

Abstract
Introduction---
Cyclic nucleotides, cAMP and cGMP, are important second messengers of intracellular signaling and play crucial roles in cardiovascular biology and diseases. Cyclic nucleotide phosphodiesterases (PDEs) control the duration, magnitude, and compartmentalization of cyclic nucleotide signaling by catalyzing the hydrolysis of cyclic nucleotides. Individual PDEs modulate distinct signaling pathways and biological functions in the cell, making it a potential therapeutic target for the treatment of different cardiovascular disorders. The clinical success of several PDE inhibitors has ignited continued interest in PDE inhibitors and in PDE-target therapeutic strategies.
Areas covered---
This review concentrates on recent research advances of different PDE isoforms with regard to their expression patterns and biological functions in the heart. The limitations of current research and future directions are then discussed. The current and future development of PDE inhibitors is also covered.
Expert opinion---
Despite the therapeutic success of several marketed PDE inhibitors, the use of PDE inhibitors can be limited by their side effects, lack of efficacy, and lack of isoform selectivity. Advances in our understanding of the mechanisms by which cellular functions are changed through PDEs may enable the development of new approaches to achieve effective and specific PDE inhibition for various cardiac therapies.
Keywords: Cyclic nucleotide, phosphodiesterase (PDE), cardiac diseases, PDE inhibitor
1. Introduction
Canonical cyclic nucleotides, cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), regulate numerous biological processes in the heart, including inotropic/chronotropic functions, metabolism and structural remodeling. cAMP can be generated through membrane adenylyl cyclases (mACs) upon the stimulation of various G-protein coupled receptors (GPCRs). cAMP can also be generated by soluble AC (sAC) in response to cellular HCO−3/CO−2/pH in the cytoplasm, nucleus, or mitochondria. cGMP can be produced by activating soluble guanylyl cyclases (sGCs) by nitric oxide (NO)/carbon monoxide (CO) or particulate guanylyl cyclases (pGCs) by natriuretic peptides (NPs). cAMP/cGMP degradation is catalyzed by a large family of PDEs with more than 100 different PDE variants derived from 21 genes and grouped into 11 broad families (PDE1-PDE11) [1]. Common cAMP and cGMP effector molecules in the heart include cAMP-dependent protein kinase (PKA), exchange protein activated by cAMP (Epac), and cGMP-dependent protein kinase (PKG). Under normal conditions, PKA-mediated phosphorylation of ion channels and contractile proteins are essential for increasing heart rate and contractility in response to fight-or-flight. For example, PKA phosphorylates L-type Ca2+ channels (LTCC) and ryanodine receptor (RYR) to induce intracellular Ca2+ elevation, thus enhancing myocyte contractility [2]. In addition, PKA phosphorylates phospholamban (PLB) and induces Ca2+ re-uptake in the sarcoplasmic reticulum (SR), leading to faster myofilament relaxation [2]. Abnormal cyclic nucleotide synthesis, degradation, and signaling have been implicated in a number of cardiac diseases including atrial fibrillation, maladaptive cardiac remodeling, and cardiac dysfunction [3].
Increasing evidence has indicated the existence of multiple functionally distinct “pools” of cyclic nucleotides. For example, catecholamines, prostaglandin 2 (PGE2), and adenosine are all able to elevate cAMP elevation in cardiomyocytes (CMs). cAMP generation through β-adrenergic receptor (β-AR) activation by catecholamine stimulates profound CM contraction, while cAMP elevation by PGE2 has a limited role in CM contractility [4]. Chronic activation of β1-AR-derived cAMP signaling elicits detrimental effects such as promoting CM hypertrophy and apoptosis [5,6], while cAMP signaling through activation of adenosine receptors is protective [7,8]. In addition, cAMP produced by adenylyl cyclase 5 (AC5) and AC6 have different cardiac effects: AC5-derived cAMP is detrimental [9,10], whereas AC6-derived cAMP is protective in pathological cardiac remodeling [11,12]. Moreover, modulation of distinct cyclic nucleotide signaling pathways via inhibition of different PDEs elicit distinct effects on CM viability: PDE1 inhibition promotes CM survival [13,14]; PDE3 inhibition promotes CM death [15]; and PDE4 inhibition has no significant effect on CM viability [15]. These differences might be due to different PDEs coupling to distinct cyclic nucleotide-dependent signalosomes. Although most studies have shown negative inotropic [16–20] and cardioprotective effects [21,22] of cGMP in the heart, cGMP from two different sources may have different mechanisms of action [23]. These lines of evidence indicate that cyclic nucleotide signaling events arising from different origins have distinct/unique or even opposing biological functions.
There has been increasing interest and effort to understand how the versatility/specificity of diverse cyclic nucleotide-mediated functions are achieved in individual cell types. In the past decade, approaches using subcellular-targeted Fluorescence Resonance Energy Transfer (FRET)-based cAMP/cGMP sensors have significantly advanced this field by demonstrating that cAMP/cGMP are not freely diffusible and multiple compartmentalized cAMP/cGMP “pools” exist in the cell [24–28] (for detailed reviews, see references [29–31]). It is believed that the versatility/specificity of cAMP/cGMP signaling is achieved through compartmentalization of diverse, discrete cAMP/cGMP pools that are associated with different multi-protein complexes each containing unique cyclases, PDEs, kinases, and other signaling molecules, leading to different biological functions (Figure 1).
Figure 1: Schematic representation of cyclic nucleotide and PDE compartmentalization in cardiac myocyte.
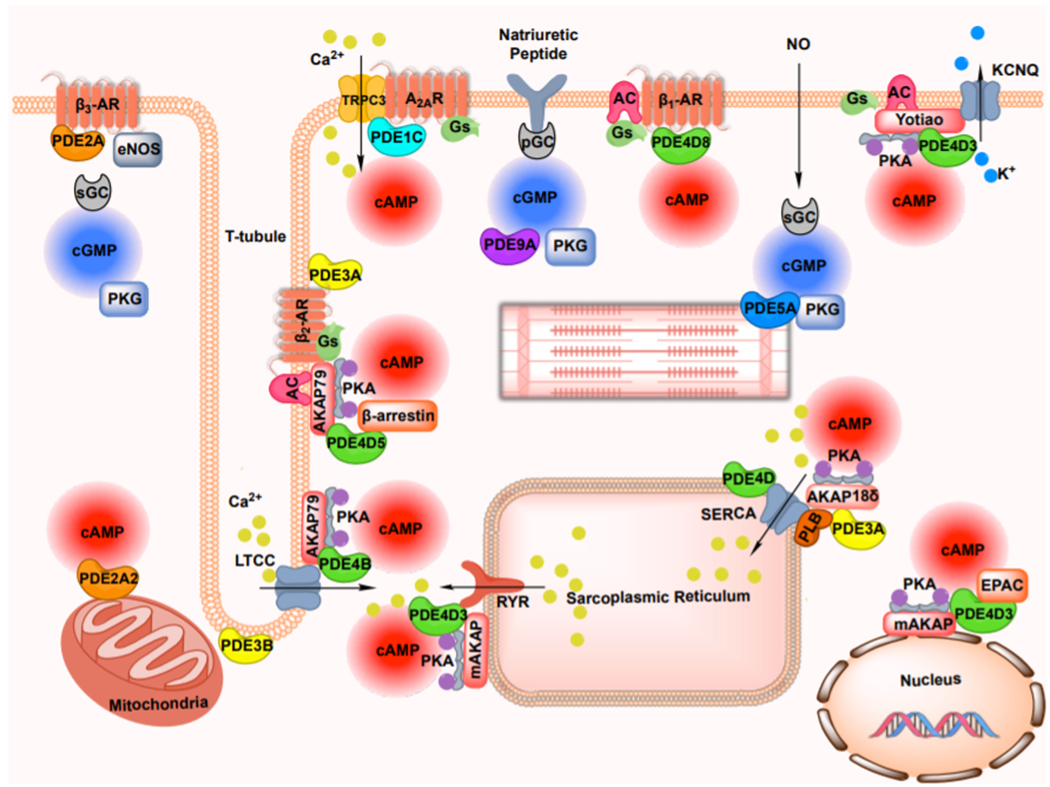
Compartmentalized cyclic nucleotide signaling is generated by individual G-protein coupled receptors, and established via the formation of multiple spatially segregated signalosomes, in which combination of cyclic nucleotide effectors, phosphodiesterase and scaffolding proteins form specific complexes via protein-protein interaction. PDE, phosphodiesterase; AR, adrenergic receptor; AC, adenylyl cyclase; sGC, soluble guanylyl cyclase; pGC, particulate guanylyl cyclase; PKA, protein kinase A; PKG, protein kinase G; cAMP, cyclic adenosine 3′,5′-monophosphate; cGMP, cyclic guanosine 3′,5′-monophosphate; LTCC, L-type calcium current; PLB, phospholamban; RYR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+ ATPase; AKAP, A-kinase anchor protein; TRPC, transient receptor potential subfamily C; A2AR, adenosine type 2A receptor; KCNQ, voltage-gated potassium channels of the KCNQ subfamily; NO, nitric oxide; EPAC, exchange protein activated by cAMP; eNOS, endothelial nitric oxide synthase.
2. Cardiac PDEs
2.1. Overview of cardiac PDEs
Among 11 PDE families, PDE1-5, and PDE8-10 have been reported in the heart [32–40]. PDE1, 2, 3, and 10 hydrolyze both cAMP and cGMP with different affinities and rates. PDE4 and 8 are cAMP-specific PDEs. PDE5 and 9 are cGMP specific PDEs. The relative expression of PDE and the contribution of PDE activity in the myocardium may be varied with species. Dysregulation in PDE expression, activation and subcellular localization affect cardiac function and have been implicated in a number of cardiac diseases. An overview of the isoforms, substrates, kinetic properties, regulatory properties and expression levels among different species of these PDEs is summarized in Table 1.
Table 1.
Biochemical Characteristics of Cardiac PDEs
|
Km(μM) |
||||||
|---|---|---|---|---|---|---|
| Family | Gene | Substrate | cAMP | cGMP | Regulators | Species-dependent expression in the heart |
| PDE1 | 1A | cAMP,cGMP | 115 | 5 | +Ca2+/CaM | • equally expressed in human, rat and mouse hearts, and both CMs an CFs • upregulated in mouse, rat and human failing hearts • upregulated in rat CMs by hypertrophic stimuli • upregulated in rat CFs by fibrotic stimuli |
| 1B | cAMP,cGMP | 24 | 2.4 | • not detected in normal human, rat and mouse hearts | ||
| 1C | cAMP,cGMP | 1 | 1 | • expressed highest in human, modest in mouse, very low in rat hearts, and in mouse CMs • increased in failing human and mouse hearts, primarily restricted in CMs |
||
| PDE2 | 2A | cAMP,cGMP | 30 | 20 | +CGMP | • detected in human, rat and mouse hearts, and both CMs and CFs, with higher expression in CFs than CMs • upregulated in human and mouse failing hearts |
| PDE3 | 3A | cAMP,cGMP | 0.15 | 0.15 | −cGMP | • expressed in human, rat, dog and mouse hearts and CMs• increase or decrease in diseased hearts, varied by studies |
| 3B | • expressed in mouse hearts and CMs | |||||
| PDE4 | 4A | cAMP | 5 | N/A | +PKA, −ERK | • expressed in human, rat and mouse hearts and CMs • decreased expression in human failing hearts and rat hypertrophic CMs |
| 4B | • expressed in human, rat and mouse hearts and CMs • decreased expression in human failing hearts and rat hypertrophic CMs |
|||||
| 4C | • not detected in normal rat heart | |||||
| 4D | • expressed in human, rat and mouse hearts and CMs • decreased expression in human and rat failing hearts and rat hypertrophic CMs |
|||||
| PDE5 | 5A | cGMP | N/A | 5 | +PKG | • expressed in human, rat and mouse hearts, and both CMs and CFs • upregulated in human, rat and mouse failing hearts, and diseased CMs |
| PDE8 | 8A | cAMP | 0.05 | N/A | • expressed in human and mouse hearts, and mouse CMs | |
| 8B | • expressed in human and mouse hearts, and mouse CMs | |||||
| PDE9 | 9A | cGMP | 0.1 | • expressed in human and mouse hearts, and CMs • upregulated in human and mouse failing hearts |
||
| PDE10 | 10A | cAMP,cGMP | 0.3 | 13 | +CAMP | • expressed in human and mouse hearts, and both CMs and CFs • upregulated in human and mouse failing hearts • upregulated in mouse CMs by hypertrophic stimuli • upregulated in mouse CFs by fibrotic stimuli |
2.1.1. Cardiac dual-substrate PDEs
Cardiac dual-substrate PDEs include PDE1, PDE2, PDE3 and PDE10. These PDEs hydrolyze both cAMP and cGMP through a catalytic domain, but some of them can be regulated through a GAF domain by either cAMP or cGMP. The PDE1 family members are encoded by three different genes (PDE1A, PDE1B and PDE1C), and activated by Ca2+/calmodulin (CaM). The PDE2 family members are encoded by one gene, PDE2A, with 3 different splice variants (PDE2A1-3). By binding to the N-terminal GAF domain, cGMP is able to stimulate PDE2 activity, thus is referred to as cGMP-stimulated PDEs. The PDE3 family members are encoded by two different genes (PDE3A and PDE3B). Three different PDE3A variants (PDE3A1-3) are defined. PDE3 binds to both cAMP and cGMP with high affinities, but the Vmax for hydrolyzing cGMP is at least 10-fold lower than that of cAMP, which allows cGMP to act as a potent competitive inhibitor of cAMP hydrolysis by PDE3. However, this inhibitory effect of cGMP is mainly for PDE3A [41,42], as PDE3B is only ≈10% as sensitive to cGMP inhibition as PDE3A [43]. Both PKA and protein kinase B (PKB) can induce a phosphorylation of PDE3 to stimulate PDE3 activity [1]. The PDE10 family members are encoded by one gene (PDE10A) with three splice variants identified (PDE10A1-3) [44–46]. It has been shown that PKA phosphorylates PDE10A and induces its translocation from membrane to cytosol [47,48]. It has also been reported that cAMP binds to the N-terminal GAF domains and increases PDE10A activity [49].
2.1.2. Cardiac cAMP-specific PDEs
PDE4 is encoded by 4 different genes (PDE4A, PDE4B, PDE4C and PDE4D), with a number of different variants [50]. PDE4 family members are subdivided into 4 groups depending on the length of the N-terminal upstream conserved regions (UCRs): long isoform; short isoform; super-short isoform and dead-short isoform [50]. UCRs mediate the homo- and hetero-dimerization of long isoform PDE4 [51]. UCRs also play a functional role in modulating the activity of PDE4 catalytic unit [50]. The long forms can be phosphorylated by PKA [1]. The catalytic domains of PDE4B, 4C, and 4D isozymes contain ERK phosphorylation motifs, and ERK phosphorylation leads to an inhibition of their activities [1]. PDE8 family members are encoded by two different genes (PDE8A and PDE8B) [52]. PDE8 includes N-terminal REC (receiver) and PAS (Per, Arnt and Sim) domains and C-terminal catalytic domain, even though REC and PAS domains are not completed in some variants [53,54]. The regulatory functions of PDE8 REC and PAS domains are not well understood. PDE8 has the highest cAMP affinity among all PDEs, and is the only PDE family insensitive to pan PDE inhibitor IBMX [1].
2.1.3. Cardiac cGMP-specific PDEs
PDE5 is encoded by a single gene PDE5A, which gives rise to 3 variants (PDE5A1-3) [55]. PDE5A variants are different in their N-terminus, which contains two GAF domains: GAF-A and GAF-B [55,56]. PDE5A is activated by cGMP binding to the GAF-A domain and cGMP-dependent kinase (PKG) phosphorylation of a serine near the GAF-A domain, which is critical for PDE5A in negative feedback regulation of cGMP signaling [55]. PDE9 is encoded by a single gene PDE9A, with more than 20 variants reported [57,58].
2.2. Regulation and function of PDEs in heart
2.2.1. PDE1
PDE1 activity is one of the major PDE activities in the human myocardium [59,60]. PDE1 inhibitor IC86340 or vinpocetine attenuates cardiac hypertrophy and fibrosis in mouse hearts induced by chronic neurohormonal stimulation with isoproterenol (ISO) or angiotensin II (Ang II), respectively [61,62]. These findings suggest a critical role of PDE1 in pathological cardiac remodeling and the therapeutic potential of targeting PDE1 in cardiac diseases associated with cardiac remodeling. Among the three PDE1 genes, the expression of PDE1A and PDE1C, but not PDE1B, has been detected in hearts from various species (Table 1) [32]. PDE1A expression appears to be consistent among human, rat and mouse hearts [32]. PDE1A expression is upregulated in hearts with hypertrophy or heart failure (HF) in various species, predominantly in CMs and activated cardiac fibroblasts (CFs) [32,61]. Differently, PDE1C expression in the heart varies with species: highest in human, modest in mouse, and very low in rat hearts [32]. PDE1C expression is further increased in failing human and mouse hearts, primarily restricted in CMs [33]. There is no PDE1C expression detected in mouse CFs under both basal and stimulated states [33]. These findings suggest different PDE1 isozymes are differentially expressed in cardiac cells.
In cultured rat neonatal or adult CMs, PDE1A expression is upregulated by hypertrophic stimuli such as Ang II or ISO [32]. CM hypertrophy stimulated by phenylephrine (PE) or Ang II is significantly alleviated by specifically knocking down Pde1a with shRNAs or PDE1 inhibitor IC86340 [32]. Although PDE1A is able to hydrolyze cAMP and cGMP in the cell-free system, PDE1A was found to primarily hydrolyze cGMP in rat CMs [32]. Consistently, PDE1A inhibition stimulates PKG activation and PKG is a critical mediator for the anti-hypertrophic effects of PDE1A inactivation [32]. These results indicate that PDE1A activation plays a critical role in CM hypertrophy by antagonizing the cGMP/PKG signaling. Interestingly, PDE1A does not regulate CM viability [33]. In addition to CMs, PDE1A upregulation was found in activated CFs within fibrotic regions of diseased hearts in vivo as well as in cultured CFs stimulated by Ang II or TGF-β in vitro [61]. Pde1a shRNA or PDE1 inhibitor IC86340 blocks Ang II or TGF-β-induced CF activation, extracellular matrix (ECM) synthesis, and pro-fibrotic gene expression in rat CFs [61]. PDE1A hydrolyzes both cAMP and cGMP in CFs. The effect of PDE1A inhibition against CF activation is dependent on cAMP/Epac1/Rap1 and cGMP/PKG signaling [61]. Despite these studies demonstrating critical roles of PDE1A in rat CMs and CFs in vitro, the role of PDE1A in cardiac disease development in vivo remains unknown. Future studies using Pde1a knockout (KO) or conditional KO mice are required to address the functional role of PDE1A in vivo. In addition, the sources of cyclic nucleotides that are regulated by PDE1A in CMs and CFs also remain to be determined. Moreover, the molecular mechanisms underlying PDE1A-mediated regulation of CM hypertrophy and CF activation remain to be characterized.
With the availability of global Pde1c KO mice [33], the roles of PDE1C in the heart have been studied in mouse cardiac disease models. For example, it has been shown that Pde1c KO alleviates cardiac hypertrophy, interstitial fibrosis, and dysfunction induced by chronic pressure overload via transverse aortic constriction (TAC) in mice [33]. In vitro, Pde1c KO and inhibition abolishes mouse adult CM hypertrophy and apoptosis [33], which appears to be dependent on cAMP/PKA signaling [33]. PDE1C does not directly regulate CF function in isolated CFs, which is consistent with the fact that PDE1C is not expressed in CFs [33]. Interestingly, the conditioned medium from Pde1c-KO CMs, but not Pde1c-WT CMs, attenuates CF activation [33], suggesting that PDE1C regulates CM-derived secreted factors that mediate CF activation and cardiac fibrosis. Recently, the mechanistic action of PDE1C in regulating CM viability has been further studied in vitro and in vivo [14]. It has been shown that in CMs, PDE1C but not PDE1A regulates CM viability [14]. PDE1C promotes CM death/apoptosis through antagonizing the adenosine type 2A receptor (A2AR)/cAMP signaling [14]. In particular, a novel multiprotein complex comprised of A2AR, PDE1C, and transient receptor potential subfamily C3 (TRPC3) has been characterized in the plasma membrane and perhaps T tubules of CMs (Figure 1) [14]. It is believed that PDE1C is activated by TRPC3-derived Ca2+ and subsequently antagonizes A2AR/cAMP signaling, thereby promoting CM death/apoptosis. Indeed, Pde1c KO or PDE1C inhibition attenuates doxorubicin-induced cardiac toxicity and dysfunction in mice, which is significantly diminished by A2AR antagonism [14]. Another study has reported the acute positive inotropic and lusitropic effects of a pan PDE1 inhibitor ITI-214 in dogs and rabbits with HF [63]. The effects of ITI-214 appear related to the cAMP signaling that is different from β-AR but related to A2BR [63]. It is believed that acute cardiac effects are mediated by inhibiting PDE1C. Taken together, these findings provide novel evidence for microdomain regulation of A2R-derived cAMP signaling by PDE1C in CMs.
2.2.2. PDE2
The expression of PDE2 has been found in different species including human, rat and mouse heart (Table 1) [64–66]. PDE2A has been detected in both CMs and CFs in the heart, with higher expression in CFs than CMs [64,67]. PDE2 expression is altered under pathological situations. For example, PDE2A protein levels are significantly increased in the left ventricular myocardium from patients with end-stage dilated cardiomyopathy or ischemic cardiomyopathy as well as in rat hearts after chronic β-AR stimulation [65].
PDE2A is important in acute cardiac contractility, CM death, and chronic cardiac remodeling. It has been reported that PDE2A modulates L-type Ca2+ current in frog ventricular CMs and human atrial CMs in vitro [68,69]. PDE2 inhibition increases the inotropic effects of β2-AR stimulation in rat left ventricular myocardium ex vivo, likely by potentiating the β2-AR/cAMP signaling [70]. NO/cGMP by activating PDE2A blunts the β-AR mediated cardiac inotropic effect [71]. Consistently, PDE2A upregulation in failing hearts desensitizes against acute β-AR responsiveness [65]. These results suggest that PDE2A regulates CM contractility by primarily regulating β-AR/cAMP signaling. Chronic inhibition of PDE2 with BAY 60-7550 has been shown to attenuate CM hypertrophic growth in vitro and TAC-induced cardiac hypertrophy in vivo, which appears to be mediated by cAMP/PKA dependent phosphorylation of nuclear factor of activated T cells (NFAT) [34]. This anti-hypertrophic effect of PDE2A inhibition appears not to take place through regulating β-AR signaling because increasing β-AR induces cardiac hypertrophy [2]. Another study showed that PDE2 inhibition reverses the cardiac remodeling and HF induced by chronic pressure overload or sympathetic hyperactivation, through promoting sGC/cGMP signaling [66]. Interestingly, a recent study reported a PDE2A2 variant in mitochondrial outer membranes of CM, where PDE2A2 regulates cAMP generated at the plasma membrane by plasma membrane AC (Figure 1) [72]. Specifically inhibiting mitochondrial PDE2A2 with genetic tools promotes PKA-dependent phosphorylation of dynamin-related protein 1 (Drp1), alters mitochondrial morphology, and thus triggers mitochondria-dependent cell death in rat neonatal CM [72]. This finding suggests that different PDE2A variants may localize and function differently in CMs.
However, using transgenic mice with CM-specific overexpression of PDE2A3 (Tg-Pde2a3) has shown inconsistent results [73]. It was found that the Tg-Pde2a3 mice have a decreased heart rate without altering the cardiac inotropic effects, thus protecting against catecholamine-induced ventricular arrhythmia [73]. CMs isolated from Tg-Pde2a3 mice reveal a remarkable reduction of Ca2+ leakage and of basal phosphorylation levels of ryanodine receptor type 2 (RyR2), which may contribute to the anti-arrhythmic effects of PDE2A expression [73]. In addition, Tg-Pde2a3 mice exhibit improved ventricular function and increased animal survival rate after myocardial infarction (MI) for 14 days without significant change of infarct sizes [73]. There are a number of possibilities for the discrepancy between PDE2A inhibition and PDE2A overexpression. One possibility could be due to the undesired effects resulted from mistargeting of overexpressed PDE2A. The second possibility could be due to multiple existing PDE2A variants that have different subcellular localizations and distinct functions. PDE2 inhibitors target all PDE2A variants while only one variant was altered in the transgenic mice. The third possibility could be that the cardiac protective effects seen in global Pde2a-KO mice are also contributed by PDE2A in non-CM of the heart such as CF, endothelial cells (ECs) and macrophages. For example, it was reported that CFs express higher levels of PDE2A compared to CMs [67]. PDE2 accelerates CF to myofibroblast phenotype conversion [67]. It was also reported that the induction of PDE2A expression by tumor necrosis factor-α contributes to increased EC permeability [74]. These non-CM effects are important in cardiac remodeling and dysfunction. Nevertheless, cardiac cell-specific Pde2a KO mice and/or genetically engineered Pde2a variant-specific mutant mice are required to further characterize the roles and cellular/molecular mechanisms of PDE2A in cardiac biology and diseases.
2.2.3. PDE3
Both PDE3A and PDE3B have been reported in heart tissue and CMs (Table 1) [35,75,76]. PDE3A is highly expressed in the myocardium of normal hearts [35]. But the changes of PDE3A expression in diseased hearts appear to be varied in different studies. For example, decreased PDE3A expression and/or activity has been reported in failing mouse and human hearts [35,77], pacing-induced failing dog hearts [78,79], and CMs isolated from rat hypertrophic hearts [35,80]. However, increased PDE3A expression and/or activity has also been described in failing rat hearts [81], failing mouse hearts [82], and ISO-induced hypertrophic mouse hearts [83]. The discrepancies may be related to the differential regulation of PDE3A expression in different cell types in the heart. PDE3A is expressed in CMs, CFs, ECs, smooth muscle cells (SMCs), immune cells, and platelets, all of which can be changed in diseased hearts.
PDE3 activity has been first known to regulate cardiac contractile function, which is the basis for using PDE3 inhibitors to treat congestive heart failure. Although PDE3 inhibitors improve cardiac function, they increase mortality in HF patients [84]. The development of Pde3a and Pde3b KO mice have helped to define PDE3A, but not PDE3B, as primarily responsible for regulating the contractile function in the heart [85,86]. Global Pde3a KO increases cardiac contractility and relaxation through cAMP-dependent elevations of Ca2+ transient amplitudes and SR Ca2+ contents [86]. This is consistent with increased PLB phosphorylation, sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity, and SR Ca2+ uptake rate by Pde3a KO [86]. The role of PDE3A in regulating SERCA2 function is further supported with the finding of a multiprotein complex in the sarcoplasmic reticulum (SR) containing PDE3A, A-kinase anchor protein 18 (AKAP18), PLB, and SERCA2 (Figure 1) [87]. In contrast to Pde3a KO, transgenic mice with myocardium-specific overexpression of PDE3A1 (Tg-Pde3a1) reveal a significant reduction of heart rate and cardiac contractile function [88].
Chronic inhibition of PDE3 activity or reduction of PDE3A expression induces CM apoptosis, which is associated with a persistent induction of inducible cAMP early repressor (ICER) [35]. ICER is a transcriptional repressor of cAMP response element-binding protein (CREB)-dependent genes including the anti-apoptotic molecule B-cell lymphoma 2 (Bcl2), thus promoting CM apoptosis. A following study further demonstrated the existence of a positive feedback loop between PDE3A and ICER, in which PDE3A reduction/inhibition activates cAMP/PKA signaling that increases ICER protein stability through PKA-dependent ICER phosphorylation [76]; ICER serves as the transcriptional repressor for PDE3A, thus down-regulating PDE3A expression. The reciprocal regulation of PDE3A and ICER levels is also confirmed in human and mouse diseased hearts [35]. Preventing PDE3A reduction, or ICER induction, is found to be able to disrupt the PDE3A-ICER feedback loop and protect CMs from apoptosis [35]. For example, overexpressing PDE3A1 in Tg-Pde3a1 mice increases CM survival and protects hearts from acute ischemia/reperfusion (I/R) injury in vivo [88]. Alternatively, activation of extracellular signal-regulated kinase 5 (ERK5) increases C-terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase activity, which promotes ICER ubiquitination and degradation, thus protecting CMs from apoptosis [89]. In addition to CM apoptosis, PDE3 inhibition by Cilostamide exerts pro-hypertrophic effect in rat neonatal CMs [34]. Consistently, Ang II-induced cardiac remodeling and dysfunction are attenuated in Tg-Pde3a1 mice [90]. These results support the conclusion that PDE3A down-regulation seen in failing hearts may play a detrimental role to the myocardium.
However, this conclusion is challenged by the results from two recent studies. For example, it has been shown that global Pde3a KO, but not Pde3b KO, reduces pathological cardiac remodeling and dysfunction in a non-ischemic heart disease mouse model by chronic pressure overload with TAC [91]. PDE3B is detected in the T-tubules in close proximity to mitochondria in CMs (Figure 1), and global Pde3b KO but not Pde3a KO protects mouse hearts from acute I/R-induced injury [75]. The discrepancies among these studies remain unknown. PDE3A and PDE3B are expressed in a large amount of different cell types that could influence cardiac remodeling and functions directly or indirectly. Therefore, tissue-specific Pde3a or Pde3b KO mice are necessary in future studies.
2.2.4. PDE4
PDE4-devoted cAMP-hydrolyzing activity in human hearts is much lower than that in rodent hearts [92]. This is primarily due to other non-PDE4 activities that are much higher in human hearts than those in rodent hearts [92]. PDE4 expression levels have been found to be consistent between human and rodent hearts (Table 1) [92]. Among four different PDE4 genes, PDE4A, PDE4B and PDE4D were reported in human and rodent hearts [36,92,93]. The expression/activity of PDE4A, PDE4B and PDE4D were found to be decreased in failing and hypertrophic hearts as well as in diseased CMs [36,80,92,94].
PDE4 activity appears to be important in the excitation-contraction coupling in rodent hearts. It has been shown that PDE4 inhibition promotes the inotropic effects in response to β-AR stimulation as well as enhanced spontaneous diastolic Ca2+ waves [95]. In CMs, different PDE4 variants appear to be localized within discrete subcellular compartments, and associated with distinct signaling complexes (Figure 1), thus yielding different biological functions [96]. For example, PDE4B was found to be associated with the CaV1.2 subunit of LTCC in response to β-AR stimulation (Figure 1) [97]. Functional studies using CM from Pde4b KO mice further showed increased contraction and Ca2+ transient, which promotes ventricular tachycardia [97]. At least four PDE4D variants were detected in human and/or rodent hearts, including PDE4D3, 4D5, 4D8 and 4D9 87. PDE4D3 was found in the complex with RyR2 channels (Figure 1) [36]. Pde4d KO mice exert hyperphosphorylated “leaky” RyR2 channels, which is associated with age-related cardiomyopathy, exercise-induced arrhythmia and accelerated myocardial infarction [36]. However, another study showed that in murine and failing human hearts, PDE4D associates with SERCA2 but not with RyR2, based on the co-immunoprecipitation study (Figure 1) [98]. PDE4D thus regulates baseline SR Ca2+ release and contractility [98]. Actually, PDE4 inhibition potentiates the phosphorylation of both RyR2 and PLB and increases both SR Ca2+ leak and SR Ca2+ load [95], suggesting both RyR2 and SERCA2 may be regulated by PDE4. Different from PDE4B, PDE4D does not regulate L-type Ca2+ current [98]. These results indicate the important roles of PDE4 in regulating cardiac functions via association of distinct multiprotein complexes.
PDE4 is also important in chronic regulation of cardiac remodeling. For example, PDE4D has been shown to directly interact with heat shock protein 20 (Hsp20) in CMs, which is important for maintaining Hsp20 in a hypo-phosphorylated state [99]. It is known that Hsp20 is cardioprotective during cardiac stress and the protective ability of Hsp20 depends on Hsp20 phosphorylation at Ser16 by PKA [100]. Therefore, PDE4 inhibition or expressing the peptide disrupting PDE4D and Hsp20 interaction results in Hsp20 phosphorylation, and protects against chronic β-AR-induced CM hypertrophy in vitro and TAC-induced cardiac hypertrophy and fibrosis in vivo [99,101]. In addition, it has been shown that PDE4D3 is targeted by the muscle-selective AKAP (mAKAP) to a signaling complex containing PKA, Epac1 and ERK5 signaling molecules at perinuclear regions (Figure 1) [102,103]. This signaling complex is important in regulating CM hypertrophy [102,103]. Since the overall PDE4 expression is decreased in failing hearts and PDE4 isoform inactivation often exerts unfavorable cardiac outcomes, it is reasonable to speculate that increasing PDE4 expression may be beneficial in heart failure. A recent study using transgenic mice with myocardium-specific overexpression of the main PDE4B isoform expressed in mouse myocardium, PDE4B3, unveils a protective role of PDE4B in ISO-induced pathological cardiac remodeling [94]. Interestingly, the moderate levels of PDE4B3 overexpression in CMs via the AAV9 vector is sufficient to protect against cardiac dysfunction induced by chronic β-AR stimulation and TAC without modifying cardiac function in healthy mice [94]. This finding suggests that cardiac gene therapy with PDE4B3 may be a new approach to treat heart failure [94]. Similarly, the cardiac protective effect has been reported for a small allosteric modulator UCR1C that activates PDE4 long isoforms [104]. PDE4 activation by the small allosteric modulator reduces nuclear PKA activity and CREB phosphorylation, hence inhibiting β-AR-stimulated cardiac myocyte hypertrophy [104]. This result suggests that PDE4 activating compound could also be useful in combatting heart failure.
2.2.5. PDE5
PDE5A is expressed in human and mouse hearts [105–108] and increases under pathological conditions (Table 1) [37,106,107,109]. PDE5A expression is detected in isolated CMs [108,110]. However, it has also been reported that PDE5A is expressed only in CFs but not CMs in mouse hypertrophied hearts [111]. The inconsistence might be due to differences in PDE5A antibodies or the variability in the severity of cardiac diseases.
In normal hearts, PDE5A appears to negatively regulate catecholamine-induced contractile function through cGMP-PKG signaling. For example, PDE5 inhibitor, sildenafil, attenuates isoproterenol-stimulated cardiac contractility [112–115]. PDE5 is detected in the Z-band of mouse CM and the localization of PDE5 to Z-bands is critical for PDE5-mediated regulation of contractility, which is dependent on endothelial nitric oxide synthase (eNOS) and PKG (Figure 1) [110]. The effect of PDE5 inhibitor on cardiac contractility is largely diminished in failing hearts, which is associated with reduced Z-band localization of PDE5A [114]. These findings implicate that PDE5 regulates highly localized cGMP pools in normal hearts.
The studies of PDE5 inhibitors in I/R injury and doxorubicin-induced cardiotoxicity have revealed important beneficial effects of PDE5 inhibition on myocardium protection. For example, PDE5 inhibition by sildenafil has been shown to protect hearts against experimental I/R injury in rabbits, when sildenafil was given prior to the ischemia [116], or sildenafil was applied at the time of reperfusion [117]. Chronic sildenafil treatment 3 days post-MI also alleviates the progression of cardiac dysfunction [118]. In addition, PDE5 inhibition has been shown to attenuate CM apoptosis and left ventricular dysfunction in a chronic doxorubicin-induced cardiomyopathy model [119]. In isolated CMs, sildenafil protects CMs from ischemia-stimulated necrosis and apoptosis [120], suggesting a critical role of PDE5 in regulating CM viability.
Because PDE5A is upregulated in failing or hypertrophic hearts, the role of PDE5A induction in disease development has been extensively investigated in different cardiac remodeling models. For example, chronic inhibition of PDE5 by sildenafil enhances cGMP level and PKG activity, thus alleviating cardiac hypertrophy and improves cardiac function induced by TAC [115,121], as well as by chronic β-AR stimulation [122]. More importantly, sildenafil is able to reverse pre-established hypertrophic heart disease [115], which supports the therapeutic significance with PDE5 inhibitors. Furthermore, the role of PDE5A elevation has been investigated in genetically modified PDE5A with inducible CM-specific overexpression of PDE5A. It was found that overexpression of PDE5A in transgenic mice suppresses PKG activation and worsens TAC-induced cardiac hypertrophy, fibrosis, and dysfunction in a dose-dependent manner [123]. The adverse effects of PDE5A overexpression are blocked by sildenafil [123]. However, the role of PKG is challenged by other studies. For example, genetic deletion of PKG in mice (except SMCs) does not worsen cardiac hypertrophy induced by TAC [124], ISO infusion [124], Ang II infusion [125], or CM-specific overexpression of Ang II type 1 receptor (AT1R) [126]. These findings do not support the role of PKG in PDE5-mediated regulation of cardiac remodeling. Additionally, PDE5 inhibition with sildenafil has moderate or no effect on Ang II-induced CM hypertrophy, but suppresses profibrotic gene expression [125]. This study suggests a potential cardioprotective effect of PDE5 derived from non-CMs. Therefore, tissue-specific Pde5a KO mice are required to precisely understand the cellular and molecular mechanisms of PDE5A in pathological cardiac remodeling and dysfunction. Another possibility is that cGMP/PKG signaling may not be the only downstream signaling that contributes to the cardiac protective effect of PDE5 inhibition. Elevation of cGMP through PDE5 inhibition may regulate the activity of other PDEs, such as inhibiting PDE3 or activating PDE2, which would lead to changes in PKA activity.
2.2.6. PDE8
Both PDE8A and PDE8B have been reported in human and mouse hearts, as well as mouse CM (Table 1) [38,53,127]. To date, cardiac-related study of PDE8 is limited to PDE8A. A study using Pde8a KO mice showed that PDE8A deficiency potentiates ISO-induced intracellular Ca2+ transient and LTCC currents [38]. However, the underlying molecular mechanism remains unknown.
2.2.7. PDE9
PDE9A has been detected in the heart and isolated CMs (Table 1) [57]. PDE9A expression is significantly increased in failing human hearts [39]. Recent evidence has indicated a pathophysiological role for PDE9 in heart diseases. PDE9A inhibition or knockdown blocks pro-hypertrophic stimulation in CMs [39]. Pde9a KO mice have less severe cardiac remodeling under pressure overload [39]. Importantly, PDE9A inhibition has been found to reverse pre-existing cardiac dysfunction, providing the clinical significance of using PED9A inhibition as potential therapeutic approach for cardiac diseases [39]. PDE9A inhibition has been shown to reverse cardiac hypertrophy or dysfunction in a natriuretic peptide receptor (NPR)/cGMP-dependent manner (Figure 1) [39], which is different from the PDE5A inhibition that is linked to the NO/cGMP pathway. The mechanistic difference between PDE5A and PDE9A is further supported by a study that shows distinct microRNA expression profiles under PDE5 and PDE9 inhibition in TAC-induced cardiac dysfunction [128]. For example, PDE5 inhibition reduces most of microRNAs associated with the disease state, whereas PDE9 inhibition has no impact [128]. MicroRNAs play important roles in regulating gene expression [129]. The different microRNA profiles elicited by PDE5 and PDE9 inhibition suggest that PDE5 or PDE9 inhibition protects against cardiac remodeling via modulating the expression of different sets of genes.
2.2.8. PDE10
PDE10A2 is the major PDE10A isoform expressed in the heart [40]. Recent study by our lab has shown that PDE10A expression is significantly increased in failing mouse and human hearts, while is decreased in mouse exercised hearts (Table 1) [40]. PDE10A directly regulates CM hypertrophy and CF activation, proliferation and ECM accumulation [40]. We have found that PDE10A inhibition or deficiency alleviates cardiac hypertrophy, cardiac fibrosis and cardiac dysfunction in both Ang II-induced and TAC-induced cardiac disease model, indicating an important role of PDE10A in pathological remodeling [40]. Moreover, inhibiting PDE10A by specific and selective PDE10A inhibitor TP-10 effectively ameliorated pre-established cardiac remodeling, suggesting a potential therapeutic effect of targeting PDE10A on pathological cardiac remodeling [40]. In addition to the cardiac role of PDE10A, it appears that PDE10A is connected to the regulation of energy homeostasis and insulin sensitivity [130–132]. PDE10A is also known to express and have a role in the brown and white adipose tissue where it is involved in the control of thermogenic expression [131,132]. Impaired thermogenesis and insulin response are associated with the development of HF [133]. Therefore, it would be intriguing to identify the metabolic contribution of adipose PDE10A in cardiac remodeling and dysfunction in the future.
3. Therapeutic application and development of PDE inhibitors in combatting cardiac diseases
There are increasing lines of evidence that support the critical roles of PDEs in regulating cardiac functions and remodeling in preclinical animal models. Therefore, increasing clinical trials on extending new indications such as cardiac disorders have been conducted with some existing marketed PDE inhibitors. Drug repurposing has apparent benefits including less safety concerns, saving time and reduced cost. In this section, we focus on the current status of marketed PDE inhibitors and PDE inhibitors under clinical trials, as well as their effects in clinical studies on cardiac diseases.
3.1. PDE3 inhibitors
PDE3 inhibition elevates cAMP in cardiac muscle and subsequently increases the rate and magnitude of cardiac contractility, which is the basis for using PDE3 inhibitors to treat congestive heart failure. Also, PDE3 inhibition stimulates vascular relaxation, thus reducing peripheral and pulmonary vascular resistance and enhances coronary blood flow. Thus, PDE3 inhibitors such as milrinone have become powerful drugs for the short-term treatment of life-threatening heart failure due to their inotropic and vasodilatory actions. Several PDE3 inhibitors are currently FDA approved and marketed inside or outside the USA. Milrinone is FDA-approved in 1987 for the treatment of acute HF or chronic HF [134,135]. Milrinone markedly improves cardiac performances including increasing cardiac contractility, cardiac relaxation, and vasodilation [135]. Concerns about its adverse effects, such as an increased risk of supraventricular and ventricular arrhythmias, and hypotension, make Milrinone to be used with special cautions [134,136]. There are continued interests and efforts in optimizing the therapeutic regiment for using PDE3 inhibitors in treating heart failure. For example, a phase II clinical trial is ongoing to test a hypothesis that carefully monitoring and optimizing Milrinone levels in children following cardiac surgery may improve clinical outcomes and reduce the duration of Milrinone infusion (ClinicalTrials.gov Identifier: NCT01841177).
3.2. PDE5 inhibitors
There are at least four PDE5 inhibitors, sildenafil, vardenafil, tadalafil, and avanafil, approved by FDA and marketed in the US, and originally recommended as the first-line treatment of erectile dysfunction [137]. The presence of abundant PDE5 in lung vascular smooth muscle makes PDE5 inhibitors an important modality for treatment of pulmonary hypertension [138]. Because of promising animal study results, there have been substantial studies attempting to test the effects of PDE5 inhibitors as therapeutics for cardiac diseases. For example, a meta-analysis in 928 patients with reduced left ventricular ejection fraction reveal the beneficial effect of PDE5 inhibitors Sildenafil or Udenafil, including improved clinical outcomes, exercise capacity and pulmonary hemodynamics [139]. Sildenafil has been tested in patients with chronic HF. A clinical study in 45 HF patients randomized to placebo or Sildenafil treatment for 1 year, shows that Sildenafil treatment improves left ventricle and diastolic function, and cardiac geometry in HF patients [140]. Moreover, Sildenafil has also been evaluated in clinical trials for its effect on diabetic cardiomyopathy. It has been found that chronic Sildenafil treatment results in improved cardiac geometry and cardiac kinetics, and reduced circulating proinflammatory chemokines in 59 diabetic men with non-ischemic and non-failing diabetic cardiomyopathy [141]. However, there are also some clinical studies that reveal disappointing results. For example, a clinical study was carried out in 52 patients with HFpEF and pulmonary hypertension and randomized to Sildenafil or placebo treatment for 12 weeks. The observation is that Sildenafil does not alter cardiac structure nor function on echocardiography compared with placebo [142]. In addition, a large scale clinical trial (the RELAX study) was conducted to test the effects of sildenafil on cardiac and pulmonary function in approximately 200 elderly patients with HFpEF. Sildenafil showed no effect on maximal or submaximal exercise capacity, clinical status, quality of life, LV remodeling, diastolic function parameters in these patients [143]. The inconsistent clinical results might be due to the variations in patient subjects with regard to the differences in etiologies of disease development, associated health problems, as well as other medications taken. Thus, clinical trial studies with more rigorous protocols are required in the future.
3.3. Other PDE inhibitors with therapeutic potential or perspective
Preclinical studies in animal models have suggested the potential therapeutic effects by targeting several other PDEs. For example, the pan PDE1 inhibitor IC86340 exerts protective effect against pathological cardiac remodeling and dysfunction in various pre-clinical mouse heart failure models, such as pressure overload-induced heart failure [33], ISO-induced cardiac hypertrophy [32], as well as doxorubicin-induced cardiac toxicity [14]. A different pan PDE1 inhibitor, ITI-214, has been shown to improve cardiac function in dog and rabbit models of heart failure [63]. Another PDE1 inhibitor, vinpocetine, attenuates pathological cardiac remodeling induced by chronic Ang II-infusion in mice [62]. Vinpocetine has been used to prevent and treat cerebrovascular disorders such as stoke and dementia, and remains widely available in dietary supplements [144]. Based on the excellent safety profile of vinpocetine in humans, targeting PDE1 might be a promising therapeutic strategy in treating cardiac diseases. Similarly, studies have found that the inhibition of PDE2 by Bay-607550 and inhibition of PDE9 by PF-04447943 all elicit improved cardiac function and reverse cardiac remodeling in pressure overload-induced heart failure in mice [34,39,66]. A recent study also showed that PDE10A inhibition by TP-10 ameliorates pre-existing pathological cardiac remodeling and dysfunction induced by chronic pressure overload in mice [40]. Importantly, several PDE10A inhibitors have been tested in humans and successfully passed phase I clinical trials (ClinicalTrials.org), suggesting PDE10A is a drug-able target. Thus, inhibiting PDE10A may represent a novel therapeutic strategy for cardiac diseases with pathological cardiac remodeling.
4. Conclusions
PDEs, as negative cAMP and/or cGMP modulators, play important roles in regulating the amplitude, duration, and compartmentalization of cyclic nucleotide signaling. PDEs exist in a superfamily with more than 20 different genes and a large number of variants (isozymes). The diversities of PDEs including substrate specificities, enzymatic properties, subcellular localizations, and expression profiles, enable individual PDEs to regulate unique cyclic nucleotide signaling pathways and specialized cellular functions. Thus, targeting a specific PDE allows selectivity, altering only one or few cyclic nucleotide signaling pathways without a global change of cellular cyclic nucleotides. Dysregulation of PDE expression/activation have been implicated in a number of cardiac diseases, and most of them appear to play causative roles in the disease development. Thus, inhibiting or activating altered PDEs may be attractive therapeutic strategies to treat cardiac diseases. Up to date, most of our knowledge of individual PDEs in cardiac biology and diseases come from studies with global KO mice or systemic PDE inhibitions in rodents. Because many PDEs are also expressed in non-cardiac organ/tissues that may have an indirect impact on the heart, the cardiac functions of individual PDEs should be evaluated carefully with considerations of non-cardiac tissue contributions. In addition, different PDE variants encoded by the same gene could have distinct cellular/subcellular localizations and play differential roles in the heart. Developing more specific genetic tools capable of targeting individual variants is needed. Moreover, differences in PDE expression and function in rodents and humans should be carefully evaluated.
5. Expert Opinion
5.1. Limitations of currently available PDE activity modulators
Currently, the major PDE activity modulators are PDE inhibitors. All PDE inhibitors developed up to date are family-specific and they do not have sufficient specificities to distinguish the isozymes within the same family. Using genetic approaches, it has been shown that different PDE isozymes in the same family could exert distinct functions in individual cells. For example, PDE1C, but not PDE1A, regulates CM viability [14,33]. PDE3A, but not PDE3B, regulates CM contractile function [85,86]. PDE4B, but not PDE4D, regulates the L-type Ca2+ current [98], while PDE4D, but not PDE4B, regulates Ca2+ release from the RyR2 [97]. Therefore, there is an urgent need to develop isozyme-selective PDE inhibitors.
In contrast, PDE activation appears to be beneficial in certain cases. For example, PDE3A1 overexpression in CMs protects CMs and mouse hearts against I/R induced cell death and cardiac injury [88]. Similarly, CM-specific overexpression of PDE4B3 in transgenic mice protects against pathological cardiac remodeling induced by chronic β-AR stimulation [94]. Interestingly, AAV9-mediated gene transfer has been used to achieve a moderate level of PDE4B3 expression in CMs, which promisingly elicits protective effects against cardiac dysfunction induced by chronic β-AR stimulation and TAC without affecting cardiac function in healthy mice [94].
5.2. Targeting multiple PDEs
Several studies have reported that administering two or more PDE inhibitors with submaximal dose could yield additive or synergistic effects, with the possibility to generate less toxicity [145–148]. For example, synergism between PDE3 and PDE4 has been found in various cell types [145–149]. Inhibition of PDE3 and PDE4 synergistically stimulate lipolysis in mouse and rat adipocytes [145], activate brown adipose tissue [147], inhibit vascular SMC migration [146], suppress effect on VCAM-1 expression and eosinophil adhesion to activated human lung microvascular ECs [149], as well as increase spontaneous firing of sinoatrial node cells [148]. Interestingly, a recent clinical trial testing dual PDE3 and PDE4 inhibitor Ensifentrine (RPL554) revealed a promising improvement and well-tolerated effect for the treatment of COPD [150].
In CMs, in vitro and in vivo, the inhibitors of PDE1, 5, and 10 are all able to suppress CM hypertrophy [33,40,121]. An additive effect was reported for PDE1 inhibition by IC86340 together with PDE5 inhibition by Sildenafil in neonatal rat CM hypertrophy [32], suggesting that targeting different PDEs concurrently have additive or synergistic effects. In CMs, PDE5 regulates the sGC/cGMP/PKG signaling [56,66], whereas PDE9A regulates pGC/cGMP/PKG signaling [39]. It would be interesting to determine the effects of inhibiting PDE5 and PDE9 together in the future. In addition, PDE2 activity is stimulated upon increasing cGMP. It would also be of value to determine the effects of inhibiting PDE2 together with PDE5 or with PDE9.
5.4. Targeting specialized signalosomes
Numerous studies have supported the notion that individual PDEs are tethered to a precise signalosome via binding partners in an isoform-specific manner, each containing unique cyclase, PDE, kinases, and/or other signaling molecules (Figure 1). The signalosomes control cyclic nucleotide signaling locally and specifically, which lead to different biological functions [27,28,34]. Promoting or disrupting isoform-specific protein-protein interactions within specific signalosome may yield a greater specificity compared to current PDE inhibitors. The disruption of protein-protein interaction is often achieved by small molecule compounds or peptides. For example, a peptide that induces the disruption of the Hsp20-PDE4D complex has been shown to promote PKA phosphorylation of Hsp20 and alleviate cardiac myocyte hypertrophy [28]. A small molecule or a peptide that disrupts the AKAP18-PKA complexes has been shown to increase cardiac myocyte contractility [28]. It remains to be examined whether targeting specialized signalosomes by disruption of protein-protein interaction might be efficacious in clinical settings.
In addition, defining the sources of cyclic nucleotides regulated by PDEs in the signalosomes could be important. For example, it has been shown that PDE1C exists in a multi-protein complex with A2AR and TRPC3 in CMs, which is critical in regulating CM viability [14]. TRPC3-derived Ca2+ activates PDE1C, which antagonizes A2AR-derived cAMP through PDE1C hydrolysis of cAMP [145]. This finding results in the hypothesis that promoting cAMP synthesis and inhibiting cAMP degradation together produce additive or synergistic effects. Indeed, it has been shown that low doses of A2R agonist combined with PDE1 inhibitor or TRPC inhibitor provide synergistic effects on CM survival [1]. PDE9A has been shown to complex with NPR in CMs [39]. It would be of great interest to examine the additive/synergistic effects from the PDE9 inhibitor in combination with NPR agonists.
Article highlights.
PDEs play an important role in regulating the amplitude, duration, and compartmentalization of cyclic nucleotide signaling.
Dysregulation of PDE expression and activation have been implicated in a number of cardiac diseases.
Increasing pre-clinical and clinical evidences support that inhibiting PDEs may be attractive therapeutic strategies to treat cardiac diseases.
It would be of value to determine the effect of targeting multiple PDEs or targeting specialized signalosomes on combating cardiac disorders in the future.
Understanding the cellular and molecular mechanisms of PDEs enable the development of new approaches to achieve effective and specific PDE inhibition for various cardiac therapies.
Acknowledgments
The authors thank Sarah Greenly for her critical reading the manuscript.
Funding:
This work was financially supported by National Institute of Health HL134910 and HL088400 (to C.Y.) and American Heart Association 17PRE33660835 (to S.C.).
Footnotes
Declaration of Interest:
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Reference
- 1.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006. September;58(3):488–520. [DOI] [PubMed] [Google Scholar]; •• An excellent and detailed review on the basic biochemical properties, cellular regulation, expression patterns, and physiological functions of PDEs
- 2.Guellich A, Mehel H, Fischmeister R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch. 2014. June;466(6):1163–75. [DOI] [PubMed] [Google Scholar]
- 3.Beavo J, Francis SH, Houslay MD. Cyclic nucleotide phosphodiesterases in health and disease. Boca Raton: CRC Press/Taylor & Francis; 2007. [Google Scholar]
- 4.Liu S, Li Y, Kim S, et al. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci U S A. 2012. April 24;109(17):6578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93(10):896–906. [DOI] [PubMed] [Google Scholar]
- 6.Feldman DS, Elton TS, Sun B, et al. Mechanisms of disease: detrimental adrenergic signaling in acute decompensated heart failure. Nat Clin Pract Cardiovasc Med. 2008. April;5(4):208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007. May;114(2):208–21. [DOI] [PubMed] [Google Scholar]
- 8.Szentmiklosi AJ, Cseppento A, Harmati G, et al. Novel trends in the treatment of cardiovascular disorders: site- and event- selective adenosinergic drugs. Curr Med Chem. 2011;18(8):1164–87. [DOI] [PubMed] [Google Scholar]
- 9.Okumura S, Takagi G, Kawabe J, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A. 2003;100(17):9986–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okumura S, Vatner DE, Kurotani R, et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation. 2007. October 16;116(16):1776–83. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi T, Tang T, Lai NC, et al. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006. August 1;114(5):388–96. [DOI] [PubMed] [Google Scholar]
- 12.Tang T, Gao MH, Lai NC, et al. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008. January 1;117(1):61–9. [DOI] [PubMed] [Google Scholar]
- 13.Knight WE, Chen S, Zhang Y, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016. October 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Knight W, Chen S, et al. Multiprotein Complex With TRPC (Transient Receptor Potential-Canonical) Channel, PDE1C (Phosphodiesterase 1C), and A2R (Adenosine A2 Receptor) Plays a Critical Role in Regulating Cardiomyocyte cAMP and Survival. Circulation. 2018. October 30;138(18):1988–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding B, Abe J, Wei H, et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005. May 17;111(19):2469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tajima M, Bartunek J, Weinberg EO, et al. Atrial natriuretic peptide has different effects on contractility and intracellular pH in normal and hypertrophied myocytes from pressure-overloaded hearts. Circulation. 1998. December 15;98(24):2760–4. [DOI] [PubMed] [Google Scholar]
- 17.Ohte N, Cheng CP, Suzuki M, et al. Effects of atrial natriuretic peptide on left ventricular performance in conscious dogs before and after pacing-induced heart failure. J Pharmacol Exp Ther. 1999. November;291(2):589–95. [PubMed] [Google Scholar]
- 18.Shah AM, MacCarthy PA. Paracrine and autocrine effects of nitric oxide on myocardial function. Pharmacol Ther. 2000. April;86(1):49–86. [DOI] [PubMed] [Google Scholar]
- 19.Pierkes M, Gambaryan S, Boknik P, et al. Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovasc Res. 2002. March;53(4):852–61. [DOI] [PubMed] [Google Scholar]
- 20.Su J, Zhang Q, Moalem J, et al. Functional effects of C-type natriuretic peptide and nitric oxide are attenuated in hypertrophic myocytes from pressure-overloaded mouse hearts. Am J Physiol Heart Circ Physiol. 2005. March;288(3):H1367–73. [DOI] [PubMed] [Google Scholar]
- 21.Hammond J, Balligand JL. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol. 2012. February;52(2):330–40. [DOI] [PubMed] [Google Scholar]
- 22.Bice JS, Burley DS, Baxter GF. Novel approaches and opportunities for cardioprotective signaling through 3’,5’-cyclic guanosine monophosphate manipulation. J Cardiovasc Pharmacol Ther. 2014. May;19(3):269–82. [DOI] [PubMed] [Google Scholar]
- 23.Su J, Scholz PM, Weiss HR. Differential effects of cGMP produced by soluble and particulate guanylyl cyclase on mouse ventricular myocytes. Exp Biol Med (Maywood). 2005. April;230(4):242–50. [DOI] [PubMed] [Google Scholar]
- 24.Fischmeister R, Castro LR, Abi-Gerges A, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006. October 13;99(8):816–28. [DOI] [PubMed] [Google Scholar]
- 25.Zaccolo M, Di Benedetto G, Lissandron V, et al. Restricted diffusion of a freely diffusible second messenger: mechanisms underlying compartmentalized cAMP signalling. Biochem Soc Trans. 2006. August;34(Pt 4):495–7. [DOI] [PubMed] [Google Scholar]
- 26.Stangherlin A, Zaccolo M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2012. January;302(2):H379–90. [DOI] [PubMed] [Google Scholar]
- 27.Lomas O, Zaccolo M. Phosphodiesterases maintain signaling fidelity via compartmentalization of cyclic nucleotides. Physiology (Bethesda). 2014. March;29(2):141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brescia M, Zaccolo M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. Int J Mol Sci. 2016. October 2;17(10). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A general review on cyclic nucleotide signaling compartmentalization
- 29.Schleicher K, Zaccolo M. Using cAMP Sensors to Study Cardiac Nanodomains. J Cardiovasc Dev Dis. 2018. March 13;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A nice review on FRET-based cAMP sensors and cAMP compartmentation
- 30.Chao YC, Surdo NC, Pantano S, et al. Imaging cAMP nanodomains in the heart. Biochem Soc Trans. 2019. October 31;47(5):1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bork NI, Nikolaev VO. cGMP Signaling in the Cardiovascular System-The Role of Compartmentation and Its Live Cell Imaging. Int J Mol Sci. 2018. March 10;19(3). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Introduce current techiniques for real-time cGMP measurement and cGMP
- 32.Miller CL, Oikawa M, Cai Y, et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res. 2009. November 6;105(10):956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knight WE, Chen S, Zhang Y, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016. November 8;113(45):E7116–E7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zoccarato A, Surdo NC, Aronsen JM, et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ Res. 2015. September 25;117(8):707–19. [DOI] [PubMed] [Google Scholar]
- 35.Ding B, Abe JI, Wei H, et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005. May 17;111(19):2469–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehnart SE, Wehrens XH, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005. October 7;123(1):25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007. July 17;116(3):238–48. [DOI] [PubMed] [Google Scholar]
- 38.Patrucco E, Albergine MS, Santana LF, et al. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J Mol Cell Cardiol. 2010. August;49(2):330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DI, Zhu G, Sasaki T, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015. March 26;519(7544):472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, Zhang Y, Lighthouse JK, et al. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation. 2020. January 21;141(3):217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shakur Y, Holst LS, Landstrom TR, et al. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–77. [DOI] [PubMed] [Google Scholar]
- 42.Movsesian M Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol Ther. 2016. July;163:74–81. [DOI] [PubMed] [Google Scholar]; • A detailed review on the role of PDE3 in cardiac diseases.
- 43.Maurice DH, Palmer D, Tilley DG, et al. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64(3):533–46. [DOI] [PubMed] [Google Scholar]
- 44.Fujishige K, Kotera J, Michibata H, et al. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem. 1999. June 25;274(26):18438–45. [DOI] [PubMed] [Google Scholar]
- 45.Loughney K, Snyder PB, Uher L, et al. Isolation and characterization of PDE10A, a novel human 3’, 5’-cyclic nucleotide phosphodiesterase. Gene. 1999. June 24;234(1):109–17. [DOI] [PubMed] [Google Scholar]
- 46.Soderling SH, Bayuga SJ, Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A. 1999. June 8;96(12):7071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Charych EI, Jiang LX, Lo F, et al. Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: implications for the treatment of schizophrenia. J Neurosci. 2010. July 7;30(27):9027–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russwurm C, Koesling D, Russwurm M. Phosphodiesterase 10A Is Tethered to a Synaptic Signaling Complex in Striatum. J Biol Chem. 2015. May 8;290(19):11936–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gross-Langenhoff M, Hofbauer K, Weber J, et al. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J Biol Chem. 2006. February 3;281(5):2841–6. [DOI] [PubMed] [Google Scholar]
- 50.Houslay MD, Baillie GS, Maurice DH. cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res. 2007. April 13;100(7):950–66. [DOI] [PubMed] [Google Scholar]
- 51.Xie M, Blackman B, Scheitrum C, et al. The upstream conserved regions (UCRs) mediate homo- and hetero-oligomerization of type 4 cyclic nucleotide phosphodiesterases (PDE4s). Biochem J. 2014. May 1;459(3):539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, Wu P, Egan RW, et al. Human phosphodiesterase 8A splice variants: cloning, gene organization, and tissue distribution. Gene. 2001. December 12;280(1–2):183–94. [DOI] [PubMed] [Google Scholar]
- 53.Soderling SH, Bayuga SJ, Beavo JA. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci U S A. 1998;95(15):8991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayashi M, Shimada Y, Nishimura Y, et al. Genomic organization, chromosomal localization, and alternative splicing of the human phosphodiesterase 8B gene. Biochem Biophys Res Commun. 2002. October 11;297(5):1253–8. [DOI] [PubMed] [Google Scholar]
- 55.Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5: expanding roles in cardiovascular regulation. Circ Res. 2007. November 26;101(11):1084–95. [DOI] [PubMed] [Google Scholar]; • A general review on the role of PDE5 in cardiovascular system.
- 56.Francis S, Zoraghi R, Kotera J, et al. Phosphodiesterase-5: molecular characteristics relating to structure, function, and regulation. Cyclic nucleotide phosphodiesterases in health and disease CRC, Boca Raton: 2006:131. [Google Scholar]
- 57.Fisher DA, Smith JF, Pillar JS, et al. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem. 1998;273(25):15559–64. [DOI] [PubMed] [Google Scholar]
- 58.Rentero C, Monfort A, Puigdomenech P. Identification and distribution of different mRNA variants produced by differential splicing in the human phosphodiesterase 9A gene. Biochem Biophys Res Commun. 2003. February 14;301(3):686–92. [DOI] [PubMed] [Google Scholar]
- 59.Vandeput F, Wolda SL, Krall J, et al. Cyclic Nucleotide Phosphodiesterase PDE1C1 in Human Cardiac Myocytes. J Biol Chem. 2007. November 9;282(45):32749–57. [DOI] [PubMed] [Google Scholar]
- 60.Chen S, Knight WE, Yan C. Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. J Cardiovasc Dev Dis. 2018. April 23;5(2):22. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A detailed review on the role of PDE1 in cardiac diseases.
- 61.Miller CL, Cai Y, Oikawa M, et al. Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol. 2011. November;106(6):1023–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu MP, Zhang YS, Xu X, et al. Vinpocetine Attenuates Pathological Cardiac Remodeling by Inhibiting Cardiac Hypertrophy and Fibrosis. Cardiovasc Drugs Ther. 2017. April;31(2):157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashimoto T, Kim GE, Tunin RS, et al. Acute Enhancement of Cardiac Function by Phosphodiesterase Type 1 Inhibition. Circulation. 2018. October 30;138(18):1974–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stephenson DT, Coskran TM, Wilhelms MB, et al. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J Histochem Cytochem. 2009. October;57(10):933–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mehel H, Emons J, Vettel C, et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J Am Coll Cardiol. 2013. October 22;62(17):1596–606. [DOI] [PubMed] [Google Scholar]
- 66.Baliga RS, Preedy MEJ, Dukinfield MS, et al. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci U S A. 2018. July 31;115(31):E7428–E7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vettel C, Lammle S, Ewens S, et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol. 2014. April 15;306(8):H1246–52. [DOI] [PubMed] [Google Scholar]
- 68.Hartzell HC, Fischmeister R. Opposite effects of cyclic GMP and cyclic AMP on Ca2+ current in single heart cells. Nature. 1986. September 18-24;323(6085):273–5. [DOI] [PubMed] [Google Scholar]
- 69.Vandecasteele G, Verde I, Rucker-Martin C, et al. Cyclic GMP regulation of the L-type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533(Pt 2):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Soler F, Fernandez-Belda F, Perez-Schindler J, et al. PDE2 activity differs in right and left rat ventricular myocardium and differentially regulates beta2 adrenoceptor-mediated effects. Exp Biol Med (Maywood). 2015. September;240(9):1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mongillo M, Tocchetti CG, Terrin A, et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006. February 3;98(2):226–34. [DOI] [PubMed] [Google Scholar]
- 72.Monterisi S, Lobo MJ, Livie C, et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife. 2017. May 2;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vettel C, Lindner M, Dewenter M, et al. Phosphodiesterase 2 Protects Against Catecholamine-Induced Arrhythmia and Preserves Contractile Function After Myocardial Infarction. Circ Res. 2017. January 6;120(1):120–132. [DOI] [PubMed] [Google Scholar]
- 74.Surapisitchat J, Jeon KI, Yan C, et al. Differential regulation of endothelial cell permeability by cGMP via phosphodiesterases 2 and 3. Circ Res. 2007. October 12;101(8):811–8. [DOI] [PubMed] [Google Scholar]
- 75.Chung YW, Lagranha C, Chen Y, et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2015. April 28;112(17):E2253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding B, Abe J, Wei H, et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005. October 11;102(41):14771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yan C, Ding B, Shishido T, et al. Activation of extracellular signal-regulated kinase 5 reduces cardiac apoptosis and dysfunction via inhibition of a phosphodiesterase 3A/inducible cAMP early repressor feedback loop. Circ Res. 2007. March 2;100(4):510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith CJ, Huang R, Sun D, et al. Development of decompensated dilated cardiomyopathy is associated with decreased gene expression and activity of the milrinone-sensitive cAMP phosphodiesterase PDE3A. Circulation. 1997;96(9):3116–23. [DOI] [PubMed] [Google Scholar]
- 79.Sato N, Asai K, Okumura S, et al. Mechanisms of desensitization to a PDE inhibitor (milrinone) in conscious dogs with heart failure. Am J Physiol. 1999;276(5 Pt 2):H1699–705. [DOI] [PubMed] [Google Scholar]
- 80.Abi-Gerges A, Richter W, Lefebvre F, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res. 2009. October 9;105(8):784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takahashi K, Osanai T, Nakano T, et al. Enhanced activities and gene expression of phosphodiesterase types 3 and 4 in pressure-induced congestive heart failure. Heart Vessels. 2002. September;16(6):249–56. [DOI] [PubMed] [Google Scholar]
- 82.Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, et al. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol. 2000. June 1;525 Pt 2:483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tse J, Brackett NL, Kuo JF. Alterations in activities of cyclic nucleotide systems and in beta-adrenergic receptor-mediated activation of cyclic AMP-dependent protein kinase during progression and regression of isoproterenol-induced cardiac hypertrophy. Biochim Biophys Acta. 1978. September 6;542(3):399–411. [DOI] [PubMed] [Google Scholar]
- 84.Packer M, Carver JR, Rodeheffer RJ, et al. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325(21):1468–75. [DOI] [PubMed] [Google Scholar]
- 85.Sun B, Li H, Shakur Y, et al. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal. 2007. August;19(8):1765–71. [DOI] [PubMed] [Google Scholar]
- 86.Beca S, Ahmad F, Shen W, et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res. 2013. January 18;112(2):289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahmad F, Shen W, Vandeput F, et al. Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2. J Biol Chem. 2015. March 13;290(11):6763–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oikawa M, Wu M, Lim S, et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2013. November;64:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Woo CH, Le NT, Shishido T, et al. Novel role of C terminus of Hsc70-interacting protein (CHIP) ubiquitin ligase on inhibiting cardiac apoptosis and dysfunction via regulating ERK5-mediated degradation of inducible cAMP early repressor. FASEB J. 2010. December;24(12):4917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iwaya S, Oikawa M, Chen Y, et al. Phosphodiesterase 3A1 protects the heart against angiotensin II-induced cardiac remodeling through regulation of transforming growth factor-beta expression. Int Heart J. 2014;55(2):165–8. [DOI] [PubMed] [Google Scholar]
- 91.Polidovitch N, Yang S, Sun H, et al. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol. 2019. July;132:60–70. [DOI] [PubMed] [Google Scholar]
- 92.Richter W, Xie M, Scheitrum C, et al. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011. March;106(2):249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kostic MM, Erdogan S, Rena G, et al. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J Mol Cell Cardiol. 1997;29(11):3135–46. [DOI] [PubMed] [Google Scholar]
- 94.Karam S, Margaria JP, Bourcier A, et al. Cardiac Overexpression of PDE4B Blunts beta-Adrenergic Response and Maladaptive Remodeling in Heart Failure. Circulation. 2020. April 8. [DOI] [PubMed] [Google Scholar]
- 95.Bobin P, Varin A, Lefebvre F, et al. Calmodulin kinase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors. Cardiovasc Res. 2016. May 1;110(1):151–61. [DOI] [PubMed] [Google Scholar]
- 96.Fertig BA, Baillie GS. PDE4-Mediated cAMP Signalling. J Cardiovasc Dev Dis. 2018. January 31;5(1). [DOI] [PMC free article] [PubMed] [Google Scholar]; • A detailed review on the role of PDE4 in cardiovascular system.
- 97.Leroy J, Richter W, Mika D, et al. Phosphodiesterase 4B in the cardiac L-type Ca(2)(+) channel complex regulates Ca(2)(+) current and protects against ventricular arrhythmias in mice. J Clin Invest. 2011. July;121(7):2651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Beca S, Helli PB, Simpson JA, et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res. 2011. October 14;109(9):1024–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sin YY, Edwards HV, Li X, et al. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the beta-agonist induced hypertrophic response in cardiac myocytes. J Mol Cell Cardiol. 2011. May;50(5):872–83. [DOI] [PubMed] [Google Scholar]
- 100.Edwards HV, Scott JD, Baillie GS. PKA phosphorylation of the small heat-shock protein Hsp20 enhances its cardioprotective effects. Biochem Soc Trans. 2012. February;40(1):210–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Martin TP, Hortigon-Vinagre MP, Findlay JE, et al. Targeted disruption of the heat shock protein 20-phosphodiesterase 4D (PDE4D) interaction protects against pathological cardiac remodelling in a mouse model of hypertrophy. FEBS Open Bio. 2014;4:923–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dodge KL, Khouangsathiene S, Kapiloff MS, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001. April 17;20(8):1921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dodge-Kafka KL, Soughayer J, Pare GC, et al. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005. September 22;437(7058):574–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang L, Burmeister BT, Johnson KR, et al. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell Signal. 2015. May;27(5):908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Corbin J, Rannels S, Neal D, et al. Sildenafil citrate does not affect cardiac contractility in human or dog heart. Curr Med Res Opin. 2003;19(8):747–52. [DOI] [PubMed] [Google Scholar]
- 106.Vandeput F, Krall J, Ockaili R, et al. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther. 2009. September;330(3):884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pokreisz P, Vandenwijngaert S, Bito V, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009. January 27;119(3):408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang M, Koitabashi N, Nagayama T, et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008. August 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lu Z, Xu X, Hu X, et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010. April 6;121(13):1474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Takimoto E, Champion HC, Belardi D, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005. January 7;96(1):100–9. [DOI] [PubMed] [Google Scholar]
- 111.Lukowski R, Rybalkin SD, Loga F, et al. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci U S A. 2010. March 23;107(12):5646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Borlaug BA, Melenovsky V, Marhin T, et al. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005. October 25;112(17):2642–9. [DOI] [PubMed] [Google Scholar]
- 113.Takimoto E, Belardi D, Tocchetti CG, et al. Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007. April 24;115(16):2159–67. [DOI] [PubMed] [Google Scholar]
- 114.Senzaki H, Smith CJ, Juang GJ, et al. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001. August;15(10):1718–26. [DOI] [PubMed] [Google Scholar]
- 115.Nagayama T, Hsu S, Zhang M, et al. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009. January 13;53(2):207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ockaili R, Salloum F, Hawkins J, et al. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol. 2002. September;283(3):H1263–9. [DOI] [PubMed] [Google Scholar]
- 117.Salloum FN, Takenoshita Y, Ockaili RA, et al. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol. 2007. February;42(2):453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chau VQ, Salloum FN, Hoke NN, et al. Mitigation of the progression of heart failure with sildenafil involves inhibition of RhoA/Rho-kinase pathway. Am J Physiol Heart Circ Physiol. 2011. June;300(6):H2272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fisher PW, Salloum F, Das A, et al. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005. April 5;111(13):1601–10. [DOI] [PubMed] [Google Scholar]
- 120.Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem. 2005. April 1;280(13):12944–55. [DOI] [PubMed] [Google Scholar]
- 121.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11(2):214–22. [DOI] [PubMed] [Google Scholar]
- 122.Hassan MA, Ketat AF. Sildenafil citrate increases myocardial cGMP content in rat heart, decreases its hypertrophic response to isoproterenol and decreases myocardial leak of creatine kinase and troponin T. BMC Pharmacol. 2005. April 6;5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang M, Takimoto E, Hsu S, et al. Myocardial remodeling is controlled by myocyte-targeted gene regulation of phosphodiesterase type 5. J Am Coll Cardiol. 2010. December 7;56(24):2021–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lukowski R, Rybalkin SD, Loga F, et al. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci U S A. March 23;107(12):5646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Patrucco E, Domes K, Sbroggio M, et al. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A. 2014. September 2;111(35):12925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Straubinger J, Schottle V, Bork N, et al. Sildenafil Does Not Prevent Heart Hypertrophy and Fibrosis Induced by Cardiomyocyte Angiotensin II Type 1 Receptor Signaling. J Pharmacol Exp Ther. 2015. September;354(3):406–16. [DOI] [PubMed] [Google Scholar]
- 127.Horvath A, Giatzakis C, Tsang K, et al. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: a novel PDE8B isoform in human adrenal cortex. Eur J Hum Genet. 2008. October;16(10):1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kokkonen-Simon KM, Saberi A, Nakamura T, et al. Marked disparity of microRNA modulation by cGMP-selective PDE5 versus PDE9 inhibitors in heart disease. JCI Insight. 2018. August 9;3(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Catalanotto C, Cogoni C, Zardo G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int J Mol Sci. 2016. October 13;17(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Porcu E, Medici M, Pistis G, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS Genet. 2013;9(2):e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hankir MK, Kranz M, Gnad T, et al. A novel thermoregulatory role for PDE10A in mouse and human adipocytes. EMBO Mol Med. 2016. July;8(7):796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nawrocki AR, Rodriguez CG, Toolan DM, et al. Genetic deletion and pharmacological inhibition of phosphodiesterase 10A protects mice from diet-induced obesity and insulin resistance. Diabetes. 2014. January;63(1):300–11. [DOI] [PubMed] [Google Scholar]
- 133.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007. July 24;116(4):434–48. [DOI] [PubMed] [Google Scholar]
- 134.Ayres JK, Maani CV. Milrinone. StatPearls; Treasure Island (FL) 2020. [Google Scholar]
- 135.Young RA, Ward A. Milrinone. A preliminary review of its pharmacological properties and therapeutic use. Drugs. 1988. August;36(2):158–92. [DOI] [PubMed] [Google Scholar]
- 136.Chong LYZ, Satya K, Kim B, et al. Milrinone Dosing and a Culture of Caution in Clinical Practice. Cardiol Rev. 2018. Jan-Feb;26(1):35–42. [DOI] [PubMed] [Google Scholar]
- 137.Dhaliwal A, Gupta M. PDE5 Inhibitor. StatPearls; Treasure Island (FL) 2020. [Google Scholar]
- 138.Corbin JD, Beasley A, Blount MA, et al. High lung PDE5: a strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem Biophys Res Commun. 2005. September 2;334(3):930–8. [DOI] [PubMed] [Google Scholar]
- 139.De Vecchis R, Cesaro A, Ariano C, et al. Phosphodiesterase-5 Inhibitors Improve Clinical Outcomes, Exercise Capacity and Pulmonary Hemodynamics in Patients With Heart Failure With Reduced Left Ventricular Ejection Fraction: A Meta-Analysis. J Clin Med Res. 2017. June;9(6):488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guazzi M, Vicenzi M, Arena R, et al. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011. January;4(1):8–17. [DOI] [PubMed] [Google Scholar]
- 141.Giannetta E, Isidori AM, Galea N, et al. Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation. 2012. May 15;125(19):2323–33. [DOI] [PubMed] [Google Scholar]
- 142.Liu LC, Hummel YM, van der Meer P, et al. Effects of sildenafil on cardiac structure and function, cardiopulmonary exercise testing and health-related quality of life measures in heart failure patients with preserved ejection fraction and pulmonary hypertension. Eur J Heart Fail. 2017. January;19(1):116–125. [DOI] [PubMed] [Google Scholar]
- 143.Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013. March 27;309(12):1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang YS, Li JD, Yan C. An update on vinpocetine: New discoveries and clinical implications. Eur J Pharmacol. 2018. January 15;819:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Snyder PB, Esselstyn JM, Loughney K, et al. The role of cyclic nucleotide phosphodiesterases in the regulation of adipocyte lipolysis. J Lipid Res. 2005. March;46(3):494–503. [DOI] [PubMed] [Google Scholar]
- 146.Palmer D, Tsoi K, Maurice DH. Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ Res. 1998;82(8):852–61. [DOI] [PubMed] [Google Scholar]
- 147.Kraynik SM, Miyaoka RS, Beavo JA. PDE3 and PDE4 isozyme-selective inhibitors are both required for synergistic activation of brown adipose tissue. Mol Pharmacol. 2013. June;83(6):1155–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vinogradova TM, Sirenko S, Lukyanenko YO, et al. Basal Spontaneous Firing of Rabbit Sinoatrial Node Cells Is Regulated by Dual Activation of PDEs (Phosphodiesterases) 3 and 4. Circ Arrhythm Electrophysiol. 2018. June;11(6):e005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abbott-Banner KH, Page CP. Dual PDE3/4 and PDE4 inhibitors: novel treatments for COPD and other inflammatory airway diseases. Basic Clin Pharmacol Toxicol. 2014. May;114(5):365–76. [DOI] [PubMed] [Google Scholar]
- 150.Singh D, Martinez FJ, Watz H, et al. A dose-ranging study of the inhaled dual phosphodiesterase 3 and 4 inhibitor ensifentrine in COPD. Respir Res. 2020. February 10;21(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]