

SUMOylation of annexin-A1 promotes an anti-inflammatory microglial phenotype and protects against cerebral ischemic injury.
Abstract
Annexin-A1 (ANXA1) has recently been proposed to play a role in microglial activation after brain ischemia, but the underlying mechanism remains poorly understood. Here, we demonstrated that ANXA1 is modified by SUMOylation, and SUMOylated ANXA1 could promote the beneficial phenotype polarization of microglia. Mechanistically, SUMOylated ANXA1 suppressed nuclear factor κB activation and the production of proinflammatory mediators. Further study revealed that SUMOylated ANXA1 targeted the IκB kinase (IKK) complex and selectively enhanced IKKα degradation. Simultaneously, we detected that SUMOylated ANXA1 facilitated the interaction between IKKα and NBR1 to promote IKKα degradation through selective autophagy. Further work revealed that the overexpression of SUMOylated ANXA1 in microglia/macrophages resulted in marked improvement in neurological function in a mouse model of cerebral ischemia. Collectively, our study demonstrates a previously unidentified mechanism whereby SUMOylated ANXA1 regulates microglial polarization and strongly indicates that up-regulation of ANXA1 SUMOylation in microglia may provide therapeutic benefits for cerebral ischemia.
INTRODUCTION
Cerebral stroke remains one of the leading causes of death and disability worldwide, with nearly 80 to 85% of cases characterized as ischemia (1). To date, the limited effective treatment for ischemic stroke is thrombolysis by intravenous administration of tissue plasminogen activator or thrombectomy within a narrow time window after stroke onset. There are no safe and available therapies for patients who have missed the optimal therapeutic time window for stroke, mostly resulting in functional disability in surviving patients; therefore, new therapeutic strategies are urgently needed (2). Recently, the postischemic inflammatory response has been found to play a crucial role in the pathophysiological progression of cerebral ischemia (3). Therefore, revealing the underlying mechanism associated with this inflammatory response would provide a novel therapeutic strategy for cerebral ischemia.
Accumulating evidence has shown that microglia, the prime resident innate immune effector cells in the central nervous system (CNS), play pivotal roles in regulating immune and inflammatory responses after various types of cerebral damage (4). Microglial activation can be classified into two major phenotypes with completely different functions, defined as “classical activation” (also termed the proinflammatory phenotype) and “alternative activation” (the anti-inflammatory phenotype) (5). There is growing evidence suggesting that active microglial cells play a double-edged role depending on shifts in the equilibrium of the proinflammatory and anti-inflammatory phenotypes during the course of ischemic brain injury (6). Classical microglial polarization typically correlates with the expression and release of multiple proinflammatory mediators, such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor–α (TNF-α), which propel the pathological process of brain ischemia. In contrast, alternative activated microglia execute an anti-inflammatory effect, promoting neuron survival and restricting brain injury after cerebral ischemic. The prime anti-inflammatory mediators, including IL-4, IL-10, and transforming growth factor–β (TGF-β), initiate the alleviation of proinflammatory responses and increase the expression of genes that are associated with tissue recovery and repair (7). Thus, the transformation of microglia from the proinflammatory phenotype to the anti-inflammatory and tissue-reparative phenotype may be a promising and effective therapeutic strategy for cerebral ischemia.
Annexin-A1 (ANXA1), a 37-kDa member of the annexin superfamily of proteins, has been found to participate in the regulation of diverse cellular functions in a variety of cell types (8). There is also overwhelming evidence for the role of ANXA1 in several anti-inflammatory processes, such as the regulation of neutrophil migration and macrophage phagocytosis (9, 10). In the CNS, ANXA1 is abundant in microglial cells. The results of an extensive study indicated that the biological functions of ANXA1 are tightly regulated by posttranslational modification (PTM), such as phosphorylation (11). For example, the phosphorylation of ANXA1 at Ser5 by TRPM7 maintains its nuclear localization and induces neuronal cell death after cerebral ischemia, while the phosphorylation of ANXA1 on Ser27 is crucial for its migration to the cell surface (12, 13). Beyond the phosphorylation modifications, Hirata et al. (14) reported that ANXA1 could also be modified by the small ubiquitin-related modifier (SUMO) protein, a processing termed SUMOylation, which is important for its helicase activity under DNA damage. However, the key SUMO modification proteins and enzymes that mediate the SUMOylation of ANXA1 and the SUMOylation sites remain poorly investigated.
SUMO (also called Sentrin) is a polypeptide of approximately 12 kDa that can be conjugated to target proteins on lysine residues by reversible covalence. Three major SUMO paralogs (SUMO1, SUMO2, and SUMO3) have been identified in mammals. Among them, SUMO1 is quite divergent, with only 47% homology with SUMO2 and SUMO3, which share 97% homology to each other and cannot be distinguished by antibodies (15). SUMOylation regulation plays a critical role in signal transduction involved in vital molecular and biological processes. This modification involves a cascade of enzymatic reactions, in a similar manner to ubiquitination. These enzymes include an E1 SUMO-activating enzyme, the E2-conjugating enzyme Ubc9, and an E3 ligase, such as the PIAS family proteins, RanBP2, and Pc2, which facilitate the transfer of SUMO from Ubc9 to specific target proteins (16). Previous data suggested that the conjugation of SUMO2 and SUMO3 is predominantly induced as an intracellular protective response to cellular stresses, such as hypoxia and inflammatory stimuli (17). There is also a series of reports showing a neuroprotective role of SUMOylation in brain lesions caused by ischemia-reperfusion injury (18). Recently, the SUMOylation of proteins that regulate the polarization of macrophages has also been reported. Wang et al. (19) reported that SUMOylation of KLF4 enhances IL-4–induced macrophage proinflammatory phenotype polarization. Thus, we examined whether SUMOylated ANXA1 regulates the polarization of microglia after brain ischemia.
In the present study, we found that ANXA1 could be modified by SUMOylation and that SUMOylation primarily occurs at three lysine residues, K113, K161, and K257. We also showed that Ubc9 and PIAS3 enhanced the SUMOylation level of ANXA1. Furthermore, SUMOylated ANXA1 promoted the anti-inflammatory phenotype polarization of microglia subjected to oxygen-glucose deprivation and reoxygenation (OGD/R) injury. A mechanistic study further revealed that SUMOylated ANXA1 acted as an adapter to facilitate an interaction between IκB kinase α (IKKα) and NBR1 to mediate IKKα degradation via selective autophagy, thus suppressing the activation of the nuclear factor κB (NF-κB) signaling pathway and the production of proinflammatory mediators in microglia challenged with OGD/R. We developed a method to manipulate gene overexpression in microglia/macrophages in the specific brain areas of animals and provided solid evidence that overexpression of SUMOylated ANXA1 in microglia/macrophages resulted in robust neuroprotection against cerebral ischemic-reperfusion injury. Together, these results indicate that up-regulation of the SUMOylation level of ANXA1 may provide a previously unidentified and potential therapeutic strategy for treating cerebral ischemia.
RESULTS
ANXA1 is modified by SUMOylation at lysine residues K113, K161, and K257
To confirm whether ANXA1 can be modified by SUMOylation, hemagglutinin (HA)–tagged ANXA1 and Flag-tagged Ubc9, along with His-tagged SUMO1, SUMO2, or SUMO3, were transfected into human embryonic kidney (HEK) 293T cells. The Ni2+-NTA (nickel–nitrilotriacetic acid) agarose precipitation results revealed that ANXA1 was modified strongly by SUMO2 and moderately by SUMO3 but weakly by SUMO1 (Fig. 1A and fig. S10A). Accordingly, we focused on the SUMO2 modification of ANXA1 in subsequent studies. Furthermore, an immunoprecipitation (IP) assay was used to verify whether SUMO2 could be covalently conjugated to ANXA1. The results suggested that a slower-migrating band of approximately 50 kDa (compared with the normal ANXA1 migrating band of 35 kDa) appeared only in the IP with anti-HA antibody but not in the IP with the immunoglobulin G control antibody (fig. S1A), which demonstrated that HA-ANXA1 was conjugated to His-SUMO2. Next, His-tagged SUMO2 together with Flag-tagged Ubc9 were transiently transduced into 293T cells, and the Ni2+-NTA pull-down results showed that endogenous ANXA1 could also be modified by SUMO2 (Fig. 1B). The SUMOylation of ANXA1 was further validated under normal and OGD/R conditions with endogenous levels of ANXA1 and SUMO2 in primary cultured microglia. The IP results indicated that endogenous ANXA1 was modified by SUMOylation, and this modification was notably weakened under OGD/R conditions (Fig. 1C). The SUMOylation of ANXA1 was also reduced in microglia/macrophages isolated from adult mice that underwent middle cerebral artery occlusion (MCAO) surgery to establish an ischemic stroke animal model (Fig. 1D). A time course study revealed that the SUMOylation levels of ANXA1 in primary cultured microglia were gradually decreased from 2 to 48 hours after OGD treatment (figs. S1B and S10L). Similarly, the results also showed that the SUMOylation of ANXA1 gradually decreased from 12 to 72 hours after the onset of ischemic stroke (figs. S1C and S10M).
Fig. 1. ANXA1 is modified by SUMO2 at K113, K161, and K257.

(A) Representative Ni2+-NTA agarose affinity pull-down assay results showing the interaction of exogenous ANXA1 with SUMO1, SUMO2, or SUMO3 in HEK293T cells. IB, immunoblotting. (B) Representative Ni2+-NTA agarose affinity pull-down assay results showing the interaction of endogenous ANXA1 with SUMO2 in HEK293T cells. (C) OGD/R down-regulates SUMOylation level of endogenous ANXA1 in microglial cells. Primary cultured microglia were challenged with OGD/R and then lysates were subjected to IP assay. IgG, immunoglobulin G. (D) SUMOylation level of endogenous ANXA1 was decreased in microglia/macrophages after ischemic stroke. Microglia/macrophages were isolated from ischemic stroke mice and then the cell lysates were subjected to IP assay. (E and F) Representative Ni2+-NTA agarose affinity pull-down assay results showing the SUMOylation of ANXA1 in HEK293T cells when Ubc9 is overexpressed (E) or knocked down by specific shRNA (F). (G) Representative Ni2+-NTA agarose affinity pull-down assay results showing the SUMOylation of ANXA1 in HEK293T cells treated with PIAS3 overexpression. (H to J) Determination of ANXA1 SUMOylation sites by mutagenesis. HEK293T cells were cotransduced with Flag-Ubc9, His-SUMO2, and either HA-tagged WT ANXA1 (HA-ANXA1-WT), HA-ANXA1 single-lysine mutant (H), HA-ANXA1 double-lysine mutant (I), or HA-ANXA1 triple-lysine mutant (J). After 24 hours, the cells were lysed and subjected to Ni2+-NTA pull-down assay. The precipitates and whole-cell lysates were detected by immunoblotting. Data represent at least three independent experiments. Mr, relative molecular mass.
As Ubc9 is the one and only E2-conjugating enzyme that is crucial for SUMOylation (20), we explored its role in ANXA1 SUMOylation. As shown in Fig. 1 (E and F), affinity pull-down assays with Ni2+-NTA agarose showed that the SUMO2 modification of ANXA1 was strongly enhanced by Ubc9 overexpression, and this modification was weakened when endogenous Ubc9 in 293T cells was knocked down by using a specific short hairpin RNA (shRNA) (fig. S1, D and E). Moreover, the IP results confirmed that HA-ANXA1 indeed interacted with Flag-Ubc9 in 293T cells (fig. S1F). Next, we transiently transfected HEK293T cells with HA-tagged ANXA1 and His-tagged SUMO2, together with a panel of green fluorescent protein (GFP)–tagged SUMO E3 ligases. As presented in Fig. 1G, the SUMOylation of ANXA1 was notably increased upon PIAS3 transfection. These results were further validated by IP assay and showed that HA-ANXA1 coimmunoprecipitated with GFP-PIAS3 (fig. S1G). These data suggest that PIAS3 functions as a SUMO E3 ligase for ANXA1. Collectively, these results conclusively confirm that ANXA1 can be modified by SUMOylation in cells.
We next attempted to ascertain the SUMO modification site(s) in ANXA1. Five lysine residues, K113, K161, K185, K257, and K312, which were predicted by the SUMOplot Analysis Program (fig. S2A), were individually mutated to arginine for SUMOylation identification. Affinity pull down with Ni2+-NTA agarose showed that the mutations K113R, K161R, and K257R of ANXA1 decreased the SUMO2 modification levels of ANXA1, while K185R and K312R had little impact on ANXA1 SUMOylation (Fig. 1H). We then generated an ANXA1 double mutant containing arginine at lysines 113, 161, and 257 (K113/161R, K113/257R, and K161/257R), and the outcome was that ANXA1 SUMOylation was markedly reduced but not completely abolished (Fig. 1I). Thus, we generated a new mutant, K113/161/257R (3KR), and found that SUMOylated bands of the ANXA1 3KR triple mutant completely impaired ANXA1 SUMOylation (Fig. 1J); these results were confirmed by IP (fig. S2B). Last, we found that the SUMOylated bands of wild-type (WT) ANXA1 were increased upon Flag-Ubc9 or GFP-PIAS3 transfection, but the SUMOylation of the ANXA1 3KR triple mutant was completely abolished even upon Flag-Ubc9 or GFP-PIAS3 transfection (fig. S2C). In addition, the corresponding lysine residues K113/161/257 of ANXA1 are highly conserved among its homologs in different species (fig. S2D). Together, these results demonstrated that K113/161/257 are the major SUMO2 modification sites of ANXA1.
SUMOylation of ANXA1 promotes microglia polarization toward an anti-inflammatory phenotype under OGD/R conditions
After identifying the SUMOylation modification of ANXA1, we then sought to determine the biological functional significance of this SUMOylation, especially its effect on ANXA1-mediated microglial activation after cerebral ischemia. To this end, ANXA1 mutants harboring mutations that affect ANXA1 SUMO modification were constructed. ANXA1 (3KR) is a SUMOylation-deficient mutant in which all of the SUMO acceptor lysine residues of ANXA1 at positions 113, 161, and 257 were mutated into arginine. In addition, we used a SUMO2 fusion-directed SUMO modification system to allow for the efficient and selective in vivo SUMOylation of ANXA1 for further investigation. To this end, SUMO2 was fused to the C terminus of WT ANXA1 (a construct named ANXA1-SUMO2) using a method that has been widely described previously (21–23). The ANXA1-SUMO2 fusion protein presented strongly enhanced SUMO modification levels (fig. S3, A and B). Next, we examined the functional significance of ANXA1 SUMOylation in microglial activation. For this purpose, adenoviruses encoding Myc-tagged WT ANXA1, ANXA1-3KR, and ANXA1-SUMO2 were used to infect primary cultured microglia. Microglial phenotypes can be differentiated by their expression of characteristic marker genes (5). Quantitative real-time polymerase chain reaction (qPCR) demonstrated that the proinflammatory phenotype marker genes (IL-1β, IL-6, TNF-α, iNOS, and CD16/32) were profoundly increased, while anti-inflammatory marker genes (Arg-1, IL-4, IL-10, TGF-β, and CD206) were significantly suppressed upon OGD/R stimulation, as previously reported. In addition, WT ANXA1 treatment had little effect on marker gene expression compared with that of the vector control. However, we found that ANXA1-SUMO2 notably restricted the up-regulation of the proinflammatory mediators and abrogated the down-regulation of the anti-inflammatory mediators induced by OGD/R stimulation. Unexpectedly, the ectopic expression of ANXA1-3KR not only enhanced the expression of inflammatory cytokines but also suppressed the expression of anti-inflammatory mediators (Fig. 2, A and B), suggesting a dominant-negative loss-of-function effect of the SUMOylation-deficient mutant. These results were confirmed by immunoblotting to determine the protein abundance of proinflammatory markers [iNOS (inducible nitric oxide synthase) and CD16/32] and anti-inflammatory markers (Arg-1 and CD206) (Fig. 2C and fig. S10B). The enzyme-linked immunosorbent assay (ELISA) also revealed that the protein secretion was consistent with the mRNA expression of proinflammatory cytokines (IL-1β, IL-6, and TNF-α) and anti-inflammatory cytokines (IL-4, IL-10, and TGF-β) (Fig. 2, D and E). Last, the expression of iNOS and Arg-1 was determined by immunofluorescence analysis. As shown in Fig. 2 (F to H), OGD/R resulted in a significant increase in the fluorescence intensity of iNOS but a decrease in the fluorescence intensity of Arg-1 in primary microglial cells. The overexpression of ANXA1-SUMO2 resulted in a decrease in iNOS expression and an increase in Arg-1 expression in primary microglial cells following OGD/R challenge, while ANXA1-3KR showed the opposite effect. Together, these results consistently indicated that SUMOylated ANXA1 could modulate microglial polarization toward the anti-inflammatory phenotype upon OGD/R stimulation.
Fig. 2. SUMOylation of ANXA1 promotes OGD/R-induced microglial anti-inflammatory phenotype polarization.
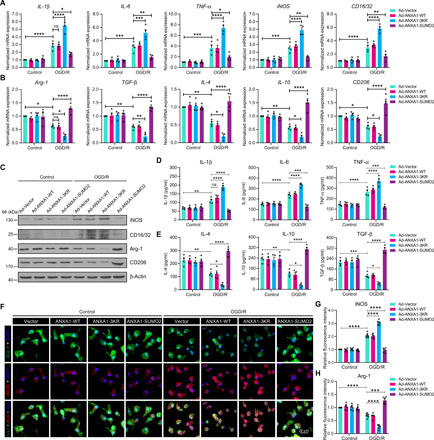
(A and B) SUMOylated ANXA1 but not the WT or the triple mutant of ANXA1 reversed OGD/R-induced mRNA expression of proinflammatory phenotype marker genes (A) and promoted mRNA expression of anti-inflammatory phenotype marker genes (B) in primary cultured microglia, as detected by qPCR. (C) The protein expression of iNOS, CD16/32, Arg-1, and CD206 in primary cultured microglia was examined by immunoblot analysis. (D and E) Expression levels of the cytokines IL-1β, IL-6, TNF-α, IL-4, IL-10, and TGF-β in microglial cell supernatants were detected by ELISA. Quantitative analysis was performed. (F) Immunofluorescence analysis of iNOS (red) and Arg-1 (green) with colabeled DAPI (blue) in primary microglia. Cells were infected with adenoviral vectors carrying WT ANXA1, ANXA1 triple mutant, or ANXA1-SUMO2 and were then treated with OGD for 1 hour and reoxygenation for 24 hours; then, iNOS and Arg-1 were detected by immunofluorescence staining. Scale bar, 20 μm. (G and H) The fluorescence intensity of iNOS (G) or Arg-1 (H) was quantified using ImageJ software. Data in (A), (B), (D), (E), (G), and (H) are presented as means ± SEM and analyzed by two-way ANOVA followed by Tukey’s post hoc test. ns for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To further investigate the role of SUMOylation of endogenous ANXA1 in regulating the phenotypic polarization of microglia, we used primary cultured microglia and induced pro- or anti-inflammatory polarization in vitro by treatment with lipopolysaccharide (LPS) plus interferon-γ (IFN-γ) or with IL-4, respectively (24). We then measured the SUMOylation level of endogenous ANXA1 under the different treatment conditions. The results revealed that the SUMO2 modification of ANXA1 was weakened under the proinflammatory phenotype but was strongly enhanced under the anti-inflammatory phenotype (figs. S3C and S10N). The down-regulation of the endogenous ANXA1 SUMOylation through the interference of the expression of Ubc9 or SUMO2/3 statistically increased the proinflammatory cytokines of primary microglia challenged with OGD/R (fig. S3, D and F). Conversely, the up-regulation of the endogenous ANXA1 SUMOylation by the overexpression of Ubc9 or SUMO2/3 notably increased anti-inflammatory cytokines (fig. S3, E and G). These results indicated that the SUMOylation of endogenous ANXA1 plays an important role in regulating the phenotypic polarization of microglia under OGD/R conditions.
SUMOylation of ANXA1 suppresses the activation of the NF-κB signaling pathway in microglial cells after OGD/R
To investigate the molecular mechanism by which SUMOylated ANXA1 mediates microglial polarization, we determined the activation of the NF-κB and mitogen-activated protein kinase-activator protein-1 (MAPK-AP-1) signaling pathways, which play crucial roles in microglial phenotype polarization, as previously described (25). Primary cultured microglia were transfected with adenoviruses expressing Myc-tagged WT ANXA1, ANXA1-3KR, and ANXA1-SUMO2 and then stimulated with OGD/R. The results demonstrated that OGD/R stimulation resulted in a strong increase in the phosphorylation level of NF-κB p65, JNK (c-Jun N-terminal kinase), ERK1/2 (extracellular signal–regulated kinase 1/2), p38 MAPK, and c-Jun. OGD/R-induced phosphorylation of p65 was suppressed by ANXA1-SUMO2 overexpression, but the phosphorylation levels of JNK, ERK1/2, p38 MAPK, and c-Jun were obviously unaffected. Conversely, ANXA1-3KR overexpression increased the phosphorylation of p65 (Fig. 3A and fig. S10C). These results suggest that SUMOylation of ANXA1 suppresses the OGD/R-induced activation of the NF-κB pathway. As expected, OGD/R treatment resulted in the same extent of signaling events in ANXA1-SUMO2–overexpressing microglia, including the phosphorylation of IKKα/β and phosphorylation and subsequent degradation of IκBα, two hallmarks of NF-κB signaling pathway activation (Fig. 3B and fig. S10D). Next, we used the dual-luciferase reporter assay to measure p65 and AP-1 transcriptional activity. The results suggest that OGD/R increased p65 and AP-1 transcriptional activation, as expected. In contrast, ANXA1-SUMO2 overexpression significantly decreased p65 transcriptional activity compared with that of the vector control, and this blocking effect was dose dependent (Fig. 3, C and D); however, ANXA1-SUMO2 overexpression had little impact on the activation of AP-1 transcriptional activity (fig. S4A). Last, we determined the effect of SUMOylated ANXA1 on the nuclear translocation of p65. As presented in Fig. 3 (E to G), cytoplasmic and nuclear fractions prepared from microglia were subjected to immunoblot analysis. The results showed that the OGD/R-induced accumulation of p65 protein in the nuclear fraction was robustly alleviated by ANXA1-SUMO2 overexpression, whereas ANXA1-3KR treatment significantly increased the OGD/R-induced nuclear translocation of p65. Consistently, the immunofluorescence assay demonstrated that SUMOylated ANXA1 inhibited OGD/R-induced p65 nuclear translocation in microglial cells (Fig. 3, H and I).
Fig. 3. SUMOylation of ANXA1 suppressed activation of the NF-κB signaling pathway in microglia.
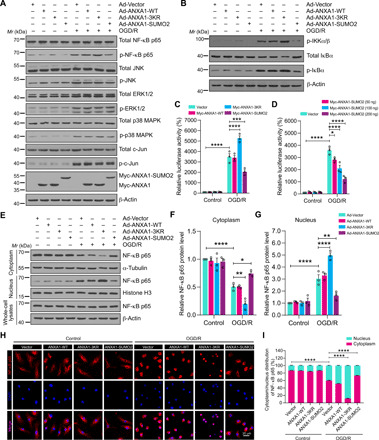
(A) Effects of WT or SUMOylation mutations of ANXA1 overexpression on the activation of the NF-κB and MAPK-AP-1 signaling pathways induced by OGD/R treatment in primary cultured microglial cells. Total protein expression and the phosphorylation levels of NF-κB p65, ERK1/2, JNK, p38 MAPK, and c-Jun were analyzed by immunoblotting. (B) Total protein expression and phosphorylation level of IKKα/β and IκBα in microglial cells were examined by immunoblotting. (C) Dual-luciferase reporter assay results show the transcriptional activity of NF-κB p65 in HEK293T cells. (D) Dual-luciferase reporter assay results show the transcriptional activity of p65 in HEK293T cells treated with increasing doses of ANXA1-SUMO2 (50, 100, and 200 ng). (E) Representative immunoblot analysis of the protein level of NF-κB p65 in cytoplasmic and nuclear extracts of microglia. (F and G) Quantification analysis of the relative amounts of NF-κB p65 protein in cytoplasmic and nuclear extracts. (H) Effects of SUMOylated ANXA1 on p65 nuclear translocation in OGD/R-stimulated microglia. Cells were infected with adenoviral vectors and were then treated with OGD for 1 hour and reoxygenation for 24 hours; then, the localization of NF-κB p65 was detected by immunofluorescence staining. (I) Quantitative analysis of NF-κB p65 nuclear translocation in (H). At least 50 cells per group were counted from three independent experiments. Data in (C), (D), (F), (G), and (I) are presented as the means ± SEM and analyzed by two-way ANOVA followed by Tukey’s post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Because ANXA1 is mostly known to act as an anti-inflammatory agent primarily through an autocrine/paracrine mechanism requiring its export from the cell and action through the formyl peptide receptor 2 (FPR2) receptor (26), we next studied the secretion of ANXA1 and its binding with FPR2. As shown in fig. S4 (B and C), the results revealed that the protein secretion of ANXA1 and its binding with FPR2 showed no significant differences between ANXA1-WT–treated, ANXA1-3KR–treated, and ANXA1-SUMO2–treated microglia under OGD/R conditions. Together, these data indicate that SUMOylation ANXA1 functions as a negative regulator to restrict NF-κB activation, thereby suppressing the expression of proinflammatory mediator genes and elevating the expression of anti-inflammatory mediator genes in OGD/R-challenged microglia.
SUMOylated ANXA1 regulates NF-κB activation through IKKα
We then explored the precise mechanisms by which SUMOylated ANXA1 restricts OGD/R-mediated NF-κB activation. It is widely accepted that NF-κB activation includes three major steps. In response to proinflammatory stimuli, adapters such as MyD88, TRAF2/6 (TNF receptor–associated factor 2/6), RIP1, or TAK1/TAB1 mediate the activation of the IKK complex, which is composed of IKKα, IKKβ, and IKKγ subunits. In turn, activated IKK induces IκBs, particularly IκBα phosphorylation and its subsequent proteasomal degradation, ultimately leading to a rapid release of NF-κB dimers, including p65/p50, which then translocate to the nucleus and activate the transcription of immune and inflammatory genes (27). We cotransfected 293T cells with plasmids carrying these major regulator molecules that participate in the NF-κB signaling pathway activation cascade, including MyD88, TRAF2, TRAF6, RIP1, TAK1/TAB1, IKKα, IKKβ, IKKγ, IκBα shRNA, and p65 along with WT ANXA1, ANXA1-3KR, or ANXA1-SUMO2, plus an NF-κB luciferase reporter vector. The results showed that activation of NF-κB by MyD88, TRAF2, TRAF6, RIP1, TAK1/TAB1, IKKα, IKKβ, and IKKγ was significantly suppressed by ANXA1-SUMO2 (Fig. 4A and fig. S5, A to J). In contrast, ANXA1-SUMO2 did not influence IκBα shRNA- and p65-mediated NF-κB signaling pathway activation, indicating that SUMOylated ANXA1 suppresses the NF-κB signaling pathway upstream of IκBα, most likely targeting the IKK complex. co-IP assay results showed that ANXA1-SUMO2 only interacted with IKKα in the cells (Fig. 4B and fig. S5K). Next, primary microglial cells were infected with adenoviruses expressing Myc-ANXA1-WT, Myc-ANXA1-3KR, or Myc-ANXA1-SUMO2 and then treated with OGD/R. The co-IP results showed that Myc-tagged ANXA1-SUMO2 interacted with endogenous IKKα, and this interaction was increased upon OGD/R challenge (Fig. 4C). We then performed an endogenous IP assay of primary cultured microglial cells and detected a stronger association between endogenous ANXA1 and IKKα when the cells were infected with adenoviruses expressing Flag-Ubc9 or His-PIAS3 (Fig. 4, D and E). Furthermore, immunofluorescence microscopic analysis further confirmed that GFP-tagged ANXA1-SUMO2 colocalized with mCherry-tagged IKKα in the cytosol (Fig. 4, F and G). Last, we found that the interaction between WT ANXA1 and IKKα was increased upon Flag-Ubc9 or GFP-PIAS3 transfection in 293T cells, but there was no interaction between Myc-ANXA1-3KR and Flag-IKKα, even upon Flag-Ubc9 or GFP-PIAS3 transfection (fig. S6, A and B). We found that IKKα overexpression could partially reverse the inhibitory effect of ANXA1-SUMO2 on the NF-κB signaling pathway activation, while the overexpression of IKKβ or IKKγ had little effect (fig. S5L). Together, these data demonstrate that SUMOylated ANXA1 targets IKKα to regulate the activation of the NF-κB signaling pathway.
Fig. 4. SUMOylated ANXA1 regulates NF-κB activation through IKKα.
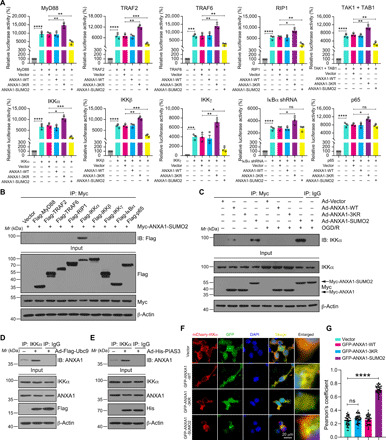
(A) Dual-luciferase reporter assay results showing the transcriptional activity of NF-κB p65. HEK293T cells were transduced with NF-κB-luc reporter; pRL-TK-luc plasmids; and plasmids encoding MyD88, TRAF2, TRAF6, RIP1, TAK1, TAB1, IKKα, IKKβ, IKKγ, ΙκBα shRNA, or p65, together with empty vector or WT ANXA1 (ANXA1-WT), triple mutant of ANXA1 (ANXA1-3KR), and constitutive SUMOylation mimic (ANXA1-SUMO2) for 24 hours and analyzed for NF-κB p65 transcriptional activity. (B) Co-IP experiments showing the interaction of SUMOylated ANXA1 with IKKα in HEK293T cells. (C) OGD/R enhances the interaction of SUMOylated ANXA1 with endogenous IKKα in primary microglia. (D and E) Ubc9 and PIAS3 enhance the binding of endogenous ANXA1 with IKKα in primary cultured microglia cells. (F) Representative immunofluorescence images present the colocalization of mCherry-IKKα with GFP-tagged WT or SUMOylation mutations of ANXA1 in HEK293T cells; scale bar, 20 μm. (G) Quantitative analysis of the colocalization was conducted by Pearson’s coefficient measurement using ImageJ software. Data represent Pearson’s coefficient mean of 50 individual cells ± SEM from at least three independent experiments. Statistical differences in (A) are determined by one-way ANOVA followed by Tukey’s post hoc test. Data in (G) are analyzed by one-way ANOVA followed by Dunnett’s post hoc test. Data are presented as means ± SEM. ns for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
SUMOylated ANXA1 mediates autophagy-dependent degradation of IKKα
Protein degradation is one of the critical ways to turn off signal transduction. In co-IP assays, we repeatedly observed that the IKKα protein level was reduced with ANXA1-SUMO2 overexpression in primary cultured microglia (Fig. 4C). These results prompted us to explore the effect of SUMOylated ANXA1 on IKKα expression. We observed that increasing doses of Myc-tagged ANXA1-SUMO2 resulted in considerably diminished Flag-IKKα protein levels (Fig. 5, A and B). qPCR results revealed that ANXA1-SUMO2 overexpression did not alter the mRNA expression of CHUK (which encodes the IKKα protein) (Fig. 5C). These results indicated that the diminished IKKα abundance occurred at the protein level. Furthermore, a cycloheximide (CHX) “chase” assay was performed to examine the time course of ANXA1-SUMO2–mediated IKKα degradation. As expected, ANXA1-SUMO2 accelerated the turnover rate of IKKα (Fig. 5, D and E). The CHX-chase analysis also revealed that ANXA1-SUMO2 overexpression led to a shorter half-life of endogenous IKKα in primary cultured microglia challenged with OGD/R compared with that of the vector control (fig. S7, A and B). We next investigated whether SUMO-modified ANXA1 regulates the endogenous IKK complex in primary microglial cells under physiological and OGD/R conditions. To this end, we found that overexpression of ANXA1-SUMO2 led to markedly reduced protein levels of IKKα but not IKKβ or IKKγ (figs. S7C and S10O). Together, these findings suggest that SUMOylated ANXA1 mediates IKKα degradation, especially under OGD/R conditions.
Fig. 5. SUMOylated ANXA1 mediates autophagy-dependent degradation of IKKα.
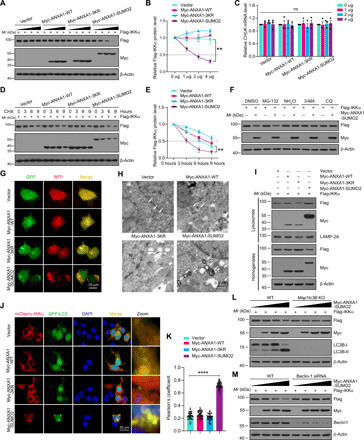
(A) Representative immunoblotting analysis of Flag-IKKα protein level in HEK293T cells transfected with SUMOylation mutations of ANXA1 in an increasing amount (0, 1, 2, and 4 μg). (B) Quantitative analysis of the protein levels of Flag-IKKα. (C) qPCR shows the mRNA levels of CHUK. (D and E) SUMOylated ANXA1 reduces the half-life of exogenous IKKα in HEK293T cells. (F) Representative immunoblots show the effects of DMSO, MG-132, NH4Cl, 3-MA, and CQ on SUMOylated ANXA1–mediated destabilization of Flag-IKKα in HEK293T cells. (G) Live-cell imaging of 293T cells transduced with tandem mRFP-GFP-LC3 plasmid for 24 hours. Scale bar, 10 μm. (H) Ultrastructural images of autophagic vacuoles in HEK293T cells were obtained by a TEM. The arrows indicate autophagic vacuoles; N represents the nucleus; scale bar, 0.5 μm. (I) Total cellular homogenates and lysosome fraction isolated from 293T cells were subjected to immunoblot analysis for the indicated proteins. (J) Representative immunofluorescence images present the colocalization of mCherry-IKKα with GFP-LC3 in 293T cells. Scale bar, 20 μm. (K) Quantitative analysis of the colocalization was conducted by Pearson’s coefficient measurement using ImageJ software. (L and M) Effects of LC3 knockout (KO) or Beclin1 knockdown on SUMOylated ANXA1–mediated IKKα degradation in HEK293T cells. Data are presented as the means ± SEM and analyzed by two-way repeated-measures ANOVA followed by Tukey’s post hoc test (B and E) or one-way ANOVA followed by Dunnett’s post hoc test (K). ns for P > 0.05, **P < 0.01, and ****P < 0.0001. siRNA, small interfering RNA.
Next, we explored the precise molecular mechanisms underlying SUMOylated ANXA1–mediated IKKα degradation. Two major systems that eukaryotic cells use for protein degradation are the ubiquitin-proteasome system and autophagy-lysosome pathways (28). Pharmacologic approaches were used to distinguish which clearance system is predominantly responsible for IKKα degradation. As shown in Fig. 5F and fig. S10E, Myc-ANXA1-SUMO2–mediated clearance of IKKα was completely abolished by the autophagy inhibitors chloroquine (CQ) or 3-methyladenine (3-MA), as well as the lysosomal inhibitor ammonium chloride (NH4Cl), but not the proteasome inhibitor MG-132 (N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal). NH4Cl, 3-MA, and CQ also inhibited ANXA1-SUMO2–induced endogenous IKKα degradation in primary microglia under OGD/R conditions (fig. S7D). Moreover, we found that autophagy induced by rapamycin or Earle’s balanced salt solution (EBSS) facilitated the clearance of IKKα, while bafilomycin A1 (Baf. A1) and NH4Cl inhibited IKKα degradation (fig. S7E).
We next examined the functions of SUMOylated ANXA1 in autophagy regulation. We transfected 293T cells with a plasmid expressing LC3 tagged in tandem with monomeric red fluorescent protein and GFP (mRFP-GFP-LC3) to determine autolysosome activation. We found that overexpression of ANXA1-SUMO2 substantially stimulated the formation of GFP-LC3 puncta and RFP-LC3 puncta (Fig. 5G). To confirm this result, ultrastructural images of autophagosomes in 293T cells transfected with ANXA1-WT, ANXA1-3KR, or ANXA1-SUMO2 plasmids were inspected by a transmission electron microscope (TEM). As expected, cells transfected with ANXA1-SUMO2 had more autophagic vacuoles than did ANXA1-WT– and ANXA1-3KR–transfected cells (Fig. 5H). These findings suggested that SUMOylated ANXA1 stimulates autophagic flux. Furthermore, we determined the accumulation of IKKα and ANXA1-SUMO2 in lysosomes by immunoblotting. Both IKKα and ANXA1-SUMO2 were found, to some extent, in the isolated lysosomes. The abundance of IKKα and ANXA1-SUMO2 in lysosomes was increased in Myc-tagged ANXA1-SUMO2–transfected 293T cells (Fig. 5I and fig. S10F). These results were consistent with the fact that ANXA1-SUMO2 protein level was decreased when Myc-ANXA1-SUMO2 was cotransfected with the Flag-IKKα plasmids, as presented in Figs. 4B and 5 (A and D) and fig. S5F. These data suggested that ANXA1-SUMO2 protein can be easily degraded also through the autophagy-lysosome pathways. We then further confirm these results by investigating endogenous SUMOylated ANXA1. As shown in fig. S7 (F and G), primary cultured microglia infected with adenoviruses expressing Flag-Ubc9 or His-PIAS3 notably enhanced the SUMOylation of endogenous ANXA1. Furthermore, in the input lanes, ANXA1 protein level was decreased in Ad-Flag-Ubc9– or Ad-His-PIAS3–infected cells; these results indicated that SUMOylation could also promote the protein degradation of endogenous ANXA1.
Next, the SUMOylated ANXA1–mediated accumulation of IKKα in lysosomes was confirmed by live-cell imaging. We examined the colocalization of GFP-IKKα and LysoTracker Red, a dye specific for live-cell lysosome labeling. As shown in fig. S7H, Myc-ANXA1-SUMO2 induced a remarkable increase in GFP-IKKα puncta, which were well colocalized with LysoTracker Red, suggesting that SUMOylated ANXA1 could mediate the accumulation of IKKα in lysosomes. In addition, immunofluorescence experiments suggested that the colocalization of mCherry-IKKα with GFP-LC3 dots was notably enhanced in cells overexpressing ANXA1-SUMO2 (Fig. 5, J and K). Consistently, the degradation of IKKα mediated by ANXA1-SUMO2 was abolished in LC3B-knockout or Beclin-1–knockdown 293T cells (Fig. 5, L and M, and fig. S10, G and H). Collectively, these data demonstrate that SUMOylated ANXA1 regulates IKKα degradation via the autophagy-lysosome pathway.
SUMOylated ANXA1 promotes NBR1-mediated selective autophagic degradation of IKKα
Motivated by the observations that SUMOylated ANXA1 specifically promoted the degradation of IKKα but not IKKβ or IKKγ, we then investigated whether SUMOylation of ANXA1 promotes degradation through selective autophagy. SUMO-modified protein has been found to facilitate autophagy-dependent degradation of specific target proteins (29). Co-IP experiments between IKKα and several major cargo receptors, including OPTN, p62/SQSTM1, NBR1, NDP52, and TOLLIP, were then performed to determine the autophagic receptor of IKKα. The results showed that IKKα specifically interacted with NBR1 but not the other receptors (Fig. 6A). Furthermore, NBR1 enhanced the degradation of IKKα induced by ANXA1-SUMO2 overexpression (Fig. 6B and fig. S10I). Further experiments demonstrated that SUMOylated ANXA1 also specifically bound NBR1 but not the other autophagic receptors (Fig. 6C). These data suggested that SUMOylated ANXA1 might bridge IKKα and NBR1. As expected, we found that SUMOylated ANXA1 enhanced the interaction between IKKα and NBR1 in a dose-dependent manner (Fig. 6D), whereas Myc-ANXA1-3KR notably inhibited the interaction between IKKα with NBR1 (Fig. 6E). We also found that the interaction between IKKα and NBR1 was substantially stronger upon OGD/R stimulation (Fig. 6F). Moreover, microscopic analysis also showed that the cytoplasmic punctate formation of IKKα-NBR1 was greatly enhanced by ANXA1-SUMO2 overexpression (Fig. 6, G and H). These results indicate that SUMOylated ANXA1 bridges IKKα and NBR1. To further substantiate these findings, we performed endogenous IP experiments of primary cultured microglial cells to examine the endogenous interaction between IKKα, NBR1, ANXA1, and SUMO2. As shown in Fig. 6I and fig. S8A, we found that IKKαNBR1, ANXA1, and SUMO2 interacted with each other under physiological conditions. However, this association was decreased under OGD/R conditions, which is consistent with the finding that the SUMO modification level of endogenous ANXA1 was decreased after OGD/R treatment in primary cultured microglia (Fig. 1C). Consistently, the interaction between IKKα, NBR1, ANXA1, and SUMO2 was attenuated in primary cultured microglia, in which endogenous ANXA1 was specifically knocked down by an ANXA1-specific shRNA (Fig. 6J). These data indicate that IKKα, NBR1, and SUMOylated ANXA1 can form a complex in which SUMOylated ANXA1 functions as a bridge to mediate the interaction.
Fig. 6. SUMOylated ANXA1 recruits IKKα to NBR1 for selective autophagic degradation.
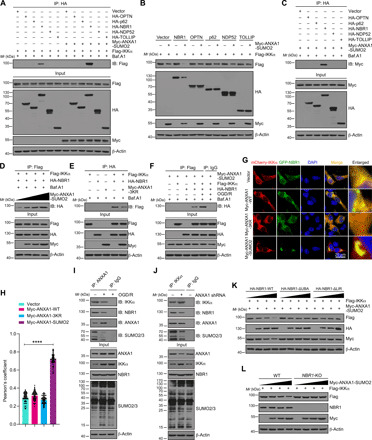
(A) Representative co-IP analysis shows the interactions of IKKα with cargo receptors in HEK293T cells. (B) Representative immunoblotting analysis shows the effects of different cargo receptors on SUMOylated ANXA1–mediated IKKα degradation in HEK293T cells. (C) Representative co-IP analysis results present the association of SUMOylated ANXA1 with NBR1 in HEK293T cells. (D) Representative co-IP analysis shows the interactions of IKKα with NBR1 in HEK293T cells. (E) Representative co-IP analysis shows the interactions of IKKα with NBR1 in HEK293T cells transfected with Myc-ANXA1-3KR. (F) Representative co-IP analysis shows the interactions of IKKα with NBR1 in HEK293T cells treated with OGD/R. (G) Immunofluorescence analysis of the colocalization between mCherry-IKKα and GFP-NBR1 in HEK293T cells. Scale bar, 20 μm. (H) Quantitative analysis of the colocalization was conducted by Pearson’s coefficient measurement using ImageJ software. (I) Representative co-IP analysis shows the interactions of ANXA1 with SUMO2, IKKα, and NBR1 in primary microglia cells treated with OGD/R. (J) Representative co-IP analysis shows the interactions of IKKα with NBR1, ANXA1, and SUMO2 in primary microglia cells treated with ANXA1 shRNA. (K) Immunoblot analysis shows the effects of WT, UBA, and LIR domain deletion mutant constructs of NBR1 on SUMOylated ANXA1–mediated Flag-IKKα degradation. (L) SUMOylated ANXA1–mediated IKKα degradation is completely abolished in the NBR1-KO cell line. WT and NBR1-knockout 293T cells were transfected with Flag-IKKα and an increasing amount of plasmid for Myc-ANXA1-SUMO2 (wedge), and cell extracts were subjected to immunoblot analysis. Data are presented as means ± SEM and analyzed by one-way ANOVA followed by Dunnett’s post hoc test. ****P < 0.0001.
It has been reported that the LC3 interacting region (LIR) of NBR1 is responsible for the recruitment of LC3 to the autophagosome, while the ubiquitin (Ub)–associated domain (UBA) directs ubiquitinated proteins to the autolysosome for selective degradation (30). Intriguingly, we observed that NBR1-ΔUBA and NBR1-ΔLIR mutants presented decreased abilities to mediate IKKα degradation (Fig. 6K and fig. S10J). Last, we generated NBR1-knockout HEK293T cells using CRISPR-Cas9 technology and detected that the degradation of IKKα caused by ANXA1-SUMO2 overexpression was completely abolished in NBR1-knockout HEK293T cells (Fig. 6L and fig. S10K). Furthermore, increasing the amount of HA-tagged NBR1 resulted in a considerable decrease in Flag-IKKα protein, but this degradation was almost completely abolished in ANXA1-knockdown HEK293T cells (figs. S8B and S10P). Collectively, these data strongly indicate that NBR1 is indispensable for SUMOylated ANXA1–mediated selective autophagic degradation of IKKα.
SUMOylated ANXA1 in microglia/macrophages protects against cerebral ischemic-reperfusion injury in vivo
Our in vitro studies suggested that SUMOylated ANXA1 promotes the anti-inflammatory phenotype polarization of microglia after OGD/R challenge, and we next investigated its protective effects against cerebral ischemic-reperfusion injury in vivo. We first developed a method to overexpress WT ANXA1 and SUMO modification mutants in the certain brain areas of ischemic stroke animal model mice. To this end, we generated an adeno-associated virus type 2/6 (AAV2/6) construct expressing SUMOylated ANXA1 only in cells expressing Cre recombinase driven by the Cx3cr1 promoter (Fig. 7A). Each AAV vector, which was able to express WT ANXA1, ANXA1-3KR, or ANXA1-SUMO2, was stereotactically injected into the hippocampus CA1 region, cerebral cortex, and striatum of adult Cx3cr1-Cre male mice (Fig. 7B). Since injection of the AAV vector will also infect infiltrating monocyte-derived macrophages and activated microglia, we used the term “microglia/macrophages” to refer to these cells collectively. Four weeks after AAV injection, the animals underwent MCAO surgery for 1 hour followed by reperfusion to establish a transient focal brain ischemia-reperfusion injury animal model. Successful ischemia was verified in all mice by monitoring the cerebral blood flow by laser Doppler flowmetry. After reperfusion, histological and behavioral studies were conducted at the indicated time points (Fig. 7C). Fluorescence microscopic analysis further confirmed the presence of SUMOylated ANXA1 in the hippocampus CA1 region, cerebral cortex, and striatum at 49 days after injection of the AAV particles in Cx3cr1-Cre mice (fig. S9A).
Fig. 7. SUMOylated ANXA1 overexpression in microglia/macrophages protects against cerebral ischemic-reperfusion injury in mice.
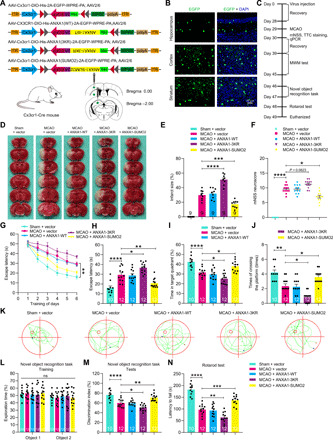
(A) Experimental design for microglia/macrophage overexpression of SUMOylated ANXA1. ITR, inverted terminal repeat; EGFP, enhanced GFP; DIO, double-flexed inverted open reading frame; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. (B) Representative images of GFP signals of Cx3cr1-Cre mice injected with the AAV vectors on day 28 after virus injection. (C) Schematic diagram presenting the experimental procedure. (D) Representative cerebral ischemic infarct size by TTC staining after 24 hours of reperfusion. Photo credit: Qian Xia, Huazhong University of Science and Technology. (E) Quantitative analysis of infarct size. (F) Neurological deficit score was assessed by the mNSS at 24 hours after reperfusion. (G to J) Latency trial (G and H) and probe trial (I and J) results in the MWM tests. (G) Escape latency to reach the hidden platform during days 1 to 6 of testing. (H) Exploration time searching for the hidden platform on day 6. (I) The percentage of time searching for the hidden platform in the target quadrant on day 7. (J) Number of times crossing over the platform location on day 7. (K) Representative swimming traces indicating the sample paths from the probe trials on day 7. (L and M) Novel object recognition tasks. (L) During the familiarization trial, the mice were scored for the time spent in exploring two identical objects. (M) During the test trial, mice were scored for the amount of time they spent in exploring the novel and familiar objects, and discrimination index is represented. (N) The time falling off the rotarod drum during the final test. Numbers in bars indicate the numbers of mice. Data are presented as means ± SEM. ns for P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. See table S3 for further details of statistical data analysis.
We first detected the ischemic infarct size using TTC (2,3,5-triphenyl-tetrazolium chloride) staining at 24 hours after reperfusion and observed that the ischemic infarct size was reduced in SUMOylated ANXA1–overexpressing mice, whereas the infarct size in ANXA1-3KR–overexpressing mice was larger than that of vector controls (Fig. 7, D and E). Next, neurological deficits were assessed on the basis of a modified neurological severity score (mNSS). We found that the neurological deficit scores of SUMOylated ANXA1–overexpressing animals were substantially lower than those of vector- or WT ANXA1–overexpressing animals (Fig. 7F).
We then performed the Morris water maze (MWM) test to determine the spatial learning and memory functions of the animals following focal ischemic injury. As shown in Fig. 7 (G to J), ANXA1-SUMO2–overexpressing mice exhibited remarkable cognitive improvement, including the latency to reach the submerged platform, the time spent in the targeting quadrant, and the times of crossing the platform, whereas the ANXA1-3KR–overexpressing mice displayed evident cognitive dysfunction. Representative swimming traces from the probe trials are presented in Fig. 7K. A novel object recognition task was then performed to confirm the cognitive improvement in SUMOylated ANXA1–treated mice. During the familiarization trial, the animals explored both identical objects equally (Fig. 7L); in the test trial, one familiar and one novel object were presented in the arena, and the ANXA1-SUMO2–treated animals displayed an evident predilection for the novel object, suggesting that they retained a good memory of the familiar object compared with that of vector control mice (Fig. 7M). Last, we performed rotarod assays to test for motor function and found that ANXA1-SUMO2–overexpressing mice displayed a moderate increase in the time to fall on the rotarod test (Fig. 7N). Collectively, these results revealed that overexpression of SUMOylated ANXA1 in microglia/macrophages significantly reduced brain ischemic infarct size, diminished neurological deficit scores, and preserved cognitive and motor function after brain ischemia.
After confirming the neuroprotective effects of SUMOylated ANXA1 against cerebral ischemic-reperfusion injury in vivo, we then examined whether the AAV-mediated introduction of SUMOylated ANXA1 could alter the phenotype polarization of microglia/macrophages in ischemic stroke mice. Microglia/macrophages were isolated from ischemic stroke animal model mice, and qPCR was used to examine the mRNA level of phenotype marker genes. As shown in fig. S9 (B and C), the proinflammatory phenotypic marker genes were notably reduced, while the anti-inflammatory phenotype marker genes were significantly increased in SUMOylated ANXA1–overexpressing mice. In contrast, ANXA1-3KR–overexpressing mice exhibited exactly the opposite trends, suggesting a dominant-negative action of the SUMOylation-deficient mutant. These data suggest that the neuroprotective effects of SUMOylated ANXA1 are relevant to its regulation of microglia polarization.
To further investigate the endogenous function of SUMOylated ANXA1 in microglia/macrophages, we injected the hippocampus CA1 region, cerebral cortex, and striatum of adult Cx3cr1-Cre male mice with the AAV, which expresses ANXA1-targeting small hairpin RNA in a Cre-dependent manner (fig. S9D). Immunoblotting results revealed reduced ANXA1 expression in isolated microglia/macrophage cells in mice injected with ANXA1 shRNA-AAV, as compared to the scramble-AAV (fig. S9, E and F), indicating successful ANXA1 knockdown in microglia/macrophage cells. The down-regulation of the endogenous ANXA1 SUMOylation through the silence of ANXA1 increased proinflammatory cytokines and attenuated anti-inflammatory cytokines in isolated microglia/macrophage cells (fig. S9, G and H). These results collectively indicated that the SUMOylation of endogenous ANXA1 plays an important role in regulating the phenotypic polarization of microglia/macrophages after cerebral ischemia.
DISCUSSION
In this study, we first confirmed that ANXA1 could be modified by SUMOylation. Next, we examined whether the SUMOylation of ANXA1 regulated the polarization of primary microglia subjected to OGD/R challenge. Intriguingly, we observed that SUMOylated ANXA1 favored microglial activation to an anti-inflammatory and tissue-reparative phenotype and restricted the expression and release of proinflammatory mediators. On the basis of this observation, we then investigated the underlying mechanism and found that SUMOylated ANXA1 suppressed NF-κB pathway activation. Further work revealed that SUMOylated ANXA1 targeted the IKK complex and specifically promoted IKKα degradation. Moreover, we found that SUMOylated ANXA1 promoted the interaction of IKKα with the autophagy receptor NBR1 by forming a complex, thereby facilitating the selective autophagic degradation of IKKα. This study also showed that overexpressing SUMOylated ANXA1 in microglia/macrophages ameliorated the neurological functional outcomes of animals subjected to cerebral ischemic injury.
SUMOylation is one of the most important PTMs regulating the various functions of proteins. It has become increasingly clear that SUMOylation plays pivotal roles in a multitude of biological functions, such as transcriptional regulation, nuclear translocation, maintaining protein stability, and signaling pathway transduction (31). Previous studies reported that ANXA1 can be modified by SUMO1 at lysine residue K257 in LNCaP cells, and the SUMOylation levels of ANXA1 were decreased by DNA-damaging agents (32). Consistent with this report, we found that ANXA1 was mainly modified by SUMO2 compared with SUMO1 and SUMO3. Moreover, in addition to the lysine residue K257, we demonstrated that ANXA1 can also be SUMOylated at lysine residues K113 and K161. Our results also suggested that the SUMOylation of ANXA1 was enhanced by the E2-conjugating enzyme Ubc9 and the E3 ligase PIAS3. Furthermore, OGD/R stimulation significantly decreased the SUMO modification level of endogenous ANXA1 in primary microglia, which suggests that the SUMOylation of ANXA1 plays an essential role in microglial activation after cerebral ischemia.
In light of this discovery, we next found that SUMOylated ANXA1 switched the activation of microglial cells from the detrimental proinflammatory phenotype to the beneficial anti-inflammatory phenotype under OGD/R conditions. Overexpression of the ANXA1-SUMO2 fusion protein in microglia increased the expression of anti-inflammatory mediators, such as Arg-1, IL-4, IL-10, TGF-β, and CD206. Overexpression also synchronously alleviated the expression and release of multiple proinflammatory mediators, such as IL-1β, IL-6, TNF-α, iNOS, and CD16/32.
We observed that there are very few differences between cells transfected with an empty vector and WT-ANXA1. These may be due to the fact that SUMOylation is a dynamic and reversible process, and it can be readily reversed by a family of sentrin/SUMO-specific proteases (33). Under the effect of SUMO-deconjugating enzymes, overexpression of WT-ANXA1 cannot up-regulate SUMOylation level of ANXA1 after OGD/R treatment. So, overexpression of WT-ANXA1 only has no obvious biological function. In contrast, overexpression of a SUMOylation-deficient mutant of ANXA1 in microglia led to the exact opposite results, which may be due to the dominant-negative action of this SUMOylation-deficient mutant. We found that overexpression of the ANXA1-3KR mutant protein markedly exacerbated microglial inflammatory responses. Although the proinflammatory and anti-inflammatory phenotype classification underestimates the complexity of microglial plasticity, the distinction nevertheless provides a useful framework for exploring the diverse functions of microglia in disease pathogenesis, such as cerebral stroke (34). In any case, this study uncovered a mechanism whereby SUMOylated ANXA1 regulates microglial activation after cerebral stroke.
Growing evidence has revealed that NF-κB is the key signaling pathway that regulates the expression of multiple proinflammatory mediators, and it is also critical for switching proinflammatory and anti-inflammatory phenotype balance in microglia (35, 36). Evidence from our study showed that SUMOylated ANXA1 significantly suppressed the activation of the NF-κB signaling pathway induced by OGD/R stimulation but had little impact on the activation of the MAPK-AP-1 signaling pathway. As the current research only shows that NF-κB can directly act as a transcription factor for the proinflammatory factors, such as IL-1β, IL-6, and TNF-α, SUMOylated ANXA1 regulating the expression of anti-inflammatory factors may be due to the fact that NF-κB can indirectly affect the expression of these genes by regulating other signaling or transcription factor pathways in the cells. Nevertheless, the precise mechanisms remain to be clarified in future studies. The IKK complex plays a critical role in NF-κB activation, and it is composed of the catalytic subunits IKKα and IKKβ and the regulatory subunit IKKγ. Numerous studies have indicated that IKKα/β-mediated phosphorylation-dependent degradation of IκB is the most critical step in canonical NF-κB signaling pathway activation, leading to the nuclear transport of the NF-κB factor and subsequent gene expression. Our results revealed that SUMOylated ANXA1 targeted the IKK complex and specifically mediated the autophagic degradation of IKKα. We found that chemical or genetic inhibition of autophagy abolished this IKKα degradation induced by SUMOylated ANXA1 overexpression. Previous studies from different groups have shown that IKKβ and IKKγ undergo autophagic degradation through different mechanisms (37, 38), but this study is the first to report that IKKα can also be degraded via selective autophagy.
Many studies have demonstrated the crucial roles of autophagy in the microglial inflammatory response and phenotype polarization (39). In the present study, we detected that the ratio of LC3-II/I was markedly enhanced by SUMOylated ANXA1 (Fig. 5L). We observed that SUMOylated ANXA1 overexpression promoted autophagosome formation and autolysosome maturation. Furthermore, SUMOylation of ANXA1 significantly facilitated the colocalization of IKKα with LC3 dots. These data collectively demonstrate that SUMOylated ANXA1 up-regulates the level of autophagy, which results in microglial anti-inflammatory phenotype polarization.
Autophagy was once considered a nonselective bulk degradation pathway of long-lived organelles and proteins; nevertheless, increasing evidence has shown that autophagic clearance of proteins mediated by cargo receptor proteins, such as p62/SQSTM1, NBR1, TOLLIP, NDP52, and OPTN, can degrade substrates in a selective manner (40). We found that SUMOylated ANXA1 formed a protein complex with IKKα and NBR1, in which SUMOylated ANXA1 functioned as a bridge to enhance the interaction between IKKα and NBR1, subsequently facilitating the selective autophagic clearance of IKKα and thereby inhibiting OGD/R-induced NF-κB signaling pathway activation (working model shown in Fig. 8). These data provide previously unkown mechanistic evidence that autophagic degradation selectively regulates NF-κB signaling pathway activation.
Fig. 8. A proposed working model of SUMOylated ANXA1–mediated microglial anti-inflammatory phenotype polarization after cerebral ischemia.
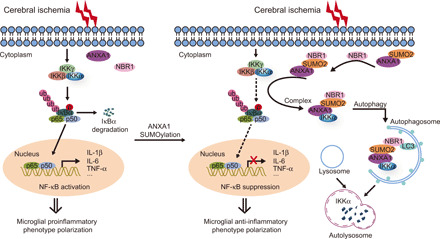
SUMOylated ANXA1 functions as a co-adapter to facilitate the interaction between IKKα and NBR1, which promotes the autophagic degradation of IKKα, thus suppressing the activation of NF-κB signaling pathway and the production of proinflammatory mediators in microglia, resulting in the anti-inflammatory phenotype polarization of microglia after brain ischemic injury.
We further examined whether up-regulated SUMOylation of ANXA1 in microglia provided protective effects in a mouse focal cerebral ischemia model. First, we developed a novel AAV-based technique to overexpress SUMOylated ANXA1 in microglia/macrophages in the hippocampus CA1 region, cerebral cortex, and striatum; nonetheless, it is not easy to manipulate gene expression in microglia/macrophage cells in a certain brain area. In this study, we demonstrated that up-regulation of ANXA1 SUMOylation in microglia/macrophage reduces the brain ischemic infarct size and diminished neurological deficit scores. Behavioral tests also showed that SUMOylated ANXA1 overexpression significantly improved the learning, memory, and motor functions of mice subjected to brain ischemia-reperfusion injury. Our results indicate that up-regulation of the SUMOylation level of ANXA1 may be a potential therapeutic strategy for brain ischemia injury. On the other hand, Cx3cr1-Cre mice express Cre recombinase under the direction of the Cx3cr1 promoter in the mononuclear phagocyte system, including the monocyte and macrophage compartments as well as microglia (41). Therefore, the injection of the AAV particles may also infect macrophages and infiltrating monocytes as well as microglia, and the specific effect of AAV particle injections on the infiltrating macrophages and monocytes remains to be determined in future studies.
In summary, our findings identified a previously unrecognized molecular mechanism by which ANXA1 participates in the phenotypic shift of microglial polarization. We have provided compelling evidence that SUMOylated ANXA1 functions as a negative regulatory factor that directs IKKα to autophagosomes for clearance in an NBR1-dependent manner, thereby exerting a powerful inhibitory effect on excessive NF-κB activation induced by brain ischemic-reperfusion injury. Overall, this study sheds light on a previously unidentified ANXA1 function and suggests that up-regulation of the SUMOylation of ANXA1 in microglia could be a novel and potential therapeutic strategy for cerebral stroke and possibly other types of neuroinflammation diseases and disorders.
MATERIALS AND METHODS
Animals
Cx3cr1-Cre mice [formally named B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J (RRID:IMSR_JAX: J025524)] in the C57BL/6 strain background were obtained from the Jackson Laboratory. WT adult C57BL/6 mice (aged 8 weeks, weighing 22 to 25 g) were purchased from Beijing Vital River Laboratory Animal Corp. Ltd. (Beijing, China). The mice were housed in groups of three to four mice per cage and kept in a 12-hour light/dark cycle at 22°C (room temperature) with water and food ad libitum. All animal experiments were approved by the Ethics Committee for Animal Experimentation of Huazhong University of Science and Technology (Wuhan, China) in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health. Efforts were made to reduce the number of mice used and their suffering.
Antibodies and reagents
The antibodies used in the present study are listed in table S1. Protein A+G agarose beads, LPS, recombinant murine IFN-γ, IL-4, and LysoTracker Red were obtained from Beyotime Biotechnology (Haimen, China), and the Ni2+-NTA agarose was purchased from QIAGEN (Dusseldorf, Germany). CHX was obtained from Calbiochem (508739; Darmstadt, Germany). Dimethyl sulfoxide (DMSO; D2650), MG-132 (C-2211), ammonium chloride (NH4Cl; A9434), 3-MA (M9281), CQ phosphate (PHR1258), and puromycin (P9620) were purchased from Sigma-Aldrich, China. Rapamycin (HY-10219) was obtained from MedChemExpress (Shanghai, China), Bafilomycin A1 (S1413) was purchased from Selleck (Houston, TX, USA), and EBSS was purchased from Gibco (Gaithersburg, MD, USA). A set of small interfering RNA of control (6568) and BECN1 (6246) was purchased from Cell Signaling Technology (Danvers, MA, USA). All other general reagents are commercially available and used as received.
Primary microglia culture and adult brain microglia isolation
Mixed glia cells were prepared from the whole brain of neonatal mice at postnatal P1 and P2 and were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 20% fetal bovine serum (FBS; Gibco) at 37°C in a 95% O2 and 5% CO2 incubator for 8 to 10 days, and the medium was changed every 3 days. Microglia were isolated from the mixed glial cultures by mild agitation at 200 rpm for 6 hours in a rotary shaker at 37°C based on the distinct adhesive features of microglia and astrocytes. The obtained microglial cells were seeded into six-well plates in high-glucose DMEM supplemented with 20% FBS at a density of 1 × 106 per well for 24 hours before further treatment. The purity of the adherent cells was verified by immunocytochemical staining, which indicated that more than 95% of the cells in the cultures were positive for the microglia-specific marker Iba-1 (1:500; Wako) (fig. S1H). A highly enriched population of microglia/macrophages was isolated from adult mice by Percoll density centrifugation using a protocol described previously (42). For the induction of a proinflammatory phenotype, LPS (100 ng/ml) and IFN-γ (20 ng/ml) were added to the microglial cultures for 24 hours. For the induction of an anti-inflammatory phenotype, IL-4 (20 ng/ml) was added to the culture for 24 hours (24).
Cell culture and transfection
HEK293T cells were purchased from the American Type Culture Collection. Cells were grown in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Sigma-Aldrich, USA) at 37°C in a humidified 5% CO2-containing atmosphere. Cells were tested and confirmed to be free of mycoplasma. Confluent cell layers were split three times per week. Transfections of the plasmids were performed by using Lipofectamine 3000 (Invitrogen, NY, USA) when the cells were 80 to 90% confluent, following the manufacturer’s instructions.
OGD/R procedure
We established an in vitro cell model of cerebral ischemia-reperfusion injury by challenging primary microglia with OGD/R as previously described (43). In brief, cells were rinsed three times and incubated in glucose-free DMEM (Gibco) prewarmed to 37°C, after which microglia were transferred to an anaerobic incubator with a 5% CO2 and 95% N2 atmosphere at 37°C for 1 hour to establish conditions of OGD. After that, the culture medium was replaced with normal DMEM containing glucose under normoxic conditions for an additional 24 hours as OGD/R.
Protein extraction and preparation
Protein extraction and subcellular fractionation were performed as we have previously described (44). For the preparation of whole-cell lysates, microglial cells were lysed in radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology) supplemented with cOmplete protease inhibitor cocktail tablets (5 mg/ml; Roche, Basel, Switzerland). Nuclear and cytoplasmic fractionations were extracted using the NE-PER Nuclear and Cytoplasmic Extraction Reagent Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the instructions provided by the manufacturer. An enhanced BCA (bicinchoninic acid) protein assay kit (Beyotime Biotechnology) was used to measure the protein concentrations of the extracts.
Enzyme-linked immunosorbent assay
The expression of inflammatory factors IL-1β, IL-6, TNF-α, IL-4, IL-10, and TGF-β in cell supernatants was determined using commercially available ELISA kits (DAKEWEI, Shenzhen, China), and ANXA1 release was measured using an ELISA kit purchased from Cloud-Clone Corp (SEE787Mu, Wuhan, China). In accordance with the manufacturer’s instructions, supernatants were stored at −80°C before measurements, and both standards and samples were run in triplicate. Samples were collected in pyrogen/endotoxin-free tubes. The optical density at 450 nm was calculated by subtracting the background, and standard curves were plotted.
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was isolated from microglia using TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions and was quantified spectrophotometrically. Afterward, complementary DNA (cDNA) was synthesized from 1 μg of total RNA using the ReverTra Ace-α-First Strand cDNA Synthesis Kit (Toyobo, Osaka, Japan). Quantitative real-time PCR was performed in a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, USA) with SYBR Green PCR Master Mix (Applied Biosystems), according to the manufacturer’s recommendations. Analysis of gene expression was performed using the 2−ΔΔCt method, and relative gene expression was normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA levels. Gene expression analyses were expressed as mRNA levels relative to controls. Primer sequences are listed in table S2.
Immunoprecipitation and immunoblot analysis
IP was performed as described previously (45). In brief, cells were washed twice with ice-cold phosphate-buffered saline (PBS) and then lysed in 2% Triton X-100 lysis buffer [20 mM tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 2% Triton X-100] supplemented with a cOmplete protease inhibitor cocktail (Roche). Protein concentration was measured by an enhanced BCA protein assay kit (Beyotime Biotechnology). Whole-cell lysates were preabsorbed for 1 hour with Protein A+G agarose beads (Beyotime Biotechnology) and then centrifuged at 2500 rpm to remove any protein adhered nonspecifically to the Protein A+G. Samples were incubated with the indicated antibodies with gentle rotation overnight at 4°C. Immunoprecipitates were extensively washed four times with IP lysis buffer and then eluted with 2× SDS–polyacrylamide gel electrophoresis (PAGE) loading buffer by boiling at 95°C for 5 min. Then, the protein samples were resolved by SDS-PAGE.
For immunoblotting detection, protein samples were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Roche Diagnostics, GA, USA). Membranes were blocked with 5% bovine serum albumin (Merck Millipore, Billerica, USA) at room temperature for 60 min and probed with the indicated antibodies overnight at 4°C. Horseradish peroxidase–conjugated secondary antibodies were used (1:20,000; Invitrogen). Immunodetection was performed with a chemiluminescence substrate kit (Thermo Pierce, Rockford, USA).
Microscopy analysis
For fluorescence microscopy detection, HEK293T cells were cultured on a glass-bottom dish and transfected with the indicated plasmids for 24 hours; then, the cells were fixed with 4% paraformaldehyde for 15 min and stained with 4′,6-diamidino-2-phenylindole (DAPI) (10 mg/ml; Sigma-Aldrich). Localization images were examined under fluorescence microscopy (IX73, Olympus, Tokyo, Japan). For LysoTracker Red staining, HEK293T cells grown on coverslips were stained with 50 nM LysoTracker Red (Beyotime Biotechnology, C1046) in prewarmed medium for 20 min at 37°C. All samples were analyzed instantaneously with a fluorescence microscope. LysoTracker Red was prevented from exposure to broadband illumination before confocal imaging to prevent photoconversion. For examination by immunofluorescence microscopy, primary cultured microglia cells grown on gelatin-coated coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 for 15 min, and blocked with 10% donkey serum for an additional 60 min. Microglial cells were then incubated with specific primary antibodies overnight at 4°C. The samples were then rinsed thoroughly three times in 0.25% Tween 20 in PBS and incubated with secondary antibodies at room temperature for 60 min. DAPI was used for counterstaining of the nuclei. The cells were examined and counted immediately under fluorescence microscopy. All the measurements were performed by an investigator who was blinded to the different treatment groups.
RNA interference experiments
The shRNA plasmids were obtained from GenePharma (Shanghai, China). The target sequences for human UBE2I (GenBank NM_003345.5, encodes the Ubc9 protein) shRNAs were as follows: no. 1, 5′-TTGGCAGTAAATC GTGTAGGCC-3′; no. 2, 5′-TATAAACACAAAGGAAGTGGCG-3′; no. 3, 5′-TTTGGTTCATTTAGAAGTTCCT-3′; and no. 4, 5′-ATTTAGAAGTTCCTGTATTCCT-3′. The shRNA sequence that targets human NFKBIA sequence (GenBank no. NM_020529.3) was designed as follows: 5′-AAGTGGAGTGGAGTCTGCTGC-3′. The shRNA sequence that targets mouse ANXA1 sequence (GenBank no. NM_010730.2) was designed as follows: 5′-GCCTCACAACCATCGTGAAGT-3′; an Scr shRNA served as a negative control.
Generation of knockout cells by the CRISPR-Cas9 system
LC3B and NBR1 knockout cells were generated using CRISPR-Cas9 technology, and the CRISPR-Cas9–based protocols for genome engineering were used as described previously (46). The sequences of the target single-guide RNA are as follows: MAP1LC3B-guide RNA: 5′-TTCAAGCAGCGCCGCACCTT-3′; NBR1-guide RNA: 5′-AGAATATGAAGAAGCGCTTA-3′.
Luciferase reporter assays
HEK293T cells were plated in 96-well plates and transfected with plasmids carrying an NF-κB or AP-1 luciferase reporter (firefly luciferase plasmid, Promega, Madison, WI, USA) and pRL-TK-luc (Renilla luciferase plasmid, Promega) together with plasmids expressing ANXA1 or the SUMOylation mutants of ANXA1. OGD/R treatment or MyD88, TRAF2, TRAF6, RIP1, IKKα, IKKβ, IKKγ, IκBα shRNA, or p65 was used as stimulators. Cells were lysed at 24 hours after transfection, and luciferase activity was detected with the Dual-Luciferase Assay kit in accordance with the manufacturer’s instructions (Promega) using a Fluoroskan Ascent FL system (Thermo Fisher Scientific). The reporter gene activity was examined by normalizing the firefly luciferase activity to Renilla luciferase activity. The experiments were performed in triplicate.
Plasmid construction
DNA fragments corresponding to the SUMO1, SUMO2, and SUMO3 coding sequences were amplified by PCR, followed by cloning into the pcDNA3.1 plasmid (Invitrogen). The GFP-tagged PIAS1, PIAS3, PIASxα, PIASxβ, PIASy, RanBP2, Pc2, LC3, NBR1, and IKKα were constructed by cloning the full-length coding sequence into pEGFP-N1 (Clontech, Mountain View, CA, USA). The Flag-tagged Ubc9, MyD88, TRAF2, TRAF6, RIP1, TAK1, TAB1, IKKα, IKKβ, IKKγ, IκBα, or p65 were constructed by cloning the full-length cDNA into p3xFLAG CMV14 (Sigma-Aldrich). The HA-tagged NBR1, TOLLIP, OPTN, and NDP52 were constructed by cloning the full-length cDNA into pcDNA4/TO (Invitrogen). HA-p62 was a gift from Q. Zhong (Addgene plasmid #28027), and the tandem mRFP-GFP-LC3 plasmid, which has previously been described, was a gift from X. Hou (Huazhong University of Science and Technology). For site-directed mutagenesis, all ANXA1 SUMOylation mutants were constructed by a QuikChange Site-Directed Mutagenesis Kit (Stratagene) using synthetic oligonucleotides containing mutations in the corresponding positions to generate lysine-to-arginine mutation. All constructs were confirmed by DNA sequencing analysis (performed by Sangon Biotechnology, Shanghai, China).
Ni2+-NTA affinity purification
SUMOylated ANXA1 in 293T cells was affinity-purified under denaturing conditions by Ni2+-NTA pull down. Briefly, HA-ANXA1 and His-SUMOs (SUMO1, SUMO2, and SUMO3) were overexpressed in HEK293T cells with Flag-tagged Ubc9. After 24 hours, cells were first washed twice with cold PBS and then lysed in 800 μl of Ni2+-NTA denaturing buffer [6 M Gu-HCl, 10 mM tris, 100 mM NaH2PO4 (pH 8.0), and 20 mM N-Ethylmaleimide (NEM)]. After shearing the DNA by sonication (2 × 20 s), the samples were cleared by centrifugation (15,000g, 10 min, 4°C). The supernatants were mixed with 50 μl of prewashed Ni2+-NTA agarose (Qiagen) and incubated for 3 hours at 4°C on a rotating wheel. The beads were then washed in successive washing steps in 1 ml of Ni2+-NTA washing buffer [8 M urea, 10 mM tris-HCl (pH 6.3), 100 mM NaH2PO4, and 0.1% Triton X-100] and lastly eluted by boiling the beads in 50 μl of 2× SDS-PAGE loading buffer containing 200 mM imidazole at 95°C for 5 min. Bound proteins were resolved by SDS-PAGE and detected by immunoblot analysis.
TEM analysis
To observe autophagy, ultrastructural analysis was performed. Briefly, HEK293T cells were fixed with ice-cold glutaraldehyde [2.5% in 0.1 M cacodylate buffer (pH 7.4)] for 30 min, postfixed in OsO4, and embedded in epoxy resin. Ultrathin sections (70 to 80 nm) were stained with uranyl acetate/lead citrate and examined in an electron microscope (Tecnai G2 20 TWIN, FEI, Hillsboro, USA). For quantification of autophagic vacuoles, the number of autophagosomes per optical field using 30 optical fields from each sample was calculated.
Viral vector transduction
Adenoviruses encompassing the Myc-tagged WT ANXA1 (ANXA1-WT), ANXA1 triple mutant (ANXA1-3KR), constitutive SUMOylated mimic of ANXA1 (ANXA1-SUMO2), Flag-tagged Ubc9, and His-tagged PIAS3 to infect primary cultured microglia were generated by Vigene Biosciences (Jinan, China). Primary cultured microglial cells were infected with the respective adenoviruses at an optimal multiplicity of infection of 100. AAV2/6 viruses were obtained from BrainVTA (Wuhan, China). These viral titers were in the range of 2 × 1012 to 3 × 1012 vector genomes (vg)/ml. Viral vectors were subdivided into aliquots stored at −80°C until use. Stereotaxic surgery was performed to deliver the AAV vectors. In brief, mice were anesthetized by an intraperitoneal injection with chloral hydrate (350 mg/kg) and immobilized on a stereotaxic apparatus (RWD Life Science, Shenzhen, China). A burr hole was used to perforate the skull, and a virus solution of 500 nl per injection site was injected into the hippocampus CA1 region, cerebral cortex, and striatum of the left hemisphere with a stepper-motorized microsyringe (Hamilton, Reno, USA) at a rate of 50 nl/min. Injections into the hippocampus CA1 were at coordinates anteroposterior (AP) −2.00 mm, lateral (L) −1.55 mm, and dorsoventral (DV) −1.55 mm; those into the cerebral cortex were at coordinates AP 0.00 mm, L −2.05 mm, and DV −1.50 mm; and those into the striatum were at coordinates AP 0.00 mm, L −2.05 mm, and DV −3.50 mm relative to the bregma and dura surface. Abbreviations of viral elements are as follows: ITR, inverted terminal repeat; DIO, double-flexed inverted open reading frame; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; Cx3cr1, chemokine (C-X3-C motif) receptor promoter.
Transient focal cerebral ischemia
Focal cerebral ischemia was induced by transient occlusion of the middle cerebral artery using the intraluminal filament technique as described previously (44). Briefly, mice were anesthetized with chloral hydrate (350 mg/kg, i.p.), and the rectal temperature was maintained at approximately 37° ± 0.5°C using a homeothermic blanket (Harvard Apparatus, Holliston, MA, USA). The left common, external, and internal carotid arteries were exposed, and the common and external carotid arteries were permanently ligated with sutures. A silicone-coated monofilament nylon suture (0.22 to 0.23 mm in diameter) was gently introduced into the internal carotid artery through the external carotid artery stump and advanced to the anterior cerebral artery until a slight resistance was felt. The distance from the tip of the suture to the bifurcation of the common cerebral artery was 10 ± 0.5 mm. Successful occlusion was verified by a laser Doppler flowmetry (PeriFlux System 5000, PERIMED, Sweden). The filament was left in place for 60 min and then withdrawn. Sham-operated mice underwent the same surgical procedure without the insertion of the filament.
TTC staining
At 24 hours after ischemic-reperfusion surgery, the mice were euthanized, and the brains were removed quickly and then frozen at −20°C for 5 min. The whole brain was cut into six serial coronal sections from the frontal tips using a mouse brain matrix (RWD Life Science) and were then stained with 2% TTC (Sigma-Aldrich, USA) at 37°C for 20 min and fixed with 4% paraformaldehyde. The ischemic infarct area of each section was measured using Image-Pro Plus image analysis software (version 6.0, Rockville, USA); the ischemic infarct size was calculated as follows: infarct size (%) = (contralateral area − ipsilateral non-infarct area)/contralateral area × 100%.
Neurological scores
At 24 hours of reperfusion after MCAO, neurological dysfunction was assessed by an mNSS, which contains motor tests (including flexion of forelimb, flexion of hindlimb, and head movement, scored 0 to 6), beam balance tests (scored 0 to 6), and reflexes absent and abnormal movements (scored 0 to 2). An accumulative score of 1 to 4, 5 to 9, or 10 to 14 indicates mild, moderate, or severe injury, respectively. Neurological performance was assessed by the blinded independent investigators.
MWM test
The spatial learning and memory of animals were detected using the MWM test as we have described previously (44). In brief, the water maze consisted of a circular tank 120 cm in diameter and 60 cm high filled with opaque water and a round platform (6 cm in diameter) that was submerged 1 cm beneath the surface of the water at a temperature of 22° ± 2°C. Different shapes were posted along the walls of the tank, which served as spatial reference cues. A digital tracking device (Xinruan Information Technology, Shanghai, China) was mounted above the maze to record the swimming traces in the water maze. Before the test, mice were habituated to the testing environment for 30 min. During the latency trials, submerged platform training was conducted in six consecutive days, and each session consisted of four trials. For each trial, the animal was released from the wall of the tank and allowed to search for and stand on the hidden platform within the 60-s trial period; if a mouse did not find the platform in the allotted time, it was guided to the platform and maintained on the platform for 10 s. A probe test was conducted 24 hours after the completion of training. In the test, the platform was removed, and the animal’s performance was recorded for 60 s. The latency of reaching the platform, the time spent in each quadrant, and the numbers of times crossing the platform area were recorded automatically for subsequent analysis.
Novel object recognition
The novel object recognition task was performed to examine the hippocampal-dependent memory. Before the test, the animals were acclimated to the testing room for 30 min in two consecutive days. During the training trial, animals were presented with two identical objects (familiar object) in an open-field apparatus (50 cm by 50 cm by 50 cm), placed at the rear left and right corners of the chamber, and allowed to freely explore the objects for 5 min. Thereafter, l hour later, one of the familiar objects was replaced by a novel object, and the animals were again placed into the arena for 5 min of free exploration. The time spent in exploring the familiar and novel objects was recorded using a digital video-tracking system (Xinruan Information Technology). Exploration was defined as contacting with the object (tail only excluded) or facing the object (distance < 2 cm). To analyze CA1-dependent cognition, a discrimination index was analyzed as the ratio of the amount of time spent in exploring the novel object over the total time spent in exploring the familiar and novel objects, and a higher preference for the novel object was rated as intact spatial recognition memory.
Rotarod test
The rotarod test was performed as we have previously described. Briefly, a rotating cylinder (6 cm in diameter) with a coarse surface for a firm grip was used to assess motor function. Before the test, animals were habituated to the testing environment for 30 min. Then, the mice were placed onto the central portion of the cylinder with the speed of the rotarod accelerated from 5 to 10 rpm and given a 30-min training session. Thereafter, the speed of the rotarod accelerated from 5 to 40 rpm over a 5-min period. The time at which each mouse falls from the rotating rod was automatically recorded by the detector. The procedure was repeated for four consecutive trials with a 30-min rest between each trial; the results were averaged to give the daily latency to fall for each mouse.
Statistical analyses
All data are presented as the means ± SEM from at least three independent experiments. The statistical analyses were performed using the software GraphPad Prism software (version 8.0.1, GraphPad Software Inc.). Comparisons among multiple groups were assessed using one- or two-way analysis of variance (ANOVA) test or repeated-measures ANOVA test, as indicated in the figure legends. Significant effects in ANOVAs were followed by Dunnett’s or Tukey’s post hoc multiple comparison tests. The discontinuous data of neurological deficit score and times of the animals crossing over the platform location during the probe trial on day 7 were analyzed by Kruskal-Wallis nonparametric test, followed by Dunnett’s post hoc test. All statistical details are shown in table S3. A value of P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns represents not significant).
Supplementary Material
Acknowledgments
We would like to thank Q. Zhong and X. Hou for providing the HA-p62 and mRFP-GFP-LC3 plasmids, respectively. Funding: This study was supported by funding from the National Natural Science Foundation of China (no. 31771126 to J.S. and no. 31800896 to X.L.) and the China Postdoctoral Science Foundation (no. 2018T110752 and no. 2017M620310 to X.L.). Author contributions: J.S. and X.L. conceived and designed the project. X.L., Q.X., and M.M. performed the experiments and conducted the data analysis. H.Z., Z.Z., and L.Y. helped with experiments. L.Z., Y.W., and Y.Z. provided valuable advice. X.L. and Q.X. wrote the manuscript with approval from all authors. J.S. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabc5539/DC1
REFERENCES AND NOTES
- 1.Benjamin E. J., Muntner P., Alonso A., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Das S. R., Delling F. N., Djousse L., Elkind M. S. V., Ferguson J. F., Fornage M., Jordan L. C., Khan S. S., Kissela B. M., Knutson K. L., Kwan T. W., Lackland D. T., Lewis T. T., Lichtman J. H., Longenecker C. T., Loop M. S., Lutsey P. L., Martin S. S., Matsushita K., Moran A. E., Mussolino M. E., O’Flaherty M., Pandey A., Perak A. M., Rosamond W. D., Roth G. A., Sampson U. K. A., Satou G. M., Schroeder E. B., Shah S. H., Spartano N. L., Stokes A., Tirschwell D. L., Tsao C. W., Turakhia M. P., VanWagner L. B., Wilkins J. T., Wong S. S., Virani S. S.; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee , Heart disease and stroke statistics—2019 update: A report from the American Heart Association. Circulation 139, e56–e528 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Moskowitz M. A., Lo E. H., Iadecola C., The science of stroke: Mechanisms in search of treatments. Neuron 67, 181–198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi K., Tian D. C., Li Z. G., Ducruet A. F., Lawton M. T., Shi F. D., Global brain inflammation in stroke. Lancet Neurol. 18, 1058–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Wright-Jin E. C., Gutmann D. H., Microglia as dynamic cellular mediators of brain function. Trends Mol. Med. 25, 967–979 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lan X., Han X., Li Q., Yang Q. W., Wang J., Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 13, 420–433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma Y., Wang J., Wang Y., Yang G.-Y., The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 157, 247–272 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Yang J., Zhao Y., Zhang L., Fan H., Qi C., Zhang K., Liu X., Fei L., Chen S., Wang M., Kuang F., Wang Y., Wu S., RIPK3/MLKL-mediated neuronal necroptosis modulates the M1/M2 polarization of microglia/macrophages in the ischemic cortex. Cereb. Cortex 28, 2622–2635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansari J., Kaur G., Gavins F. N. E., Therapeutic potential of annexin A1 in ischemia reperfusion injury. Int. J. Mol. Sci. 19, 1211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugimoto M. A., Vago J. P., Teixeira M. M., Sousa L. P., Annexin A1 and the resolution of inflammation: Modulation of neutrophil recruitment, apoptosis, and clearance. J. Immunol. Res. 2016, 8239258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan X., Krahling S., Smith D., Williamson P., Schlegel R. A., Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol. Biol. Cell 15, 2863–2872 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Acunto C. W., Gbelcova H., Festa M., Ruml T., The complex understanding of Annexin A1 phosphorylation. Cell. Signal. 26, 173–178 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y., Wang J., Jiang H., Yu Z., Li X., Shi J., Following OGD/R, annexin 1 nuclear translocation and subsequent induction of apoptosis in neurons are assisted by myosin IIA in a TRPM7 kinase-dependent manner. Mol. Neurobiol. 51, 729–742 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Solito E., Christian H. C., Festa M., Mulla A., Tierney T., Flower R. J., Buckingham J. C., Post-translational modification plays an essential role in the translocation of annexin A1 from the cytoplasm to the cell surface. FASEB J. 20, 1498–1500 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirata F., Thibodeau L. M., Hirata A., Ubiquitination and SUMOylation of annexin A1 and helicase activity. Biochim. Biophys. Acta 1800, 899–905 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Martin S., Wilkinson K. A., Nishimune A., Henley J. M., Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat. Rev. Neurosci. 8, 948–959 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiss-Friedlander R., Melchior F., Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Lao Y., Yang K., Wang Z., Sun X., Zou Q., Yu X., Cheng J., Tong X., Yeh E. T. H., Yang J., Yi J., DeSUMOylation of MKK7 kinase by the SUMO2/3 protease SENP3 potentiates lipopolysaccharide-induced inflammatory signaling in macrophages. J. Biol. Chem. 293, 3965–3980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Datwyler A. L., Lättig-Tünnemann G., Yang W., Paschen W., Lee S. L., Dirnagl U., Endres M., Harms C., SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J. Cereb. Blood Flow Metab. 31, 2152–2159 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K., Zhou W., Cai Q., Cheng J., Cai R., Xing R., SUMOylation of KLF4 promotes IL-4 induced macrophage M2 polarization. Cell Cycle 16, 374–381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y. J., Hallenbeck J. M., SUMO and ischemic tolerance. Neuromolecular Med. 15, 771–781 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Qiu C., Wang Y., Zhao H., Qin L., Shi Y., Zhu X., Song L., Zhou X., Chen J., Zhou H., Zhang H., Tellides G., Min W., Yu L., The critical role of SENP1-mediated GATA2 deSUMOylation in promoting endothelial activation in graft arteriosclerosis. Nat. Commun. 8, 15426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wen D., Wu J., Wang L., Fu Z., SUMOylation promotes nuclear import and stabilization of polo-like kinase 1 to support its mitotic function. Cell Rep. 21, 2147–2159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raman N., Weir E., Muller S., The AAA ATPase MDN1 acts as a SUMO-targeted regulator in mammalian pre-ribosome remodeling. Mol. Cell 64, 607–615 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Qin C., Fan W. H., Liu Q., Shang K., Murugan M., Wu L. J., Wang W., Tian D.-S., Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke 48, 3336–3346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim B. W., Koppula S., Hong S. S., Jeon S. B., Kwon J. H., Hwang B. Y., Park E. J., Choi D. K., Regulation of microglia activity by glaucocalyxin-A: Attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-κB and p38 MAPK signaling pathways. PLOS ONE 8, e55792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machado M. G., Tavares L. P., Souza G. V. S., Queiroz-Junior C. M., Ascencao F. R., Lopes M. E., Garcia C. C., Menezes G. B., Perretti M., Russo R. C., Teixeira M. M., Sousa L. P., The Annexin A1/FPR2 pathway controls the inflammatory response and bacterial dissemination in experimental pneumococcal pneumonia. FASEB J. 34, 2749–2764 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Mussbacher M., Salzmann M., Brostjan C., Hoesel B., Schoergenhofer C., Datler H., Hohensinner P., Basilio J., Petzelbauer P., Assinger A., Schmid J. A., Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front. Immunol. 10, 85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dikic I., Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 86, 193–224 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Lorente M., Garcia-Casas A., Salvador N., Martinez-Lopez A., Gabicagogeascoa E., Velasco G., Lopez-Palomar L., Castillo-Lluva S., Inhibiting SUMO1-mediated SUMOylation induces autophagy-mediated cancer cell death and reduces tumour cell invasion via RAC1. J. Cell Sci. 132, jcs234120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rozenknop A., Rogov V. V., Rogova N. Y., Lohr F., Guntert P., Dikic I., Dotsch V., Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J. Mol. Biol. 410, 477–487 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Yang Y., He Y., Wang X., Liang Z., He G., Zhang P., Zhu H., Xu N., Liang S., Protein SUMOylation modification and its associations with disease. Open Biol. 7, 170167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caron D., Maaroufi H., Michaud S., Tanguay R. M., Faure R. L., Annexin A1 is regulated by domains cross-talk through post-translational phosphorylation and SUMOYlation. Cell. Signal. 25, 1962–1969 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Yeh E. T., SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol. Chem. 284, 8223–8227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun D., Yu Z., Fang X., Liu M., Pu Y., Shao Q., Wang D., Zhao X., Huang A., Xiang Z., Zhao C., Franklin R. J., Cao L., He C., LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep. 18, 1801–1816 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taetzsch T., Levesque S., McGraw C., Brookins S., Luqa R., Bonini M. G., Mason R. P., Oh U., Block M. L., Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia 63, 423–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J., Sun Z., Jin M., Tu Y., Wang S., Yang X., Chen Q., Zhang X., Han Y., Pi R., Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-κB pathway. J. Neuroimmunol. 305, 108–114 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Liu K., Zhang L., Zhao Q., Zhao Z., Zhi F., Qin Y., Cui J., SKP2 attenuates NF-κB signaling by mediating IKKβ degradation through autophagy. J. Mol. Cell Biol. 10, 205–215 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y., Guo X., Yan W., Chen Y., Ke M., Cheng C., Zhu X., Xue W., Zhou Q., Zheng L., Wang S., Wu B., Liu X., Ma L., Huang L., Huang K., ANGPTL8 negatively regulates NF-κB activation by facilitating selective autophagic degradation of IKKγ. Nat. Commun. 8, 2164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin M.-M., Wang F., Qi D., Liu W. W., Gu C., Mao C.-J., Yang Y.-P., Zhao Z., Hu L.-F., Liu C.-F., A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front. Aging Neurosci. 10, 378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirkin V., History of the selective autophagy research: How did it begin and where does it stand today? J. Mol. Biol. 43, 3–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yona S., Kim K. W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J.-K., Tansey M. G., Microglia isolation from adult mouse brain. Methods Mol. Biol. 1041, 17–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia Q., Li X., Zhou H., Zheng L., Shi J., S100A11 protects against neuronal cell apoptosis induced by cerebral ischemia via inhibiting the nuclear translocation of annexin A1. Cell Death Dis. 9, 657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X., Zheng L., Xia Q., Liu L., Mao M., Zhou H., Zhao Y., Shi J., A novel cell-penetrating peptide protects against neuron apoptosis after cerebral ischemia by inhibiting the nuclear translocation of annexin A1. Cell Death Differ. 26, 260–275 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X., Zhao Y., Xia Q., Zheng L., Liu L., Zhao B., Shi J., Nuclear translocation of annexin 1 following oxygen-glucose deprivation-reperfusion induces apoptosis by regulating Bid expression via p53 binding. Cell Death Dis. 7, e2356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabc5539/DC1