 8i, which show sub-micromolar STAT3-inhibitory activity in vitro (IC50 values of 0.52, 0.38, and 0.34 μM, respectively). To improve membrane permeability, we further derived analogues containing carboxylic acid surrogates, 7e, 7f, 7g, and 9k, which at 1 μM or less strongly inhibited the viability, anchorage-dependent and independent growth, and colony formation of MDA-MB-231 or MDA-MB-468 human breast cancer cells that harbor aberrantly active STAT3.
Results and Discussion
Progression from the Proline Linker: SAR of Early Azetidine Analogues Shows Nanomolar Potency in Disrupting STAT3 DNA-Binding Activity In Vitro
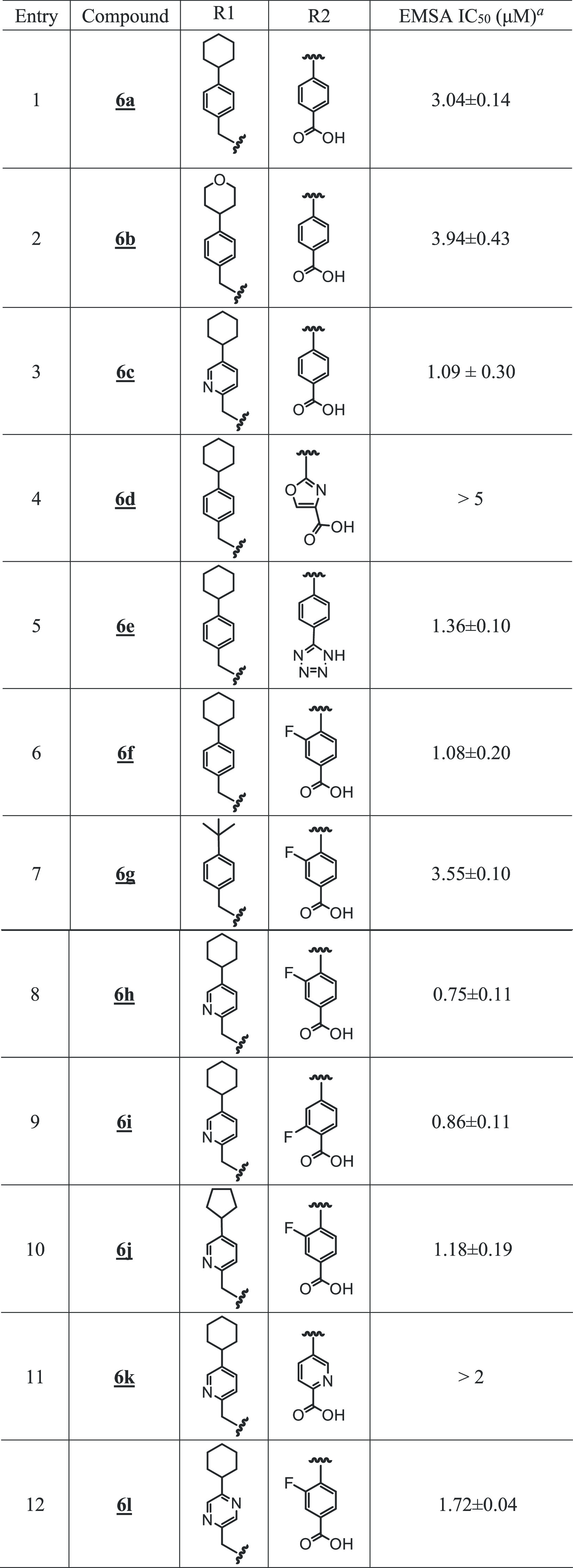
Progression from the previously reported proline linker (Supporting Information Figure S1, 3)30 into other cyclic amino acid linkers led us to discover more potent inhibitors of STAT3 activity, as measured by DNA-binding activity/electrophoretic mobility shift assay (EMSA). In this assay, nuclear extracts containing active STAT3:STAT3 prepared from cancer cells are preincubated with increasing concentrations of the compounds at room temperature for 30 min, prior to incubation with the radiolabeled high-affinity sis-inducible element (hSIE) probe (from the c-fos promoter) that binds active STAT3 and subjecting to EMSA analysis.23,24,26 The basic premise is that the binding of the high-affinity compounds to STAT3 inhibits STAT3 DNA-binding activity, as shown in the past.13,14,23,24,26,30 The bands corresponding to the DNA-bound STAT3 are scanned, quantified by ImageJ, and represented as percent of control (100%), which are plotted against the concentration of the compounds. Representative plots are shown in the Supporting Information Figure S2. Although changing the 5-membered proline analogue, 3, to the corresponding 6-membered, pipecolamide analogue, 4, decreased STAT3-inhibitory potency from EMSA IC50 2.4 μM for 3 to IC50 5.4 μM for 4 (Supporting Information Figure S3), changing to the 4-membered azetidine-2-carboxamide analogue, 5a (Supporting Information Figure S3), gave over a 4-fold boost in potency in vitro over proline, 3, against STAT3 DNA-binding activity (Figure 1A). The concentration at which there is 50% inhibition of STAT3 DNA-binding activity relative to the dimethyl sulfoxide (DMSO)-treated control in the EMSA analysis, IC50, is 0.52 μM for 5a. This result not only represented over a log-order improvement in potency from the corresponding glycine-based analogue, BP-1-102, but also represented one of the first cases of small-molecule direct inhibitor of STAT3 DNA-binding activity in vitro, with sub-micromolar potency. To confirm that the DNA-bound protein is STAT3, we performed supershift analysis.31−33 The presence of the specific anti-STAT3 antibody with the nuclear extract sample blocked and/or caused a shift in the band for DNA:STAT3 complex (Supporting Information Figure S4A). We also tested napabucasin (BBI-608), the stem cell inhibitor, which is also purported to inhibit STAT3 function,34 and another purported STAT3 inhibitor, C188-9,35 both of which showed a minimal direct effect on STAT3 activity up to 10 μM in the DNA-binding assay/EMSA analysis (Supporting Information Figure S4B), suggesting that these compounds have very little direct effects on STAT3 DNA-binding activity. As previously observed with other chiral inhibitors, such as 2 and 3 (Supporting Information Figure S1),30 the (R)-enantiomer was more potent than the (S)-enantiomer (EMSA IC50 of 0.52 μM for 5a vs 2.22 μM for 5b; Figures 1A and 2), and changing the azetidine core from the azetidine-2-carboxamide to the azetidine-3-carboxamide (5c) (Figures 1A and 2) resulted in loss of activity. Our structure–activity exploration thus focused on (R)-azetidine-2-carboxamides.
Figure 1.

Azetidine analogues inhibit STAT3 DNA-binding activity in vitro. Nuclear extracts of equal total protein prepared from NIH3T3/v-Src fibroblasts containing activated STAT3 were preincubated with increasing concentrations of the designated versions of azetidines (A) salicylic acids, (B) benzoic acids, (C) methyl esters, (D) hydroxamic acids/methylamines, or (E) heterocycles for 30 min at room temperature prior to incubating with the radiolabeled hSIE probe that binds STAT3 and performing EMSA analysis; bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined. Positions of STAT3:DNA complexes in gel are labeled; control lanes (0) represent nuclear extracts pretreated with 10% DMSO. Data are representative of two to three independent determinations.
Figure 2.

Structures of initial azetidine-based STAT3 inhibitors.
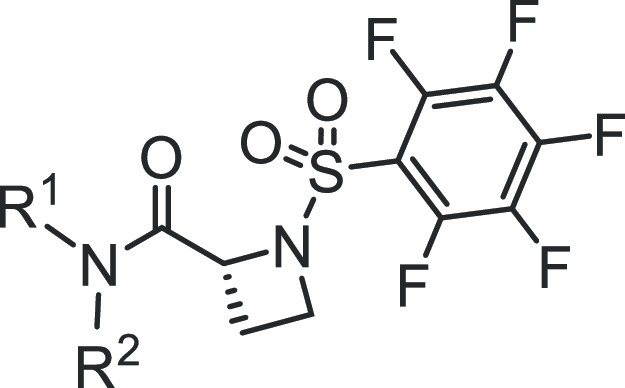
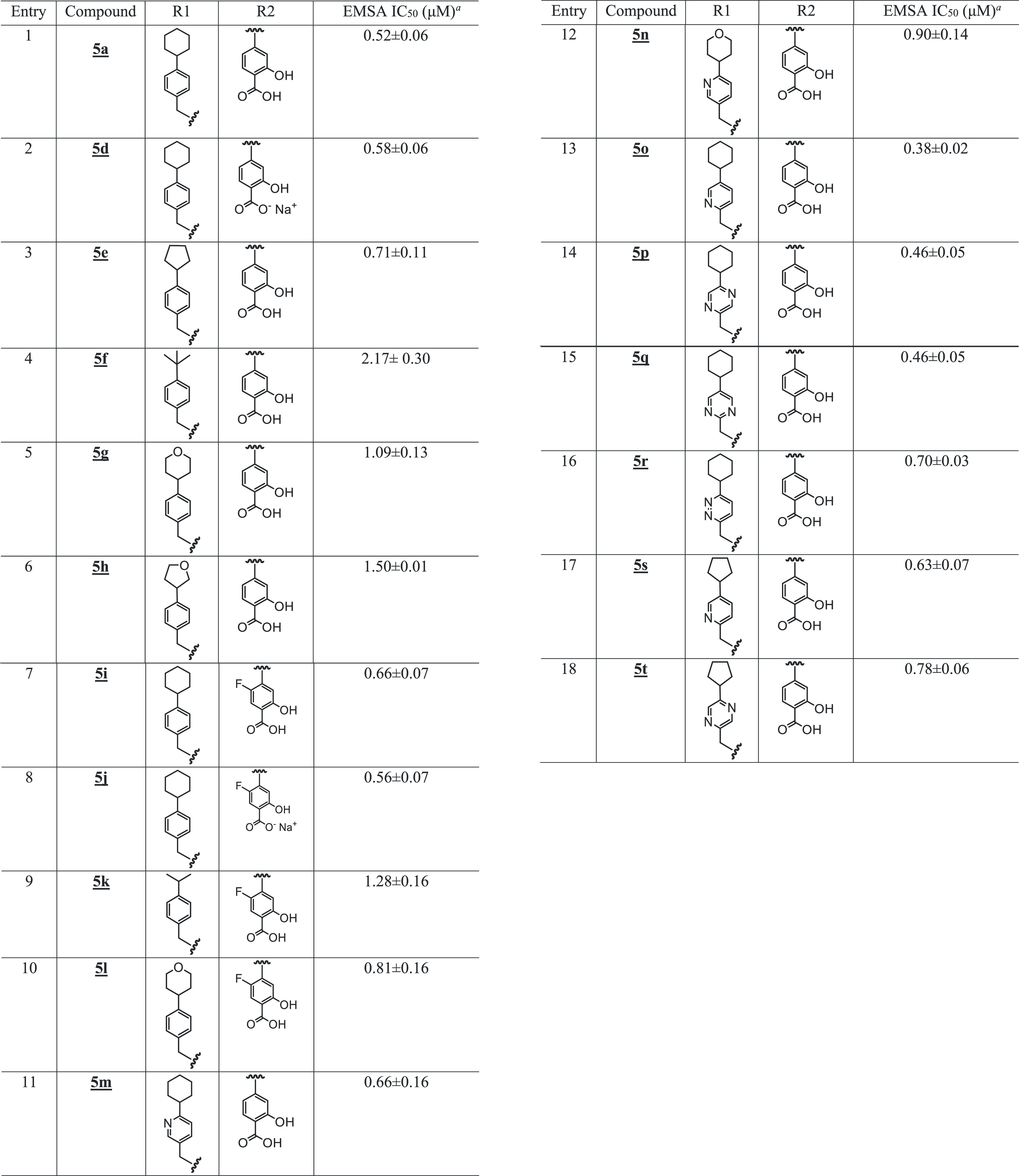
We took an iterative medicinal chemistry approach to design and synthesize small-molecule STAT3 inhibitors of improved physicochemical properties and that are potent and selective against tumor cells harboring constitutively active STAT3. Based on our success in the previous work with the alanine and proline analogues,30 we systematically varied the benzoic acid and cyclohexylbenzyl moieties of the lead compound to optimize potency and physicochemical properties (Figure 1 and Tables 1 and 2). Changes in the cyclohexyl group to decrease lipophilicity were tolerated but with a variable effect on potency (Table 1, entries: 3–6), suggesting that the increased potency provided by the azetidine scaffold could allow for a balancing of physicochemical properties while maintaining sufficient potency. However, cyclohexyl remained the optimum. Introduction of polarity by changing the phenyl ring of the benzyl portion of the molecule to a heterocycle was successful. Although replacement of the phenyl in the cyclohexylbenzyl moiety with a 3-pyridyl resulted in a slight decrease in potency (EMSA IC50 of 0.66 μM for 5m vs IC50 of 0.52 μM for 5a; Figure 1A and Table 1, entry 11), its replacement with the 2-pyridyl analogue provided 50% boost in potency (EMSA IC50 of 0.38 μM for 5o; Figure 1A and Table 1, entry 13), potentially indicating an additional binding interaction with the STAT3 protein. Moreover, increasing the polarity at this very lipophilic region (i.e., the cyclohexylbenzyl moiety) results in better molecular polarity distribution and thus may improve the drug-like properties. Other modifications of the phenyl ring to pyrazine, pyrimidine, or pyridazine (e.g., 5p, 5q, and 5r with IC50 values of 0.46, 0.46, and 0.70 μM, respectively; Figure 1A and Table 1) resulted in compounds retaining high affinity. In this heterocyclic series as well, replacement of the cyclohexyl with the less lipophilic cyclopentyl or tetrahydropyranyl resulted in slightly less potent compounds (e.g., 5n, 5s, and 5t; Table 1). Similar results were seen with the 5-fluorosalicylates (Table 1, entries 7–10).
Table 1. SAR of Salicylic Acid-Based Analogues.
Bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined.
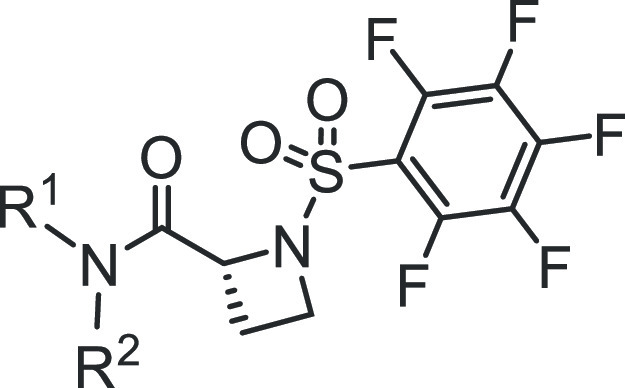
Table 2. SAR of Benzoic Acid-Based Analogues.
Bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined.
Benzoic acids, other than salicylic acids, led to less potent compounds (Table 2). However, in the cyclohexylpyridylmethyl subseries, the STAT3 potency was regained when the benzoic acid group was substituted with fluorine at the 2- or 3-position (Figure 1B and Table 2; compounds 6h and 6i with IC50 values 0.75 and 0.86 μM, respectively). Replacement of the benzoic acid by a 4-oxazolecarboxylic acid or a 2-pyridinecarboxylic acid led to compounds with weaker activity (Table 2, entries 4 and 11).
Analogues with Carboxylic Acid Motif Have Low Cellular Activities
Given that constitutively active STAT3 promotes tumor cell proliferation and survival,27,36 we tested the aforementioned, most active azetidine analogues for their effects on the growth of the human breast cancer MDA-MB-231 and MDA-MB-468 cells that harbor active STAT3.26,30 Despite their sub-micromolar potency in the in vitro cell-free STAT3 DNA-binding/EMSA assay (Tables 1 and 2), they showed weak activity against the breast cancer cells up to 10 μM (Supporting Information Figure S5A,B). This is presumably due to poor cell membrane permeability afforded by the ionized polar carboxylate group.37 Even tetrazole 6e (Table 2), which still partially ionizes at physiological pH, showed weak cellular activity at 10 μM. In this case, the concentration at which there is loss of 50% of DMSO-treated control cell numbers relative to the untreated, control cell numbers, EC50, is greater than 10 μM against MDA-MB-231 cells.
Carboxylate Methyl Esters, Phthalide, and Methyl Amide Versions Improve Cellular Potency of the Azetidine Analogues
To test whether the carboxylate group was responsible for the low cellular activity, methyl esters were prepared and evaluated (Figure 1C and Table 3). Not surprisingly, the cell-free STAT3-inhibitory potencies of the methyl ester versions were lower than their corresponding acid analogues (Figure 1C and Table 3), highlighting the importance of the acid motif for inhibiting STAT3 activity.23,24 By contrast, the ester versions generally showed stronger cellular activities (Figure 3A and Table 3), presumably by facilitating cell membrane permeability and functioning as prodrugs inside cells. Thus, 7a, the methyl ester of salicylic acid 5a, had cell-free EMSA potency of >4 μM (Figure 1C and Table 3), as compared to 0.52 μM for 5a (Figure 1A and Table 1). However, 7a showed good cellular activity against breast cancer cells following treatment for 72 h, with EC50 values of 2.7 and 2.5 μM against MDA-MB-231 and MDA-MB-468 viable cells, respectively (Supporting Information Figure S5C and Table 3), as compared with 5a, which showed no activity up to 10 μM (Supporting Information Figure S5A). Similar results were seen when comparing the methyl ester 7b with its corresponding acid, 6f (EC50 against MDA-MB-231 cells after 72 h treatment was 4.4 μM for 7b vs > 10 μM for 6f) (Supporting Information Figure S5B,C). The esters of the more active cyclohexylpyridylmethyl analogues, 7c and 7d, also showed weaker STAT3-inhibitory activities in the cell-free EMSA assay (Figure 1C and Table 3, entries 3 and 4) than their corresponding acids (Table 1, entries 11 and 13), whereas they showed good activities against MDA-MB-231 viable cells, with EC50 of 2.0 μM for 7c and 1.8 μM for 7d (Supporting Information Figure S5C and Table 3).
Table 3. SAR of Ester Analogues.

Bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined.
Figure 3.

In vitro cell viability and growth studies for effects of active azetidine analogs. (A) Human breast cancer MDA-MB-231 and MDA-MB-468 cells harboring aberrantly active STAT3 and (B) normal breast epithelial MCF-10A or breast cancer MCF-7 cells that do not and growing in 96-well culture were treated once with 0–10 μM of the indicated STAT3 inhibitors, or (C) human breast cancer cells in 96-well culture were first treated with 1 μM 7g for 6 h followed by treatment with 0–100 nM docetaxel (Doc) or 0–20 μM cisplatin (Cis), or the cells were treated with docetaxel or cisplatin alone. After 72-h culture, cells were harvested and subjected to CyQuant cell proliferation assay for the number of viable cells, which are plotted as % cell viability against concentration from which EC50 values were derived; or (D) human breast cancer MDA-MB-231 or MCF-7 cells in a 6-well culture plate were untreated or treated once with 2 μM 9k and every 24 h, cells were harvested and subjected to trypan blue exclusion/phase-contrast microscopy for viable cell counts, which were plotted against the duration of treatment. Values are mean ± SEM of two to three studies each in three replicates.
The interesting phthalide 7g showed one of the best activities against MDA-MB-231 and MDA-MB-468 viable cells, with EC50 of 0.9 and 1.0 μM, respectively (Figure 3A and Tables 3 and 6). The esters 7e (EC50 1.4 and 1.4 μM, respectively) and 7f (EC50 1.6 and 1.6 μM, respectively) also showed relatively better cellular activities than their carboxylic acid versions against the viable cell numbers of MDA-MB-231 and MDA-MB-468 cells (Figure 3A and Tables 3 and 6). Notably, combined treatment with 7g and docetaxel38 or cisplatin,39 both chemotherapeutic agents used to treat TNBC, showed a strong shift of the dose–response curves to the left indicative of an enhanced response (Figure 3C). We note the apparent weaker cell-free EMSA potencies of the methyl esters, which reflect their prodrug properties, and is the reason the esters are more active in cells than outside of cells. Under conditions that promote hydrolysis, as is the case inside cells, the esters will be converted to carboxylates. Moreover, the in vitro activities of these compounds compare favorably to the parent lead compounds, BP-1-102, SH5-07, and SH4-54, with IC50 of 6.8, 3.9, and 4.7 μM, respectively, and cellular activities at 10–20, 3.8, and 4.5 μM, respectively.24,26
Table 6. EMSA IC50 and EC50 Values of Select Esters and Heterocycles.

With these promising results, our efforts continued also on other bioisosteric replacements of the benzoic acid or salicylic acid moieties that would enable penetration into cells. We first concentrated our attention on benzohydroxamic acids, which had shown promise in our previous work (e.g., SH5-07).26 The first benzohydroxamic acids we prepared, 8a to 8d (Table 4), showed only moderate inhibitory potencies against both the STAT3 DNA-binding activity in the cell-free EMSA assay (Figure 1D and Table 4) and the viable cell numbers of MDA-MB-231 and MDA-MB-468 cells (Table 4). However, by introducing the novel cyclohexylpyridylmethyl group, the corresponding benzohydroxamic acid analogue 8i was found to have the most potent STAT3-inhibitory activity in the whole series in the EMSA assay, with an IC50 value of 0.34 μM (Figure 1D and Table 4, entry 9), though it did not have good cellular activity (EC50 6.2 μM against MDA-MB-468; Table 4, entry 9). Presumably, the increased polar surface area (PSA, 140 Å2) mainly imparted by both the pyridyl ring in one vector and the benzohydroxamic acid in the other vector had an impact on the sluggish cell membrane permeability. Likewise, the benzohydroxamic acid-pyrazine analogue 8s gave a similar result (Figure 1D, Supporting Information Figure S5D, and Table 4, entry 19). The O-methyl hydroxamic acid 8j (PSA 129 Å2, EMSA assay IC50 0.51 μM; Table 4, entry 10) was similarly weak in the assay for viable cell numbers (Supporting Information Figure S5D and Table 4, entry 10).
Table 4. SAR of Benzohydroxamic Acid and Salicylamide Analogues.
Bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined. nd: not determined.
Variations of the sub-micromolar 3-fluorobenzoic acid analogue, 6h, were explored. The 3-fluorobenzohydroxamic acid, 8l, was weakly active against breast cancer cells (Supporting Information Figure S5D and Table 4) while maintaining the good STAT3-inhibitory potency in the EMSA assay (Figure 1D, IC50 of 0.89 μM; Table 4, entry 12). We next sought to replace the polar hydroxamic acid group while maintaining favorable PSA and cLogP. The cutoff values that we followed, in accordance with the literature, were PSA < 120 Å2 37 and cLogP ≤ 5.40 The methyl salicylamide, 8q (PSA 119 Å2), showed good activity against breast cancer cells in both viability assays, with EC50 1.8 and 1.8 μM against MDA-MB-231 cells and MDA-MB-468 cells, respectively, and trypan blue exclusion/phase-contrast microscopy cell growth assay (Supporting Information Figure S5C,E and Table 4, entry 17), and it also showed a high STAT3-inhibitory potency (IC50 0.77 μM; Figure 1D and Table 4, entry 17). The more polar primary salicylamide 8p (PSA 133 Å2) showed relatively modest activity against MDA-MB-231 and MDA-MB-468 cells, with EC50 2.9 and 2.6 μM, respectively (Table 4 and Supporting Information Figure S5C), while strongly inhibiting STAT3 DNA-binding activity in EMSA (IC50 0.66 μM).
Altogether, the replacement of the salicylic or benzoic acid system with a methyl salicylate or benzoate (7a, 7b, 7c, 7d, 7e, 7f), phthalide (7g) (Table 3, Figure 3A, and Supporting Information Figure S5C), or salicylamide (8p, 8q) (Table 4 and Supporting Information Figure S5C) gave analogues that showed variable cellular activities. Notably, 7e, 7f, and 7g represent the best in the group to possess high cellular activities, with an EC50 of 0.9–1.6 μM (Figure 3A and Table 6). Comparatively, 7e, 7f, and 7g showed relatively weaker activities against the normal human breast epithelial MCF-10A, with an EC50 of 3.8–4.6 μM, and against the breast cancer MCF-7 cells that do not harbor constitutively active STAT3, with an EC50 of 4.6–8.9 μM (Figure 3B and Table 6).
Additional specificity studies were conducted for the inhibitors 7a, 7b, 7c, 8p, and 8q. The results showed that these compounds are relatively weaker in inhibiting the number of viable cells that do not harbor aberrantly active STAT3, with EC50 4.6 and 7.2 μM (8p), and 3.8 and 4.6 μM (8q) against MCF-7 or MCF-10A cells, respectively, and 4.3 μM (7a), 6.0 μM (7b) and 3.6 μM (7c) against MCF-10A cells that do not harbor aberrantly active STAT3 (Supporting Information Figure S5F and Table 4). Notably, treatment with 2 μM of 8q showed no effect on the growth of MCF-7 cells as measured by trypan blue exclusion/phase-contrast microscopy (Supporting Information Figure S5E). These inhibitors therefore show varied relative preferences against STAT3-dependent tumor cells over cells that do not depend on STAT3 activity. Notably, the carboxylic acid bioisosteres provided the first evidence that strong cellular activity could be achieved.
Isosteric Replacement of Salicylic Acid Moiety with Benzo-Fused N-Heterocyclic Systems Retained the In Vitro Activity of Analogues and Greatly Improved Their Cellular Activity
A concurrent approach was to incorporate benzo-fused N-heterocyclic systems, which were also successful in providing analogues with good cellular potency (Figure 1E and Table 5). The benzo-fused N-heterocyclic replacement for the salicylic acids allowed for further fine tuning of the cLogP and especially PSA to achieve the desirable physicochemical properties while maintaining potency. Initial results from the isoquinolinone analogues were encouraging (Tables 5 and 6); for example, compound 9b, with a PSA value of 99 Å2 showed cellular activity against MDA-MB-231 cells, with an EC50 of 1.2 μM, while retaining STAT3-inhibitory potency (IC50 0.79 μM) (Figure 1E and Table 5, entry 2). The corresponding quinazolinone analogues were moderately active against MDA-MB-231 and MDA-MB-468 cells (Table 5). This was especially the case for the N-methyl variant 9g that was moderately active against MDA-MB-231 and MDA-MB-468 cells (EC50 2.1 and 2.2 μM, respectively) while also maintaining good STAT3-inhibitory activity, with an IC50 of 1.15 μM (Figure 1E and Table 5, entry 7). As for the analogues with benzo-fused 5-membered N-heterocycles, while the cellular activity of benzotriazole 9r against MDA-MB-231 and MDA-MB-468 was moderate (EC50 2.1 and 2.3, respectively), that of 9q was rather low (EC50 of 6.6 and 5.1 μM, respectively), despite both having relatively good PSA values (107 and 119 Å2, respectively; Table 5), a reflection perhaps of the more basic pyridine over the pyrazine systems. The STAT3-inhibitory potencies for both 9q and 9r, however, were high (IC50 0.61 and 0.63 μM, respectively; Figure 1E and Table 5, entries 17 and 18). On the other hand, results for the isoindolinones 9i and 9j showed moderate cellular activities (EC50 2.0 μM against MDA-MB-231 and PSA 99 Å2 for both compounds), while 9i exhibited better potency than 9j against STAT3 DNA-binding activity (IC50 0.64 and 1.09 μM, respectively; Table 5, entries 9 and 10; Supporting Information Table S1). On the other hand, phthalimide 9k presented good inhibitory activities in the cell-free STAT3 DNA-binding/EMSA (IC50 1.18 μM) (PSA 116 Å2; Table 5, entry 11) and in the cell viability assays (EC50 1.7 and 1.9 μM against MDA-MB-231 and MDA-MB-468 cells, respectively) (Figure 3A). Moreover, trypan blue exclusion/phase-contrast microscopy showed that 2 μM 9k treatment of MDA-MB-231 cells strongly inhibited cell growth (Figure 3D). By contrast, the test of 9k on MCF-10A and MCF-7 cells that do not harbor constitutively active STAT3 showed weaker effects on both cell viability, with EC50 of 8.1 and 7.0 μM, respectively (Figure 3B, Table 5, entry 11, and Table 6). Further, trypan blue exclusion/phase-contrast microscopy showed that treatment with 2 μM 9k of MCF-7 cells had no effect on cell growth (Figure 3D). Furthermore, 72 h treatment of MCF-7 and MCF-10A cells with 9a or 9d showed only moderate effects on viable cell numbers (Table 5 and Supporting Information Figure S5F, Table S1). Other systems such as indazole (9l, 9m, 9n, and 9o) or benzimidazole (9p) did not look as promising (Table 5). Comparatively, our inhibitors show better selectivity against tumor cells harboring aberrantly active STAT3 than the purported STAT3 inhibitors, napabucasin (BBI-608)34 or C188-9.35 Treatments with increasing inhibitor concentrations showed weak effects for C188-9 (EC50 25.7 μM) against MDA-MB-231 cells but 2-fold stronger activity (EC50 13.75 μM) against MCF-7 cells that do not harbor constitutively active STAT3, while napabucasin showed strong effects against both MDA-MB-231 (EC50 1.8 μM) and MCF-7 cells (EC50 1.49 μM) (Supporting Information Figure S5G,H) suggesting the lack of specificity for either inhibitor.
Table 5. SAR of Benzo-Fused N-Heterocyclic Analogues.


Bands corresponding to STAT3:DNA complexes in gel were quantified using ImageJ and represented as a percent of control and plotted against the concentration of compounds, from which IC50 values were determined. nd: not determined.
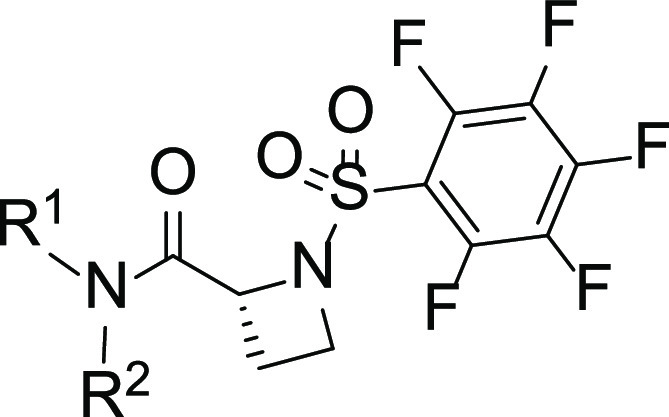
Isothermal Titration Calorimetry (ITC) Studies of Inhibitor Binding to STAT3
Given the enhanced potency of the azetidine-based inhibitors against STAT3, we were interested to determine their level of binding to the target in vitro. We performed isothermal titration calorimetry (ITC) studies, as previously described.41 The binding isotherm from the integrated thermogram fit using the one-site model in the PEAQ-ITC software generated from the titration of the representative inhibitors, 7g (red) and 9k (blue), into STAT3 shows KD of 880 and 960 nM, respectively (Figure 4A). The signature plots showing the thermodynamics parameters for each titration reveal ΔH = −22.7 kJ/mol, ΔG = −34.6 kJ/mol, and −TΔS = −11.9 kJ/mol for 7g and ΔH = −20.8 kJ/mol, ΔG = −34.4 kJ/mol, and −TΔS = −13.6 kJ/mol for 9k (Figure 4B). Results together show that the azetidine inhibitors directly bind with high affinity to STAT3.
Figure 4.
Isothermal titration calorimetry (ITC) measurements of 7g, and 9k during incubation with STAT3. (A) Binding isotherm from the integrated thermogram fit using the one-site model in the PEAQ-ITC software generated from the titration of the inhibitors, 7g (red) and 9k (blue), into STAT3. The KD was 880 and 960 nM, respectively, and (B) the signature plots showing the thermodynamics parameters for each titration reveal ΔH = −22.7 kJ/mol, ΔG = −34.6 kJ/mol, and −TΔS = −11.9 kJ/mol for 7g and ΔH = −20.8 kJ/mol, ΔG = −34.4 kJ/mol, and −TΔS = −13.6 kJ/mol for 9k. Data are representative of three independent experiments.
Comparison of the Inhibition of STAT3 DNA-Binding Activity over That of STAT1 and STAT5 In Vitro
To further establish the specificity of select new analogues with potent activity against STAT3, we investigated their effects on other STAT family members, including STAT1 and STAT5 DNA-binding activities in vitro, as previously reported.13,23,24,26 Nuclear extracts were prepared from epidermal growth factor (EGF)-stimulated fibroblasts overexpressing the EGF receptor (NIH3T3/EGFR) containing active STAT1, STAT3, and STAT5. Extracts of equal total protein were incubated with increasing or a single concentration of the azetidine compounds prior to incubation with the radiolabeled hSIE probe that binds STAT1 and STAT3 or the mammary gland factor element (MGFe) that binds STAT1 and STAT5 and performing the EMSA analysis. Results show that select azetidine analogues had minimum effects on both STAT5 and STAT1 DNA-binding activities (Figure 5 and Supporting Information Figure S6). Excluding the esters of the carboxylic acids that in principle are prodrugs, we selected and evaluated the selectivity of the representative analogues from the other subgroups. All compounds tested showed preferentially potent disruption of the DNA-binding activity of STAT3:STAT3 homodimers ahead of STAT1:STAT3 heterodimers, which were inhibited ahead of STAT1:STAT1 homodimers or STAT5:STAT5 homodimers, with potencies (IC50) of 0.52, 2.61, 12.0, and 9.3 μM (5a), 0.38, 1.46, >20, and >20 μM (5o), 1.08, 4.92, >20, and 17.5 μM (6f), 0.77, 3.14, >20, and >20 μM (8q), and 1.18, 4.71, >20, and >20 μM (9k) (Figure 5). Other compounds, including 5e, 5g, 5i, 5m, 5n, 7a, 7b, 8d, 8f, 8q, and 9k, also showed minimal effect on the DNA-binding activity of STAT5:STAT5 or STAT1:STAT1 homodimers (Supporting Information Figure S6). Altogether, these data show that active azetidine inhibitors tested have preferential effects on the DNA-binding activity of STAT3 over that of STAT1 or STAT5.
Figure 5.

Comparing the effect of new analogues on STAT1, STAT3, or STAT5 DNA-binding activity in vitro. Nuclear extracts of equal total protein prepared from epidermal growth factor-stimulated NIH3T3/EGFR fibroblasts containing activated STAT1, STAT3, and STAT5 were preincubated with increasing concentrations of the designated compounds for 30 min at room temperature prior to incubating with the radiolabeled hSIE probe that binds STAT1 and STAT3 (upper panel) or the MGFe probe that binds STAT5 (bottom panel) and performing EMSA analysis. Positions of STAT:DNA complexes in gel are labeled; control lanes (0) represent nuclear extracts preincubated with 10% DMSO. Data are representative of two to three independent determinations.
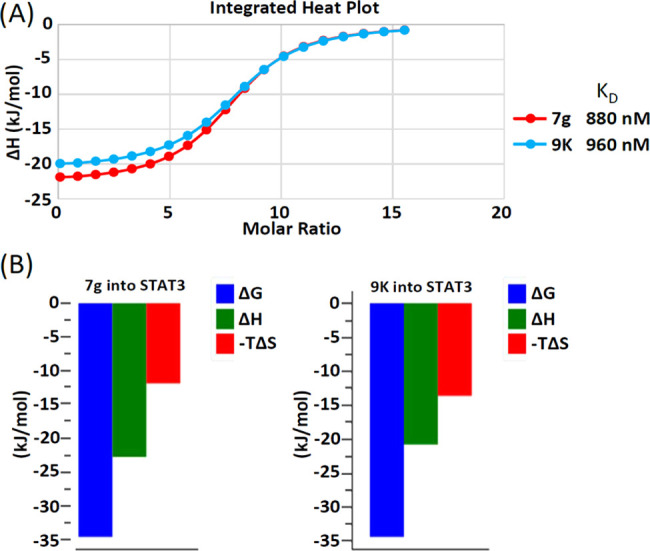
Analogues Inhibited Constitutive STAT3 Phosphorylation and DNA-Binding Activity in Human Breast Cancer Cells
The human breast cancer MDA-MB-231 and MDA-MB-468 cells harbor aberrantly active STAT327,42 and are sensitive to the azetidine inhibitors (Figure 3A). We were interested to determine the effect of select active azetidine analogues on the constitutive STAT3 signaling in the breast cancer cells. Cells were treated with inhibitors, including 7g, 8q, and 9k, at 1–5 μM for 0–24 h. Nuclear extracts were prepared and subjected to DNA-binding activity/EMSA analysis, while whole-cell lysates were prepared for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)/Western blotting analysis to determine effects on intracellular STAT3 activity.23,24,26 Results showed that STAT3 DNA-binding activity was inhibited to variable degrees in MDA-MB-468 cells by 7g, 8q, and 9k and in a time-dependent manner and as early as 5 min for 7g and 9k and 6 h for 8q (Figure 6A and Supporting Information Figure S7A). Similarly, treatment with increasing concentrations of 7g, 8q, and 9k inhibited pY705STAT3 in dose- and time-dependent manner in the breast cancer cells (Figure 6B and Supporting Information Figure S7B). For the 9k treatment condition, the pYSTAT3 bounced back by 24 h (Figure 6B-iii). Comparatively, treatment of MDA-MB-468 and MDA-MB-231 cells for 2–3 h with the purported STAT3 inhibitor, BBI-608,34 at 0.5–5 μM showed weak to moderate effects on STAT3 DNA-binding activity or pY705STAT3 (Supporting Information Figure S7C,D). For nonspecific effects, immunoblotting analysis showed that treatment of MDA-MB-231 cells with 1 or 3 μM 7g had no measurable effect on the tyrosine kinases, EGFR, JAK2, and Src, or on AKT and ERK1/2 (Supporting Information Figure S7E). These results show that select cell-permeable azetidine inhibitors are active at 1–3 μM against constitutive STAT3 induction in human breast cancer cells.
Figure 6.
Effects of compounds on constitutive STAT3 activation in tumor cells. (A) Nuclear extracts of equal total protein prepared from the human breast cancer, MDA-MB-468 cells untreated (DMSO, 0) or treated with 5 μM of the indicated analogues for 1–3 h were subjected to STAT3 DNA-binding assay using the hSIE probe that binds STAT3, and (B) immunoblotting analysis of whole-cell lysates of equal total protein prepared from (i) and (ii) MDA-MB-231 cells untreated (DMSO, 0) or treated with 1 or 3 μM of 7g for 3–24 h or (iii) MDA-MB-468 cells untreated (DMSO, 0) or treated with 3 μM of 9k for 3–24 h and probing for pY705STAT3, STAT3, or tubulin. Positions of STAT3:DNA complex or proteins in gel are shown; control (0 or Con) lane represents whole-cell lysates or nuclear extracts prepared from 0.05% DMSO-treated cells. Data are representative of two to three independent determinations.
Azetidines Inhibited the Colony Survival of Human Breast Cancer Cells
Human MDA-MB-231 breast cancer cells in a single-cell culture were treated once with analogues 7e, 7f, 7g, 8q, or 9k at 0.5–1 μM and allowed to culture until colonies were visible, which were stained and imaged. The results indicate that at the concentration of 0.5 μM, 7g shows significant inhibition of colony formation; however, 7e, 7f, and 9k only show minimal to moderate inhibition of colony formation (Figure 7). Moreover, at the concentration of 1 μM, the results show a complete inhibition of colony formation for 7g, a near-complete inhibition for 7e, and a significant inhibition for 7f and 9k (Figure 7), whereas, 8q does not show any inhibitory effect up to 1 μM (Supporting Information Figure S8). These results indicate that selected azetidine-based STAT3 inhibitors attenuate the survival of cancer cells that harbor constitutively active STAT3 at concentrations that inhibit STAT3 activity.
Figure 7.

Compounds 7e, 7f, 7g, and 9k inhibit the colony survival of human breast cancer cells in vitro. Human breast cancer MDA-MB-231 cells were seeded as single-cell culture and treated once with 0–1 μM of the indicated compounds and allowed to culture until large colonies were visible, which were stained with crystal violet and imaged. Data are representative of three independent determinations.
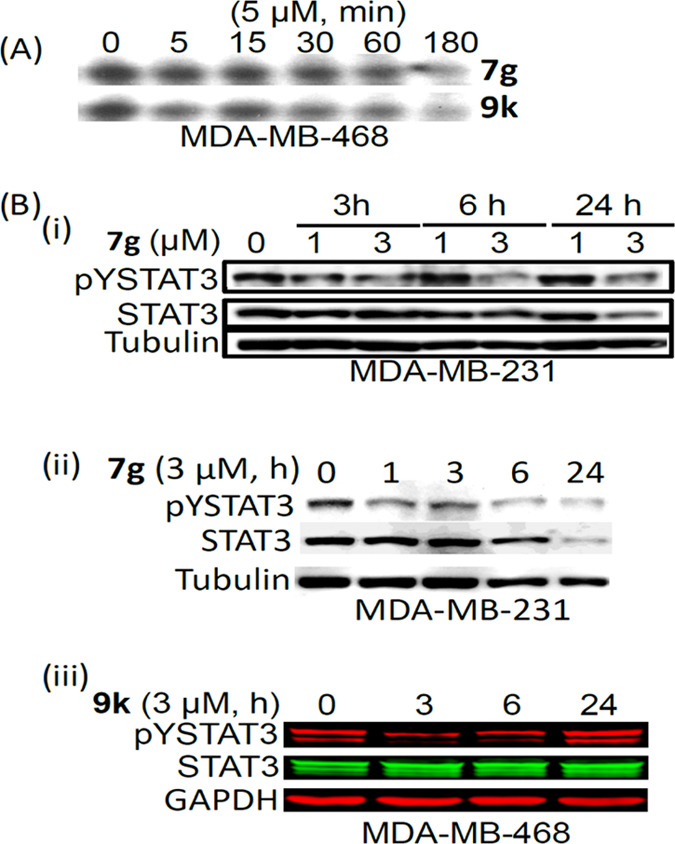
Inhibition of STAT3-Regulated Genes and Induction of Apoptosis
Consistent with the dysregulation of genes that promote tumor cell growth, survival, and malignant phenotype,27 treatment of breast cancer cells with 1 or 3 μM 7g for 3–24 h inhibited the expression of STAT3 target genes, c-Myc, vascular endothelial growth factor (VEGF), Bcl-2, and survivin (Figure 8) and induced poly (ADP-ribose) polymerase (PARP) cleavage in parallel with the inhibition of pY705STAT3 (Figure 9).
Figure 8.
Inhibition of STAT3 target gene expression in breast cancer cells. Immunoblotting analysis of whole-cell lysates of equal total protein prepared from MDA-MB-231 cells untreated (DMSO, C) or treated with 1 or 3 μM of 7g for 3–24 h and probing for c-Myc Bcl-2, VEGF, survivin, or tubulin. Positions of proteins in gel are shown; control (C) lane represents whole-cell lysates prepared from 0.05% DMSO-treated cells. Data are representative of two to three independent determinations.
Figure 9.

Induction of apoptosis of human breast cancer cells. Human breast cancer, MDA-MB-231 and MDA-MB-468 cells in culture were treated with 1 or 3 μM 7g for 0–24 h, whole-cell lysates were prepared, and samples of equal total protein were subjected to SDS/PAGE–Western blotting analysis probing for pYSTAT3, STAT3, full-length PARP, cleaved PARP, and tubulin. Positions of proteins in gel are shown; control (0) lane represents whole-cell lysates prepared from 0.05% DMSO-treated cells. Data are representative of two independent determinations.
Initial Evaluation of Solubility and Metabolic Characteristics of the Azetidine Compounds
The in vivo activity of a drug is influenced by parameters, such as solubility, permeability, and metabolism,43−45 which are dependent on its physicochemical characteristic. With medicinal chemistry effort focused on the optimization of the physicochemical features, we conducted an industry-standard evaluation of solubility and metabolism assays46 through the contract research organization (CRO), Eurofins-CEREP. In general, compounds show good aqueous solubility, as found by simulated gastric fluid (SGF) and simulated intestinal fluid (SIF), above the standard cutoff of 60 μg/mL.47,48 For example, promising phthalide 7g has a solubility at SGF of 116 μg/mL and at SIF of 200 μg/mL (Table S2). Preliminary results from a human HLM MetID study of the phthalide 7g showed both the parent compound and its hydroxy-acid metabolite (Supporting Information Figure S9), which is the result of hydrolytic lactone opening, and a presumably more potent version in terms of directly inhibiting STAT3 DNA-binding activity, suggesting that 7g and its corresponding hydroxy-acid version exist in equilibrium. Additional metabolites of 7g were also identified (Supporting Information Figure S9).
Conclusions
Despite the strong validation of STAT3 as a target, as a transcription factor, it has presented significant challenges for drug discovery/development research due to the flat protein surface that lacks deep pockets to target and design high-affinity binders. Much of the earlier efforts to develop inhibitors of STAT3 focused on targeting the SH2 domain to disrupt the interactions with pTyr-containing binding sites and the STAT3:STAT3 dimerization event.8,9 These efforts have generated a number of small-molecule inhibitors,11−26,35 though the clinical development of these inhibitors has been hampered due to their low potency, unclear mechanisms of inhibition of STAT3 signaling, and pharmacokinetic limitations among others.27 New methods are required to design unique potent small-molecule binders that can potentially overcome the limitations. The nanomolar potent PROTAC-STAT3 degraders, including SD-36, are a new class of inhibitors, and the proof-of-concept studies in which SD-36 inhibited the growth of xenograft models of leukemia and lymphomas show that they hold promise.28,29 In the current study, we have taken a different strategy to address the challenge of potency by creating the new azetidine class of inhibitors. The azetidine series marks a significant advancement in the study of small-molecule STAT3 inhibitors. With the change from R-proline-amides to R-azetidine-2-carboxamides, the first analogues have been realized with sub-micromolar potency in the STAT3 DNA-binding activity/EMSA, i.e., salicylate 5a (EMSA IC50 0.55 μM) among many others. Optimization of the salicylate series provided the 5-cyclohexyl-2-pyridinylmethyl analogues that led to the potent salicylate analogue, 5o (EMSA IC50 0.38 μM), which represented a log-order improvement in potency over earlier published analogues.24,26 Further elaboration of the salicylic acid portion of the molecule provided benzamides and benzo-fused N-heterocycles, which maintained the sub-micromolar potency in the in vitro STAT3 DNA-binding/EMSA assay and furnished the most potent analogue in the whole azetidine series, i.e., benzohydroxamic acid 8i (EMSA IC50 0.34 μM). Having overcome the potency barrier, we focused the medicinal chemistry efforts on optimizing the physicochemical properties while maintaining potency. We were able to trade some potency for improved cell permeability, and several compounds showed improved activity in cell-based assays over previous analogues. In particular, ester versions of the carboxylic acid, including 7e and 7f, or the lactone, 7g, or compounds that contained carboxylic acid bioisosteres, such as salicylamides (8p and 8q) and some of the benzo-fused N-heterocycle analogues (9b, 9f, and 9k), showed good cellular activity. Notably, 7e, 7f, 7g, and 9k have the best cellular activities against breast tumor cells that harbor aberrantly active STAT3, including inhibiting cell growth, suppressing STAT3 target gene expression, and inducing apoptosis. The current azetidine series of compounds therefore are significantly improved over their leads, BP-1-102, SH5-07, and SH4-54, which had EMSA IC50 of 6.8, 3.9, and 4.7 μM, respectively, and cellular activities at 10–20, 3.8, and 4.5 μM, respectively. They also compare more favorably to napabucasin (BBI-608) and C188-9. The azetidine compounds therefore represent promising new chemical entities in the search for suitable small-molecule, direct STAT3 inhibitors for further clinical development into new anticancer agents either as standalone or in combination. Their utility in combination therapy with chemotherapy, including docetaxel or cisplatin, is demonstrated herein.
Experimental Section
Cell Lines and Reagents
The human breast cancer MDA-MB-468 and MCF-7 cell lines and the normal breast epithelial line MCF-10A have been reported previously,24,26,31,32 and MDA-MB-231 was obtained from the National Cancer Institute on December 2, 2015. MCF-10A cells were grown in Dulbecco’s modified Eagle’s medium, DMEM/F12 with 5% horse serum plus EGF (20 ng/mL), insulin (10 μg/mL), hydrocortisone (0.5 mg/mL), and 100 ng/mL cholera toxin. All other cells were grown in DMEM plus 10% heat-inactivated fetal bovine serum (FBS). Cell line authentication was done in December 2015 by American Type Culture Collection (ATCC) for MDA-MB-468, which was found to be authentic. Mycoplasma test was conducted on MDA-MB-231 and MCF-10A IDEXX BioAnalytics (Columbia, MO) in Dec 2019. Both lines are negative, while the test has not been done on MCF-7 and MDA-MB-468. Antibodies against STAT3, pY705STAT3, pY1173EGFR, EGFR, pY1007/1008JAK2, JAK2, pY416Src, Src, pS473AKT, AKT, pT202/Y204ERK1/2 (p44/42), ERK1/2, full-length poly (ADP-ribose) polymerase (PARP), cleaved PARP, tubulin, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Cisplatin and docetaxel were purchased from Sigma-Aldrich (St. Louis, MO).
Nuclear Extract Preparation, Gel Shift Assays, and Densitometric Analysis
Nuclear extract preparations and DNA-binding activity/electrophoretic mobility shift assay (EMSA) were carried out as previously described.13,14,23,24,26 The 33P-labeled oligonucleotide probes used were hSIE (high-affinity sis-inducible element from the c-fos gene, m67 variant, 5′-AGCTTCATTTCCCGTAAATCCCTA) that binds STAT1 and STAT3 and MGFe (mammary gland factor element from the bovine β-casein gene promoter, 5′-AGATTTCTAGGAATTCAA) for STAT1 and STAT5 binding. Except where indicated, nuclear extracts of equal total protein were preincubated with compound for 30 min at room temperature prior to incubation with the radiolabeled probe for 30 min at 30 °C before subjecting to EMSA analysis. Where appropriate, bands corresponding to STAT3:DNA complexes were scanned and quantified for each concentration of compound using ImageJ and plotted as a percent of control (DMSO) against the concentration of compound, from which the IC50 values were derived.
Immunoblotting Analysis
Whole-cell lysate preparation and immunoblotting analysis were performed as previously reported.23,24 Briefly, cultured cells treated or not were harvested and whole-cell lysates were prepared in RIPA buffer. Samples of equal total protein were subjected to SDS-PAGE and immunoblotting analysis. Primary antibodies used were anti-STAT3, pY705STAT3, PARP, c-Myc, Bcl-2, VEGF, survivin, tubulin, and GAPDH. All antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA), except GAPDH from Santa Cruz Biotechnology (Dallas, Texas).
Cell Proliferation and Viability Assays
Studies were performed as previously reported.23,24,26 Briefly, cultured cells in 6-well or 96-well plates were treated with or without compounds for the indicated concentrations, and cells were harvested every 24 h up to 96 h for viable cell count by trypan blue exclusion phase-contrast microscopy, or after 72 h, cells were subjected to CyQuant cell proliferation assay following the manufacturer’s instructions (Invitrogen/ThermoFisher Scientific). In the case of the combination treatment, cells in culture were first treated with azetidine inhibitor, 7g, for 6 h followed by treatment with docetaxel or cisplatin and then harvested after a total of 72 h treatment for CyQuant assay. Cell viability was normalized to the percentage of the control groups.
Clonogenic Survival Assays
Colony survival assay was performed as previously reported.24,26 Briefly, cells were seeded as single-cell cultures in 6-well plates (250 cells per well), treated once the next day with compounds at the indicated concentrations and allowed to culture until large colonies were visible. Colonies were stained with crystal violet for 4 h and photographed.
Isothermal Titration Calorimetry (ITC)
The ITC experiment was carried out as previously described41 with some modification using Malvern Panalytical MicroCal PEAQ-ITC (United Kingdom). Studies were done at 25 °C. Briefly, STAT3 inhibitors, previously suspended in 100% DMSO, were diluted in 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 150 mM KCl buffer, so the final DMSO was 5%. To avoid buffer mismatch, STAT3 in HEPES buffer was diluted in HEPES buffer with 5% DMSO final concentration. Three hundred microliter volumes (300 μL) of 3.0 μM STAT3 were placed in the cell and titrated with 250 μM inhibitors. Titrations took place by injecting 2 μL inhibitor in a 2.5 min injection for the titration peak to return to the baseline. The KD was calculated using the MicroCal PEAQ-ITC analysis software, as well as Prism GraphPad software, using the one-site model. Control experiments were carried out by titration of the inhibitors into buffer, buffer into STAT3, and buffer into buffer. The three controls were used as a composite for the ITC experiment to subtract the heat of dilution and background noise from the measurements.
General Methods for Chemistry
All reagents and solvents were purchased from commercial sources and used without further purification. All moisture-sensitive reactions were performed under a static atmosphere of nitrogen or argon in oven-dried glassware. Tetrahydrofuran (THF), dichloromethane (DCM), diethyl ether (Et2O), toluene, and dimethylformamide (DMF) used in the reactions were dried by being passed through an SPS system. Other anhydrous solvents were purchased from commercial sources. Thin-layer chromatography (TLC) was performed on glass plates, 250–1000 μm. Flash column chromatography was performed on silica gel, 200–400 mesh. 1H NMR spectra were obtained as CDCl3, CD3OD, or (CD3)2SO solutions using an Agilent 300MHz NMR spectrometer with an Agilent DD2 console, and chemical shifts were expressed in ä (ppm) using residual solvent (CDCl3, 7.26 ppm; CD3OD, 3.31 ppm; and (CD3)2SO, 2.50 ppm) as the reference standard. When peak multiplicities are reported, the following abbreviations are used: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br-s (broadened singlet), dd (doublet of doublets), and dt (doublet of triplets). Coupling constants, when reported, are reported in hertz (Hz). All compounds were analyzed by LC/MS (liquid chromatography/mass spectrometry) using an Agilent Triple Quad 640 LC/MS. Ionization was generally achieved via electron spray (ESI) unless otherwise indicated. The LC fraction detection consisted of a variable wavelength detector, and all tested compounds had purity greater than 95%. High-resolution mass spectral (HRMS) data was obtained for all tested compounds using either an Agilent 6200 LC/MSD TOF or an Agilent 6545 Q-TOF LC/MS, and reported exact masses were calculated based on an algorithm using MS (ESI) m/z for [M + H]+ and [M + Na]+ adducts and were within 5 ppm of the expected target mass. Chiral molecules were analyzed by chiral HPLC using Chiralpak AD-H or OD-H columns (4.6 mm × 250 mm, UV detection at 254 or 261nm); eluents used were hexane and i-PrOH.
Acknowledgments
We thank all colleagues and members of our laboratory for the stimulating discussions and Dr. Joel Kawakami and Mabel Bernaldez of Chaminade University of Honolulu for their contributions to the design of the Cover Art. This work was supported by NIH/NCI R01 CA208851 (J.T.), LEIDOS Biomedical Research/NCI Contract 19X122Q (J.T.), and Cedars-Sinai Start-up funds (JT).
Glossary
Abbreviations
- STAT
signal transducer and activator of transcription
- EMSA
electrophoretic mobility shift assay
- PARP
poly (ADP-ribose) polymerase
- VEGF
vascular endothelial growth factor
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01705.
Supplementary results and discussion; chemistry; early leads (Figure S1); dose–response curves for the cell-free EMSA analysis for the most active compounds (Figure S2); EMSA IC50 and cell viability EC50 values for select esters and heterocyclics (Table S1) (PDF)
Molecular formula strings (CSV)
Qualitative Analysis Report: Compound 5a (PDF)
Qualitative Analysis Report: Compound 5o (PDF)
Qualitative Analysis Report: Compound 7e (PDF)
Qualitative Analysis Report: Compound 7f (PDF)
Qualitative Analysis Report: Compound 7g (PDF)
Qualitative Analysis Report: Compound 8i (PDF)
Qualitative Analysis Report: Compound 9k (PDF)
Author Contributions
# C.B.-P. and P.Y. authors contributed equally to this work.
The authors declare the following competing financial interest(s): JT is a co-founder of Novella, LLC, which has licensed STAT3 IP. The remaining authors have no competing interests.
Supplementary Material
References
- Darnell J. E. Jr. Reflections on Stat3, Stat5, and Stat6 as fat STATs. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 6221–6224. 10.1073/pnas.93.13.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. E. Jr. The Jak-STAT pathway: summary of initial studies and recent advances. Recent Prog. Horm. Res. 1996, 51, 391–403. [PubMed] [Google Scholar]
- Darnell J. E. Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Bromberg J.; Darnell J. E. Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- Darnell J. E. Jr; Kerr I. M.; Stark G. R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Bromberg J. F.; Horvath C. M.; Besser D.; Lathem W. W.; Darnell J. E. Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. 10.1128/MCB.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell J. E. Validating Stat3 in cancer therapy. Nat. Med. 2005, 11, 595–596. 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets 2004, 8, 409–422. 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- Turkson J.; Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- Darnell J. E. Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Turkson J.; Ryan D.; Kim J. S.; Zhang Y.; Chen Z.; Haura E.; Laudano A.; Sebti S.; Hamilton A. D.; Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- Turkson J.; Kim J. S.; Zhang S.; Yuan J.; Huang M.; Glenn M.; Haura E.; Sebti S.; Hamilton A. D.; Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004, 3, 261–269. [PubMed] [Google Scholar]
- Siddiquee K.; Zhang S.; Guida W. C.; Blaskovich M. A.; Greedy B.; Lawrence H. R.; Yip M. L.; Jove R.; McLaughlin M. M.; Lawrence N. J.; Sebti S. M.; Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7391–7396. 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee K. A.; Gunning P. T.; Glenn M.; Katt W. P.; Zhang S.; Schrock C.; Sebti S. M.; Jove R.; Hamilton A. D.; Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem. Biol. 2007, 2, 787–798. 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- Song H.; Wang R.; Wang S.; Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4700–4705. 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray J. S. A new small-molecule Stat3 inhibitor. Chem. Biol. 2006, 13, 1123–1124. 10.1016/j.chembiol.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Ren Z.; Cabell L. A.; Schaefer T. S.; McMurray J. S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003, 13, 633–636. 10.1016/S0960-894X(02)01050-8. [DOI] [PubMed] [Google Scholar]
- McMurray J. S.; Mandal P. K.; Liao W. S.; Ren Z.; Chen X. Inhibition of Stat3 by cell-permeable peptidomimetic prodrugs targeted to its SH2 domain. Adv. Exp Med. Biol. 2009, 611, 545–546. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Liao W. S.; McMurray J. S. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009, 11, 3394–3397. 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Bai L.; Bernard D.; Nikolovska-Coleska Z.; Gomez C.; Zhang J.; Yi H.; Wang S. Structure-based design of conformationally constrained, cell-permeable Stat3 inhibitors. ACS Med. Chem. Lett. 2010, 1, 85–89. 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Nikolovska-Coleska Z.; Yang C.-Y.; Gomez C.; Gao W.; Krajewski K.; Jiang S.; Roller P.; Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of Stat3 via ‘click chemistry’. Bioorg. Med. Chem. Lett. 2007, 17, 3939–3942. 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- Leung K. H.; Liu L. J.; Lin S.; Lu L.; Zhong H. J.; Susanti D.; Rao W.; Wang M.; Che W.; Chan D. S.; Leung C. H.; Chan P. W.; Ma D. L. Discovery of a small-molecule inhibitor of Stat3 by ligand-based pharmacophore screening. Methods 2015, 71, 38–43. 10.1016/j.ymeth.2014.07.010. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yue P.; Fletcher S.; Zhao W.; Gunning P. T.; Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem. Pharmacol. 2010, 79, 1398–409. 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Yue P.; Page B. D.; Li T.; Zhao W.; Namanja A. T.; Paladino D.; Zhao J.; Chen Y.; Gunning P. T.; Turkson J. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9623–9628. 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Sun Y.; Pireddu R.; Yang H.; Urlam M. K.; Lawrence H. R.; Guida W. C.; Lawrence N. J.; Sebti S. M. A novel inhibitor of Stat3 homodimerization selectively suppresses Stat3 activity and malignant transformation. Cancer Res. 2013, 73, 1922–1933. 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P.; Lopez-Tapia F.; Paladino D.; Li Y.; Chen C.-H.; Hilliard T.; Chen Y.; Tius M.; Turkson J. Hydroxamic acid and benzoic acid-based Stat3 inhibitors suppress human glioma and breast cancer phenotypes in vitro and in vivo. Cancer Res. 2016, 76, 652–663. 10.1158/0008-5472.CAN-14-3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy G.; Hilliard T. S.; Turkson J. Therapeutic modulators of Stat signaling for human diseases. Nat. Rev. Drug Discovery 2013, 12, 611–629. 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Bai L.; Xu R.; Zhao Y.; Chen J.; McEachern D.; Chinnaswamy K.; Wen B.; Dai L.; Kumar P.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Meagher J. L.; Yi H.; Sun D.; Stuckey J. A.; Wang S. Structure-based discovery of SD-36 as a potent, selective, and efficacious protac degrader of Stat3 protein. J. Med. Chem. 2019, 62, 11280–11300. 10.1021/acs.jmedchem.9b01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L.; Zhou H.; Xu R.; Zhao Y.; Chinnaswamy K.; McEachern D.; Chen J.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Jiang H.; Wen B.; Kumar P.; Meagher J. L.; Sun D.; Stuckey J. A.; Wang S. A potent and selective small-molecule degrader of Stat3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511.e17. 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Tapia F.; Brotherton-Pleiss C.; Yue P.; Murakami H.; Costa Araujo A. C.; Reis dos Santos B.; Ichinotsubo E.; Rabkin A.; Shah R.; Lantz M.; Chen S.; Tius M. A.; Turkson J. Linker variation and structure-activity relationship analyses of car-boxylic acid-based small molecule Stat3 inhibitors. ACS Med. Chem. Lett. 2018, 9, 250–255. 10.1021/acsmedchemlett.7b00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia R.; Yu C. L.; Hudnall A.; Catlett R.; Nelson K. L.; Smithgall T.; Fujita D. J.; Ethier S. P.; Jove R. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Diff. 1997, 8, 1267–1276. [PubMed] [Google Scholar]
- Garcia R.; Bowman T. L.; Niu G.; Yu H.; Minton S.; Muro-Cacho C. A.; Cox C. E.; Falcone R.; Fairclough R.; Parson S.; Laudano A.; Gazit A.; Levitzki A.; Kraker A.; Jove R. Constitutive activation of Stat3 by the Src and Jak tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- Turkson J.; Bowman T.; Garcia R.; Caldenhoven E.; De Groot R. P.; Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. 10.1128/MCB.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi S.; Chen K.; Feng L.; Fu G.; Yang Q.; Deng M.; Zhao H.; Li Z.; Yu L.; Fang Z.; Xu B. Napabucasin (BBI608) eliminate AML cells in vitro and in vivo via inhibition of Stat3 pathway and induction of DNA damage. Eur. J. Pharmacol. 2019, 855, 252–261. 10.1016/j.ejphar.2019.05.020. [DOI] [PubMed] [Google Scholar]
- Lewis K. M.; Bharadwaj U.; Eckols T. K.; Kolosov M.; Kasembeli M. M.; Fridley C.; Siller R.; Tweardy D. J. Small-molecule targeting of signal transducer and activator of transcription (Stat) 3 to treat non-small cell lung cancer. Lung Cancer 2015, 90, 182–190. 10.1016/j.lungcan.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P.; Turkson J. Targeting STAT3 in cancer: how successful are we?. Expert Opin. Invest. Drugs 2009, 18, 45–56. 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder J.; Grootenhuis P. D.; Bayada D. M.; Delbressine L. P.; Ploemen J. P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. 10.1023/A:1015040217741. [DOI] [PubMed] [Google Scholar]
- Sharma P.; López-Tarruella S.; García-Saenz J. A.; Ward C.; Connor C. S.; Gómez H. L.; Prat A.; Moreno F.; Jerez-Gilarranz Y.; Barnadas A.; Picornell A. C.; Del Monte-Millán M.; Gonzalez-Rivera M.; Massarrah T.; Pelaez-Lorenzo B.; Palomero M. I.; González Del Val R.; Cortes J.; Fuentes Rivera H.; Bretel Morales D.; Márquez-Rodas I.; Perou C. M.; Wagner J. L.; Mammen J. M.; McGinness M. K.; Klemp J. R.; Amin A. L.; Fabian C. J.; Heldstab J.; Godwin A. K.; Jensen R. A.; Kimler B. F.; Khan Q. J.; Martin M. Efficacy of neoadjuvant carboplatin plus docetaxel in triple-negative breast cancer: combined analysis of two cohorts. Clin. Cancer Res. 2017, 23, 649–657. 10.1158/1078-0432.CCR-16-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X. C.; Zhang J.; Xu B. H.; Cai L.; Ragaz J.; Wang Z. H.; Wang B. Y.; Teng Y. E.; Tong Z. S.; Pan Y. Y.; Yin Y. M.; Wu C. P.; Jiang Z. F.; Wang X. J.; Lou G. Y.; Liu D. G.; Feng J. F.; Luo J. F.; Sun K.; Gu Y. J.; Wu J.; Shao Z. M. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–46. 10.1016/S1470-2045(15)70064-1. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Alonso-Valenteen F.; Pacheco S.; Srinivas D.; Rentsendorj A.; Chu D.; Lubow J.; Sims J.; Miao T.; Mikhael S.; Hwang J. Y.; Abrol R.; Medina Kauwe L. K. HER3-targeted protein chimera forms endosomolytic capsomeres and self-assembles into stealth nucleocapsids for systemic tumor homing of RNA interference in vivo. Nucleic Acids Res. 2019, 47, 11020–11043. 10.1093/nar/gkz900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; H L.; Herrmann A.; Buettner R.; Jove R. Revisiting Stat3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer. 2014, 14, 736–746. 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- Avdeef A. Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem. 2001, 1, 277–351. 10.2174/1568026013395100. [DOI] [PubMed] [Google Scholar]
- Martinez M. N.; Amidon G. L. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J. Clin. Pharmacol. 2002, 42, 620–43. 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E.. Drug-like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization, 2nd ed.; Elsevier: Cambridge, MA, 2016. [Google Scholar]
- Chaturvedi P. R.; Decker C. J.; Odinecs A. Prediction of pharmacokinetic properties using experimental approaches during early drug discovery. Curr. Opin. Chem. Biol. 2001, 5, 452–463. 10.1016/S1367-5931(00)00228-3. [DOI] [PubMed] [Google Scholar]
- Guha R.; Dexheimer T. S.; Kestranek A. N.; Jadhav A.; Chervenak A. M.; Ford M. G.; Simeonov A.; Roth G. P.; Thomas C. J. Exploratory analysis of kinetic solubility measurements of a small molecule library. Bioorg. Med. Chem. 2011, 19, 4127–4134. 10.1016/j.bmc.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.; Carter G. T. In vitro solubility assays in drug discovery. Curr. Drug Metab. 2008, 9, 879–885. 10.2174/138920008786485100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.