

Abstract
Introduction:
Antipsychotic drugs that target the dopamine system have been central to the treatment of schizophrenia, but their limitations in efficacy, tolerability, and precision necessitate improved treatment strategies. Multiple lines of research have implicated glutamatergic dysfunction in the hippocampus as an early source of pathophysiology in schizophrenia. Novel compounds have been designed to treat glutamatergic dysfunction, but they have produced inconsistent results in clinical trials.
Areas Covered:
This review discusses how the hippocampus is thought to drive psychotic symptoms through its influence on the dopamine system. It offers the reader an evaluation of proposed treatment strategies including direct modulation of GABA or glutamate neurotransmission or reducing the deleterious impact of stress on circuit development. Finally, we offer a perspective on aspects of future research that will advance our knowledge and may create new therapeutic opportunities. PubMed was searched for relevant literature between 2010–2020 and related studies.
Expert Opinion:
Targeting the aberrant excitatory-inhibitory functioning observed in the hippocampus and its related circuits has the potential to both alleviate symptoms and reduce the risk of transition to psychosis if implemented as an early intervention. Longitudinal multimodal brain imaging combined with mechanistic theories generated from animal models can be used to better understand the progression of hippocampal-dopamine circuit dysfunction and factors related to heterogeneity in treatment response.
Keywords: Schizophrenia, Antipsychotic, Dopamine, Glutamate, GABA, Hippocampus, D2, Early Intervention
1.0. Introduction
Schizophrenia is most characteristically defined by psychotic symptoms, which include hallucinations and delusions. Psychosis in schizophrenia typically emerges in late adolescence and early adulthood (1). The first episode of psychosis (FEP) is often preceded by a prodromal period as sub-threshold symptoms develop, including attenuated psychotic symptoms, and deterioration in social and cognitive functioning (2). Some patients experience sustained symptom remittance with treatment and fully recover functional capability following FEP, However, many patients remain chronically ill with exacerbations and remissions of symptoms over time (3–5).
Current antipsychotic drugs are generally effective at dampening psychotic symptoms, but they often leave a high level of disease burden (6, 7), and numerous side effects that contribute to treatment nonadherence (8). Considerable heterogeneity exists in symptom profiles and treatment response. Up to 30% of patients display persistent symptoms despite multiple trials of antipsychotic drugs, referred to as treatment resistance (9, 10). There are few options available to patients with treatment resistance because all antipsychotic drugs produce their therapeutic effect via a common mechanism, by blocking D2 receptors (11). Blockade of D2 receptors reduces dopamine (DA) transmission, in line with the DA hypothesis, which postulates that increased presynaptic DA transmission in the striatum underlies the expression of psychosis (12, 13). However, it has been proposed that patients who do not respond to current drugs may either not display this hyperdopaminergic phenotype, which would be apparent from the start of treatment, or they may develop DA supersensitivity over time following prolonged D2 antagonist treatment (14–16). These situations highlight the need for alternative treatment strategies.
Although DA hyperactivity plays a critical role in schizophrenia, it is not the only neurotransmitter involved in the development of psychosis and multiple lines of research have demonstrated that it is secondary to dysregulation of excitatory and inhibitory neurotransmission (17, 18). In addition, current DA antagonist drugs fail to effectively address cognitive and negative symptoms of this disorder (19). Numerous novel target mechanisms have been tested in clinical trials, yet none have proven sufficiently effective to be approved for clinical practice (20, 21). This review will focus on promising strategies for treating DA dysfunction in schizophrenia through targeting upstream pathophysiology in the hippocampus, with a focus on targeting excitatory-inhibitory dysregulation through 1) drugs that act therapeutically to regulate glutamate, GABA, or inflammation 2) approaches that are preventative in nature, such as mitigating the effects of stress or early behavioral intervention (Table 1).
Table 1.
A summary of potential targets for the treatment of psychosis.
| Approach | Therapeutic/Preventative | Putative Clinical Effect | Examples |
|---|---|---|---|
| GABA receptors | Therapeutic | Increase GABA signaling to increase inhibition of pyramidal neurons |
|
| NMDA receptors | Therapeutic | Enhance NMDA-mediated interneuron function to increase inhibition of pyramidal neurons |
|
| mGluR2/3 receptors | Therapeutic | Normalize glutamate release to reduce pyramidal neuron hyperactivity |
|
| Inflammation | Both | Prevent oxidative stress to protect PV+ interneurons |
|
| Stress | Preventative | Protect hippocampal circuits from environmental stressors/excessive HPA activation |
|
2.0. Neurotransmitter Dysregulation in Schizophrenia
2.1. Dopamine
Neuroimaging studies have demonstrated that psychotic symptoms are associated with increased presynaptic DA signaling in the striatum, including measures of synthesis capacity and release (22–27). Higher DA synthesis capacity is also observed in clinically high risk (CHR) individuals, which correlates with greater severity of prodromal symptoms (28, 29). DA synthesis capacity progressively increases as CHR individuals transition to psychosis and it may be able to predict likelihood of conversion (28–31). These findings support the hypothesis that presynaptic DA dysfunction in subcortical projections, particularly to the associative striatum (26), is closely linked to the onset and expression of psychotic symptoms. The DA hypothesis has held up to decades of research, but it has also been modified based on evidence that the nature of DA dysregulation varies based on brain region (13). Specifically, blunted DA release has been reported in prefrontal cortical regions in patients with schizophrenia, which is suggested to contribute to cognitive symptoms (32). Potential therapeutic options for modifying cortical DA transmission have been discussed elsewhere (33, 34), and this review will focus on treatment of psychosis associated with the subcortical DA dysregulation and other target structures.
All current antipsychotics target the DA system by blocking D2 receptors (11). Animal research has shown that D2 antagonists reduce the number of spontaneously active DA neurons (i.e. population activity) (35–37). Blockade of both presynaptic and postsynaptic D2 receptors results in overexcitation-induced depolarization block, leading to a broad reduction in DA signaling (38). The majority of antipsychotic drugs act as D2 antagonists; however, D2 partial agonists, such as aripiprazole are also available. D2 partial agonists produce a “normalizing” effect on DA neuron population activity depending on the state of the DA system (39, 40). In contrast to D2 antagonists, D2 partial agonists do not induce depolarization block in vivo, and instead may functionally act as an agonist on presynaptic D2 receptors to downregulate DA neuron activity (41). Although current antipsychotics directly target the DA system, there is little evidence for dysfunction within the DA system itself. Its hyperresponsiveness is instead proposed to be a consequence of excitatory-inhibitory imbalance in afferent structures (42).
2.2. Glutamate and GABA
One strategy to address DA dysfunction in schizophrenia is to modulate the upstream abnormalities in glutamate and GABA neurotransmission. Schizophrenia is associated with altered excitatory-inhibitory transmission in several brain regions, and some studies have shown these abnormalities are present in CHR individuals (43). The hippocampus displays a hypermetabolic state in patients with schizophrenia (44–49). Increased perfusion has also been observed in CHR individuals in the CA1 region (50–52), which spreads to the subiculum, the predominant output structure of the hippocampus, upon onset of psychosis (48). Elevated hippocampal glutamate levels have been observed in patients diagnosed with schizophrenia (43, 53–55), and reported in CHR individuals in some studies (50, 53), although not all (56). There is evidence that glutamate levels in CHR individuals may predict conversion to psychosis (53, 57), although this is also not a consistent finding (50, 56). It is possible that these changes in the hippocampus relate to a general psychosis spectrum that may or may not reach diagnostic criteria for schizophrenia, which may contribute to inconsistencies in longitudinal studies looking at CHR individuals. Additionally, there are different indices of glutamate functioning, including glutamate itself, its metabolite, glutamine, and Glx (glutamate + glutamine). Some studies have found increases in glutamate, whereas others have reported increases in glutamine, Glx (43, 58), or the ratio of glutamine to glutamate (59, 60), which has been suggested to be a more sensitive measure of glutamate release (59)
The increased metabolism of the hippocampus is associated with a loss of local GABAergic parvalbumin (PV+) interneurons that normally regulate pyramidal neuron activity, and local atrophy, which is believed to reflect loss of the interneurons and neuropil (61–63). More research is needed to understand the relationship between these pathophysiological findings, how they relate to vulnerability to psychosis in the high-risk state, and their relative progression over time. Ultimately, the resulting loss of GABA interneuron function is proposed to lead dysregulation of excitatory neurotransmission within the hippocampus, and thereby a disruption of a polysynaptic circuit that regulates DA transmission.
The methyazoxymethanol acetate (MAM) neurodevelopmental rodent model (64, 65) has demonstrated that increased pyramidal neuron activity in the hippocampus leads to an increased number of spontaneously active DA neurons available for phasic DA release. The offspring of pregnant females that receive an injection of MAM at gestational day (GD) 17 develop adult phenotypes relevant to schizophrenia, in contrast to the offspring of pregnant females that receive a saline (SAL) injection (64, 66, 67). Similar to findings in schizophrenia patients (17), MAM rats show loss of PV+ interneurons in the hippocampus (63), resulting in an increase in pyramidal cell activity and a baseline hyperactive state (68). The subiculum of the hippocampus extends glutamatergic projections to the nucleus accumbens, which in turn inhibits the ventral pallidum. (69, 70). The ventral pallidum holds a variable proportion of DA neurons in the VTA in a hyperpolarized state. As a consequence of hippocampal hyperactivity, greater inhibition of GABAergic neurons in the ventral pallidum results in reduced inhibitory hold on DA neurons in the VTA and an increase in the number of spontaneously active DA neurons compared to control rats (68). This circuit is normally adaptive to environmental stimuli to set the gain of DA neurotransmission. However, with abnormally increased activity of the hippocampus, the DA neuron population activity is proposed to be set at a high gain state, allowing for increased release of DA in the striatum, consistent with clinical studies in patients with schizophrenia (42, 63, 68) (Figure 1).
Figure 1. The hippocampus regulates DA neuron population activity through a polysynaptic circuit.
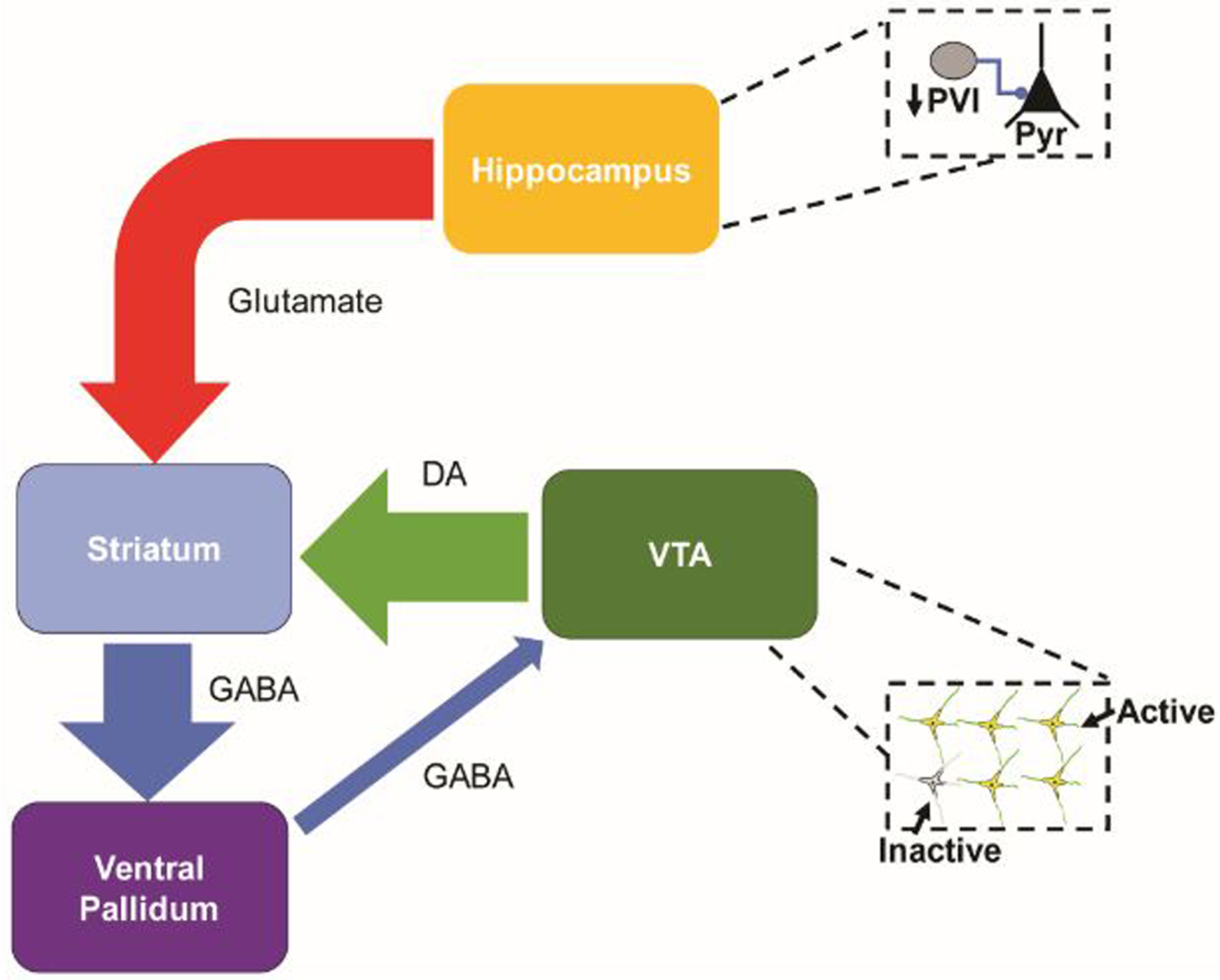
Pyramidal neurons (Pyr) in the subiculum of the hippocampus send excitatory projections to neurons in the nucleus accumbens that, in turn, inhibit activity in the ventral pallidum. The ventral pallidum holds a variable subset of DA neurons in the ventral tegmental area (VTA) in an inhibited state. Through this circuit, activation of the hippocampus modulates the number of spontaneously active DA neurons. In schizophrenia, hyperactivity of Pyr in the hippocampus, following a loss of PV+ interneurons, drives an increase in DA neuron population activity. The increased gain results in increased phasic striatal DA release.
3.0. Promising Antipsychotic Drug Targets
Numerous novel target compounds for the treatment of schizophrenia have shown promise in preclinical research, but to date, none have shown sufficient efficacy in phase 3 clinical trials to enter clinical practice (34). However, the increased striatal DA signaling targeted by current antipsychotics is a consequence of upstream anomalies that are implicated in both the development and expression of psychotic symptoms (71). Indeed, the induction of DA supersensitivity by prior treatment with current antipsychotic drugs may interfere with the efficacy of such novel target agents (72). These targets are derived from a well-supported framework and still show promise, but future studies are required to better understand their mechanisms of action and optimal methods for clinical trial design and ultimately treatment.
3.1. GABA
Compounds that target the GABA system aim to restore normal patterns of PV+ interneuron activity. Indeed, transplants of GABA interneuron precursor cells into the hippocampus of MAM rats has been shown to normalize hippocampal pyramidal neuron activity and DA neuron population activity, demonstrating that restoring GABA function is sufficient to normalize the aberrant hippocampus-DA circuit (73). Increased GABA signaling can also be accomplished through direct modulation of GABA receptors. Broad action GABA modulators, such as benzodiazepines are problematic due to their sedative actions, risk of dependency and inferiority in reducing psychotic symptoms compared to current antipsychotics in patients with chronic schizophrenia (74), but other, more localized options, have been studied for their potential therapeutic value.
One target that has shown promise is the α5 subtype of the GABAA (α5GABAA) receptor, which is highly expressed in limbic brain regions, including the hippocampus (75). α5GABAA receptors are involved in generating a tonic inhibitory current to pyramidal neurons, which may help coordinate spike timing (76–78). A positive allosteric modulator (PAM) that acts at α5GABAA receptors has been tested in the MAM model, and reduced DA neuron population activity and amphetamine-induced hyperlocomotion to control levels when administered either systemically or directly infused into the ventral hippocampus. α5GABAA PAM administration also reduced evoked responses in hippocampal neurons (79). The α5GABAA receptor may be an effective antipsychotic target to normalize hippocampal activity, but it has yet to be tested in patients.
The mood stabilizer valproate has also been studied as a potential antipsychotic that affects the GABA system. Valproate inhibits histone deacetylation and can increase GABA synthesis at high concentrations (80–83). As a treatment for schizophrenia, some clinical trials found that valproate could enhance the rate of improvement of symptoms when used as an adjunct to current antipsychotics, but results were inconsistent (84–86). More recently, valproate has been studied for its potential in reinstating critical periods of plasticity in adulthood (87–89), and thus might provide additional value in conjunction with other therapies.
3.2. Glutamate
Evidence from preclinical and clinical studies have supported the role of glutamate system dysfunction in the pathophysiology of schizophrenia, including elevated levels of glutamate in the hippocampus of unmedicated patients (43). Thus, another promising option for the treatment of schizophrenia is inhibition of glutamate release.
Numerous glutamate-targeting compounds have been evaluated in clinical trials. NMDA receptor co-agonists, including D-cycloserine, D-serine, and glycine, were among the earliest studied in an effort to enhance NMDA-mediated interneuron function (90), thereby increasing inhibition of pyramidal neurons. Despite initial promise in clinical trials, none of these compounds passed phase 2 or phase 3 clinical trials as either a monotherapy or adjunct to current treatments (91, 92). Selective glycine transporter 1 (GlyT1) inhibitors, such as sarcosine and biopertin have also been tested as an alternative method to increase the availability of glycine at NMDA receptors (93). Sarcosine has demonstrated success in clinical trials as an adjunct treatment in early clinical trials in improving positive, negative, and cognitive symptoms (94, 95) with some evidence for greater efficacy than NMDA receptor agonists (96). However, not all trials have shown significant results with GlyT1 inhibitors, such as when added to clozapine or tested as a monotherapy (97–99). D-amino acid oxidase (DAAO) inhibition with compounds such as sodium benzoate, has also recently been explored as a method of enhancing NMDA receptor activation by blocking D-amino acid metabolism. Sodium benzoate has produced promising results as an adjunctive therapy in early clinical trials (100–102). Larger clinical trials are needed to better assess the potential benefits of NMDA receptor-targeting drugs.
Group II metabotropic glutamate receptor (mGluR2/3) agonists were another target that garnered interest as a novel treatment for schizophrenia. mGluR2/3 are expressed in limbic brain regions and localized presynaptically on glutamatergic terminals to negatively regulate glutamate release (103). Preclinical research produced extensive support for mGluR2/3 agonists and PAMs (104–109). The mGluR2/3 agonist, pomagluemtad methionil, was shown to reduce hippocampal pyramidal neuron hyperactivity in the MAM model, resulting in the downstream normalization of DA neuron population activity and improvement in a hippocampal-dependent task, which were not observed in normal rats (110).However, while pomaglumetad methionil demonstrated efficacy in a phase 2 clinical trial (111) it subsequently failed to show efficacy in other phase 2 trials as a monotherapy or adjunct therapy (112–114) and as a monotherapy in a phase 3 trial (115). It has been suggested that it may be most effective early in the disease (116) or that the dose used in clinical trials was not adequate (117). Another possibility is that withdrawal from current D2 antagonist drugs leaves a state of DA receptor supersensitivity, impeding the efficacy of non-D2 drugs at reversing the hyperdopaminergic state, as suggested in animal models (72). This could also account for why patients that had not been treated as long with D2 antagonists show greater efficacy. Additional trials are needed in FEP and CHR populations for clarification and further research is needed to understand why certain groups may respond better to glutamate-targeting drugs.
3.3. Inflammation
Another potential means of early intervention in schizophrenia is through targeting neuroinflammation and immune system dysregulation (118). Prenatal inflammatory events, such as infections, illicit maternal immune activation that may cause deleterious effects on neurodevelopment. In combination with other risk factors, it may lead to disrupted synaptic plasticity and impaired development of PV+ interneurons from oxidative stress that leave the brain vulnerable to additional hits of stress (119, 120). In animal models, early life immune activation, such as through injecting double-stranded RNA poly(I:C) or bacterial lipopolysaccharide (LPS), alter cytokine levels in the placenta, amniotic fluid and fetal brain (121–125). This leads to increased vulnerability to stress-induced increases in inflammatory markers in adulthood (126), consistent with findings in patients with schizophrenia (127, 128). Risk of transition to psychosis is also associated with increases in inflammatory markers (129, 130). Some anti-inflammatory compounds have demonstrated prophylactic effects in maternal immune activation rodent models, including N-acetylcysteine and sulforaphane (131, 132). Antioxidants that reduce inflammation, such as omega-3 fatty acids (133) have produced mixed success in reducing transition rates in patients (2, 134, 135), but investigation of other anti-inflammatory compounds may be warranted. For example, some studies have demonstrated a deficit in glutathione in patients with schizophrenia, which is a prominent cellular antioxidant. Glutamatergic dysfunction and subsequent atrophy may be most apparent in the presence of insufficient glutathione, and it has been proposed that interventions that increase glutathione may be beneficial in preventing the detrimental effects of oxidative stress (136, 137).
3.4. Stress
Based on the diathesis-stress model, early disruption in brain development can lead to heightened vulnerability to stress (138, 139). Increased vulnerability to stress implies that there does not need to be a difference in the acute stress exposure, but in vulnerable individuals stress may trigger the first episode and precede relapses (140, 141). Dysregulated hypothalamic-pituitary-adrenal (HPA) axis activity is implicated the development of schizophrenia (142). In CHR individuals, salivary cortisol levels are correlated with severity of anxiety, suspiciousness, and impaired stress tolerance (142, 143) and also higher in individuals who transition to schizophrenia compared to those who do not (144). It is specifically distressing psychotic-like experiences in adolescence that are most indicative of CHR state and greater risk of developing a psychotic disorder (145). Compared to healthy controls, patients with active psychosis and those at CHR demonstrate greater stress-induced DA release, associated with an elevated cortisol response to the stressor (146).Together, these findings suggest a close link between stress sensitivity and the development of DA dysfunction.
The hippocampus is highly susceptible to stress (147, 148) and the influence of the HPA axis on hippocampal circuits may serve as a critical mediating link between environmental stressors and the development of psychosis, as these systems can act synergistically to stimulate subcortical DA (149). Early neurodevelopmental insults and other factors that increase response to stress early in life may increase the vulnerability of the hippocampus to HPA activation. The prelimbic prefrontal cortex (plPFC) normally limits the impacts of stress on basolateral amygdala activation in rats (150, 151), which holds a robust modulatory influence on DA neuron activity (152). For example, plPFC lesions performed in naïve rats shortly post-weaning (postnatal day 25) caused the rats to show increased anxiety-like behaviors as adults. When exposed to stressors during puberty do not result in increased DA neuron population activity in adulthood in control rats, the plPFC-lesioned rats exhibit the hyperdopaminergic phenotype as adults (153). However, a combination of prepubertal stressors can lead to increased DA neuron population activity, increased hippocampal pyramidal neuron activity, and a loss of hippocampal PV+ interneurons even in naïve rats that have not experienced plPFC lesions (87, 153). Similarly, MAM rats exhibit a greater response to stress and amygdala hyperactivity pre-pubertally prior to the emergence of the hyperdopaminergic state in adulthood (154, 155). Prepubertal environmental enrichment was sufficient to prevent DA hyperresponsivity in MAM rats through normalizing ventral hippocampal pyramidal neuron activity; however, it was not sufficient to reduce anxiety-like behavior in an elevated plus maze nor hyperactivity of the basolateral amygdala (156). Thus, treatments that reduce stress, and more specifically, protect PV+ interneurons in the hippocampus, may be particularly effective when administered during adolescence.
Taken together, these studies suggest that severe stress or heightened stress responsivity during adolescence may exacerbate the impact of inflammation, leading to impaired development of PV+ interneurons in the hippocampus, and ultimately impaired DA system function that may reciprocally impact stress (157, 158). The hippocampus is a particularly promising target for reducing the effects of stress because PV+ interneurons in the hippocampus continue to mature through late adolescence (159), creating a potential window of opportunity for intervention. The interaction between stress and neurobiological changes indicates that psychological treatments, such as cognitive remediation, cognitive behavioral therapy, or psychosocial therapies, have the potential to be disease modifying. Psychotherapy can be implemented to mitigate stress reactivity and promote social integration, which could have beneficial effects in protecting circuits from disruption. Hybrid treatments with the use of psychotherapy alongside pharmacotherapy may be the most successful in reducing the impact of stress on the neurodevelopment schizophrenia and related disorders (160–162).
4.0. Timing of Treatment
The prodromal phase of schizophrenia is characterized by a period of functional decline, including the emergence of sub-clinical psychotic symptoms that generally begin after puberty and progress in severity (163). It is unclear to what extent schizophrenia is progressive following the prodromal period (164, 165), due to confounding variables such as antipsychotic medication (166–170). The duration of untreated psychosis is a critical factor in determining prognosis, suggesting that active psychosis may be progressively detrimental until treated and alterations in brain structure and neurophysiology may already be established by the time of diagnosis (163). Early interventions may thus provide greater benefits (171).
The developmental changes that occur during puberty have been suggested to be a critical period surrounding the onset of psychosis, such that targeting pathophysiological processes during this time may provide long-term amelioration of symptoms (158). This period of plasticity to allow for maturation may contribute to the vulnerability of the developing brain to environmental factors, including psychological stress, social isolation, and drug abuse, that can lead to the emergence of psychiatric disorders (126, 172). Critical period closure following puberty is marked by the development of perineuronal nets that surround PV+ interneurons to stabilize synapses (173). Oxidative stress may disrupt the formation of perineuronal nets, leading to impaired PV+ interneuron function (87, 174). Antioxidants and glutamate modulating agents have been shown to prevent damage to the perineuronal nets in rodent studies (175). Pharmacological treatments administered during puberty in the MAM model have also been shown to prevent PV+ interneuron loss and circumvent the emergence of schizophrenia-relevant phenotypes in adulthood. Furthermore, peripubertal administration of the anxiolytic drug diazepam prevented the increase in DA neuron population activity, increased anxiety-like behavior and the higher neuronal firing rates within the basolateral amygdala normally present in adult MAM rats, compared to adult SAL rats (154, 155). Long term administration of benzodiazepines is not a feasible intervention due to issues including dependency (74), but these findings indicate that alleviating anxiety and abnormal stress responsivity during puberty may preserve PV+ interneurons and prevent the progression of abnormalities associated with psychosis.
5.0. Heterogeneity in Treatment Response
Due to the substantial variability in symptoms and treatment response, in addition to overlap with other disorders, schizophrenia is not likely to be a distinct disease entity. Its clinical presentation displays better conformity with the concept of a syndrome that lies upon a spectrum and potentially encompass multiple subtypes (176, 177). The heterogeneity is likely reflected in individual differences in the underlying neurobiology that are not adequately treated by current antipsychotics.
PET studies have demonstrated that some patients with treatment resistant schizophrenia do not display striatal hyperdopaminergic activity that is observed in treatment-responsive patients. In contrast, treatment resistant patients displayed increased levels of glutamate in the anterior cingulate, which is present from the first episode (14, 15, 178–180). Differences in glutamate have also been observed in patients who respond to clozapine, the antipsychotic that currently has the highest efficacy for refractory symptoms (181). Clozapine-responsive patients displayed higher levels of glutamate and glutamine in the putamen compared to patients who respond to first-line treatment and those who are resistant to clozapine (“ultra-treatment resistant”) (182). Whether this is indicative of a different disease pathophysiology or differences in system responsivity is unclear. For example, rapid antipsychotic drug-induced induction of depolarization block of DA neurons requires the presence of a hyper-excited DA system at baseline. If the DA system function is attenuated, addition of antipsychotics may be subthreshold for inducing depolarization block and preventing abnormal DA system activation (35, 183). Further research is needed to determine whether treatment resistance from the first psychotic episode lies on a spectrum or represents multiple distinct subtypes. A greater understanding of how individual patterns of network dysfunction contribute to differences in symptoms and clinical outcomes is critical to developing personalized treatments.
6.0. Summary
Current D2-targeting antipsychotics alleviate certain symptoms in patients, but given their central role in the pathophysiology of schizophrenia (17, 63, 68, 184), GABAergic and glutamatergic targets still hold promise as an effective therapy to regulate glutamate activity and downstream dopamine hyperactivity. Additionally, they may provide options for individuals who are resistant to current antipsychotics and display glutamate hyperactivity, as demonstrated by neuroimaging studies. Early interventions that protect excitatory-inhibitory circuits, such as by reducing stress and neuroinflammation, have the potential to modify illness progression. Additional research on the development of psychosis and factors that contribute to heterogeneity in treatment response is needed to best implement these novel strategies.
7.0. Expert Opinion: A Framework for Future Research
The findings discussed in this review support the hypothesis that targeting prodromal and early phase schizophrenia has the potential to limit the pathophysiological progression that leads to a chronic illness state. The implementation of early intervention strategies for CHR individuals requires reliable biomarkers to predict disease course and treatment response. Potential candidates include striatal DA concentrations, metabolic activity and glutamate and GABA concentrations in regions including the hippocampus and anterior cingulate, resting state connectivity, salivary cortisol, and serologic markers of oxidative stress and inflammation (10, 163, 185–188). More research is needed to understand the relative reliability of these options, how different trajectories progress, whether current clinical trial designs are effective at identifying novel non-D2 agents, and whether treating general psychopathology at the CHR state is beneficial across psychiatric disorders (189). It is also imperative to accurately predict the risk of transition to schizophrenia to avoid consequences, such as stigma, unnecessary treatment, and related adverse effects (190). Treatment of CHR individuals may reduce their risk of transition to schizophrenia, but they may remain at risk for other psychiatric disorders, such as depression or anxiety (189). A greater understanding of the neurobiology underlying the heterogeneity in schizophrenia will help to determine which therapeutic strategies are most effective for preventing or minimizing psychotic symptoms for an individual. We propose that the most promising avenues of research for novel antipsychotics and early intervention strategies are compounds that normalize excitatory-inhibitory transmission and either reduce stress or mitigate its effects.
Targeting excitatory-inhibitory processes, particularly compounds that regulate hippocampal activity, may avoid the side effects associated with D2 antagonist treatment and may also alleviate more symptoms of the disorder, including negative and cognitive symptoms. Indeed, a hyperactive hippocampus can impact more than the DA system; its dysrhythmia may also interfere with functions in its other targets related to cognition and affect/negative symptoms (119). Clinical trials of excitatory-inhibitory-targeting agents have yielded disappointing results, but further research is needed to understand why these compounds failed, what impact prior treatments may have had on novel compound response, whether there are additional variables that must be considered, and how to more effectively target symptoms. There is evidence to suggest that these compounds may not be effective in patients with chronic schizophrenia who have already received prior D2 antagonist antipsychotic drug treatment (72). However, some of these treatments may be effective in appropriate circumstances, such as in antipsychotic-naïve patients or those who are early in the disease (116). In addition to their prophylactic potential, treatments that normalize glutamate in the hippocampus may provide much-needed options for patients resistant to current antipsychotics (10, 14).
Additional longitudinal studies in clinical populations and neurodevelopmental animal models are needed to characterize the pathophysiological progression of the hippocampus-DA circuit over time and its relationship with changes in symptoms. Additional time points in large samples may provide clarity to the interplay between hippocampal glutamate levels, increased metabolism, and atrophy and how these factors relate to changes in the DA system (191). A timeline of whether a strategy may be most effective in the high-risk state, and to what extent it remains beneficial following the first episode could provide valuable insight into early intervention strategies.
Compounds that treat schizophrenia closer to the site of pathology by modulating glutamate and GABA transmission and/or mitigating the effects of stress hold promise for improving outcomes in patients with schizophrenia. Future research must focus on improving our understanding of the heterogeneity of the disease to determine which patients are most likely to respond to a given intervention. Additionally, a more detailed timeline of the progression of pathophysiological changes involved in schizophrenia is required to determine the optimal window for treatment. Targeted research questions from clinical studies and animal models that move toward the goal of a more reliable and individualized process of treatment, driven by a more informed understanding of the variables at hand, will ultimately help break the status quo and improve clinical outcomes.
Article Highlights.
Developmental animal models relevant to schizophrenia are an effective approach to examine how neurotransmitter systems interact and change over time, which can provide valuable information about potential targets for early intervention and treatment.
Animal models have demonstrated that stress during puberty can impair perineuronal nets around interneurons and have long-lasting impacts on dopamine neuron activity.
Modulating excitatory-inhibitory dysregulation is promising as an alternative therapeutic strategy to D2-targeting antipsychotic drugs, and merits further research as a potential early intervention.
Targeting hippocampal pathophysiology upstream of dopamine neuron dysfunction has the potential to both normalize dopamine neuron activity and alleviate other symptoms of schizophrenia.
Future research must focus on categorizing subtypes to determine which patients may benefit most from a treatment, adapting clinical trial design for non-dopaminergic agents, and employing animal models to study changes that may not be apparent in normal rodents.
Funding
The research of the authors is supported by grants from US National Institutes of Health (MH57440 to AAG)
Reviewer disclosures
One reviewer reports serving on the Aristada Schizophrenia Advisory Board for Alkermes and the MedinCell Psychiatry Advisory Board. They have conducted clinical research supported by the NIMH, Sunovion, the Stanley Foundation, Takeda, Taisho, Lundbeck, Boehringer Ingelheim, NeuroRX, Teva and Lilly within the last 24 months and were a co-investigator on a study that receives lumeteperone and reimbursement for safety testing for an investigator-initiated research from Intra-Cellular Therapies Inc. He owns a small number of shares of common stock from GSK. One reviewer has received manuscript or speaker’s fees from Astellas, Dainippon Sumitomo Pharma, Eisai, Eli Lilly, Elsevier Japan, Janssen Pharmaceuticals, Kyowa Yakuhin, Meiji Seika Pharma, Mitsubishi Tanabe Pharma, MSD, Novartis, Otsuka Pharmaceutical, Shionogi, Shire, Tsumura, Wiley Japan, and Yoshitomi Yakuhin, and research grants from Eisai, Mochida Pharmaceutical, Meiji Seika Pharma and Shionogi. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose
Footnotes
Declaration of Interest
AA Grace has received consulting fees from Alkermes, Lundbeck, Takeda, Roche, Lyra, Concert, and research funding from Lundbeck. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophrenia bulletin. 2009;35(3):528–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larson MK, Walker EF, Compton MT. Early signs, diagnosis and therapeutics of the prodromal phase of schizophrenia and related psychotic disorders. Expert review of neurotherapeutics. 2010;10(8):1347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegarty JD, Baldessarini RJ, Tohen M, Waternaux C, Oepen G. One hundred years of schizophrenia: a meta-analysis of the outcome literature. American Journal of psychiatry. 1994;151(10):1409–16. [DOI] [PubMed] [Google Scholar]
- 4.Harrow M, Jobe T, Faull R. Does treatment of schizophrenia with antipsychotic medications eliminate or reduce psychosis? A 20-year multi-follow-up study. Psychological Medicine. 2014;44(14):3007–16. [DOI] [PubMed] [Google Scholar]
- 5.Zipursky RB, Menezes NM, Streiner DL. Risk of symptom recurrence with medication discontinuation in first-episode psychosis: a systematic review. Schizophrenia Research. 2014;152(2–3):408–14. [DOI] [PubMed] [Google Scholar]
- 6.Eack SM, Newhill CE. Psychiatric symptoms and quality of life in schizophrenia: a meta-analysis. Schizophrenia bulletin. 2007;33(5):1225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kennedy JL, Altar CA, Taylor DL, Degtiar I, Hornberger JC. The social and economic burden of treatment-resistant schizophrenia: a systematic literature review. International clinical psychopharmacology. 2014;29(2):63–76. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl j Med. 2005;2005(353):1209–23. [DOI] [PubMed] [Google Scholar]
- 9.Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS medicine. 2005;2(5):e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lally J, Ajnakina O, Di Forti M, Trotta A, Demjaha A, Kolliakou A, et al. Two distinct patterns of treatment resistance: clinical predictors of treatment resistance in first-episode schizophrenia spectrum psychoses. Psychological Medicine. 2016;46(15):3231–40. [DOI] [PubMed] [Google Scholar]
- 11.Kapur S, Remington G. Dopamine D 2 receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biological psychiatry. 2001;50(11):873–83. [DOI] [PubMed] [Google Scholar]
- 12.Van Rossum J The significance of dopamine-receptor blockade for the action of neuroleptic drugs. Neuropsychopharmacology. Proceedings 5th Collegium Internationale Neruropsychopharmacologicum, 1967. 1967. [Google Scholar]
- 13.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophrenia bulletin. 2009;35(3):549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demjaha A, Lappin J, Stahl D, Patel M, MacCabe J, Howes O, et al. Antipsychotic treatment resistance in first-episode psychosis: prevalence, subtypes and predictors. Psychological Medicine. 2017:1–9. [DOI] [PubMed] [Google Scholar]
- 15.Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biological psychiatry. 2014;75(5):e11–e3. [DOI] [PubMed] [Google Scholar]
- 16.Chouinard G, Samaha A-N, Chouinard V-A, Peretti C-S, Kanahara N, Takase M, et al. Antipsychotic-Induced Dopamine Supersensitivity Psychosis: Pharmacology, Criteria, and Therapy. Psychotherapy and Psychosomatics. 2017;86(4):189–219. [DOI] [PubMed] [Google Scholar]
- 17.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25(1):1–27. [DOI] [PubMed] [Google Scholar]
- 18.McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19(1):15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–23. [DOI] [PubMed] [Google Scholar]
- 20.Marek G When is a Proof-of-Concept (POC) not a POC? Pomaglumetad (LY2140023) as a Case Study for Antipsychotic Efficacy. Current pharmaceutical design. 2015;21(26):3788–96. [DOI] [PubMed] [Google Scholar]
- 21.Bespalov A, Steckler T, Altevogt B, Koustova E, Skolnick P, Deaver D, et al. Failed trials for central nervous system disorders do not necessarily invalidate preclinical models and drug targets. Nature Reviews Drug Discovery. 2016;15(7):516. [DOI] [PubMed] [Google Scholar]
- 22.Laruelle M, Abi-Dargham A, Van Dyck CH, Gil R, D’Souza CD, Erdos J, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proceedings of the National Academy of Sciences. 1996;93(17):9235–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. American Journal of Psychiatry. 1998;155(6):761–7. [DOI] [PubMed] [Google Scholar]
- 24.Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biological psychiatry. 1999;46(1):56–72. [DOI] [PubMed] [Google Scholar]
- 25.Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Archives of general psychiatry. 2012;69(8):776–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Archives of general psychiatry. 2010;67(3):231–9. [DOI] [PubMed] [Google Scholar]
- 27.McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophrenia bulletin. 2017;44(6):1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howes OD, Montgomery AJ, Asselin M-C, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Archives of general psychiatry. 2009;66(1):13–20. [DOI] [PubMed] [Google Scholar]
- 29.Howes OD, Bose SK, Turkheimer F, Valli I, Egerton A, Valmaggia LR, et al. Dopamine synthesis capacity before onset of psychosis: a prospective [18F]-DOPA PET imaging study. American Journal of Psychiatry. 2011;168(12):1311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Molecular psychiatry. 2011;16(9):885–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egerton A, Chaddock CA, Winton-Brown TT, Bloomfield MA, Bhattacharyya S, Allen P, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biological psychiatry. 2013;74(2):106–12. [DOI] [PubMed] [Google Scholar]
- 32.Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA psychiatry. 2015;72(4):316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnsten AF, Girgis RR, Gray DL, Mailman RB. Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biological psychiatry. 2017;81(1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girgis RR, Zoghbi AW, Javitt DC, Lieberman JA. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: a critical and comprehensive review. Journal of psychiatric research. 2019;108:57–83. [DOI] [PubMed] [Google Scholar]
- 35.Valenti O, Cifelli P, Gill KM, Grace AA. Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. Journal of Neuroscience. 2011;31(34):12330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grace A, Bunney B. Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. Journal of Pharmacology and Experimental Therapeutics. 1986;238(3):1092–100. [PubMed] [Google Scholar]
- 37.Chiodo LA, Bunney BS. Typical and atypical neuroleptics: differential effects of chronic administration on the activity of A9 and A10 midbrain dopaminergic neurons. Journal of Neuroscience. 1983;3(8):1607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grace AA, Bunney BS, Moore H, Todd CL. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends in neurosciences. 1997;20(1):31–7. [DOI] [PubMed] [Google Scholar]
- 39.Shapiro DA, Renock S, Arrington E, Chiodo LA, Li-Xin L, Sibley DR, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28(8):1400. [DOI] [PubMed] [Google Scholar]
- 40.Burris K, Molski T, Xu C, Ryan E, Tottori K, Kikuchi T, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. The Journal of pharmacology and experimental therapeutics. 2002;302(1):381–9. [DOI] [PubMed] [Google Scholar]
- 41.Sonnenschein SF, Gill KM, Grace AA. State-dependent effects of the D 2 partial agonist aripiprazole on dopamine neuron activity in the MAM neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2019;44(3):572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends in pharmacological sciences. 2011;32(9):507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poels E, Kegeles L, Kantrowitz J, Slifstein M, Javitt D, Lieberman J, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Molecular psychiatry. 2014;19(1):20–9. [DOI] [PubMed] [Google Scholar]
- 44.Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Archives of general psychiatry. 2009;66(9):938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talati P, Rane S, Kose S, Blackford JU, Gore J, Donahue MJ, et al. Increased hippocampal CA1 cerebral blood volume in schizophrenia. NeuroImage: Clinical. 2014;5:359–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11(5):543–50. [DOI] [PubMed] [Google Scholar]
- 47.Malaspina D, Harkavy-Friedman J, Corcoran C, Mujica-Parodi L, Printz D, Gorman JM, et al. Resting neural activity distinguishes subgroups of schizophrenia patients. Biological psychiatry. 2004;56(12):931–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78(1):81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McHugo M, Talati P, Armstrong K, Vandekar SN, Blackford JU, Woodward ND, et al. Hyperactivity and reduced activation of anterior hippocampus in early psychosis. American Journal of Psychiatry. 2019;176(12):1030–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Provenzano FA, Guo J, Wall MM, Feng X, Sigmon HC, Brucato G, et al. Hippocampal Pathology in Clinical High-Risk Patients and the Onset of Schizophrenia. Biological psychiatry. 2019. [DOI] [PubMed] [Google Scholar]
- 51.Allen P, Azis M, Modinos G, Bossong MG, Bonoldi I, Samson C, et al. Increased resting hippocampal and basal ganglia perfusion in people at ultra high risk for psychosis: replication in a second cohort. Schizophrenia bulletin. 2017;44(6):1323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Modinos G, Egerton A, McMullen K, McLaughlin A, Kumari V, Barker GJ, et al. Increased resting perfusion of the hippocampus in high positive schizotypy: A pseudocontinuous arterial spin labeling study. Human brain mapping. 2018;39(10):4055–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bossong MG, Antoniades M, Azis M, Samson C, Quinn B, Bonoldi I, et al. Association of hippocampal glutamate levels with adverse outcomes in individuals at clinical high risk for psychosis. JAMA psychiatry. 2019;76(2):199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraguljac NV, White DM, Hadley J, Reid MA, Lahti AC. Hippocampal‐parietal dysconnectivity and glutamate abnormalities in unmedicated patients with schizophrenia. Hippocampus. 2014;24(12):1524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kraguljac NV, White DM, Reid MA, Lahti AC. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA psychiatry. 2013;70(12):1294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howes OD, Bonoldi I, McCutcheon RA, Azis M, Antoniades M, Bossong M, et al. Glutamatergic and dopaminergic function and the relationship to outcome in people at clinical high risk of psychosis: a multi-modal PET-magnetic resonance brain imaging study. Neuropsychopharmacology. 2019:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biological psychiatry. 2010;68(7):599–602. [DOI] [PubMed] [Google Scholar]
- 58.Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in schizophrenia: a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA psychiatry. 2016;73(7):665–74. [DOI] [PubMed] [Google Scholar]
- 59.Bustillo J, Rowland L, Mullins P, Jung R, Chen H, Qualls C, et al. 1 H-MRS at 4 tesla in minimally treated early schizophrenia. Molecular psychiatry. 2010;15(6):629–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto K, Engberg G, Shimizu E, Nordin C, Lindström LH, Iyo M. Elevated glutamine/glutamate ratio in cerebrospinal fluid of first episode and drug naive schizophrenic patients. BMC psychiatry. 2005;5(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophrenia research. 2011;131(1):165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heckers S, Konradi C. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophrenia research. 2015;167(1):4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. Journal of Neuroscience. 2009;29(8):2344–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biological psychiatry. 2006;60(3):253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnston M, Carman A, Coyle J. Effects of fetal treatment with methylazoxymethanol acetate at various gestational dates on the neurochemistry of the adult neocortex of the rat. Journal of neurochemistry. 1981;36(1):124–8. [DOI] [PubMed] [Google Scholar]
- 66.Modinos G, Allen P, Grace AA, McGuire P. Translating the MAM model of psychosis to humans. Trends in neurosciences. 2015;38(3):129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flagstad P, Mørk A, Glenthøj BY, Van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29(11):2052. [DOI] [PubMed] [Google Scholar]
- 68.Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. Journal of Neuroscience. 2007;27(42):11424–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Floresco SB, Todd CL, Grace AA. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. Journal of Neuroscience. 2001;21(13):4915–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31(7):1356. [DOI] [PubMed] [Google Scholar]
- 71.Sonnenschein SF, Gomes FV, Grace AA. Dysregulation of Midbrain Dopamine System and the Pathophysiology of Schizophrenia. Frontiers in Psychiatry. 2020;11:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gill KM, Cook JM, Poe MM, Grace AA. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophr Bull. 2014;40(2):341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Molecular psychiatry. 2013;18(11):1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sim F, Sweetman I, Kapur S, Patel MX. Re-examining the role of benzodiazepines in the treatment of schizophrenia: a systematic review. Journal of Psychopharmacology. 2015;29(2):212–23. [DOI] [PubMed] [Google Scholar]
- 75.Stefanits H, Milenkovic I, Mahr N, Pataraia E, Hainfellner JA, Kovacs GG, et al. GABAA receptor subunits in the human amygdala and hippocampus: Immunohistochemical distribution of 7 subunits. Journal of Comparative Neurology. 2018;526(2):324–48. [DOI] [PubMed] [Google Scholar]
- 76.Glykys J, Mann EO, Mody I. Which GABAA receptor subunits are necessary for tonic inhibition in the hippocampus? Journal of Neuroscience. 2008;28(6):1421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, et al. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing γ-aminobutyric acid type A receptors. Proceedings of the National Academy of Sciences. 2004;101(10):3662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonin RP, Martin LJ, MacDonald JF, Orser BA. α5GABAA receptors regulate the intrinsic excitability of mouse hippocampal pyramidal neurons. Journal of neurophysiology. 2007;98(4):2244–54. [DOI] [PubMed] [Google Scholar]
- 79.Gill KM, Lodge DJ, Cook JM, Aras S, Grace AA. A novel α5GABAAR-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology. 2011;36(9):1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biggs CS, Pearce BR, Fowler LJ, Whitton PS. The effect of sodium valproate on extracellular GABA and other amino acids in the rat ventral hippocampus: an in vivo microdialysis study. Brain research. 1992;594(1):138–42. [DOI] [PubMed] [Google Scholar]
- 81.Löscher W Valproate enhances GABA turnover in the substantia nigra. Brain research. 1989;501(1):198–203. [DOI] [PubMed] [Google Scholar]
- 82.Tremolizzo L, Doueiri M-S, Dong E, Grayson DR, Davis J, Pinna G, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biological psychiatry. 2005;57(5):500–9. [DOI] [PubMed] [Google Scholar]
- 83.Tremolizzo L, Carboni G, Ruzicka W, Mitchell C, Sugaya I, Tueting P, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proceedings of the National Academy of Sciences. 2002;99(26):17095–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Basan A, Kissling W, Leucht S. Valproate as an adjunct to antipsychotics for schizophrenia: a systematic review of randomized trials. Schizophrenia research. 2004;70(1):33–7. [DOI] [PubMed] [Google Scholar]
- 85.Tseng P-T, Chen Y-W, Chung W, Tu K-Y, Wang H-Y, Wu C-K, et al. Significant effect of valproate augmentation therapy in patients with schizophrenia: a meta-analysis study. Medicine. 2016;95(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Y, Xia J, Helfer B, Li C, Leucht S. Valproate for schizophrenia. Cochrane Database of Systematic Reviews. 2016(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gomes FV, Zhu X, Grace AA. The pathophysiological impact of stress on the dopamine system is dependent on the state of the critical period of vulnerability. Molecular psychiatry. 2019:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Silingardi D, Scali M, Belluomini G, Pizzorusso T. Epigenetic treatments of adult rats promote recovery from visual acuity deficits induced by long‐term monocular deprivation. European Journal of Neuroscience. 2010;31(12):2185–92. [DOI] [PubMed] [Google Scholar]
- 89.Baroncelli L, Scali M, Sansevero G, Olimpico F, Manno I, Costa M, et al. Experience affects critical period plasticity in the visual cortex through an epigenetic regulation of histone post-translational modifications. Journal of Neuroscience. 2016;36(12):3430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. Journal of Neuroscience. 2007;27(43):11496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsai GE, Lin P-Y. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Current pharmaceutical design. 2010;16(5):522–37. [DOI] [PubMed] [Google Scholar]
- 92.Tuominen HJ, Tiihonen J, Wahlbeck K. Glutamatergic drugs for schizophrenia: a systematic review and meta-analysis. Schizophrenia research. 2005;72(2–3):225–34. [DOI] [PubMed] [Google Scholar]
- 93.Javitt DC. Glycine transport inhibitors in the treatment of schizophrenia Novel Antischizophrenia Treatments: Springer; 2012. p. 367–99. [DOI] [PubMed] [Google Scholar]
- 94.Tsai G, Lane H-Y, Yang P, Chong M-Y, Lange N. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biological psychiatry. 2004;55(5):452–6. [DOI] [PubMed] [Google Scholar]
- 95.Lane H-Y, Chang Y-C, Liu Y-C, Chiu C-C, Tsai GE. Sarcosine or D-serine add-on treatment for acute exacerbation of schizophrenia: a randomized, double-blind, placebo-controlled study. Archives of General Psychiatry. 2005;62(11):1196–204. [DOI] [PubMed] [Google Scholar]
- 96.Lane H-Y, Lin C-H, Huang Y-J, Liao C-H, Chang Y-C, Tsai GE. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. International Journal of Neuropsychopharmacology. 2010;13(4):451–60. [DOI] [PubMed] [Google Scholar]
- 97.Lane H-Y, Liu Y-C, Huang C-L, Chang Y-C, Liau C-H, Perng C-H, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biological psychiatry. 2008;63(1):9–12. [DOI] [PubMed] [Google Scholar]
- 98.Bugarski-Kirola D, Wang A, Abi-Saab D, Blättler T. A phase II/III trial of bitopertin monotherapy compared with placebo in patients with an acute exacerbation of schizophrenia–results from the CandleLyte study. European Neuropsychopharmacology. 2014;24(7):1024–36. [DOI] [PubMed] [Google Scholar]
- 99.Lane H-Y, Huang C-L, Wu P-L, Liu Y-C, Chang Y-C, Lin P-Y, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to clozapine for the treatment of schizophrenia. Biological psychiatry. 2006;60(6):645–9. [DOI] [PubMed] [Google Scholar]
- 100.Lane H-Y, Lin C-H, Green MF, Hellemann G, Huang C-C, Chen P-W, et al. Add-on Treatment of Benzoate for Schizophrenia: A Randomized, Double-blind, Placebo-Controlled Trial of d-Amino Acid Oxidase Inhibitor. JAMA Psychiatry. 2013;70(12):1267–75. [DOI] [PubMed] [Google Scholar]
- 101.Lin C-H, Lin C-H, Chang Y-C, Huang Y-J, Chen P-W, Yang H-T, et al. Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Biological psychiatry. 2018;84(6):422–32. [DOI] [PubMed] [Google Scholar]
- 102.Lin C-Y, Liang S-Y, Chang Y-C, Ting S-Y, Kao C-L, Wu Y-H, et al. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: A randomised, double-blind, placebo-controlled trial. The World Journal of Biological Psychiatry. 2017;18(5):357–68. [DOI] [PubMed] [Google Scholar]
- 103.Nicoletti F, Bockaert J, Collingridge G, Conn P, Ferraguti F, Schoepp D, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60(7–8):1017–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I, et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H] JNJ-40068782. Journal of Pharmacology and Experimental Therapeutics. 2013;346(3):514–27. [DOI] [PubMed] [Google Scholar]
- 105.Rorick-Kehn LM, Johnson BG, Knitowski KM, Salhoff CR, Witkin JM, Perry KW, et al. In vivo pharmacological characterization of the structurally novel, potent, selective mGlu2/3 receptor agonist LY404039 in animal models of psychiatric disorders. Psychopharmacology. 2007;193(1):121–36. [DOI] [PubMed] [Google Scholar]
- 106.Mezler M, Geneste H, Gault L, Marek GJ. LY-2140023, a prodrug of the group II metabotropic glutamate receptor agonist LY-404039 for the potential treatment of schizophrenia. Curr Opin Investig Drugs. 2010;11(7):833–45. [PubMed] [Google Scholar]
- 107.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature reviews Drug discovery. 2009;8(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pehrson AL, Moghaddam B. Impact of metabotropic glutamate 2/3 receptor stimulation on activated dopamine release and locomotion. Psychopharmacology. 2010;211(4):443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349–52. [DOI] [PubMed] [Google Scholar]
- 110.Sonnenschein SF, Grace AA. The mGluR2/3 agonist pomaglumetad methionil normalizes aberrant dopamine neuron activity via action in the ventral hippocampus. Neuropsychopharmacology. 2020:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nature medicine. 2007;13(9):1102–7. [DOI] [PubMed] [Google Scholar]
- 112.Adams DH, Kinon BJ, Baygani S, Millen BA, Velona I, Kollack-Walker S, et al. A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC psychiatry. 2013;13(1):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stauffer VL, Millen BA, Andersen S, Kinon BJ, LaGrandeur L, Lindenmayer J, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophrenia research. 2013;150(2–3):434–41. [DOI] [PubMed] [Google Scholar]
- 114.Downing AM, Kinon BJ, Millen BA, Zhang L, Liu L, Morozova MA, et al. A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC psychiatry. 2014;14(1):351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adams DH, Zhang L, Millen BA, Kinon BJ, Gomez J-C. Pomaglumetad methionil (LY2140023 monohydrate) and aripiprazole in patients with schizophrenia: a phase 3, multicenter, double-blind comparison. Schizophrenia research and treatment. 2014;2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78(11):754–62. [DOI] [PubMed] [Google Scholar]
- 117.Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS, et al. Proof of mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: RCTs of pomaglumetad and TS-134 on ketamine-induced psychotic symptoms and pharmacoBOLD in healthy volunteers. Neuropsychopharmacology. 2020:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Howes O, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Translational psychiatry. 2017;7(2):e1024–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nature reviews Neuroscience. 2016;17(8):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Do KQ, Cuenod M, Hensch TK. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophrenia bulletin. 2015;41(4):835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cai Z, Pan Z-L, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatric research. 2000;47(1):64-. [DOI] [PubMed] [Google Scholar]
- 122.Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophrenia research. 2001;47(1):27–36. [DOI] [PubMed] [Google Scholar]
- 123.Gilmore JH, Jarskog LF, Vadlamudi S. Maternal poly I: C exposure during pregnancy regulates TNFα, BDNF, and NGF expression in neonatal brain and the maternal–fetal unit of the rat. Journal of neuroimmunology. 2005;159(1–2):106–12. [DOI] [PubMed] [Google Scholar]
- 124.Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi G. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Molecular psychiatry. 2006;11(1):47–55. [DOI] [PubMed] [Google Scholar]
- 125.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. Journal of Neuroscience. 2007;27(40):10695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339(6123):1095–9. [DOI] [PubMed] [Google Scholar]
- 127.Müller N, Myint A-M, Krause D, Weidinger E, Schwarz MJ. Anti-inflammatory treatment in schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2013;42:146–53. [DOI] [PubMed] [Google Scholar]
- 128.Tomasik J, Rahmoune H, Guest PC, Bahn S. Neuroimmune biomarkers in schizophrenia. Schizophrenia Research. 2016;176(1):3–13. [DOI] [PubMed] [Google Scholar]
- 129.Chan MK, Krebs M, Cox D, Guest P, Yolken RH, Rahmoune H, et al. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Translational psychiatry. 2015;5(7):e601–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Perkins DO, Jeffries CD, Addington J, Bearden CE, Cadenhead KS, Cannon TD, et al. Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: preliminary results from the NAPLS project. Schizophrenia bulletin. 2015;41(2):419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lanté F, Meunier J, Guiramand J, De Jesus Ferreira MC, Cambonie G, Aimar R, et al. Late N‐acetylcysteine treatment prevents the deficits induced in the offspring of dams exposed to an immune stress during gestation. Hippocampus. 2008;18(6):602–9. [DOI] [PubMed] [Google Scholar]
- 132.Matsuura A, Ishima T, Fujita Y, Iwayama Y, Hasegawa S, Kawahara-Miki R, et al. Dietary glucoraphanin prevents the onset of psychosis in the adult offspring after maternal immune activation. Scientific reports. 2018;8(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Amminger GP, McGorry PD. Update on omega-3 polyunsaturated fatty acids in early-stage psychotic disorders. Neuropsychopharmacology. 2012;37(1):309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McGorry PD, Killackey E, Yung A. Early intervention in psychosis: concepts, evidence and future directions. World psychiatry. 2008;7(3):148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sommer IE, van Westrhenen R, Begemann MJ, de Witte LD, Leucht S, Kahn RS. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophrenia bulletin. 2014;40(1):181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dempster K, Jeon P, MacKinley M, Williamson P, Théberge J, Palaniyappan L. Early treatment response in first episode psychosis: a 7-T magnetic resonance spectroscopic study of glutathione and glutamate. Molecular Psychiatry. 2020:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tsugawa S, Noda Y, Tarumi R, Mimura Y, Yoshida K, Iwata Y, et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. Journal of Psychopharmacology. 2019;33(10):1199–214. [DOI] [PubMed] [Google Scholar]
- 138.Corcoran C, Gallitano A, Leitman D, Malaspina D. The neurobiology of the stress cascade and its potential relevance for schizophrenia. Journal of Psychiatric Practice®. 2001;7(1):3–14. [DOI] [PubMed] [Google Scholar]
- 139.Thompson JL, Pogue-Geile MF, Grace AA. Developmental pathology, dopamine, and stress: a model for the age of onset of schizophrenia symptoms. Schizophrenia bulletin. 2004;30(4):875–900. [DOI] [PubMed] [Google Scholar]
- 140.Nuechterlein KH, Dawson ME. A heuristic vulnerability/stress model of schizophrenic episodes. Schizophrenia bulletin. 1984;10(2):300. [DOI] [PubMed] [Google Scholar]
- 141.van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature. 2010;468(7321):203. [DOI] [PubMed] [Google Scholar]
- 142.Holtzman C, Trotman H, Goulding S, Ryan A, Macdonald A, Shapiro D, et al. Stress and neurodevelopmental processes in the emergence of psychosis. Neuroscience. 2013;249:172–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Corcoran C, Smith C, McLaughlin D, Auther A, Malaspina D, Cornblatt B. HPA axis function and symptoms in adolescents at clinical high risk for schizophrenia. Schizophrenia research. 2012;135(1–3):170–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Walker EF, Trotman HD, Pearce BD, Addington J, Cadenhead KS, Cornblatt BA, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biological psychiatry. 2013;74(6):410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kline E, Thompson E, Bussell K, Pitts SC, Reeves G, Schiffman J. Psychosis-like experiences and distress among adolescents using mental health services. Schizophrenia research. 2014;152(2–3):498–502. [DOI] [PubMed] [Google Scholar]
- 146.Mizrahi R, Addington J, Rusjan PM, Suridjan I, Ng A, Boileau I, et al. Increased stress-induced dopamine release in psychosis. Biological psychiatry. 2012;71(6):561–7. [DOI] [PubMed] [Google Scholar]
- 147.Sapolsky RM, Uno H, Rebert CS, Finch CE. Hippocampal damage associated with prolonged glucocorticoid exposure in primates. Journal of Neuroscience. 1990;10(9):2897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sapolsky RM. A mechanism for glucocorticoid toxicity in the hippocampus: increased neuronal vulnerability to metabolic insults. Journal of Neuroscience. 1985;5(5):1228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Walker EF, Diforio D. Schizophrenia: a neural diathesis-stress model. Psychological review. 1997;104(4):667. [DOI] [PubMed] [Google Scholar]
- 150.Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. Journal of Neuroscience. 2002;22(1):324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rosenkranz JA, Grace AA. Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. Journal of Neuroscience. 2001;21(11):4090–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Belujon P, Grace AA. Regulation of dopamine system responsivity and its adaptive and pathological response to stress. Proceedings of the Royal Society B: Biological Sciences. 2015;282(1805):20142516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gomes FV, Grace AA. Prefrontal cortex dysfunction increases susceptibility to schizophrenia-like changes induced by adolescent stress exposure. Schizophrenia bulletin. 2017;43(3):592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Du Y, Grace AA. Peripubertal diazepam administration prevents the emergence of dopamine system hyperresponsivity in the MAM developmental disruption model of schizophrenia. Neuropsychopharmacology. 2013;38(10):1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Du Y, Grace AA. Amygdala hyperactivity in MAM model of schizophrenia is normalized by peripubertal diazepam administration. Neuropsychopharmacology. 2016;41(10):2455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhu X, Grace AA. Prepubertal environmental enrichment prevents dopamine dysregulation and hippocampal hyperactivity in MAM schizophrenia model rats. Biological Psychiatry. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gomes FV, Zhu X, Grace AA. Stress during critical periods of development and risk for schizophrenia. Schizophrenia research. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gomes FV, Rincón-Cortés M, Grace AA. Adolescence as a period of vulnerability and intervention in schizophrenia: insights from the MAM model. Neuroscience & Biobehavioral Reviews. 2016;70:260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Caballero A, Diah KC, Tseng KY. Region‐specific upregulation of parvalbumin‐, but not calretinin‐positive cells in the ventral hippocampus during adolescence. Hippocampus. 2013;23(12):1331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rector NA, Beck AT. Cognitive behavioral therapy for schizophrenia: an empirical review. The Journal of nervous and mental disease. 2001;189(5):278–87. [DOI] [PubMed] [Google Scholar]
- 161.Patterson TL, Leeuwenkamp OR. Adjunctive psychosocial therapies for the treatment of schizophrenia. Schizophrenia research. 2008;100(1–3):108–19. [DOI] [PubMed] [Google Scholar]
- 162.Howes OH, Kaar SJ. Antipsychotic drugs: challenges and future directions. World Psychiatry. 2018;17(2):170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, et al. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Archives of general psychiatry. 2012;69(3):220–9. [DOI] [PubMed] [Google Scholar]
- 164.Woods BT. Is schizophrenia a progressive neurodevelopmental disorder? Toward a unitary pathogenetic mechanism. American Journal of Psychiatry. 1998;155(12):1661–70. [DOI] [PubMed] [Google Scholar]
- 165.Zipursky RB, Reilly TJ, Murray RM. The myth of schizophrenia as a progressive brain disease. Schizophrenia bulletin. 2013;39(6):1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ho B-C, Andreasen NC, Ziebell S, Pierson R, Magnotta V. Long-term antipsychotic treatment and brain volumes: a longitudinal study of first-episode schizophrenia. Archives of general psychiatry. 2011;68(2):128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Konopaske GT, Dorph-Petersen K-A, Sweet RA, Pierri JN, Zhang W, Sampson AR, et al. Effect of chronic antipsychotic exposure on astrocyte and oligodendrocyte numbers in macaque monkeys. Biological psychiatry. 2008;63(8):759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Konopaske GT, Dorph-Petersen K-A, Pierri JN, Wu Q, Sampson AR, Lewis DA. Effect of chronic exposure to antipsychotic medication on cell numbers in the parietal cortex of macaque monkeys. Neuropsychopharmacology. 2007;32(6):1216. [DOI] [PubMed] [Google Scholar]
- 169.Dorph-Petersen K-A, Pierri JN, Perel JM, Sun Z, Sampson AR, Lewis DA. The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: a comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology. 2005;30(9):1649. [DOI] [PubMed] [Google Scholar]
- 170.Lieberman JA, Tollefson GD, Charles C, Zipursky R, Sharma T, Kahn RS, et al. Antipsychotic drug effects on brain morphology in first-episode psychosis. Archives of general psychiatry. 2005;62(4):361–70. [DOI] [PubMed] [Google Scholar]
- 171.McGorry PD, Nelson B, Amminger GP, Bechdolf A, Francey SM, Berger G, et al. Intervention in individuals at ultra-high risk for psychosis: a review and future directions. The Journal of clinical psychiatry. 2009. [DOI] [PubMed] [Google Scholar]
- 172.Burke AR, McCormick CM, Pellis SM, Lukkes JL. Impact of adolescent social experiences on behavior and neural circuits implicated in mental illnesses. Neuroscience & Biobehavioral Reviews. 2017;76:280–300. [DOI] [PubMed] [Google Scholar]
- 173.Fawcett JW, Oohashi T, Pizzorusso T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature reviews Neuroscience. 2019;20(8):451–65. [DOI] [PubMed] [Google Scholar]
- 174.Murthy S, Kane GA, Katchur NJ, Mejia PSL, Obiofuma G, Buschman TJ, et al. Perineuronal nets, inhibitory interneurons, and anxiety-related ventral hippocampal neuronal oscillations are altered by early life adversity. Biological psychiatry. 2019;85(12):1011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cabungcal J-H, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proceedings of the National Academy of Sciences. 2013;110(22):9130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Heckers S, Barch DM, Bustillo J, Gaebel W, Gur R, Malaspina D, et al. Structure of the psychotic disorders classification in DSM‐5. Schizophrenia research. 2013;150(1):11–4. [DOI] [PubMed] [Google Scholar]
- 177.Gillespie AL, Samanaite R, Mill J, Egerton A, MacCabe JH. Is treatment-resistant schizophrenia categorically distinct from treatment-responsive schizophrenia? A systematic review. BMC psychiatry. 2017;17(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Demjaha A, Murray RM, McGuire PK, Kapur S, Howes OD. Dopamine synthesis capacity in patients with treatment-resistant schizophrenia. American Journal of Psychiatry. 2012;169(11):1203–10. [DOI] [PubMed] [Google Scholar]
- 179.Mouchlianitis E, Bloomfield MA, Law V, Beck K, Selvaraj S, Rasquinha N, et al. Treatment-resistant schizophrenia patients show elevated anterior cingulate cortex glutamate compared to treatment-responsive. Schizophrenia bulletin. 2015;42(3):744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology. 2012;37(11):2515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic: a double-blind comparison with chlorpromazine. Archives of general psychiatry. 1988;45(9):789–96. [DOI] [PubMed] [Google Scholar]
- 182.Goldstein ME, Anderson VM, Pillai A, Kydd RR, Russell BR. Glutamatergic Neurometabolites in Clozapine-Responsive and -Resistant Schizophrenia. International Journal of Neuropsychopharmacology. 2015;18(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Grace AA, Gomes FV. The circuitry of dopamine system regulation and its disruption in schizophrenia: insights into treatment and prevention. Schizophrenia bulletin. 2019;45(1):148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends in neurosciences. 2012;35(1):57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sarpal DK, Argyelan M, Robinson DG, Szeszko PR, Karlsgodt KH, John M, et al. Baseline striatal functional connectivity as a predictor of response to antipsychotic drug treatment. American Journal of Psychiatry. 2016;173(1):69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sarpal DK, Lencz T, Malhotra AK. In support of neuroimaging biomarkers of treatment response in first-episode schizophrenia. American Journal of Psychiatry. 2016;173(7):732–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dazzan P, Arango C, Fleischacker W, Galderisi S, Glenthøj B, Leucht S, et al. Magnetic resonance imaging and the prediction of outcome in first-episode schizophrenia: a review of current evidence and directions for future research. Schizophrenia Bulletin. 2015;41(3):574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mondelli V, Ciufolini S, Belvederi Murri M, Bonaccorso S, Di Forti M, Giordano A, et al. Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophrenia bulletin. 2015;41(5):1162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Millan MJ, Andrieux A, Bartzokis G, Cadenhead K, Dazzan P, Fusar-Poli P, et al. Altering the course of schizophrenia: progress and perspectives. Nature Reviews Drug Discovery. 2016;15(7):485. [DOI] [PubMed] [Google Scholar]
- 190.Haroun N, Dunn L, Haroun A, Cadenhead KS. Risk and protection in prodromal schizophrenia: ethical implications for clinical practice and future research. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lieberman JA, Small SA, Girgis RR. Early detection and preventive intervention in schizophrenia: from fantasy to reality. American Journal of Psychiatry. 2019;176(10):794–810. [DOI] [PubMed] [Google Scholar]