

During passage through the human gastrointestinal tract, enterohemorrhagic Escherichia coli (EHEC) is exposed to membrane-damaging bile in the small intestine. We previously reported that EHEC treatment with a physiological bile salt mixture upregulates basRS, encoding a two-component system, and arnBCADTEF, encoding the aminoarabinose lipid A modification pathway (J.
KEYWORDS: antimicrobial resistance, bile salts, defensins, enterohemorrhagic E. coli, gastrointestinal infection, host defense peptides
ABSTRACT
During passage through the human gastrointestinal tract, enterohemorrhagic Escherichia coli (EHEC) is exposed to membrane-damaging bile in the small intestine. We previously reported that EHEC treatment with a physiological bile salt mixture upregulates basRS, encoding a two-component system, and arnBCADTEF, encoding the aminoarabinose lipid A modification pathway (J. V. Kus, A. Gebremedhin, V. Dang, S. L. Tran, A. Serbanescu, and D. Barnett Foster, J Bacteriol 193: 4509–4515, 2011, https://doi.org/10.1128/JB.00200-11). The present study examined the effect of bile salt mix (BSM) treatment on EHEC resistance to three human gastrointestinal defense peptides—HD-5, HNP-1, and LL-37—as well as the role of basRS and arnT in the respective responses. After BSM treatment, EHEC resistance to HD-5 and HNP-1 was significantly increased in a BSM-, defensin dose-dependent manner. The resistance phenotype was dependent on both basRS and arnT. However, the BSM treatment did not alter EHEC resistance to LL-37, even when the ompT gene, encoding an LL-37 cleavage protease, was disrupted. Interestingly, enteropathogenic E. coli, a related pathogen that infects the small intestine, showed a similar BSM-induced resistance phenotype. Using a model of EHEC infection in Galleria mellonella, we found significantly lower survival rates in wax moth larvae infected with BSM-treated wild-type EHEC than in those infected with a BSM-treated basS mutant, suggesting that treatment with a physiological BSM enhances virulence through a basS-mediated pathway. The results of this investigation provide persuasive evidence that bile salts typically encountered during transit through the small intestine can serve as an environmental cue for EHEC, enhancing resistance to several key host defense peptides.
INTRODUCTION
Enterohemorrhagic Escherichia coli (EHEC) is a foodborne pathogen that can cause severe infections, including bloody diarrhea that can lead to life-threatening disease, including hemolytic uremic syndrome (HUS) (1–3). The prelude to EHEC colonization of the human large intestine involves an arduous journey through the stomach and the small intestine, where the pathogen encounters acute acid stress and membrane-disrupting bile (4–8). The major components of human hepatic bile, the bile salts, range in concentration from 0.2% to 2% (wt/vol) in the small intestine (9, 10) and have been shown to disrupt bacterial membrane lipids and integral membrane proteins (11), resulting in leakage of intracellular material and bacterial killing (12, 13).
However, enteric pathogens are quite capable of resisting the deleterious effects of bile. In fact, Salmonella, Helicobacter, E. coli, and Campylobacter have all been isolated from the gallbladders of humans or animals (14–16). Bile tolerance requires, but is not limited to, the proteins that maintain cell envelope composition and structure and those that preserve intracellular homeostasis by extruding bile (9). The best-characterized mechanism of bile resistance in Gram-negative bacteria depends on the expression of efflux pumps, including AcrAB, which actively expel bile (9, 17). Pathogens also employ enzymatic action to modify or transform bile salts to a less harmful form (5).
A number of pathogens have also been reported to use bile salts as cues to enhance fitness and modulate virulence factor expression (18–25). In some cases, pathogens downregulate selected virulence genes, including the example of bile salt-induced downregulation of invasion genes in Salmonella (22). In other cases, virulence gene expression is upregulated in response to bile salt mix (BSM) treatment, as in the case of Vibrio cholerae bile salt-induced upregulation of flagellar genes and motility (24–26), which may benefit the pathogen by promoting transit toward the preferred colonization site.
We showed previously, by using transcriptome analysis, that exposure of EHEC to a physiological BSM upregulates acrAB, encoding an efflux pump, as well as basRS (also known as pmrAB), encoding a two-component system, and genes involved in the modification of lipid A with aminoarabinose (arnBCADTEF and ugd) (27). We also found that BSM treatment enhances EHEC resistance to the bacterial antimicrobial lipopeptide polymyxin B (PMB) in a basS- and arnT-dependent manner. That study was the first to suggest that EHEC “senses” bile and responds with both protective and virulence strategies.
The mechanism of killing of Gram-negative pathogens by cationic antimicrobials like PMB is not fully understood, but it is thought that PMB initially binds to the anionic lipopolysaccharide, which allows the PMB to access to the bacterial inner membrane (28). Modifications of lipid A that neutralize the anionic charge, including modification with 4-amino-4-deoxy-l-arabinose (l-Ara4N) have been shown to increase resistance of EHEC and other Gram-negative pathogens to PMB (27, 29). Using a similar mode of action, host defense peptides (HDPs) play an important role in the human innate immune system by exhibiting broad-spectrum antimicrobial activity against both Gram-positive and Gram-negative bacteria (30). HDPs are peptides of 12 to 40 amino acids with a net positive charge ranging from +2 to +7 that are thought to bind to bacterial membranes through electrostatic interaction, resulting in membrane disruption and bacterial killing (31). Resistance to HDPs include alteration of bacterial surfaces, activation of efflux pumps, proteolytic degradation of HDPs, stimulation of bacterial regulatory networks, and even alteration of host processes (9, 32). Studies have shown that modification of lipid A with l-Ara4N in Pseudomonas, Salmonella, and E. coli increases resistance to common HDPs and PMB (32–36). OmpT protease, known to be expressed in EHEC and marginally in enteropathogenic E. coli (EPEC) and uropathogenic E. coli, has been shown to cleave the cathelicidin LL-37, a cationic antimicrobial protein (CAMP) encountered primarily in the large intestinal lumen (37, 38). In fact, OmpT-mediated proteolysis of LL-37 is considered one of the primary resistance mechanisms of EHEC against LL-37 (39). (40).
Induction of protective mechanisms against HDPs has been shown for a number of enteric pathogens to be triggered by exposure to HDPs themselves as well as changes in divalent cation concentrations and in culture pH (9, 41, 42). While BSM induce EHEC resistance to PMB, the physiological relevance of EHEC’s BSM-induced PMB resistance for human infection is not clear. The present study examined the effect of BSM treatment on EHEC resistance to several human HDPs, including human defensins HD-5, and HNP-1, as well as cathelicidin LL-37. The results demonstrate that a physiological BSM induces an arnT- and basRS-dependent EHEC resistance to HD-5 and HNP-1 but not to LL-37. This phenotype is preserved for two different strains of EHEC as well as EPEC. We also evaluated the role of basS and BSM treatment in a Galleria mellonella model of EHEC infection.
RESULTS
BSM treatment enhances resistance of EHEC and EPEC to HD-5 and HNP-1.
Radial diffusion assays demonstrate that treatment with a 1.5% BSM significantly enhances resistance of the wild-type (WT) EHEC strains 86-24 and EDL933 to HD-5 (Fig. 1A and B). As the HD-5 concentration was increased, the size of the clear zone increased for the untreated EHEC as expected, due to the increased sensitivity at higher HD-5 concentrations. However, for the BSM-treated EHEC strain, the size of the clear zone is significantly lower than that for the corresponding untreated EHEC strain at both 0.11 μg/μl and 0.33 μg/μl HD-5, indicating that BSM treatment induces resistance to HD-5. Results also indicate that EHEC resistance is dependent on the concentration of BSM (data not shown). A similar BSM-induced phenotype was achieved with EHEC 86-24 and another small-intestinal CAMP, HNP-1 (Fig. 1C). Finally, we were also able to achieve the same results with the related attaching and effacing pathogen EPEC E2348/69 with both HD-5 and HNP-1 (Fig. 1D and E, respectively).
FIG 1.

Bile salt treatment enhances resistance of EHEC and EPEC to HD-5 and HNP-1. Radial diffusion assays show resistance as a function of clear-zone size with various concentrations of host defense peptides. Clear zones show extent of killing of EHEC 86-24 with HD-5 (A), EHEC EDL933 with HD-5 (B), EHEC 86-24 with HNP-1 (C), EPEC E2348/69 with HD-5 (D), and EPEC E2348/64 with HNP-1 (E), where bacteria are cultured in the absence (black bars) or the presence (gray bars) of 1.5% BSM. Results are expressed as means and standard errors of the means (3 biological replicates, 3 technical replicates). Statistical analysis is 2× ANOVA with post hoc Tukey's multiple-comparison test. **, P < 0.01; *, P < 0.05.
The two-component system BasRS plays a key role in BSM-induced HD-5 and HNP-1 resistance.
Since the two-component system BasRS has been shown to play a role in BSM-induced resistance of EHEC to PMB (27), we wanted to assess the role of these proteins in the HD-5 and HNP-1 resistance phenotypes. Disruption of either basS or basR resulted in loss of the BSM-induced HD-5 resistance phenotype, and complementation with basS or basR restored it, suggesting that basS and basR play key roles in the mediating the BSM-induced HD-5 resistance (Fig. 2A). The same results were also evident for the HNP-1 resistance phenotype (Fig. 2B), confirming the importance of the roles of basS and basR in mediating the BSM-induced resistance to HNP-1.
FIG 2.
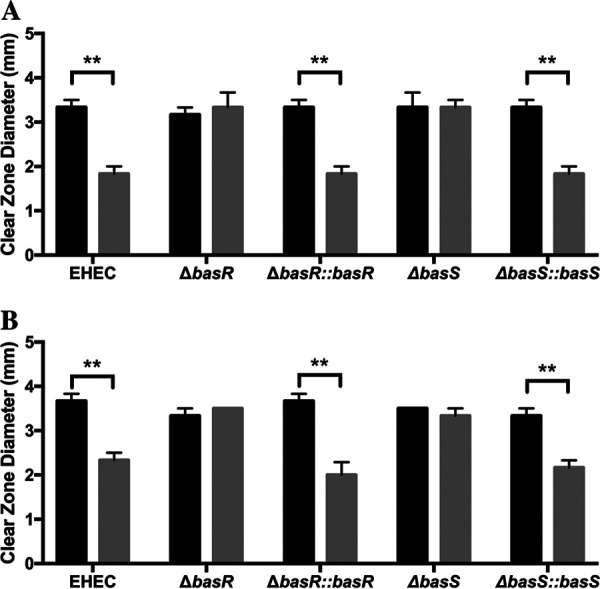
BSM-induced resistance to HD-5 and HNP-1 depends on basS and basR. Clear-zone sizes show the extent of HD-5 (0.11 μg/μl) (A) and HNP-1 (0.11 μg/μl) (B) killing of EHEC 86-24, EHEC ΔbasS, EHEC ΔbasR, and complemented strains cultured in the absence (black bars) or the presence (gray bars) of 1.5% BSM. Results are expressed as means and standard errors of the means (3 biological replicates, 3 technical replicates). Statistical analysis was 2× ANOVA with post hoc Tukey's multiple-comparison test. **, P < 0.01.
Aminoarabinose modification of lipid A is involved in the BSM-induced resistance phenotype.
Lipid A modification with aminoarabinose is mediated by enzymes encoded in the arn operon, with the final enzyme encoded by arnT. Transcriptome analysis of BSM-treated versus untreated EHEC previously showed significant upregulation of arnT after BSM treatment and arnT was found to play a role in the BSM-induced resistance of EHEC to PMB (27). Consequently, we asked whether arnT could also play a role in EHEC resistance to HD-5. When arnT was disrupted, the BSM-induced resistance phenotype was lost in an HD-5 dose-dependent manner and was restored in the arnT-complemented arnT mutant (Fig. 3A). Similarly when arnT was disrupted, the BSM-induced resistance to HNP-1 was also lost, and complementation restored the WT phenotype (Fig. 3B). Finally, we also demonstrated that killing of the arnT mutant was dependent on the concentration of HDP, regardless of the induction with BSM (Fig. 3C). These data support the integral role of arnT in BSM-induced resistance of EHEC to HD-5 and HNP-1.
FIG 3.

BSM-induced resistance to HD-5 and HNP-1 is lost in the ΔarnT strain and restored in the complemented strain. Clear-zone sizes show the extent of HD-5 (0.11 μg/μl) (A) and HNP-1 (0.11 μg/μl) killing of EHEC 86-24, ΔarnT, and ΔarnT::arnT and HNP-1 dose-dependent killing of EHEC ΔarnT (C). All strains were cultured in the absence (black bars) or the presence (gray bars) of 1.5% BSM. Results are expressed as means and standard errors of the means (3 biological replicates, 3 technical replicates). Statistical analysis is 2× ANOVA with post hoc Tukey's multiple-comparison test. *, P < 0.05.
BSM treatment does not enhance EHEC resistance to LL-37.
In contrast to the HD-5 and HNP-1 results, BSM treatment of EHEC did not enhance the resistance to the human cathelicidin, LL-37 (Fig. 4A). This was confirmed for two different concentrations of LL-37, namely, 0.5 and 1.0 μg/ml (the latter shown in Fig. 4A). When arnT was disrupted, there was a significant decrease in resistance of the arnT mutant relative to WT but only for the higher concentration of LL-37 tested, and as with the WT, there was no change in the resistance of the arnT mutant after BSM treatment (Fig. 4A). Complementation of arnT in the arnT mutant restored the level of EHEC WT resistance, and similar to what was seen with the WT, BSM induction did not increase resistance of the complemented mutant to LL-37. These results indicate, for the first time, that arnT plays a role in resistance of untreated EHEC to LL-37 but does not offer any additional resistance after bile salt treatment.
FIG 4.

BSM treatment does not induce resistance to LL-37. Clear-zone sizes show extent of LL-37 (1.0 μg/ml) killing of EHEC 86-24 WT, ΔarnT, and ΔarnT::arnT (A) and EHEC EDL933, the ompT mutant (EDL 933), and EPEC E2348/69 (B), where bacteria were cultured in the absence (black bars) or the presence (gray bars) of 1.5% BSM. Results are expressed as means and standard errors of the means (at least 3 biological replicates, 3 technical replicates). Statistical analysis was 2× ANOVA with post hoc Tukey's multiple-comparison test, *, P < 0.05; ns, not significant.
Since OmpT has been reported to play a significant role in EHEC resistance to LL-37 (39), we considered the possibility that OmpT-mediated resistance masked an increase in resistance afforded by the BSM treatment. However, when ompT was disrupted, while there was a decrease in resistance to LL-37 in the untreated ompT mutant, BSM treatment did not alter the level of resistance (Fig. 4B), indicating that even in the absence of ompT, the BSM treatment does not enhance EHEC resistance to LL-37. It is possible that the BSM treatment is not able to induce sufficient modification of lipopolysaccharide (LPS) to register a measurable change in LL-37 resistance. Interestingly, the same phenotype was observed for EPEC (Fig. 4B).
Role of basS in BSM-mediated virulence in a Galleria mellonella model of infection.
Using a Galleria mellonella model of EHEC infection, we evaluated the role of basS and the impact of BSM treatment in EHEC virulence as measured by survival of Galleria mellonella larvae after infection. When we compared infection with WT EHEC versus the isogenic basS mutant, there was a significantly higher survival of larvae infected with the mutant but only at the highest inoculum, suggesting that basS plays a partial role in the virulence of untreated EHEC in this model (Fig. 5). However, when we compared infection with BSM-treated WT EHEC to infection with BSM-treated basS mutant, there was a dramatic and significant difference across all times points and for two levels of inoculum (Fig. 6). These results suggest that basS plays a critical role in BSM-mediated induction of EHEC virulence during infection and is consistent with the in vitro findings of its importance in resistance to several HDPs. Interestingly, there was no significant survival difference between larvae infected with untreated EHEC and those infected with BSM-treated EHEC (results not shown). These results could suggest either that the differences were too small to be evident in this model or that the model may not be optimal for assessing these differences in virulence associated with changes in resistance to host defense peptides.
FIG 5.

Survival of Galleria mellonella larvae after injection with PBS or suspensions of 108 (A), 107 (B), and 106 (C) cells of EHEC 86-24 or the ΔbasS mutant. Results are representative of 3 biological replicates. Statistical analysis of Kaplan-Meier curves used the log-rank test. *, P < 0.05 for WT versus mutant.
FIG 6.

Survival of Galleria mellonella larvae after injection with PBS or suspensions of 108 (A), 107 (B), and 106 (C) BSM-treated bacterial cells. EHEC 86-24 and EHEC ΔbasS were grown in M9 medium and subcultured in the same medium with 1.5% BSM. Results are representative of 3 biological replicates. Statistical analysis of Kaplan-Meier curves used the log-rank test. ****, P < 0.0001, and ***, P < 0.001, for WT versus mutant.
DISCUSSION
Enteric pathogens encounter membrane-damaging bile salts upon entering the small intestine, and research now suggests that a number of them use bile salts as cues to enhance fitness and modulate virulence factor expression (18–25). The results of the present study suggest that bile salts trigger a protective mechanism in EHEC against another local antimicrobial assault by CAMPs within the same microenvironment. These findings demonstrate that physiologically relevant mixes of bile salts, typically encountered during transit through the small intestine, enhance EHEC O157:H7 resistance to the human alpha defensins HD-5 and HNP-1. HD-5 is typically expressed in Paneth cells and some villous epithelial cells in normal human duodenum, jejunum, and ileum, while HNP-1 is expressed in sparse lamina propria neutrophils (43). The resistance phenotype is dependent on the BSM-induced expression of the two-component system genes basRS and their regulon, the arn operon, which mediates modification of lipid A with l-Ara4N (27). The BSM-induced resistance was evident for two different strains of EHEC O157:H7 (namely, 86-24 and EDL933) and also for the related attaching and effacing pathogen EPEC E2348/69.
Interestingly, we did not see increased resistance of BSM-treated EHEC to the cathelicidin LL-37. This was initially surprising, since electrostatic repulsion of CAMPs through modification of LPS charge has long been thought to be a primary pathogen resistance mechanism. However, recent research suggests that a myriad of different mechanisms, including other modifications of lipid A (acylation and lipooligosaccharide core heptose 1 addition), modulation of capsule expression, and omptin protease-mediated cleavage of CAMPs, can enhance HDP resistance (35, 39, 40, 44). Since ompT has already been shown to make an important contribution to EHEC resistance against LL-37 (39), we also assessed BSM-induced resistance of the ompT-disrupted isogenic mutant. While we were able to demonstrate that ompT played a role in EHEC resistance to LL-37, there was no BSM-induced increase in LL-37 resistance in the mutant.
Since we know that the arn operon is upregulated after BSM treatment of EHEC (27) and that arnT plays a role in PMB resistance (45), we expected that it could play a role in EHEC resistance to LL-37 and might also serve to enhance resistance after bile salt treatment. Our findings reveal that arnT does play a role in LL-37 resistance but that bile salt treatment does not alter that resistance, suggesting that the BSM-induced modification of lipid A with l-Ara4N is not sufficient to alter resistance to LL-37. Nevertheless, it is sufficient to enhance resistance to the both HD-5 and HNP-1. Interestingly, there are significant differences in the structure and net charge of HD-5 and HNP-1 relative to LL-37. HD-5 and HNP-1 have net charges of +4 and +3, respectively, while LL-37 has a net charge of +6, and unlike HD-5 and HNP-1, LL-37 presents its positive charges along one face of its alpha-helical structure (31). This difference in net charge and structure may play a role in the differences in BSM-induced resistance. However, it is also possible that other differences in resistance mechanisms may be involved. Interestingly, when mice deficient for CRAMP (a cathelicidin-related murine CAMP) were challenged with Ara4N-deficient Salmonella enterica serovar Typhimurium, there was no increase in attenuation relative to the wild-type mice, suggesting that CRAMP alone is not the major mediator of clearance of Ara4N-deficient S. Typhimurium (46). However, it is clear from our data that arnT is important for full LL-37 resistance, a novel finding for EHEC.
EHEC resistance to human defensins provides an important contribution to survival and fitness during transit through the human gastrointestinal (GI) tract. The employment of bile salt exposure which occurs during EHEC transit through the small intestine as a cue for upregulation of mechanisms to enhance resistance to human defensins also encountered in this same environment is a rational strategy for this pathogen. Since we have shown that basS plays an important role in the enhancement of the BSM-induced resistance phenotype of EHEC against both PMB (27) and HD-5 and HNP-1 (this study), we expected that it would play a role in an in vivo model. The results from the Galleria mellonella infection model study support this interpretation to the extent that there is a significant, dramatic difference in survival of larvae infected with BSM-treated WT EHEC versus a BSM-treated basS mutant. However, future studies to extend and confirm these findings in a small-animal model of infection would be most valuable.
In conclusion, this study clearly demonstrates that exposure of EHEC to a physiologically relevant mix of bile salts enhances resistance to the human defensins HD-5 and HNP-1 in a basS- and arnT-dependent manner and suggests that this phenotype plays an important role in survival and fitness during infection.
MATERIALS AND METHODS
Strains and culture conditions.
A list of bacterial strains used in this study is provided in Table 1. Bacteria were maintained as glycerol stocks (stored at −80°C) and were routinely streaked onto Luria-Bertani (LB) agar plates (tryptone, 1% [wt/vol]; yeast, 0.5% [wt/vol]; sodium chloride, 0.5% [wt/vol]; agar 1.5% [wt/vol]) with appropriate antibiotics where necessary and grown in a 37°C static incubator for 16 to 18 h. For all experimental work, bacteria were grown overnight either in LB broth or N minimal medium with 0.2% glucose and 1 mM MgCl2 with the appropriate antibiotics at 37°C for 16 to 18 h at 200 rpm and then subcultured in the same culture media statically at 37°C with 5% CO2 to mid-log phase. The subculture growth conditions are intended to represent physiologically relevant microaerobic conditions (47).
TABLE 1.
Bacterial strains used in this study
| Strain | Descriptiona | Source or reference |
|---|---|---|
| 86-24 | WT EHEC O157:H7 strain 86-24 | 49 |
| 86-24 ΔbasS | 86-24 with Kanr disruption of basS gene; Kanr50 | 27 |
| 86-24 ΔbasS::basS | 86-24 with Kanr disruption of basS gene transformed with pBADGr containing basS gene; Kanr50 Genr20 | This study |
| 86-24 ΔbasR | 86-24 with Kanr disruption of basR gene; Kanr50 | This study |
| 86-24 ΔbasR::basR | 86-24 with Kanr disruption of basR gene transformed with pBADGr containing basR gene; Kanr50 Genr20 | This study |
| 86-24 ΔarnT | 86-24 with Kanr disruption of arnT gene; Kanr50 | 27 |
| 86-24 ΔarnT::arnT | 86-24 transformed with vector pBADGr containing arnT gene; Kanr50 Genr20 | This study |
| EDL 933 | WT EHEC O157:H7 strain EDL 933 | 50 |
| EDL 933 ΔompT | EDL 933 ΔompT mutant created by sacB gene-based allelic exchange | 39 |
| EPEC E2348/69 | WT typical EPEC strain (O127:H6) | 51 |
Kanr50, kanamycin resistance at 50 μg/ml; Genr20, gentamycin resistance at 20 μg/ml.
BSM and peptide suspensions and concentrations.
Bile salt mixture (BSM) (B-3426; Sigma-Aldrich) at a final concentration of either 0.15% or 1.5% (wt/vol) was used as a treatment in all assays, as previously described (27). Human defensins HD-5 and HNP-1, generously provided by Wuyuan Lu of the University of Maryland, were solubilized in 0.01% acetic acid and 0.2% bovine serum albumin in 0.01% acetic acid, respectively. LL-37 (Anaspec Peptide) was suspended in water to a stock concentration of 64 μg/ml. Stock concentrations of peptides were stored at −20°C.
Radial diffusion assay.
A modified version of a radial diffusion assay was used to determine the effect of BSM on the survival of EHEC 86-24 when challenged with antimicrobial peptide (48). Briefly, overnight cultures grown in LB with or without 1.5% BSM with shaking were diluted into fresh medium with or without BSM and incubated at 37°C and 5% CO2 under static conditions to mid-log phase. Subcultures were washed and resuspended in 10 mM sodium phosphate buffer (NAPB), pH 7.4. A portion (4 × 106 CFU) of each sample was inoculated into 10 ml of low-nutrient agarose (10 mM NAPB, 0.03% [wt/vol] tryptic soy broth [Sigma], 1% [wt/vol] agarose type I [low electroendosmosis; A-6013; Sigma-Aldrich], 0.2% [vol/vol] Tween [Sigma]), dispersed with gentle vortexing, and poured into 100- by 25-mm petri dishes. A 3-μl sample of antimicrobial peptide was allowed to diffuse from a center well in the agarose at 37°C for 3 h. Duplicate wells were made for each concentration of peptide. The lower agarose layer of each plate was then covered with 10 ml of overlay agar, which was allowed to solidify. Plates were inverted and incubated at 37°C for no more than 16 h. The zone of clearing around each well was measured, providing a measure of the extent of killing of the bacteria with the specific antimicrobial peptide.
Galleria mellonella model of infection.
Galleria mellonella larvae were obtained from The Worm Lady. All larvae were used the same day. Bacteria were routinely cultured M9 medium with 0.4% glucose and 200 μM MgSO4 with the appropriate antibiotics at 37°C for 16 to 18 h at 200 rpm and then subcultured in the same culture media. For infections, 1 ml of each culture was harvested by centrifugation and washed three times in 1× phosphate-buffered saline (PBS) (pH 7.4). The final, washed bacterial pellet was resuspended in 1 ml 1× PBS (pH 7.4) and adjusted to the designated number of CFU, which was confirmed by plate counting for each sample. Larvae were infected with one selected strain by injection into the hemolymph via the hindmost right proleg. For all experiments, larvae were incubated at 37°C in standard petri dishes for up to 72 h. All experiments were conducted at least in triplicate. Control larvae were injected with PBS (pH 7.4) to measure any lethal effects of the injection process. All organisms were monitored for survival for up to 72 h. Larvae were considered dead when they did not show any response to touch. Ten worms each were injected with the same sample, and each treatment result was based on at least three biological replicates.
Statistical analysis.
Results are presented as means and standard errors of the means. To test statistical significance among the groups, one- or two-way analysis of variance (ANOVA) was used, followed by post hoc comparisons with Tukey’s method. Statistical analyses for G. mellonella infections were carried out with Kaplan-Meier curves using the log-rank test.
ACKNOWLEDGMENTS
This work was supported by an NSERC Discovery Grant (no. 05220) to Debora Barnett Foster.
Wuyuan Lu of the School of Medicine, University of Maryland is acknowledged for the generous donation of human defensin HD-5 and HNP-1. Charles Bevin of the Department of Medical Microbiology and Immunology, University of California, is acknowledged for his advice on the radial diffusion assays.
REFERENCES
- 1.Karpman D, Ståhl A. 2014. Enterohemorrhagic Escherichia coli pathogenesis and the host response, p 403–417. In Sperandio V, Hovde CJ (ed), Enterohemorrhagic Escherichia coli and other Shiga toxin-producing E. coli. ASM Press, Washington, DC. [DOI] [PubMed] [Google Scholar]
- 2.Page AV, Liles WC. 2013. Enterohemorrhagic Escherichia coli infections and the hemolytic-uremic syndrome. Med Clin North Am 97:681–695. doi: 10.1016/j.mcna.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Karmali MA. 2004. Infection by Shiga toxin-producing Escherichia coli: an overview. Mol Biotechnol 26:117–122. doi: 10.1385/MB:26:2:117. [DOI] [PubMed] [Google Scholar]
- 4.Barnett Foster D. 2013. Modulation of the enterohemorrhagic E. coli virulence program through the human gastrointestinal tract. Virulence 4:315–323. doi: 10.4161/viru.24318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Begley M, Gahan CG, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Griffin PM. 1995. Escherichia coli O157:H7 and other enterohemorrhagic Escherichia coli, p 739–761. In Blaser MJ, Smith PD, Ravdin JI, Greenberg HB, Guerrant RL (ed), Infections of the gastrointestinal tract. Raven Press, Ltd, New York, NY. [Google Scholar]
- 7.Jubelin G, Desvaux M, Schüller S, Etienne-Mesmin L, Muniesa M, Blanquet-Diot S. 2018. Modulation of enterohaemorrhagic Escherichia coli survival and virulence in the human gastrointestinal tract. Microorganisms 6:115. doi: 10.3390/microorganisms6040115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.House B, Kus JV, Prayitno N, Mair R, Que L, Chingcuanco F, Gannon V, Cvitkovitch DG, Barnett Foster D. 2009. Acid-stress-induced changes in enterohaemorrhagic Escherichia coli O157:H7 virulence. Microbiology (Reading) 155:2907–2918. doi: 10.1099/mic.0.025171-0. [DOI] [PubMed] [Google Scholar]
- 9.Gunn JS. 2000. Mechanisms of bacterial resistance and response to bile. Microbes Infect 2:907–913. doi: 10.1016/s1286-4579(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 10.Pumbwe L, Skilbeck CA, Nakano V, Avila-Campos MJ, Piazza RM, Wexler HM. 2007. Bile salts enhance bacterial co-aggregation, bacterial-intestinal epithelial cell adhesion, biofilm formation and antimicrobial resistance of Bacteroides fragilis. Microb Pathog 43:78–87. doi: 10.1016/j.micpath.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Coleman R, Lowe PJ, Billington D. 1980. Membrane lipid composition and susceptibility to bile salt damage. Biochim Biophys Acta 599:294–300. doi: 10.1016/0005-2736(80)90075-9. [DOI] [PubMed] [Google Scholar]
- 12.Fujisawa T, Mori M. 1997. Influence of various bile salts on beta-glucuronidase activity of intestinal bacteria. Lett Appl Microbiol 25:95–97. doi: 10.1046/j.1472-765x.1997.00180.x. [DOI] [PubMed] [Google Scholar]
- 13.Noh DO, Gilliland SE. 1993. Influence of bile on cellular integrity and beta-galactosidase activity of Lactobacillus acidophilus. J Dairy Sci 76:1253–1259. doi: 10.3168/jds.S0022-0302(93)77454-8. [DOI] [PubMed] [Google Scholar]
- 14.Prouty AM, Brodsky IE, Manos J, Belas R, Falkow S, Gunn JS. 2004. Transcriptional regulation of Salmonella enterica serovar Typhimurium genes by bile. FEMS Immunol Med Microbiol 41:177–185. doi: 10.1016/j.femsim.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Fox JG, Otto G, Murphy JC, Taylor NS, Lee A. 1991. Gastric colonization of the ferret with Helicobacter species: natural and experimental infections. Rev Infect Dis 13:S671–S680. doi: 10.1093/clinids/13.Supplement_8.S671. [DOI] [PubMed] [Google Scholar]
- 16.Flores C, Maguilnik I, Hadlich E, Goldani LZ. 2003. Microbiology of choledochal bile in patients with choledocholithiasis admitted to a tertiary hospital. J Gastroenterol Hepatol 18:333–336. doi: 10.1046/j.1440-1746.2003.02971.x. [DOI] [PubMed] [Google Scholar]
- 17.Prouty AM, Brodsky IE, Falkow S, Gunn JS. 2004. Bile-salt-mediated induction of antimicrobial and bile resistance in Salmonella typhimurium. Microbiology (Reading) 150:775–783. doi: 10.1099/mic.0.26769-0. [DOI] [PubMed] [Google Scholar]
- 18.Prouty AM, Gunn JS. 2000. Salmonella enterica serovar typhimurium invasion is repressed in the presence of bile. Infect Immun 68:6763–6769. doi: 10.1128/iai.68.12.6763-6769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malik-Kale P, Parker CT, Konkel ME. 2008. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J Bacteriol 190:2286–2297. doi: 10.1128/JB.01736-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pace JL, Chai TJ, Rossi HA, Jiang X. 1997. Effect of bile on Vibrio parahaemolyticus. Appl Environ Microbiol 63:2372–2377. doi: 10.1128/AEM.63.6.2372-2377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torres AG, Tutt CB, Duval L, Popov V, Nasr AB, Michalski J, Scaletsky IC. 2007. Bile salts induce expression of the afimbrial LDA adhesin of atypical enteropathogenic Escherichia coli. Cell Microbiol 9:1039–1049. doi: 10.1111/j.1462-5822.2006.00850.x. [DOI] [PubMed] [Google Scholar]
- 22.Eade CR, Hung C-C, Bullard B, Gonzalez-Escobedo G, Gunn JS, Altier C. 2016. Bile acids function synergistically to repress invasion gene expression in Salmonella by destabilizing the invasion regulator HilD. Infect Immun 84:2198–2208. doi: 10.1128/IAI.00177-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pope LM, Reed KE, Payne SM. 1995. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect Immun 63:3642–3648. doi: 10.1128/IAI.63.9.3642-3648.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65:1131–1134. doi: 10.1128/IAI.65.3.1131-1134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wibbenmeyer JA, Provenzano D, Landry CF, Klose KE, Delcour AH. 2002. Vibrio cholerae OmpU and OmpT porins are differentially affected by bile. Infect Immun 70:121–126. doi: 10.1128/iai.70.1.121-126.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nesper J, Lauriano CM, Klose KE, Kapfhammer D, Kraiss A, Reidl J. 2001. Characterization of Vibrio cholerae O1 El tor galU and galE mutants: influence on lipopolysaccharide structure, colonization, and biofilm formation. Infect Immun 69:435–445. doi: 10.1128/IAI.69.1.435-445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kus JV, Gebremedhin A, Dang V, Tran SL, Serbanescu A, Foster DB. 2011. Bile salts induce resistance to polymyxin in enterohemorrhagic Escherichia coli O157:H7. J Bacteriol 193:4509–4515. doi: 10.1128/JB.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaara M, Viljanen P. 1985. Binding of polymyxin B nonapeptide to gram-negative bacteria. Antimicrob Agents Chemother 27:548–554. doi: 10.1128/aac.27.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maria-Neto S, de Almeida KC, Macedo MLR, Franco OL. 2015. Understanding bacterial resistance to antimicrobial peptides: from the surface to deep inside. Biochim Biophys Acta 1848:3078–3088. doi: 10.1016/j.bbamem.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 30.De Smet K, Contreras R. 2005. Human antimicrobial peptides: defensins, cathelicidins and histatins. Biotechnol Lett 27:1337–1347. doi: 10.1007/s10529-005-0936-5. [DOI] [PubMed] [Google Scholar]
- 31.Jenssen H, Hamill P, Hancock REW. 2006. Peptide antimicrobial agents. Clin Microbiol Rev 19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moskowitz SM, Ernst RK, Miller SI. 2004. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol 186:575–579. doi: 10.1128/jb.186.2.575-579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raetz CRH, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem 76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan A, Guan Z, Raetz CRH. 2007. An undecaprenyl phosphate-aminoarabinose flippase required for polymyxin resistance in Escherichia coli. J Biol Chem 282:36077–36089. doi: 10.1074/jbc.M706172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunn JS. 2008. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol 16:284–290. doi: 10.1016/j.tim.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Kim S-H, Jia W, Parreira VR, Bishop RE, Gyles CL. 2006. Phosphoethanolamine substitution in the lipid A of Escherichia coli O157: H7 and its association with PmrC. Microbiology (Reading) 152:657–666. doi: 10.1099/mic.0.28692-0. [DOI] [PubMed] [Google Scholar]
- 37.Thomassin JL, Brannon JR, Kaiser J, Gruenheid S, Le Moual H. 2012. Enterohemorrhagic and enteropathogenic Escherichia coli evolved different strategies to resist antimicrobial peptides. Gut Microbes 3:556–561. doi: 10.4161/gmic.21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brannon JR, Burk DL, Leclerc J-M, Thomassin J-L, Portt A, Berghuis AM, Gruenheid S, Le Moual H. 2015. Inhibition of outer membrane proteases of the omptin family by aprotinin. Infect Immun 83:2300–2311. doi: 10.1128/IAI.00136-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomassin JL, Brannon JR, Gibbs BF, Gruenheid S, Le Moual H. 2012. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect Immun 80:483–492. doi: 10.1128/IAI.05674-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomassin J-L, Lee MJ, Brannon JR, Sheppard DC, Gruenheid S, Le Moual H. 2013. Both group 4 capsule and lipopolysaccharide O-antigen contribute to enteropathogenic Escherichia coli resistance to human α-defensin 5. PLoS One 8:e82475. doi: 10.1371/journal.pone.0082475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Withee J, Williams M, Disney T, Schlosser W, Bauer N, Ebel E. 2009. Streamlined analysis for evaluating the use of preharvest interventions intended to prevent Escherichia coli O157:H7 illness in humans. Foodborne Pathog Dis 6:817–825. doi: 10.1089/fpd.2008.0255. [DOI] [PubMed] [Google Scholar]
- 42.Bauer ME, Shafer WM. 2015. On the in vivo significance of bacterial resistance to antimicrobial peptides. Biochim Biophys Acta 1848:3101–3111. doi: 10.1016/j.bbamem.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunliffe RN. 2003. Alpha-defensins in the gastrointestinal tract. Mol Immunol 40:463–467. doi: 10.1016/s0161-5890(03)00157-3. [DOI] [PubMed] [Google Scholar]
- 44.Nishino K, Hsu F-F, Turk J, Cromie MJ, Wosten MMSM, Groisman EA. 2006. Identification of the lipopolysaccharide modifications controlled by the Salmonella PmrA/PmrB system mediating resistance to Fe(III) and Al(III). Mol Microbiol 61:645–654. doi: 10.1111/j.1365-2958.2006.05273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrera CM, Hankins JV, Trent MS. 2010. Activation of PmrA inhibits LpxT-dependent phosphorylation of lipid A promoting resistance to antimicrobial peptides. Mol Microbiol 76:1444–1460. doi: 10.1111/j.1365-2958.2010.07150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strandberg KL, Richards SM, Tamayo R, Reeves LT, Gunn JS. 2012. An altered immune response, but not individual cationic antimicrobial peptides, is associated with the oral attenuation of Ara4N-deficient Salmonella enterica serovar Typhimurium in mice. PLoS One 7:e49588. doi: 10.1371/journal.pone.0049588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaines JM, Carty NL, Colmer-Hamood JA, Hamood AN. 2005. Effect of static growth and different levels of environmental oxygen on toxA and ptxR expression in the Pseudomonas aeruginosa strain PAO1. Microbiology (Reading) 151:2263–2275. doi: 10.1099/mic.0.27754-0. [DOI] [PubMed] [Google Scholar]
- 48.Lehrer RI, Rosenman M, Harwig SS, Jackson R, Eisenhauer P. 1991. Ultrasensitive assays for endogenous antimicrobial polypeptides. J Immunol Methods 137:167–173. doi: 10.1016/0022-1759(91)90021-7. [DOI] [PubMed] [Google Scholar]
- 49.Karmali MA, Mascarenhas M, Shen S, Ziebell K, Johnson S, Reid-Smith R, Isaac-Renton J, Clark C, Rahn K, Kaper JB. 2003. Association of genomic O island 122 of Escherichia coli EDL 933 with verocytotoxin-producing Escherichia coli seropathotypes that are linked to epidemic and/or serious disease. J Clin Microbiol 41:4930–4940. doi: 10.1128/JCM.41.11.4930-4940.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riley LW, Remis RS, Helgerson SD, McGee HB, Wells JG, Davis BR, Hebert RJ, Olcott ES, Johnson LM, Hargrett NT, Blake PA, Cohen MC. 1983. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N Engl J Med 308:681–685. doi: 10.1056/NEJM198303243081203. [DOI] [PubMed] [Google Scholar]
- 51.Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S. 1978. Escherichia coli strains that cause diarrhea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i:1119–1122. doi: 10.1016/s0140-6736(78)90299-4. [DOI] [PubMed] [Google Scholar]