

Abstract
Background
The NIH Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability (STRIDES) initiative provides NIH-funded researchers cost-effective access to commercial cloud providers, such as Amazon Web Services (AWS) and Google Cloud Platform (GCP). These cloud providers represent an alternative for the execution of large computational biology experiments like transcriptome annotation, which is a complex analytical process that requires the interrogation of multiple biological databases with several advanced computational tools. The core components of annotation pipelines published since 2012 are BLAST sequence alignments using annotated databases of both nucleotide or protein sequences almost exclusively with networked on-premises compute systems.
Findings
We compare multiple BLAST sequence alignments using AWS and GCP. We prepared several Jupyter Notebooks with all the code required to submit computing jobs to the batch system on each cloud provider. We consider the consequence of the number of query transcripts in input files and the effect on cost and processing time. We tested compute instances with 16, 32, and 64 vCPUs on each cloud provider. Four classes of timing results were collected: the total run time, the time for transferring the BLAST databases to the instance local solid-state disk drive, the time to execute the CWL script, and the time for the creation, set-up, and release of an instance. This study aims to establish an estimate of the cost and compute time needed for the execution of multiple BLAST runs in a cloud environment.
Conclusions
We demonstrate that public cloud providers are a practical alternative for the execution of advanced computational biology experiments at low cost. Using our cloud recipes, the BLAST alignments required to annotate a transcriptome with ∼500,000 transcripts can be processed in <2 hours with a compute cost of ∼$200–$250. In our opinion, for BLAST-based workflows, the choice of cloud platform is not dependent on the workflow but, rather, on the specific details and requirements of the cloud provider. These choices include the accessibility for institutional use, the technical knowledge required for effective use of the platform services, and the availability of open source frameworks such as APIs to deploy the workflow.
Background
The NIH Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability (STRIDES) initiative [1] permits NIH-supported scientists to explore the use of cloud environments and provides cost-effective access to industry-leading commercial cloud providers. The NIH's STRIDES cloud provider partners, at the time of this study, were Amazon Web Services (AWS; Seattle, WA, USA) and Google Cloud Platform (GCP; Mountain View, CA, USA). Cloud computing offers an on-demand model where a user can dynamically allocate “unlimited” compute resources and then release them as soon as the analysis is complete [2]. These services offer a reduced cost of compute resources and a friendly user interface that makes cloud computing accessible for large computational biology experiments.
As part of the STRIDES initiative, NIH-funded institutions began to upload and compute data in the cloud. Public biological databases such as the SRA [3] and computational tools such as BLAST [4], from the NCBI, were migrated and are available for public use on AWS and GCP. In addition, NIH-funded researchers are contributing to the NIH's STRIDE initiative not only by migrating data analysis workflows to the cloud but also by disseminating information about the suitability of cloud computing for computational biology experiments.
The annotation of RNA transcripts with functional and biological processes is an important step in developing an understanding of the biological complexity of an organism. Annotation is a challenging process that requires the integration of multiple biological databases and several computational tools to accurately assign a function to an RNA product. Available public information on a target organism is the main limitation of the annotation of non-model organisms. The NCBI Genome database, for instance, contains 54,049 genome-sequencing projects by organism [5]. This includes 12,204 eukaryotes' genomes for >1,000 species or strains at different assembly levels (95 complete genomes, 1,872 chromosomes, 7,743 scaffolds, and 2,494 contigs [6]). Although these data include an important group of organisms, there is a lack of annotation of several species that have significant public health and economic importance. Significantly, in the plant kingdom, Viridiplantae, only 3 complete genomes, 331 chromosomes, 625 scaffolds, and 394 contigs are annotated. Advances in next-generation sequencing technologies and the decrease in the cost of sequencing a complete transcriptome are driving a new era in which annotation will be increasing, important, and productive.
A review of published articles since 2012 [7–15] reveals that many developed pipelines have a common core component and use the NCBI BLAST tools [16] to align assembled transcriptomes against annotated databases of nucleotides or proteins to identify similarity and infer function. After an assembly, these alignments are the initial step to identify close and/or distant homologous genes, proteins, and functional domains that could be cross-referenced with other public databases, such as Gene Ontology [17], to generate new annotations of query sequences. As the number of transcripts assembled per study increases, the computing power and storage required to align these transcripts to the BLAST databases also increases. On-premises computer infrastructures (including server farms) have been used mainly for the computation of sequence alignments using BLAST. Many laboratories, however, are not equipped with the compute power required for the analysis of increased transcriptome sequencing results. Although a minimum infrastructure could be easy to build and maintain, it may be unnecessary and less financially burdensome with the advent of cloud computing and its use in computational biology.
The use of cloud environments for computational biology experiments is increasing [18–21]. However, little has been published estimating cloud costs and implementation best practices. A recent work published by Ohta at al. [22] presents a tool named CWL-metrics that collects run time metrics of Docker containers and workflow metadata to analyze workflow resource requirements. This study presents a cost estimation for the execution on the cloud for AWS EC2 instances but does not mention the cloud batch system for users to submit thousands of jobs to the cloud.
Modern cloud providers offer “unlimited” compute resources that can be accessed on-demand. An “instance," as the virtual machines are named in the cloud environment, is deployed using a variety of operating systems such as GNU/Linux or Microsoft Windows. Users pay only for the time that the instance is running plus the cost of other resources such as network egress and/or the size of network storage devices. A workflow can be deployed on a manually created instance, but this is not cost efficient because the instance will need to be manually reconfigured with workflow dependencies. It will also remain active once the analysis is completed, which wastes resources.
Private genomic cloud providers, e.g., DNAnexus [23], DNAstar [24], Seven Bridges [25], SciDAP (scidap.com), and others, also offer cloud-based genomics frameworks. These commercial cloud providers make the execution of computational biology experiments easier by offering command line and web-based interfaces designed for genomic data analysis.
Most cloud providers offer a batch system that can do the configuration automatically for users to submit several parallel jobs. The batch system makes the process of instance creation, set-up, and termination fully automatic.
Batch processing is a technique for processing data as a single large collection of iterative steps instead of individually. It reduces user interactivity to process submissions by automating the remaining steps. Modern cloud providers offer a batch system that can be personalized to process many different workflows. Figure 1 shows the components of a generic cloud batch system. It is composed of a “batch queue" to which users submit the “tasks." Each task uses a “job definition" to create a “job" where all computational resources and the workflow steps are outlined. Then, an instance is automatically created with the resources requested by the job. Because all the data for the analysis are in the cloud, the instance downloads the input data from the “cloud storage system" and, after successfully completing the workflow, uploads the results, releasing all computational resources.
Figure 1:

Basic components in a cloud-based batch system.
In this article, we present a comparative study of multiple BLAST searches and alignments required to annotate transcriptome data. This study aims to establish an estimation of the cost and time needed for the execution of multiple BLAST searches on the cloud. Our recommendation on best practices for deploying computational biology workflows in the cloud is also presented.
Methods
Transcriptome annotation workflow
This study focuses only on the many BLAST alignments that are the most compute-demanding core of a transcriptome annotation process. BLAST alignments require considerable compute resources, which generate intermediate results that are used to complete the annotation process. The remaining part of the annotation pipeline is excluded from our study because it can be executed on a workstation and does not require extensive use of the cloud.
The input for the workflow is a transcriptome in FASTA format. First, TransDecoder (TransDecoder, RRID:SCR_017647) [26] is executed to generate all open reading frames (ORFs) from the input file. Then, BLASTP and RPS-BLAST are executed on the TransDecoder output files, generating a list of homologous proteins and conserved protein domains (BLASTP uses the BLAST nr database, and RPS-BLAST uses the NCBI Conserved Domain Database [CDD] [27]). The transcriptome files are also used as inputs for BLASTN and RPST-BLASTN, which are executed using the BLAST nt database and the NCBI CDD database, respectively. These processes generate a list of homologous genes and a list of conserved domains (see Fig. 2). The workflow was implemented using the Common Workflow Language (CWL) [28] and is freely available at [29].
Figure 2:
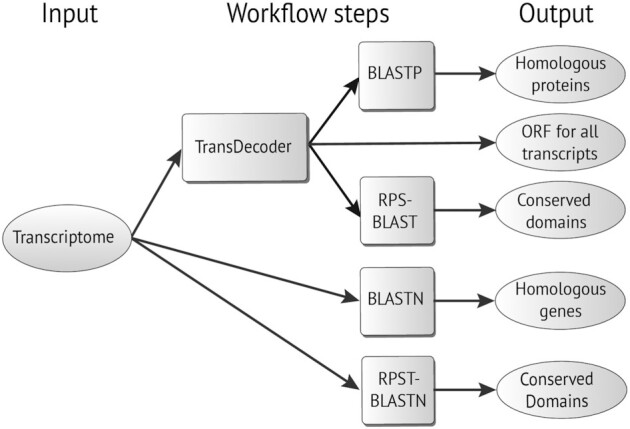
Schema of the transcriptome annotation workflow.
The workflow uses as input a FASTA file, which we named “query," and includes multiple transcripts to be processed. The number of transcripts to be included in a query is another parameter that merits analysis. The size of the query affects the workflow processing time because a complete transcriptome could comprise thousands to hundreds of thousands of transcripts assembled from a next-generation sequencing experiment [30].
Our analysis is based on the execution of the workflow with a batch system provided by each cloud platform. This approach keeps the compute time, and therefore the cost, to a minimum. It also limits user interaction with the jobs to only the submission step.
Containerized workflows
Containerizing a workflow involves bundling it with all its dependencies and configuration files so that it can be executed across different computing environments. The workflow dependencies in the container use the same version and compiled libraries when executed in any computing infrastructure, which makes the process highly reproducible. In this study, we use Docker as the container engine. Docker permits the creation of container images that can be used on a personal laptop or on a cloud platform. The workflow container image generated is freely available from the Google Container registry [31] with name gcr.io/cbb-research-dl/transannot-cloud-cmp. All files used to generate this image are available at [32].
Common Workflow Language
CWL [28] is an open standard workflow language used to describe and implement complex pipelines, which uses interchangeable blocks. The resulting product is portable and scalable. It can be executed across a variety of hardware environments as dissimilar as personal laptops or the cloud.
Workflow managers are tools that simplify the execution of workflows in multiple computational environments. Some have been developed to manage and execute CWL workflows, like Toil [33], CWL-Airflow [34], Arvados [35], and reana [36]. Others, however, use their own workflow languages, like Nextflow [37] and SnakePipes [38]. All provide a unified interface to users to choose the compute environment to process jobs. Users can configure the workflow manager to submit jobs to a high-performance compute cluster or to a cloud provider. Nevertheless, all these workflow managers use the cloud batch system to submit jobs for computing in the cloud.
In this study, we aim to estimate the minimum cost of executing a transcriptome annotation pipeline in the cloud. We selected CWL because it is the workflow language with many available workflow managers. Also, CWL provides a reference implementation runner: cwltool [39] (cwltool, RRID:SCR_015528). This runner can be executed on the command line inside a GCP or AWS job definition, minimizing all dependencies for processing a workflow. We intentionally avoided the use of workflow managers so as to be able to quantify run time for the workflow steps as precisely as possible.
Fig. 3 shows the scheme of the transcriptome annotation workflow used in this study.
Figure 3:

Transcriptome annotation workflow schema [40].
Google Cloud Platform
The GCP offers a batch system specifically designed for life sciences, the Cloud Life Sciences [41]. This system was initially Google Genomics but has evolved to allow the scientific community to process biomedical data at scale.
Cloud Life Sciences offers an API implemented for users to develop their own workflow in JSON format using 3 main attributes: actions, environments, and resources. “Actions" are the list of “commands" to execute using a defined container image. They also include statements to mount local solid-state drives (SSDs) or network storage devices, defined in “resources." “Environments" define the environment variables available inside the container. Finally, “resources" define the instance type and the local SSD or network storage devices.
The API, using the JSON described in Box 1, automatically creates instances on-demand, following the requirements defined in the resources section of the job JSON file. GCP also provides a customized container image where the instance interacts with other GCP products such as Google Storage where data are stored. In addition, GCP creates the instances using a customized Linux operating system that formats and mounts the instance local disks, making them available for the jobs.
Box 1: Brief extract of the GCP pipeline definition JSON file.

Box 1 shows a brief extract of the pipeline used in GCP. We show only the main activity where the “command" attribute defines the command line to execute the CWL workflow. The “ImageUri" attribute defines the container image used to run the command—in this case, our previously created Docker image. Finally, the “mounts" attribute defines the paths in the container to mount the disks created in the resources attribute.
The “VirtualMachine" attribute defines the resources used to create the job instance. In this attribute, users can define instance boot disk size, operating system, extra disks, and the machine type. The complete JSON file is available at [42].
AWS
AWS Batch [43] is the batch system provided by AWS. It comprises compute environments, job queues, and job definitions. The “compute environment" defines the computational resources to be used by the batch jobs. It is connected to the Amazon Elastic Container Service (ECS), which is a fully managed service that creates and manages computer clusters inside the Amazon cloud environment. The resources defined by the compute environment are used by the ECS to create and set up instances in which the workload is distributed. “Job queues" are used as an intermediate service to associate submitted jobs with the compute environments. Last, the jobs use a “job definition," in JSON format, which defines specific information for the job, e.g., container images, commands, number of vCPUs, RAM, environment variables, and local or remote folder to mount on the container.
Box 2 shows a brief extract of the job definition JSON script used in AWS. The “containerProperties" attribute defines the job properties. “Image" defines the container image, in this case our Docker image. “Command" defines the command to be executed inside the container. In the case of AWS, a single command can be outlined in the job definition; thus, complex pipelines with multiple steps can be encapsulated in a BASH script. This script can be stored inside the container image, or the container can download it at run time. For simplicity, we have included this script inside the Docker image.
Box 2: Brief extract of the AWS job definition JSON file.

The AWS Batch system automatically creates all infrastructure, network components, and compute instances, following the requirements of the compute environments. The default configuration of the Amazon Machine Image (AMI) used for the instances, however, is not configured to use local SSD disks available on certain machine types. This limits the default options on the AWS Batch system to certain types of workflows. Workflows that use intensive disk IO operations will have improved performance and efficiency if local SSD disks are used. Thus, a modified AMI capable of using the instance local disks is required for our study. We create a customized AMI for our study that is freely available in the AWS zone us-east1 with ID: ami-0dac0383cac1dc96e. This AMI creates an array with the local SSD disks in the instance using the Linux utility mdadm. The array is formatted with XFS filesystem and mounted in a folder named “/data."
To improve the default AWS Batch options, Amazon offers a Virtual Private Cloud (VPC) that provides an extra layer of isolation for the resources used by the AWS Batch system. This VPC logically isolates all resources used in a defined virtual network, improving the security. It is customizable for each compute problem.
The templates used in our study to create all the components of the AWS Batch system are available at [44]. All resources are created in the Jupyter notebook in “02–AWS-Batch.”
GCP and AWS batch system limitations
Bioinformatics best practices for pipeline execution require the containerization of each tool included in the analysis. Projects such as Bioconda [45] and Biocontainers [46] provide standard containerized images for thousands of bioinformatics tools. However, the batch system for both tested cloud providers requires that all tools used in the workflow be included in a single container. Each action in the cloud job definition is associated with a single Docker image that is used to execute the action task. Docker-in-Docker, the process to execute Docker containers inside another Docker container, is not permitted in either GCP or AWS. This limitation constrains users to containerize all tools involved in a workflow into a single Docker image. Hence, knowledge of how to create Docker images is a requirement for the migration of workflows to the cloud.
GCP and AWS transitory instances
Both GCP and AWS offer access to transitory instances, which are spare compute capacity at a reduced cost. These instances are called “SPOT" in AWS and “Preemptible" in GCP. The transitory instances at reduced cost results from the fact that the cloud provider might terminate the instance at any time. Preemptible prices are fixed in GCP but not in AWS. The cost of the SPOT instances has a minimum but can be increased to the normal EC2 price if the demand for resources increases.
Transitory instances for workflow execution require extra processing steps to identify terminated jobs for resubmission. This is a reasonable option to reduce the cost of the analysis but requires a flexible timeframe to complete all analyses. Users need to be aware of this caveat.
Jupyter notebooks
Jupyter notebooks are an open source web application framework for the creation and sharing of documents that contain live code (Jupyter Notebook, RRID:SCR_018315) [47]. They are a standard way to share scientific code for ease of reproducibility and reuse [48]. The implementation of our study was fully developed in Jupyter notebooks. Readers can reproduce our results and figures using the notebooks that are available at the project GitHub repository. The notebooks create all cloud resources and submit the jobs to the batch systems. They also retrieve the job logs in JSON format and create the figures automatically from those logs. Each notebook includes a description about its purpose and is named using a numeric prefix to highlight the execution order.
The notebooks implemented in this study are designed to be executed on a local laptop or a workstation. Both interact asynchronously with the cloud providers using the command line APIs provided. In the case of GCP, we used the Google Cloud SDK [49]. For AWS, we used the AWS Command Line Interface [50]. In these notebooks, the workflow input files are created, these are uploaded to each cloud provider storage space, the cloud batch systems are configured, the jobs are submitted, and the results are retrieved. The notebooks interact with the cloud batch system to process jobs and retrieve results and logs stored in the results/PRJNA320545 folder.
Results and Discussion
In this study, we present an analysis of the complexity, cost, and best practices for executing the core components of a transcriptome annotation workflow in the cloud. For our experiments, we used the 2 cloud provider partners of the NIH's STRIDES Initiative: GCP and AWS. For each cloud provider, similar compute instances were tested using 16, 32, and 64 vCPUs. The machine types and their resources are described in Table 1. We used the transcriptome assembled from a public BioProject with ID PRJNA320545 for the organism Opuntia streptacantha (prickly pear cactus). The transcriptome includes 474,563 transcripts generated with Trinity [51] and is available in data/PRJNA320545/transcriptome.fasta.gz. The transcriptome length distribution and statistical metrics are available in the 01–Data Partitioning notebook.
Table 1:
Machine types with resources in each cloud
| Machine type | vCPUs | Memory (GB) | Instance Local SSD (GB) | Network bandwidth (Gbit/s) | Region | Last used date | Cost (USD/hour) |
|---|---|---|---|---|---|---|---|
| AWS | |||||||
| m5d | 16 | 64 | 2 × 300 | ≤10 | us-east-1 | 12 Nov 2020 | 0.904 |
| 32 | 128 | 2 × 600 | 10 | us-east-1 | 12 Nov 2020 | 1.808 | |
| 64 | 256 | 4 × 600 | 20 | us-east-1 | 12 Nov 2020 | 3.616 | |
| m5dn | 16 | 64 | 2 × 300 | ≤25 | us-east-1 | 12 Nov 2020 | 1.088 |
| 32 | 128 | 2 × 600 | 25 | us-east-1 | 12 Nov 2020 | 2.176 | |
| 64 | 256 | 4 × 600 | 75 | us-east-1 | 12 Nov 2020 | 4.352 | |
| GCP | |||||||
| n1 | 16 | 60 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 0.861 |
| 32 | 120 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 1.393 | |
| 64 | 240 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 2.475 | |
| n2 | 16 | 64 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 0.951 |
| 32 | 128 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 1.572 | |
| 64 | 256 | 24 × 375 | 32 | us-east1-c | 30 Oct 2020 | 2.816 |
Prices and instance type may change in the future as is common practice among cloud providers.
From the O. streptacantha pool of transcripts, we analyzed 3 sizes of query files: 2,000, 6,000, and 10,000 transcripts in each input query file. Two experiments were executed. First 20 FASTA files (input files for the workflow) for each query size were randomly created (see notebook “01–Data Partitioning”). Each of these files was submitted independently as a job to the batch system on each cloud provider. For the second experiment, 120,000 transcripts were randomly selected and then partitioned in files with 2,000, 6,000, and 10,000 transcripts to analyze the relationship between query size, run time, and cost.
Jobs were submitted to each cloud platform using the notebooks “02–Google Cloud Platform” and “02–AWS-Batch.” In each notebook, the input files created for each experiment were copied to the respective cloud storage system, followed by job submissions for each configuration of machine type/CPU.
Four times were collected from the jobs:
the total run time
the time to transfer the BLAST databases to the instance local SSD disk
the time executing the CWL workflow
the time for creation, set-up, and release of the instance
Figure 4 shows the collected times for the 10,000-query size. Figure 4a shows the total run time for each input file (each containing 10,000 transcripts) for a total of 200,000 transcripts processed for each cloud provider, machine type, and the number of vCPUs.
Figure 4:

Time and cost for the 10,000 query size files. (a) Total time for each input file for each configuration (cloud provider/machine type/vCPUs). The total cost of processing the 20 input files (200,000 transcripts in total) is at the top of each box using normal and transitory instances. The cost of processing 1 transcript is at the bottom of each box. (b) Time and percent of the total cost for instance creation, set-up, and release. (c) Time and percent of the cost for transferring the BLAST databases to the instance from the cloud storage bucket (S3 in AWS and Cloud Storage in GCP). (d) Time and percent of the cost for the CWL workflow execution. Input files in all plots can be identified by the coloring especified in the top plot legend.
In addition, each box in Fig. 4a shows the total cost for the 20 files using normal and transitory instances (top) and the cost of processing 1 transcript (bottom). Figure 4b–d shows the remaining 3 times collected from the jobs.
The total running times for the 10,000-query sized files are similar for the same number of vCPUs notwithstanding the cloud provider. Furthermore, this example shows how the running time can be reduced by more than half by increasing the number of vCPUs. Unfortunately, this time reduction does not decrease the total cost of the project because the price per hour for machines with more vCPUs increases as well.
The AWS platform is more efficient than the GCP during instance creation, set-up, and release (see Fig. 4b). This step is only 0.1% of the total cost despite the Docker image used in the study being hosted in the Google Container Registry. The GCP cost for this stage goes from 1.5% to 4.5% on bigger machines. The differences are due to the Amazon ECS, which allocates new jobs on existing instances as soon as the instance gets free without releasing them, whereas GCP creates, sets up, and releases an instance for each job.
Transferring the BLAST databases from each cloud storage (S3 in AWS and Cloud Storage in GCP) bucket (Fig. 4c; current size is 342 GB) to the instance local SSD disk is a crucial step in reducing the cost of the analysis. Initially, we tested the default parameters in both cloud providers, which use network storage devices taking an average of 1 hour, which is ∼30% of the total cost of the analysis and takes more time than the CWL workflow execution. After customizing both batch systems to use the instance local SSD disks, the time was reduced to a range of 4–11% of the total cost in the 10,000-query size.
As expected, the CWL workflow execution time is the most time-consuming part of the job (Fig. 4d). All configurations show similar times for executing the CWL workflow. The GCP N1 machine type spent more time on the CWL workflow than the other machine types in all configurations because the GCP N1 is the Google first-generation machine type with slower vCPUs.
Figure 5 shows the time and cost of processing 120,000 transcripts using second-generation 64-vCPU instances on each cloud provider. Reducing the number of transcripts per input file reduces the total run time but will also increase the cost of the analysis because more instances will be used. BLAST databases are transferred to more instances, spending, on average, 10 minutes for each instance. For example, our experiment with the 10,000-query size processes all transcripts in ∼105 minutes with a total cost of 59.37 USD using 12 instances (GCP, N2, 64 vCPUs). Processing the same number of transcripts with a query size of 2,000 costs 122.36 USD with all transcripts processed in 43 minutes using 60 instances (GCP, N2, 64 vCPUs).
Figure 5:

Left (a): Total processing time for 120,000 transcripts using different query sizes. Right (b): Total cost using normal compared to transitory instances.
We have determined that a transcriptome with ∼500,000 transcripts can be processed in <2 hours with a compute cost ranging from 200 to 250 USD using normal instances. For transitory instances (SPOT in AWS and preemptible in GCP) the total cost could be reduced to 50 USD for the complete analysis. However, the processing of all transcripts requires a flexible timeframe owing to the availability of the transitory instances and the number of terminated jobs that require resubmission. In our opinion, these are reasonable costs that make the transcriptome annotation process in the cloud accessible to any genomic laboratory without access to an on-premise computational infrastructure.
Best practices
Our recommendations for best practices using public cloud providers for computational biology experiments are as follows:
-
For reproducibility, write the pipeline using a workflow language.
We recommend CWL because the resulting product is portable and scalable, and it can be executed across a variety of computational environments as dissimilar as personal laptops or the cloud. As mentioned above, CWL is the workflow language with many workflow managers available and they can be directly executed in a container using the cwltool runner.
-
Containerize the CWL workflow with Docker.
Use Conda/Bioconda to install all Bioinformatics tools in the container image.
Use Jupyter Notebooks for coding and documenting each step during experiments.
Use the cloud provider batch system for deploying jobs.
-
Cloud computing behaves differently than local workstations or on-premise clusters.
Users should define and execute small tests with their data and workflow before submitting large jobs. Testing different cloud services and configurations could help to reduce the run time and cost for the whole analysis.
Use the instance local disks for computing instead of the default network devices.
Use transitory instances to reduce the cost only if there are no timeframe restrictions for completing the analysis.
Conclusion
Despite differences in the configuration and set-up of batch systems between GCP and AWS, the cost and processing time are similar for the type of workflow we designed for our experiment. In our opinion, for BLAST-based workflows, the choice of a cloud platform is not dependent on the workflow but, rather, on the specific details of the cloud provider. These specific details are related to the accessibility of each cloud platform for institutional use, the technical knowledge of the specific platform services, and/or the availability of open source frameworks to deploy the workflows on a specific cloud provider.
We found that GCP is easier to use because it only requires a JSON file for batch processing whereas AWS needs a complete set-up of all batch system components. GCP is more suitable for daily data analysis work in research laboratories. On the other hand, AWS, once properly configured, is more efficient in terms of machine creation, set-up, and release. The ECS can reuse instances, reducing the cost for large data analysis projects. AWS is more suitable for large data analysis groups to establish a set of queues and compute environments for multiple pipelines.
Availability of Supporting Source Code and Requirements
Project name: Cloud comparison for Transcript-Annotation data analysis pipeline
Project home page: https://github.com/ncbi/cloud-transcriptome-annotation
Operating system(s): Linux and MacOS
Programming languages: Python, BASH
Other requirements: Conda/Bioconda, Jupyter Notebook
RRID: RRID:SCR_019268
Data Availability
Snapshots of the GitHub archive are available in the GigaScience GigaDB repository [52].
Abbreviations
AMI: Amazon Machine Images (AMI); API: application program interface; AWS: Amazon Web Services; BLAST: Basic Local Alignment Search Tool; CDD: Conserved Domain Database; CPU: central processing unit; CWL: Common Workflow Language; ECS: Elastic Container Service; GCP: Google Cloud Platform; JSON: JavaScript Object Notation; NCBI: National Center for Biotechnology Information; NIH: National Institutes of Health; ORF: open reading frame; RAM: random access memory; SRA: Sequence Read Archive; SSD: solid-state drive; STRIDES: Science and Technology Research Infrastructure for Discovery, Experimentation, and Sustainability; VPC: Virtual Private Cloud.
Competing Interests
The authors declare that they have no competing interests.
Funding
This work was supported by the Intramural Research Program of the National Library of Medicine, National Center for Biotechnology Information, at the National Institutes of Health.
Authors’ Contributions
All authors contributed to the design of the annotation workflow and the manuscript preparation. R.V.A. designed, implemented, and executed all cloud environments, configurations, and experiments. All authors read and approved all versions of the manuscript.
Supplementary Material
Tazro Ohta -- 7/27/2020 Reviewed
Tazro Ohta -- 12/4/2020 Reviewed
Lucas Carey -- 9/14/2020 Reviewed
Lucas Carey -- 11/17/2020 Reviewed
Saskia Hiltemann -- 9/17/2020 Reviewed
ACKNOWLEDGEMENTS
We thank NCBI BLAST Group: Christiam Camacho, Vadim Zalunin, Greg Boratyn, Ryan Connor, and Tom Madden for their support with BLAST; and NCBI Cloud and System Group: Al Graeff, Brian Koser, Andrew Arensburger, Brad Plecs, Ron Patterson, and Dima Beloslyudtsev for their support with the cloud platforms.
Contributor Information
Roberto Vera Alvarez, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, NIH, 9000 Rockville Pike, Bethesda, MD 20890, USA.
Leonardo Mariño-Ramírez, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, NIH, 9000 Rockville Pike, Bethesda, MD 20890, USA.
David Landsman, Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, NIH, 9000 Rockville Pike, Bethesda, MD 20890, USA.
References
- 1. NIH STRIDES Initiative. https://cloud.cit.nih.gov/.Accessed 19 January, 2021 [Google Scholar]
- 2. Langmead B, Nellore A. Cloud computing for genomic data analysis and collaboration. Nat Rev Genet. 2018;19(4):208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. SRA in the Cloud. https://www.ncbi.nlm.nih.gov/sra/docs/sra-cloud/. Accessed 19 January, 2021 [Google Scholar]
- 4. Official NCBI BLAST+ Docker Image Documentation. https://github.com/ncbi/blast_plus_docs. Accessed 19 January, 2021 [Google Scholar]
- 5. Sayers EW, Agarwala R, Bolton EE, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020;48(D1):D9–D16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Genome information by organism https://www.ncbi.nlm.nih.gov/genome/browse/#!/eukaryotes/. Accessed 30 June 2020. [Google Scholar]
- 7. Al-Qurainy F, Alshameri A, Gaafar A-R, et al. Comprehensive Stress-based de novo transcriptome assembly and annotation of guar (Cyamopsis tetragonoloba (L.) Taub.): an important industrial and forage crop. Int J Genomics. 2019;2019:7295859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chabikwa TG, Barbier FF, Tanurdzic M, et al. De novo transcriptome assembly and annotation for gene discovery in avocado, macadamia and mango. Sci Data. 2020;7(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ji P, Liu G, Xu J, et al. Characterization of common carp transcriptome: sequencing, de novo assembly, annotation and comparative genomics. PLoS One. 2012;7(4):e35152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Torre S, Tattini M, Brunetti C, et al. RNA-seq analysis of Quercus pubescens leaves: de novo transcriptome assembly, annotation and functional markers development. PLoS One. 2014;9(11):e112487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carruthers M, Yurchenko AA, Augley JJ, et al. De novo transcriptome assembly, annotation and comparison of four ecological and evolutionary model salmonid fish species. BMC Genomics. 2018;19(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bryant DM, Johnson K, DiTommaso T, et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017;18(3):762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vera Alvarez R, Medeiros Vidal N, Garzón-Martínez GA, et al. Workflow and web application for annotating NCBI BioProject transcriptome data. Database (Oxford). 2017;2017, doi: 10.1093/database/bax008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gamez RM, Rodríguez F, Vidal NM, et al. Banana (Musa acuminata) transcriptome profiling in response to rhizobacteria: Bacillus amyloliquefaciens Bs006 and Pseudomonas fluorescens Ps006. BMC Genomics. 2019;20(1):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altschul SF, Gish W, Miller W, et al. Basic Local Alignment Search Tool. J Mol Biol. 1990;215(3):403–10. [DOI] [PubMed] [Google Scholar]
- 17. Ashburner M, Ball CA, Blake JA The Gene Ontology Consortium, et al. , The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peters K, Bradbury J, Bergmann S, et al. PhenoMeNal: processing and analysis of metabolomics data in the cloud. Gigascience. 2019;8(2), doi: 10.1093/gigascience/giy149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Belyeu JR, Nicholas TJ, Pedersen BS, et al. SV-plaudit: A cloud-based framework for manually curating thousands of structural variants. Gigascience. 2018;7(7), doi: 10.1093/gigascience/giy064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kiar G, Gorgolewski KJ, Kleissas D, et al. Science in the cloud (SIC): a use case in MRI connectomics. Gigascience. 2017;6(5), doi: 10.1093/gigascience/gix013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hiltemann S, Mei H, de Hollander M, et al. CGtag: complete genomics toolkit and annotation in a cloud-based Galaxy. Gigascience. 2014;3(1), doi: 10.1186/2047-217X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ohta T, Tanjo T, Ogasawara O. Accumulating computational resource usage of genomic data analysis workflow to optimize cloud computing instance selection. Gigascience. 2019;8(4), doi: 10.1093/gigascience/giz052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DNAnexus. www.dnanexus.com. Accessed 19 January, 2021. [Google Scholar]
- 24. DNASTAR. www.dnastar.com. Accessed 19 January, 2021. [Google Scholar]
- 25. SevenBridges. www.sevenbridges.com. Accessed 19 January, 2021. [Google Scholar]
- 26. Haas B, Papanicolaou A. TransDecoder (Find Coding Regions Within Transcripts). https://github.com/TransDecoder/TransDecoder/wiki. Accessed 7 December 2020. [Google Scholar]
- 27. Yang M, Derbyshire MK, Yamashita RA, et al. NCBI's conserved domain database and tools for protein domain analysis. Curr Protoc Bioinformatics. 2020;69(1):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peter A, Crusoe MR, Tijanić N, et al. Common Workflow Language, v1.0. 2016. https://www.commonwl.org/. Accessed 7 December 2020. [Google Scholar]
- 29. NCBI cloud transcriptome annotation https://github.com/ncbi/cloud-transcriptome-annotation/blob/master/bin/cwl-ngs-workflows-cbb/workflows/Annotation/transcriptome_annotation.cwl. Accessed 19 January, 2021 [Google Scholar]
- 30. Pertea M. The human transcriptome: an unfinished story. Genes (Basel). 2012;3(3):344–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Container Registry. https://cloud.google.com/container-registry. Accessed 19 January, 2021. [Google Scholar]
- 32. NCBI cloud transcriptome annotation https://github.com/ncbi/cloud-transcriptome-annotation/tree/master/config/gcp/docker . Accessed 19 January, 2021. [Google Scholar]
- 33. Vivian J, Rao AA, Nothaft FA, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35(4):314–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kotliar M, Kartashov AV, Barski A. CWL-Airflow: a lightweight pipeline manager supporting Common Workflow Language. Gigascience. 2019;8(7), doi: 10.1093/gigascience/giz084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arvados. https://arvados.org/. Accessed 19 January, 2021. [Google Scholar]
- 36. Reana Reproducible research data analysis platform. http://reanahub.io/. Accessed 19 January, 2021. [Google Scholar]
- 37. Di Tommaso P, Chatzou M, Floden EW, et al. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316–9. [DOI] [PubMed] [Google Scholar]
- 38. Bhardwaj V, Heyne S, Sikora K et al. snakePipes: facilitating flexible, scalable and integrative epigenomic analysis. Bioinformatics. 2019;35:4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Common-workflow-language/cwltool https://github.com/common-workflow-language/cwltool. Accessed 19 January, 2021. [Google Scholar]
- 40. Workflow:transcriptome_annotation. https://view.commonwl.org/workflows/github.com/ncbi/cloud-transcriptome-annotation/blob/master/bin/cwl-ngs-workflows-cbb/workflows/Annotation/transcriptome_annotation.cwl. Accessed 19 January, 2021. [Google Scholar]
- 41. Cloud Life Sciences. https://cloud.google.com/life-sciences. Accessed 19 January, 2021 [Google Scholar]
- 42. NCBI cloud-transcriptome-annotation GCP. https://github.com/ncbi/cloud-transcriptome-annotation/blob/master/config/gcp/pipeline.json. Accessed 19 January, 2021 [Google Scholar]
- 43. AWS Batch. https://aws.amazon.com/batch/. Accessed 19 January, 2021. [Google Scholar]
- 44. NCBI cloud-transcriptome-annotation AWS. https://github.com/ncbi/cloud-transcriptome-annotation/tree/master/config/aws. Accessed 19 January, 2021. [Google Scholar]
- 45. Grüning B, Dale R, Sjödin A, et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018;15(7):475–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. da Veiga Leprevost F, Grüning BA, Alves Aflitos S, et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017;33(16):2580–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shen H. Interactive notebooks: sharing the code. Nature. 2014;515(7525):151–2. [DOI] [PubMed] [Google Scholar]
- 48. Perkel JM. Why Jupyter is data scientists' computational notebook of choice. Nature. 2018;563(7729):145–6. [DOI] [PubMed] [Google Scholar]
- 49. Cloud SDK. https://cloud.google.com/sdkAccessed 19 January, 2021. [Google Scholar]
- 50. AWS Command Line Interface. https://aws.amazon.com/cli/Accessed 19 January, 2021. [Google Scholar]
- 51. Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vera-Alvarez R, Mariño-Ramírez L, Landsman D. Supporting data for “Transcriptome annotation in the cloud: complexity, best practices and cost.". GigaScience Database. 2020, 10.5524/100847. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Vera-Alvarez R, Mariño-Ramírez L, Landsman D. Supporting data for “Transcriptome annotation in the cloud: complexity, best practices and cost.". GigaScience Database. 2020, 10.5524/100847. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Tazro Ohta -- 7/27/2020 Reviewed
Tazro Ohta -- 12/4/2020 Reviewed
Lucas Carey -- 9/14/2020 Reviewed
Lucas Carey -- 11/17/2020 Reviewed
Saskia Hiltemann -- 9/17/2020 Reviewed
Data Availability Statement
Snapshots of the GitHub archive are available in the GigaScience GigaDB repository [52].