

Abstract
The escalating burden of type 2 diabetes (T2D) and its related complications, has become a major public health challenge worldwide. Substantial evidence indicates that T2D is one of the culprits for the high prevalence of Alzheimer’s disease (AD) in diabetic subjects. This study aimed to investigate the possible mitochondrial alterations in the pancreas induced by hyperglycemia in diabetes. We used a diabetic TallyHO/JngJ (TH), and non-diabetic, SWR/J mice strains. The diabetic and non-diabetic status in animals was assessed by performing intraperitoneal glucose tolerance test at four-time points i.e., 4-, 8-, 16- and 24-weeks of age. We divided 24 weeks old TH and SWR/J mice into 3 groups: Controls, diabetic TH mice, and diabetic TH mice treated with SS31 peptide. After the treatment of male TH mice with SS31, intraperitoneally, for 4 weeks, we studied mitochondrial dynamics, biogenesis, and function. The mRNA and protein expression levels of mitochondrial proteins were evaluated using qPCR and immunoblot analysis. The diabetic mice after 24 weeks of age, showed overt pancreatic injury as demonstrated by disintegration, and atrophy of β-cells with vacuolization and reduced islet size. Mitochondrial dysfunction was observed in TH mice, as evidenced by significantly elevated H2O2 production, lipid peroxidation, and reduced ATP production. Furthermore, mRNA expression and immunoblot analysis of mitochondrial dynamics genes were significantly affected in diabetic mice, compared to controls. However, treatment of animals with SS31 reduced mitochondrial dysfunction and restored most of the mitochondrial functions, and mitochondrial dynamics processes to near normal in TH mice. In conclusion, mitochondrial dysfunction is established as one of the molecular events that occur in the pathophysiology of T2D. Further, SS31 treatment may confer protection against the mitochondrial alterations induced by hyperglycemia in diabetic TallyHO/JngJ mice.
Keywords: Type 2 diabetes, TallyHO/JngJ mice, Alzheimer’s disease, Mitochondrial dysfunction, Oxidative stress
Introduction
Type 2 diabetes (T2D) is a condition in which a high level of blood glucose results in increased hepatic glucose production, impaired insulin production by pancreatic β-cells, and insulin resistance [1]. T2D is associated with a variety of genetic and environmental risk factors, including age, family history of diabetes, poor diet, obesity, and physical inactivity. Current global estimates by International Diabetes Federation (IDF) demonstrated that 463 million people were having T2D in 2019, worldwide, and this number is expected to rise to 700 million by 2045, with a 51% increase [2] Although diabetes affects the people of all ages. However, a higher prevalence of diabetes is reported in older adults aged 60–69 years. The recent global facts demonstrated diabetes prevalence of 9.3% in 2019, which is expected to rise to 10.2% (578 million) by 2030 and 10.9% (700 million) by 2045 [2]. Over the past 3 decades, a significant number of epidemiological studies demonstrated a strong link between T2D and a higher risk of developing Alzheimer’s Disease (AD) [3–7]. Besides, T2D is known to increase the risk of dementia by 60% compared with individuals without diabetes [8]. Further, T2D-related conditions, including obesity, hyperinsulinemia, insulin resistance, and metabolic syndrome, also increase the risk of cognitive impairment and AD [5,9,10]. Despite substantial epidemiological evidence linking AD and diabetes, the underlying molecular mechanisms remain to be elucidated.
The male TallyHo/Jng (TH) mouse is a polygenic inbred model of T2D, which shows similar characteristics as humans [11] in which both genetic and environmental factors are known to interplay in T2D pathology [12]. Animal genetic models have been served as a valuable resource for the study of disease pathophysiology. The TallyHo/Jng (TH) mouse is a polygenic inbred model for human type 2 diabetes and obesity [12]. This mice model may be most suitable for the study of molecular events that happen in the progression of type 2 diabetes to Alzheimer’s disease [13]. This albino strain of mice was originated from the progeny of diabetic mice discovered in an outbred colony of Theiler’s original mice in the United Kingdom, which were then imported to The Jackson Laboratory for further research in 1992 [14]. The TH mice model shows several phenotypic characteristics demonstrated in the pathophysiology of type 2 diabetes in humans. The most common parameters are obesity, hyperleptinemia, hyperinsulinemia, insulin resistance, glucose intolerance, hyperlipidemia, and hyperglycemia [14]. Kim et al. studied the genetic basis of obesity and type 2 diabetes in TH mice using several outcross experiments with normal strains, which further led to the identification of multiple quantitative trait loci linked to adiposity and hyperglycemia [14]. The TH mice have become available only quite recently, so this diabesity model is not that well known to the diabetes research community. Recently, several studies have been reported in studying this mouse model for the identification and development of therapeutic targets in obesity and T2D [15–20]. Clearly, much more research on this model is warranted.
Recently, several mitochondria-targeted molecules have been developed and are currently being tested in several laboratories [21–23]. SS31 is one of the tetra-peptide molecules developed with other SS peptides that are shown to play a protective role against mitochondrial insult by inhibiting mitochondrial swelling, oxidative damage, and reperfusion injury in several pathological conditions [24], including islet cell transplantation [25], myocardial infarction [26] and ALS [27].
Based on the neuroprotection capacity of SS31, the Reddy group [28–32] evaluated the therapeutic potential of SS31 in various cell models of neurodegenerative diseases including AD and Huntington’s disease (HD). In Amyloid beta (Aβ)-treated mouse neuroblastoma cells and primary neurons from AβPP mice [29], we found that SS31 treatment protects the cells and primary neurons against Aβ toxicity by modulating the mitochondrial fission and fusion processes, mitochondrial functions, and synaptic activities [28]. In another study, our research findings suggest that mitochondria-targeted molecules SS31 are protective against mutant Htt-induced mitochondrial and synaptic damage in HD neurons [32].
In a recent study, TH mice reported exhibiting mitochondrial dysfunction [33]. Therefore, in the current study, we treat TH mice with SS31 and study the beneficial effects against mitochondrial dysfunction in TH mice.
The purpose of the present study was 1) to characterize the TH mouse model; 2) To study the mitochondrial alteration in diabetes; and 3) to investigate the ameliorative role of a small peptide, SS31, on the molecular alterations induced by hyperglycemia in the mitochondria of diabetic mice, compared with a non-diabetic SWR/J mouse strain.
Material and Methods
Animals
Diabetic inbred TallyHo/JngJ (TH) and non-diabetic SWR/J mouse strains used in the present study were procured from The Jackson Laboratory. Both TH and SWR/J mice strains shared 86.8% of their genetic makeup [34]. That’s why SWR/J may be one of the most frequently used non-diabetic and non-obese strains in research as it does not develop diabetes when fed a standard laboratory chow diet. All the animals were housed in clean polypropylene cages and fed a standard chow diet ad libitum with free access to water under controlled temperature and humidity with a 12-h light/dark cycle. All the experiments were performed according to the guidelines for use and care of laboratory animals and were approved by the institutional animal care & use committee (IACUC) of Texas Tech University Health Sciences Center, Lubbock, TX, United States.
Characterization of animals
To establish the obesity in TH and SWR/J mice (both the sexes), body weight (bw) of all the mice groups was continuously measured at 4, 8, 12, 16, 20 and 24 weeks of their age. Similarly, to ascertain the diabetic status/insulin resistance in TH and SWR/J mice (both the sexes), an intraperitoneal glucose tolerance test (IPGTT) was performed in all the animals at different time points i.e., 4, 8, 16, and 24 weeks of age. Briefly, mice were fasted overnight, and the baseline blood glucose level was measured in blood using a portable glucometer (AlphaTRAK 2 blood glucose monitoring system kit) via tail nick. Animals were injected intraperitoneally with glucose (1 mg/g bw) in normal saline. Blood glucose levels were then measured at 0, 15, 30, 60, and 120 min after injection. For the insulin tolerance test, all the animals were fasted for 4 hours and then injected insulin intraperitoneally. Glucose was then measured in blood obtained via tail nick at 0, 15, 30, 60, and 120 min.
SS31
The SS31 peptide, a mitochondria-targeted antioxidant, was purchased from Biogenix, Inc., CA. The SS31 tetra-peptide was originally synthesized by Dr. Hazel H. Szeto in collaboration with Dr. Peter W. Schiller. Drs. Szeto and Schiller designed and synthesized four different peptides (SS31, SS02, SS20, and SS19) with the amino acids Dmt, D-Arg, Phe, and Lys, and with the Dmt residue [23].
Treatment of animals
After confirming diabetic status at 24 weeks of age, male TH diabetic mice and age and sex matched non-diabetic SWR/J mice were then divided into three experimental groups:
Control group: Non-diabetic, SWR/J mice were administered normal saline (i.p.) 4 times per week for 4 weeks.
TH group: Diabetic TH mice were administered normal saline (i.p.) 4 times per week for 4 weeks.
TH+SS31 group: Diabetic TH mice were administered with SS31 (5mg/kg body weight) (i.p.) 4 times per week for 4 weeks.
At the end of 4 weeks of treatment, animals were sacrificed by cervical dislocation following an overnight fast. Blood and other tissues (pancreas, liver, skeletal muscles, and brain) were collected and stored in the deep freezer for further assays.
Mitochondrial Functional Assays
Hydrogen Peroxide
Hydrogen peroxide (H2O2) is one of the key reactive oxygen species generated via respiratory chain cascade as well as byproducts of cellular metabolism, including protein folding. The H2O2 generation was measured in the pancreas tissue of all the three animal groups using an Amplex® Red H2O2 Assay Kit (Molecular Probes, Eugene, OR, USA). In brief, the reaction mixture consists of mitochondrial proteins (μg/μl), Amplex Red reagents (50 μM), horseradish peroxidase (0.1 U/ml), and a reaction buffer (1X). The mixture was incubated at room temperature for 30 minutes, followed by spectrophotometer readings of fluorescence at 570 nm. Finally, H2O2 production was estimated using a standard curve equation, expressed in nmol/μg mitochondrial protein. The protein concentration was measured in all the animal groups by a BCA Protein Assay Kit (Pierce Biotechnology). All the measurements were done according to the instructions provided by the manufacturers of Assay kits.
Lipid Peroxidation Assay
Lipid peroxidation is an indicator of oxidative stress. The final product of lipid peroxidation is 4-hydroxy-2-nonenol (HNE), which was measured in the pancreas tissue of TH and control mice groups using HNE-His ELISA Kit (Cell BioLabs, Inc., San Diego, CA, USA). Briefly, the freshly prepared protein was added to a 96-well protein binding plate and incubated overnight at 4°C. The plate was then washed 3 times with a buffer. After the last wash, the anti-HNE-His antibody was added to the protein in the wells, which was then incubated for 2 hours at room temperature and then washed again 3 times. Further, the samples were incubated with a secondary antibody conjugated with peroxidase for 2 hours at room temperature, followed by incubation with an enzyme-substrate. Optical density was then measured at 450nm to quantify the level of HNE. Lipid peroxidation levels were compared among control, untreated TH, and SS31 treated TH mice groups.
Determination of ATP Levels
ATP levels were measured in mitochondria isolated from tissues of TH and control mice using an ATP determination kit (Molecular Probes, USA). The bioluminescence assay is based on the reaction of ATP with recombinant firefly luciferase and its substrate luciferin. Luciferase catalyzes the formation of light from ATP and luciferin. It is the emitted light that is linearly related to the ATP concentration, which is measured with a luminometer. ATP levels were measured from mitochondrial pellets using a standard curve method.
Hematoxylin and Eosin (H&E) Staining
Mice of all the three experimental groups were perfused through the heart with 10 ml saline, using a pump at 5 ml/min, to clear the required tissues of blood, then followed with cold 4% paraformaldehyde in sodium phosphate buffer, pH 7.4 for approximately 15 minutes. Providing the perfusion was successful, and tissues were cleared of blood. The tissues were removed, cut into 2–3 mm blocks, and placed in 30% sucrose for cryoprotection. Blocks of the pancreas tissue were snap-frozen in liquid nitrogen and stored in −80° C. Frozen sections were cut on a Reichert-Jung 1800 cryostat at 10 μm, mounted directly on super frost slides (Fisher Scientific, Pittsburgh PA), and air-dried for 10–30 min before processing for staining. H&E staining was used to study the histopathological damage in tissues. For this purpose, the slide-mounted 8–12 um cryostat sections were dehydrated through absolute alcohol and rehydrated to water. Slides of all the groups were placed in hematoxylin stain for 4 minutes, rinsed in water, differentiated in 70% alcohol, and stained in 0.01% eosin Y for 2 seconds, rinsed with 95% ethanol, dehydrated with absolute ethanol, and cleared in xylenes for 15 minutes before coverslipping.
Quantitative Real-Time PCR (qPCR)
For the gene expression studies, total RNA was extracted from the tissues of control, TH mice, and SS31 treated TH mice using TriZol reagent (Invitrogen, ST, USA). Total RNA (5μg) was reverse transcribed to cDNA using the Superscript III First-strand synthesis kit (Invitrogen, Carlsbad, USA) in a final volume of 20μl. The sample was incubated at 65°C for 5mins and then chilled on ice for 1 minute. 10μl of cDNA mix containing 50μM oligo 10X RT-buffer, 25mM Mgcl2, 0.1M DTT, RNase out, 20U Superscript III reverse transcriptase was added. The sample was incubated at 50°C for 50 minutes, and then the reaction was terminated at 85°C for 5 minutes. Using primer express software (Applied Biosystems, Carlsbad, CA, USA), we designed the oligonucleotide primers for the housekeeping genes β-actin, GAPDH, mitochondrial structural genes, fission genes (Drp1, Fis1), fusion genes (MFN1, MFN2, Opa1) as described in our previous study [35]. The primer sequence for the above-mentioned genes are listed in Table 1. mRNA expression levels of the above-mentioned genes were quantified using SYBR Green based real-time RT-PCR (Applied bioscience, 7900HT r\fast Real-time PCR system). All reactions were carried out as follows: 50°C for 2 mins, followed by 40 cycles of 95°C for 10mins, 95°C for 15 s, and 60°C for 1 min. The mRNA transcript was normalized against b-actin. The standard curve was then normalized mRNA transcript level plotted against log cDNA input (ng). Briefly, the CT method involved averaging duplicates samples, which were taken as the CT values for β-actin, and mitochondrial genes. β-Actin normalization was used for determining mitochondrial dynamics, biogenesis genes because the β-actin CT values were similar for the SS31-treated mice and the untreated Tally-HO mice. The ΔCT-value was obtained by subtracting the average β-actin CT value from the average CT value of the mitochondrial genes. The ΔCT of the untreated mice was used as the calibrator. Fold change was calculated according to the formula 2−nΔΔCT, where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value. To determine the statistical significance of mRNA expression, the CT value differences between the untreated TH mice and other lines of mice were used in relation to β-actin normalization.
Table 1:
Summary of quantitative real-time RT-PCR oligonucleotide primers used in measuring mRNA expression in mitochondrial dynamics, mitochondrial biogenesis, and housekeeping genes in Control SWR/J, diabetic TH and SS31 treated TH mice.
| Gene | DNA sequence (5′−3′) | Product size |
|---|---|---|
| Mitochondrial dynamics genes | ||
| Drp1 | Forward primer ATGCCAGCAAGTCCACAGAA | 86 |
| Reverse primer TGTTCTCGGGCAGACAGTTT | ||
| Fis1 | Forward primer CAAAGAGGAACAGCGGGACT | 95 |
| Reverse primer ACAGCCCTCGCACATACTTT | ||
| MFN1 | Forward primer GCAGACAGCACATGGAGAGA | 83 |
| Reverse primer GATCCGATTCCGAGCTTCCG | ||
| MFN2 | Forward primer TGCACCGCCATATAGAGGAAG | 78 |
| Reverse primer TCTGCAGTGAACTGGCAATG | ||
| Opa1 | Forward primer ACCTTGCCAGTTTAGCTCCC | 82 |
| Reverse primer TTGGGACCTGCAGTGAAGAA | ||
| Housekeeping genes | ||
| β-Actin | Forward primer AGAAGCTGTGCTATGTTGCTCTA | 91 |
| Reverse primer TCAGGCAGCTCATAGCTCTTC | ||
| GAPDH | Forward primer TTCCCGTTCAGCTCTGGG | 59 |
| Reverse primer CCCTGCATCCACTGGTGC | ||
Western Blotting
To determine the impact of hyperglycemia and SS31 treatment on the protein levels of mitochondrial dynamics and the biogenesis genes (only those showing altered mRNA expression), we performed immunoblotting analyses of protein lysates from the pancreas of experimental groups as described in our previous study [35]. Protein lysate was prepared using the RIPA buffer (Thermo Scientific MA, USA). Sixty micrograms of the protein lysate were resolved on a 10% SDS PAGE gel. The resolved proteins are transferred on to nylon membrane (Novax Inc., San Diego, CA, USA) and incubated with blocking buffer (5% milk in TBST) for one hour at room temperature. The PVDF membrane was then incubated overnight with the primary antibodies (Table 2). The membranes were washed with a TBST buffer three times at 10-min intervals and were then incubated for 2 h with appropriate secondary antibodies, followed by three additional washes at 10-min intervals. Proteins were detected with chemiluminescence reagents (Thermo Scientific, IL, USA), and the bands from the immunoblots were quantified on a Kodak Scanner (ID Image Analysis Software (Kodak Digital Science, Kennesaw, GA, USA). Briefly, image analysis was used to analyse gel images captured with a Kodak Digital Science CD camera to define the positions and specific regions of the bands.
Table 2:
Summary of antibody dilutions and conditions used in the immunoblotting analysis of mitochondrial dynamics and mitochondrial biogenesis proteins in Control, TH, and TH+SS31 groups.
| Markers | Primary antibody (species and dilution) | Supplier | Secondary antibody (species and dilution) | Purchased from company, city & state |
|---|---|---|---|---|
| Drp1 | Rabbit polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 | Rabbit polyclonal 1:400 | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Fis1 | Rabbit polyclonal 1:500 | Protein Tech Group, Inc., Chicago, IL | Donkey Anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 | Rabbit polyclonal 1:400 | Abcam, Cambridge, MA | Donkey Anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| GAPDH | Mouse monoclonal 1:500 | Sigma-Aldrich, St Luis, MO | Sheep Anti-mouse HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
Statistical analysis
Values were expressed as Mean ± Standard Deviation. Data were analysed using one way ANOVA between the treatment groups. Values with p<0.05 (two-tailed) were considered as statistically significant. Statistical analysis was performed using the Statistical Package of Social Sciences (SPSS) for Windows, version 20 (SPSS, Inc., Chicago, IL).
Results
Characterization of diabetic and non-diabetic animals
Body Weight
Figure 1 shows the gain in body weight of TH and SWR/J mice (both male and female) at 4 different time points i.e., 4, 8, 16, and 24 weeks of age. These results indicate that the bodyweight of TH mice of both sexes at 4 weeks of age was significantly higher than the age- and sex-matched SWR/J control mice. This higher mean body weight in TH mice of both sexes were maintained throughout the study period of 8, 16, and 24 weeks (Fig. 1) and shows moderate obesity at a normal chow diet. However, few mice lost their body weight and became lean after developing diabetes at the age of 16–24 weeks of age. We also observed reduced locomotor activity in both male and female TH mice, compared to age and sex-matched SWR/J mice without any diet differences.
Figure 1.
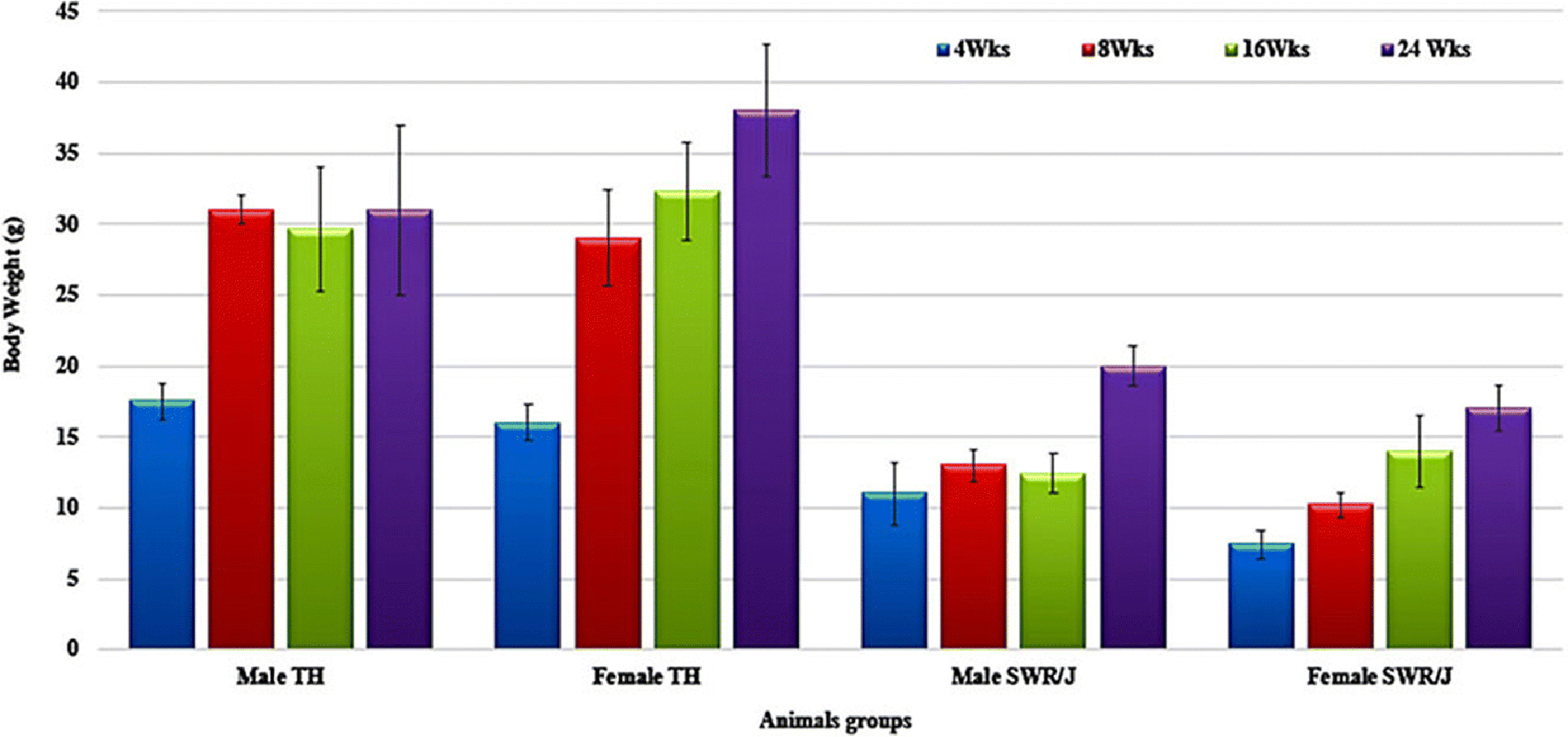
Bodyweight changes in TH and SWR/J mice (Male and female) at 4-, 8-, 16- and 24-weeks of age. Data are Means±SD. (n=6 for each group)
Hyperglycemia
As shown in Fig. 2, both pre-pubertal male and female TH mice were normoglycemic at 4 weeks. The development of diabetes started in post-pubertal male TH mice and exhibited a prominent increase in the blood glucose levels between 8 to 16 weeks of age, reaching full-blown diabetic levels, which further worsen at 24 weeks of age (280–500 mg/dl, fasting). Although TH males are consistently hyperglycemic, the range and age of onset of diabetes vary from litter to litter. On the other hand, female TH mice were normoglycemic throughout this study period. Hyperglycemia was not observed in SWR/J mice of both the sexes at any time point of this study.
Figure 2.
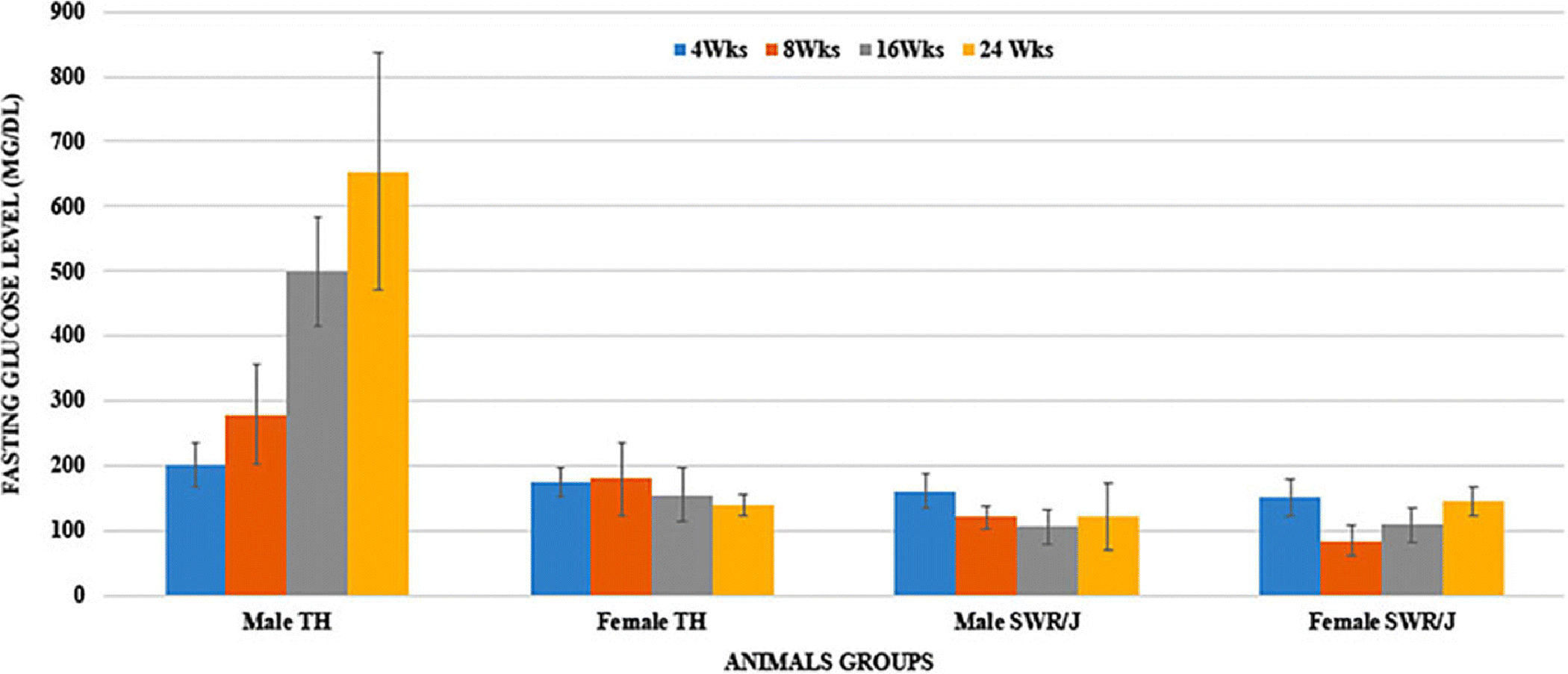
Fasting blood glucose levels in TH and SWR/J mice (Male and female) at 4-, 8-, 16- and 24-weeks of age. Data are Means±SD. (n=6 for each group)
Insulin Resistance
To understand the progression from normoglycemic state to glucose intolerance/ insulin resistance and then the development of diabetes in TH mice, an intraperitoneal glucose tolerance test (IPGTT) was performed at all the study time points, i.e., 8, 16, and 24 weeks of age. Fig. 3 (A–C) shows the results of IPGTT in TH and SWR/J mice of both the sexes at all the time points. It exhibited impaired glucose tolerance in male TH mice at 8 weeks that further worsened at 16 and 24 weeks of age. Our data revealed that male TH mice were obese, hyperinsulinemic, and hyperglycemic by 4 weeks of age and rapidly developed diabetes by 8 weeks of age. Contrary, female TH mice showed normal glucose tolerance at all the time points. On the other hand, as expected, SWR/J mice (both the sexes) did not show glucose intolerance or hyperglycemia at any point of time.
Figure 3.
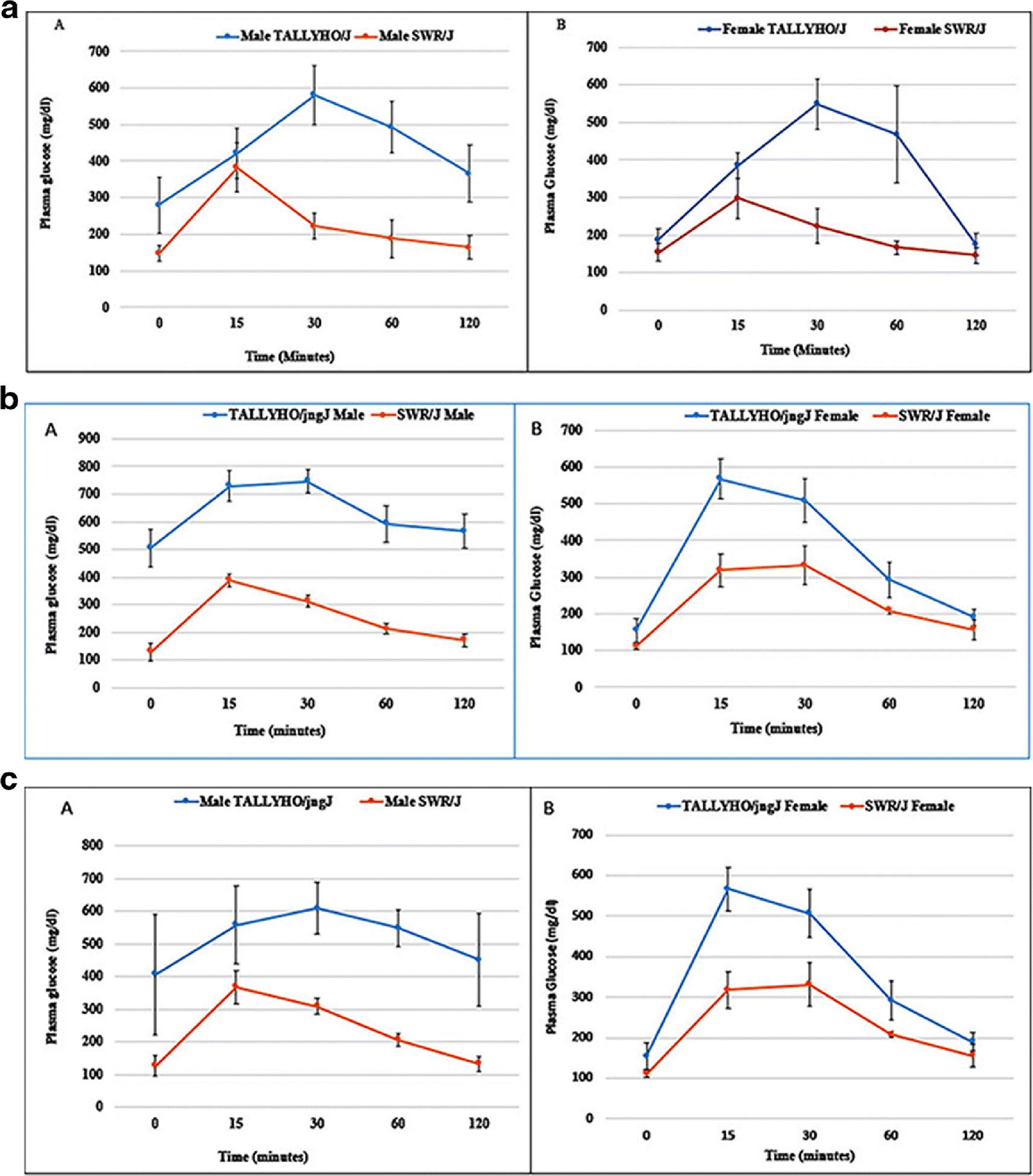
(A): Intraperitoneal glucose tolerance test in TH and SWR/J mice, male (A) and female (B) at 8 weeks of age. Data are Means±SD. (n=6 for each group).
(B): Intraperitoneal glucose tolerance test in TH and SWR/J mice, male (A) and female (B) at 16 weeks of age. Data are Means±SD. (n=6 for each group).
(C): Intraperitoneal glucose tolerance test in TH and SWR/J mice, male (A) and female (B) at 24-Weeks of age. Data are Means±SD. (n=6 for each group).
Impact of Hyperglycemia and SS31on mitochondrial functions
The results of the present study indicate that only male TH mice developed diabetes and moderate obesity at 16–24 weeks of age, whereas female TH mice did not develop diabetes at any time point. So for further studies on the impact of hyperglycemia and SS31 on mitochondria, we included only male diabetic TH mice with age and sex-matched SWR/J controls (24 weeks of age) for further studies. We studied biochemical, microscopic, and molecular changes in the mitochondrial functioning due to persistent hyperglycemia and ameliorative action of SS31 in TH mice in comparison to SWR/J and untreated TH groups as described in the treatment section. Mitochondrial functions were evaluated by assessing the levels of hydrogen peroxide (H2O2), lipid peroxidation, and mitochondrial ATP generation in the pancreas of SWR/J, TH, and TH+SS31 groups at 24 weeks of age.
H2O2 levels
Fig. 4A shows the effect of hyperglycemia and treatment with SS31 on hydrogen peroxide levels in control, TH, and TH+SS31 groups. Significantly higher values of H2O2 were observed in diabetic male TH mice, relative to age and sex-matched control group (p<0.05), indicating that hyperglycemia increases H2O2 levels in type 2 diabetes. However, the treatment of male TH mice with SS31 for one month significantly abolished the impact of hyperglycemia on H2O2 levels.
Figure 4.
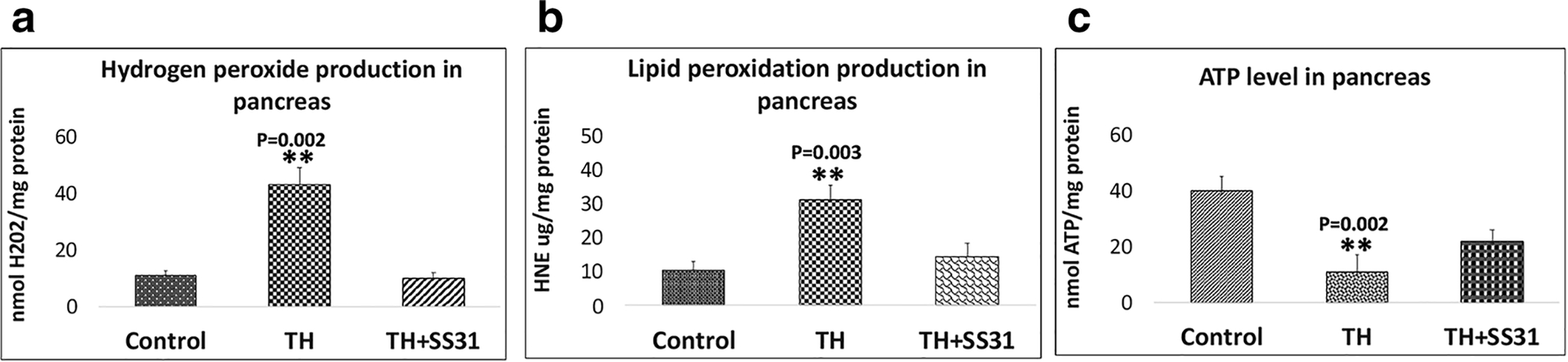
Mitochondrial functional assay in the pancreas of Control, TH mice and SS31 treated TH mice. Data are Means±SD. (n=6 for each group).
Lipid peroxidation
Fig. 4B shows the hyperglycemia-induced levels of HNE, an indicator of lipid peroxidation in the pancreas of TH mice. These results indicate a significant increase in the levels of lipid peroxidation that may be induced by persistent hyperglycemia in TH mice (p<0.05), compared to age and sex-matched control group animals. Though, treatment of male TH mice with SS31 for 4 weeks significantly reduced the lipid peroxidation, as shown in Fig. 5B.
Figure 5. (A-C):
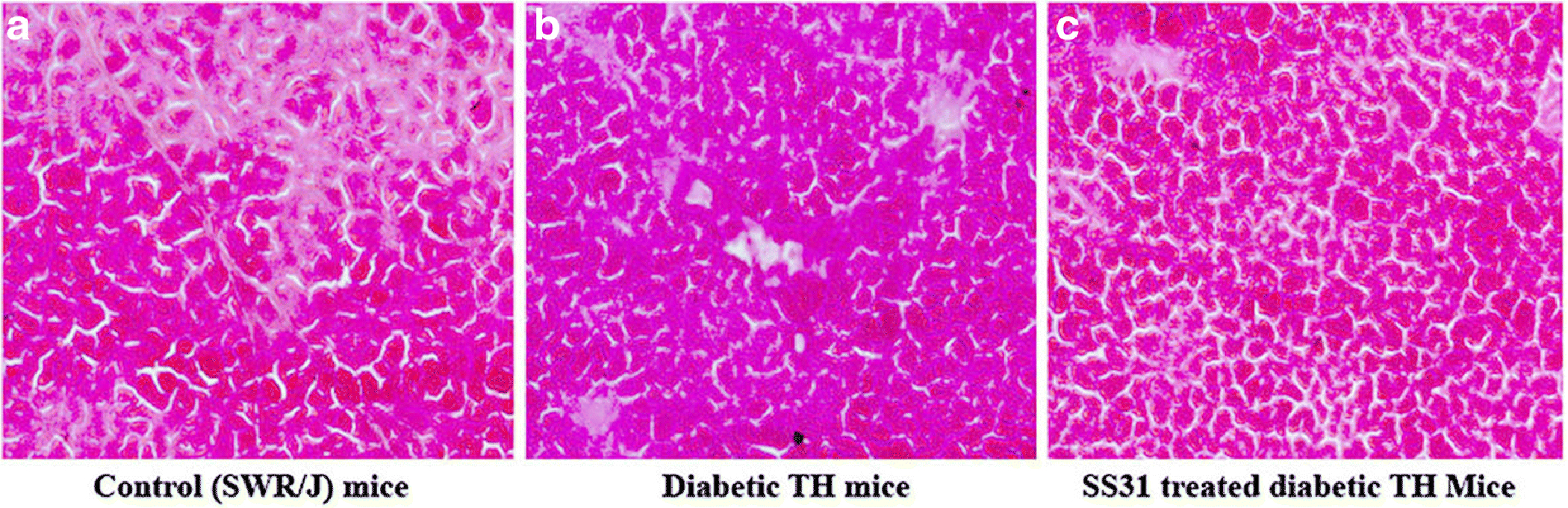
H&E staining of the pancreas tissue of control SWR/j, diabetic TH, and SS31 treated diabetic TH mice.
Determination of adenine triphosphate (ATP) levels
As shown in Fig.4C, significantly decreased levels of mitochondrial ATP were observed in TH mice compared to the control group (p<0.05), indicating that hyperglycemia reduces the mitochondrial ATP generation in male TH mice. However, this decrease in mitochondrial ATP generation was somewhat compensated in SS31 treated diabetic TH mice.
Histological Examinations
Histological examination of pancreatic tissue in control mice showed normal appearance of pancreatic beta cells (Fig. 5A). Sections from diabetic TH pancreatic tissue showed disintegration of beta cells, atrophy of beta cells with vacuolization, and reduced islet size (Fig. 5B). Whereas, the section from SS31 treated pancreatic tissue showed normal architecture of the beta cells similar to the control mice (Fig. 5C).
Impact of SS31 treatment on diabetes-induced alterations in mRNA expression
Mitochondrial dynamic genes
We compared gene expression data of TH mice with age and sex-matched SWR/J controls in order to understand diabetes-induced alterations in mRNA levels of mitochondrial genes. As shown in Table 3, the mitochondrial structural genes were significantly affected by prolonged hyperglycemia in male TH mice, and treatment of SS31 significantly reduced the mRNA expression of the Drp1 gene by 2.7 folds (P=0.001) and Fis1 gene by 2.5 fold (p=0.001) whereas, it significantly enhanced the mRNA level of the fusion genes, Mfn 1 gene by 3 folds (P=0.001); Mfn2 gene by 3.65 folds (P=0.001) and Opa1 gene by 1.8 folds (P=0.04) in the pancreas of SS31 treated TH mice, compared to untreated TH mice (Table 3).
Table 3:
mRNA fold changes of mitochondrial structural genes in the pancreas of SS31 treated Tally-HO mice relative to untreated Tally-HO mice.
| Mitochondrial Genes | mRNA fold change in TH+SS31 | |
|---|---|---|
| Mitochondrial structural genes | Drp1 | −2.7** |
| Fis1 | −2.5** | |
| Mfn1 | 3.0** | |
| Mfn2 | 3.5** | |
| OPA1 | 1.8* |
These observations indicate SS31 reduces the fission activity and enhances the fusion activity in diabetic TallyHO mice.
Immunoblot analysis
As shown in Fig 6, the levels of mitochondrial fission proteins, Drp1 (P=0.041) and Fis1 (P=0.032) were found to be elevated in the pancreas of untreated TH mice relative to the control mice. Whereas, in the case of SS31 treated TH mice, the expression levels of Drp1 (P=0.020) and Fis1 (P=0.014) were lowered relative to the untreated TH mice and were similar to the control mice. Contrary, the mitochondrial fusion proteins, Mfn1 (P=0.044) and Mfn2 were lowered in the pancreas of untreated TH mice relative to control mice, statistical significance was reached only for Mfn1 (P=0.044), and in the case of SS31 treated TH mice, the expression levels of Mfn1 (P=0.011) and Mfn2 were enhanced, compared to the untreated TH mice and visibly similar to control mice. These findings are in agreement with the mRNA expression levels of the mitochondrial fission and fusion genes that were quantified with real-time RT-PCR analysis.
Figure 6 (A-B).
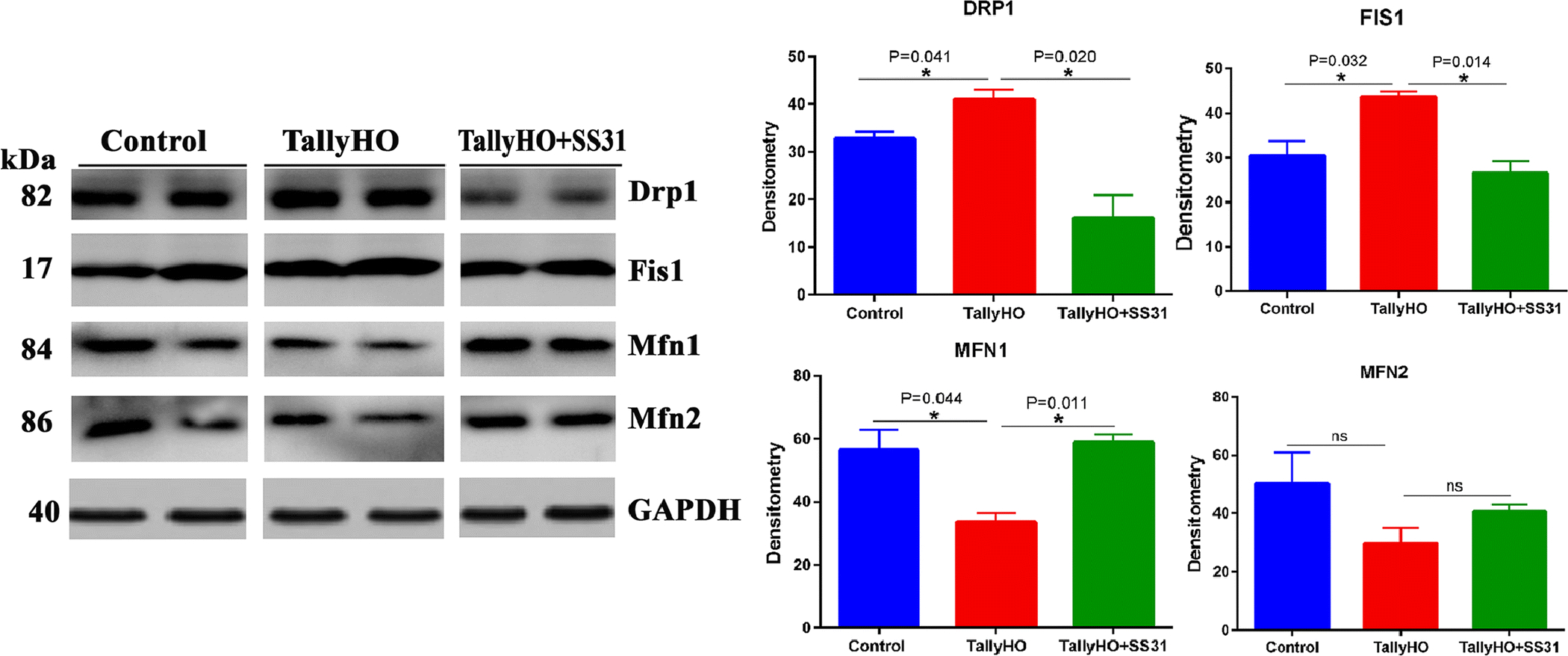
Immunoblot and densitometric analysis of mitochondrial fission in the Pancreas of the wild-type, SS31 treated Tally-HO mice and untreated Tally-HO mice.
These observations suggest that SS31 may modulate the hyperglycemia-induced alterations mitochondrial dynamics in the pancreas of TH mice.
Discussion
Several epidemiological studies have established that T2D is one of the risk factors for the development of AD. However, the underlying mechanisms have not been well elucidated [8,3–7,36]. Impaired glucose tolerance leads to insulin resistance, which is a well-established risk factor for the development of T2D in human populations [37]. The high risk of developing T2D correlates with reduced β-cell function relative to the degree of insulin sensitivity [38]. The present study was planned to establish the molecular events that take place in T2D that may lead to the development of AD in the later stage of life. This study may help us in connecting the high prevalence of AD with diabetes by investigating the underlying molecular mechanisms and potential therapeutic targets.
To investigate the molecular events in diabetes pathology, we used a polygenic, natural model of humanized T2D, TallyHO/JngJ mice, because this mouse model may be most suitable for the identification of possible therapeutic targets in diabetes, obesity, and AD [13,39]. Preliminary results of this study indicated higher body weight in both the sexes of TH mice at all the four different time points i.e., 4-, 8-, 16- and 24 weeks, compared to age and sex-matched control mice. However, the obesity in male TH mice was not as severe as that is found in female TH mice. We studied the progression of TH mice from normoglycemic state to glucose intolerance/insulin resistance and then the development of diabetes at different time points of age, indicated by fasting glucose levels and intraperitoneal glucose tolerance test at these time-points. The present study exhibited the development of diabetes in male TH mice at 8 weeks of age, which further worsen at 16- and 24-weeks of age. Our results indicate that only male TH mice developed diabetes with mild obesity, whereas female TH mice remain non-diabetic and obese at these time-points. These findings were in agreement with the earlier studies done in TH mice, which exhibited mild obesity and insulin resistance/diabetes in male TH mice only [40,14,41]. Similar to a previous study, the reduced locomotor activity levels in TH mice, compared with SWR/J mice without diet differences, may not directly correlate with increased body mass in response to high-fat diets in TH mice [42]. Substantial evidence indicates that the interaction of various genes and other environmental determinants, including diet, obesity, dyslipidemia, etc. play a critical role in the pathophysiology of diabetes in male TH mice [43–48].
Our current study observations demonstrating diabetes-induced alterations in the mitochondrial functions evident by enhanced levels of hydrogen peroxide, and lipid peroxidation, and reduced mitochondrial ATP production in diabetic mice, are in agreement with the earlier studies [49,50]. The state of insulin resistance in diabetic conditions leads to excessive generation of ROS that induces oxidative stress and tissue damage [51]. The excess of ROS generated during oxidative phosphorylation in mitochondria may primarily trigger mitochondrial dysfunction by interacting with mitochondrial and cellular components such as DNA, proteins, and lipids [52,53]. Several factors are involved in causing mitochondrial dysfunction, of which aging, hyperglycemia, and excess of fatty acids are key contributors to mitochondrial dysfunction in diabetic mice models. Oxidative damage to islet β cells has also been reported in human type 2 diabetes. In this study, the overt pancreatic injury in TH mice was demonstrated by the disintegration of β-cells, and atrophy with vacuolization and reduced islet size. These observations might be due to the enhanced lipid peroxidation and overproduction of reactive oxygen species, as reported in previous studies [51].
Mitochondrial dynamics is a delicate physiological balance between fission and fusion in mitochondria, which is essential for their maintenance in the growing cells, regulation of cell death pathway, and removal of damaged mitochondria [54,55]. In the current study, hyperglycemia-induced abnormal mitochondrial dynamics i.e., increased fission, decreased fusion, and biogenesis, have been reported in the pancreatic tissue of diabetic TH mice. Similar mitochondrial changes were reported in the literature [56,57]. In a previous study [30], we administered SS31(IP) to an AD mouse model (APP) for 6 weeks and studied various parameters including mitochondrial dynamics, biogenesis, and mitochondrial function (H2O2 production, lipid peroxidation, cytochrome c oxidase activity, and mitochondrial ATP). We observed reduced levels of mRNA expression and reduced protein levels of fission genes, and increased levels of mitochondrial fusion, biogenesis, and synaptic genes in SS31-treated APP mice relative to SS31-untreated APP mice. These mitochondrial abnormalities were observed to be modulated by the SS31 treatment indicating that SS31 is protective against mitochondrial and synaptic toxicities in disease progression in APP transgenic mice [30]. Similarly, in the SS31-treated mutant Htt neurons, fission genes Drp1 and Fis1 were down-regulated, and fusion genes Mfn1, Mfn2, and Opa1 were up-regulated relative to untreated neurons, suggesting that mitochondria-targeted molecules reduce fission activity [32].
Earlier studies have also reported a significant increase in the levels of mitochondrial fission modulator, dynamin-related protein 1, which supports hyperglycemia-induced apoptosis in pancreatic β-cells [58]. The activation of AMPK, SIRT1/3, and PGC-1α increased the mitochondrial capacity for oxidative phosphorylation, restoration of physiologic mitochondrial superoxide production, which is beneficial for insulin secretion by pancreatic β cells, insulin sensitivity in skeletal muscle and liver, and prevention of micro- and macro-vascular complications in diabetes [59]. Mitochondrial damage can also contribute to the development of age-dependent insulin resistance [60]. Previous studies demonstrated that T2D is associated with increased myocardial oxidative stress, and mitochondrial dysfunction, regardless of body mass index [61,62]. It is well-established that structurally damaged mitochondria are present in AD neurons and in the primary neurons from AD mice, particularly at nerve terminals [63]. AD is reported to begin 20 years or more before symptoms arise with small changes in the brain that are unnoticeable to the person affected [64].
We observed increased levels of mitochondrial fission proteins and reduced levels of mitochondrial fusion proteins in 24-weeks old diabetic TH mice, indicating the abnormal mitochondrial dynamics in TH mice. Mitochondrial dynamics enable the maintenance of a metabolically efficient mitochondrial population, and disruption of either fusion or fission alters mitochondrial morphology and functionality. Mitochondrial fission and fusion processes occur continually, and along with the removal of damaged mitochondria by mitophagy, these processes maintain the mitochondrial health and physiological state of the cell [65,66]. Prolonged hyperglycemia in diabetes may be responsible for impaired mitochondrial dynamics (increased fission and decreased fusion) and mitochondrial structural abnormalities in TH mice, as agreed in previous studies [67]. Our present study findings agree with a previous study of diabetic/obese rats [68]. Alterations in mitochondrial dynamics proteins and the balance between mitochondrial fusion and fission have been shown to contribute to several diseases [69,70]. Several studies have documented the role of mitochondrial dysfunction in the pathophysiology of T2DM [71–77]. Reduced mitochondrial respiration, ATP production, and mitochondrial density and mRNA have been reported in the insulin resistance and diabetes [78–83]. The findings of the current study demonstrated mitochondrial dysfunction as a connecting link between the T2D and AD pathology.
In the current study, administration of a mitochondria-targeted small molecule, SS31 reduced excessive mitochondrial fragmentation and enhanced mitochondrial fusion in the pancreas of Tally-HO mice by decreasing Drp1 and Fis1 expression levels and increasing the expression of Mf1, Mfn2, and Opa1. In agreement with the mRNA data, the protein data also showed reduced levels of Drp1 and Fis1 in the pancreas of SS31 treated TH mice. Also, SS31 administration of SS31 improved the pancreatic beta-cell architecture in Tally-HO mice. Several recent studies demonstrated that SS31 plays a protective role by modulating mitochondrial functions in a wide range of pathologic conditions [84–86,31,87]. A previous study demonstrated that SS31 is associated with the increase in the expression of fusion genes, decrease H2O2 production, increase ATP levels, and may protect the mitochondria and neurons from aging and amyloid beta-induced oxidative stress [88]. Elevated glycemia and insulin resistance observed in TH mice induce mitochondrial structural and functional changes that may play a vital role in the development of AD in aged diabetic mice. Also, several in vivo and in vitro experimental studies revealed that the insulin signalling pathway is impaired in AD [89–93].
Conclusions
The TallyHO/JngJ mouse is a polygenic model of early onset of T2D that displays key clinical features mimicking human T2D, including hyperglycemia with obesity and hyperinsulinemia. The structural and functional modifications in mitochondria, inflicted by prolonged hyperglycemia in TH mice, may play a vital role in the development of AD-like pathological alterations at an older age. Together, these observations point to insulin resistance and oxidative stress leading to mitochondrial dysfunctioning as a common molecular mechanism connecting type 2 diabetes and AD. Our findings suggest that SS31 can reduce diabetes-induced mitochondrial dysfunction, and maintains mitochondrial dynamics. Additionally, TallyHO/JngJ mouse is established as an important mouse model for understanding the underlying mechanisms linking T2D and AD in future studies.
Acknowledgments
The research presented in this article was supported by NIH grants AG042178, AG047812, NS105473, AG060767, AG069333 and AG066347 (to PHR). JSB was financially supported by the University Grants Commission, Govt. of India, under Raman Post-Doctoral Fellowship in the USA.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
None declared
References
- 1.Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetic medicine : a journal of the British Diabetic Association 15 (7):539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 2.Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, Shaw JE, Bright D, Williams R (2019) Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Research and Clinical Practice 157:107843. doi: 10.1016/j.diabres.2019.107843 [DOI] [PubMed] [Google Scholar]
- 3.Li X, Song D, Leng SX (2015) Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment. Clinical interventions in aging 10:549–560. doi: 10.2147/CIA.S74042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P (2006) Risk of dementia in diabetes mellitus: a systematic review. The Lancet Neurology 5 (1):64–74. doi: 10.1016/S1474-4422(05)70284-2 [DOI] [PubMed] [Google Scholar]
- 5.Vieira MNN, Lima-Filho RAS, De Felice FG (2017) Connecting Alzheimer’s disease to diabetes: Underlying mechanisms and potential therapeutic targets. Neuropharmacology. doi: 10.1016/j.neuropharm.2017.11.014 [DOI] [PubMed] [Google Scholar]
- 6.Davis WA, Zilkens RR, Starkstein SE, Davis TME, Bruce DG (2017) Dementia onset, incidence and risk in type 2 diabetes: a matched cohort study with the Fremantle Diabetes Study Phase I. Diabetologia 60 (1):89–97. doi: 10.1007/s00125-016-4127-9 [DOI] [PubMed] [Google Scholar]
- 7.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53 (9):1937–1942 [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee S, Peters SA, Woodward M, Mejia Arango S, Batty GD, Beckett N, Beiser A, Borenstein AR, Crane PK, Haan M, Hassing LB, Hayden KM, Kiyohara Y, Larson EB, Li CY, Ninomiya T, Ohara T, Peters R, Russ TC, Seshadri S, Strand BH, Walker R, Xu W, Huxley RR (2016) Type 2 Diabetes as a Risk Factor for Dementia in Women Compared With Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes care 39 (2):300–307. doi: 10.2337/dc15-1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon S, Cho H, Kim J, Lee DW, Kim GH, Hong YS, Moon S, Park S, Lee S, Lee S, Bae S, Simonson DC, Lyoo IK (2017) Brain changes in overweight/obese and normal-weight adults with type 2 diabetes mellitus. Diabetologia 60 (7):1207–1217. doi: 10.1007/s00125-017-4266-7 [DOI] [PubMed] [Google Scholar]
- 10.Pal K, Mukadam N, Petersen I, Cooper C (2018) Mild cognitive impairment and progression to dementia in people with diabetes, prediabetes and metabolic syndrome: a systematic review and meta-analysis. Social Psychiatry and Psychiatric Epidemiology 53 (11):1149–1160. doi: 10.1007/s00127-018-1581-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Triggle CR (2006) Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. Canadian journal of physiology and pharmacology 191 (2):437–446. doi: 10.1139/y07-010 [DOI] [Google Scholar]
- 12.Leiter EH, Strobel M, O’Neill A, Schultz D, Schile A, Reifsnyder PC (2013) Comparison of Two New Mouse Models of Polygenic Type 2 Diabetes at the Jackson Laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. Journal of diabetes research 2013:165327. doi: 10.1155/2013/165327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamarai K, Bhatti JS, Reddy PH (2019) Molecular and cellular bases of diabetes: Focus on type 2 diabetes mouse model-TallyHo. Biochim Biophys Acta Mol Basis Dis. doi: 10.1016/j.bbadis.2019.05.004 [DOI] [PubMed] [Google Scholar]
- 14.Kim JH, Saxton AM (2012) The TALLYHO mouse as a model of human type 2 diabetes. Methods in molecular biology 933:75–87. doi: 10.1007/978-1-62703-068-7_6 [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Guo L, Zhang Y, Walzem RL, Pendergast JS, Printz RL, Morris LC, Matafonova E, Stien X, Kang L, Coulon D, McGuinness OP, Niswender KD, Davies SS (2014) Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. The Journal of clinical investigation 124 (8):3391–3406. doi: 10.1172/JCI72517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neschen S, Scheerer M, Seelig A, Huypens P, Schultheiss J, Wu M, Wurst W, Rathkolb B, Suhre K, Wolf E, Beckers J, Hrabe de Angelis M (2015) Metformin supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by suppressing endogenous glucose production in diabetic mice. Diabetes 64 (1):284–290. doi: 10.2337/db14-0393 [DOI] [PubMed] [Google Scholar]
- 17.Thrailkill KM, Bunn RC, Uppuganti S, Ray P, Popescu I, Kalaitzoglou E, Fowlkes JL, Nyman JS (2020) Canagliflozin, an SGLT2 inhibitor, corrects glycemic dysregulation in TallyHO model of T2D but only partially prevents bone deficits. Bone:115625. doi: 10.1016/j.bone.2020.115625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fluitt MB, Shivapurkar N, Kumari M, Singh S, Li L, Tiwari S, Ecelbarger CM (2020) Systemic inhibition of miR-451 increases fibrotic signaling and diminishes autophagic response to exacerbate renal damage in Tallyho/Jng mice. Am J Physiol Renal Physiol 319 (3):F476–F486. doi: 10.1152/ajprenal.00594.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q, Tsuji-Hosokawa A, Willson C, Watanabe M, Si R, Lai N, Wang Z, Yuan JX, Wang J, Makino A (2020) Chloroquine differentially modulates coronary vasodilation in control and diabetic mice. Br J Pharmacol 177 (2):314–327. doi: 10.1111/bph.14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franko A, Neschen S, Rozman J, Rathkolb B, Aichler M, Feuchtinger A, Brachthauser L, Neff F, Kovarova M, Wolf E, Fuchs H, Haring HU, Peter A, Hrabe de Angelis M (2017) Bezafibrate ameliorates diabetes via reduced steatosis and improved hepatic insulin sensitivity in diabetic TallyHo mice. Mol Metab 6 (3):256–266. doi: 10.1016/j.molmet.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy MP, Smith RA (2007) Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110 [DOI] [PubMed] [Google Scholar]
- 22.Reddy PH (2006) Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J Biomed Biotechnol 2006 (3):31372. doi: 10.1155/JBB/2006/31372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szeto HH (2006) Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J 8 (3):E521–531. doi: 10.1208/aapsj080362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 279 (33):34682–34690. doi: 10.1074/jbc.M402999200 [DOI] [PubMed] [Google Scholar]
- 25.Thomas DA, Stauffer C, Zhao K, Yang H, Sharma VK, Szeto HH, Suthanthiran M (2007) Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J Am Soc Nephrol 18 (1):213–222. doi: 10.1681/ASN.2006080825 [DOI] [PubMed] [Google Scholar]
- 26.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK (2007) Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis 18 (3):215–220. doi: 10.1097/01.mca.0000236285.71683.b6 [DOI] [PubMed] [Google Scholar]
- 27.Petri S, Kiaei M, Damiano M, Hiller A, Wille E, Manfredi G, Calingasan NY, Szeto HH, Beal MF (2006) Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem 98 (4):1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x [DOI] [PubMed] [Google Scholar]
- 28.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH (2011) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet 20 (23):4515–4529. doi: 10.1093/hmg/ddr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH (2010) Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis 20 Suppl 2:S609–631. doi: 10.3233/JAD-2010-100564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy PH, Manczak M, Kandimalla R (2017) Mitochondria-targeted small molecule SS31: a potential candidate for the treatment of Alzheimer’s disease. Hum Mol Genet 26 (8):1597. doi: 10.1093/hmg/ddx129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy PH, Manczak M, Yin X, Reddy AP (2018) Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease. J Alzheimers Dis 62 (4):1549–1565. doi: 10.3233/JAD-170988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin X, Manczak M, Reddy PH (2016) Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum Mol Genet 25 (9):1739–1753. doi: 10.1093/hmg/ddw045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter CA, Kartal F, Koc ZC, Murphy T, Kim JH, Denvir J, Koc EC (2019) Mitochondrial oxidative phosphorylation is impaired in TALLYHO mice, a new obesity and type 2 diabetes animal model. Int J Biochem Cell Biol 116:105616. doi: 10.1016/j.biocel.2019.105616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathews CE, Leiter EH (2005) Rodent models for the study of diabetes In: Kahn CR, Weir GC, King GL, Jacobson AM, Moses AC, Smith RJ (eds) Joslin’s Diabetes Mellitus. 14 edn. Lippincott Williams, Willkins, Philadelphia, pp 292–328 [Google Scholar]
- 35.Manczak M, Reddy PH (2015) Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington’s disease. Human molecular genetics 24 (25):7308–7325. doi: 10.1093/hmg/ddv429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, Wang H, Pi C, Shi Y, He X (2020) Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. Journal of diabetes research 2020:4981814. doi: 10.1155/2020/4981814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeFronzo RA (2004) Pathogenesis of type 2 diabetes mellitus. Medical Clinics of North America 88 (4):787–835. doi: 10.1016/j.mcna.2004.04.013 [DOI] [PubMed] [Google Scholar]
- 38.Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, Neifing JL, Ward WK, Beard JC, Palmer JP, et al. (1993) Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42 (11):1663–1672 [DOI] [PubMed] [Google Scholar]
- 39.Ramasubramanian B, Reddy PH (2019) Are TallyHo Mice A True Mouse Model for Type 2 Diabetes and Alzheimer’s Disease? Journal of Alzheimer’s disease : JAD 72 (s1):S81–s93. doi: 10.3233/jad-190613 [DOI] [PubMed] [Google Scholar]
- 40.Warren SM (2012) TallyHo diabetic phenotype limited to male mice: female mice provide obese, nondiabetic mouse model. Wound Repair Regen 129 (4):727e. doi:10.1111/j.1524-475X.2012.00803.x10.1111/j.1524-475X.2012.00803.x10.1097/PRS.0b013e318245eaff10.1097/PRS.0b013e318245eaff [DOI] [PubMed] [Google Scholar]
- 41.Kim JH, Stewart TP, Soltani-Bejnood M, Wang L, Fortuna JM, Mostafa OA, Moustaid-Moussa N, Shoieb AM, McEntee MF, Wang Y, Bechtel L, Naggert JK (2006) Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. The Journal of endocrinology 191 (2):437–446. doi: 10.1677/joe.1.06647 [DOI] [PubMed] [Google Scholar]
- 42.Parkman J, Mao X, Dillon K, Gudivada A, Moustaid-Moussa N, Saxton A, Kim J (2016) Genotype-dependent Metabolic Responses to Semi-Purified High-Sucrose High-Fat Diets in the TALLYHO/Jng vs. C57BL/6 Mouse during the Development of Obesity and Type 2 Diabetes. Experimental and Clinical Endocrinology & Diabetes [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhee SD (2005) Type 2 diabetes mouse model TallyHo carries an obesity gene on chromosome 6 that exaggerates dietary obesity. Biochemical and biophysical research communications 22 (2):171–181. doi: 10.1016/j.bbrc.2005.10.160 [DOI] [PubMed] [Google Scholar]
- 44.Reifsnyder PC (2012) Subcongenic analysis of tabw2 obesity QTL on mouse chromosome 6. Journal of diabetes research 13:81. doi:10.1155/2013/16532710.1155/2013/16532710.1186/1471-2156-13-8110.1186/1471-2156-13-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naggert JK (2005) Glucose intolerance in young TallyHo mice is induced by leptin-mediated inhibition of insulin secretion. The Journal of endocrinology 338 (4):1779–1787. doi: 10.1677/joe.1.06647 [DOI] [PubMed] [Google Scholar]
- 46.Naggert JK (2001) Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Physiological genomics 74 (3):273–286. doi: 10.1152/physiolgenomics.00197.2004 [DOI] [PubMed] [Google Scholar]
- 47.Mustoe TA (2011) Obesity of TallyHO/JngJ mouse is due to increased food intake with early development of leptin resistance. Experimental cell research 119 (4):243–251. doi:10.1016/j.yexcr.2011.07.00410.1016/j.yexcr.2011.07.00410.1055/s-0030-126720210.1055/s-0030-1267202 [DOI] [PubMed] [Google Scholar]
- 48.Bae MA (2010) Genetic and genomic analysis of hyperlipidemia, obesity and diabetes using (C57BL/6J x TALLYHO/JngJ) F2 mice. PloS one 11:713. doi:10.1371/journal.pone.001816810.1371/journal.pone.001816810.1186/1471-2164-11-71310.1186/1471-2164-11-713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green K, Brand MD, Murphy MP (2004) Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 53 Suppl 1:S110–118. doi: 10.2337/diabetes.53.2007.s110 [DOI] [PubMed] [Google Scholar]
- 50.Busik JV, Mohr S, Grant MB (2008) Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 57 (7):1952–1965. doi: 10.2337/db07-1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhatti JS, Bhatti GK, Reddy PH (2017) Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochimica et biophysica acta 1863 (5):1066–1077. doi: 10.1016/j.bbadis.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harper ME, Bevilacqua L, Hagopian K, Weindruch R, Ramsey JJ (2004) Ageing, oxidative stress, and mitochondrial uncoupling. Acta physiologica Scandinavica 182 (4):321–331. doi: 10.1111/j.1365-201X.2004.01370.x [DOI] [PubMed] [Google Scholar]
- 53.Hu F, Liu F (2011) Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cellular signalling 23 (10):1528–1533. doi: 10.1016/j.cellsig.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hales KG (2004) The machinery of mitochondrial fusion, division, and distribution, and emerging connections to apoptosis. Mitochondrion 4 (4):285–308. doi: 10.1016/j.mito.2004.05.007 [DOI] [PubMed] [Google Scholar]
- 55.Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337 (6098):1062–1065. doi: 10.1126/science.1219855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wada J, Nakatsuka A (2016) Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med Okayama 70 (3):151–158. doi: 10.18926/AMO/54413 [DOI] [PubMed] [Google Scholar]
- 57.Bhatti JS, Bhatti GK, Reddy PH (2017) Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis 1863 (5):1066–1077. doi: 10.1016/j.bbadis.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Men X, Wang H, Li M, Cai H, Xu S, Zhang W, Xu Y, Ye L, Yang W, Wollheim CB, Lou J (2009) Dynamin-related protein 1 mediates high glucose induced pancreatic beta cell apoptosis. The international journal of biochemistry & cell biology 41 (4):879–890. doi: 10.1016/j.biocel.2008.08.031 [DOI] [PubMed] [Google Scholar]
- 59.Sharma K (2015) Mitochondrial hormesis and diabetic complications. Diabetes 64 (3):663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science (New York, NY) 300 (5622):1140–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B (2014) Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation 130 (7):554–564. doi: 10.1161/CIRCULATIONAHA.113.008476 [DOI] [PubMed] [Google Scholar]
- 62.Bhatti JS, Kumar S, Vijayan M, Bhatti GK, Reddy PH (2017) Therapeutic Strategies for Mitochondrial Dysfunction and Oxidative Stress in Age-Related Metabolic Disorders. Progress in molecular biology and translational science 146:13–46. doi: 10.1016/bs.pmbts.2016.12.012 [DOI] [PubMed] [Google Scholar]
- 63.Wang X, Wang W, Li L, Perry G, Lee H-g, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1842 (8):1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. New England Journal of Medicine 367 (9):795–804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nature reviews Molecular cell biology 11 (12):872–884 [DOI] [PubMed] [Google Scholar]
- 66.Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125 (7):1241–1252 [DOI] [PubMed] [Google Scholar]
- 67.Rovira-Llopis S, Banuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM (2017) Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox biology 11:637–645. doi: 10.1016/j.redox.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi J, Chandrasekaran K, Demarest TG, Kristian T, Xu S, Vijaykumar K, Dsouza KG, Qi NR, Yarowsky PJ, Gallipoli R (2014) Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Annals of clinical and translational neurology 1 (8):589–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stiles L, Shirihai OS (2012) Mitochondrial dynamics and morphology in beta-cells. Best practice & research Clinical endocrinology & metabolism 26 (6):725–738. doi: 10.1016/j.beem.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaufman BA, Li C, Soleimanpour SA (2015) Mitochondrial regulation of β-cell function: Maintaining the momentum for insulin release. Molecular aspects of medicine 42:91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Irving BA, Nair KS (2007) Aging and diabetes: mitochondrial dysfunction. Current diabetes reports 7 (4):249–251 [DOI] [PubMed] [Google Scholar]
- 72.Abdul-Ghani MA, DeFronzo RA (2008) Mitochondrial dysfunction, insulin resistance, and type 2 diabetes mellitus. Current diabetes reports 8 (3):173–178 [DOI] [PubMed] [Google Scholar]
- 73.Mulder H, Ling C (2009) Mitochondrial dysfunction in pancreatic beta-cells in Type 2 diabetes. Molecular and cellular endocrinology 297 (1–2):34–40. doi: 10.1016/j.mce.2008.05.015 [DOI] [PubMed] [Google Scholar]
- 74.Schrauwen-Hinderling VB, Roden M, Kooi ME, Hesselink MK, Schrauwen P (2007) Muscular mitochondrial dysfunction and type 2 diabetes mellitus. Current opinion in clinical nutrition and metabolic care 10 (6):698–703. doi: 10.1097/MCO.0b013e3282f0eca9 [DOI] [PubMed] [Google Scholar]
- 75.Maassen JA (2006) Mitochondrial dysfunction in adipocytes: the culprit in type 2 diabetes? Diabetologia 49 (4):619–620. doi: 10.1007/s00125-006-0165-z [DOI] [PubMed] [Google Scholar]
- 76.Parish R, Petersen KF (2005) Mitochondrial dysfunction and type 2 diabetes. Current diabetes reports 5 (3):177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lowell BB, Shulman GI (2005) Mitochondrial dysfunction and type 2 diabetes. Science (New York, NY) 307 (5708):384–387. doi: 10.1126/science.1104343 [DOI] [PubMed] [Google Scholar]
- 78.Mogensen M, Sahlin K, Fernstrom M, Glintborg D, Vind BF, Beck-Nielsen H, Hojlund K (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56 (6):1592–1599. doi: 10.2337/db06-0981 [DOI] [PubMed] [Google Scholar]
- 79.Razak F, Anand SS (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. N Engl J Med 2004; 350: 664–71. Vascular medicine 9 (3):223–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, Pypaert M, Shulman GI (2005) Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. The Journal of clinical investigation 115 (12):3587–3593. doi: 10.1172/JCI25151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morino K, Petersen KF, Sono S, Choi CS, Samuel VT, Lin A, Gallo A, Zhao H, Kashiwagi A, Goldberg IJ, Wang H, Eckel RH, Maegawa H, Shulman GI (2012) Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes 61 (4):877–887. doi: 10.2337/db11-1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America 100 (14):8466–8471. doi: 10.1073/pnas.1032913100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics 34 (3):267–273. doi: 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- 84.Hou Y, Shi Y, Han B, Liu X, Qiao X, Qi Y, Wang L (2018) The antioxidant peptide SS31 prevents oxidative stress, downregulates CD36 and improves renal function in diabetic nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 33 (11):1908–1918. doi: 10.1093/ndt/gfy021 [DOI] [PubMed] [Google Scholar]
- 85.Lee FY, Shao PL, Wallace CG, Chua S, Sung PH, Ko SF, Chai HT, Chung SY, Chen KH, Lu HI, Chen YL, Huang TH, Sheu JJ, Yip HK (2018) Combined Therapy with SS31 and Mitochondria Mitigates Myocardial Ischemia-Reperfusion Injury in Rats. International journal of molecular sciences 19 (9). doi: 10.3390/ijms19092782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reddy PH, Manczak M, Kandimalla R (2017) Mitochondria-targeted small molecule SS31: a potential candidate for the treatment of Alzheimer’s disease. Human molecular genetics 26 (8):1483–1496. doi: 10.1093/hmg/ddx052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang J, Li X, Li M, Li J, Xiao W, Ma W, Chen X, Liang X, Tang S, Luo Y (2013) Mitochondria-targeted antioxidant peptide SS31 protects the retinas of diabetic rats. Current molecular medicine 13 (6):935–945 [DOI] [PubMed] [Google Scholar]
- 88.Lim S, Rashid MA, Jang M, Kim Y, Won H, Lee J, Woo JT, Kim YS, Murphy MP, Ali L, Ha J, Kim SS (2011) Mitochondria-targeted antioxidants protect pancreatic beta-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 28 (5):873–886. doi: 10.1159/000335802 [DOI] [PubMed] [Google Scholar]
- 89.Steen E, Terry BM, J Rivera E, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? Journal of Alzheimer’s Disease 7 (1):63–80 [DOI] [PubMed] [Google Scholar]
- 90.Liu Y, Liu F, Grundke‐Iqbal I, Iqbal K, Gong CX (2011) Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. The Journal of pathology 225 (1):54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deng Y, Li B, Liu Y, Iqbal K, Grundke-Iqbal I, Gong C-X (2009) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. The American journal of pathology 175 (5):2089–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gasparini L, Netzer WJ, Greengard P, Xu H (2002) Does insulin dysfunction play a role in Alzheimer’s disease? Trends in pharmacological sciences 23 (6):288–293 [DOI] [PubMed] [Google Scholar]
- 93.De Felice FG, Ferreira ST (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63 (7):2262–2272 [DOI] [PubMed] [Google Scholar]