

Abstract
DNA polymerases are today used throughout scientific research, biotechnology, and medicine, in part for their ability to interact with unnatural forms of DNA created by synthetic biologists. Here especially, natural DNA polymerases often do not have the “performance specifications” needed for transformative technologies. This creates a need for science-guided rational (or semi-rational) engineering to identify variants that replicate unnatural base pairs (UBPs), unnatural backbones, tags, or other evolutionarily novel features of unnatural DNA. In this review, we provide a brief overview of the chemistry and properties of replicative DNA polymerases and their evolved variants, focusing on the Klenow fragment of Taq DNA polymerase (Klentaq). We describe comparative structural, enzymatic, and molecular dynamics studies of WT and Klentaq variants, complexed with natural or noncanonical substrates. Combining these methods provides insight into how specific amino acid substitutions distant from the active site in a Klentaq DNA polymerase variant (ZP Klentaq) contribute to its ability to replicate UBPs with improved efficiency compared with Klentaq. This approach can therefore serve to guide any future rational engineering of replicative DNA polymerases.
Keywords: biotechnology, computer modeling, directed evolution, DNA polymerase, enzyme mechanism, enzyme structure, molecular dynamics, nucleoside/nucleotide analogue, protein-DNA interaction, synthetic biology, X-ray crystallography, expanded genetic alphabets
DNA polymerases (1), RNA polymerases (2), and reverse transcriptases (3) are enzymes central to terran biology and permit life to propagate and exploit its genetic inheritance. DNA polymerases are also workhorses in biotechnology (4, 5), where they often must manage DNA molecules having unusual structural features. For example, some WT bacterial polymerases can replicate DNA-containing UBPs that interact by steric complementarity without any interbase hydrogen bonding (6) (Fig. 1). This finding has been exploited to obtain engineered strains of Escherichia coli that can express proteins built from noncanonical amino acids with improved efficiency (15, 16). This capability was a driver of the recent $2.5 billion acquisition of Synthorx by Sanofi-Aventis.
Figure 1.

Chemical structures of nucleobase pairs. R indicates the point of covalent attachment to either deoxyribose or ribose in DNA or RNA, respectively. A, Watson–Crick nucleobase pairs (7). B, isosteric UBPs developed by Kool and co-workers (8). C, hydrophobic UBPs developed by Romesberg and co-workers (9, 10). D, hydrogen-bonding UBPs used in hachimoji DNA developed by Benner and co-workers (11, 12). E, hydrophobic UBPs developed by Hirao and co-workers (13, 14), where R1 can be a variety of substituents.
DNA polymerase variants are also playing key roles at other scientific frontiers. For example, NextGen sequencing (17) has transformed medicine (18), but only because it was enabled by the use of DNA polymerase variants capable of replicating “Watson–Crick” (WC) deoxynucleoside triphosphates (7) “tagged” with fluorescent markers and carrying blocking groups appended to their 3′-positions (19).
Expanded genetic alphabets and engineered DNA polymerases
These prior studies, however, only “scratch the surface” of the possible modified nucleotides that have scientific, biotechnological, and medical value. For example, DNA can have many more independently replicable groups than A, T, G, or C (Fig. 1). Scientifically, these groups can be varied to learn whether WC DNA represents the only molecular solution for the long-term storage and replication of biological information (11, 20–22). Alexander Rich was likely the first to recognize the importance of this question, suggesting in 1962 that the isocytidine:isoguanine (S:B) pair (Fig. 1) might be used as an additional information storing unit in DNA (23).
Further, many have asked whether the sugar and phosphate components of the DNA backbone might be modified. Synthetic biologists have therefore synthesized numerous such modified DNA molecules to delineate molecular changes that can (and cannot) be tolerated by duplex DNA (24–26).
The extent to which the core concept of WC pairing can be changed has also been explored by synthetic biologists. For example, the laboratories of Eric Kool (8), Floyd Romesberg (9, 10), and Ichiro Hirao (13, 14) have attempted to dispense with interbase hydrogen bonding (Fig. 1).
Developing a different approach, Steven Benner and co-workers (11, 12) synthesized UBPs that exploit orthogonal patterns of hydrogen bonding. This led to multiple discoveries about the molecular changes that can (and cannot) be tolerated in replicating units, especially how the fidelity of replication can be influenced by protonation, deprotonation, and multiple tautomeric forms of the nucleobases (27). These efforts led to the preparation of “hachimoji” DNA (28), an artificially expanded genetic system (AEGIS) in which A, G, T, and C are augmented by two unnatural pyrimidine analogs, pseudocytidine (or isocytidine) (S) and 6-amino-3-(2′-deoxyribo-furanosyl)-5-nitro-1H-pyridin-2-one (Z), and their size and hydrogen bond complementary partners, the purine analogs, isoguanine (B), and 2-amino-8-(1-β-d-2′-deoxyribofuranosyl)imidazo[1,2-a]-1,3,5-triazin-[8H]-4-one- (P) (27, 28) (Fig. 1). The additional P:Z and B:S base pairs form three interbase hydrogen bonds, as does the natural G:C. Work in this area has also been driven by commercial factors; the use of AEGIS UBPs as components of diagnostic tests has generated lifetime sales in excess of $1 billion (29).
Future disruptive technologies exploiting these and other expanded genetic alphabets will almost certainly require engineered DNA polymerase variants having expanded ranges of biophysical and catalytic properties. Unfortunately, and despite extensive research into the structure and mechanism of DNA polymerases from a wide range of organisms (30–36), rationally predicting how specific amino acid replacements in these enzymes will impact the incorporation of any particular kind of UBP into duplex DNA remains almost impossible. This reflects a lack of fundamental understanding about how multiple factors act synergistically to control UBP incorporation efficiency and fidelity (30, 37, 38). These factors include nucleobase pair complementarity, nucleobase tautomerism, variations in hy-drogen bond free energies, and the conformational dynamics of the polymerase itself as incorporation proceeds through the catalytic cycle. Directed evolution methods (39) are therefore viewed as the best approach to identifying DNA polymerase variants that can incorporate UBPs with high efficiency and fidelity (40–42).
These facts make it timely to review how X-ray crystallography, computational modeling, and directed evolution strategies can be combined to obtain DNA polymerases capable of replicating artificial DNA containing UBPs. Given the recent publication of a comprehensive discussion about the replication of UBP-containing DNA by unengineered WT polymerases (43), we draw on work from our laboratories, the only studies (to our knowledge) that employ a combined experimental and computational approach.
Generally speaking, enzymes that replicate DNA must manage three contradictory demands as they catalyze their critical reactions. First, this class of polymerases must be highly specific, making not much more than one mistake every billion turnovers (44). Mistakes mean somatic mutations potentially leading to cancer, germ line mutations that create inherited genetic disease, and, ultimately, an error catastrophe that leads to the death of the organism. Typical replicative polymerases achieve specificity at both the elongation step and a proofreading step, the latter in the form of 3′–5′ exonuclease domains (see below). Other replicative DNA polymerases, such as reverse transcriptases (45), lack proofreading and are therefore more error-prone. DNA polymerases that function primarily in DNA repair, including those important for genome stability and trans-lesion synthesis, also often have lower fidelity (46, 47).
Second, DNA polymerases must accept four different substrates: (i) template dG and dCTP, (ii) template dC and dGTP, (iii) template dA and dTTP, and (iv) template dT and dATP. Most other enzymes accept only one.
Finally, DNA polymerases must work rapidly. An E. coli cell replicates its entire genome by copying 4000 nucleotides each second. Even though many replication forks are used, this rate is as fast as that of many enzymes using only one substrate in pathways able to tolerate mistakes.
Managing these demands is especially difficult because the four substrates have few molecular features in common to present to the polymerase (Fig. 1). For example, the four nucleobases present different functionalities to the major groove, a methyl group for T, a hydrogen for C, two hydrogen bond acceptors for G, and one donor and one acceptor for A.
One feature that all four substrates have in common is their relative size, leading to the rule that large purines must pair with small pyrimidines. Further, the functional groups located on the sides of all four bases in the minor groove of the DNA duplex share a common feature: electron density presented by the exocyclic C=O groups of T and C and the N-3 nitrogen in the purine rings of A and G. Thus, the “minor groove scanning hypothesis” proposes that polymerases donate hydrogen bonds to this electron density in the template, primer, and incoming triphosphate as a way of enforcing Watson–Crick geometry, and hence fidelity, on base pair recognition (48).
These hydrogen bond contacts and size constraints are well-represented in over two dozen crystal structures solved to date (32, 43). The electronic constraint in the minor groove also appears to be important for nonstandard base pairing, even pairing that, as in the Romesberg pair (Fig. 1), involves no interbase hydrogen bonding (22). Moreover, the easiest AEGIS UBP to incorporate into DNA by polymerase-catalyzed reactions has been Z:P (Fig. 1), which is the only pair of those with shuffled hydrogen-bonding patterns for which both components present electron density to the minor groove (49).
Although the coupling reaction catalyzed by DNA polymerases seems superficially straightforward, this simplicity hides many details that remain controversial (30, 34, 50–52). DNA synthesis proceeds in the 5′ to 3′ direction in a primer-dependent reaction requiring Mg2+ as a cofactor. In the first step, the enzyme forms an initial binary complex with a DNA duplex composed of a template strand and a shorter, complementary primer strand possessing a 3′-hydroxyl group. dNTPs then bind to the binary complex to give a ternary (or “pre-incorporation”) complex (Fig. 2). Correctly matched dNTPs reside longer in the active site, allowing the enzyme to adopt a “closed” conformation in which the 3′-hydroxyl group in the primer can attack the α-phosphate of the bound dNTP (Fig. 2A). This reaction requires the presence of two (53, 56), or possibly three (30, 54, 55), Mg2+ ions within the active site of human polymerase β (Fig. 2C) or Klentaq (Fig. 2D). Subsequent release of PPi generates a binary (or “post-incorporation”) complex in which the primer has been elongated by one nucleobase, which then serves as the substrate for the next templated addition of dNTP. These steps are repeated until no unpaired nucleobases remain in the template strand.
Figure 2.

A, kinetic scheme for the incorporation of matched dNTPs by T7 DNA polymerase (38, 53). When the incoming dNTP is correct, the enzyme undergoes a rapid conformational change from the open to closed form of the pre-incorporation complex. B, kinetic scheme for the incorporation of mis-matched dNTPs by T7 DNA polymerase (38, 53). When the incoming dNTP is incorrect, the rate constant for the chemical step becomes smaller than that for the conformational change back to the open form of the pre-incorporation complex. C, chemical mechanism of primer elongation for the 2-Mg (54) and 3-Mg models (55). The location of the third Mg2+ ion together with its water ligands is shown in red. Residue numbering is for human DNA polymerase β. D, chemical mechanism of primer elongation in the active site of T. aquaticus DNA polymerase, which lacks one of the carboxylate ligands present in human DNA polymerase β.
Replicative DNA polymerases, especially those for which DNA binding is the rate-limiting step, exhibit a property referred to as processivity (57). Processivity is a measure of the number of dNTPs incorporated into the growing strand before the template-primer duplex dissociates from the enzyme (58). Processivity, of course, permits these DNA polymerases to achieve high rates of nucleotide incorporation. This process requires translocation of the enzyme along the template-primer or the template-primer along the enzyme (depending on your point of view) during polymerization. We discuss the conformational changes that occur during dNTP incorporation below.
When mismatched dNTPs are present in the ternary complex, kinetic evidence shows that the active site remains in an “open” conformation so that dissociation of the incoming nucleotide can take place prior to formation of the new P–O bond (37, 38) (Fig. 2B). Alternatively, should a mismatched dNTP be incorporated, most replicative polymerases include a 3′–5′ exonuclease domain that can remove the mismatch. In this case, the 3′-hydroxyl end of the primer shifts from the polymerase to the exonuclease active site for removal of the mismatched nucleotide (59, 60). This proofreading step is essential for ensuring fidelity during replication by DNA polymerases, which typically make errors every 104 to 105 nucleobases. The 3′–5′ exonucleolytic activity improves this error rate by 2–3 orders of magnitude (61).
A variety of assays exist to assess the kinetics of nucleobase incorporation and processivity. These have been extensively reviewed (34, 62), but here we give two examples used in our work to determine either incorporation efficiency (63) or fidelity (64) when DNA polymerases and their variants are challenged with AEGIS UBPs (Fig. 3). For example, incorporation efficiency can be measured by nested PCR using primers tagged at the 5′-end with multiple consecutive UBPs. In this assay, amplicons are formed in the reaction mixture only if the polymerase is capable of replicating the UBPs (Fig. 3A).
Figure 3.
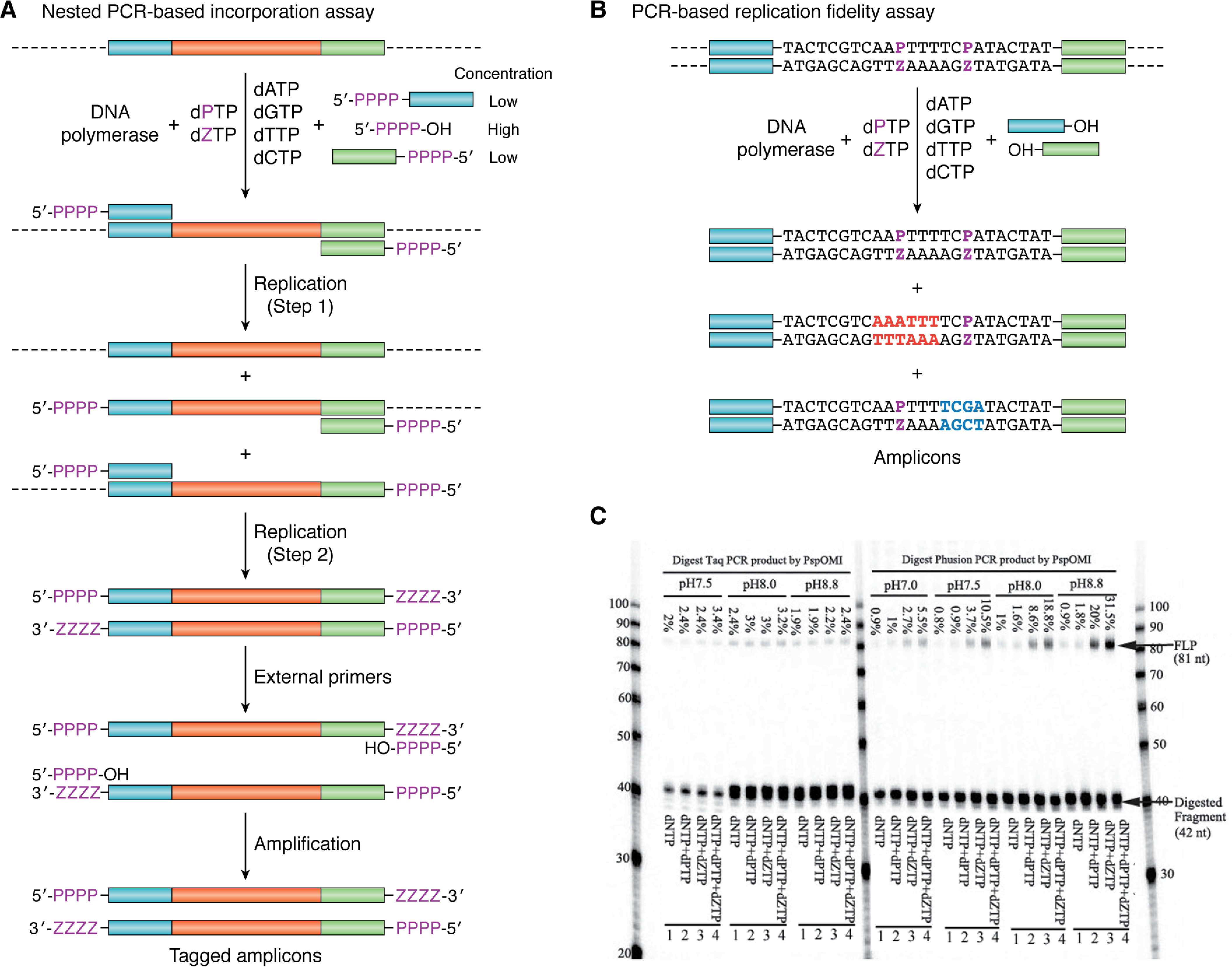
A, nested PCR-based incorporation assay for DNA containing Z:P nucleobase pairs using a target DNA sequence (cyan-red-green) composed of only WC base pairs. Replication is initiated by chimeric primers that can bind to the original gene but have UBPs at the 5′-end. These chimeric primers are present at low concentration (Step 1). After consumption of the chimeric primers (Step 2), amplification is continued by external 5′-PPPP-OH primers (containing only UBPs), which are present at high concentration. As a result, full-length amplicons are produced only if the DNA polymerase can efficiently incorporate UBPs (based on Fig. 2 of Ref. 65). B, PCR-based replication fidelity assay for DNA containing Z:P nucleobase pairs. If the UBP is lost to a T:A base pair, then a Dra restriction site (red) is generated in the amplicons. Similarly, if the UBP is lost to a C:G base pair, then an Alu restriction site (blue) is produced. C, gel showing misincorporation rates in the PCR amplification of template DNA in the presence of dZTP and/or dPTP components using WT Klentaq and WT Phusion DNA polymerases at different pH values (64). Four parallel PCRs were performed followed by digestion with PspOMI for 16 h. The ratio between the amount of full-length product (FLP) and all oligonucleotides gives the misincorporation rates shown in the figure. Lane 1, negative control PCR amplification of template DNA in the presence of WC dNTPs (200 mm each), followed by digestion with PspOMI. Lane 2, five-letter PCR amplification of template DNA in the presence of WC dNTPs (200 mm each) and dZTP (25 mm), followed by digestion with PspOMI. Lane 3, five-letter PCR amplification of template DNA in the presence of WC dNTPs (200 mm each) and dPTP (25 mm), followed by digestion with PspOMI. Lane 4, six-letter PCR amplification of template DNA in the presence of WC dNTPs (200 mm each), dZTP (25 mm), and dPTP (25 mm), followed by digestion with PspOMI. Taken from Ref. 64 and used with permission.
Assaying fidelity requires the DNA polymerase to replicate a sequence in which UBPs are present (64) (Fig. 3B). In the event that the UBP is lost during PCR cycles, two possible unique restriction sites are created in the amplicons, depending on whether the UBP is replaced by T:A or C:G. Incubating the PCR products with two restriction endonucleases therefore gives cleaved products for the amplicons from which the UBP has been lost in both possible transition mutations (Fig. 3C). This assay thereby reports on the extent to which polymerase fidelity deviates from 100%. In addition, performing the PCR with different concentrations of the UBP components permits a quantitative assessment of fidelity.
Obtaining DNA polymerases engineered for altered substrate specificity
Some, albeit limited, success has been reported in modifying the substrate preferences of DNA polymerases by structure-guided mutagenesis studies (66, 67). The complexity of the conformational changes undergone by the polymerase during each catalytic cycle seems to require that variants contain multiple residue replacements. Most successful efforts to obtain DNA polymerases with altered catalytic and/or biophysical properties have therefore selected suitable variants from large libraries (40, 68–72). Examples include several Taq polymerase variants evolved for specific functions (Table 1). The fact that several of the amino acid substitutions are found in more than one of these Taq variants may ultimately inform rationale design of variants with new functional properties in the future. To date, however, only one of these published variants (ZP Klentaq) has been subjected to structural and dynamic characterizations to determine the impact of each of the various substitutions on the properties of the variant polymerase (see below).
Table 1.
Examples of evolved Taq DNA polymerase variants
Residues in boldface/italic type are found in two or more of the reported Taq DNA polymerase variants. The second column indicates whether variants were generated for specific substrates, such as C2′ modified nucleic acid, or for a specific property, such as thermostability.
| Taq variant | Substrate/Property | Amino acid substitutions | Reference |
|---|---|---|---|
| SFM4-3 | C2′ modification | I614E/E615G/N583S/D655N/E681K/E742Q/M747R | 70 |
| AA40 | RNA, DNA | E602V/A608V/I614M/E615G | 73 |
| M1 | 3′ mismatches | G84A/D144G/K314R/E520G/F598L/A608V/E742G | 74 |
| M4 | 3′ mismatches | D58G, R74P, A109T, L245R, R343G, G370D, E520G, N583S, E694K, A743P | 74 |
| 5D4 | Hydrophobic bases | V62I, Y78H, T88S, P114Q, P264S, E303V, G389V, E424G, E432G, E602G, A608V, I614M, M761T, M775T | 75 |
| H15 | Thermostability | K225E/E388V/K540R/D578G/N583S/M747R | 76 |
| RT-KTQ | m6-A sequencing | L459M/S515R/I638F/M747K | 69 |
| ZP | Z:P substrates | M444V/P527A/E551E/E832V | 65 |
Of the plethora of directed evolution strategies, compartmentalized self-replication (CSR) (76, 77) or one of its modifications (40, 68) is often used to obtain engineered DNA polymerases. In CSR, water droplets containing appropriate primers and dNTPs are formed in a water/oil emulsion. Droplets that also contain a single E. coli cell carrying a plasmid encoding a single variant of a thermophilic DNA polymerase, such as Klentaq, can then be used to select enzymes with desired properties. In our work (65), we employ the nested PCR strategy described above (Fig. 3A) to identify DNA polymerase variants that can replicate UBPs orthogonal to WC nucleobases, such as Z and P, because only these will replicate copies of their gene in the plasmid (Fig. 4). Those variants that cannot incorporate UBPs efficiently will not replicate their genes. Thus, genes encoding DNA polymerases with the desired substrate specificity become represented more in the mixture of DNA amplicons obtained when the emulsion is broken at the end of several rounds of PCR (Fig. 4). The resulting DNA amplicons are inserted into a new set of plasmids that are used to transform E. coli for use in a subsequent selection round. After a defined number of rounds, genes encoding DNA polymerase variants with the ability to incorporate UBPs become highly enriched in the library. The set of enriched genes can be sequenced to identify the molecular changes that permit the polymerase variant to replicate the UBP. Standard methods can then be used to express and purify individual variants prior to any detailed evaluation of their fidelity and incorporation efficiencies (63, 64).
Figure 4.
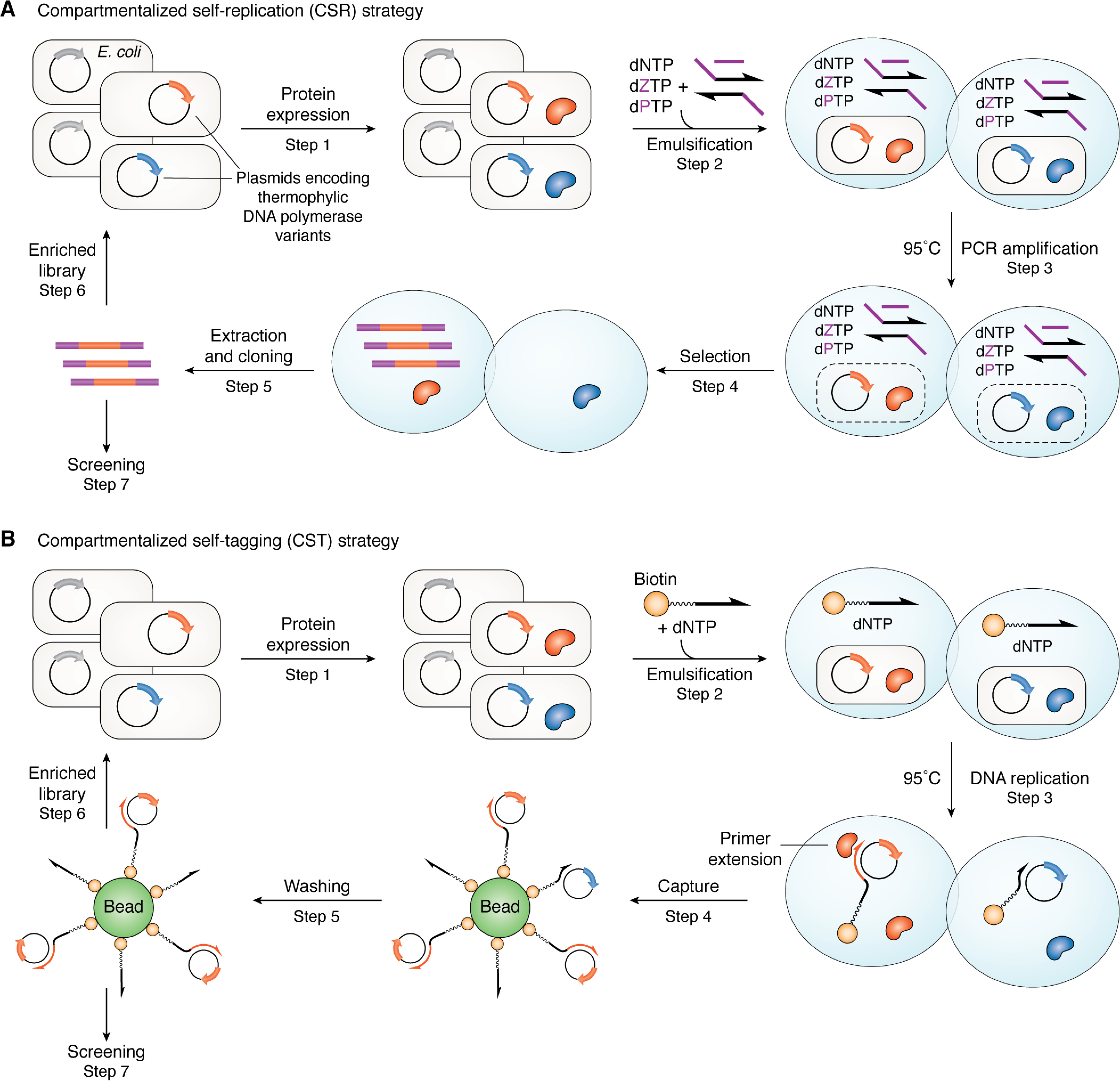
A, overview of a modified CSR strategy for identifying DNA polymerase variants capable of replicating AEGIS UBPs from a library. Each E. coli cell carries a thermophilic DNA polymerase variant (Step 1) and is emulsified with dNTPs and nested primers within a single water droplet. Heating lyses the cells and inactivates endogenous mesophilic polymerases (Step 2). PCR amplification is then performed (Step 3). Polymerases that can replicate UBPs (red) amplify their respective genes, enriching the pool of variant genes in the pool; those that cannot replicate UBPs fail to do so, so their genes are lost (Step 4). The amplified genes encoding polymerase variants that can replicate UBPs are extracted after breaking the emulsion and used to create a new library of plasmids (Step 5) that can be subjected to a further selection round (Step 6). After an appropriate number of selection rounds, the pool of enriched genes can be sequenced (Step 7) and used to express polymerase variants that can be characterized using standard in vitro methods. Based on Fig. 3 of Ref. 65. B, In CST, each E. coli cell carries a thermophilic DNA polymerase variant encoded on a plasmid (Step 1), which also contains a sequence containing one or more UBPs complementary to a single biotin-linked (“tagged”) primer and is emulsified with dNTPs and the tagged primer within a single water droplet. Heating lyses the cells and inactivates endogenous mesophilic polymerases (Step 2). Subsequent DNA replication (Step 3) by the thermophilic DNA polymerase variant results in a “biotin-tagged” form of the plasmid containing the gene (red) encoding the polymerase variant that can replicate the UBP(s). Polymerases that cannot replicate the UBP (blue) will only give weakly stable primer-plasmid complexes. After breaking the emulsion, tagged plasmids are captured onto a bead (Step 4) in proportion to the stability of the extended primer-plasmid complex (Step 5). The recovered plasmid DNA is then amplified and used in a new round of selection (Step 6) or screened (Step 7). Based on Fig. S1 of Ref. 78).
The original CSR strategy, however, requires the replication of the entire polymerase gene, which contains more than 2000 nucleobases. Thus, only the most active variants are recovered. Many technical aspects, including the appropriate choice of oil or surfactant containing the water droplets (79, 80), must be optimized if reproducible results are to be obtained (81).
Improved variations of this selection method have therefore been reported by Philip Holliger and co-workers (78, 82), including compartmentalized self-tagging (CST) (Fig. 4B). In CST, the positive feedback loop depends on the polymerase “tagging” a plasmid containing its encoding gene by extension of a biotinylated oligonucleotide. As a result, plasmids containing genes that encode DNA polymerases with altered substrate selectivity can be enriched by selective capture onto an affinity column. Clearly, the UBP(s) must be located within a segment of plasmid DNA that is replicated with high efficiency and fidelity by the DNA polymerase variant. In addition, the sensitivity of the assay is higher because CST does not require multiple self-replications of the complete gene encoding the polymerase variant.
Insights from structural studies of WT and evolved Klentaq DNA polymerase complexes
In contrast to some other critical cellular enzymes and machines, DNA polymerases have been reinvented throughout evolution (32). As a consequence, several distinct structural classes of polymerases provide multiple opportunities to identify an enzyme that will efficiently and faithfully replicate an unnatural base pair. Replicative DNA polymerases have been classified as families A–D, with D being the most recently identified DNA polymerase family from Archaea (32).
To date, naturally occurring family A and B DNA polymerases have been used to successfully replicate single UBPs. Family A polymerases are found primarily in bacteria; their peptide fold is described as including fingers, palm, and thumb domains associated with polymerase activity (Fig. 5A). They also have 3′–5′ exonuclease and 5′–3′ exonuclease domains, the second removing the RNA primers required for lagging strand synthesis (32). In Klentaq, the Thermus aquaticus DNA polymerase (83), corresponding to the Klenow or large fragment of the enzyme (84), the N-terminal 5′–3′ exonuclease domain is absent, and the 3′–5′ exonuclease domain lacks activity. In contrast, family B DNA polymerases, found in all archaea, include a 3′–5′ exonuclease domain and a polymerase domain, which is related to that in family A polymerases (32).
Figure 5.
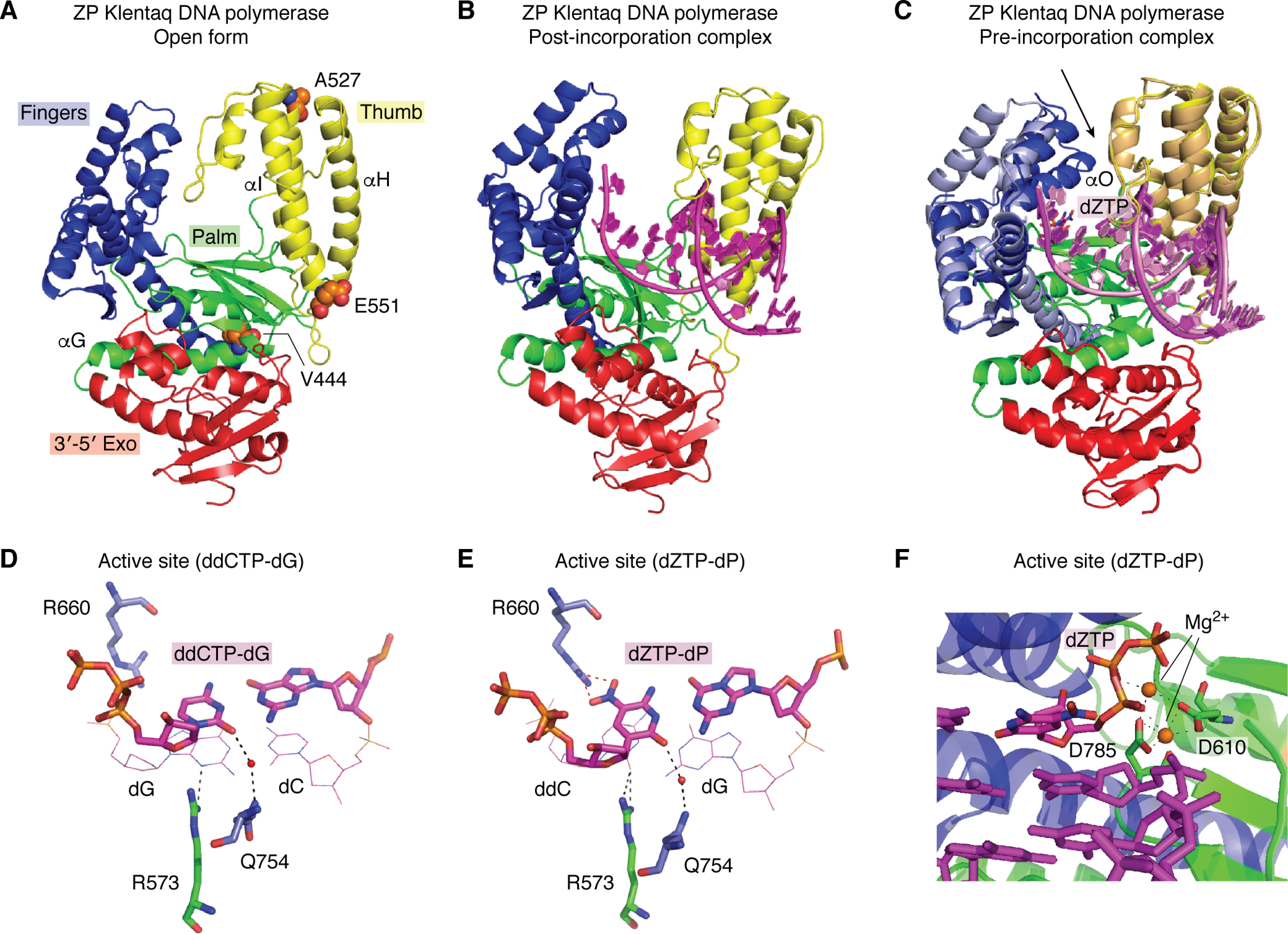
Structural properties of Klentaq DNA polymerase. A, the open form of ZP Klentaq (from PDB entry 5W6Q), shown in a cartoon rendering, includes fingers (blue), palm (green), thumb (yellow), and 3′–5′ exonuclease (red) domains. This architecture is conserved in all family A polymerases. Variant amino acid substitutions are shown as sphere models (orange, carbon; blue, nitrogen; red, oxygen), M444V, P527A, and D551E. (The fourth amino acid substitution, E832V, is disordered in the structures). α-Helices G, H, and I contribute to dynamic motion in ZP Klentaq. B, a similar rendering of ZP Klentaq in the binary, post-incorporation complex (PDB entry 5W6Q) is shown with template-primer, including P:Z in the active site (magenta cartoon rendering). C, the pre-incorporation complex of ZP Klentaq (PDB entry 5W6K) is shown superimposed on the post-incorporation complex from B. The black arrow indicates the most significant conformational change associated with the fingers domain (light blue for the initial and post-incorporation and blue for the pre-incorporation complex). The O helix closes down, forming the active site in the ternary complex with bound dZTP (magenta stick rendering). The thumb domains show a slight shift in position (light orange for post-incorporation and yellow for pre-incorporation complex). The palm and exonuclease domains show no significant differences in overall position in the two complexes. The substrate nucleic acid is shown as a cartoon rendering (pink in the post-incorporation and magenta in the post-incorporation complex). Close-ups of hydrogen-bonding interactions in the active site for the WT Klentaq bound to ddCTP-dG and dZTP-dP are shown in D and E, respectively. In each case, the dNTP is positioned through a water-mediated hydrogen-bonding interaction involving O2 (minor groove edge of the nucleobase) with Gln-754. Similarly, the penultimate primer nucleobase presents either N3 or O2 for hydrogen-bonding to Arg-573 (again a minor groove interaction). In the case of the ZP Klentaq ternary complex (E), Arg-660 hydrogen-bonds to the NO2 group present on the major groove edge of Z. F, the active site including template-primer and dZTP (magenta stick rendering), Mg2+ ions (orange spheres), and coordinating residues Asp-610 and Asp-785 (green stick rendering) are shown with a ribbon rendering of the enzyme, palm in green and fingers in blue.
Hydrophobic nonhydrogen-bonding pairs developed by Romesberg (NAM:TPT3) (8, 9) and Hirao (Ds:Px) (10, 11) have been replicated with selectivities exceeding 99% using DeepVent DNA polymerase (family B) (85) or a combination of DeepVent DNA polymerase and Taq DNA polymerase (family A) (14), respectively. It has proven much more difficult, however, to replicate templates containing consecutive hydrophobic nucleobases. The problem appears to be caused, in part, by disruptions to the DNA double helix resulting from the presence of hydrophobic UBPs (86). For example, the dNaM:d5SICS pair adopts an intercalative stacking mode in duplex DNA, as seen in both NMR structures and binary template-primer Klentaq complexes (87, 88). At least 6 natural base pairs must separate Ds-Px pairs if replication is to be successful (89); as yet there is no report of DNA containing consecutive NaM:PTP3 (or structurally related hydrophobic UBPs) being replicated. Of course, the necessity of replicating more than one UBP, either consecutive or separted by standard WC nucleobases depends on the biotechnological application (15).
A truly artificial genome would presumably include consecutive UBPs just as natural genomes include runs of sequential A:T or G:C pairs; cells possessing such a genome would require a DNA polymerase that can replicate through these regions of sequence. We have shown that up to 4 consecutive Z:P pairs can be incorporated by WT Klentaq polymerase (63); these UBP tetrads do not adversely impact the ability of duplex DNA to adopt A- and B-conformations (90, 91). Unfortunately, the efficiency of Z:P incorporation by WT Klentaq is only 99.8% per theoretical PCR cycle (63), meaning that the UBP is easily lost during PCR-based applications. An engineered Klentaq variant (ZP Klentaq), exhibiting improved fidelity and Z:P incorporation efficiency was, however, obtained using CSR (Fig. 4A) (65). Remarkably, ZP Klentaq contains only four amino acid substitutions, M444V, P527A, D551E, and E832V, all of which are located distal from the active site (Fig. 5A).
In family A DNA polymerases, a large conformational change in the fingers domain occurs upon binding of the complementary dNTP, resulting in the formation of a closed complex that facilitates incorporation (Fig. 5). Of the structurally characterized DNA polymerases related to Klentaq at the sequence level (Table 2), only three have been captured in both pre- and post-incorporation complexes: ZP Klentaq (92), WT Klentaq (6), and WT Geobacillus kaustophilus DNA polymerase (93). Incorporation of nucleotides by the Geobacillus enzyme exhibits a fingers domain closure angle of 37°. In contrast to the Geobacillus enzyme, incorporation of natural dNTPs by WT Klentaq involves a much larger conformational change in which the fingers domain rotates by ∼59° (Fig. 5, B and C). In the closed conformation of the polymerase, correctly paired nucleotides are selected by hydrogen bonding and size complementarity. Despite retaining these features, Z:P pairs are more efficiently and faithfully incorporated by ZP Klentaq, in which the fingers close down by ∼64° (Table 3). Protein-nucleic acid interactions in both pre- and post-incorporations for ZP Klentaq are similar to those observed in the analogous WT complexes, suggesting that the evolved polymerase closely mimics the WT enzyme in this regard (Fig. 5, D and E).
Table 2.
Structure-based alignments of Taq-related DNA polymerases
Binary and apo structures were superimposed with ZP Klentaq binary complex (5W6Q:CEF, chain identifiers); ternary complexes were superimposed with ZP Klentaq ternary complex (5W6K). RMSD, root mean square deviation
| PDB | Type | Source | No. of aligned Cα atoms | RMSD | Sequence identity |
|---|---|---|---|---|---|
| Å | % | ||||
| 5DKU | Apo | Plasmodium | 418/521 | 2.8 | 24.6 |
| 4DQS | Binary | G. kaustophilus | 486/521 | 1.8 | 42.8 |
| 1KFD | Apo | E. coli | 429/521 | 1.9 | 42.7 |
| 2BPD | Binary | Geobacilllus stearothermophilus | 488/521 | 1.8 | 43.4 |
| 4XVI | Binary | Homo sapiens (Pol ν) | 439/521 | 2.5 | 31.9 |
| 6VDE | Apo | Mycobacterium smegmatis | 487/521 | 2.1 | 40.0 |
| 4 × 0Q | Ternary | H. sapiens (Pol θ) | 443/522 | 2.4 | 29.6 |
Table 3.
Comparisons of binary and ternary complexes
ZP tern, the ternary complex of ZP Klentaq; ZP Bin C, the binary complex of ZP Klentaq. *, Klenow fragment of G. kaustophilus DNA polymerase I. All other structures correspond to the Klentaq, the Klenow fragment of Thermus aquaticus DNA polymerase.
| PDB file 1 | PDB file 2 | Fixed domain | Moving domain | Rotation angle | Translation | Closure | Bending residues |
|---|---|---|---|---|---|---|---|
| degrees | Å | % | |||||
| 5W6K ZP tern | 5W6Q ZP Bin C | 299–637 | 638–671 | 63.6 | −1.7 | 88.8 | 632–638 |
| 672–829 | 670–704 | ||||||
| 3RTV | 3SZ2 | 295–637 | 638–671 | 59.6 | −1.5 | 79.4 | 626-638 |
| 672–830 | 669–696 | ||||||
| 3KTQ | 4KTQ | 296–636 | 637–670 | 58.2 | −1.9 | 67.8 | 626–637 |
| 671–829 | 670–686 | ||||||
| 4DQQ* | 4DQS* | 300–679 | 680–716 | 36.6 | −0.7 | 86.6 | 676–680 |
| 717–874 | 712–744 |
The post-incorporation ZP Klentaq complex (PDB entry 5W6Q) (92) also exhibits increased thumb domain motion (∼4°) relative to the analogous WT Klentaq complex (PDB entry 3SZ2) (6). Although Z:P pairs are readily accommodated in both B- and A-form DNA duplexes (90, 91) and retain standard features (groove widths, base stacking parameters, etc.) consistent with these helical forms, modeling of a post-incorporation complex of P:Z bound to the WT Klentaq suggested that some structural alterations within the active site would be required to accommodate the UBP (92). The two base pairs closest to the site of incorporation of the next dNTP exhibit A-form, which is significantly wider than B-form. This feature of the active site may allow WT Klentaq to accommodate a single hydrophobic UBP, which is wider than hydrogen-bonded pairs (6, 94).
Of the amino acid substitutions in ZP Klentaq, M444V was considered to be the best candidate for providing access to increased relative domain motion as compared with the WT enzyme. Residue 444 resides within the hydrophobic core of the palm domain, which serves as the command center for the enzyme. Both the fingers and thumb domains are connected to the palm domain. In addition, the essential catalytic residues Asp-610 and Asp-785, which coordinate Mg2+ and the incoming dNTP in the active site of the enzyme, reside in the palm domain (Fig. 5F). Substitution of a Val for Met at this position creates open space within the core that could potentially translate into increased motion of the fingers and thumb domains.
Augmenting crystallography with molecular dynamics simulations of natural and evolved DNA polymerases
Molecular dynamics (MD) simulations are a well-validated method of studying protein dynamics (95) and have yielded important information about conformational changes undergone by DNA polymerases (96, 97). Our recent work also demonstrates that MD simulations can be used to understand how substitutions far from the active site might alter substrate selectivity (98). For example, microsecond trajectories have shown how the dynamic motions of ZP Klentaq differ from those of WT Klentaq in the binary complex containing template-primer DNA duplexes (Fig. 6A). They can also highlight correlated motions in networks of residues that are critical to altered domain motions in the variant DNA polymerase (Fig. 6B) (98), which permit the engineered enzyme to bind WC and Z:P-containing template-primer duplexes in an equivalent fashion, thereby increasing the efficiency of UBP incorporation. This seems to be a consequence of replacing Met-444 and Asp-551 at the base of the thumb domain by valine and glutamate, respectively, which allows the fingers, palm, and thumb domains in ZP Klentaq to move into position about the AEGIS template-primer duplex. Thus, replacing Met-444 by valine in the hydrophobic core changes the populated interactions with adjacent residues (Phe-564, Met-779, and Leu-780) in the hydrophobic core (Fig. 6C) with the consequence that the G helix becomes more flexible in the variant compared with the WT polymerase (98).
Figure 6.
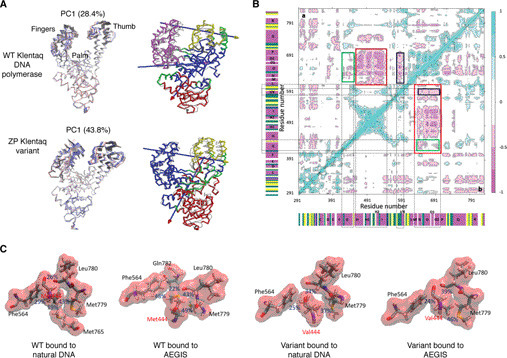
A, two complementary cartoon representations of motions in the binary complexes of WT Klentaq (top) and ZP Klentaq variant (bottom). Left, the major principal component (PC1) (99) at early (red), intermediate (white), and late (blue) stages of the sampled MD trajectory, showing that the dynamics of the fingers and palm domains are altered when WC and AEGIS template-primer strands are bound for the two DNA polymerases. Right, domain motions of the polymerase complexed to AEGIS DNA computed using DynDom (100). For reference, the WT Klentaq/WC DNA binary complex (PDB entry 3SZ2) is shown in gray. Each axis of domain motion is indicated by an arrow, the head of which is colored to represent the moving domain (red, yellow, or purple). The fixed domain (blue) and regions that bend during the motion are shown in green. B, dynamic cross-correlation maps computed with bio3d (101) for the AEGIS DNA-containing binary complex of WT Klentaq (top triangle) and ZP Klentaq (bottom triangle). Correlated (range: 0.25–1) and anti-correlated (range: −0.25 to −1) motions are colored from light to dark blue and pink, respectively. Areas rendered in white correspond to noncorrelated motions (range: −0.25 to 0.25). Secondary structural elements are also included on the map. Colored boxes show interconnected residue networks with altered motions in the two polymerases. C, altered interactions between residue 444 and surrounding side chains in the hydrophobic core observed in the MD trajectories of the WT Klentaq/Watson–Crick DNA binary complex (left), WT Klentaq/AEGIS DNA binary complex (middle left), ZP Klentaq/Watson–Crick DNA binary complex (middle right), and ZP Klentaq/AEGIS DNA binary complex (right). The percentages of occurrence of specific interactions in the MD trajectory are also shown. Based on Fig. 7 of Ref. 98).
As well as modeling how amino acid substitutions might impact long time-scale motions, such as domain reorientations, MD simulations can illuminate specific molecular changes that have the greatest impact on the altered catalytic biophysical and catalytic properties of the DNA polymerase variant. In the case of ZP Klentaq, the role of each of the substitutions (M444V, P527A, D551E, or E832V) was determined using MD simulations of the four single-point Klentaq variants (98). These calculations showed that replacing Met-444 in the hydrophobic core of the palm domain with valine makes a significant contribution to altering dynamic motions in ZP Klentaq. In addition, changing Asp-551 to glutamate alters the interactions of the H and I helices at the base of the thumb domain, which affects G helix flexibility (Fig. 6B) (98).
Care must be taken to perform MD simulations on the correct form of the enzyme. For example, introducing valine in place of Glu-832, which breaks a salt bridge with Arg-596, has little impact on the dynamics of the ZP Klentaq/DNA binary complex. However, it does alter protein/DNA interactions. This substitution may therefore exert its effects in the pre-incorporation (ternary) complex during dNTP binding and incorporation.
MD simulations have also provided interesting insights into the “closing mechanism” of DNA polymerase I prior to the incorporation of WC nucleobases (102) and the temperature-dependent activity of a Klentaq variant in which Ile-707, located 20 Å away from the active site, is replaced by leucine (103). This I707L Klentaq variant exhibits low activity at 37 °C but efficiently replicates DNA at 68 °C, permitting improved PCR amplification of difficult sequences. MD simulations show that the I707L Klentaq variant is almost immobile at 37 °C but exhibits increased flexibility at 68 °C. In contrast, the WT enzyme exhibits less pronounced dynamic changes in simulations performed at 37 and 68 °C. Thus, it is likely that the reduced activity of the I707L Klentaq variant is associated with the enzyme remaining in its closed conformation at lower temperatures (i.e. dNTP binding is blocked) and that the pre-incorporation complex cannot form. As observed for ZP Klentaq (98), the molecular origins of this behavior could be traced to changes in the hydrophobic core; replacing isoleucine by leucine results in an alternate conformation of an adjacent phenylalanine (Phe-749), thereby repositioning the O and O1 helices. As a consequence, the active site becomes filled with nucleobases in the template overhang, thereby blocking entry of the incoming dNTP.
Outlook
Realizing the full potential of UBPs in the creation of new research, diagnostic, and therapeutic tools will ultimately depend on our ability to identify DNA polymerase variants that can faithfully and efficiently replicate those UBPs. Many of these applications will necessarily require enzymes that are active at the high temperatures used in PCR, limiting the pool of candidate DNA polymerases primarily to family A and family B members. For other applications, such as using dNaM-dTPT3 (Fig. 1) in semi-synthetic organisms that introduce noncanonical amino acids into specific proteins or the isothermal amplification of AEGIS UBPs (104), DNA polymerases that function at 37 °C, such as those in E. coli, will be adequate as starting points for structure-based engineering or library construction and selection.
To date, progress in engineering DNA polymerases has been encouraging (105). In our work aimed at improving the ability of these enzyme to replicate UBPs (65, 92, 98), ZP Klentaq takes advantage of increased domain flexibility to improve the replication efficiency of Z:P pairs, even though both WT Klentaq and ZP Klentaq grip the template-primer duplex in a similar manner prior to and following incorporation of the AEGIS UBP.
Combining X-ray crystallography with MD simulations yields two important conclusions. First, not all substituted residues impact the properties of the evolved enzyme equally. Second, substitution of a key residue, such as Met-444, can result in increased relative domain motion, even in an enzyme that exhibits a very large conformational change during catalysis. It is this altered motion that allows ZP Klentaq to incorporate UBPs more readily than the WT enzyme.
CSR, CST, and related strategies for the directed evolution of DNA polymerases, which typically produce variants containing several amino acid substitutions, might therefore be improved by targeting fewer residues outside the active site for variation. Coupling structure determinations, MD simulations, and enzymatic characterization will provide guiding principles for CSR library design and facilitate rational approaches to optimize family A DNA polymerases to replicate multiple UBPs. Such work can be guided by understanding amino acid substitutions already identified in Klentaq variants (Table 1); for example, Asn-583, Ile-614, and Met-747 are substituted in three of eight variant enzymes, whereas Glu-520, Glu-602, Ala-609, Glu-615, and Glu-742 are substituted in two of eight variants. None of these residues are involved in direct hydrogen-bonding interactions with the substrate template-primer or the dNTP in the active site (Fig. 7). The past evolutionary history of DNA polymerases may also prove useful for selecting specific sites for variation (42, 106).
Figure 7.

A cartoon rendering showing two views of the ternary complex of Klentaq (fingers, blue; palm, green; thumb, yellow; exonuclease domain, red) with amino acid substitutions common to evolved Klentaq variants shown in van der Waals sphere renderings (carbon, orange; oxygen, red; nitrogen, blue; sulfur, yellow). None of the side chains of these residues are involved in direct hydrogen-bonding interactions with the template-primer or dNTP in the active site. Most of these residues are distant from substrate template-primer and dNTP, although the main-chain NH of Glu-615 hydrogen-bonds to the 3′-OH of the dNTP deoxyribose ring, and Asn-583 makes a water-mediated hydrogen bond to the backbone of the template strand. In addition, Ile-614 and Glu-615 are within van der Waals contact distance of the dNTP bound to the active site, and Met-747 is within contact distance of the template strand.
We also note that whereas most engineering efforts to date have focused exclusively on improving the ability of DNA polymerases to incorporate UBPs with little consideration for exonuclease activity, a Thermococcus gorgonarius DNA polymerase variant selected to replicate xeno-nucleic acids contains two amino acid substitutions that inactivate the 3′–5′ exonuclease site (107). In theory, incorporation of UBPs that slow down polymerization would allow the substrate to be positioned within the exonuclease active site for proofreading. Whether the exonuclease would efficiently process the newly incorporated unnatural nucleobase or not has yet to be fully investigated. It is possible that engineered DNA polymerases may have to possess optimized exonuclease function as well as improved efficiencies for UBP incorporation to maintain artificial genomes.
In summary, the next generation of useful DNA polymerases must (i) replicate more than one UBP with rates and fidelities similar to those observed for WT DNA polymerases when replicating WC dNTPs and (ii) exhibit no significant pausing following UBP incorporation to avoid invoking proofreading mechanisms. These properties will permit the fidelity of these engineered enzymes to approach 99.99%, allowing them to generate full-length replication products in applications involving multiple rounds of PCR. We suggest a strategy for obtaining these DNA polymerase variants that builds on studies of ZP Klentaq, in which a combination of X-ray crystallography and computer simulations showed the importance of a few key residues for conferring increased dynamic motion in the thumb and fingers domains, thereby improving UBP incorporation efficiency. In this approach, altering the properties of an evolved DNA polymerase variant by introducing additional amino acid substitutions will take advantage of a fundamental, structure-based understanding of how dynamic changes in residue “networks” impact kinetic properties (108, 109).
Funding and additional information—This work was supported by Biotechnology and Biological Sciences Research Council, UK, Grant P/018017/1 (to N. G. J. R.). This review is also based on work supported by the National Science Foundation under Grant MCB-1939086 (to M. M. G. and S. A. B.). Research reported in this publication was also supported by the National Institutes of Health under Director's Award R01GM128186 (to S. A. B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—S. A. B. owns intellectual property associated with AEGIS nucleobases. Some of the compounds mentioned in this article are sold by Firebird Biomolecular Sciences, LLC, which is owned by S. A. B.
- UBP
- unnatural base pair
- Taq
- Thermus aquaticus DNA polymerase
- Klentaq
- the Klenow fragment of Taq DNA polymerase
- ZP Klentaq
- an evolved Klentaq DNA polymerase variant
- WC
- Watson-Crick
- AEGIS
- artificially expanded genetic information system
- CSR
- compartmentalized self-replication
- CST
- compartmentalized self-tagging
- MD
- molecular dynamics
- PDB
- Protein Data Bank.
References
- 1. Goodman, M., and Tippin, B. (2000) The expanding polymerase universe. Nat. Rev. Mol. Cell Biol. 1, 101–109 10.1038/35040051 [DOI] [PubMed] [Google Scholar]
- 2. Young, R. A. (1991) RNA polymerase II. Annu. Rev. Biochem. 60, 689–715 10.1146/annurev.bi.60.070191.003353 [DOI] [PubMed] [Google Scholar]
- 3. Autexier, C., and Lue, N. F. (2006) The structure and function of telomerase reverse transcriptase. Annu. Rev. Biochem. 75, 493–517 10.1146/annurev.biochem.75.103004.142412 [DOI] [PubMed] [Google Scholar]
- 4. Aschenbrenner, J., and Marx, A. (2017) DNA polymerases and biotechnological applications. Curr. Opin. Biotechnol. 48, 187–195 10.1016/j.copbio.2017.04.005 [DOI] [PubMed] [Google Scholar]
- 5. Gardner, A. F., Jackson, K. M., Boyle, M. M., Buss, J. A., Potapov, V., Gehring, A. M., Zatopek, K. M., Corrêa, I. R., Jr., Ong, J. L., and Jack, W. E. (2019) Therminator DNA polymerase: modified nucleotides and unnatural substrates. Front. Mol. Biosci. 6, 28 10.3389/fmolb.2019.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Betz, K., Malyshev, D. A., Lavergne, T., Welte, W., Diederichs, K., Dwyer, T. J., Ordoukhanian, P., Romesberg, F. E., and Marx, A. (2012) Klentaq polymerase replicates unnatural base pairs by inducing a Watson-Crick geometry. Nat. Chem. Biol. 8, 612–614 10.1038/nchembio.966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watson, J. D., and Crick, F. H. C. (1953) Molecular structure of nucleic acids. A structure for deoxyribose nucleic acid. Nature 171, 737–738 10.1038/171737a0 [DOI] [PubMed] [Google Scholar]
- 8. Kool, E. T. (2002) Replacing the nucleobases in DNA with designer molecules. Acc. Chem. Res. 35, 936–943 10.1021/ar000183u [DOI] [PubMed] [Google Scholar]
- 9. Feldman, A. W., and Romesberg, F. E. (2018) Expansion of the genetic alphabet: a chemist's approach to synthetic biology. Acc. Chem. Res. 51, 394–403 10.1021/acs.accounts.7b00403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malyshev, D. A., and Romesberg, F. E. (2015) The expanded genetic alphabet. Angew. Chem. Int. Ed. Engl. 54, 11930–11944 10.1002/anie.201502890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Piccirilli, J. A., Krauch, T., Moroney, S. E., and Benner, S. A. (1990) Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 343, 33–37 10.1038/343033a0 [DOI] [PubMed] [Google Scholar]
- 12. Benner, S. A., Karalkar, N. B., Hoshika, S., Laos, R., Shaw, R. W., Matsuura, M., Fajardo, D., and Moussatche, P. (2016) Alternative Watson-Crick synthetic genetic systems. Cold Spring Harb. Perspect. Biol. 8, a023770 10.1101/cshperspect.a023770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirao, I., Kimoto, M., and Yamashige, R. (2012) Natural versus artificial creation of base pairs in DNA: origin of nucleobases from the perspective of unnatural base pair studies. Acc. Chem. Res. 45, 2055–2065 10.1021/ar200257x [DOI] [PubMed] [Google Scholar]
- 14. Yamashige, R., Kimoto, M., Takezawa, Y., Sato, A., Mitsui, T., Yokoyama, S., and Hirao, I. (2012) Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 40, 2793–2806 10.1093/nar/gkr1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer, E. C., Hashimoto, K., Zhang, Y., Feldman, A. W., Dien, V. T., Karadeema, R. J., Adhikary, R., Ledbetter, M. P., Krishnamurthy, R., and Romesberg, F. E. (2020) New codons for efficient production of unnatural proteins in a semisynthetic organism. Nat. Chem. Biol. 16, 570–576 10.1038/s41589-020-0507-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu, C. C., and Schultz, P. G. (2010) Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444 10.1146/annurev.biochem.052308.105824 [DOI] [PubMed] [Google Scholar]
- 17. Metzker, M. L. (2010) Sequencing technologies—the next generation. Nat. Rev. Genet. 11, 31–46 10.1038/nrg2626 [DOI] [PubMed] [Google Scholar]
- 18. McCombie, W. R., McPherson, J. D., and Mardis, E. R. (2019) Next-generation sequencing technologies. Cold Spring Harb. Perspect. Med. 9, a036798 10.1101/cshperspect.a036798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leary, J. J., Brigati, D. J., and Ward, D. C. (1983) Rapid and sensitive colorimetric method for visualizing biotin-labeled DNA probes hybridized to DNA or RNA immobilized on nitrocellulose: Bio-blots. Proc. Natl. Acad. Sci. U. S. A. 80, 4045–4049 10.1073/pnas.80.13.4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Orgel, L. E. (1992) Molecular replication. Nature 358, 203–209 10.1038/358203a0 [DOI] [PubMed] [Google Scholar]
- 21. Robertson, M. P., and Joyce, G. F. (2012) The origins of the RNA world. Cold Spring Harb. Perspect. Biol. 4, a003608 10.1101/cshperspect.a003608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Powner, M. W., Gerland, B., and Sutherland, J. D. (2009) Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 10.1038/nature08013 [DOI] [PubMed] [Google Scholar]
- 23. Rich, A. (1962) Problems of evolution and biochemical information transfer. In Horizons in Biochemistry (Kasha, M., and Pullman, B., eds) pp. 103–126, Academic Press, Inc., New York [Google Scholar]
- 24. Herdewijn, P. (2010) Nucleic acids with a six-membered carbohydrate mimic in the backbone. Chem. Biodivers. 7, 1–59 10.1002/cbdv.200900185 [DOI] [PubMed] [Google Scholar]
- 25. Taylor, A. I., Houlihan, G., and Holliger, P. (2019) Beyond DNA and RNA: the expanding toolbox of synthetic genetics. Cold Spring Harb. Perspect. Biol. 11, a032490 10.1101/cshperspect.a032490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hutter, D., Blaettler, M. O., and Benner, S. A. (2002) From phosphate to bis(methylene)sulfone: non-ionic backbone linkers. Helv. Chim. Acta 85, 2777–2806 [DOI] [Google Scholar]
- 27. Eberlein, L., Beierlein, F. R., Hommes, N. R. V., Radadiya, A., Heil, J., Benner, S. A., Clark, T., Kast, S. M., and Richards, N. G. J. (2020) Tautomeric equilibria of nucleobases in the Hachimoji expanded genetic alphabet. J. Chem. Theory Comput. 16, 2766–2777 10.1021/acs.jctc.9b01079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoshika, S., Leal, N. A., Kim, M.-J., Kim, M.-S., Karalkar, N. B., Kim, H.-J., Bates, A. M., Watkins, N. E., Jr., SantaLucia, H. A., Meyer, A. J., DasGupta, S., Piccirilli, J. A., Ellington, A. D., SantaLucia, J., Jr., Georgiadis, M. M., et al. (2019) Hachimoji DNA and RNA: a genetic system with eight building blocks. Science 363, 884–887 10.1126/science.aat0971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glushakova, L. G., Alto, B. W., Kim, M.-S., Hutter, D., Bradley, A., Bradley, K. M., Burkett-Cadena, N. D., and Benner, S. A. (2019) Multiplexed kit based on Luminex technology and achievements in synthetic biology discriminates Zika, chikungunya and dengue viruses in mosquitoes. BMC Infect. Dis. 19, 418 10.1186/s12879-019-3998-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Raper, A. T., Reed, A. J., and Suo, Z. (2018) Kinetic mechanism of DNA polymerases: contributions of conformational dynamics and a third metal ion. Chem. Rev. 118, 6000–6025 10.1021/acs.chemrev.7b00685 [DOI] [PubMed] [Google Scholar]
- 31. Wu, W.-J., Yang, W., and Tsai, M.-D. (2017) How DNA polymerases catalyse replication and repair with contrasting fidelity. Nat. Rev. Chem. 1, 68 10.1038/s41570-017-0068 [DOI] [Google Scholar]
- 32. Raia, P., Delarue, M., and Sauguet, L. (2019) An updated structural classification of replicative DNA polymerases. Biochem. Soc. Trans. 47, 239–249 10.1042/BST20180579 [DOI] [PubMed] [Google Scholar]
- 33. Steitz, T. A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 274, 17395–17398 10.1074/jbc.274.25.17395 [DOI] [PubMed] [Google Scholar]
- 34. Johnson, K. A. (2010) The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048 10.1016/j.bbapap.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kiefer, J. R., Mao, C., Braman, J. C., and Beese, L. S. (1998) Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature 391, 304–307 10.1038/34693 [DOI] [PubMed] [Google Scholar]
- 36. Kropp, H. M., Dürr, S. L., Peter, C., Diederichs, K., and Marx, A. (2018) Snapshots of a modified nucleotide moving through the confines of a DNA polymerase. Proc. Acad. Natl. Acad. Sci. U. S. A. 115, 9992–9997 10.1073/pnas.1811518115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joyce, C. M., and Benkovic, S. J. (2004) DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry 43, 14317–14324 10.1021/bi048422z [DOI] [PubMed] [Google Scholar]
- 38. Kunkel, T. A., and Bebenek, K. (2000) DNA replication fidelity. Annu. Rev. Biochem. 69, 497–529 10.1146/annurev.biochem.69.1.497 [DOI] [PubMed] [Google Scholar]
- 39. Arnold, F. H. (2019) Innovation by evolution: bringing new chemistry to life. Angew. Chem. Int. Ed. Engl. 58, 14420–14426 10.1002/anie.201907729 [DOI] [PubMed] [Google Scholar]
- 40. Houlihan, G., Arangundy-Franklin, S., and Holliger, P. (2017) Exploring the chemistry of genetic information storage and propagation through polymerase engineering. Acc. Chem. Res. 50, 1079–1087 10.1021/acs.accounts.7b00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen, T., and Romesberg, F. E. (2014) Directed polymerase evolution. FEBS Lett. 588, 219–229 10.1016/j.febslet.2013.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laos, R., Thomson, J. M., and Benner, S. A. (2014) DNA polymerases engineered by directed evolution to incorporate non-standard nucleotides. Front. Microbiol. 5, 565 10.3389/fmicb.2014.00565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Betz, K., and Marx, A. (2020) The structural basis for processing of unnatural base pairs by DNA polymerases. Chem. Eur. J. 26, 3446–3463 10.1002/chem.201903525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fowler, R. G., Degnen, G. E., and Cox, E. C. (1974) Mutational specificity of a conditional Escherichia coli mutator, mutD5. Mol. Gen. Genet. 133, 179–191 10.1007/BF00267667 [DOI] [PubMed] [Google Scholar]
- 45. Yu, H., and Goodman, M. F. (1992) Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888–10896 [PubMed] [Google Scholar]
- 46. Brambati, A., Barry, R. M., and Sfeir, A. (2020) DNA polymerase theta (Polθ)—an error-prone polymerase necessary for genome stability. Curr. Opin. Genet. Dev. 60, 119–126 10.1016/j.gde.2020.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Du, H., Wang, P., Wu, J., He, X., and Wang, Y. (2020) The roles of polymerases ν and θ in replicative bypass of O6- and N2-alkyl-2′-deoxyguanosine lesions in human cells. J. Biol. Chem. 295, 4556–4562 10.1074/jbc.RA120.012830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hendrickson, C., Devine, K., and Benner, S. A. (2004) Probing minor groove recognition contacts by DNA polymerases and reverse transcriptases using 3-deaza-2′-deoxyadenosine. Nucleic Acids Res. 32, 2241–2250 10.1093/nar/gkh542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benner, S. A. (2004) Understanding nucleic acids using synthetic chemistry. Acc. Chem. Res. 37, 784–797 10.1021/ar040004z [DOI] [PubMed] [Google Scholar]
- 50. Joyce, C. M., and Steitz, T. A. (1994) Function and structure relationships in DNA polymerases. Annu. Rev. Biochem. 63, 777–822 10.1146/annurev.bi.63.070194.004021 [DOI] [PubMed] [Google Scholar]
- 51. Kuchta, R. D., Benkovic, P., and Benkovic, S. J. (1988) Kinetic mechanism whereby DNA polymerase-I (Klenow) replicates DNA with high fidelity. Biochemistry 27, 6716–6725 10.1021/bi00418a012 [DOI] [PubMed] [Google Scholar]
- 52. Steitz, T. A. (1993) DNA- and RNA-dependent DNA polymerases. Curr. Opin. Struct. Biol. 3, 31–38 10.1016/0959-440X(93)90198-T [DOI] [Google Scholar]
- 53. Tsai, Y. C., and Johnson, K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9687 10.1021/bi060993z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steitz, T. A. (1998) A mechanism for all polymerases. Nature 391, 231–232 10.1038/34542 [DOI] [PubMed] [Google Scholar]
- 55. Gao, Y., and Yang, W. (2016) Capture of a third Mg2+ is essential for catalyzing DNA synthesis. Science 352, 1334–1337 10.1126/science.aad9633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoon, H., and Warshel, A. (2017) Simulating the fidelity and the three Mg mechanism of pol η and clarifying the validity of transition state theory in enzyme catalysis. Proteins 85, 1446–1453 10.1002/prot.25305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bambara, R. A., Uyemura, D., and Choi, T. (1978) On the processive mechanism of DNA polymerase I: quantitative assessment of processivity. J. Biol. Chem. 253, 413–423 [PubMed] [Google Scholar]
- 58. Korangy, F., and Julin, D. A. (1992) A mutation in the consensus ATP-binding sequence of the RecD subunit reduces the processivity of the RecBCD enzyme from Escherichia coli. J. Biol. Chem. 267, 3088–3095 [PubMed] [Google Scholar]
- 59. Donlin, M. J., Patel, S. S., and Johnson, K. A. (1991) Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30, 538–546 10.1021/bi00216a031 [DOI] [PubMed] [Google Scholar]
- 60. Fidalgo da Silva, E., and Reha-Krantz, L. J. (2007) DNA polymerase proofreading: active site switching catalyzed by the bacteriophage T4 DNA polymerase. Nucleic Acids Res. 35, 5452–5463 10.1093/nar/gkm591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bebenek, A., and Ziuzia-Graczyk, I. (2018) Fidelity of DNA replication—a matter of proof-reading. Curr. Genet. 64, 985–996 10.1007/s00294-018-0820-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berdis, A. J. (2009) Mechanisms of DNA polymerases. Chem. Rev. 109, 2862–2879 10.1021/cr800530b [DOI] [PubMed] [Google Scholar]
- 63. Yang, Z., Chen, F., Alvarado, J. B., and Benner, S. A. (2011) Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 133, 15105–15112 10.1021/ja204910n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen, F., Yang, Z. Y., Yang, M. C., Alvarado, J. B., Wang, G. G., and Benner, S. A. (2011) Recognition of an expanded genetic alphabet by type II restriction endonucleases and their application to analyze polymerase fidelity. Nucleic Acids Res. 39, 3949–3961 10.1093/nar/gkq1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Laos, R., Shaw, R., Leal, N. A., Gaucher, E., and Benner, S. (2013) Directed evolution of polymerases to accept nucleotides with non-standard hydrogen-bond patterns. Biochemistry 52, 5288–5294 10.1021/bi400558c [DOI] [PubMed] [Google Scholar]
- 66. Astatke, M., Ng, K., Grindley, N. D., and Joyce, C. M. (1998) A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 95, 3402–3407 10.1073/pnas.95.7.3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gardner, A. F., and Jack, W. E. (2002) Acyclic and dideoxy terminator preference denote divergent sugar recognition by archaeon and Taq DNA polymerases. Nucleic Acids Res. 30, 605–613 10.1093/nar/30.2.605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Coulther, T. A., Stern, H. R., and Beuning, P. J. (2019) Engineering polymerases for new functions. Trends Biotechnol. 37, 1091–1103 10.1016/j.tibtech.2019.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Aschenbrenner, J., Werner, S., Marchand, V., Adam, M., Motorin, Y., Helm, M., and Marx, A. (2018) Engineering of a DNA polymerase for direct m6A sequencing. Angew. Chem. Int. Ed. 57, 417–421 10.1002/anie.201710209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen, T., Hongdilokkul, N., Liu, Z., Adhikary, R., Tsuen, S. S., and Romesberg, F. E. (2016) Evolution of thermophilic DNA polymerases for the recognition and amplification of C2′-modified DNA. Nat. Chem. 8, 556–562 10.1038/nchem.2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schultz, H. J., Gochi, A. M., Chia, H. E., Ogonowsky, A. L., Chiang, S., Filipovic, N., Weiden, A. G., Hadley, E. E., Gabriel, S. E., and Leconte, A. M. (2015) Taq DNA polymerase mutants and 2′-modified sugar recognition. Biochemistry 54, 5999–6008 10.1021/acs.biochem.5b00689 [DOI] [PubMed] [Google Scholar]
- 72. Ellefson, J. W., Gollihar, J., Shroff, R., Shivram, H., Iyer, V. R., and Ellington, A. D. (2016) Synthetic evolutionary origin of a proofreading reverse transcriptase. Science 352, 1590–1593 10.1126/science.aaf5409 [DOI] [PubMed] [Google Scholar]
- 73. Ong, J. L., Loakes, D., Jaroslawski, S., Too, K., and Holliger, P. (2006) Directed evolution of DNA polymerase, RNA polymerase and reverse transcriptase activity in a single polypeptide. J. Mol. Biol. 361, 537–550 10.1016/j.jmb.2006.06.050 [DOI] [PubMed] [Google Scholar]
- 74. Ghadessy, F. J., Ramsay, N., Boudsocq, F., Loakes, D., Brown, A., Iwai, S., Vaisman, A., Woodgate, R., and Holliger, P. (2004) Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution. Nat. Biotechnol. 22, 755–759 10.1038/nbt974 [DOI] [PubMed] [Google Scholar]
- 75. Loakes, D., Gallego, J., Pinheiro, V. B., Kool, E. T., and Holliger, P. (2009) Evolving a polymerase for hydrophobic base analogues. J. Am. Chem. Soc. 131, 14827–14837 10.1021/ja9039696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ghadessy, F. J., Ong, J. L., and Holliger, P. (2001) Directed evolution of polymerase function by compartmentalized self-replication. Proc. Natl. Acad. Sci. U. S. A. 98, 4552–4557 10.1073/pnas.071052198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tawfik, D., and Griffiths, A. D. (1998) Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 16, 652–656 10.1038/nbt0798-652 [DOI] [PubMed] [Google Scholar]
- 78. Pinheiro, V. B., Taylor, A. I., Cozens, C., Abramov, M., Renders, M., Zhang, S., Chaput, J. C., Wengel, J., Peak-Chew, S. Y., McLaughlin, S. H., Herdewijn, P., and Holliger, P. (2012) Synthetic genetic polymers capable of heredity and evolution. Science 336, 341–344 10.1126/science.1217622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lu, W.-C., and Ellington, A. D. (2013) In vitro selection of proteins via emulsion compartments. Methods 60, 75–80 10.1016/j.ymeth.2012.03.008 [DOI] [PubMed] [Google Scholar]
- 80. Davidson, E. A., Diugosz, P. J., Levy, M., and Ellington, A. D. (2009) Directed evolution of proteins in vitro using compartmentalization in emulsions. Curr. Protoc. Mol. Biol. 24, 24–26 10.1002/0471142727.mb2406s87 [DOI] [PubMed] [Google Scholar]
- 81. Miller, O. J., Bernath, K., Agresti, J. J., Amitai, G., Kelly, B. T., Mastrobattista, E., Taly, V., Magdassi, S., Tawfik, D. S., and Griffiths, A. D. (2006) Directed evolution by in vitro compartmentalization. Nat. Methods 3, 561–570 10.1038/nmeth897 [DOI] [PubMed] [Google Scholar]
- 82. Pinheiro, V. B., Arangundy-Franklin, S., and Holliger, P. (2014) Compartmentalized self-tagging for in vitro-directed evolution of XNA polymerases. Curr. Protoc. Nucleic Acid Chem. 57, 1–18 10.1002/0471142700.nc0909s57 [DOI] [PubMed] [Google Scholar]
- 83. Kim, Y., Eom, S. H., Wang, J., Lee, D.-S., Suh, S. W., and Steitz, T. A. (1995) Crystal structure of Thermus aquaticus DNA polymerase. Nature 376, 612–616 10.1038/376612a0 [DOI] [PubMed] [Google Scholar]
- 84. Ollis, D. L., Brick, P., Hamlin, R., Xuong, N. G., and Steitz, T. A. (1985) Structure of large fragment of Escherichia coli DNA polymerase-I complexed with dTMP. Nature 313, 762–766 10.1038/313762a0 [DOI] [PubMed] [Google Scholar]
- 85. Malyshev, D. A., Dhami, K., Lavergne, T., Chen, T., Dai, N., Foster, J. M., Corrèa, I. R., Jr., and Romesberg, F. E. (2014) A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388 10.1038/nature13314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Negi, I., Kathuria, P., Sharma, P., and Wetmore, S. D. (2017) How do hydrophobic nucleobases differ from natural DNA nucleobases? Comparison of structural features and duplex properties from QM calculations and MD simulations. Phys. Chem. Chem. Phys. 19, 16365–16374 10.1039/c7cp02576a [DOI] [PubMed] [Google Scholar]
- 87. Malyshev, D. A., Pfaff, D. A., Ippoliti, S. I., Hwang, G. T., Dwyer, T. J., and Romesberg, F. E. (2010) Solution structure, mechanism of replication, and optimization of an unnatural base pair. Chem. Eur. J. 16, 12650–12659 10.1002/chem.201000959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Betz, K., Malyshev, D. A., Lavergne, T., Welte, W., Diederichs, K., Romesberg, F. E., and Marx, A. (2013) Structural insights into DNA replication without hydrogen bonds. J. Am. Chem. Soc. 135, 18637–18643 10.1021/ja409609j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kimoto, M., Kawai, R., Mitsui, T., Yokoyama, S., and Hirao, I. (2009) An unnatural base pair system for efficient PCR amplification and functionalization of DNA molecules. Nucleic Acids Res. 37, e14 10.1093/nar/gkn956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Georgiadis, M. M., Singh, I., Kellett, W. F., Hoshika, S., Benner, S. A., and Richards, N. G. J. (2015) Structural basis for a six nucleotide genetic alphabet. J. Am. Chem. Soc. 137, 6947–6955 10.1021/jacs.5b03482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Molt, R. W., Jr., Georgiadis, M. M., and Richards, N. G. J. (2017) Consecutive non-natural PZ nucleobase pairs in DNA impact helical structure as seen in 50 µs molecular dynamics simulations. Nucleic Acids Res. 45, 3643–3653 10.1093/nar/gkx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Singh, I., Laos, R., Hoshika, S., Benner, S. A., and Georgiadis, M. M. (2018) Snapshots of an evolved DNA polymerase pre- and post-incorporation of an unnatural nucleotide. Nucleic Acids Res. 46, 7977–7988 10.1093/nar/gky552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang, W., Wu, E. Y., Hellinga, H. W., and Beese, L. S. (2012) Structural factors that determine selectivity of a high fidelity DNA polymerase for deoxy-, dideoxy-, and ribonucleotides. J. Biol. Chem. 287, 28215–28226 10.1074/jbc.M112.366609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Betz, K., Kimoto, M., Diederichs, K., Hirao, I., and Marx, A. (2017) Structural basis for expansion of the genetic alphabet with an artificial nucleobase pair. Angew. Chem. Int. Ed. Engl. 56, 12000–12003 10.1002/anie.201704190 [DOI] [PubMed] [Google Scholar]
- 95. Dror, R. O., Dirks, R. M., Grossman, J. P., Xu, H., and Shaw, D. E. (2012) Biomolecular simulation: a computational microscope for molecular biology. Annu. Rev. Biophys. 41, 429–452 10.1146/annurev-biophys-042910-155245 [DOI] [PubMed] [Google Scholar]
- 96. Walker, A. R., and Cisneros, G. A. (2017) Computational simulations of DNA polymerases: detailed insights on structure/function/mechanism from native proteins to cancer variants. Chem. Rex. Toxicol. 30, 1922–1935 10.1021/acs.chemrestox.7b00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Meli, M., Sustarsic, M., Craggs, T. D., Kapanidis, A. N., and Colombo, G. (2016) DNA polymerase conformational dynamics and the role of fidelity-conferring residues: insights from computational simulations. Front. Mol. Biosci. 3, 20 10.3389/fmolb.2016.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ouaray, Z., Singh, I., Georgiadis, M. M., and Richards, N. G. J. (2020) Building better enzymes: molecular basis of improved non-natural nucleobase incorporation by an evolved DNA polymerase. Protein Sci. 29, 455–468 10.1002/pro.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Skjaerven, L., Martinez, A., and Reuter, N. (2011) Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins 79, 232–243 10.1002/prot.22875 [DOI] [PubMed] [Google Scholar]
- 100. Taylor, D., Cawley, G., and Hayward, S. (2014) Quantitative method for the assignment of hinge and shear mechanism in protein domain movements. Bioinformatics 30, 3189–3196 10.1093/bioinformatics/btu506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grant, B. J., Rodrigues, A. P. C., El Sawy, K. M., McCammon, J. A., and Caves, L. S. D. (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22, 2695–2696 10.1093/bioinformatics/btl461 [DOI] [PubMed] [Google Scholar]
- 102. Miller, B. R., 3rd, Beese, L. S., Parish, C. A., and Wu, E. Y. (2015) The closing mechanism of DNA polymerase I at atomic resolution. Structure 23, 1609–1620 10.1016/j.str.2015.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Modeste, E., Mawby, L., Miller, B., III, Wu, E., and Parish, C. A. (2019) A molecular dynamics investigation of the thermostability of cold-sensitive I707L Klentaq1 DNA polymerase and its wild-type counterpart. J. Chem. Inf. Model. 59, 2423–2431 10.1021/acs.jcim.9b00022 [DOI] [PubMed] [Google Scholar]
- 104. Yang, Z., Kim, H.-J., Le, J. T., McLendon, C., Bradley, K. M., Kim, M.-S., Hutter, D., Hoshika, S., Yaren, O., and Benner, S. A. (2018) Nucleoside analogs to manage sequence divergence in nucleic acid amplification and SNP detection. Nucleic Acids Res. 46, 5902–5910 10.1093/nar/gky392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nikoomanzar, A., Chim, N., Yik, E. J., and Chaput, J. C. (2020) Engineering polymerases for applications in synthetic biology. Q. Rev. Biophys. 53, e8 10.1017/S0033583520000050 [DOI] [PubMed] [Google Scholar]
- 106. Chen, F., Gaucher, E. A., Leal, N. A., Hutter, D., Havemann, S. A., Govindarajan, S., Ortlund, E. A., and Benner, S. A. (2010) Reconstructed evolutionary adaptive paths give polymerases accepting reversible terminators for sequencing and SNP detection. Proc. Natl. Acad. Sci. U. S. A. 107, 1948–1953 10.1073/pnas.0908463107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Houlihan, G., Arangundy-Franklin, S., Porebski, B. T., Subramanian, N., Taylor, A. I., and Holliger, P. (2020) Discovery and evolution of RNA and Xna reverse transcriptase function and fidelity. Nat. Chem. 12, 683–690 10.1038/s41557-020-0502-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hu, S. S., Offenbacher, A. R., Lu, E. D., and Klinman, J. P. (2019) Comparative kinetic isotope effects on first- and second-order rate constants of soybean lipoxygenase variants uncover a substrate-binding network. J. Biol. Chem. 294, 18069–18076 10.1074/jbc.RA119.010826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hu, S. S., Offenbacher, A. R., Thompson, E. M., Gee, C. L., Wilcoxen, J., Carr, C. A. M., Prigozhin, D. M., Yang, V., Alber, T., Britt, R. D., Fraser, J. S., and Klinman, J. P. (2019) Biophysical characterization of a disabled double mutant of soybean lipoxygenase: the “undoing” of precise substrate positioning relative to metal cofactor and an identified dynamical network. J. Am. Chem. Soc 141, 1555–1567 10.1021/jacs.8b10992 [DOI] [PMC free article] [PubMed] [Google Scholar]