

Abstract
FST and kinship are key parameters often estimated in modern population genetics studies in order to quantitatively characterize structure and relatedness. Kinship matrices have also become a fundamental quantity used in genome-wide association studies and heritability estimation. The most frequently-used estimators of FST and kinship are method-of-moments estimators whose accuracies depend strongly on the existence of simple underlying forms of structure, such as the independent subpopulations model of non-overlapping, independently evolving subpopulations. However, modern data sets have revealed that these simple models of structure likely do not hold in many populations, including humans. In this work, we analyze the behavior of these estimators in the presence of arbitrarily-complex population structures, which results in an improved estimation framework specifically designed for arbitrary population structures. After generalizing the definition of FST to arbitrary population structures and establishing a framework for assessing bias and consistency of genome-wide estimators, we calculate the accuracy of existing FST and kinship estimators under arbitrary population structures, characterizing biases and estimation challenges unobserved under their originally-assumed models of structure. We then present our new approach, which consistently estimates kinship and FST when the minimum kinship value in the dataset is estimated consistently. We illustrate our results using simulated genotypes from an admixture model, constructing a one-dimensional geographic scenario that departs nontrivially from the independent subpopulations model. Our simulations reveal the potential for severe biases in estimates of existing approaches that are overcome by our new framework. This work may significantly improve future analyses that rely on accurate kinship and FST estimates.
Author summary
Kinship coefficients and FST, which measure relatedness and population structure, respectively, are important quantities needed to accurately perform various analyses on genetic data, including genome-wide association studies and heritability estimation. However, existing estimators require restrictive assumptions of independence that are not met by real human and other datasets. In this work we find that existing estimators can be severely biased under reasonable scenarios, first by theoretically determining their properties, and then using an admixture simulation to illustrate our findings. In particular, we find that existing FST estimators are downwardly biased, and that existing kinship matrix estimators have related biases that are on average downward and of similar magnitude but vary for every pair of individuals. These insights led us to a new estimation framework for kinship and FST that is practically unbiased for any population structure, as demonstrated by theory and simulations. Our new approaches—available as open-source R packages—are easy to use and are more widely applicable than existing approaches, and they are likely to improve downstream analyses that require accurate kinship and FST estimates.
Introduction
In population genetics studies, one is often interested in characterizing structure, genetic differentiation, and relatedness among individuals. Two quantities often considered in this context are FST and kinship. FST is a parameter that measures structure in a subdivided population, satisfying FST = 0 for an unstructured population and FST = 1 if every locus has become fixed for some allele in each subpopulation. More generally, FST is the probability that alleles drawn randomly from a subpopulation are “identical by descent” (IBD) relative to an ancestral population [1, 2]. The kinship coefficient is a measure of relatedness between individuals defined in terms of IBD probabilities, and it is closely related to FST, since the mean kinship of the parents in a subpopulation is the FST of the following generation [1].
This work focuses on the estimation of FST and kinship from biallelic single-nucleotide polymorphism (SNP) data. Existing estimators can be classified into parametric estimators (methods that require a likelihood function) and non-parametric estimators (such as the method-of-moments estimators we focus on, which only require low-order moment equations). There are many likelihood approaches that estimate FST and kinship, but these are limited by assuming independent subpopulations or Normal approximations for FST [3–11] or totally outbred individuals for kinship [12, 13]. Additionally, more complete likelihood models such as that of Jacquard [14] are underdetermined for biallelic loci [15]. Non-parametric approaches such as those based on the method of moments are considerably more flexible and computationally tractable [16], so they are the natural choice to study arbitrary population structures.
The most frequently-used FST estimators are derived and justified under the “independent subpopulations model,” in which non-overlapping subpopulations evolved independently by splitting all at the same time from a common ancestral population. The Weir-Cockerham (WC) FST estimator assumes subpopulations of differing sample sizes and equal per-subpopulation FST relative to the common ancestral population [17]. The Weir-Hill FST estimator generalized WC for subpopulations with different FST values, and first considered arbitrary coancestry between subpopulations, resulting in estimates of a linearly-transformed FST, namely (where is the unknown mean coancestry value between subpopulations) [4, 18, 19]. Weir-Hill has further evolved into the Weir-Goudet approach, incorporating relatedness for subpopulations and individuals based on allele matching, also estimating a linearly-transformed FST [20–22]. Note that the Weir-Hill and Weir-Goudet approaches intended to estimate such linearly-transformed quantities, which may be negative, and they did not aim to estimate IBD probabilities [4, 18–22]; in contrast, our goal is to estimate IBD probabilities, which must be non-negative and valid probabilities. The “Hudson” FST estimator [23] assumes two subpopulations with different FST values. All of the previous FST estimators are ratio estimators derived using the method of moments to have unbiased numerators and denominators, which gives approximately unbiased ratio estimates when their assumptions are met [4, 17, 23]. We also evaluate BayeScan [10], which estimates population-specific FST values using a Bayesian model and the Dirichlet-Multinomial likelihood function—thus representing non-method-of-moments approaches—but which like other existing FST estimators also assumes that subpopulations are non-overlapping and evolve independently. These FST estimators are important contributions, used widely in the field.
Kinship coefficients are now commonly calculated in population genetics studies to capture structure and relatedness. Kinship is utilized in principal components analyses and linear-mixed effects models to correct for structure in Genome-Wide Association Studies (GWAS) [16, 24–30] and to estimate genome-wide heritability [31, 32]. Often absent in previous models is a clear identification and role of the ancestral population T that sets the scale of the kinship estimates used. Omission of T makes sense when kinship is estimated on an unstructured population (where only a few individual pairs are closely related; there T is the current population). Our more complete notation brings T to the fore and highlights its key role in kinship estimation and its applications. The most commonly-used kinship estimator [16, 27, 30–36] is also a method-of-moments estimator whose operating characteristics are largely unknown in the presence of structure. We show here that this popular estimator is accurate only when the average kinship is zero, which implies that the population must be unstructured.
The goal of our work is to consistently estimate IBD probabilities, namely kinship coefficients and FST, for which there are currently no consistent estimators under general relatedness. Estimation of these as probabilities, as opposed to linearly-transformed quantities that may be negative, is important since the probabilistic definition of these parameters was required to derive their fundamental connections to many applications in genetics, including allele fixation [1, 2, 37], DNA forensics [3], and heritability [38, 39]. Although IBD probabilities are not absolute, but rather depend on the choice of ancestral population [40], their values become fixed upon agreeing to estimate them in terms of the Most Recent Common Ancestor (MRCA) population, which has long been the choice for models of FST [17, 23, 41] and kinship estimation from pedigrees [42, 43] or markers [12, 13].
Recent genome-wide studies have revealed that humans and other natural populations are structured in a complex manner that break the assumptions of the above estimators. Such complex population structures has been observed in several large human studies, such as the Human Genome Diversity Project [44, 45], the 1000 Genomes Project [46], Human Origins [47–49], and other contemporary [50–54] and archaic populations [55, 56]. We have also demonstrated that the global human population has a complex kinship matrix and no independent subpopulations [57–59]. Therefore, there is a need for innovative approaches designed for complex population structures. To this end, we reveal the operating characteristics of these frequently-used FST and kinship estimators in the presence of arbitrary forms of structure, which leads to a new estimation strategy for FST and kinship.
Here, we study existing FST and kinship method-of-moments estimators in models that allow for arbitrary population structures (see Fig 1 for an overview of the results). First, in section The generalized FST for arbitrary population structures we present the generalized definition of FST for arbitrary population structures [57]. In section The kinship and coancestry models we review the kinship model for genotype covariance [1, 14] and the coancestry model for individual-specific allele frequencies [57, 60, 61]. In section Assessing the accuracy of genome-wide ratio estimators we obtain new strong convergence results for a family of ratio estimators that includes the most common FST and kinship estimators. Next, we calculate the convergence values of these FST (section FST estimation based on the independent subpopulations model) and kinship (section Characterizing a kinship estimator and its relationship to FST) estimators under arbitrary population structures, where we find biases that are not present under their original assumptions about structure (panels “Indep. Subpop. FST Estimator” and “Existing Kinship Estimator” in Fig 1). We characterize the limit of the standard kinship estimator, identifying complex biases or distortions, in agreement with recent work [21, 62].
Fig 1. Accuracy of FST and kinship estimators: Overview of models and results.

Our analysis is based on the generalized FST definition (section The generalized FST for arbitrary population structures) and two parallel models: the “Coancestry Model” for individual-specific allele frequencies (πij), and the “Kinship Model” for genotypes (xij). The “Coancestry in Terms of Kinship” panel connects kinship (, ) and coancestry () parameters (section The kinship and coancestry models). We use these models to study the accuracy of FST and kinship method-of-moment estimators under arbitrary population structures. The “Indep. Subpop. FST Estimator” panel shows the bias resulting from the misapplication of FST estimators for independent subpopulations () to arbitrary structures (section FST estimation based on the independent subpopulations model), as calculated under the coancestry model. The “Existing Kinship Estimator” panel shows the bias in the standard kinship model estimator () and its resulting plug-in FST estimator (; section Characterizing a kinship estimator and its relationship to FST), as calculated under the kinship model. The “New Kinship Estimator” panel presents a new statistic Ajk that estimates kinship with a uniform bias, which together with a consistent estimator of its minimum value () results in our new kinship () and FST () estimators, which are consistent under arbitrary population structure (section A new approach for kinship and FST estimation).
In section A new approach for kinship and FST estimation we introduce a new approach for kinship and FST estimation for arbitrary population structures, and demonstrate the improved performance using a simple implementation of these estimators (panel “New Kinship Estimator” in Fig 1). There are two key innovations. First, based on the method of moments, we derive a statistic that estimates kinship coefficients up to a shared unknown scaling factor. Second, we propose a new condition, the identification of unrelated individual pairs in the data, which yields the value of the unknown scaling factor and enables the consistent estimation of kinship matrices and FST. We present a simple implementation of this second estimator, based on taking the minimum average statistic value between subpopulations, which in section Simulations evaluating FST and kinship estimators is shown to perform well under some misspecification, namely in an admixture scenario that does not actually have subpopulations [63–65]. Elsewhere, we analyze the Human Origins and 1000 Genomes Project datasets with our novel kinship and FST estimation approach, where we demonstrate its coherence with the African Origins model, and illustrate the shortcomings of previous approaches in these complex data [59]. In summary, we identify a new approach for unbiased estimation of FST and kinship, and we provide new estimators that are nearly unbiased.
Results
The generalized FST for arbitrary population structures
The existing FST definition requires individuals to belong to discrete, non-overlapping subpopulations, so it must be generalized in order to apply to arbitrary population structures (such as the admixture model with individual-specific ancestry proportions considered in our simulations). Our generalized FST can be understood as a two-step strategy: (1) we define FST on a per-individual basis, and (2) we define FST for a group of individuals as a weighted average of the per-individual FST values [57].
The inbreeding coefficient of an individual j relative to an ancestral population T is defined as the probability that the two alleles at a random locus are identical by descent (IBD) [37]. Note that the ancestral population T determines what is IBD: only relationships since T count toward IBD. This total inbreeding coefficient () is the individual analog of Wright’s total inbreeding coefficient FIT, the latter of which is the mean over a group of individuals [2]. Wright partitioned total inbreeding (FIT) into local (FIS) and structural (FST) coefficients defined by a subpopulation S that contains all individuals in question and evolved from the ancestral population T, so that FIS is the inbreeding of individuals relative to S (as opposed to T) and FST is inbreeding of the subpopulation S relative to T, and these coefficients satisfy (1 − FIT) = (1 − FIS)(1 − FST) [2]. In our generalized definitions for one individual j, we restrict the subpopulation of interest (S) to be Lj, called the local subpopulation of j, which is the most recent subpopulation from which j drew its alleles. In this case, is the local inbreeding coefficient of j (always relative to its local subpopulation Lj), and is the structural inbreeding coefficient of j (equal to the inbreeding of the subpopulation Lj relative to T), and being a special case of Wright’s equation, they also satisfy [57]
| (1) |
Now we discuss estimating the three quantities we just introduced. First, the total inbreeding coefficient () should be estimated from the variance of genotypes, using the practically unbiased approach we introduce in this work. Second, note that the local inbreeding coefficient () corresponds to (non-population) family relatedness, so it can be taken to be the inbreeding calculated from a pedigree if it is available [42]. Note that estimation of the various inbreeding coefficients from pedigrees was the only approach available to Wright when he studied cattle and defined inbreeding and FST [2, 37]. Alternatively, in the absence of pedigrees, local inbreeding can be estimated from inferred self-IBD blocks or unusually-large runs of homozygosity [66–68]. Lastly, since the structural inbreeding coefficient () is given by the previous two quantities (solving from Eq (1)) by
| (2) |
then we propose estimating using this equation, from the above estimates of and .
As a toy example, suppose we estimate a total inbreeding coefficient of for a given individual whose parents are first cousins, then the pedigree expectation for its local inbreeding is , and the structural inbreeding (i.e. the FST of this individual) using Eq (2) is . However, if in the same example () the individual instead had parents who were second cousins, then , then the structural estimate becomes , which is much closer to the total inbreeding value. Thus, when total inbreeding estimates are much larger than local inbreeding estimates, correcting for the latter via Eq (2) may not change the numerical estimate of structural inbreeding by a meaningful amount. Conversely, as the local inbreeding coefficient is reduced exponentially with the degree of relatedness of the parents ( for n-th cousins), and as local inbreeding is required to be recent (to exclude population-level inbreeding), then sufficiently-accurate estimates of structural inbreeding can be obtained by estimating non-zero local inbreeding only for individuals with the most related parent pairs (above a certain degree of relatedness).
We define the generalized FST across n individuals as the weighted average of the per-individual structural inbreeding coefficients (i.e., individual FST values) [57],
| (3) |
where wj is the weight of individual j and the weights are required to sum to one and be non-negative. The above is a straightforward generalization of Wright’s FST: if every individual j has Lj = S as its local subpopulation, then Eq (3) becomes , where is the inbreeding coefficient of subpopulation S relative to T, so it has the same meaning as Wright’s FST (the exact weights here do not matter as long as , as required). Moreover, if each individual j belongs to one of K subpopulations Su (u ∈ {1, …, K}) and if subpopulations are weighted equally ( for every Su), then Eq (3) becomes , so it equals the (unweighted) average subpopulation-specific FST (i.e., ), which is the FST definition for multiple subpopulations prevalent in modern work [4, 21, 23]. The last case illustrates the need for weights, which above downweights individuals that belong to subpopulations with greater numbers of observations. In general, weights allow adjustment for skewed or unbalanced samples. However, in complicated scenarios without subpopulations and no obvious sampling biases, for simplicity we recommend using uniform weights () for the target generalized FST.
In terms of total and local inbreeding coefficients (using Eq (2)), the generalized FST equals
which immediately suggests the estimation strategy when estimates of the total and local inbreeding coefficients are available. For simplicity, in the remainder of this work we shall consider only locally-outbred individuals ( for all j), for which the generalized FST simply equals the weighted mean total inbreeding coefficient:
| (4) |
This greatly simplifies our discussion of bias for all of the FST estimators we analyzed; determining the statistical properties of local inbreeding estimators is beyond the scope of this work. Moreover, the assumption of locally-outbred individuals is satisfied in all of the simulations presented in this work.
The kinship and coancestry models
The generalized FST above is given solely in terms of inbreeding coefficients. In order to establish our results and framework, it is necessary to consider kinship coefficients as well. The kinship coefficient is the extension of the inbreeding coefficient for a pair of individuals: the kinship coefficient of two individuals j and k relative to an ancestral population T is the probability that two alleles, chosen at random from each individual at a random locus, are IBD [1]. Note that the self-kinship coefficient is related to the inbreeding coefficient by [16].
Kinship coefficients determine the covariance structure of genotypes, which is the key to estimating kinship and FST from genotype data. We shall concentrate on biallelic variants, which include single-nucleotide polymorphisms, and are the dominant data from genotyping microarrays and whole-genome sequencing studies. We shall also restrict our attention to diploid organisms in this present work. Genotypes are encoded into variables xij for each locus i and individual j that count the number of alleles (dosage) of a given reference type, so for diploid organisms xij = 2 is homozygous for the reference allele, xij = 0 is homozygous for the alternative allele, and xij = 1 is heterozygous. Based on the definition of the IBD probabilities, the kinship model determines the mean and covariance structure of the genotype random variables at neutral loci [1, 2, 14, 16, 37]:
| (5) |
where is the allele frequency at locus i in the ancestral population T and .
The coancestry model resembles the kinship model, but it is formulated in terms of allele frequencies, which simplifies our analysis of FST estimators for subpopulations as well as yielding kinship coefficients under the admixture model we simulate from in this work. Let πij be the individual-specific allele frequency (IAF) at locus i for individual j, which is a real number between zero and one [60, 61]. Our coancestry model assumes that [57]
| (6) |
where is the coancestry coefficient between individuals j and k relative to the ancestral population T. This model is inspired by coancestry models for subpopulations common in the FST literature [4, 5, 21, 23], and exactly equals those models when subpopulation sizes go to infinity, in which case j and k index subpopulations rather than individuals, and πij is interpreted as the true allele frequency at locus i for subpopulation j.
The coancestry model connects to the kinship model under the additional assumption that the alleles of an individual j are drawn independently from its IAF,
| (7) |
In this case, marginalizing the intermediate IAF random variables (πij) and matching the resulting genotype moments results in the following equivalence [57]:
| (8) |
The coancestry coefficient equals the kinship coefficient between two different individuals, but the self-coancestry coefficient equals the inbreeding coefficient (rather than the self-kinship coefficient). However, since in the coancestry model alleles are drawn independently conditional on the IAF in Eq (7), then the only structure present is the population structure, so these coancestry models cannot generate family structures, unlike the more general kinship model that also encompasses pedigrees. Therefore, despite Eq (8), the kinship and coancestry are not equivalent models except under the more restrictive assumptions of the coancestry model. Thus, individuals drawn from this model are always locally-outbred, so also equals the structural inbreeding coefficient, and the generalized FST under the coancestry model is therefore
| (9) |
which also generalizes previous definitions of FST under coancestry for subpopulations [4, 5, 21, 23]. The kinship and coancestry models, and their connection, is included in the overview Fig 1.
Assessing the accuracy of genome-wide ratio estimators
In this section we change gears to focus on theoretical convergence properties of two broad estimator families. The resulting theory will be applied repeatedly to various FST and kinship estimators of interest in later sections.
Many FST and kinship coefficient method-of-moments estimators are ratio estimators, a general class of estimators that tends to be biased and to have no closed-form expectation [69]. In the FST literature, the expectation of a ratio is frequently approximated with a ratio of expectations [4, 17, 23]. Specifically, ratio estimators are often called “unbiased” if the ratio of expectations is unbiased, even though the ratio estimator itself may be biased [69]. Here we characterize the behavior of two ratio estimator families calculated from genome-wide data, known as “ratio-of-means” and “mean-of-ratios” estimators [23], detailing conditions where the previous approximation is justified and providing additional criteria to assess the accuracy of such estimators.
Ratio estimators
The general problem of forming ratio estimators involves random variables ai and bi calculated from genotypes at each locus i, such that E[ai] = Aci and E[bi] = Bci and the goal is to estimate . A and B are constants shared across loci (given by FST or ), while ci depends on the ancestral allele frequency and varies per locus. The problem is that the single-locus estimator is biased, since , which applies to ratio estimators in general [69]. Below we study two estimator families that combine large numbers of loci to better estimate .
Convergence
The solution we recommend is the “ratio-of-means” estimator , where , and , which is common for FST estimators [4, 17, 19, 23, 70]. Note that and , where . We will assume bounded terms (|ai|, |bi| ≤ C for some finite C), a convergent , and Bc ≠ 0, which are satisfied by common estimators. Given independent loci, we prove almost sure convergence to the desired quantity (S1 Text),
| (10) |
a strong result that implies , justifying previous work [4, 17, 23]. Moreover, the error between these expectations scales with (S1 Text), just as for standard ratio estimators [69]. Although real loci are not independent due to genetic linkage, their dependence is very localized, so this estimator will perform well if the effective number of independent loci is large.
In order to test if a given ratio-of-means estimator converges to its ratio of expectations as in Eq (10), the following three conditions can be tested. (i) The expected values of each term ai, bi must be calculated and shown to be of the form E[ai] = Aci and E[bi] = Bci for some A and B shared by all loci i and some ci that may vary per locus i but must be shared by both E[ai], E[bi]. In the estimators we study, A and B are functions of IBD probabilities such as and FST, while ci is a function of only. (ii) The mean ci must converge to a non-zero value for infinite loci. (iii) Both |ai|, |bi| ≤ C must be bounded for all i by some finite C (the estimators we study usually have C = 1 or C = 4). If these conditions are satisfied, then Eq (10) holds for independent loci and the A and B found in the first step. See the next section for an example application of this procedure to an FST estimator.
Another approach is the “mean-of-ratios” estimator , used often to estimate kinship coefficients [16, 27, 30–35] and FST [46]. If each is biased, their average across loci will also be biased, even as m → ∞. However, if for all loci i = 1, …, m as the number of individuals n → ∞, and is bounded, then
| (11) |
Therefore, mean-of-ratios estimators must satisfy more restrictive conditions than ratio-of-means estimators, as well as large n (in addition to the large m needed by both estimators), to estimate well. We do not provide a procedure to test whether a given mean-of-ratios estimator converges as shown above.
FST estimation based on the independent subpopulations model
Now that we have detailed how ratio estimators may be evaluated for their accuracy, we turn to existing estimators and assess their accuracy under arbitrary population structures. We study the FST estimators Weir-Cockerham (WC) [17], Weir-Hill [4], “Hudson” [23], and Weir-Goudet (equals HudsonK below for biallelic loci; S1 Text) [21]. The panel “Indep. Subpop. FST Estimator” in Fig 1 provides an overview of our results, which we detail in this section.
The FST estimator for independent subpopulations and infinite subpopulation sample sizes
The WC, Weir-Hill, and Hudson method-of-moments estimators have small sample size corrections that remarkably make them consistent (as the number of independent loci m goes to infinity) for finite numbers of individuals. However, these small sample corrections also make the estimators unnecessarily cumbersome for our purposes (see Methods, section Previous FST estimators for the independent subpopulations model for complete formulas). In order to illustrate clearly how these estimators behave, both under the independent subpopulations model and for arbitrary structure, here we construct simplified versions that assume infinite sample sizes per subpopulation (Methods, section Previous FST estimators for the independent subpopulations model). This simplification corresponds to eliminating statistical sampling, leaving only genetic sampling to analyze [71]. Note that our simplified estimator nevertheless illustrates the general behavior of the WC, Weir-Hill, and Hudson estimators under arbitrary structure, and the results are equivalent to those we would obtain under finite sample sizes of individuals. While the Hudson FST estimator compares two subpopulations [23], based on that work we derive a generalized “HudsonK” estimator for more than two subpopulations in Methods, section Generalized HudsonK FST estimator. Note that HudsonK, first derived in [58], also equals the Weir-Goudet FST estimator for subpopulations [21] when loci are biallelic, which was derived independently using allele matching (S1 Text).
Under infinite subpopulation sample sizes, the allele frequencies at each locus and every subpopulation are known. Let j ∈ {1, …, n} index subpopulations rather than individuals and πij be the true allele frequency in subpopulation j at locus i. Note that πij are not estimated allele frequencies, but rather true subpopulation allele frequencies; this abstraction does not result in a practical estimation approach, but it greatly simplifies understanding of bias for subpopulations in a setting where there there is no statistical sampling. Although in this analysis of FST estimators the πij values are applied to subpopulations, for coherence with our previous work we shall call them “individual-specific allele frequencies” (IAF) [60, 61]. Whether for individuals or subpopulations, the key assumption is that IAFs satisfy the coancestry model of Eq (6). In this special case of infinite subpopulation sample sizes, all of WC, Weir-Hill, and HudsonK simplify to the following FST estimator for independent subpopulations (“indep”; derived in Methods, section Previous FST estimators for the independent subpopulations model):
| (11) |
| (12) |
| (13) |
The goal is to estimate , which is the special case of Eq (9) that weighs every subpopulation j equally ().
FST estimation under the independent subpopulations model
Under the independent subpopulations model for j ≠ k, where T is the most recent common ancestor (MRCA) population of the set of subpopulations. Note that the estimator in Eq (13) can be derived directly from Eq (6) and these assumptions using the method of moments (ignoring the existence of previous FST estimators; S1 Text). The expectations of the two recurrent terms in Eq (13) are
Eliminating and solving for FST in this system of equations recovers the estimator in Eq (13).
Before applying the convergence result in Eq (10), we test that the three conditions listed in section Assessing the accuracy of genome-wide ratio estimators are met. Condition (i): The locus i terms are and , which satisfy E[ai] = Aci and E[bi] = Bci with A = FST, B = 1, and . Condition (ii): over the distribution across loci. Condition (iii): Since , then and , and since n ≥ 2, C = 1 bounds both |ai| and |bi|. Therefore, for independent loci,
FST estimation under arbitrary coancestry
Now we consider applying the independent subpopulations FST estimator to dependent subpopulations. The key difference is that now for every (j, k) will be assumed in our coancestry model in Eq (6), and now T may be either the MRCA population of all subpopulations or a more ancestral population. In this general setting, (j, k) may index either subpopulations or individuals. The two terms of now satisfy
where is the mean coancestry with uniform weights. There are two equations but three unknowns: FST, , and . The independent subpopulations model satisfies , which allows for the consistent estimation of FST. Therefore, the new unknown precludes consistent FST estimation without additional assumptions. As shown later, our additional assumption is that we can identify unrelated individuals in the data, which determines all unknowns. We defer our complete solution to this problem until kinship and its estimation challenges have been presented.
The FST estimator for independent subpopulations converges more generally to
| (14) |
(the conclusion of panel “Indep. Subpop. FST Estimator” in Fig 1), where
is the average of all between-subpopulation coancestry coefficients, in agreement with related calculations regarding the WC and Weir-Hill estimators [4, 21]. Therefore, under arbitrary structure the independent subpopulations estimator’s bias is due to the coancestry between subpopulations. While the limit in Eq (14) appears to vary depending on the choice of T, it is in fact a constant with respect to T (proof in S1 Text).
Since (S1 Text), this estimator has a downward bias in the general setting: it is asymptotically unbiased () only when , while bias is maximal when , where . For example, if so the MRCA population T is fixed, but n is large and for most pairs of subpopulations, then as well, and . Therefore, the magnitude of the bias of is unknown if is unknown, and small estimates may arise even if FST is very large.
Coancestry estimation as a method of moments
Since the generalized FST is given by coancestry coefficients in Eq (9), a new FST estimator could be derived from estimates of . Here we attempt to define a method-of-moments estimator for , and find an underdetermined estimation problem, just as for FST. This is consistent with IBD parameters in general requiring a reference population to be determined [40], whereas in this subsection this reference population is unspecified.
Given IAFs and the coancestry model of Eq (6), the first and second moments that average across loci are
| (15) |
| (16) |
where , , and is as before.
Suppose first that only are of interest. There are n estimators given by Eq (16) with j = k, each corresponding to an unknown . However, all these estimators share two nuisance parameters: and . While can be estimated from Eq (15), there are no more equations left to estimate , so this system is underdetermined. The estimation problem remains underdetermined if all estimators in Eq (16) are considered rather than only the j = k cases. Therefore, we cannot estimate coancestry coefficients consistently using only the first two moments without additional assumptions.
Characterizing a kinship estimator and its relationship to FST
Given the biases we see for under arbitrary structures in the previous section, we now turn to the generalized definition of FST and pursue an estimate of it. Recall that our generalized FST in Eq (3) is defined in terms of inbreeding coefficients, which are a special case of the kinship coefficient. Kinship coefficients also determine the bias of in Eq (14) (since coancestry and kinship coefficients are closely related: see panel “Coancestry in Terms of Kinship” in Fig 1). Therefore, we will consider estimates of kinship and inbreeding in this section. Estimating kinship is also important for GWAS approaches that control for population structure [16, 24–35, 72, 73].
In this section, we focus on a standard kinship method-of-moments estimator and calculate its limit for the first time (panel “Existing Kinship Estimator” in Fig 1). We study estimators that use genotypes or IAFs, and construct FST estimators from their kinship estimates. We find biases comparable to those of in the previous section, and define unbiased FST estimators that require knowing the mean kinship or coancestry, or its proportion relative to FST. The results of this section directly motivate and help construct our new kinship and FST estimation approach in the following section.
Characterization of the standard kinship estimator
Here we analyze a standard kinship estimator that is frequently used [16, 27, 30–36]. We generalize this estimator to use weights in estimating the ancestral allele frequencies, and we write it as a ratio-of-means estimator due to the favorable theoretical properties of this format as detailed in the earlier section Assessing the accuracy of genome-wide ratio estimators:
| (17) |
| (18) |
The estimator in Eq (18) resembles the sample covariance estimator applied to genotypes, but centers by locus i rather than by individuals j and k, and normalizes using estimates of . We derive Eq (18) directly using the method of moments in S1 Text. The weights in Eq (17) must satisfy wj > 0 and , so that and .
Utilizing the kinship model for genotypes from Eq (5), we find that Eq (18) converges to
| (19) |
where and , which agrees with related derivations [21, 62]. (This is the conclusion of panel “Existing Kinship Estimator” in Fig 1; see S1 Text for intermediate calculations that lead to Eq (19).) Therefore, the bias of varies per pair of individuals j and k. Analogous distortions have been observed for sample covariances of genotypes [74]. The limit of in Eq (19) is constant with respect to T (proof in S1 Text). Similarly, inbreeding coefficient estimates derived from Eq (18) converge to
| (20) |
The difference between the bias of for j ≠ k in Eq (19) and in Eq (20) is visible in the kinship estimates shown toward the end of the results section. The limits of the ratio-of-means versions of two more estimators [32] are, if uses Eq (17),
| (21) |
The estimators in Eqs (18) and (21) are unbiased when is replaced by [16, 32, 36], and are consistent when is consistent [60]. Surprisingly, in Eq (17) is not consistent (it does not converge almost surely to ) for arbitrary population structures, which is at the root of the bias in Eqs (19) to (21). In particular, although is unbiased, its variance (S1 Text, and some special cases shown elsewhere, e.g., [19]),
| (22) |
may be asymptotically non-zero as n → ∞, since is fixed and may take on any value between zero and one for arbitrary population structures. Further, as n → ∞ if and only if for almost all pairs of individuals (j, k). These observations hold for any weights such that . An important consequence is that the plug-in estimate of is biased (S1 Text),
which is present in all estimators we have studied.
Estimation of coancestry coefficients from IAFs
Here we form a coancestry coefficient estimator analogous to Eq (18) but using IAFs. Assuming the moments in Eq (6), this estimator and its limit are
| (23) |
| (24) |
where and are analogous to and . Eq (23) generalizes Eq (11) for arbitrary weights. Thus, use of IAFs does not ameliorate the estimation problems we have identified for genotypes. Like Eq (22), in Eq (23) is not consistent because may not converge to zero for arbitrary population structures, which causes the bias observed in Eq (24).
FST estimator based on the standard kinship estimator
Since the generalized FST is defined as a mean inbreeding coefficient in Eq (3), here we study the FST estimator constructed as where is the inbreeding estimator derived from the standard kinship estimator. Although is biased, we nevertheless plug it into our definition of FST so that we may study how bias manifests. Note that we do not recommend utilizing this FST estimator in practice, but we find these results informative for identifying how to proceed in deriving new estimators in the following section.
Remarkably, the three estimators in Eqs (20) and (21) give exactly the same plug-in if the weights in FST and in Eq (17) match, namely
| (25) |
where the limit assumes locally-outbred individuals so Eq (4) holds. The analogous FST estimator for IAFs and its limit are
| (26) |
The estimators in Eqs (25) and (26) for individuals and their limits resemble those of classical FST estimators for populations of the form [4, 5]. in Eq (26) for subpopulations j with uniform weight and one locus is also GST for two alleles [75]. Compared to in Eq (13), in Eq (26) admits arbitrary weights and, by forgoing bias correction under the independent subpopulations model, is a simpler target of study.
Like in Eq (13), in Eqs (25) and (26) are downwardly biased since . in Eq (26) may converge arbitrarily close to zero since can be arbitrarily close to FST (S1 Text). Moreover, although for large n (see Eq (8) and panel “Coancestry in Terms of Kinship” in Fig 1), in extreme cases can exceed FST under the coancestry model (where ) and also under extreme local kinship, where in Eq (25) converges to a negative value.
Adjusted consistent oracle FST estimators and the “bias coefficient”
Here we explore two adjustments to from IAFs in Eq (26) that rely on having minimal additional information needed to correct its bias. These “oracle” approaches require information that is not known in practice, but this exercise helps us understand the problem more deeply and finds further connections between the various FST estimators.
If is known, the bias in Eq (26) can be reversed, yielding the consistent estimator
| (27) |
Consistent estimates are also possible if a scaled version of is known, namely
| (28) |
which we call the “bias coefficient” and which has interesting properties. The bias coefficient quantifies the departure from the independent subpopulations model by comparing the mean coancestry () to the mean inbreeding coefficient (), and given FST > 0 satisfies 0 < sT ≤ 1 (S1 Text). The limit in Eq (26) in terms of sT is
| (29) |
Treating the limit as equality and solving for FST yields the following consistent estimator:
| (30) |
| (31) |
Note that and from Eqs (12) and (13) are the special case of Eqs (30) and (31) for uniform weights and ; hence, generalizes .
Lastly, using either Eqs (26) or (29), the relative error of converges to
| (32) |
which is approximated by sT if FST ≪ 1, hence the name “bias coefficient”. Note sT varies depending on the choice of T, which is necessary since FST (and hence the relative bias of from FST) depends on the choice of T.
A new approach for kinship and FST estimation
Here, we propose a new estimation approach for kinship coefficients that has properties favorable for obtaining nearly unbiased estimates (panel “New Kinship Estimator” in Fig 1). These new kinship estimates yield an improved FST estimator. We present the general approach and implement a simple version of one key estimator that results in the complete proof-of-principle estimator that is evaluated in the next section and applied to human data in [59]. We also compare our approach to a related estimator of non-IBD linearly-transformed kinship values [20–22] that was proposed concurrently to ours [58].
General approach
In this subsection we develop our new estimator in two steps. First, we compute a new statistic Ajk that is proportional in the limit of infinite loci to times a nuisance factor vT. Second, we estimate and remove vT to yield the proposed estimator . —an estimator of the limit of the minimum Ajk—yields vT if the least related pair of individuals in the data has , which sets T to the MRCA population of all the individuals in the data. The new kinship estimator immediately results in new inbreeding () and FST () estimators. This general approach leaves the implementation of open; the simple implementation applied in this work is described in subsection Proof-of-principle kinship estimator using subpopulation labels, but our method can be readily improved by substituting in a better in the future.
Applying the method of moments to Eq (5), we derive the following statistic (S1 Text), whose expectation is proportional to :
| (33) |
Compared to the standard kinship estimator in Eq (19), which has a complex asymptotic bias determined by n parameters ( for each j ∈ {1, …, n}), the Ajk statistics estimate kinship with a bias controlled by the sole unknown parameter shared by all pairs of individuals. The key to estimating is to notice that if then . Thus, assuming , which sets T to the MRCA population, then the minimum Ajk yields the nuisance parameter. However, we recommend using a more stable estimate than the minimum Ajk to unbias all Ajk, such as the estimator presented in the next subsection.
In general, suppose is a consistent estimator of the limit of the minimum E[Ajk|T], or equivalently,
along with the assumption that for some vT ≠ 0. Our new kinship estimator follows directly from replacing with and solving for in Eq (33), which results in a consistent kinship estimator (given the convergence proof of section Assessing the accuracy of genome-wide ratio estimators):
| (34) |
The resulting new inbreeding coefficient estimator is
| (35) |
and the new FST estimator is consistent for locally-outbred individuals (estimates Eq (4)):
| (36) |
Thus, only the implementation of is left unspecified from this general estimation approach of kinship and FST. The implementation of used in the analyses in this work is given in the next subsection.
Proof-of-principle kinship estimator using subpopulation labels
To showcase the potential of the new estimators, we implement a simple proof-of-principle version of needed for our new kinship estimator ( in Eq (34)). This relies on an appropriate partition of the n individuals into K subpopulations (denoted Su for u ∈ {1, …, K}), where the only requirement is that the kinship coefficients between pairs of individuals across the two most unrelated subpopulations is zero, as detailed below. Note that, unlike the the independent subpopulations model of section FST estimation based on the independent subpopulations model, these K subpopulations need not be independent nor unstructured. The desired estimator is the minimum average Ajk over all subpopulation pairs:
| (37) |
This consistently estimates the limit of the minimum Ajk if for the least related pair of subpopulations Su, Sv.
This estimator should work well for individuals truly divided into subpopulations, but may be biased for a poor choice of subpopulations, in particular if the minimum mean between subpopulations is far greater than zero. For this reason, inspection of the kinship estimates is required and careful construction of appropriate subpopulations may be needed. See our analysis of human data for detailed examples [59]. Future work could focus on a more general that circumvents the need for subpopulations of our proof-of-principle estimator.
Comparison to the Weir-Goudet kinship estimator for individuals
Here we analyze the Weir-Goudet (WG) kinship estimator for individuals [20–22]. This has connections to our new estimator but differs in having the goal of estimating linearly-transformed kinship values. In our framework, the WG estimator is given by
Therefore, this estimator differs from our proposal [58] by replacing our with . Under the kinship model, the expectation of is
Therefore, the limit of this estimator is
| (38) |
which agrees with calculations in the original WG work [20–22]. Note that, assuming that kinship coefficients must be non-negative, the above estimator recovers the kinship IBD probabilities if and only if which holds if and only if for every pair of individuals j ≠ k. The resulting WG inbreeding coefficient estimator is
which estimates linearly-transformed inbreeding values [21]. Therefore, the resulting WG FST estimator (for individuals) also targets a linearly-transformed FST value (under locally-outbred individuals, where FST is given by Eq (4)), namely
The WG authors also briefly consider a variant of their kinship estimator that is normalized using the minimum kinship value as we did, developed concurrently with our approach [58], but was largely dismissed as an unnecessary correction [21, 76]. See S1 Text for a detailed proof that the general estimator framework we propose here (Eqs (33) and (34)) is algebraically equivalent to our original formulation in [58].
Note that the original WG does not estimate FST from individuals as considered above; instead, FST is estimated from coancestry estimates for subpopulations (which equals our HudsonK for biallelic loci, S1 Text) [20–22]. For completeness, we consider both kinds of FST estimates in the evaluations that follow.
Simulations evaluating FST and kinship estimators
Overview of simulations
We simulate genotypes from two models to illustrate our results when the true population structure parameters are known. Both simulations have clearly-defined IBD probability parameters in terms of the MRCA population. The first simulation satisfies the independent subpopulations model that existing FST estimators assume. The second simulation is from an admixture model with no independent subpopulations and pervasive kinship designed to induce large downward biases in existing kinship and FST estimators (Fig 2). This admixture scenario resembles the population structure we estimated for Hispanics in the 1000 Genomes Project [59]: compare the simulated kinship matrix (Fig 2B) and admixture proportions (Fig 3C) to our estimates on the real data [59]. Both simulations have n = 1000 individuals, m = 300, 000 loci, and K = 10 subpopulations or intermediate subpopulations. These simulations have FST = 0.1, comparable to previous estimates between human populations (in 1000 Genomes, the estimated FST between CEU (European-Americans) and CHB (Chinese) is 0.106, between CEU and YRI (Yoruba from Nigeria) it is 0.139, and between CHB and YRI it is 0.161 [23]).
Fig 2. Coancestry matrices of simulations.
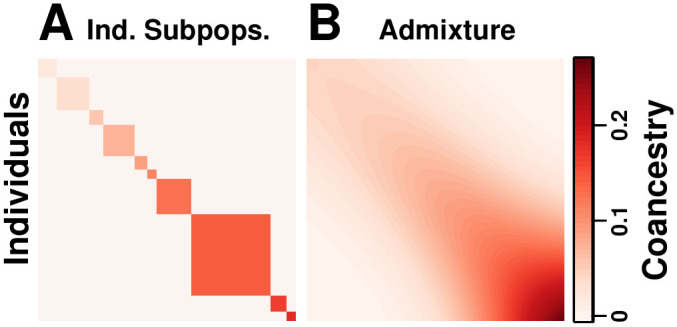
Both panels have n = 1000 individuals along both axes, K = 10 subpopulations (final or intermediate), and FST = 0.1. Color corresponds to between individuals j and k (equal to off-diagonal, along the diagonal). (A) The independent subpopulations model has between subpopulations, and varying per subpopulation, resulting in a block-diagonal coancestry matrix. (B) Our admixture scenario models a 1D geography with extensive admixture and intermediate subpopulation differentiation that increases with distance, resulting in a smooth coancestry matrix with no independent subpopulations (no between blocks). Individuals are ordered along each axis by geographical position.
Fig 3. 1D admixture scenario.
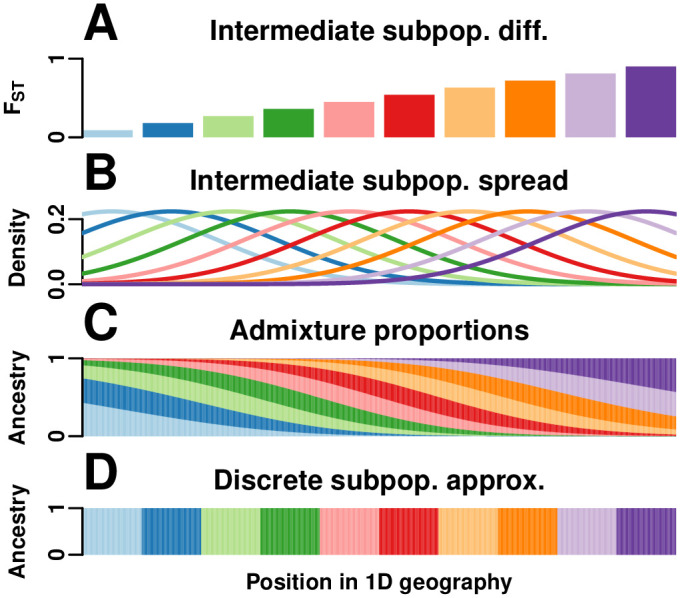
We model a 1D geography population that departs strongly from the independent subpopulations model. (A) K = 10 intermediate subpopulations, evenly spaced on a line, evolved independently in the past with FST increasing with distance, which models a sequence of increasing founder effects (from left to right) to mimic the global human population. (B) Once differentiated, individuals in these intermediate subpopulations spread by random walk modeled by Normal densities. (C) n = 1000 individuals, sampled evenly in the same geographical range, are admixed proportionally to the previous Normal densities. Thus, each individual draws most of its alleles from the closest intermediate subpopulation, and draws the fewest alleles from the most distant populations. Long-distance random walks of intermediate subpopulation individuals results in kinship for admixed individuals that decays smoothly with distance in Fig 2B. (D) For FST estimators that require a partition of individuals into subpopulations, individuals are clustered by geographical position (K = 10).
The independent subpopulations simulation satisfies the HudsonK and BayeScan estimator assumptions: each independent subpopulation Su has a different FST value of relative to the MRCA population T (Fig 2A). Ancestral allele frequencies are drawn uniformly between 0.01 and 0.5. Allele frequencies for Su and locus i are drawn independently from the Balding-Nichols (BN) distribution [3] with parameters and . Every individual j in subpopulation Su draws alleles randomly with probability . Subpopulation sample sizes are drawn randomly (Methods, section Simulations).
The admixture simulation corresponds to a “BN-PSD” model [6, 27, 34, 60, 77]: the intermediate subpopulations are independent subpopulations that draw from the BN model, then each individual j constructs its allele frequencies as , which is a weighted average of the subpopulation allele frequencies with the admixture proportions qju of individual j and subpopulation u as weights (which satisfy ), as in the Pritchard-Stephens-Donnelly (PSD) admixture model [63–65]. We constructed qju that model admixture resulting from spread by random walk of the intermediate subpopulations along a one-dimensional geography, as follows. Intermediate subpopulations Su are placed on a line with differentiation that grows with distance, which corresponds to a serial founder effect (Fig 3A). Upon differentiation, individuals in each Su spread by random walk, a process modeled by Normal densities (Fig 3B). Admixed individuals derive their ancestry proportional from these Normal densities, resulting in a genetic structure governed by geography (Figs 3C and 2B) and departing strongly from the independent subpopulations model (Fig 3D). The amount of spread—which sets the mean kinship across all individuals—was chosen to give a bias coefficient of , which by Eq (32) results in a large downward bias for (in contrast, the independent subpopulations simulation has sT = 0.1). The true coancestry and FST parameters of this simulation are given by the values of the intermediate subpopulations and the admixture coefficients qju of the individuals via the following equations [57]:
| (39) |
The first equation above connecting coancestry to admixture proportions was derived independently in other work [62], but the FST for the admixed individuals was absent and instead follows from our generalized FST definition given in Eq (9). See Methods, section Simulations for additional details regarding these simulations.
Evaluation of FST estimators
Our admixture simulation illustrates the large biases that can arise if existing FST estimators that require independent subpopulations or FST estimates derived from existing kinship estimators are misapplied to arbitrary population structures to estimate the generalized FST, and demonstrate the higher accuracy of our new FST estimator ( given by the combination of Eqs (36) and (37)). The WC FIT (total inbreeding) estimator was also evaluated.
First, we test these estimators in our independent subpopulations simulation. The HudsonK (Methods, section Generalized HudsonK FST estimator) and BayeScan FST estimators are consistent in this simulation, since their assumptions are satisfied (Fig 4A). The WC FST estimator assumes that for all subpopulations Su, which does not hold; nevertheless, WC has only a small bias (Fig 4A). The WC FIT estimator arrives at similar estimates, as it should since there is no local inbreeding, so the true FIT also equals FST. The Weir-Hill estimator permits different values per subpopulation, but assigns equal weight to individuals rather than subpopulations (Methods, section The Weir-Hill FST estimator), resulting in a slightly different target FST (we verified that these estimates are unbiased for this FST). For comparison, we show the standard kinship-based in Eq (25) (weights from Methods, section Simulations) and based on the Weir-Goudet kinship estimates for individuals, both of which do not have corrections that would make them consistent under the independent subpopulations model. Since the number of subpopulations K is large, has a small relative bias of about (Fig 4A); greater bias is expected for smaller K. Our new FST estimator has a very small bias in this simulation resulting from estimating the minimum kinship from the smallest kinship between subpopulations (see Eq (37)) rather than their average as HudsonK does implicitly (Fig 4A).
Fig 4. Evaluation of FST estimators.

The Weir-Cockerham, Weir-Hill, Weir-Goudet (for individuals), HudsonK (equal to Weir-Goudet for subpopulations, S1 Text), BayeScan, in Eq (25) derived from the standard kinship estimator, and our new FST estimator in Eqs (34) and (37), are evaluated on simulated genotypes from our two models (Fig 2). The Weir-Cockerham FIT estimator was also included to show that estimation of total inbreeding behaves similarly to FST estimators. (A) The independent subpopulations model required by the Weir-Hill, HudsonK, and BayeScan FST estimators. All but standard kinship () and Weir-Goudet (for individuals) recover the target FST IBD probability in Eq (9) (red line) with small errors. (B) Our admixture scenario, which has no independent subpopulations, was constructed so . Only our new estimates are accurate. The rest of these estimators give values smaller than the target FST IBD probability, which result from treating kinship as zero between every subpopulations imposed by geographic clustering (or between individuals for Standard Kinship and Weir-Goudet). The estimator limit in Eq (14) (green dotted line) overlaps the true FST (red line) in (A) but not (B). Estimates (blue) include 95% prediction intervals (often too narrow to see) from 39 independently-simulated genotype matrices for each model (Methods, section Prediction intervals).
Next we test these estimators in our admixture simulation. To apply the FST estimators that require subpopulations to the admixture model, individuals are clustered into subpopulations by their geographical position (Fig 3D). We find that estimates of all existing methods are smaller than the true FST by nearly half, as predicted by the limit of in Eq (14) (Fig 4B). The WC FIT estimator obtains slightly larger estimates than the WC FST estimator, but overall remains as biased as the other FST estimators, showing that the use of a total inbreeding estimator for independent subpopulations displays the same bias as the corresponding FST estimator. By construction, the kinship-based also has a large relative bias of about sT = 50%; remarkably, all existing FST estimators for subpopulations suffer from comparable biases. Thus, the corrections for independent subpopulations present in the WC, Weir-Hill and HudsonK estimators, or the Bayesian likelihood modeling of BayeScan, are insufficient for accurate estimation of the target generalized FST (Eq (9)) in this admixture scenario. Only our new FST estimator achieves accurate estimates of the generalized FST in the admixture simulation (Fig 4B).
Evaluation of kinship estimators
Our admixture simulation illustrates the distortions of the standard kinship estimator in Eq (18), the linearly-transformed kinship values given by the Weir-Goudet estimator, and demonstrates the improved accuracy of our new kinship estimator given by the combination of Eqs (34) and (37). Kinship matrix estimates and their limits are visualized as heatmaps in Fig 5, whereas estimator accuracy is shown directly in Fig 6. The limit of the standard estimator in Eq (18) would have had a uniform bias if held for all individuals j. For that reason, our admixture simulation has varying differentiation per intermediate subpopulation Su (Fig 3A), which causes large differences in per individual j and therefore large distortions in . The Weir-Goudet approach estimates the linearly-transformed kinship values calculated in Eq (38).
Fig 5. Evaluation of kinship estimators.

Observed accuracy for two existing kinship coefficient estimators is illustrated in our admixture simulation and contrasted to the nearly unbiased estimates of our new estimator. Plots show n = 1000 individuals along both axes, and color corresponds to between individuals j ≠ k and to along the diagonal ( is in the same scale as for j ≠ k; plotting , which have a minimum value of , would result in a discontinuity in this figure). (A) True kinship matrix. (B) Estimated kinship using our new estimator in Eqs (34) and (37) from simulated genotypes recovers the true kinship matrix with high accuracy. (C) Theoretical limit of in Eq (19) as the number of independent loci goes to infinity demonstrates the accuracy of our bias predictions under the kinship model. (D) Standard kinship estimates given by Eq (18) from simulated genotypes are downwardly biased on average and distorted by pair-specific amounts. (E) Theoretical limit of the Weir-Goudet kinship estimator given by Eq (38). (F) Weir-Goudet kinship estimates from the same simulated genotypes agree with our calculated limit.
Fig 6. Accuracy of kinship estimators.

Here the estimated kinship values are directly compared to their true values, in the same admixture simulation data (n = 1000 individuals) shown in the previous figure. (A) Kinship between different individuals (excluding inbreeding). The new estimator has practically no bias in this evaluation (falls on the 1-1 dashed gray line). The standard estimator has a complex, non-linear bias that covers a large area of errors. (B) Inbreeding comparison, shows the bias of the standard estimate follows a different pattern for inbreeding compared to kinship between individuals. To better visualize and compare data across panels, a random subset of n points (out of the original n(n − 1)/2 unique individual pairs) were plotted in (A), matching the number of individuals (number of points in (B)).
Our new kinship estimator (Fig 5B) recovers the true kinship matrix of this complex population structure (Fig 5A), with an RMSE of 2.83% relative to the mean (Fig 6). In contrast, estimates using the standard estimator have a large overall downward bias (Fig 5C), resulting in an RMSE of 115.72% from the true relative to the mean (Fig 6). Additionally, estimates from are very distorted, with an abundance of cases—some of which are negative estimates (blue in Fig 5C)—but remarkably also cases with (top left corner of Figs 5C and 6).
Now we compare the convergence of the ratio-of-means and mean-of-ratios versions of the standard kinship estimator to their biased limit we calculated in Eq (19) (Fig 5D). The ratio-of-means estimate (Fig 5C) has an RMSE of 2.14% from its limit relative to the mean . In contrast, the mean-of-ratios estimates that are prevalent in the literature have a greater RMSE of 10.77% from the same limit in Eq (19). Thus, as expected from our theoretical results in section Assessing the accuracy of genome-wide ratio estimators, the ratio-of-means estimate is much closer to the desired limit than the mean-of-ratio estimate. The distortions are similar for the estimator that uses IAFs in Eq (24), with reduced RMSEs from its limit of 0.32% and 8.82% for the ratio-of-means and mean-of-ratios estimates, respectively.
Evaluation of oracle-adjusted FST estimators
Here we verify additional calculations for the bias of the standard kinship-based estimator and the unbiased adjusted “oracle” FST estimators that require the true mean kinship or the bias coefficient sT to be known. Note that in Eq (36) is related but not identical to these oracle estimators. We tested both IAF (Fig 7A) and genotype (Fig 7B) versions of these estimators. The unadjusted in Eq (26) is severely biased (blue in Fig 7) by construction, and matches the calculated limit for IAFs and genotypes (green lines in Fig 7, which are close because ). In contrast, the two consistent adjusted estimators and in Eqs (27) and (31) estimate FST quite well (blue predictions overlap the true FST red line in Fig 7). However, and are oracle methods, since they require parameters (, , sT) that are not known in practice.
Fig 7. Evaluation of standard and adjusted FST estimators.

The convergence values we calculated for the standard kinship plug-in and adjusted FST estimators are validated using our admixture simulation. All adjusted estimators are unbiased but are “oracle” methods, since the mean kinship (), mean coancestry (), or bias coefficient ( for IAFs, replaced by for genotypes) are usually unknown. (A) Estimation from individual-specific allele frequencies (IAFs): is the standard coancestry plug-in estimator in Eq (26); “Adj. ” is in Eq (27); “Adj. s” is in Eq (31). (B) For genotypes, is given in Eq (25), and the adjusted estimators use rather than . Lines: true FST (red line), limits of biased estimators (green lines, which differ slightly per panel). Estimates (blue) include 95% prediction intervals (too narrow to see) from 39 independently-simulated genotype matrices for our admixture model (Methods, section Prediction intervals).
Prediction intervals were computed from estimates over 39 independently-simulated IAF and genotype matrices (Methods, section Prediction intervals). Estimator limits are always contained in these intervals because the number of independent loci (m = 300, 000) is sufficiently large. Estimates that use genotypes have wider intervals than estimates from IAFs; however, IAFs are not known in practice, and use of estimated IAFs might increase noise. Genetic linkage, not present in our simulation, will also increase noise in real data.
Discussion
We studied analytically the most commonly-used estimators of FST and kinship, which can be derived using the method of moments. We determined the estimation limits of convergence of these approaches under two models of arbitrary population structure (Fig 1). We found that no existing approaches estimate the generalized FST (an IBD probability) accurately (but note that some of these approaches intended to estimate a linearly-transformed FST quantity and not the IBD probability). We also showed that the standard kinship estimator is biased on structured populations (particularly when the average kinship is comparable to the kinship coefficients of interest), and this bias varies for each pair of individuals. These results led us to a new kinship estimator, which is consistent if the minimum kinship is estimated consistently (Fig 1). We presented an implementation of this approach, which is practically unbiased in our simulations. Our kinship and FST estimates in human data are consistent with the African Origins model while suggesting that human differentiation is considerably greater than previously estimated [59].
Estimation of FST in the correct scale is crucial for its interpretation as an IBD probability, for obtaining comparable estimates in different datasets and across species, as well as for DNA forensics [3, 7, 19, 20, 78–80]. Our framework results in a new unbiased genome-wide FST estimator. However, our findings may not have direct implications for single-locus FST estimate approaches where only the relative ranking matters, such as for the identification of loci under selection [8, 10, 81–86], assuming that the bias of the genome-wide estimator carries over uniformly to all single-locus estimates. Our convergence calculations in section Assessing the accuracy of genome-wide ratio estimators require large numbers of loci, so they do not apply to single-locus estimates. Moreover, various methods for single-locus FST estimation for multiple alleles suffer from a strong dependence to the maximum allele frequency and heterozygosity [83–85, 87–90] that suggests that a more complicated bias is present in these single-locus FST estimators.
We have shown that the misapplication of existing FST estimators for independent subpopulations may lead to downwardly-biased estimates that can approach zero even when the true generalized FST is large. Weir-Cockerham [17], Weir-Hill [4], HudsonK (which generalizes the Hudson pairwise FST estimator [23] to K independent populations; also equals the Weir-Goudet approach for subpopulations [21]; S1 Text), and BayeScan [10]FST estimates in our admixture simulation are all smaller than the FST target by nearly a factor of two (Fig 4B), and differ from our new FST estimates in humans by nearly a factor of three [59]. To be accurate, existing FST estimators require independent subpopulations, so the observed biases arise from their misapplication to subpopulations that are neither independent not homogeneous. Nevertheless, natural populations—particularly humans—often do not adhere to the independent subpopulations model [59, 91–95].
The standard kinship coefficient estimator we investigated is often used to control for population structure in GWAS and to estimate genome-wide heritability [16, 27, 30–35]. While this estimator was known to be biased [16, 35], no closed-form limit had been calculated until very recently [21, 62]. These kinship estimates are biased downwards on average, but bias also varies for each pair of individuals (Figs 1 and 5). Thus, the use of these distorted kinship estimates may be problematic in GWAS or for estimating heritability, but the extent of the problem remains to be determined.
We developed a theoretical framework for assessing genome-wide ratio estimators of FST and kinship. We proved that common ratio-of-means estimators converge almost surely to the ratio of expectations for infinite independent loci (S1 Text). Our result justifies approximating the expectation of a ratio-of-means estimator with the ratio of expectations [4, 17, 23]. However, mean-of-ratios estimators may not converge to the ratio of expectations for infinite loci. Mean-of-ratios estimators are potentially asymptotically unbiased for infinite individuals, but it is unclear which estimators have this behavior. We found that the ratio-of-means kinship estimator had much smaller errors from the ratio of expectations than the more common mean-of-ratios estimator, whose convergence value is unknown. Therefore, we recommend ratio-of-means estimators, whose asymptotic behavior is well understood.
Our new framework enables accurate FST estimation in more complex datasets than before, but challenges remain. One challenge is the estimation of local inbreeding coefficients, which are required for estimating the generalized FST when not all individuals are locally outbred. To this end, we suggest employing existing approaches that infer inbreeding from large runs of homozygosity or related strategies [66–68], particularly when such self-IBD blocks are much larger than observed between individuals in the same subpopulation. A streamlined approach for jointly estimating total and local inbreeding is desirable, but will require an appropriate evaluation featuring realistic simulation of local inbreeding in a complex population structure. Another challenge is the estimation of the minimum kinship value without the use of subpopulation labels, so that accurate FST estimates can be obtained with even less user supervision. A more general unsupervised method could better ensure accuracy under extreme cases, such as when there are few unrelated individual pairs. These challenges can be overcome with the estimators we have presented, although supervision is needed to ensure that local inbreeding and the minimum kinship are estimated correctly.
We have demonstrated the need for new models and methods to study complex population structures, and have proposed a new approach for kinship and FST estimation that provides nearly unbiased estimates in this setting. Extending our implementation to deliver consistent accuracy in arbitrary population structures will require further innovation, and the results provided here may be useful in leading to more robust estimators in the future.
Methods
Previous FST estimators for the independent subpopulations model
Here we summarize the previous Weir-Cockerham, Weir-Hill, and Hudson FST estimators for independent subpopulations and derive the generalized HudsonK estimator for more than two subpopulations (which also equals the recent Weir-Goudet FST estimator for subpopulations under biallelic loci; S1 Text). We show that each of these estimators reduces, under infinite subpopulation sizes, to in Eqs (11) to (13) that was studied in the results. In this section, let i index the m loci, j index the n subpopulations, nj be the number of individuals sampled from subpopulation j, and be the sample reference allele frequency at locus i in subpopulation j.
The Weir-Cockerham FST estimator
The Weir-Cockerham (WC) FST estimator [17] estimates the coancestry parameter θT shared by each of the n independent subpopulation in consideration. Let denote the fraction of heterozygotes in subpopulation j for locus i. The ratio-of-means WC FST estimator and its limit for independent subpopulations ( for j ≠ k) with equal differentiation () is
Note that above weighs every individual equally by weighing subpopulation j proportional to its sample size nj, so it equals the estimator in Eq (17) with uniform weights.
Now we simplify this estimator as the sample size of every subpopulation becomes infinite. First set the sample size of every subpopulation nj equal to their mean , which implies C2 = 0 and
Now we take the limit as the sample size , which results in sample allele frequencies converging to the true subpopulation allele frequencies for every subpopulation j and locus i, and
which matches the in Eqs (11) to (13) as desired. Note the number of subpopulations n remains finite, and the sample heterozygosity is not needed in the limit.
The Weir-Hill FST estimator
Weir and Hill developed new estimators for subpopulation-specific FST values and considered the effects of non-independent subpopulations [4]. However, these estimators target linearly-transformed FST values, and recover the FST defined in Eq (9) only when subpopulations are independent [4], so we group them here with other estimators that strictly assume independent subpopulations. For simplicity, here we only consider the global FST estimator; the estimators of the coancestry matrix of the subpopulations was found to have the same overall linear transformation [4]. In the limit of infinite subpopulation sizes, this estimator also converges to the asymptotic FST estimator for independent subpopulations () discussed in the main text.
The Weir-Hill (WH) FST estimator, simplified here for biallelic loci but extended to average over loci, and its limit, are given by
where the target FST and both weigh individuals (rather than subpopulations) equally [4]:
For equal sample sizes nj = nS∀j, we have , , and the estimator becomes
Therefore, as sample sizes per subpopulation go to infinity (nS → ∞, which results in for every (i, j)), we again recover the desired limiting FST estimator for independent subpopulations ( in Eqs (11) to (13)).
The Hudson FST estimator
The Hudson pairwise FST estimator [23] measures the differentiation of two subpopulations (j, k). The estimator and its limit for two independent subpopulations () is
| (40) |
Generalized HudsonK FST estimator
Here we derive the “HudsonK” estimator (first made available in [58]), which generalizes the Hudson pairwise FST estimator in Eq (40) to n independent subpopulations. This estimator also equals the recent Weir-Goudet FST estimator for subpopulations [21] (for biallelic loci; S1 Text). Note that for independent subpopulations, the FST of all the subpopulations equals the mean pairwise FST of every pair of subpopulations:
For that reason, averaging numerators and denominators of the pairwise estimator in Eq (40) before computing the ratio, we obtain the generalized estimator and a limit under independent subpopulations of
Note that unlike the WC and Weir-Hill estimators, above weighs every subpopulation equally, so every individual is weighed inversely proportional to the sample sizes nj of their subpopulation j.
Like WC and Weir-Hill, simplifies to in Eqs (11) to (13) in the limit of infinite sample sizes nj → ∞, where for every (i, j).
Simulations
Construction of subpopulation allele frequencies
We simulate K = 10 subpopulations Su and m = 300, 000 independent loci. Every locus i draws We set where τ ≤ 1 tunes FST. For the independent subpopulations model, so gives the desired FST (τ ≈ 0.18 for FST = 0.1). For the admixture model, τ is found numerically (τ ≈ 0.90 for FST = 0.1; see last subsection). Lastly, values are drawn from the Balding-Nichols distribution,
which results in subpopulation allele frequencies that obey the coancestry model of Eq (6), with and [3], as desired.
Random subpopulation sizes
We randomly generate sample sizes r = (ru) for K subpopulations and individuals, as follows. First, draw x ∼ Dirichlet (1, …, 1) of length K and r = round(n x). While , draw a new r, to prevent small subpopulations (they do not occur in real data). Due to rounding, may not equal n as desired. Thus, while , a random u is updated to ru ← ru + sgn(δ), which brings δ closer to zero at every iteration. Weights for individuals j in Su are so the generalized FST matches from the independent subpopulations model (see section The generalized FST for arbitrary population structures), which HudsonK estimates.
Admixture proportions from 1D geography
We construct qju from random-walk migrations along a one-dimensional geography. Let xu be the coordinate of intermediate subpopulation u and yj the coordinate of a modern individual j. We assume qju is proportional to f(|xu − yj|), or
where f is the Normal density function with μ = 0 and tunable σ. The Normal density models random walks, where σ sets the spread of the populations (Fig 5). Our simulation uses xu = u and , so the intermediate subpopulations span between 1 and K and individuals span between and . For the FST estimators that require subpopulations, individual j is assigned to the nearest subpopulation Su (the u that minimizes |xu − yj|; Fig 3D); these subpopulations have equal sample size, so is appropriate.
Choosing σ and τ
Here we find values for σ (controls qjk) and τ (scales ) that give and FST = 0.1 in the admixture model. In our simulation, and , so applying those parameters to Eq (39) gives and . Therefore,
depends only on σ. A numerical root finder finds that σ ≈ 1.78 gives . For fixed qju,
FST = 0.1 is achieved with τ ≈ 0.901.
Prediction intervals
Prediction intervals with α = 95% correspond to the range of n = 39 independent FST estimates. In the general case, n independent statistics are given in order X(1) < … < X(n). Then I = [X(j), X(n+1−j)] is a prediction interval with confidence [96]. In our case, j = 1 and n = 39 gives α = 0.95, as desired. Each estimate was constructed from simulated data with the same dimensions and structure as before (fixed and qju; fixed sample sizes for the independent subpopulations model), but with drawn separately for each estimate.
BayeScan and Weir-Goudet implementations
Weir-Goudet (WG) kinship estimates [20–22] were calculated using the function snpgdsIndivBeta in the R package SNPRelate 1.20.1 available on Bioconductor and GitHub. We found identical estimates using the function beta.dosage in the R package hierfstat 0.4.30 available on GitHub. WG (individuals) FST estimates were computed from the kinship estimates as described in section Comparison to the Weir-Goudet kinship estimator for individuals.
BayeScan 2.1 was downloaded from http://cmpg.unibe.ch/software/BayeScan/. To estimate FST, first the per-subpopulation FST values were estimated across loci assuming no selection, then the global FST was given by the mean FST across subpopulations.
Software
An R package called popkin, which implements the kinship and FST estimation methods proposed here, is available on the Comprehensive R Archive Network (CRAN) at https://cran.r-project.org/package=popkin and on GitHub at https://github.com/StoreyLab/popkin.
An R package called bnpsd, which implements the BN-PSD admixture simulation, is available on CRAN at https://cran.r-project.org/package=bnpsd and on GitHub at https://github.com/StoreyLab/bnpsd.
An R package called popkinsuppl, which implements memory-efficient algorithms for the Weir-Cockerham, Weir-Hill, and HudsonK FST estimators, and the standard kinship estimator, is available on GitHub at https://github.com/OchoaLab/popkinsuppl.
Public code reproducing these analyses are available at https://github.com/StoreyLab/human-differentiation-manuscript.
Supporting information
Includes mathematical proofs and other calculations, including proof of convergence of ratio-of-means estimators, proof that the Weir-Goudet FST estimator for subpopulations equals HudsonK, derivation of existing method-of-moment estimators, proof that FST and kinship estimator limits are constants with respect to the ancestral population T, mean coancestry bounds, moments of estimator building blocks, the derivation of our new kinship estimator, and proof that our estimator from our original 2016 manuscript is algebraically equivalent to the one presented here.
(PDF)
Data Availability
The data and computer code for these manuscripts can be found at https://github.com/StoreyLab/human-differentiation-manuscript.
Funding Statement
This research was supported in part by National Institutes of Health, National Human Genome Research Institute grant R01 HG006448 (JDS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Malécot G. Mathématiques de l’hérédité. Masson et Cie; 1948. [Google Scholar]
- 2.Wright S. The genetical structure of populations. Ann Eugen. 1951;15(4):323–354. [DOI] [PubMed] [Google Scholar]
- 3.Balding DJ, Nichols RA. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica. 1995;96(1-2):3–12. 10.1007/BF01441146 [DOI] [PubMed] [Google Scholar]
- 4.Weir BS, Hill WG. Estimating F-Statistics. Annual Review of Genetics. 2002;36(1):721–750. 10.1146/annurev.genet.36.050802.093940 [DOI] [PubMed] [Google Scholar]
- 5.Nicholson G, Smith AV, Jónsson F, Gústafsson O, Stefánsson K, Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. Journal of the Royal Statistical Society: Series B (Statistical Methodology). 2002;64(4):695–715. 10.1111/1467-9868.00357 [DOI] [Google Scholar]
- 6.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balding DJ. Likelihood-based inference for genetic correlation coefficients. Theoretical Population Biology. 2003;63(3):221–230. 10.1016/S0040-5809(03)00007-8 [DOI] [PubMed] [Google Scholar]
- 8.Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Molecular Ecology. 2004;13(4):969–980. 10.1111/j.1365-294X.2004.02125.x [DOI] [PubMed] [Google Scholar]
- 9.Foll M, Gaggiotti O. Identifying the Environmental Factors That Determine the Genetic Structure of Populations. Genetics. 2006;174(2):875–891. 10.1534/genetics.106.059451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180(2):977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coop G, Witonsky D, Rienzo AD, Pritchard JK. Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics. 2010;185(4):1411–1423. 10.1534/genetics.110.114819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson EA. The estimation of pairwise relationships. Ann Hum Genet. 1975;39(2):173–188. 10.1111/j.1469-1809.1975.tb00120.x [DOI] [PubMed] [Google Scholar]
- 13.Milligan BG. Maximum-likelihood estimation of relatedness. Genetics. 2003;163(3):1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacquard A. Structures génétiques des populations. Paris: Masson et Cie; 1970. [Google Scholar]
- 15.Csűrös M. Non-identifiability of identity coefficients at biallelic loci. Theor Popul Biol. 2014;92:22–29. 10.1016/j.tpb.2013.11.001 [DOI] [PubMed] [Google Scholar]
- 16.Astle W, Balding DJ. Population Structure and Cryptic Relatedness in Genetic Association Studies. Statist Sci. 2009;24(4):451–471. 10.1214/09-STS307 [DOI] [Google Scholar]
- 17.Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population Structure. Evolution. 1984;38(6):1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- 18.Weir BS, Cardon LR, Anderson AD, Nielsen DM, Hill WG. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 2005;15(11):1468–1476. 10.1101/gr.4398405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckleton J, Curran J, Goudet J, Taylor D, Thiery A, Weir BS. Population-specific FST values for forensic STR markers: A worldwide survey. Forensic Science International: Genetics. 2016;23:91–100. 10.1016/j.fsigen.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weir B, Zheng X. SNPs and SNVs in forensic science. Forensic Science International: Genetics Supplement Series. 2015;5(Dec):e267–e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weir BS, Goudet J. A Unified Characterization of Population Structure and Relatedness. Genetics. 2017;206(4):2085–2103. 10.1534/genetics.116.198424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goudet J, Kay T, Weir BS. How to estimate kinship. Mol Ecol. 2018;27(20):4121–4135. 10.1111/mec.14833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhatia G, Patterson N, Sankararaman S, Price AL. Estimating and interpreting FST: the impact of rare variants. Genome Res. 2013;23(9):1514–1521. 10.1101/gr.154831.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie C, Gessler DD, Xu S. Combining different line crosses for mapping quantitative trait loci using the identical by descent-based variance component method. Genetics. 1998;149(2):1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38(2):203–208. 10.1038/ng1702 [DOI] [PubMed] [Google Scholar]
- 26.Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177(1):577–585. 10.1534/genetics.107.075614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 28.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178(3):1709–1723. 10.1534/genetics.107.080101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, et al. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–354. 10.1038/ng.548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44(7):821–824. 10.1038/ng.2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rakovski CS, Stram DO. A kinship-based modification of the armitage trend test to address hidden population structure and small differential genotyping errors. PLoS ONE. 2009;4(6):e5825 10.1371/journal.pone.0005825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thornton T, McPeek MS. ROADTRIPS: case-control association testing with partially or completely unknown population and pedigree structure. Am J Hum Genet. 2010;86(2):172–184. 10.1016/j.ajhg.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Speed D, Balding DJ. Relatedness in the post-genomic era: is it still useful? Nat Rev Genet. 2015;16(1):33–44. 10.1038/nrg3821 [DOI] [PubMed] [Google Scholar]
- 36.Wang B, Sverdlov S, Thompson E. Efficient Estimation of Realized Kinship from SNP Genotypes. Genetics. 2017; p. genetics.116.197004. 10.1534/genetics.116.197004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright S. Systems of Mating. V. General Considerations. Genetics. 1921;6(2):167–178. 10.1093/genetics/6.2.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lush JL. Heritability of Quantitative Characters in Farm Animals. Hereditas. 1949;35(S1):356–375. 10.1111/j.1601-5223.1949.tb03347.x [DOI] [Google Scholar]
- 39.Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4th ed Harlow: Pearson; 1996. [Google Scholar]
- 40.Thompson EA. Identity by descent: variation in meiosis, across genomes, and in populations. Genetics. 2013;194(2):301–326. 10.1534/genetics.112.148825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slatkin M. Inbreeding coefficients and coalescence times. Genetics Research. 1991;58(2):167–175. 10.1017/S0016672300029827 [DOI] [PubMed] [Google Scholar]
- 42.Emik LO, Terrill CE. Systematic procedures for calculating inbreeding coefficients. J Hered. 1949;40(2):51–55. 10.1093/oxfordjournals.jhered.a105986 [DOI] [PubMed] [Google Scholar]
- 43.García-Cortés LA. A novel recursive algorithm for the calculation of the detailed identity coefficients. Genetics Selection Evolution. 2015;47(1):33 10.1186/s12711-015-0108-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, et al. Genetic Structure of Human Populations. Science. 2002;298(5602):2381–2385. 10.1126/science.1078311 [DOI] [PubMed] [Google Scholar]
- 45.Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci U S A. 2005;102(44):15942–15947. 10.1073/pnas.0507611102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Consortium TGP. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513(7518):409–413. 10.1038/nature13673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lazaridis I, Nadel D, Rollefson G, Merrett DC, Rohland N, Mallick S, et al. Genomic insights into the origin of farming in the ancient Near East. Nature. 2016;536(7617):419–424. 10.1038/nature19310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skoglund P, Posth C, Sirak K, Spriggs M, Valentin F, Bedford S, et al. Genomic insights into the peopling of the Southwest Pacific. Nature. 2016;538(7626):510–513. 10.1038/nature19844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, et al. The Genetic Structure and History of Africans and African Americans. Science. 2009;324(5930):1035–1044. 10.1126/science.1172257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moreno-Estrada A, Gravel S, Zakharia F, McCauley JL, Byrnes JK, Gignoux CR, et al. Reconstructing the Population Genetic History of the Caribbean. PLOS Genetics. 2013;9(11):e1003925 10.1371/journal.pgen.1003925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno-Estrada A, Gignoux CR, Fernández-López JC, Zakharia F, Sikora M, Contreras AV, et al. The genetics of Mexico recapitulates Native American substructure and affects biomedical traits. Science. 2014;344(6189):1280–1285. 10.1126/science.1251688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leslie S, Winney B, Hellenthal G, Davison D, Boumertit A, Day T, et al. The fine-scale genetic structure of the British population. Nature. 2015;519(7543):309–314. 10.1038/nature14230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baharian S, Barakatt M, Gignoux CR, Shringarpure S, Errington J, Blot WJ, et al. The Great Migration and African-American Genomic Diversity. PLoS Genet. 2016;12(5):e1006059 10.1371/journal.pgen.1006059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522(7555):207–211. 10.1038/nature14317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allentoft ME, Sikora M, Sjögren KG, Rasmussen S, Rasmussen M, Stenderup J, et al. Population genomics of Bronze Age Eurasia. Nature. 2015;522(7555):167–172. 10.1038/nature14507 [DOI] [PubMed] [Google Scholar]
- 57.Ochoa A, Storey JD. FST and kinship for arbitrary population structures I: Generalized definitions. bioRxiv. 2016; 10.1101/083915 [DOI] [PMC free article] [PubMed]
- 58.Ochoa A, Storey JD. FST and kinship for arbitrary population structures II: Method of moments estimators. bioRxiv. 2016; 10.1101/083923 [DOI]
- 59.Ochoa A, Storey JD. New kinship and FST estimates reveal higher levels of differentiation in the global human population. bioRxiv. 2019; 10.1101/653279 [DOI]
- 60.Thornton T, Tang H, Hoffmann TJ, Ochs-Balcom HM, Caan BJ, Risch N. Estimating kinship in admixed populations. Am J Hum Genet. 2012;91(1):122–138. 10.1016/j.ajhg.2012.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao W, Song M, Storey JD. Probabilistic models of genetic variation in structured populations applied to global human studies. Bioinformatics. 2016;32(5):713–721. 10.1093/bioinformatics/btv641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng X, Weir BS. Eigenanalysis of SNP data with an identity by descent interpretation. Theoretical Population Biology. 2016;107:65–76. 10.1016/j.tpb.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol. 2005;28(4):289–301. 10.1002/gepi.20064 [DOI] [PubMed] [Google Scholar]
- 65.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browning BL, Browning SR. A Fast, Powerful Method for Detecting Identity by Descent. The American Journal of Human Genetics. 2011;88(2):173–182. 10.1016/j.ajhg.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gazal S, Sahbatou M, Perdry H, Letort S, Génin E, Leutenegger AL. Inbreeding Coefficient Estimation with Dense SNP Data: Comparison of Strategies and Application to HapMap III. HHE. 2014;77(1-4):49–62. [DOI] [PubMed] [Google Scholar]
- 68.Joshi PK, Esko T, Mattsson H, Eklund N, Gandin I, Nutile T, et al. Directional dominance on stature and cognition in diverse human populations. Nature. 2015;523(7561):459–462. 10.1038/nature14618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cochran WG. Sampling techniques. 3rd ed Wiley; 1977. [Google Scholar]
- 70.Reynolds J, Weir BS, Cockerham CC. Estimation of the Coancestry Coefficient: Basis for a Short-Term Genetic Distance. Genetics. 1983;105(3):767–779. 10.1093/genetics/105.3.767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weir BS. Genetic data analysis II Methods for discrete population genetic data. Sunderland, USA: Sinauer Associates; 1996. [Google Scholar]
- 72.Bourgain C, Hoffjan S, Nicolae R, Newman D, Steiner L, Walker K, et al. Novel case-control test in a founder population identifies P-selectin as an atopy-susceptibility locus. Am J Hum Genet. 2003;73(3):612–626. 10.1086/378208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi Y, Wijsman EM, Weir BS. Case-Control Association Testing in the Presence of Unknown Relationships. Genet Epidemiol. 2009;33(8):668–678. 10.1002/gepi.20418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nei M. Analysis of Gene Diversity in Subdivided Populations. PNAS. 1973;70(12):3321–3323. 10.1073/pnas.70.12.3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weir BS, Goudet J. A unified characterization of population structure and relatedness. bioRxiv. 2016; p. 088260. [DOI] [PMC free article] [PubMed]
- 77.Raj A, Stephens M, Pritchard JK. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014;197(2):573–589. 10.1534/genetics.114.164350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nelis M, Esko T, Mägi R, Zimprich F, Zimprich A, Toncheva D, et al. Genetic Structure of Europeans: A View from the North–East. PLOS ONE. 2009;4(5):e5472 10.1371/journal.pone.0005472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silva NM, Pereira L, Poloni ES, Currat M. Human Neutral Genetic Variation and Forensic STR Data. PLOS ONE. 2012;7(11):e49666 10.1371/journal.pone.0049666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steele CD, Court DS, Balding DJ. Worldwide FST Estimates Relative to Five Continental-Scale Populations. Annals of Human Genetics. 2014;78(6):468–477. 10.1111/ahg.12081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cavalli-Sforza LL. Population Structure and Human Evolution. Proceedings of the Royal Society of London Series B, Biological Sciences. 1966;164(995):362–379. [DOI] [PubMed] [Google Scholar]
- 82.Lewontin RC, Krakauer J. Distribution of Gene Frequency as a Test of the Theory of the Selective Neutrality of Polymorphisms. Genetics. 1973;74(1):175–195. 10.1093/genetics/74.1.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beaumont MA, Nichols RA. Evaluating Loci for Use in the Genetic Analysis of Population Structure. Proceedings of the Royal Society of London B: Biological Sciences. 1996;263(1377):1619–1626. 10.1098/rspb.1996.0237 [DOI] [Google Scholar]
- 84.Vitalis R, Dawson K, Boursot P. Interpretation of Variation Across Marker Loci as Evidence of Selection. Genetics. 2001;158(4):1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12(12):1805–1814. 10.1101/gr.631202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Porter AH. A test for deviation from island-model population structure. Molecular Ecology. 2003;12(4):903–915. 10.1046/j.1365-294X.2003.01783.x [DOI] [PubMed] [Google Scholar]
- 87.Bowcock AM, Kidd JR, Mountain JL, Hebert JM, Carotenuto L, Kidd KK, et al. Drift, admixture, and selection in human evolution: a study with DNA polymorphisms. PNAS. 1991;88(3):839–843. 10.1073/pnas.88.3.839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hedrick PW. A Standardized Genetic Differentiation Measure. Evolution. 2005;59(8):1633–1638. 10.1111/j.0014-3820.2005.tb01814.x [DOI] [PubMed] [Google Scholar]
- 89.Jakobsson M, Edge MD, Rosenberg NA. The Relationship Between FST and the Frequency of the Most Frequent Allele. Genetics. 2013;193(2):515–528. 10.1534/genetics.112.144758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Edge MD, Rosenberg NA. Upper bounds on FST in terms of the frequency of the most frequent allele and total homozygosity: the case of a specified number of alleles. Theor Popul Biol. 2014;97:20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lewontin RC. The Apportionment of Human Diversity. Evolutionary Biology. 1972;6:381–398. [Google Scholar]
- 92.Barbujani G, Magagni A, Minch E, Cavalli-Sforza LL. An apportionment of human DNA diversity. PNAS. 1997;94(9):4516–4519. 10.1073/pnas.94.9.4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, et al. Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. 10.1038/nature07331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coop G, Pickrell JK, Novembre J, Kudaravalli S, Li J, Absher D, et al. The Role of Geography in Human Adaptation. PLoS Genet. 2009;5(6):e1000500 10.1371/journal.pgen.1000500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192(3):1065–1093. 10.1534/genetics.112.145037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beran R, Hall P. Interpolated Nonparametric Prediction Intervals and Confidence Intervals. Journal of the Royal Statistical Society Series B (Methodological). 1993;55(3):643–652. 10.1111/j.2517-6161.1993.tb01929.x [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Includes mathematical proofs and other calculations, including proof of convergence of ratio-of-means estimators, proof that the Weir-Goudet FST estimator for subpopulations equals HudsonK, derivation of existing method-of-moment estimators, proof that FST and kinship estimator limits are constants with respect to the ancestral population T, mean coancestry bounds, moments of estimator building blocks, the derivation of our new kinship estimator, and proof that our estimator from our original 2016 manuscript is algebraically equivalent to the one presented here.
(PDF)
Data Availability Statement
The data and computer code for these manuscripts can be found at https://github.com/StoreyLab/human-differentiation-manuscript.