

Abstract
Mammalian Asn-linked glycans are extensively processed as they transit the secretory pathway to generate diverse glycans on cell surface and secreted glycoproteins. Additional modification of the glycan core by α-1,6-fucose addition to the innermost GlcNAc residue (core fucosylation) is catalyzed by an α-1,6-fucosyltransferase (FUT8). The importance of core fucosylation can be seen in the complex pathological phenotypes of FUT8 null mice, which display defects in cellular signaling, development, and subsequent neonatal lethality. Elevated core fucosylation has also been identified in several human cancers. However, the structural basis for FUT8 substrate specificity remains unknown.
Here, using various crystal structures of FUT8 in complex with a donor substrate analog, and with four distinct glycan acceptors, we identify the molecular basis for FUT8 specificity and activity. The ordering of three active site loops corresponds to an increased occupancy for bound GDP, suggesting an induced-fit folding of the donor-binding subsite. Structures of the various acceptor complexes were compared with kinetic data on FUT8 active site mutants and with specificity data from a library of glycan acceptors to reveal how binding site complementarity and steric hindrance can tune substrate affinity. The FUT8 structure was also compared with other known fucosyltransferases to identify conserved and divergent structural features for donor and acceptor recognition and catalysis. These data provide insights into the evolution of modular templates for donor and acceptor recognition among GT-B fold glycosyltransferases in the synthesis of diverse glycan structures in biological systems.
Keywords: glycosyltransferase, substrate recognition, enzyme mechanism, glycobiology, glycoprotein biosynthesis, glycoprotein structure, N-linked glycosylation
Glycan structures on cell surface and secreted glycoproteins contribute to numerous interactions with the extracellular environment (1–3). Nonreducing terminal glycan structures commonly serve as binding sites for protein interactions (4) and, in some instances, modifications to the glycan core structure can also modulate protein function by influencing interactions with binding partners or altering biological dynamics and function (5–17).
The contributions of fucose (Fuc) residues are unique among the various glycan epitopes that influence glycoprotein function (18). In mammalian cells, Fuc residues are found in four discrete contexts based on linkages to either peptide domains or glycan structures (18) and each is generated by a separate enzyme family that is distinguished by its CAZy classification (19). Fuc residues can be attached directly to Ser/Thr side chains on EGF or thrombospondin repeat domains through the action of POFUT1 (CAZY GT65) and POFUT2 (GT68), respectively (18, 20), which influence protein folding and quality control of the domains in the endoplasmic reticulum (designations here and below employ human gene terminology). Modifications of N- and O-glycoproteins and glycolipids with Fuc-α1,2-Gal linkages by FUT1 and FUT2 (GT11) can create H-antigens as precursors for ABO blood group structures (18, 21–23). Terminal Fuc-α1,3/4-GlcNAc linkages are generated by GT10 fucosyltransferases to form Lewis antigen structures important in immune and inflammatory responses and vertebrate development (24, 25). Finally, N-glycan structures can be modified by α1,6-Fuc addition to the innermost GlcNAc residue (core fucosylation) catalyzed by FUT8 (GT23) (18, 26–28). Two additional fucosyltransferase families have also been identified in nonmammalian systems (CAZy families GT37 and GT74 (29–31)). Structures for several of the FUT family members have been determined (20, 29, 30, 32–42) and all are GT-B fold catalytic domains (32, 43) comprised of two adjacent Rossmann-folds with active sites found in the cleft between the two domains.
The roles of core fucosylation by FUT8 are diverse. FUT8 gene disruption in mice causes postnatal semi-lethality with emphysema-like changes in the lungs and extracellular matrix destruction (44, 45), severe growth retardation (14), defects in antigen presentation and immune response (5), aberrant B-cell development (9), defects in T-cell receptor signaling (10, 46) impaired synaptic plasticity (47), schizophrenia-like symptoms (48), and enhanced neuroinflammation (49). Patients with FUT8-CDG harboring defects in the FUT8 gene also present a similar spectrum of clinical symptoms (50, 51).
An increase in core fucosylation has also been reported in numerous cancers (52–63) and secreted glycoprotein cancer biomarkers containing elevated core fucosylation have been developed (64–66). Numerous approaches for generation of nonfucosylated antibodies are also being pursued through glycoengineering to improve IgG-FcγR interactions as next-generation therapeutics (67, 68).
Given the high impact of core fucosylation in animal systems, we have pursued structural and kinetic studies to determine the molecular basis for substrate recognition and catalysis by FUT8. Numerous FUT8 substrate specificity studies have been performed (69–78), including substrate profiling with libraries of acceptor compounds and glycoprotein acceptors, but these studies only identified relative activities rather than performing detailed kinetic analysis (79–82). Kinetic studies were performed using a GlcNAc2Man3GlcNAc2-Asn (A2-Asn, see Fig. 2 for substrate nomenclature) glycan structure as acceptor (71), but the enzyme recognizes numerous additional N-glycan processing intermediates with varied affinities, which has resulted in conflicting data for some substrates (77, 78).
Figure 2.

Structure of the FUT8:GDP complex. The structure of the human FUT8:GDP dimer. The interface is formed from an extended 4-helix bundle with contributions from two helices of each chain and likely represents the biological dimer in solution. A, representation of the homodimer where discrete domains of the proteins are colored differently. Chain B is comprised of an N-terminal helix pair (tan) followed by the GT-B domain (yellow), and the C-terminal SH3 domain (magenta). For chain C, the N-terminal helical pair (green) is followed by the GT-B domain (cyan) and the SH3 domain (blue). Two rotations of the structure are shown illustrating the 4-helix bundle and the interactions between the SH3 domains and the side of the helical bundle. B, zoom-in representation of hydrophobic side chain interactions within the 4-helix bundle shown as a side view and end-on view. C, cartoon representation of the full-length biological dimer of FUT8 as a transmembrane, Golgi-localized enzyme in vivo with a 77-amino acid linker “stem region” between the transmembrane span and the catalytic domain (green line). Helices of chain B (red) and chain C (cyan) are shown in cartoon representation, β strands are shown as yellow cartoons and GDP molecules are shown as white sticks. The N-terminal ends of the respective chains in the crystal structure are shown as purple spheres.
Initial structural studies on FUT8 over a decade ago determined the overall protein-fold (36), but the absence of bound ligands or substrates provided little insight into substrate recognition. Recently, structures were published for a FUT8–GDP–A2-Asn acceptor complex (41, 42) that identified many aspects of substrate recognition. In the present paper we describe several FUT8 structures, including complexes showing that the occupancy of GDP, a sugar donor analog, correlates with the ordering of specific loop regions important for an induced-fit conformation change upon donor binding. We also describe four glycan acceptor complexes that illustrate the structural basis for FUT8 substrate specificity and the roles of acceptor-binding site complementarity and steric hindrance in acceptor substrate selection. Detailed kinetic analysis using a library of glycan acceptors and a collection of FUT8 active site mutants identify the key interactions required for high affinity substrate recognition and catalysis. We also compare the FUT8 structure with several GT-B fucosyltransferases in other CAZy families to identify far more structural similarities between these enzymes than previously appreciated. Despite the strong structural similarities, each enzyme employs a different strategy for acceptor substrate recognition and catalysis. Overall, the data provide a framework for understanding the evolution of donor- and acceptor-binding template structures used by related fucosyltransferases, and by extension other GT-B fold enzymes, in selective substrate recognition and synthesis of diverse glycan structures in biological systems.
Results
Expression, purification, and enzymatic characterization of recombinant human FUT8
The soluble catalytic domain of human FUT8 was expressed as a secreted and N-terminally His-tagged GFP fusion protein in HEK293 cells as previously described for the production and biochemical characterization of numerous human glycosyltransferases (Fig. S1A) (83). Purification of the enzyme from the conditioned media, fusion tag cleavage, and further purification led to an enzyme preparation amenable to enzymatic and structural studies (Fig. S1B). The oligomeric structure of FUT8 was examined by size exclusion-multiangle light scattering (SEC-MALS) revealing a single peak eluting with a predicted molecular mass of ∼132 kDa consistent with a predominately dimeric form for the ∼62.6 kDa expression product with tags removed (Fig. S1C).
Enzymatic characterization of WT FUT8 with a library of glycan acceptors
Prior studies have focused on kinetic and inhibition studies using various donor substrates and analogs that profiled the contributions of the respective nucleoside, diphosphate, and sugar components (71). Acceptor specificities have also been profiled using a collection of N-glycoprotein (69, 75, 78), glycopeptide (72, 73, 75, 76, 84), and glycan-Asn substrates (71, 74, 78), or glycan arrays (41, 81) of acceptors, but analysis of acceptor kinetics has been restricted to fluorophore-tagged or glycopeptide-linked A2-Asn. Here, we have employed a panel of Asn-linked glycan substrates to assess activity and kinetic constants using GDP-Glo assays. Of the various substrates, the A1-Asn and A2-Asn substrates were shown to display the optimal catalytic efficiencies (kcat/Km) (Table S1 and Fig. 1), which is consistent with prior data demonstrating a requirement for the full GlcNAc-β1,2-Man-α1,3-Man- arm for high affinity substrate recognition (73). The Km value for the A2-Asn substrate was higher (52 μm) than values previously reported for the fluorophore-tagged (12 μm (42)) or glycopeptide-linked A2-Asn (13 μm (71)) substrates consistent with the prior data indicating that extensions to the peptide backbone increases the affinity of the acceptor substrate (77, 78). The addition of a β1,4-GlcNAc residue to the GlcNAc-β1,2-Man-α1,3-Man arm (A3'-Asn) reduced catalytic efficiency by 4.1-fold (Fig. 1). Similarly, extension of the GlcNAc-β1,2-Man-α1,6-Man arm with a β1,4-Gal residue (G1-Asn) reduced catalytic efficiency by 4.8-fold, whereas additional branching to form A3-Asn or incomplete mannose trimming to form NM5N2-Asn reduced catalytic efficiency by 19.8- and 8.3-fold, respectively (Fig. 1). Substrates lacking the β1,2-GlcNAc on the Man-α1,3-Man arm (M5N2-Asn and M3N2-Asn intermediates) had drastically reduced catalytic efficiencies (1590-fold reduction and no detectable activity, respectively). Finally, a tetra-antennary substrate (A4-Asn) had no detectable activity as acceptor. These data, in combination with previous results on relative acceptor specificities (71, 73, 79–82), indicate that the unmodified GlcNAc-β1,2-Man-α1,3-Man arm in both A1-Asn and A2-Asn are the critical determinants for high affinity substrate recognition, and additional β1,4-GlcNAc branching (A3'-Asn) on the subterminal Man residues or modifications on the GlcNAc-β1,2-Man-α1,6-Man arm (A3-Asn, A4-Asn, NM5N2-Asn, or G1-Asn) interferes with acceptor recognition (Fig. 1). Thus, FUT8 acceptor specificity exhibits a stringent exclusion of substrates containing terminal glycan branching beyond the A2-Asn core structure.
Figure 1.

Enzymatic characterization of WT FUT8 with a library of glycan acceptors. The upper panel shows a graphical representation of of kcat/Km values for enzyme activity using WT FUT8 from Table S1 and the respective glycan acceptor structures shown in the lower panel. The nomenclature abbreviation is shown below each structure and the cartoon representations employ standard symbol nomenclature for glycans (116). Asterisk associated with the M5N2-Asn* structures indicates that this substrate contains an Fmoc modification (see “Experimental Procedures”).
Crystallization and structure of the FUT8:GDP complex
Structural studies on FUT8 followed similar approaches that have been taken with other recent glycosyltransferase structures (32, 83, 85, 86) and other recent structures of human FUT8:A2-Asn complexes (41, 42). The soluble catalytic domain of FUT8 was initially crystallized in the presence of 5 mm GDP. X-ray diffraction data were collected to 2.25 Å resolution and the structure was solved by molecular replacement using the prior apo FUT8 structure (PDB 2DE0 (36)) as a search model. There were four chains in the asymmetric unit of the FUT8:GDP complex. The overall structure was comprised of a GT-B–fold but, as noted in the prior FUT8 structures (41, 42, 71), the enzyme has three large helical segments N-terminal to the GT-B–fold (two involved in homodimerization, see below) and an SH3 domain near the C terminus (Fig. 2).
Dimeric structure of FUT8
The SEC-MALS data for the purified enzyme indicated that FUT8 is a dimer in solution (Fig. S1). Similar to recent analysis of FUT8 structures (42, 87), the asymmetric unit of the present FUT8:GDP complex shows that chains B and C form a dimer with an interface that buries 3192 Å2 of surface area (Fig. 2). Chains A and D form analogous dimers with their crystallographically related symmetry mates. PISA analysis indicates that the interface is stable, with a favorable solvation free-energy gain (ΔiG) of −29.8 kcal mol−1 and a p value of 0.057, which corresponds to a high likelihood that the surface is interaction-specific (88) The dimer interface is formed from the first two N-terminal α-helices of FUT8, which fold into a coiled-coil helix structure that associates with the corresponding coiled-coil pairs from adjacent subunits to form a 4-helix bundle, which buries heptad repeat residues in the hydrophobic core (Fig. 2, A and B) (89). Although this 4-helix bundle interaction was recognized in the recent FUT8:GDP:A2-Asn structures (41, 42, 87), it is notable that this dimer interface is also stabilized by extensive H-bonding and van der Waals interactions between the exposed face of the helical hairpin from one chain and the SH3 domain and residues from the GT-B–fold (residues 388-396) from the adjacent chain (Fig. 2A). Interestingly, the interactions between the SH3 domain and the lateral face of the 4-helix do not involve prolines, despite the fact the interface is formed from same loop regions that canonical SH3 domains use to recognize proline-rich ligand sequences (90) (Fig. 2A). The N termini of the two chains are adjacent at the base of the 4-helix bundle, but ∼77 amino acids of N-terminal “stem region” (91) were not resolved in the present structure and would be linked to the type 2 transmembrane segment in the full-length FUT8 enzyme (Fig. 2C). The program DISOPRED3 (92) predicts that the linker region of human FUT8 will most likely be intrinsically disordered.
GDP occupancy in the FUT8:GDP complex and the role of flexible loop conformations
Although the overall fold of the FUT8:GDP complex was similar to the prior apo structure (PDB 2DE0 (36)) and the recent FUT8:GDP:A2 complexes (41, 42), there were notable differences in conformations of several loops between each of the four chains in the GDP complex and also with the previously published structures. The loop regions are comprised of Loop 1 (residues 245-273), Loop 2 (residues 365-378), and Loop 3 (residues 436-443) (Fig. 3B). These three flexible loops enclose the GDP-binding subsite. Chains A and B of the FUT8:GDP complex contained unambiguous electron density for a well-ordered GDP, chain C contains a weakly ordered GDP, and chain D contains no density for the nucleotide (Fig. 3, A and B). The conformations and ordering of the three loops are strongly correlated with the occupancy of bound ligand. The fact that the crystals were grown in saturating GDP suggests that variable occupancy of the nucleotide is due to the crystal lattice selecting for loop conformations that weakened binding of the donor. Thus, in solution it is likely that these loops are flexible and undergo an induced fit folding upon binding of the donor substrate (Fig. 3D). The structures of each of the chains will be discussed separately below.
Figure 3.

Varied structures and ligand occupancy between chains in the FUT8:GDP complex. The four chains in the FUT8:GDP asymmetric unit A, exhibited varied GDP occupancy between the respective chains and different loop conformations (chains A, B, C, and D in the crystal lattice are labeled and boxed regions represent the GDP-binding sites shown in panel B). Chains A and B show full occupancy of the bound GDP, whereas chain C shows partial occupancy, and chain D shows no occupancy as indicated by the difference density map (Fo − Fc) for the donor analog contoured at 3σ. B, zoom-in representations of the GDP-binding sites. Upper panels, zoom-in of the difference density maps (Fo − Fc) for the donor analog as indicated by the boxed regions in panel A with cartoon representations of the respective protein regions. Middle panels, chains A-D and the apo-FUT8 structure (PDB 2DE0 (36)) are shown in cartoon representation (chain A, tan; chain B, green; chain C, slate; chain D, yellow; Apo-FUT8 2DE0, orange). Significant differences in conformation were observed in three loops (Loop 1 (residues 245-273, green), Loop 2 (residues 365-378, red), and Loop 3 (residues 436-443, cyan)) with labels in the respective color. In chains C and D, disorder in Loops 2 and 3 were observed and the ends of the respective ordered regions are indicated by the red (Loop 2) or cyan (Loop 3) spheres, respectively. The 2DE0 structure also had a disordered region for Loop 2 indicated by the red spheres. GDP (yellow sticks) is modeled in the binding site of each chain for reference. Lower panels, surface representation of the four chains in FUT8:GDP and the apo-FUT8 (2DE0) structures with GDP (yellow sticks) modeled in the binding site of each chain for reference. Coloring is as shown in the middle panels. C, the progressive conformational changes in Loops 1-3 are illustrated by an overlay of the loops from chains A-D and the PDB 2DE0 structure. A fully extended “flipped-out” conformation is represented by the 2DE0 structure and progressive closure of Loop 2 and Loop 3 and repositioning of Loop 1 in chains C and D leads to partial occupancy in the GDP-binding site for chain C. Further closure of Loops 1-3 in chains A and B leads to full occupancy of the donor analog and completion of the induced fit donor interactions.
Chains A and B of the FUT8:GDP complex exhibit full occupancy for the bound GDP and Loops 1, 2, and 3 interact with the bound nucleotide (Fig. 3, A and B). The loop conformations of chain A are in analogous positions to the previously described FUT8:GDP:A2-Asn complexes (41, 42). Loop 2 harbors Arg365 and Lys369, which have direct ionic interactions with the β-phosphate oxygen of the nucleotide and help to enclose the bound nucleotide (Fig. 4, A and B). Minor differences in Loop 2 conformations are seen between chains A and B and are likely due to crystal packing (Fig. 3C). Loop 3 adopts a single conformation in chains A and B, and contributes significant packing interactions to the nucleotide and ribose through Ala436 and Arg441, respectively. In addition, Arg441 forms a complex ion pairing interaction with Asp368 in Loop 2, which in turn bridges to Arg365 in Loop 2 to completely enclose the bound nucleotide (the “Arg365-Asp368-Arg441 cage” (Fig. 4C). Finally, Tyr250 in Loop 1 forms an H-bond with the O2 ribose hydroxyl of the bound GDP in both chains A and B (Fig. 5, A and B). The β-phosphate has additional interactions with the side chain hydroxyl of Ser469 and the backbone nitrogen of Gln470, whereas the α-phosphate has polar interactions with the backbone nitrogen atoms of Gly221 and Cys222. The extensive electrostatic interactions with the β-phosphate likely explains why GDP is a much more potent inhibitor of FUT8 (3.6 μm Ki) than GMP (2.3 mm Ki) (71). The ribose O3 hydroxyl also H-bonds with the peptide nitrogen of Tyr220, and the purine ring of the nucleoside interacts with His363, Thr408, and Asp453 (Fig. 4, A and B). There are additional minor differences in conformation for solvent-exposed side chains between chains A and B, but interactions with the bound GDP are conserved.
Figure 4.
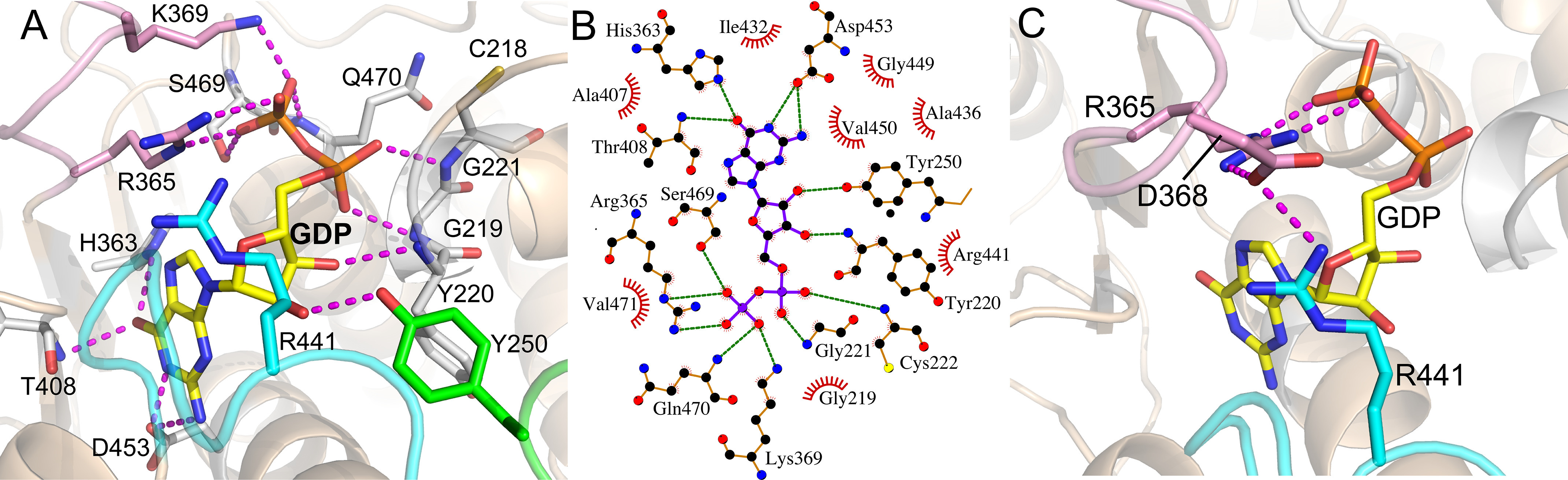
FUT8 interactions with GDP. Interactions between FUT8 and the GDP donor analog are illustrated in stick and cartoon representation (A) with hydrogen bonds indicated by magenta dotted lines. Interacting residues arising from Loop 2 are shown in pink stick representation, whereas residues coming from Loops 1 and 3 are shown as green and cyan sticks, respectively. B, Ligplot (117) representation depicting packing interactions (red, feathered lines) and hydrogen bonds (green, dashed lines) of GDP (highlighted in yellow) in the FUT8 active site (orange ball and stick with atomic colors). C, the Arg365-Asp368-Arg441 cage formed by the closing of Loops 2 and 3 to enclose the GDP-binding site results in a series of ionic interactions between Arg365 (Loop 2), Asp368 (Loop 2), and Arg441 (Loop 3). Only Arg365 directly interacts with the GDP (pair of ionic interactions with the β-phosphate), whereas the other two residues bridge Loops 2 and 3 to complete the enclosure of the donor nucleotide.
Figure 5.
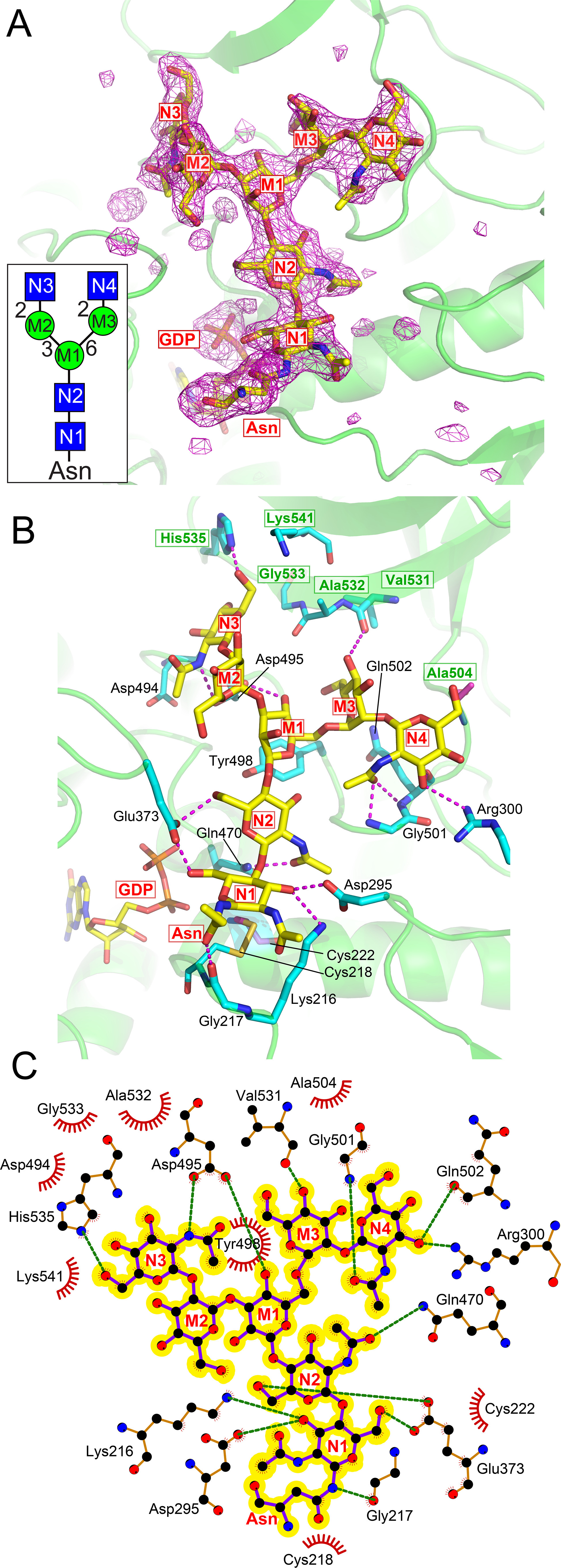
FUT8 interactions with the A2 acceptor. A, the structure of the FUT8:A2-Asn acceptor complex is shown as a difference density map (Fo – Fc) of the acceptor contoured at 3.5 σ before modeling the GlcNAc2Man3GlcNAc2-Asn structure (yellow sticks). The inset shows a cartoon representation (see Fig. 2 for cartoon nomenclature) of the A2-Asn glycan with labeling of each monosaccharide inside each respective symbol and the corresponding labeling is also employed in the density map. B, the FUT8:A2-Asn complex is shown in yellow stick and cartoon representation with interacting residues shown in cyan stick representation and hydrogen bonds as magenta dashed lines. Monosaccharide residues of the A2-Asn structure are labeled as in Panel A. GDP is shown in yellow stick representation. Residues arising from the SH3 domain are indicated by green residue labeling enclosed in green boxes. C, Ligplot (117) representation depicting packing interactions (red, feathered lines) and hydrogen bonds (green, dashed lines) of A2-Asn (highlighted in yellow) in the FUT8 active site (orange ball and stick with atomic colors).
In contrast, chain C has significantly weaker electron density for the bound GDP (Fig. 3, A and B) and a significant portion of Loops 2 and 3 are completely disordered (indicated by red and blue spheres in Fig. 3B, upper panels). Only residues 365-366 and 375-376 are ordered in Loop 2 and residues 430-432 are ordered in Loop 3. The conformation of Loop 1 in chain C is similar to chains A and B with Tyr250 hydrogen bonding with the ribose O2 hydroxyl of the bound GDP. The disorder of Loops 2 and 3 opens the Arg365-Asp368-Arg441 cage and exposes the GDP to solvent (Fig. 3B). The rest of the binding site retains the other H-bonding and ionic interactions between the GDP and the polypeptide similar to chains A and B. Thus, the loss of the Arg365-Asp368-Arg441 cage and the associated electrostatic and packing interactions likely explains the lower occupancy of the nucleotide.
Chain D has no visible density for GDP in the binding site (Fig. 3, A and B). The disorder for Loop 2 was similar to chain C, but residues 430-435 in Loop 3 are positioned to indicate that this peptide segment is extended away from the GDP-binding site reminiscent of the position of the same loop in the PDB 2DE0 structure. A similar loss of binding residues is found for Loops 2 and 3 compared with chain C. In addition, Loop 1 in chain D is altered and does not position Tyr250 within H-bonding distance of the ribose O2 hydroxyl in the nucleotide. Thus, loss of interactions with all three loops explains the absence of a bound GDP in chain D.
Finally, a comparison with PDB 2DE0 shows that the loops in the unliganded structure are similar in flexibility to those in chain D (Fig. 3B). In PDB 2DE0, Loop 2 extends away from the active site with residues 373-376 being disordered. Although Loop 3 is ordered in PDB 2DE0, it has moved >25 Å away from the GDP-binding site. The conformation of Loop 1 is similar to that observed in the FUT8:GDP chain D. These altered conformations are additional evidence that all three loops are likely disordered in solution, and supports an induced-fit folding upon nucleotide binding (Fig. 3D).
Structure of the FUT8:GDP:A2-Asn complex
To examine the acceptor substrate interactions, we crystallized a FUT8:GDP:A2-Asn complex. The structure was solved at 2.4 Å resolution in space group P6522 with two chains in the asymmetric unit. Application of crystallographic symmetry reveals the same dimer that we previously observed. The two chains are essentially the same (rmsd 0.20 Å for 464 Cα atoms) with the exception of minor surface loops and solvent-exposed side chain differences. The structure is similar to chain A of the FUT8:GDP (rmsd 0.34 Å for 459 Cα atoms), with the exception that the conformation of Loop 2 is significantly different from the rest of the chains in the FUT8:GDP complex.
Both chains in the FUT8:GDP:A2-Asn complex display well-defined electron density for GDP and the entire A2-Asn molecule, which binds in a complementary surface groove on the face of the GT-B–fold and extends from a position adjacent to the GDP-binding site toward the SH3 domain (Fig. 5A). The FUT8:GDP:A2-Asn structure and its ligand interactions (Fig. 5, B and C) are essentially identical to those identified in the recently published FUT8:GDP:A2 complexes (41, 42) (the rmsd range from 0.45 to 0.54 Å for 450-467 corresponding Cα atoms in PDB 6TKV (41)). Briefly, GlcNAc residue N1 (Fig. 5) is positioned adjacent to the GDP-binding site with the O6 hydroxyl facing toward the GDP ligand through H-bonding interactions with Glu373. This residue is positioned in Loop 2 and is fully ordered in its interaction with the hydroxyl nucleophile of the acceptor. Glu373 also interacts through a salt bridge to Lys369 and subsequently to the β-phosphate oxygen of the GDP. These interactions suggest that Glu373 acts as the catalytic base to deprotonate the N1 O6 nucleophilic hydroxyl and then in turn protonate the phosphate of the leaving GDP product (see “Catalytic Mechanism” below). Additional interactions include H-bonds from the N1 O3 hydroxyl to Asp295 and Lys216, the Asn amide nitrogen with Gly217, and hydrogen bonds between Gln470 and the N2 N-acetyl glycosidic oxygen. A further matrix of hydrogen bonding interactions extends throughout the length of the A2-Asn structure all the way to the nonreducing terminal GlcNAc residues (N3 and N4). Many of the interactions with the terminal A2-Asn residues are contributed by the SH3 domain (Fig. 5B, green residue labeling with green boxes). All of the monosaccharides in the acceptor have at least one H-bond to the enzyme surface with the exception of the internal Man residue in the GlcNAc-β1,2-Man-α1,3-Man arm (M2 residue). The details of the acceptor interactions are shown in Fig. 5, B and C. Most notable among these interactions is the position of the SH3 domain, which sterically blocks the linear extension of the A2 glycan beyond the β-linked Man residue (M1) and leads to a bifurcation of the two terminal glycan branches in opposite directions across the surface of the SH3 domain. Each branch has interactions with the SH3 domain including residues N3, M3, and N4. The close interaction of the M1 residue with the SH3 domain surface also precludes binding of glycans containing a bisecting GlcNAc residue.
Enzymatic characterization of FUT8 active site mutants
To confirm the contributions of binding site amino acids to acceptor glycan interactions we mutated each of the respective amino acids to alanine, tested the activity, and performed kinetic analysis on the active mutants (Fig. 6C). A subset of the residues in the GDP-binding site had previously been tested for enzyme activity (36) and we confirmed that mutation of these and other residues that directly interact with the GDP lead to complete enzyme inactivation (Table S3 and Fig. 6, B and C). In addition, we probed residues that contribute to acceptor glycan interactions and found that mutation of three residues, Asp295 that H-bonds with the O3 hydroxyl of N1, Tyr498 that provides van der Waals packing interactions with the hydrophobic face of the β-Man residue (M1), and Asp495 that H-bonds with M1 and the terminal N3 residue, all lead to complete loss of enzyme activity (Fig. 6A). Mutations of other interacting residues result in significantly decreased catalytic efficiency, but not complete inactivation (Fig. 6C). These reductions in kcat/Km include Lys216 that H-bonds with N1 (160-fold reduction in kcat/Km), Gln470 that H-bonds with N2 (80-fold reduction), Asp494 adjacent to N3 (800-fold reduction), His535 that H-bonds with N3 (11-fold reduction), and Gln502 that H-bonds with N4 (16-fold reduction). A similar reduction in activity was previously seen for mutants in the SH3 domain including H535A as well as H535A/Lys541A and T550A/L552A double mutants in prior studies (93) and a collection of residues conserved between human FUT8 and chicken c-Src (87) demonstrating the critical role of the SH3 domain in providing acceptor substrate specificity and affinity. Thus, whereas the donor-binding site is exceptionally sensitive to mutational perturbation, the acceptor-binding site has fixed points of essential interaction and other regions that contribute incremental binding energy to catalytic efficiency.
Figure 6.
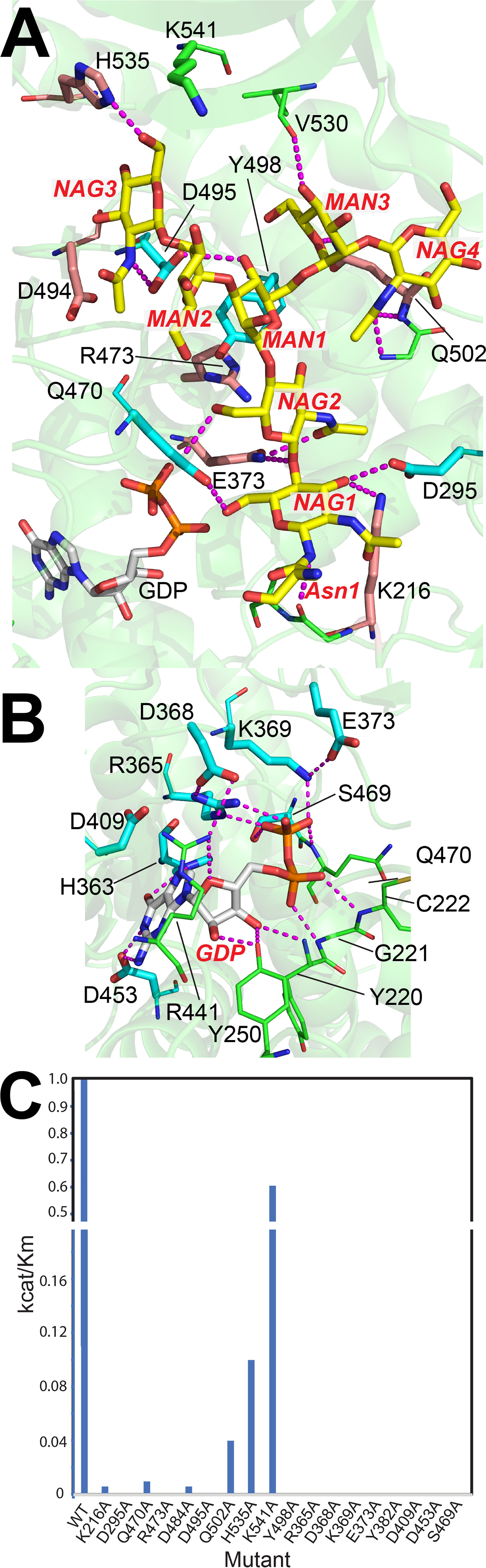
Structural representation of the FUT8 active site mutants depicting the effects on enzyme kinetics. Residues in the FUT8 acceptor-binding site (A) or donor site (B) were mutated to Ala, expressed, purified, and assayed for enzyme activity using the A2-Asn acceptor substrate (Table S3). C, kcat/Km values for each of the respective mutants are shown. In A and B, residues that led to enzyme inactivation when mutated to Ala are indicated by cyan stick representations. Residues that reduced activity are indicated by tan stick representation. The K541A mutation had a minor impact on kcat/Km and is indicated in green stick representation.
Structures of FUT8:GDP:A3'-Asn FUT8:GDP:A3-Asn, FUT8:GDP:NM5N2-Asn complexes and implications for recognition of alternative substrates
The kinetic studies with varied acceptor substrate structures indicated a preference for glycans containing an unmodified GlcNAc-β1,2-Man-α1,3-Man arm (A1-Asn and A2-Asn structures), and reduced activities for substrates that harbor additional modifications to the core A2-Asn structure. The impacts of these modifications on kcat/Km are highlighted in Fig. 7A. To examine substrate interactions with alternative acceptor structures we performed additional co-crystallization studies with A3'-Asn (Fig. 7B), A3-Asn (Fig. 7C), and NM5N2-Asn (Fig. 7D) acceptors. In each case we obtained well-defined electron density for at least a portion of the corresponding acceptor glycan.
Figure 7.
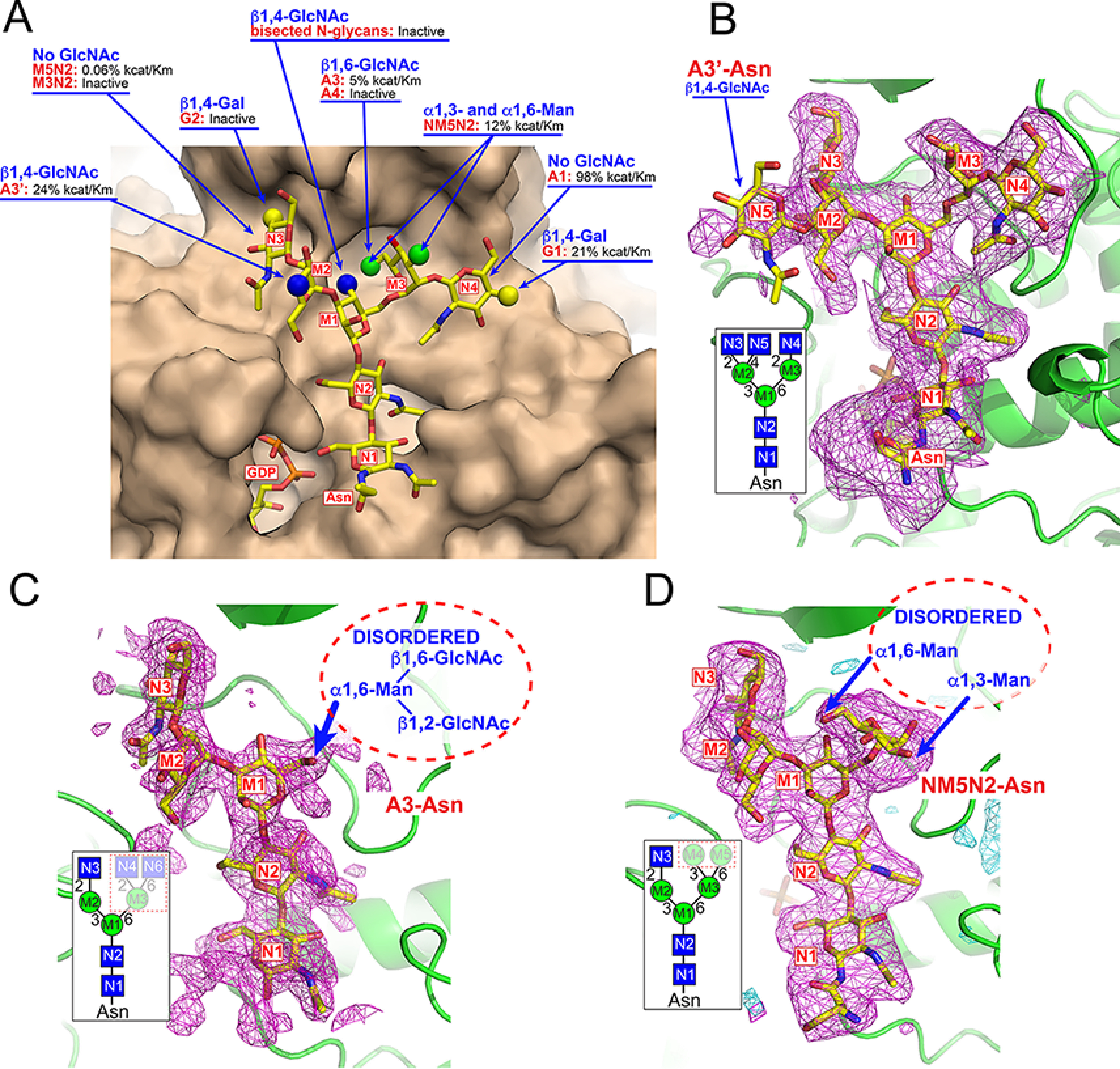
Impact of acceptor modifications on enzyme activity and structures of acceptor complexes. Modifications to the acceptor glycan structure impacts catalytic efficiency in enzyme assays (A). Surface representation of the FUT8:A2-Asn complex (tan) with the A2-Asn structure are displayed in yellow stick representation. Alternative acceptor structures are indicated by spherical representations at positions where they differ from the A2-Asn structure. For each modification, the respective glycan structure is shown in red text and the effect on enzyme activity is listed based on enzyme assay data from Table S1. In cases where the respective acceptor was listed as “inactive” there was no activity detected in the in vitro enzyme assays. Values for kcat/Km are listed by comparison to activity using the A2-Asn substrate. The lack of activity toward the G2-Asn substrate or the glycans with bisected GlcNAcs were based on prior assays from other groups (73, 75, 82). The Polder maps (115) (magenta mesh and contoured at 2.8 σ and 3.5 σ, respectively) were obtained from the crystal structures of three additional acceptor complexes and are shown in B–D. The Polder maps (115) for A3'-Asn, A3-Asn, and NM5N2-Asn acceptors were calculated following a modified procedure to reduce model bias (see “Experimental procedures”). B, the structure of the FUT8:GDP:A3'-Asn complex was essentially the same as the FUT8:GDP:A2-Asn complex except for the additional β1,4-GlcNAc residue (N5) extending from the Man-α1,3- residue (M2). This residue extends into the solvent and has no additional interactions with the enzyme surface. The inset in B indicates the cartoon representation of the A3'-Asn glycan with residue labeling that is also used to label the residues in the difference density (Polder) map. C, the structure of the FUT8:GDP:A3-Asn complex retained the density for the GlcNAc-β1,2Man-α1,3Man-β1,4GlcNAc-β1,4GlcNAc-βAsn region of the A2-Asn complex. However, the entire extension from the Man-α1,6- arm (residues M3, N4, and N6) was disordered and not resolved in the structure (whited out region in the dotted box for the inset; structure and dotted oval in the Polder map (115)). D, the structure of the FUT8:GDP:NM5N2-Asn complex also retained the electron density for the GlcNAc-β1,2Man-α1,3Man-β1,4GlcNAc-β1,4GlcNAc-βAsn region similar to the A2-Asn complex along with density for the Man-α1,6- (M3) residue extending from the Man-β1,4- residue (M1). However, the additional terminal Man-α1,3- (M4) and Man-α1,6- (M5) residues in the NM5N2-Asn structure were disordered and not resolved in the structure (whited out region in dotted box for the inset structure and dotted oval in the Polder map (115).
The structure of the FUT8:GDP:A3'-Asn complex was solved at 3.3 Å resolution, in the same space group as FUT8:GDP:A2-Asn. FUT8:GDP:A3'-Asn has two chains in the asymmetric unit and both chains display unambiguous electron density for the GDP and for all residues of the A3'-Asn acceptor. The overall enzyme structure was similar to the two chains of the FUT8:GDP:A2-Asn complex (rmsd 0.22-0.29 Å for 465 Cα atoms between the corresponding chains) and the donor and acceptor interactions within the respective binding subsites were identical to the FUT8:GDP:A2-Asn complex. The major difference was the presence of an extra β1,4-GlcNAc residue (N5) on the GlcNAc-β1,2-Man-α1,3-Man arm that distinguishes the A3'-Asn structure from A2-Asn (Fig. 7B and inset). In both chains, the N5 residue faces into the solvent and does not contribute directly to acceptor-binding interactions.
The structure of the FUT8:GDP:A3-Asn complex was determined at 2.47 Å resolution in space group P65 with eight chains in the asymmetric unit. All eight chains display unambiguous electron density for the GDP, but density for the acceptor was evident only in chains A, D, E, and H. In each case where the acceptor density was present, only a single distal arm and the core of the glycan structure (GlcNAc-β1,2-Man-α1,3-Man-β1,4-GlcNAc-β1,4-GlcNAc-β-Asn) was resolved (Fig. 7C). The Man-α1,6- (M3) branching residue from the core β-Man (M1) was disordered along with the two distal GlcNAc residues (N4 and N6) extending from this arm (Fig. 7C and inset). The disorder for these three monosaccharides in the acceptor complex indicates that the predicted steric hindrance (Fig. 7A) for the additional β1,6-GlcNAc residue (N6) altered the positioning of the entire branch.
The structure of the FUT8:GDP:NM5N2-Asn complex was determined at 3.2 Å resolution. Similar to FUT8:GDP:A3-Asn, this structure was also in space group P65 with eight chains in the asymmetric unit. For this complex, the electron density for both the GDP and the GlcNAc-β1,2-Man-α1,3-[Man-α1,6-]Man-β1,4-GlcNAc-β1,4-GlcNAc-Asn portion of the acceptor was visible in all eight chains, although the order of the GlcNAc-Asn region of the acceptor varied between chains (Fig. 7D and inset). The electron density for the M3 branch extending from the core β-Man (M1) is well-defined. However, density for the M4 and M5 residues was weak and they were left unmodeled.
The steric constraints of the acceptor-binding site clearly indicate that the 1,6-substituents on the M3 residue in the A3-Asn and NM5N2:Asn complexes are not compatible with the same binding geometry as the A2-Asn structure (GlcNAc-β1,6- (N6) for A3-Asn and Man-α1,6- (M5) for NM5N2-Asn). These modifications to the M3 residue lead to rotation or disorder of the more flexible Man-α1,6- linkage between M3 and M1 to accommodate the bulky branched substituents and likely account for the reduced catalytic efficiencies for these glycan acceptors.
Sequence and structural alignment of FUT8 with other related GTs
Previous sequence (94), HCA (95), and structural (42) alignments of fucosyltransferases have compared α-1,2- and α-1,6-fucosyltransferases with mammalian protein-O-fucosyltransferases (POFUTs) and identified three conserved sequence motifs that are common among the sequence families (Fig. S2, red boxes), including residues involved in binding the nucleotide portion of the sugar donor (42). Structures are now available for mouse (38), human (37), and Caenorhabditis elegans (33) POFUT1 (GT), C. elegans (39), and human (40) POFUT2 (GT68), Arabidopsis FUT1 (GT37 (29, 30)), NodZ (GT23, (35, 96)), and human FUT8 (GT28) and structural alignment using the PROMALS3D server (97) allowed us to make a more comprehensive comparison between these enzymes to identify additional conserved structural elements. The resulting alignment revealed extensive structural similarity far beyond the three previously identified motifs (Fig. S2, blue boxes). Visualization of these conserved elements (Fig. S3A, Conserved) indicated the core of both Rossmann-folds were highly conserved among these enzymes including more than eight helical segments and four of the β-strands within the Rossmann-fold associated with the donor-binding site. Six β-strands of the second Rossmann-fold are less conserved in position, but still maintain a high degree of structural similarity among the collection of enzymes (Fig. S3, A and B). The degree of structural similarity is striking considering pairwise primary sequence similarities were <15%. The PROMALS3D alignment also identified 13 amino acids that are most conserved among the structures, some of which were noted in a more focused comparative analysis of the sugar donor-binding site (42). Displaying these amino acids within the framework of the FUT8 structure shows that 9 of the 13 amino acids are clustered within the base of the GDP-binding site proximal to the Rossmann-fold (Fig. S3C) and either directly interact with the donor or contribute to the architecture of the donor site. The other four residues are distributed more widely within the FUT8 structure and do not interact with substrates or contribute to catalysis. For FUT8, the conserved binding site residues are not a part of Loops 1-3 that undergo conformational changes upon donor binding and, surprisingly, these three dynamic loops are not conserved in position or sequence among the other fucosyltransferase structures.
In contrast to the similarity in the underlying Rossmann-fold and donor-binding subsite, each of the enzymes are entirely different in their modes of interactions with their respective acceptor molecules (Fig. S3). Although the positions of acceptor interactions are approximately similar in the clefts between the two Rossmann-folds, each enzyme employs unique loop insertions into the Rossmann-fold scaffold to assemble the acceptor binding subsites (Fig. S3). For the POFUTs that recognize larger protein domain structures, the interactions are assembled through the generation of a deep cleft between the two subdomains. For the glycan-directed fucosyltransferases (FUT8 and AtFUT1), the interactions are through a more flattened face of the cleft and, for FUT8, the recruitment of an additional SH3 domain insertion near the C terminus. This clearly indicates the presence of evolutionary divergence in the respective loop regions to provide unique acceptor binding specificities, whereas the core scaffold of the GT-B structure and donor interactions are more conserved.
An even more striking observation from the aligned structures is the similarities in positions for the acceptor nucleophiles in the respective structures (Fig. 8B). FUT8 and AtFUT1 (29, 30) structures have been solved as extended glycan acceptor complexes, whereas POFUT1 (38) and POFUT2 (39) have been solved in complex with EGF and thrombospondin repeat domain acceptors, respectively. In each instance, the corresponding acceptor hydroxyl has been identified in a position for nucleophilic attack. Surprisingly, despite the differences in the respective acceptor structures, the nucleophilic hydroxyls are positioned similarly. The nucleophilic hydroxyls for the two glycan-directed enzymes (FUT8 and AtFUT1) superimpose almost exactly. Similarly, the nucleophilic Ser/Thr hydroxyls for the POFUTs also superimpose with each other at a position <2 Å from the nucleophiles for the glycan-directed enzymes.
Figure 8.

Proposed catalytic mechanism of FUT8 and other fucosyltransferases. A, the proposed mechanism for FUT8 is illustrated based on the alignment of the FUT8:GDP:A2-Asn complex with the human POFUT2:GDP-Fuc complex (PDB 4AP6 (40)). The aligned structures overlay the nucleotide and ribose (green sticks for GDP bound to FUT8 and white sticks for GDP-Fuc bound to POFUT2) in both structures and place the Fuc (cyan sticks) residue within the FUT8 active site in an appropriate position for an in-line SN2 inverting mechanism. The O6 GlcNAc nucleophile (magenta sphere) in the A2-Asn acceptor (green sticks) is positioned appropriately relative to the Fuc C1 (yellow sphere) and the Glu373 catalytic base (pink sticks). The trajectory for the catalytic base and nucleophilic attack is shown with yellow dotted lines. The Glu373 base is also in proximity to Lys369 in Loop 2 (pink cartoon), which shuttles the proton from Glu373 to the β-phosphate of the GDP leaving group (magenta dotted lines). The position of the GlcNAc is also stabilized by an H-bond between O4 and Asp368 in Loop 2 (gray dotted line). B, the structures FUT8:GDP:A2-Asn, POFUT1:GDP-Fuc:EGF domain complex, C. elegans POFUT2:GDP:thrombospondin repeat complex, human POFUT2:GDP-Fuc complex, NodZ:GDP complex, and Arabidopsis FUT1:GDP–xylo-oligosaccahride complex were aligned as described in Fig. S3. The acceptor monosaccharide residues for the respective glycans (FUT8 (green sticks) and AtFUT1 (light blue sticks)) or protein domains (POFUT1 (purple sticks) and POFUT2 (gray sticks)) are shown as single monosaccharides or amino acids (full Ser side chain modeled for mouse Factor VII EGF1 domain from an mutant Ala residue in the mouse POFUT1:GDP-Fuc:EGF domain structure (38)). The respective hydroxyl nucleophiles are shown as magenta spheres. The donor analogs (GDP for all structures except POFUT1) are shown in white stick representation. The GDP-Fuc structure from the POFUT1:GDP-Fuc complex is shown in white stick representation except for the Fuc residue, which is shown as cyan sticks with C1 as a yellow sphere. The proposed catalytic base for FUT8 (Glu373 in Loop 2) is shown in pink stick representation. Yellow dotted lines represent the deprotonation trajectory of the nucleophilic hydroxyl by Glu373 and the subsequent SN2 nucleophilic attack of the GDP-Fuc sugar donor as proposed in the FUT8 catalytic mechanism. The proposed catalytic base for POFUT2 (E52) is shown in white stick representation. Yellow dotted lines represent the deprotonation trajectory of the nucleophilic Ser hydroxyl by Glu52 and the subsequent SN2 nucleophilic attack of the GDP-Fuc sugar donor in the POFUT2 catalytic mechanism. The two catalytic bases interact from the opposite faces of their respective nucleophilic hydroxyls and emerge from completely different regions of the conserved core structures of their respective enzymes.
FUT8 catalytic mechanism
Among the six aligned fucosyltransferases in Fig. S3, only two have a clearly identified catalytic base (FUT8 and POFUT2 (39)). POFUT1 and AtFUT1 have no apparent ionizable side chain in proximity to the acceptor hydroxyl and are proposed to use a SN1 mechanism employing a proton shuttle (20, 30, 33, 38). NodZ was not solved as an acceptor complex and its catalytic base was not identified (35, 96).
To identify the catalytic mechanism of FUT8 we superimposed the aligned structure of the intact GDP-Fuc from the POFUT2:GDP-Fuc complex (40) in the donor-binding site (Fig. 8A). This resulted in the placement of the Fuc residue in the FUT8 active site without steric hindrance and predicted H-bonds for the fucose O2 with the backbone nitrogen of Gly219 and for O4 with Asp368 (from Loop 2). This latter interaction is consistent with the loss of FUT8 activity for the D368A mutant (Table S3). In addition, C1 of the Fuc residue is directly in line for nucleophilic attack by the O6 hydroxyl of N1 (Fig. 8A). Thus, the structure of the FUT8:GDP:A2-Asn complex and the modeled donor confirm the inverting catalytic mechanism of the base-catalyzed SN2 nucleophilic attack of the sugar nucleotide donor by the N1 O6 hydroxyl.
Although both FUT8 and POFUT2 perform similar SN2 in-line nucleophilic attacks of their respective GDP-Fuc donors, the catalytic base for POFUT2 (E52 (39)) originates from a completely different region of the protein structure compared with FUT8 (Fig. 8B). The catalytic base (Glu373) for FUT8 is on the opposite side of its acceptor nucleophile relative to Glu52 in POFUT2, whereas POFUT1 and AtFUT1 do not have ionizable side chains at either position. Clearly, whereas the enzymes conserve residues for donor and nucleophile binding, the constraints for positioning the catalytic base and enzymatic mechanism are not conserved among these enzymes.
Discussion
Prior studies on FUT8 examined the range of glycan substrates that can be recognized (69–78) and how these modifications modulate a diverse array of biological functions (6, 8–10, 12, 14, 15, 17, 44–68, 98, 99). Our detailed kinetic analysis demonstrates the importance of the N-glycan GlcNAc-β1,2-Man-α1,3-Man arm as a key determinant for high affinity substrate recognition. Glycans that have not been modified by MGAT1 (e.g. Man5-3N2-Asn) were ineffective substrates for human FUT8 even though recombinant expression data previously indicated these structures can be modified when mannose trimming is inhibited or FUT8 has been overexpressed (77, 78, 100). The A1-Asn and A2-Asn processing intermediates exhibited the highest catalytic efficiencies, while A3'-Asn, A3-Asn, G1, or incomplete processing to NM5N2-Asn significantly reduced catalytic efficiency, and extension to A4-Asn abolished activity (Fig. 7A). Thus, FUT8 exhibits a highly constrained active site that restricts steric access to all but a few structures despite the fact that all N-glycan processing intermediates contain the same core Man3GlcNAc2-Asn acceptor structure.
Structures of FUT8 donor complexes
To address the restricted substrate recognition, we first examined structures of FUT8 in complex with the donor analog, GDP. Loop regions were identified that were extended away from the donor-binding site when no GDP was bound and a progressive increase in GDP density was found as the loops flipped in to enclose the donor analog. Thus, catalysis proceeds by initial GDP-Fuc interaction with the donor-binding site predominately through hydrogen bonding between the nucleotide and the base of the donor-binding pocket. The highly flexible Loops 1-3 progressively become ordered while enclosing the sugar nucleotide and locking the donor in place through the Arg365-Asp368-Arg441 cage. Binding of the glycan acceptor with its GlcNAc O6 hydroxyl positioned as nucleophile leads to deprotonation by Glu373, in-line SN2 attack of the donor C1, and shuttling of the proton from Glu373 through Lys369 to the β-phosphate of the leaving group (Fig. 8A). The importance of each of these donor interactions was demonstrated through binding site mutants that all individually led to enzyme inactivation.
Structures of FUT8 acceptor complexes
The structures of several high and low affinity FUT8 acceptor complexes also allowed mapping of steric contributions to substrate binding affinity and specificity. Initial structural studies on the FUT8:GDP:A2-Asn complex identified analogous interactions found in prior structural studies on equivalent complexes (41, 42). The Man-β1,4GlcNAc-β1,4-βAsn acceptor core bound within the cleft between the two Rossmann-folds, whereas the C-terminal SH3 domain presented a flattened surface and steric barrier for branched substrate interactions that established acceptor glycan specificity. Our kinetic analysis of site directed mutants also probed the roles of individual amino acids in substrate binding and catalysis to validate the structural model.
The studies were then extended by examining additional acceptors complexes with reduced binding affinities. The A3'-Asn structure extended the additional β1,4-GlcNAc into solvent relative to the A2-Asn complex (Fig. 7B). A similar solvent-exposed β1,4-Gal extension was predicted for the G1 structure based on comparison with the A2-Asn complex. Both acceptor modifications reduce catalytic efficiencies by 3-4–fold likely as a result of entropic penalties from the solvent-exposed sugar rather than steric restrictions.
In contrast, modifications to the M3 residue, especially at the O6 position, led to a steric clash with the enzyme surface (Fig. 7A). As a result, both the NM5N2-Asn (α1,6-Man on M3) and A3-Asn (β1,6-GlcNAc on M3) had significant disorder for residues on the M3 branch, whereas the remainder of the acceptor structure was well resolved. This disorder is consistent with reduced catalytic efficiencies for these acceptors (8.4- and 20-fold reduction in kcat/Km, respectively) and a displacement of the M3 residue as a consequence of the O6 modification. NM5N2-Asn had slightly better catalytic efficiency than A3-Asn, consistent with the well-defined electron density for the M3 residue in the crystal structure and the reduced size of the Man-α1,6- substituent versus GlcNAc-β1,6- for A3-Asn. These comparisons of catalytic efficiencies and acceptor complex structures highlight the role of the SH3 domain as a critical binding interface for tuning branch-specific acceptor affinity through both complementarity interactions and steric restrictions.
Conserved and distinctive structural elements for substrate recognition among FUTs
The FUT8 structure also allowed a comparison with other known GT-B fold fucosyltransferases to identify conserved and divergent structural features. These alignments revealed a far more extensive similarity within the core Rossmann-folds and previously noted donor-binding site residues (42) and other regions identified in sequence alignments (95, 101). Surprisingly, no conservation in position or sequence for the three dynamic loops that enclose the FUT8 donor-binding site were identified indicating that the mechanism of induced-fit donor binding was not preserved among the other fucosyltransferases.
Despite the similarities in GT-B fold structure and sugar donor-binding interactions, no structural similarity was observed for the loops involved in acceptor recognition (Fig. S3). POFUT1 and POFUT2 employ deep clefts between the two Rossmann-folds for recognition of their respective protein substrate domains, but use completely different sets of loop interactions (38, 39). AtFUT1 (30) and FUT8 employ a more flattened face of the enzyme surface for acceptor interactions, but also use completely different loop structures and an additional SH3 domain for FUT8.
Catalytic mechanism and evolution of modular substrate recognition by glycosyltransferases
The use of unique acceptor-binding loop insertions in GT-B fold fucosyltransferases is highly reminiscent of hypervariable substrate-binding loops in the evolution of GT-A fold glycosyltransferases (32, 102, 103). In contrast to the two Rossmann domains in GT-B fold enzymes, GT-A fold glycosyltransferases have a single, conserved Rossmann-fold core and employ proximal residues for donor interactions, whereas more divergent extended loop insertions are free to rapidly evolve and diversify novel acceptor specificities (32, 102, 103). This rapid evolution of the loop insertions is constrained by the necessity of positioning the acceptor nucleophile for attack and access to a catalytic base for nucleophile activation relative to the C1 of the sugar donor. GT-A fold inverting glycosyltransferases have highly conserved positions for the catalytic base, hydroxyl nucleophile, and generally use a conserved DXD motif for metal binding and sugar nucleotide positioning (32). For GT-A fold retaining enzymes the position for the hydroxyl nucleophile is shifted to no longer use of the enzyme-associated catalytic base, and instead a donor substrate-assisted SN1 catalytic mechanism is employed (32).
The present studies on the GT-B fold fucosyltransferases indicate an analogous use of Rossmann-fold core structures, conserved proximal residues for donor interactions, and highly divergent loop regions for distinctive acceptor recognition. Surprisingly, whereas the fucosyltransferases all have inverting catalytic mechanisms, their strategies for use of a catalytic base are quite distinct. AtFUT1 has no discernable catalytic base and water-mediated proton shuttle mechanism has been invoked (30). For POFUT1 the donor β-phosphate oxygen has been proposed as the catalytic base with the possible involvement of a water-mediated proton shuttle (20, 33, 38). FUT8 and POFUT2 (40) both have clearly identified catalytic bases, but they originate from different regions of their protein structures and act from opposite faces in deprotonation of their respective nucleophilic hydroxyls. Thus, despite the highly similar core structures, positions of donor sugars and acceptor nucleophiles among the fucosyltransferases, their respective catalytic base mechanisms are entirely different.
Although a general understanding of GT-A fold glycosyltransferase evolution is emerging (32, 102, 103), a parallel understanding of GT-B fold enzymes has lagged behind largely because of their more complex domain architectures and fewer determined protein structures in complex with substrate analogs. However, evolutionary parallels between the two protein-fold classes are now evident and further insights into selective substrate recognition among the broader collection of GT-B glycosyltransferases will expand our understanding of how diverse glycan structures are elaborated in biological systems.
Experimental procedures
Cloning, expression, fusion tag cleavage, and purification of FUT8 in HEK293F cells
The expression construct encoding the catalytic domain of human FUT8 (α1,6-fucosyltransferase, Uniprot Q9BYC5, residues 41-575) was generated by amplification of a Mammalian Gene Collection clone and transferring it into a pDONR221 vector through Gateway recombination (83). The PCR product, containing the Gateway att1 recombination sites, was subcloned into a pGEN2-DEST mammalian expression vector that employs a cytomegalovirus promoter and encodes an NH2-terminal sequence followed by an His8 tag, an AviTag recognition site, “superfolder” GFP, the 7-amino acid recognition sequence of TEV protease (83), and the coding region of human FUT8. The protein was expressed through transient transfection in either WT HEK293F (Freestyle 293F, ThermoFisher Scientific) cells or mutant HEK293S (GnTl−) cells (ATCC, catalog number CRL-3022). Briefly, cells were maintained in suspension culture at 1-3 × 106 cells/ml in a humidified CO2 shaker incubator (37 °C, 150 rpm). Transient transfection was performed at a cell density of 3-3.5 × 106 cells/ml in media containing 9 parts of FreestyleTM 293 Expression Medium (Life Technologies) and 1 part of Ex-Cell 293 serum-free medium (Sigma) using polyethyleneimine (linear 25 kDa PEI, Polysciences, Inc., PA) at a concentration of 5 μg/ml and a plasmid DNA concentration of 4 μg/ml. At 24 h post-transfection the suspension cultures were diluted 1:1 with culture media containing valproic acid (2.2 mm final concentration, Sigma). Cultures containing the secreted GFP-tagged protein were harvested 6 days post-transfection. For evaluation of enzyme kinetics, alanine mutants were generated using Q5 site-directed mutagenesis kit (New England Biolabs) and sequences were confirmed for the desired mutations. Both WT and mutant forms were expressed in the pGEN2-DEST vector and expressed as a soluble secreted form.
The WT and mutant forms of the protein were purified as described previously (Table S4) (83, 86). The harvested supernatant was filtered through 0.8-μm filter membrane to remove particulate matter and then loaded onto nickel-nitrilotriacetic acid resin (Qiagen) pre-equilibrated with 20 mm HEPES, pH 7.4, 300 mm NaCl, 20 mm imidazole, and 5% glycerol. After washing steps, the GFP-FUT8 fusion protein was eluted using 300 mm imidazole and further pooled and concentrated for determination of enzyme kinetics. Removal of the GFP tag was performed by treating the concentrated protein with TEV protease at a ratio of 1:5 relative to the fusion protein and incubating the sample for 16 h at 4 °C. The sample was then loaded to a nickel-nitrilotriacetic acid resin to remove the cleaved GFP and His-tagged TEV protease leading to a purified untagged protein in the flow-through fraction. For structural studies, the protein was further purified using a Superdex 75 column (GE Healthcare) pre-equilibrated with 20 mm HEPES, pH 7.4, 150 mm NaCl, and 60 mm imidazole and the peak fractions were collected.
SEC-MALS
The oligomerization of FUT8 was analyzed by injecting the enzyme (20 μl, 1 mg/ml) on an analytical grade Superdex 75 column pre-equilibrated with 25 mm HEPES, pH 7.4, 150 mm NaCl, 0.02% NaN3. Light scattering and deferential refractive index were measured using a MiniDAWN TREOS detector (Wyatt Tech.) and Optilab rEx detector (Wyatt Tech.) respectively. Data were analyzed using the ASTRA 6.0 software (Wyatt Tech.)
Alkaline phosphatase treatment of GDP-fucose
For enzyme kinetics, GDP-fucose (Carbosynth) was used as donor at a final concentration of 0.2 mm. Prior to performing the assay, GDP-fucose was treated with calf intestinal alkaline phosphatase (CIAP, Promega) to remove free GDP. Briefly, a reaction mix of 3 units/µl of CIAP/50 µl of 50 mm GDP-fucose in 1× CIAP buffer (5 µl) was prepared in a microcentrifuge tube. Multiple doses of CIAP (3 units/µl) were added every 2 h and incubated for a total of 6 h at 37 °C with gentle shaking (∼100-150 rpm). After 6 h, the concentration of GDP-fucose in the reaction volume was adjusted to 20 mm. The entire reaction mix was filtered through a Microcon 10-kDa centrifugal filter unit (Millipore) with Ultracel-10 membrane by centrifuging at 14,000 × g for 30 min to remove CIAP. The filtrate was recovered and incubated with another 3 units of CIAP for 16 h at 37 °C with gentle shaking (∼100-150 rpm). Finally, the reaction mix was further filtered through a Microcon 10-kDa centrifugal filter unit as mentioned above. The filtrate was stored at −20 °C and used as the stock solution of GDP-fucose. The CIAP-treated GDP-fucose gave lower background and improved signal to noise ratio in enzyme kinetic assays compared with untreated GDP-fucose.
FUT8 kinetic analysis
Enzyme kinetics for the WT and mutant form of the enzyme were determined using the GDP-GloTM Glycosyltransferase assay (Promega) that detects GDP as a released product of the glycosyltransferase reaction. Assays were performed in a 10-μl reaction volume consisting of a universal buffer (200 mm each of Tris, MES, MOPS, pH 7.5), 0.2 mm GDP-fucose (CIAP-treated, see above) in reactions containing varied concentrations of the respective glycan substrates: A1-Asn, A2-Asn, A3-Asn, A3'-Asn, A4-Asn, G1-Asn, NM5N2-Asn, M3N2-Asn, and M5N2-Asn-Fmoc. Reactions were carried out using purified GFP-fusion enzyme (WT or alanine mutants) at 37 °C for 30 min. For acceptor titration, assays were performed using the desired substrate at a concentration range of 0-1 mm with 0.2 mm GDP-fucose as final concentration of donor. Similarly, for donor titration, reactions were carried out using GDP-fucose (0-0.2 mm) with 0.5-1 mm A2-Asn as final concentration. Reactions were stopped using 5 μl of GDP-detection reagent at a 1:1 ratio in a white, opaque 384-well–plate and incubated in dark for 1 h at room temperature. The released GDP product was detected based on luminescence using a GloMax Multidetection plate reader (Promega). The steady state parameters of Km, kcat, and Vmax values were calculated using a GDP standard curve and nonlinear curve fitting in GraphPad Prism 6 software.
Synthesis of oligosaccharide acceptors
The glycan acceptors (A1-Asn, G1-Asn, A2-Asn, A3-Asn, A3'-Asn, and A4-Asn) were synthesized employing established chemoenzymatic methods for N-glycan synthesis (104, 105). The human MGAT1, MGAT4, and MGAT5 glycosyltransferases used in the synthesis were expressed in the HEK293 cell expression system as previously described (83). Briefly, a sialylglycopeptide (Neu5Ac2Gal2GlcNAc2Man3GlcNAc2-peptide) isolated from hen egg yolk powder (106) was treated with Clostridium perfringens neuraminidase (New England Biolabs) to produce the G2-peptide, and further treated with Aspergillus niger β-gal (Megazyme) to generate the A2-peptide. The glycopeptide was then exhaustively treated with Pronase from Streptomyces griseus (Sigma-Aldrich) to trim the peptide to a single Asn residue and generate A2-Asn (GlcNAc2Man3GlcNAc2-Asn). Treatment of A2-Asn with human MGAT4 or MGAT5 in the presence of UDP-GlcNAc produced the two acceptors, A3'-Asn and A3-Asn, respectively, with the composition of GlcNAc3Man3GlcNAc2-Asn. Treating the A2-Asn with both MGAT4 and MGAT5 provided acceptor A4-Asn (GlcNAc4Man3GlcNAc2-Asn).
Treatment of A2-Asn with Streptococcus pneumoniae N-acetylglucosaminidase (New England Biolabs) generated Man3-Asn, which was then treated with MGAT1 in the presence of UDP-GlcNAc to generate A1-Asn (GlcNAcMan3GlcNAc2-Asn). G1-Asn (GalGlcNAc2Man3GlcNAc2-Asn) was obtained by treating G2-peptide with Escherichia coli β-gal (Sigma-Aldrich), which preferentially cleaves the galactose on the MGAT1 branch (105, 107), followed by Pronase treatment.
NM5N2-Asn (GlcNAcMan5GlcNAc2-Asn) and Man5N2-Asn-Fmoc (Man5GlcNAc2-Asn-Fmoc) were prepared from Man9GlcNAc2-Asn isolated from soybean agglutinin (108). The crude soybean agglutinin isolated from soybean flour was digested with Pronase (Sigma Aldrich) and purified to afford Man9GlcNAc2-Asn, which was subsequently treated with Bacteriodes thetaiotaomicron α1,2-mannosidase to provide Man5N2-Asn. Installation of the Fmoc group produced Man5N2-Asn-Fmoc, which was subsequently treated with human MGAT1 in the presence of UDP-GlcNAc, followed by Fmoc removal to provide NMan5N2-Asn (GlcNAcMan5GlcNAc2-Asn).
Crystallization and data collection
The catalytic domain of FUT8 (10 mg/ml in a storage buffer containing 20 mm HEPES, pH 7.5, 50 mm NaCl, 300 mm betaine) was mixed with GDP or with GDP plus acceptor and screened for crystallization conditions using a TTP Labtech Mosquito Crystal robot and optimized using hanging drop vapor diffusion with 2-μl drops (1:1 protein:reservoir ratio). For the FUT8:GDP complex, crystals containing 5 mm GDP grew in less than 1 week from a reservoir of 1.4 m ammonium sulfate, 0.6 m l-proline, 0.1 m HEPES, pH 7.5. Crystals were transferred to the reservoir solution supplemented with 5 mm GDP, and 20% cryoprotectant (1:1:1 ethylene glycol, DMSO, and glycerol). FUT8:GDP crystallized in space group P65 and diffracted to 2.25 Å (Table S2).
For the FUT8:GDP:A2-Asn complex, 10 mg/ml of protein was prepared in storage buffer containing 5 mm GDP and 5 mm A2-Asn and crystals grew from a reservoir solution of 1 m lithium sulfate, 10 mm nickel chloride, and 0.1 m Tris, pH 8.5. Crystals were transferred to the reservoir solution supplemented with 5 mm GDP, 5 mm A2-Asn, and 20% cryoprotectant (1:1:1 ethylene glycol, DMSO, and glycerol). FUT8:GDP:A2-Asn crystallized in space group P6522 and diffracted to 2.4 Å (Table S2).
For the FUT8:GDP:A3'-Asn complex, 10 mg/ml of protein was prepared in storage buffer containing 10 mm GDP and 10 mm A3'-Asn and crystals grew from a reservoir solution of 1.2 m lithium sulfate, 12 mm nickel chloride, and 0.1 m Tris, pH 8.5. Crystals were transferred to the reservoir solution supplemented with 10 mm GDP, 10 mm A3'-Asn. A 40:60 mixture of paratone and paraffin was used as the cryoprotectant. FUT8:GDP:A3'-Asn crystallized in space group P6522 and diffracted to 3.3 Å (Table S2).
For the FUT8:GDP:A3-Asn complex, 10 mg/ml of protein was prepared in storage buffer containing 10 mm GDP and 10 mm A3-Asn and crystals grew overnight from a reservoir solution of 0.2 m l-proline, 10% PEG3350, and 0.1 m HEPES, pH 7.2. Crystals were transferred to the reservoir solution supplemented with 10 mm GDP, 10 mm A3'-Asn, and 18% cryoprotectant (1:1:1 ethylene glycol, DMSO, and glycerol) supplemented with 0.2 m sodium ascorbate. FUT8:GDP:A3-Asn crystallized in space group P65 and diffracted to 2.47 Å (Table S2).
For the FUT8:GDP:NM5M2-Asn complex, 10 mg/ml of protein in storage buffer containing 10 mm GDP and 10 mm NM5M2-Asn was prepared and crystals grew from a reservoir solution of 0.2 m ammonium chloride, 10% PEG3350. Crystals were transferred to the reservoir solution supplemented with 10 mm GDP, 10 mm NM5M2-Asn, and 18% cryoprotectant (1:1:1 ethylene glycol, DMSO, and glycerol). FUT8:GDP:NM5N2-Asn crystallized in space group P65 and diffracted to 3.2 Å (Table S2).
All cryo-protected crystals were flash cooled in liquid nitrogen and X-ray data were collected at the SER-CAT 22-BM beamline using a MAR-CCD detector (FUT8:GDP and FUT8:GDP:A2-Asn) or at the 22-ID beamline using an Eiger-16M detector (FUT8:GDP:A3-Asn, FUT8:GDP:A3'-Asn, and FUT8:GDP:NM5N2-Asn) at the Argonne National Laboratory. The data were processed using XDS (109). Five percent of the data were set aside for cross-validation.
Phasing and refinement
The crystal structures of FUT8:GDP and FUT8:GDP:A2-Asn were solved using molecular replacement with the apo-FUT8 structure (PDB 2DE0 (36)) as the search model. The NH2-terminus (residues 41-107) and the C-terminal residue Lys575 were disordered and left unmodeled. Successive rounds of automated refinement in Phenix (110) and iterative manual fitting using Coot (111) produced the final models (Table S2). The B-factors were refined using TLS (112).
The crystal structures of FUT8:GDP:A3'-Asn, FUT8:GDP:A3-Asn, and FUT8:NM5N2-Asn were solved using molecular replacement with FUT8:A2-Asn as the search model. In FUT8:GDP:A3'-Asn, the NH2 terminus (residues 41-107) and C terminus (residues 574-575) were disordered and left unmodeled. Similarly, in FUT8:GDP:A3-Asn, the NH2 terminus (residues 41-107 in chains A, B, C, D, and F, and 41-108 in chains E, G, and H) and C terminus (residue 575 in all chains) were left unmodeled. In FUT8:NM5N2-Asn, the NH2 terminus (residues 41-107) and C terminus (residue 575) were not modeled. All three structural models were refined like FUT8:GDP and their respective statistics are reported in Table S2. Data intensity statistics from Xtriage (110) for FUT8:GDP:A3-Asn and FUT8:GDP:NM5N2-Asn indicated the presence of merohedral twinning with the twin operator (h, -h-k, -l). The twin fraction ki refined to 0.25 for FUT8:GDP:A3-Asn and to 0.15 for the FUT8:GDP:NM5N2-Asn datasets, respectively.
During structural refinement, geometrical restraints were used to restrain monosaccharides with weak electron density in the lowest energy chair conformation (4C1) (110, 113). The linkage torsion angles for the glycosidic bonds (ϕ, ψ) in FUT8:A2-Asn, FUT8:A3'-Asn, FUT8:A3-Asn, and FUT8:NM5N2-Asn complexes were determined using the Carbohydrate Ramachandran Plot (CaRP) and fall into the energetically preferred areas of the GlyTorsion plot (114).
FUT8 loop residues Pro299, Pro305, and Pro358 form cis-peptide bonds, none of which are conserved in the NodZ:GDP complex (PDB code 3SIX (35)), the human POFUT2:GDP:Fuc complex (PDB 4AP6 (40)), the C. elegans POFUT2:GDP:thrombospondin repeat complex (PDB 5FOE (39)) or the AtFUT1:GDP:xylo-oligosaccahride complex (PDB 5KOR (29)). However, the mouse POFUT1:GDP-Fuc:EGF domain complex (PDB 5KY3 (38)) conserves one of the cis-peptide bonds (H357:P358) as R237:P238 in the sequence.
Structural analysis
Structure based sequence alignment was performed using default parameters on the PROMALS3D server (97) using structures of the human FUT8:GDP:A2-Asn complex, mouse POFUT1:GDP-Fuc:EGF domain complex (PDB 5KY3 (38)), the C. elegans POFUT2:GDP:thrombospondin repeat complex (PDB 5FOE (39)), the human POFUT2:GDP-Fuc complex (PDB 4AP6 (40)), the NodZ:GDP complex (PDB 3SIX (35)), and the AtFUT1:GDP:xylo-oligosaccahride complex (PDB 5KOR (29)). Alignment of the structural models was performed in Coot (111) followed by manual adjustment in PyMOL and displayed using PyMOL (Schrödinger). Structural elements identified in the PROMALS3D analysis were extracted from the full protein alignment for display as an overlay of the respective structures. The difference density maps (Fo − Fc) shown in the figures for the bound GDP and the A2 acceptor were calculated subsequent to structure solution and initial restrained refinement of the polypeptide, but prior to the modeling of the respective ligands. In all cases, the final refined coordinates were used to depict the ligand model. Polder maps (115) for the A3'-Asn, A3-Asn, and NM5N2-Asn acceptors were calculated following a modified procedure to reduce model bias (103). Briefly, prior to calculating the Polder map, the acceptor was omitted and the remaining model subjected to Cartesian simulated annealing at 5000 K. The acceptor coordinates were then used to calculate a reduced-bias Polder map.
Data availability
All atomic coordinates and structure factors were deposited in the RCSB Protein Data Bank (PDB) under accession codes 6X5H (FUT8:GDP), 6X5R (FUT8:GDP:A2), 6X5T (FUT8:GDP:A3), 6X5S (FUT8:GDP:A3') and 6X5U (FUT8:GDP:NM5N2).
Supplementary Material
Acknowledgments
We are grateful for the technical support of Rosemary Kim.
This article contains supporting information.
Author contributions—B. M. B., R. K., Z. A. W., and K. W. M. conceptualization; B. M. B., R. K., L. L., A. R., C. L., G. Z., G. P. B., J-Y. Y., L-X. W., G-J. B., Z. A. W., and K. W. M. resources; B. M. B., R. K., Z. A. W., and K. W. M. data curation; B. M. B., R. K., Z. A. W., and K. W. M. formal analysis; B. M. B., R. K., L-X. W., G-J. B., Z. A. W., and K. W. M. validation; B. M. B., R. K., Z. A. W., and K. W. M. investigation; B. M. B., R. K., L. L., A. R., C. L., G. Z., G. P. B., J-Y. Y., L-X. W., G-J. B., Z. A. W., and K. W. M. methodology; B. M. B., R. K., Z. A. W., and K. W. M. writing-original draft; B. M. B., R. K., Z. A. W., and K. W. M. writing-review and editing; L-X. W., G-J. B., Z. A. W., and K. W. M. supervision; Z. A. W. and K. W. M. funding acquisition; Z. A. W. and K. W. M. project administration; K. W. M. visualization.
Funding and additional information—This work was supported by National Institutes of Health Grants R01GM130915 (to K. W. M., and Z. A. W.), P41GM103390 (to K. W. M., and G. J. B.), P01GM107012 (to K. W. M., and G. J. B.), and R01GM080374 (to L-X. W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- EGF
- epidermal growth factor
- SEC-MALS
- size exclusion-multiangle light scattering
- POFUT
- protein-O-fucosyltransferase
- rmsd
- root mean square deviation
- SH3
- Src homology 3
- CIAP
- calf intestinal alkaline phosphatase
- TEV
- tobacco etch virus
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl.
References
- 1. Barb, A. W., Meng, L., Gao, Z., Johnson, R. W., Moremen, K. W., and Prestegard, J. H. (2012) NMR characterization of immunoglobulin G Fc glycan motion on enzymatic sialylation. Biochemistry 51, 4618–4626 10.1021/bi300319q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Varki, A. (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3, 97–130 10.1093/glycob/3.2.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Varki, A. (2017) Biological roles of glycans. Glycobiology 27, 3–49 10.1093/glycob/cww086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cummings, R. D., and Pierce, J. M. (2014) The challenge and promise of glycomics. Chem. Biol. 21, 1–15 10.1016/j.chembiol.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li, W., Yu, R., Ma, B., Yang, Y., Jiao, X., Liu, Y., Cao, H., Dong, W., Liu, L., Ma, K., Fukuda, T., Liu, Q., Ma, T., Wang, Z., Gu, J., et al. (2015) Core fucosylation of IgG B cell receptor is required for antigen recognition and antibody production. J. Immunol. 194, 2596–2606 10.4049/jimmunol.1402678 [DOI] [PubMed] [Google Scholar]
- 6. Shields, R. L., Lai, J., Keck, R., O'Connell, L. Y., Hong, K., Meng, Y. G., Weikert, S. H., and Presta, L. G. (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcγ RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 277, 26733–26740 10.1074/jbc.M202069200 [DOI] [PubMed] [Google Scholar]
- 7. Subedi, G. P., Falconer, D. J., and Barb, A. W. (2017) Carbohydrate-polypeptide contacts in the antibody receptor CD16A identified through solution NMR spectroscopy. Biochemistry 56, 3174–3177 10.1021/acs.biochem.7b00392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iijima, J., Kobayashi, S., Kitazume, S., Kizuka, Y., Fujinawa, R., Korekane, H., Shibata, T., Saitoh, S. I., Akashi-Takamura, S., Miyake, K., Miyoshi, E., and Taniguchi, N. (2017) Core fucose is critical for CD14-dependent Toll-like receptor 4 signaling. Glycobiology 27, 1006–1015 10.1093/glycob/cwx075 [DOI] [PubMed] [Google Scholar]
- 9. Li, W., Ishihara, K., Yokota, T., Nakagawa, T., Koyama, N., Jin, J., Mizuno-Horikawa, Y., Wang, X., Miyoshi, E., Taniguchi, N., and Kondo, A. (2008) Reduced alpha4beta1 integrin/VCAM-1 interactions lead to impaired pre-B cell repopulation in alpha1,6-fucosyltransferase deficient mice. Glycobiology 18, 114–124 10.1093/glycob/cwm107 [DOI] [PubMed] [Google Scholar]
- 10. Liang, W., Mao, S., Sun, S., Li, M., Li, Z., Yu, R., Ma, T., Gu, J., Zhang, J., Taniguchi, N., and Li, W. (2018) Core fucosylation of the T cell receptor is required for T cell activation. Front. Immunol. 9, 78 10.3389/fimmu.2018.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matsumoto, K., Yokote, H., Arao, T., Maegawa, M., Tanaka, K., Fujita, Y., Shimizu, C., Hanafusa, T., Fujiwara, Y., and Nishio, K. (2008) N-Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 99, 1611–1617 10.1111/j.1349-7006.2008.00847.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakayama, K., Wakamatsu, K., Fujii, H., Shinzaki, S., Takamatsu, S., Kitazume, S., Kamada, Y., Takehara, T., Taniguchi, N., and Miyoshi, E. (2019) Core fucose is essential glycosylation for CD14-dependent Toll-like receptor 4 and Toll-like receptor 2 signalling in macrophages. J. Biochem. 165, 227–237 10.1093/jb/mvy098 [DOI] [PubMed] [Google Scholar]
- 13. Pinho, S. S., Seruca, R., Gärtner, F., Yamaguchi, Y., Gu, J., Taniguchi, N., and Reis, C. A. (2011) Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol. Life Sci. 68, 1011–1020 10.1007/s00018-010-0595-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang, X., Gu, J., Ihara, H., Miyoshi, E., Honke, K., and Taniguchi, N. (2006) Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J. Biol. Chem. 281, 2572–2577 10.1074/jbc.M510893200 [DOI] [PubMed] [Google Scholar]
- 15. Zhao, Y., Itoh, S., Wang, X., Isaji, T., Miyoshi, E., Kariya, Y., Miyazaki, K., Kawasaki, N., Taniguchi, N., and Gu, J. (2006) Deletion of core fucosylation on alpha3beta1 integrin down-regulates its functions. J. Biol. Chem. 281, 38343–38350 10.1074/jbc.M608764200 [DOI] [PubMed] [Google Scholar]
- 16. Manabe, Y., Marchetti, R., Takakura, Y., Nagasaki, M., Nihei, W., Takebe, T., Tanaka, K., Kabayama, K., Chiodo, F., Hanashima, S., Kamada, Y., Miyoshi, E., Dulal, H. P., Yamaguchi, Y., Adachi, Y., et al. (2019) The core fucose on an IgG antibody is an endogenous ligand of Dectin-1. Angew. Chem. Int. Ed. Engl. 58, 18697–18702 10.1002/anie.201911875 [DOI] [PubMed] [Google Scholar]
- 17. Falconer, D. J., Subedi, G. P., Marcella, A. M., and Barb, A. W. (2018) Antibody fucosylation lowers the FcγRIIIa/CD16a affinity by limiting the conformations sampled by the N162-glycan. ACS Chem. Biol. 13, 2179–2189 10.1021/acschembio.8b00342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider, M., Al-Shareffi, E., and Haltiwanger, R. S. (2017) Biological functions of fucose in mammals. Glycobiology 27, 601–618 10.1093/glycob/cwx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lira-Navarrete, E., and Hurtado-Guerrero, R. (2018) A perspective on structural and mechanistic aspects of protein O-fucosylation. Acta Crystallogr. F Struct. Biol. Commun. 74, 443–450 10.1107/S2053230X18004788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scharberg, E. A., Olsen, C., and Bugert, P. (2016) The H blood group system. Immunohematology 32, 112–118 [PubMed] [Google Scholar]
- 22. Stowell, C. P., and Stowell, S. R. (2019) Biologic roles of the ABH and Lewis histo-blood group antigens part I: infection and immunity. Vox Sang. 114, 426–442 10.1111/vox.12787 [DOI] [PubMed] [Google Scholar]
- 23. Lowe, J. B. (1993) The blood group-specific human glycosyltransferases. Baillieres Clin. Haematol. 6, 465–492 10.1016/S0950-3536(05)80155-6 [DOI] [PubMed] [Google Scholar]
- 24. Costache, M., Cailleau, A., Fernandez-Mateos, P., Oriol, R., and Mollicone, R. (1997) Advances in molecular genetics of alpha-2- and alpha-3/4-fucosyltransferases. Transfus. Clin. Biol. 4, 367–382 10.1016/S1246-7820(97)80042-0 [DOI] [PubMed] [Google Scholar]
- 25. Ma, B., Simala-Grant, J. L., and Taylor, D. E. (2006) Fucosylation in prokaryotes and eukaryotes. Glycobiology 16, 158R–184R 10.1093/glycob/cwl040 [DOI] [PubMed] [Google Scholar]
- 26. Ihara, H., Tsukamoto, H., Gu, J., Miyoshi, E., Taniguchi, N., and Ikeda, Y. (2014) Fucosyltransferase 8: GDP-fucose N-glycan core α6-fucosyltransferase (FUT8). in Handbook of Glycosyltransferases and Related Genes (Taniguchi, N., Honke, K., Fukuda, M., Narimatsu, H., Yamaguchi, Y., and Angata, T., eds) 2nd Ed., pp. 581–596, Springer, Tokyo, Japan [Google Scholar]
- 27. Kizuka, Y., and Taniguchi, N. (2016) Enzymes for N-glycan branching and their genetic and nongenetic regulation in cancer. Biomolecules 6, 25 10.3390/biom6020025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagae, M., Yamaguchi, Y., Taniguchi, N., and Kizuka, Y. (2020) 3D structure and function of glycosyltransferases involved in N-glycan maturation. Int. J. Mol. Sci. 21, 437 10.3390/ijms21020437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rocha, J., Ciceron, F., de Sanctis, D., Lelimousin, M., Chazalet, V., Lerouxel, O., and Breton, C. (2016) Structure of Arabidopsis thaliana FUT1 reveals a variant of the GT-B class fold and provides insight into Xyloglucan fucosylation. Plant Cell 28, 2352–2364 10.1105/tpc.16.00519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urbanowicz, B. R., Bharadwaj, V. S., Alahuhta, M., Peña, M. J., Lunin, V. V., Bomble, Y. J., Wang, S., Yang, J. Y., Tuomivaara, S. T., Himmel, M. E., Moremen, K. W., York, W. S., and Crowley, M. F. (2017) Structural, mutagenic and in silico studies of xyloglucan fucosylation in Arabidopsis thaliana suggest a water-mediated mechanism. Plant J. 91, 931–949 10.1111/tpj.13628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Der Wel, H., Fisher, S. Z., and West, C. M. (2002) A bifunctional diglycosyltransferase forms the Fucα1,2Galβ1,3-disaccharide on Skp1 in the cytoplasm of dictyostelium. J. Biol. Chem. 277, 46527–46534 10.1074/jbc.M208824200 [DOI] [PubMed] [Google Scholar]
- 32. Moremen, K. W., and Haltiwanger, R. S. (2019) Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 15, 853–864 10.1038/s41589-019-0350-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lira-Navarrete, E., Valero-González, J., Villanueva, R., Martínez-Júlvez, M., Tejero, T., Merino, P., Panjikar, S., and Hurtado-Guerrero, R. (2011) Structural insights into the mechanism of protein O-fucosylation. PLoS ONE 6, e25365 10.1371/journal.pone.0025365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun, H. Y., Lin, S. W., Ko, T. P., Pan, J. F., Liu, C. L., Lin, C. N., Wang, A. H., and Lin, C. H. (2007) Structure and mechanism of Helicobacter pylori fucosyltransferase: a basis for lipopolysaccharide variation and inhibitor design. J. Biol. Chem. 282, 9973–9982 10.1074/jbc.M610285200 [DOI] [PubMed] [Google Scholar]
- 35. Brzezinski, K., Dauter, Z., and Jaskolski, M. (2012) Structures of NodZ α1,6-fucosyltransferase in complex with GDP and GDP-fucose. Acta Crystallogr. D Biol. Crystallogr. 68, 160–168 10.1107/S0907444911053157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ihara, H., Ikeda, Y., Toma, S., Wang, X., Suzuki, T., Gu, J., Miyoshi, E., Tsukihara, T., Honke, K., Matsumoto, A., Nakagawa, A., and Taniguchi, N. (2007) Crystal structure of mammalian alpha1,6-fucosyltransferase, FUT8. Glycobiology 17, 455–466 10.1093/glycob/cwl079 [DOI] [PubMed] [Google Scholar]
- 37. McMillan, B. J., Zimmerman, B., Egan, E. D., Lofgren, M., Xu, X., Hesser, A., and Blacklow, S. C. (2017) Structure of human POFUT1, its requirement in ligand-independent oncogenic Notch signaling, and functional effects of Dowling-Degos mutations. Glycobiology 27, 777–786 10.1093/glycob/cwx020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li, Z., Han, K., Pak, J. E., Satkunarajah, M., Zhou, D., and Rini, J. M. (2017) Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol. 13, 757–763 10.1038/nchembio.2381 [DOI] [PubMed] [Google Scholar]
- 39. Valero-Gonzalez, J., Leonhard-Melief, C., Lira-Navarrete, E., Jimenez-Oses, G., Hernandez-Ruiz, C., Pallares, M. C., Yruela, I., Vasudevan, D., Lostao, A., Corzana, F., Takeuchi, H., Haltiwanger, R. S., and Hurtado-Guerrero, R. (2016) A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 12, 240–246 10.1038/nchembio.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen, C. I., Keusch, J. J., Klein, D., Hess, D., Hofsteenge, J., and Gut, H. (2012) Structure of human POFUT2: insights into thrombospondin type 1 repeat fold and O-fucosylation. EMBO J. 31, 3183–3197 10.1038/emboj.2012.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. García-García, A., Ceballos-Laita, L., Serna, S., Artschwager, R., Reichardt, N. C., Corzana, F., and Hurtado-Guerrero, R. (2020) Structural basis for substrate specificity and catalysis of alpha1,6-fucosyltransferase. Nat. Commun. 11, 973 10.1038/s41467-020-14794-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Järva, M. A., Dramicanin, M., Lingford, J. P., Mao, R., John, A., Jarman, K. E., Grinter, R., and Goddard-Borger, E. D. (2020) Structural basis of substrate recognition and catalysis by fucosyltransferase 8. J. Biol. Chem. 295, 6677–6688 10.1074/jbc.RA120.013291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lairson, L. L., Henrissat, B., Davies, G. J., and Withers, S. G. (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 10.1146/annurev.biochem.76.061005.092322 [DOI] [PubMed] [Google Scholar]
- 44. Wang, X., Inoue, S., Gu, J., Miyoshi, E., Noda, K., Li, W., Mizuno-Horikawa, Y., Nakano, M., Asahi, M., Takahashi, M., Uozumi, N., Ihara, S., Lee, S. H., Ikeda, Y., Yamaguchi, Y., et al. (2005) Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 102, 15791–15796 10.1073/pnas.0507375102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang, X., Gu, J., Miyoshi, E., Honke, K., and Taniguchi, N. (2006) Phenotype changes of Fut8 knockout mouse: core fucosylation is crucial for the function of growth factor receptor(s). Methods Enzymol. 417, 11–22 10.1016/S0076-6879(06)17002-0 [DOI] [PubMed] [Google Scholar]
- 46. Fujii, H., Shinzaki, S., Iijima, H., Wakamatsu, K., Iwamoto, C., Sobajima, T., Kuwahara, R., Hiyama, S., Hayashi, Y., Takamatsu, S., Uozumi, N., Kamada, Y., Tsujii, M., Taniguchi, N., Takehara, T., et al. (2016) Core fucosylation on T cells, required for activation of T-cell receptor signaling and induction of colitis in mice, is increased in patients with inflammatory bowel disease. Gastroenterology 150, 1620–1632 10.1053/j.gastro.2016.03.002 [DOI] [PubMed] [Google Scholar]
- 47. Gu, W., Fukuda, T., Isaji, T., Hang, Q., Lee, H. H., Sakai, S., Morise, J., Mitoma, J., Higashi, H., Taniguchi, N., Yawo, H., Oka, S., and Gu, J. (2015) Loss of α1,6-fucosyltransferase decreases hippocampal long term potentiation: implications for core fucosylation in the regulation of AMPA receptor heteromerization and cellular signaling. J. Biol. Chem. 290, 17566–17575 10.1074/jbc.M114.579938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fukuda, T., Hashimoto, H., Okayasu, N., Kameyama, A., Onogi, H., Nakagawasai, O., Nakazawa, T., Kurosawa, T., Hao, Y., Isaji, T., Tadano, T., Narimatsu, H., Taniguchi, N., and Gu, J. (2011) α1,6-Fucosyltransferase-deficient mice exhibit multiple behavioral abnormalities associated with a schizophrenia-like phenotype: importance of the balance between the dopamine and serotonin systems. J. Biol. Chem. 286, 18434–18443 10.1074/jbc.M110.172536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu, X., Zhang, D., Shoji, H., Duan, C., Zhang, G., Isaji, T., Wang, Y., Fukuda, T., and Gu, J. (2019) Deficiency of α1,6-fucosyltransferase promotes neuroinflammation by increasing the sensitivity of glial cells to inflammatory mediators. Biochim. Biophys. Acta 1863, 598–608 10.1016/j.bbagen.2018.12.008 [DOI] [PubMed] [Google Scholar]
- 50. Ng, B. G., Dastsooz, H., Silawi, M., Habibzadeh, P., Jahan, S. B., Fard, M. A. F., Halliday, B. J., Raymond, K., Ruzhnikov, M. R. Z., Tabatabaei, Z., Taghipour-Sheshdeh, A., Brimble, E., Robertson, S. P., Faghihi, M. A., and Freeze, H. H. (2020) Expanding the molecular and clinical phenotypes of FUT8-CDG. J. Inherit. Metab. Dis. 43, 871–879 10.1002/jimd.12221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ng, B. G., Xu, G., Chandy, N., Steyermark, J., Shinde, D. N., Radtke, K., Raymond, K., Lebrilla, C. B., AlAsmari, A., Suchy, S. F., Powis, Z., Faqeih, E. A., Berry, S. A., Kronn, D. F., and Freeze, H. H. (2018) Biallelic mutations in FUT8 cause a congenital disorder of glycosylation with defective fucosylation. Am. J. Hum. Genet. 102, 188–195 10.1016/j.ajhg.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen, C. Y., Jan, Y. H., Juan, Y. H., Yang, C. J., Huang, M. S., Yu, C. J., Yang, P. C., Hsiao, M., Hsu, T. L., and Wong, C. H. (2013) Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 630–635 10.1073/pnas.1220425110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Höti, N., Yang, S., Hu, Y., Shah, P., Haffner, M. C., and Zhang, H. (2018) Overexpression of α(1,6)-fucosyltransferase in the development of castration-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 21, 137–146 10.1038/s41391-017-0016-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hutchinson, W. L., Du, M. Q., Johnson, P. J., and Williams, R. (1991) Fucosyltransferases: differential plasma and tissue alterations in hepatocellular carcinoma and cirrhosis. Hepatology 13, 683–688 10.1002/hep.1840130412 [DOI] [PubMed] [Google Scholar]
- 55. Ito, Y., Miyauchi, A., Yoshida, H., Uruno, T., Nakano, K., Takamura, Y., Miya, A., Kobayashi, K., Yokozawa, T., Matsuzuka, F., Taniguchi, N., Matsuura, N., Kuma, K., and Miyoshi, E. (2003) Expression of α1,6-fucosyltransferase (FUT8) in papillary carcinoma of the thyroid: its linkage to biological aggressiveness and anaplastic transformation. Cancer Lett. 200, 167–172 10.1016/S0304-3835(03)00383-5 [DOI] [PubMed] [Google Scholar]
- 56. Liu, Y. C., Yen, H. Y., Chen, C. Y., Chen, C. H., Cheng, P. F., Juan, Y. H., Chen, C. H., Khoo, K. H., Yu, C. J., Yang, P. C., Hsu, T. L., and Wong, C. H. (2011) Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 11332–11337 10.1073/pnas.1107385108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Muinelo-Romay, L., Vazquez-Martín, C., Villar-Portela, S., Cuevas, E., Gil-Martín, E., and Fernández-Briera, A. (2008) Expression and enzyme activity of α(1,6)fucosyltransferase in human colorectal cancer. Int. J. Cancer 123, 641–646 10.1002/ijc.23521 [DOI] [PubMed] [Google Scholar]
- 58. Shao, K., Chen, Z. Y., Gautam, S., Deng, N. H., Zhou, Y., and Wu, X. Z. (2016) Posttranslational modification of E-cadherin by core fucosylation regulates Src activation and induces epithelial-mesenchymal transition-like process in lung cancer cells. Glycobiology 26, 142–154 10.1093/glycob/cwv089 [DOI] [PubMed] [Google Scholar]
- 59. Tada, K., Ohta, M., Hidano, S., Watanabe, K., Hirashita, T., Oshima, Y., Fujnaga, A., Nakanuma, H., Masuda, T., Endo, Y., Takeuchi, Y., Iwashita, Y., Kobayashi, T., and Inomata, M. (2020) Fucosyltransferase 8 plays a crucial role in the invasion and metastasis of pancreatic ductal adenocarcinoma. Surg. Today 50, 767–777 10.1007/s00595-019-01953-z [DOI] [PubMed] [Google Scholar]
- 60. Takahashi, T., Ikeda, Y., Miyoshi, E., Yaginuma, Y., Ishikawa, M., and Taniguchi, N. (2000) α1,6-Fucosyltransferase is highly and specifically expressed in human ovarian serous adenocarcinomas. Int. J. Cancer 88, 914–919 [DOI] [PubMed] [Google Scholar]
- 61. Tu, C. F., Wu, M. Y., Lin, Y. C., Kannagi, R., and Yang, R. B. (2017) FUT8 promotes breast cancer cell invasiveness by remodeling TGF-β receptor core fucosylation. Breast Cancer Res. 19, 111 10.1186/s13058-017-0904-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang, X., Chen, J., Li, Q. K., Peskoe, S. B., Zhang, B., Choi, C., Platz, E. A., and Zhang, H. (2014) Overexpression of α(1,6)-fucosyltransferase associated with aggressive prostate cancer. Glycobiology 24, 935–944 10.1093/glycob/cwu051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang, Y., Fukuda, T., Isaji, T., Lu, J., Im, S., Hang, Q., Gu, W., Hou, S., Ohtsubo, K., and Gu, J. (2015) Loss of α1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 29, 3217–3227 10.1096/fj.15-270710 [DOI] [PubMed] [Google Scholar]
- 64. Aoyagi, Y., Isokawa, O., Suda, T., Watanabe, M., Suzuki, Y., and Asakura, H. (1998) The fucosylation index of α-fetoprotein as a possible prognostic indicator for patients with hepatocellular carcinoma. Cancer 83, 2076–2082 [DOI] [PubMed] [Google Scholar]
- 65. Flores, A., and Marrero, J. A. (2014) Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clin. Med. Insights Oncol. 8, 71–76 10.4137/CMO.S9926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Saldova, R., Fan, Y., Fitzpatrick, J. M., Watson, R. W., and Rudd, P. M. (2011) Core fucosylation and α-3 sialylation in serum N-glycome is significantly increased in prostate cancer comparing to benign prostate hyperplasia. Glycobiology 21, 195–205 10.1093/glycob/cwq147 [DOI] [PubMed] [Google Scholar]
- 67. Luo, C., Chen, S., Xu, N., Wang, C., Sai, W. B., Zhao, W., Li, Y. C., Hu, X. J., Tian, H., Gao, X. D., and Yao, W. B. (2017) Glycoengineering of pertuzumab and its impact on the pharmacokinetic/pharmacodynamic properties. Sci. Rep. 7, 46347 10.1038/srep46347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Satoh, M., Iida, S., and Shitara, K. (2006) Non-fucosylated therapeutic antibodies as next-generation therapeutic antibodies. Expert Opin. Biol. Ther. 6, 1161–1173 10.1517/14712598.6.11.1161 [DOI] [PubMed] [Google Scholar]
- 69. Bosmann, H. B., Hagopian, A., and Eylar, E. H. (1968) Glycoprotein biosynthesis: the characterization of two glycoprotein: fucosyl transferases in HeLa cells. Arch Biochem. Biophys. 128, 470–481 10.1016/0003-9861(68)90053-2 [DOI] [PubMed] [Google Scholar]
- 70. Ihara, H., Hanashima, S., Okada, T., Ito, R., Yamaguchi, Y., Taniguchi, N., and Ikeda, Y. (2010) Fucosylation of chitooligosaccharides by human α1,6-fucosyltransferase requires a nonreducing terminal chitotriose unit as a minimal structure. Glycobiology 20, 1021–1033 10.1093/glycob/cwq064 [DOI] [PubMed] [Google Scholar]
- 71. Ihara, H., Ikeda, Y., and Taniguchi, N. (2006) Reaction mechanism and substrate specificity for nucleotide sugar of mammalian α1,6-fucosyltransferase: a large-scale preparation and characterization of recombinant human FUT8. Glycobiology 16, 333–342 10.1093/glycob/cwj068 [DOI] [PubMed] [Google Scholar]
- 72. Kamińska, J., Glick, M. C., and Kościelak, J. (1998) Purification and characterization of GDP-L-Fuc:N-acetyl beta-d-glucosaminide alpha1→6-fucosyltransferase from human blood platelets. Glycoconj. J. 15, 783–788 [DOI] [PubMed] [Google Scholar]
- 73. Longmore, G. D., and Schachter, H. (1982) Product-identification and substrate-specificity studies of the GDP-l-fucose:2-acetamido-2-deoxy-beta-d-glucoside (FUC goes to Asn-linked GlcNAc) 6-alpha-l-fucosyltransferase in a Golgi-rich fraction from porcine liver. Carbohydr. Res. 100, 365–392 10.1016/S0008-6215(00)81049-6 [DOI] [PubMed] [Google Scholar]
- 74. Shao, M. C., Sokolik, C. W., and Wold, F. (1994) Specificity studies of the GDP-(l)-fucose: 2-acetamido-2-deoxy-beta-d-glucoside (Fuc→Asn-linked GlcNAc) 6-alpha-l-fucosyltransferase from rat-liver Golgi membranes. Carbohydr. Res. 251, 163–173 10.1016/0008-6215(94)84283-3 [DOI] [PubMed] [Google Scholar]
- 75. Voynow, J. A., Kaiser, R. S., Scanlin, T. F., and Glick, M. C. (1991) Purification and characterization of GDP-l-fucose-N-acetyl β-d-glucosaminide α1→6-fucosyltransferase from cultured human skin fibroblasts: requirement of a specific biantennary oligosaccharide as substrate. J. Biol. Chem. 266, 21572–21577 [PubMed] [Google Scholar]
- 76. Wilson, J. R., Williams, D., and Schachter, H. (1976) The control of glycoprotein synthesis: N-acetylglucosamine linkage to a mannose residue as a signal for the attachment of l-fucose to the asparagine-linked N-acetylglucosamine residue of glycopeptide from alpha1-acid glycoprotein. Biochem. Biophys. Res. Commun. 72, 909–916 10.1016/S0006-291X(76)80218-5 [DOI] [PubMed] [Google Scholar]
- 77. Yang, Q., and Wang, L. X. (2016) Mammalian α-1,6-fucosyltransferase (FUT8) is the sole enzyme responsible for the N-acetylglucosaminyltransferase I-independent core fucosylation of high-mannose N-glycans. J. Biol. Chem. 291, 11064–11071 10.1074/jbc.M116.720789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yang, Q., Zhang, R., Cai, H., and Wang, L. X. (2017) Revisiting the substrate specificity of mammalian α1,6-fucosyltransferase reveals that it catalyzes core fucosylation of N-glycans lacking β1,3-arm GlcNAc. J. Biol. Chem. 292, 14796–14803 10.1074/jbc.M117.804070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Calderon, A. D., Liu, Y., Li, X., Wang, X., Chen, X., Li, L., and Wang, P. G. (2016) Substrate specificity of FUT8 and chemoenzymatic synthesis of core-fucosylated asymmetric N-glycans. Org. Biomol. Chem. 14, 4027–4031 10.1039/C6OB00586A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li, L., Liu, Y., Ma, C., Qu, J., Calderon, A. D., Wu, B., Wei, N., Wang, X., Guo, Y., Xiao, Z., Song, J., Sugiarto, G., Li, Y., Yu, H., Chen, X., et al. (2015) Efficient chemoenzymatic synthesis of an N-glycan isomer library. Chem. Sci. 6, 5652–5661 10.1039/C5SC02025E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Serna, S., Yan, S., Martin-Lomas, M., Wilson, I. B., and Reichardt, N. C. (2011) Fucosyltransferases as synthetic tools: glycan array based substrate selection and core fucosylation of synthetic N-glycans. J. Am. Chem. Soc. 133, 16495–16502 10.1021/ja205392z [DOI] [PubMed] [Google Scholar]
- 82. Tseng, T. H., Lin, T. W., Chen, C. Y., Chen, C. H., Lin, J. L., Hsu, T. L., and Wong, C. H. (2017) Substrate preference and interplay of fucosyltransferase 8 and N-acetylglucosaminyltransferases. J. Am. Chem. Soc. 139, 9431–9434 10.1021/jacs.7b03729 [DOI] [PubMed] [Google Scholar]
- 83. Moremen, K. W., Ramiah, A., Stuart, M., Steel, J., Meng, L., Forouhar, F., Moniz, H. A., Gahlay, G., Gao, Z., Chapla, D., Wang, S., Yang, J. Y., Prabhakar, P. K., Johnson, R., Rosa, M. D., et al. (2018) Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 14, 156–162 10.1038/nchembio.2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yang, Q., An, Y., Zhu, S., Zhang, R., Loke, C. M., Cipollo, J. F., and Wang, L. X. (2017) Glycan remodeling of human erythropoietin (EPO) through combined mammalian cell engineering and chemoenzymatic transglycosylation. ACS Chem. Biol. 12, 1665–1673 10.1021/acschembio.7b00282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kadirvelraj, R., Yang, J. Y., Sanders, J. H., Liu, L., Ramiah, A., Prabhakar, P. K., Boons, G. J., Wood, Z. A., and Moremen, K. W. (2018) Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc. Natl. Acad. Sci. U.S.A. 115, 4637–4642 10.1073/pnas.1716988115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Meng, L., Forouhar, F., Thieker, D., Gao, Z., Ramiah, A., Moniz, H., Xiang, Y., Seetharaman, J., Milaninia, S., Su, M., Bridger, R., Veillon, L., Azadi, P., Kornhaber, G., Wells, L., et al. (2013) Enzymatic basis for N-glycan sialylation: structure of rat α2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 288, 34680–34698 10.1074/jbc.M113.519041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ihara, H., Okada, T., Taniguchi, N., and Ikeda, Y. (2020) Involvement of the alpha-helical and Src homology 3 domains in the molecular assembly and enzymatic activity of human α1,6-fucosyltransferase, FUT8. Biochim. Biophys. Acta 1864, 129596 10.1016/j.bbagen.2020.129596 [DOI] [PubMed] [Google Scholar]
- 88. Krissinel, E., and Henrick, K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 89. Pratap, J. V., Luisi, B. F., and Calladine, C. R. (2013) Geometric principles in the assembly of α-helical bundles. Philos. Trans. A Math. Phys. Eng. Sci. 371, 20120369 10.1098/rsta.2012.0369 [DOI] [PubMed] [Google Scholar]
- 90. Desrochers, G., Cappadocia, L., Lussier-Price, M., Ton, A. T., Ayoubi, R., Serohijos, A., Omichinski, J. G., and Angers, A. (2017) Molecular basis of interactions between SH3 domain-containing proteins and the proline-rich region of the ubiquitin ligase Itch. J. Biol. Chem. 292, 6325–6338 10.1074/jbc.M116.754440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Paulson, J. C., and Colley, K. J. (1989) Glycosyltransferases: structure, localization, and control of cell type-specific glycosylation. J. Biol. Chem. 264, 17615–17618 [PubMed] [Google Scholar]
- 92. Jones, D. T., and Cozzetto, D. (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31, 857–863 10.1093/bioinformatics/btu744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tomida, S., Takata, M., Hirata, T., Nagae, M., Nakano, M., and Kizuka, Y. (2020) The SH3 domain in the fucosyltransferase FUT8 controls FUT8 activity and localization and is essential for core fucosylation. J. Biol. Chem. 295, 7992–8004 10.1074/jbc.RA120.013079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Oriol, R., Mollicone, R., Cailleau, A., Balanzino, L., and Breton, C. (1999) Divergent evolution of fucosyltransferase genes from vertebrates, invertebrates, and bacteria. Glycobiology 9, 323–334 10.1093/glycob/9.4.323 [DOI] [PubMed] [Google Scholar]
- 95. Martinez-Duncker, I., Mollicone, R., Candelier, J. J., Breton, C., and Oriol, R. (2003) A new superfamily of protein-O-fucosyltransferases, alpha2-fucosyltransferases, and alpha6-fucosyltransferases: phylogeny and identification of conserved peptide motifs. Glycobiology 13, 1C–5C 10.1093/glycob/cwg113 [DOI] [PubMed] [Google Scholar]
- 96. Brzezinski, K., Stepkowski, T., Panjikar, S., Bujacz, G., and Jaskolski, M. (2007) High-resolution structure of NodZ fucosyltransferase involved in the biosynthesis of the nodulation factor. Acta Biochim. Pol. 54, 537–549 10.18388/abp.2007_3227 [DOI] [PubMed] [Google Scholar]
- 97. Pei, J., Kim, B. H., and Grishin, N. V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 10.1093/nar/gkn072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liu, D., Gao, Z., and Yue, L. (2019) Fucosyltransferase 8 deficiency suppresses breast cancer cell migration by interference of the FAK/integrin pathway. Cancer Biomark 25, 303–311 10.3233/CBM-190209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shinkawa, T., Nakamura, K., Yamane, N., Shoji-Hosaka, E., Kanda, Y., Sakurada, M., Uchida, K., Anazawa, H., Satoh, M., Yamasaki, M., Hanai, N., and Shitara, K. (2003) The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 278, 3466–3473 10.1074/jbc.M210665200 [DOI] [PubMed] [Google Scholar]
- 100. Crispin, M., Harvey, D. J., Chang, V. T., Yu, C., Aricescu, A. R., Jones, E. Y., Davis, S. J., Dwek, R. A., and Rudd, P. M. (2006) Inhibition of hybrid- and complex-type glycosylation reveals the presence of the GlcNAc transferase I-independent fucosylation pathway. Glycobiology 16, 748–756 10.1093/glycob/cwj119 [DOI] [PubMed] [Google Scholar]
- 101. Breton, C., Oriol, R., and Imberty, A. (1998) Conserved structural features in eukaryotic and prokaryotic fucosyltransferases. Glycobiology 8, 87–94 10.1093/glycob/8.1.87 [DOI] [PubMed] [Google Scholar]
- 102. Taujale, R., Venkat, A., Huang, L. C., Zhou, Z., Yeung, W., Rasheed, K. M., Li, S., Edison, A. S., Moremen, K. W., and Kannan, N. (2020) Deep evolutionary analysis reveals the design principles of fold A glycosyltransferases. Elife 9, e54532 10.7554/eLife.54532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kadirvelraj, R., Yang, J. Y., Sanders, J. H., Liu, L., Ramiah, A., Prabhakar, P. K., Boons, G. J., Wood, Z. A., and Moremen, K. W. (2018) Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc. Natl. Acad. Sci. U.S.A. 115, 4637–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li, T., Liu, L., Wei, N., Yang, J. Y., Chapla, D. G., Moremen, K. W., and Boons, G. J. (2019) An automated platform for the enzyme-mediated assembly of complex oligosaccharides. Nat. Chem. 11, 229–236 10.1038/s41557-019-0219-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu, L., Prudden, A. R., Capicciotti, C. J., Bosman, G. P., Yang, J. Y., Chapla, D. G., Moremen, K. W., and Boons, G. J. (2019) Streamlining the chemoenzymatic synthesis of complex N-glycans by a stop and go strategy. Nat. Chem. 11, 161–169 10.1038/s41557-018-0188-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Liu, L., Prudden, A. R., Bosman, G. P., and Boons, G. J. (2017) Improved isolation and characterization procedure of sialylglycopeptide from egg yolk powder. Carbohydr. Res. 452, 122–128 10.1016/j.carres.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. van den Eijnden, D. H., Blanken, W. M., and van Vliet, A. (1986) Branch specificity of beta-d-galactosidase from Escherichia coli. Carbohydr. Res. 151, 329–335 10.1016/S0008-6215(00)90352-5 [DOI] [PubMed] [Google Scholar]
- 108. Trastoy, B., Du, J. J., Klontz, E. H., Li, C., Cifuente, J. O., Wang, L. X., Sundberg, E. J., and Guerin, M. E. (2020) Structural basis of mammalian high-mannose N-glycan processing by human gut Bacteroides. Nat. Commun. 11, 899 10.1038/s41467-020-14754-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kabsch, W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Urzhumtsev, A., Afonine, P. V., and Adams, P. D. (2013) TLS from fundamentals to practice. Crystallogr. Rev. 19, 230–270 10.1080/0889311X.2013.835806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Agirre, J., Iglesias-Fernández, J., Rovira, C., Davies, G. J., Wilson, K. S., and Cowtan, K. D. (2015) Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 10.1038/nsmb.3115 [DOI] [PubMed] [Google Scholar]
- 114. Lutteke, T., Frank, M., and von der Lieth, C. W. (2004) Carbohydrate Structure Suite (CSS): analysis of carbohydrate 3D structures derived from the PDB. Nucleic Acids Res. 33, D242–D246 10.1093/nar/gki013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liebschner, D., Afonine, P. V., Moriarty, N. W., Poon, B. K., Sobolev, O. V., Terwilliger, T. C., and Adams, P. D. (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73, 148–157 10.1107/S2059798316018210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Neelamegham, S., Aoki-Kinoshita, K., Bolton, E., Frank, M., Lisacek, F., Lütteke, T., O'Boyle, N., Packer, N. H., Stanley, P., Toukach, P., Varki, A., and Woods, R. J., SNFG Discussion Group (2019) Updates to the symbol nomenclature for glycans guidelines. Glycobiology 29, 620–624 10.1093/glycob/cwz045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wallace, A. C., Laskowski, R. A., and Thornton, J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 10.1093/protein/8.2.127 [DOI] [PubMed] [Google Scholar]
- 118. Diederichs, K., and Karplus, P. A. (1997) Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 10.1038/nsb0497-269 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All atomic coordinates and structure factors were deposited in the RCSB Protein Data Bank (PDB) under accession codes 6X5H (FUT8:GDP), 6X5R (FUT8:GDP:A2), 6X5T (FUT8:GDP:A3), 6X5S (FUT8:GDP:A3') and 6X5U (FUT8:GDP:NM5N2).