

Abstract
The Rossmann-like fold is the most prevalent and diversified doubly-wound superfold of ancient evolutionary origin. Rossmann-like domains are present in a variety of metabolic enzymes and are capable of binding diverse ligands. Discerning evolutionary relationships among these domains is challenging because of their diverse functions and ancient origin. We defined a minimal Rossmann-like structural motif (RLM), identified RLM-containing domains among known 3D structures (20%) and classified them according to their homologous relationships. New classifications were incorporated into our Evolutionary Classification of protein Domains (ECOD) database. We defined 156 homology groups (H-groups), which were further clustered into 123 possible homology groups (X-groups). Our analysis revealed that RLM-containing proteins constitute approximately 15% of the human proteome. We found that disease-causing mutations are more frequent within RLM domains than within non-RLM domains of these proteins, highlighting the importance of RLM-containing proteins for human health.
Graphical Abstract
Introduction
The Rossmann fold [1,2] is an ancient and structurally diverse fold initially discovered in a wide range of nucleotide-binding proteins that bind diphosphate-containing cofactors such as NAD(H). The Rossmann fold belongs to the doubly-wound superfold, which is one of the most prevalent superfolds in nature [3]. The core of these proteins consists of a three-layered α/β/α sandwich topology, with two sets of β-α-β-α-β units forming a single parallel β-sheet (321456 topology) flanked by α-helices. A defining structural feature of this fold is a crossover between β-strands 3 and 4 that creates a pocket capable of adapting to various ligands [4,5]. A minimal Rossmann-like motif (RLM), which we are targeting in this study, is defined to maintain the doubly-wound three-layer α/β/α sandwich topology and to contain the crossover between the second and third strands (Fig 1) [5]. The RLM occurs in a large number of α/β/α three-layered sandwiches, many of which were suggested to have evolved prior to the Last Universal Common Ancestor (LUCA) from a primordial generic nucleotide-binding domain [6]. The extant Rossmann-like domains are linked to a large variety of metabolic enzymes, DNA/RNA binding, and regulatory proteins and are capable of binding various ligands and small compounds vital for their functions [5,7]. Rossmann-fold domains discovered in CRISPR-Cas systems play important ligand-binding roles in the cyclic oligoadenylate (cOA)-mediated signal transduction pathway [8,9].
Fig 1. Minimal Rossmann-like motif (RLM) definition.

(A) RLM SSEs adapted from 5-formly-3-hydroxy-2-methylpyridine 4-carboxylic acid (FHMPC) 5-dehydrogenase (PDB: 4om8) are numbered and colored in rainbow, with a magenta loop (usually catalytic) between the first β-strand - element I (β1) and the first α-helix - element II (α1). The second α-helix - element IV (α2) forms crossover between the second β-strand - element III (β2) and the third β-strand - element V (β3). The crossover loop is a loop at the N-terminal part of α2. Element IV can be α-helix, β-strand, or loop. The unlabeled SSEs (colored in slate) are considered as an insertion to the RLM, which can occur between element III (β2) and element IV (α2) or in any of the loops connecting the RLM SSEs. (B) An interaction matrix defines RLM search strategy using ProSMoS program [34]. Interaction type “T” considers the angle between vectors corresponding to particular RLM elements. (C) RLM 2D topology diagram. All colors correspond to the panel (A).
Many proteins originated by gene duplication, recombination, and divergence [6], however there are cases when convergent evolution has occurred and a fold has emerged in parallel many times in evolution [10]. RLM-containing proteins may be one such example [5]. However, discrimination between divergent and convergent evolution remains a challenge in evolutionary biology in general and in protein evolution in particular [11]. Being one of the most prevalent folds, the Rossmann fold plays an important role in a large variety of processes [12]. Rossmann fold enzymes account for almost 40% of reference metabolic reactions [5]. Closely related RLM enzymes are able to catalyze different chemical reactions using a similar topologies and can diverge to bind different ligands in their similar binding sites. Conversely, non-homologous RLM domains can converge to catalyze the same reaction or to bind the same ligand using different binding modes [5]. Moreover, some enzymes can undergo functional convergence after evolutionary divergence [13]. In this scenario, two homologous enzymes with distinct functions are both duplicated, and their copies diverge to evolve new functions, which are, however, identical to each other [13]. This evolutionary path has also been suggested for pyridine nucleotide disulfide oxidoreductases, which include a RLM-containing FAD-binding domain [14]. Many proteins are functioning as multidomain conglomerates. Combination of domains’ homologous relationship in such conglomerates may vary [15]. It was shown that domains from superfamilies A and B can be found in combinations AB or BA, but very rarely in both combinations [15]. Rossmann-like domains are an example of this rare exception [7].
Being linked to a large variety of metabolic enzymes, dysfunction of RLM-containing proteins can lead to various diseases [5,16]. For example, renalase is a highly expressed flavoprotein in the kidney and heart that metabolizes catecholamines and catecholamine-like substances. Its FAD-binding domain contains an RLM [17]. There is an association between renalase and stroke risk among patients with type 2 diabetes [18]. Also, SNPs in the renalase gene result in increased risk of hypertension and stroke, as well as unfavorable effects in coronary disease [18]. Another example are the sirtuins, which possess histone deacetylase or mono-ribosyltransferase activity and regulate important pathways in bacteria, archaea, and eukaryotes. Human sirtuin has a two-domain structure composed of a large RLM-containing domain and a smaller zinc-binding motif. Sirtuin takes part in DNA repair and also influences transcriptional repression [19]. Sirtuin also plays an important role in maintenance of metabolic homeostasis, thereby having an impact on several pathways in cancer, metabolism, and aging [20]. Malaria parasite Plasmodium falciparum tryptophanyl-tRNA synthetase (Pf-WRS) belongs to the class I tRNA synthetases, characterized by an RLM-containing catalytic domain [21]. Inhibiting parasite aminoacyl-tRNA synthetases is considered to be a novel approach in antimalarial drug development [22]. Dwivedi et al. showed that DNA-processing protein A (DprA) of Helicobacter pylori, whose DNA-binding domain adopts an RLM topology, was found to be sufficient for binding with either ssDNA or dsDNA and to play a crucial role in the process of natural transformation [23]. Honbou et al. showed that DJ-1 protein, a flavodoxin containing an RLM, participates in pathways related to cell transformation, male fertility, oxidative stress response, and Parkinson’s disease. However, the molecular mechanism by which DJ-1 contributes to these multiple functions remains unknown [24]. Each of these examples demonstrate the significance of targeting RLM proteins for treatment of a wide range of diseases.
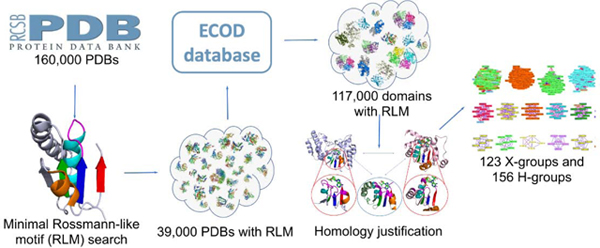
In this study, we identified all RLM-containing domains among known 3D structures from the Protein Data Bank (PDB) and classified them into possible homology groups (X-groups) and homology groups (H-groups), as defined by the Evolutionary Classification of protein Domains (ECOD) database [25,26]. ECOD is a protein classification of homologous domains with a five-level hierarchy: architecture (A), possible homology (X), homology (H), topology (T), and family (F). Recently [5] we discussed the differences between ECOD and other domain classification databases, such as SCOP [27] and CATH [28]. Here we also compared classification of RLM-containing domains in ECOD and new version of SCOP database, i.e. SCOP2 [29] (Supplementary Table 1 and 2, online supplement: http://prodata.swmed.edu/rossmann_fold/scop2_vs_ecod.html). In spite of minimal Rossmann-like motif being the structural core of the majority of RLM-containing domains, manual consideration revealed F-groups in which RLM has deteriorated in their members. Nevertheless, such evolutionary exceptions should be considered as Rossmann-like, due to their homologous relationships with different F-groups that contain the RLM. We showed that RLM-containing proteins constitute approximately 15% of the human proteome. Considering the distribution of RLM domains in the four largest taxonomic groups (archaea, bacteria, eukaryotes and viruses), our analysis revealed homologous domains that belong solely to a single taxonomic group (e.g., bacteria, eukaryotes or viruses). However, we did not find exclusively archaeal RLM-containing H-groups. Analyzing disease-causing mutations (DCMs) in RLM-containing proteins, we found that the overall fraction of DCMs within RLM domains is higher than within non-RLM domains of these proteins. Moreover, we discovered that RLM proteins are linked with a large variety of diseases including, but not limited to different types of cancer [16,20,30], diabetes [18], malaria [21], Parkinson’s disease [24], epilepsy [31], Noonan syndrome [32] among many others. These examples emphasize the significance of targeting RLM proteins for treatment of a wide range of diseases. According to our current classification in ECOD, the large number of possible homology groups (X-groups) suggest that RLM folds have arisen multiple times in evolution. To help explain this observation, we found the contact order of Rossmann-like domains with a repeating α/β topology is significantly lower than that of another superfold, Ig-like, that exhibits non-repetitive topology and has considerably fewer possible homology groups. Thus, the multiple origins of the RLM fold could have arisen in part by an ease of folding RLM domains into their repeating α/β topology.
Results and Discussion
Definition and evolutionary classification of RLM-containing proteins
A minimal RLM folding unit [5,33] is defined as a three-layer α/β/α sandwich, with at least three parallel β-strands and a crossover between the second (element III corresponds to β2, Fig 1) and third β-strands (element V corresponds to β3, Fig 1) of the motif. We refer to the secondary structure elements (SSEs) that comprise the RLM using “β” for β-strands and “α” for α-helices, together with the Arabic numeral of the motif element outlined in Figure 1. For example, element I represents the first β-strand and is designated “β1”. Element IV (helix α2) can be also represented as a β-strand or a linker (loop) without regular secondary structure, whereas the helical cap of element II takes part in binding a variety of ligands (mostly phosphate containing) and was thus represented as α-helix only [5]. The majority of RLM domains contain an α-helix as the fourth element of the RLM (Fig S1). Only three ECOD topology groups (T-groups) contain domains with β-strand as the fourth element of the RLM: FAD/NAD(P)-binding domain (2003.1.2), Nucleotide-binding domain (2003.1.3), and OmpH-like (5094.1.1). Four T-groups contain domains with a linker as the fourth element: DNA-specific exonuclease RecJ C-terminal domain (7511.1.1), STIV B116-like (4259.1.1), Dipeptide transport protein (7535.1.1), and PRTase-like (7573.1.1). Using this RLM definition, more than 117,000 (about 50,000 non-redundant by sequence) RLM domains were detected in the ECOD database. These domains were found in 38,685 (more than 20% of all known protein structures) PDB structures as of June 2020.
Rossmann fold proteins likely arose at the early stages of protein evolution and have frequently diverged since that time, adopting a large variety of functions [5]. Therefore, their classification based on evolutionary relationships is complicated. Protein homology is defined as originating from a common ancestor and is usually identified using sequence and structure similarities [35]. We used these similarities to cluster 117,000 identified RLM domains, and treated these clusters as homologous (see Material and Methods). However, sometimes the overall similarities of sequences and structures are insufficient to detect distant homologs that diverged in the ancient past. In these cases, we considered additional functional justifications for homology defined by the conservation of residues in the active site, as well as the orientation and type of ligands found there. Such justifications are often found transitively, by comparing an intermediate RLM domain with two apparently unrelated ones (as defined by automated clustering).
Our clustering approach, together with additional functional justifications (exemplified below), resulted in 156 homology groups (H-groups). These were further clustered into 123 possible homology groups (X-groups) – the highest level in our classification (the whole dataset: http://prodata.swmed.edu/rossmann_fold/files_fin/clusters_table.html). Our newly obtained classification of RLM-containing domains was incorporated into the ECOD database (beginning with version v275) [25,26]. All changes made to RLM domains classification in ECOD are reflected in Supplementary Table 3. In this table, we defined three types of reclassification. a) Expanded H-group (H-group was expanded by moving to it several H-groups from old version). These are mostly large H-groups containing a large number of RLM domains, for example Rossmann-related (2003.1) and P-loop domains-related (2004.1). This type of reclassification expanded 6 H-groups. b) H-group moved. There are two subtypes of this reclassification. (b1) All domains from this H-group were moved to an expanded H-group. In the current version of classification this H-group no longer exists as an independent unit. (b2) H-group was moved to another X-group and still exists as independent H-group. 41 H-groups belong to (b1) and 2 belong to (b2). One H-group (AtpF-like) encountered both (b1) and (b2) as part of it was moved to an existing H-group and part of it was moved to another X-group and still exists as an H-group. c) New X-group (H-group was reclassified as a new independent X-group). This type of reclassification was mostly applied for H-groups within former “Other Rossmann-like domains” X-group, and overall it was applied for 104 H-groups. Thus, the most significant changes and most affected cases in current ECOD classification of RLM domains are linked to elimination of the largest RLM X-group in previous versions of ECOD, namely “Other Rossmann-like domains”. Among the 104 new X-groups, five are no longer considered as RLM-containing and removed from our RLM dataset and analysis.
Reclassification of the former “Other Rossmann-like domains” X-group provided 102 new X-groups, which constitute about 4% (102 out of 2460) of overall X-groups number in new ECOD v275. Moreover, 44 H-groups changed their location and were moved or merged to another H-groups, representing 22% (44 out of 202) of initial number of RLM-containing H-groups in old versions of ECOD. The majority of 44 H-groups transitive cases are small H-groups, which distant homology relationship to other groups is difficult to detect using automatic methods. There are several general reasons for that. First, the small number of known domain 3D structures, which belong to a particular H-group, may not be sufficient to detect distant homology relationship to other groups. Some of merged H-groups contained only one known domain structure. Second, in addition to the small number of known structures, the poor quality of 3D structures is another obstacle for automatic homology detection. Third, viral proteins are capable of evolving very fast and, in many cases, they adopt unique sequence and structure. Viral proteins are a special cohort in evolutionary classification of domains, which brings significant difficulties in defining the right place in classification for these proteins. Justification of function and active site consideration might help to reveal homologous relationship in all cases discussed above (but not always), which again underlines their importance in evolutionary domains classification.
Possible homologs (i.e. members of the same X-group) are required to have some similarity in architecture and topology that suggests a linkage between these structures. However, significant evidence for homology of domains within an X-group is often lacking. Improved homology detection tools, as well as determination of new protein structures, may allow us to detect more accurate evidence of homology in the future. The lack of experimentally-determined protein structures leading to missing links in evolutionary classification is also supported by the fact that most X-groups in our dataset contain a single homology (H-group) and topology group (T-group) (shown with white background, Supplementary Fig S1 http://prodata.swmed.edu/rossmann_fold/tgr_cons/) that often contains only one family (F-group) (marked with green asterisk Fig S1).
Figure 2A shows distribution of RLM X-groups sizes, for all X-groups that contain more than two F-groups. The top three RLM X-groups that are most populated by families are Rossmann-like (ECOD id: 2003), P-loop domains-like (ECOD id: 2004) and Flavodoxin-like (ECOD id: 2007). For each revised RLM homology group, we outlined a complete set of F-group level representative domains, specifying domain classification details, protein name, Pfam database accession number [36], Pfam seed alignment depth, and signature sequence conserved motif (for example: http://prodata.swmed.edu/rossmann_fold/files_fin/2003.1_reps.html). The largest X-groups mentioned above are mostly represented by the following largest homology groups (H-groups) in our dataset: Rossmann-related (ECOD id: 2003.1), P-loop domains-related (ECOD id: 2004.1), and Class I glutamine amidotransferase-like (ECOD id: 2007.1) groups. The Rossmann-related H-group includes different nucleotide-binding Rossmann-fold domains with a Gly-rich loop (GxGxxG with variations) [37,38] between RLM β1 and α1, and a D/E motif at the C-terminus of β2 [39]. The Rossmann-related H-group also includes domains that have degraded signature motifs, but retain high structure similarity to other members of the group (http://prodata.swmed.edu/rossmann_fold/files_fin/2003.1_reps.html). The P-loop domain-related H-group includes domains with the following signature motifs: phosphate binding Walker A or P-loop motif (G-x(4)-GK-[TS]) between RLM β1 and α1, Walker B motif essential for ATP hydrolysis (G-x(3)-LhhhD[E], h stands for hydrophobic amino acid) at the C-terminus of β3 of second RLM [40], DEAD/DEAH motif in helicases at the C-terminus of β3 of second RLM [41], beta-CASP domains [42], as well as domains with signature motifs degraded [43] or reduced [44] (http://prodata.swmed.edu/rossmann_fold/files_fin/2004.1_reps.html). Class I glutamine amidotransferase-like H-group includes RLM-containing glutamine amidotransferase-like domains with catalytic triad Ser-His-Glu of aspartyl dipeptidases [45], chelatase-like domains [46], periplasmic binding-like domains [47], CheY-like domains [48], etc.
Fig 2.

Distributions of (A) RLM X-groups sizes, for all X-groups which contain more than two F-groups; (B) relative contact order (CO) values for RLM-containing domains (red), Ig-like domains (green) and OB-like domains (blue).
Additionally, we generated heatmaps comparing HHpred homology probability [49], TMalign [50] structure similarity score, and a TM/HH weighted average score (average of HHpred homology probability and TMalign structure similarity score) for representative domains within each homology group, as well as distributions of each of these parameters for representative domains’ comparison both inside and outside of a particular homology group (see Materials and Methods). The total distribution of average score for the representative domains comparison inside and outside of all homology groups illustrates that overall sequence and structure similarity between two domains is not always sufficient to substantiate homology (Fig 3), and that additional justification is necessary (e.g. justification of functional similarity and active site consideration) as discussed below.
Fig 3.

Distributions of average score for representative domains (comparison inside and outside of all homology groups) show a need for functional consideration and manual curation.
To explain the relatively large number of possible homology groups representing RLM domains, we sought to compare characteristics of their folds to those of another superfold, Immunoglobulin-like, that may be less prone to convergent evolution. The topology of RLM domains includes repeating β/α subunits, whose 3-dimensional contacts are expected to exhibit a relatively low average separation in their sequences, or a low contact order (CO). In contrast, Ig-like β-sandwich domains include seven strands in two sheets with a non-repetitive Greek-key topology. A comparison of CO [51] distributions for RLM-containing domains (123 X-groups), immunoglobulin-like domains (one X-group) and OB-like domains (one X-group) revealed significant differences between pairs of distributions RLM vs Ig-like and RLM vs OB-like (Mann-Whitney test P-value < 0.0001, Fig 2B), with the CO of RLM domains shifted towards lower values. Previous studies for two-state folding have suggested the rate of folding decreases with increasing CO, pointing to an important contribution of topological complexity to folding and the transition state [52,53]. Potentially, the multiple independent origins of RLM domains could represent an ease of folding in the transition state, whereby the sequence-local contacts within RLM repeating units can serve as a successful nucleation cores in multiple different native topologies. As such, well-studied Flavodoxin-like proteins are thought to fold through transition states involving different regions of the molecule [54], while the unfolding pathway tends to be similar for IG-like proteins [55]. Similar to the Ig-like domains, the OB-fold is also less prone to convergent evolution [56]. Thus, the variability of transition states gained through ease of folding alternate RLM repeating units might explain the large number of possible homology groups (X-groups) in the evolutionary classification of RLM domains.
Our updated classification of RLM-containing domains revised and eliminated the largest RLM X-group in previous versions of ECOD, namely “Other Rossmann-like domains”, which was an assembly of possibly homologous domains that did not fit elsewhere. Manual consideration, based on topology and sequence similarity, did not substantiate uniting more than 100 of the homology groups within the previous “Other Rossmann-like domains” X-group. The remaining 24 H-groups were found to have possible homology links to other large X-groups in our classification and were moved there accordingly. For example, the pyruvate-ferredoxin oxidoreductase (PFOR), domain III POR family previously formed an independent homology group within the “Other Rossmann-like domains” X-group, but is now classified with other P-loop domains (see discussion below). However, most of the homology groups that were previously placed in the “Other Rossmann-like domains” X-group are now classified as independent ECOD X-groups.
The importance of justification of function and active site consideration can be revealed by the following example. Sequence profile similarity is low (HHpred prob. = 0%) [49,57] between DNA-adenine methyltransferase (Fig 4A) and the classic NAD(P)-binding Rossmann-fold domain from 12-hydroxydehydrogenase/15-Oxo-prostaglandin 13-reductase (Fig 4B) and their structure similarity (Dali Z-score = 4.4) is insufficient for confident homology inference (HHpred probability greater than or equal to 99%, see Materials and Methods). Although the overall topology of both domains is Rossmann-like, the methyltransferase contains an additional antiparallel β-strand (β7) inserted into the sheet, which is a signature structural feature of this topology [58]. This unique insertion in the sheet, combined with the presence of several helical extensions to the core Rossmann-like topology, prevent detection of the overall sequence and structure similarity between these two individual domains.
Fig 4. Example of distant homology that can be detected by active site similarity.

RLM SSEs are colored in rainbow with active site residues shown in magenta. (A) DNA-adenine methyltransferase (PDB: 1yf3) binds SAH. (B) 12-hydroxydehydrogenase /15-Oxo-prostaglandin 13-reductase (PDB: 1v3v) binds NADP. (C) Caffeoyl-CoA O-methyltransferase (PDB: 1sus) binds SAH. Sequence homology probability was calculated using HHpred [49], structure similarity score was calculated using DALI [59]. (D) HHpred sequence alignment of caffeoyl-CoA O-methyltransferase (PDB: 1sus) and 12-hydroxydehydrogenase / 15-Oxo-prostaglandin 13-reductase (PDB: 1v3v). Gly residues from Gly-rich loop are colored by magenta. (E) HHpred sequence alignment of caffeoyl-CoA O-methyltransferase (PDB: 1sus) and DNA-adenine methyltransferase (PDB: 1yf3). Gly residues from Gly-rich loop are colored by magenta.
However, functional considerations reflected in the similarity of their active sites, as well as a transitive relationship to another methyltransferase (which also was not automatically clustered with these domains), supports homology between these two domains. Caffeoyl-CoA O-methyltransferase (Fig 4C) has a common methyltransferase topology that includes the signature antiparallel β-strand and has high structure similarity to the DNA-adenine methyltransferase (Dali Z-score = 9.8). Both domains bind S-adenosyl-L-homocysteine (SAH) at their active sites and are clear homologs. The sequence similarity between the caffeoyl-CoA O-methyltransferase and 12-hydroxydehydrogenase, which binds NADP, suggests homology (HHpred Prob. = 96%). Comparison of the ligand positions in all three proteins revealed similarity of binding modes (Fig 4, bottom pictures). Equivalent elements of the RLM (magenta residues) position the adenine rings of the methyltransferase SAH and the 12-hydroxydehydrogenase NADP in a similar orientation. Moreover, all of these three domains contain a Gly-rich loop located between β1 (dark blue) and α1 (cyan), which contributes to the protein active site. The consensus sequences of caffeoyl-CoA O-methyltransferase (PDB: 1sus) and 12-hydroxydehydrogenase / 15-Oxo-prostaglandin 13-reductase (PDB: 1v3v) each contain three Gly residues (Fig 4D), whereas DNA-adenine methyltransferase (PDB: 1yf3) has only two (Fig 4E). While the composition of the Gly-rich regions in these proteins is slightly different in sequence (Fig 4D–E, Gly residues colored by magenta), they superimpose in their tertiary structures. Gly residues that localize in the N-terminal part of RLM helix α1 (cyan) usually coordinate phosphate [39,60], as is the case for 12-hydroxydehydrogenase /15-Oxo-prostaglandin 13-reductase binding to NADP. The Gly-rich loop from the methyltransferases allows binding of the peptide portion of SAH instead. Taken together, this evidence suggests that these domains are homologs [61–63].
Deteriorations and rearrangements of RLM domains hindering their recognition
The RLM is the most conserved region of Rossmann-like domains [5]. It participates in ligand-binding for many of the RLM domains we studied and is therefore essential for function. However, some Rossmann-like structures lack key secondary structure components that contribute to the RLM. One example of these exceptions resides in pyruvate-ferredoxin oxidoreductase (PFO), which is required for the transfer of electrons from pyruvate to ferredoxin using coenzyme A (CoA), iron-sulfur clusters, and thiamine pyrophosphate [64]. This multi-domain protein typically consists of six domains, including two duplicated RLM-containing thiamin-diphosphate-binding domains, one RLM-containing TK C-terminal domain, and one three-layer α/β sandwich without an identified RLM. Thiamin-diphosphate-binding domains adopt a six-stranded parallel β-sheet (order 213465) flanked by six helices, whereas TK C-terminal domains adopt a six-stranded β-sheet with antiparallel β1 (order 132456) flanked by four helices. These considerable structural differences in topology lead to defining the thiamin-diphosphate-binding and TK C-terminal domains as two independent possible homology groups (X-groups) in our classification, implying a lack of evidence for their homologous relationship to other Rossmann-like proteins. Thus, these domains cannot necessarily be considered as duplications in PFO. They could have arisen by recombination of convergent RLM domains. Similarly, the function and evolutionary relationship of the non-RLM domain from PFO (domain III, Fig 5A) was unclear due to its unique sequence and structural features. Although PFO domain III lacks an RLM, its mainly α/β topology, together with the presence of other RLM domains in the structure, suggested that PFO domain III could be related to RLM domains. PFO domain III has an antiparallel β-strand inserted between the β1 and β2 of the presumed RLM (Fig 5A, colored yellow). This insertion makes this domain topologically unique and lacking significant structural similarity to domains outside its family. Positional conservation in PFO domain III highlights Gly-rich loops between β1 and α1, which are usually considered a signature motif for classic nucleotide-binding Rossmann-like domains [65]. Composition of the Gly-rich region varies between members of the PFO domain III family (Pfam: PF01558). Such diversity of this sequence region can also occur for Rossmann-like domains [44]. Recently, a structure of PFO domain III bound to CoA suggested it plays an important role in the functioning of this protein [44]. The presence of a Gly-rich loop in PFO domain III might suggest a similar CoA binding mode to that of nucleotide-binding Rossmann folds. However, comparison of their CoA binding revealed significant differences. As we showed recently, a common binding mode for CoA by Rossmann-like domains positions the adenine nucleotide ring of CoA in a pocket formed by the conserved Gly-rich motif loop and loops from the crossover, with phosphates coordinated by the N-terminal part of RLM α1 [5]. Alternatively, the binding mode of CoA in PFO reveals the adenine ring coordinated by the N-terminal part of the helix α1. Such a binding mode is inherent for RLM-containing P-loop domains [40,66]. A search for closest structures to PFO domain III outside of its ECOD family identified P-loop domains (Dali Z-scores between 7.6 and 4.9). Indeed, similarity was found both between the active site structure constituents and the positions of the phosphate and adenine ring in PFO domain III and in an identified structure UDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase (Fig. 5B–C). Conserved glycines in the active sites of ligase (Walker A motif GxxGK) and PFO (Gly-rich loop GxGxxG) are aligned (Fig 5B–C, magenta). Binding of CoA is an unusual function for P-loop domains. No domains containing a classical Walker A or P-loop sequence motif (G-x(4)-GK-TS) exist in our dataset that bind CoA as substrate or cofactor. Assuming domain III of PFO evolved from a P-loop domain (due to the similarity of binding site), the domain must have undergone significant structural (insertion of an antiparallel β-strand) and partial sequence changes (it lacks conserved residues K, T and S from the Walker A motif) to adopt a new CoA-binding function. Moreover, some members of the PFO domain III family (PF01558) have entirely lost the Walker A motif (oxalate oxidoreductases, Fig 5C). Oxalate oxidoreductase (OOR) subunit delta has a nearly identical structure to domain III of PFO with an additional “plug loop” at the C-terminal part of the domain (shown in magenta, Fig 5C). The “plug loop” is present only in OOR subunit delta among the members of the POR family (PF01558) and coordinates opening and closing of the binding site to moderate oxalate decarboxylation [44]. Despite this lack of Walker A in OOR, oxalate oxidoreductase subunit delta and PFO domain III should be considered Rossmann-like domains (despite lacking the RLM), since they are homologous to P-loop domains.
Fig 5. POR family domains.

(A) Domain III of pyruvate:ferredoxin oxidoreductase (PDB: 6ciq) with CoA bound. Gly residues from Gly-rich loop colored by magenta. (B) Binding site superposition of pyruvate:ferredoxin oxidoreductase’s domain III (PDB: 6ciq, binds CoA), shown in beige, and UDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase (PDB: 2jfg, binds ATP), shown in slate green. Adenine rings of ATP and CoA are colored in salmon, ribose rings in olive. Walker A motif is shown by magenta. (C) Sequence alignment of pyruvate:ferredoxin oxidoreductase’s domain III (PDB: 6ciq) and UDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase (PDB: 2jfg). Active site residues shown in magenta. (D) Oxalate oxidoreductase (OOR) subunit delta (PDB: 5c4i). “Plug loop” is colored by magenta. (A-C) Presumed RLM elements are colored by rainbow, and the antiparallel β-strands inserted between β1 and β2 of presumed RLM are colored by yellow.
Another example of unusual Rossmann-like domains that lack the RLM is a family of lipases (Lipase_3). Lipases, which catalyze the hydrolysis of acylglycerols, adopt a canonical α/β hydrolase fold whose evolution has been previously discussed [67]. The lipase α/β hydrolase homology group is Rossmann-like and usually includes an RLM. However, members of the Lipase_3 family (PF01764) (e.g, Thermomyces lanuginose lipase, Fig 6A) do not contain an RLM, which is important for functioning of most Rossmann-like enzymes. Members of this family adopt a three-layer α/β/α sandwich with sequentially ordered β-strands in the middle layer lacking the required RLM crossover (Fig 6A). In contrast, other lipase families include the RLM crossover (e.g., a human lipase, Fig 6B). With respect to the Lipase_3 family structure, other lipases have an additional β-strand/α-helix (blue and cyan, Fig 6B) following the core β-strand of the common fold (slate, Fig 6A and B) completing the RLM. The signature conserved sequence motif for all lipases is the catalytic triad (Ser, Asp, His, Fig 6A–E, shown by magenta sticks) [68]. Location of the catalytic triad is similar for Lipase_3 and other Lipase family members (Fig 6A–B), and all three catalytic residues are aligned to each other (Fig 6E, shown in magenta). Within RLM-containing lipase domains, the catalytic triad is located outside of the RLM, and the first catalytic residue Ser152 is located within the loop following the last RLM β-strand β4 (Fig 6B). Another signature structural feature of all lipases is the lid or flap (shown in yellow, Fig 6A–D), which is an α-helix that covers the catalytic triad and interacts with the substrate [68–70]. Lipase_3 (PF01764) family members form the lid following strand β2 (shown in green, Fig 6A), whereas the location of the lid for Lipase (PF00151) family members is shifted to the C-terminal section of the domain, following the strand β7 (shown in brown, Fig 6B). These two lipases bind the same substrate, diundecyl phosphatidylcholine, but use different modes. In the human lipase (Lipase family), RLM elements take part in coordination of the substrate, namely the loop between RLM β1 (dark blue) and α1 (cyan) and helix α2 (orange) (Fig 6B, D). The RLM β1-α1 loop in the human lipase occupies the lid position in T.lanuginose lipase (Lipase_3 family) (Fig 6C–D), however this loop is insufficient for coverage of the catalytic triad and for fully substituting for the function of the lid. The substrate binding mode and lid location difference between members of the Lipase_3 and Lipase families might eliminate evolutionary pressure for maintaining the RLM in the Lipase_3 family. However, they still catalyze the same reaction of triacylglycerol hydrolysis (EC: 3.1.1.3) using the same catalytic triad [69,70]. The existence of cases where the RLM motif had deteriorated within H-groups suggests that in very distant homologues divergence could disrupt this motif to accommodate adaptation to different functionals. Moreover, small H-groups provide additional difficulties in identification of possible homology between them, while many structures remain to be solved that might help in this distinction.
Fig 6. Homologous lipase domains may lack the RLM.

The catalytic triad is shown in magenta sticks. The RLM is colored in rainbow (blue, cyan, green, orange, red). The lid is colored yellow. Diundecyl phosphatidylcholine (PDB: PLC) is shown in sticks and colored by elements. (A) Lipase_3 family (PF01764) member Thermomyces lanuginose lipase (PDB: 1einA), which lacks an RLM, is colored light orange. β-strands that correspond to (B) Lipase family (PF00151) human lipase (PDB: 1lpaB) are colored in corresponding colors. Active site of (C) T.lanuginose lipase (PDB: 1einA) and (D) human lipase (PDB: 1lpaB). (E) Dali structure-based sequence alignment of T.lanuginose lipase (PDB: 1einA) and the human lipase (PDB: 1lpaB). Catalytic triad shown by magenta. Secondary structure elements are shown by the same colors as in (A-B).
An unusual crossing loop unifies RLM structures of functionally diverse homologs.
The peptidyl-tRNA hydrolase-like ECOD homology group (ECOD id: 2011.2) contains seven families that cannot be classified automatically due to a lack of sequence similarity between families (http://prodata.swmed.edu/rossmann_fold/files_fin/2011.2_heatmap_prob.html). Nevertheless, all members possess an unusual crossing loop between RLM β3 and the next antiparallel β-strand (Fig 7), as well as overall similarity in topology of secondary structural elements. Additionally, two of the families share a 3(10)-helix extending the N-terminus of the α1 helix as a structure feature, which takes part in ligand binding. Hydrogenase maturating endopeptidase (HYBD, PDB: 1cfz) exemplifies the fold (Fig 7A). This protein forms a three-layer α/β sandwich with a five-stranded β-sheet (order 21354) flanked by five α-helices. The sheet includes an antiparallel strand β4 formed by an unusual crossing loop between β3 and β4 (shown in red and salmon respectively, Fig 7A). HYBD binds a metal cation [71] using a residue from the unusual 3(10)-helix (yellow in Fig 7A), as well as from the RLM elements β3 and an α-helix C-terminal to the RLM [72]. The RLM helix α2 (shown in orange Fig 7A) also adopts a 3(10)-helix that is specific to the HYBD family (HycI in ECOD). The transition from a 3(10)-helix to an α-helix in α1 of HYBD causes a distinctive bending of the RLM element (Fig 7B).
Fig 7. Transition between 3(10)- and α-helices results in bending of α-helical part.

(A) Hydrogenase maturating endopeptidase from E.coli (PDB: 1cfz). RLM is shown in rainbow. 3(10)-helix is colored in yellow. Metal-binding residues are shown as sticks. Cd2+ is shown as sphere. (B) Main chain sticks representation of RLM elements β1 (C-atoms are colored in blue) and α1 (3(10)-helix part is colored in yellow, α-helix (alpha) part – in cyan) of HYBD. Hydrogen bonds are shown as orange dashed lines.
Archaeal proteasome activator (PDB: 3vr0) is a PAC2 family member of the Peptidyl-tRNA hydrolase-like homology group with a similar domain core as in HYBD. PAC2 includes several additional SSEs including an N-terminal strand extension, a β-hairpin insertion following RLM helix α1, and a β/α insertion C-terminal to the RLM (shown in wheat, marine blue and pink respectively, Fig 8B).
Fig 8. Probable evolutionary path between RLM-containing domains from Peptidyl-tRNA hydrolase-like homology group.

(A) Hydrogenase maturating endopeptidase from E.coli (PDB: 1cfz); (B) archaeal proteasome activator from Pyrococcus furiosus (PDB: 3vr0); (C) PH0006 protein from Pyrococcus horikoshii (PDB: 2gfq); (D) purine nucleoside phosphorylase from E.coli (PDB: 4ts3), formycin A is shown by sticks and colored by elements. RLM is shown in rainbow. 3(10)-helix is colored in yellow in A and B. Metal-binding residues are shown by sticks in A, B. Residues near metal-binding site are shown by sticks in C.
This protein forms a homotetramer that binds to the mature 20S proteasome as a cap, functioning as a proteasome activator [73]. Its 3D structure contains an Au+ atom that interacts with the 3(10)-helix and strand β3, which represents a similar binding mode as seen in HYBD. Helix α2 (Fig 8B, shown orange) is represented by a canonical α-helix in this protein family, whereas HYBD has a 3(10)-helix instead. The middle layer of the domain core of archaeal proteasome activator and HYBD is formed by a β-sheet with strands order 21354 with antiparallel strand β4 and signature crossing loop between β3 and β4 (shown in red and salmon respectively, Fig 8B).
Peptidyl-tRNA hydrolase-like domain of PH0006 protein from P. horikoshii (Fig 8C), a member of D-aminoacyl-tRNA deacylase ECOD family, has a similar topology and middle layer β-strand order (21354) as the two examples described above. This protein has no 3(10)-helices in its structure and its Mg2+ binding site is shifted to the C-terminal segment of the domain (Fig 8C, shown in sticks). This family contains four conserved sequence motifs, which take part in forming the metal binding site: SxH, HxxG, ExxHHxP, and ExG (Fig 8C, H and E residues shown in magenta) [74]. Helix α2 (Fig 8C, shown orange) is also represented by a canonical α-helix, however the angle between α2 and β1 is about 90 degrees, which is uncommon for RLM proteins. PH0006 protein shares similar insertions with archaeal proteasome activator, namely a β-hairpin insertion following RLM helix α1, and a β/α insertion C-terminal to the RLM, as well as the signature crossing loop between β3 and antiparallel β4. With the shifting of the binding site, PH0006 protein underwent a fold change. The α-helix (Fig 8B, shown grey in archaeal proteasome activator) following the antiparallel β-strand (Fig 8B–C, shown in pink) deteriorated into a loop in PH0006 that coordinates a Mg2+ cation (Fig 8C).
Finally, purine nucleoside phosphorylase (PNP) (Fig 8D), being homologous to the three protein families described above, also has a similar topology. PNP catalyzes the reversible phosphorolytic cleavage of the glycosidic bond of purine nucleosides using phosphate [75]. This domain does not contain the aforementioned 3(10)-helices, and its crossing loop between β3 and antiparallel β4 of the domain’s core forms a β-barrel together with the antiparallel β-strand from the β/α insertion C-terminal to the RLM (Fig 8D, shown pink) and β5 of the core (Fig 8D, shown olive). One additional β-strand takes part in the formation of the β-barrel (Fig 8D, shown deep purple). The ligand binding site is also shifted to the C-terminal section of the domain compared to HYBD, however the longer β3 in PNP still interacts with the ligand, whereas α1 helix no longer takes part in binding. The initial catalytic step involves breaking an α-helix α8 into two segments, with one segment moving toward the binding pocket (Fig 8D, dark green α-helix) [75]. This movement positions catalytic residues (Fig 8D, shown in magenta sticks) to interact with the substrate. All this evidence suggests that the probable evolutionary path of the fold between these homologous families shown in Fig 8: α1 3(10)-helix of HYBD and PAC2 families binds functional metal; elimination of the 3(10)-helix and formation of additional β-strands (β-hairpin insertion following RLM helix α1, and a β/α insertion C-terminal to the RLM); the binding site was shifted to the C-terminal section of the domain with the deterioration of the α-helix to a loop that coordinates Mg binding in PH0006 protein; and additional β-strands allowed the formation of the β-barrel, whose elements take part in ligand binding in purine nucleoside phosphorylase.
Glycoside hydrolase minimal RLM domain trimer reveals β-strand deletion.
A putative glycoside hydrolase from Bacteroides thetaiotaomicron (PDB: 3sgg) consists of a C-terminal TIM-barrel domain responsible for hydrolase activity, and an N-terminal trimeric repeat of three RLM domains. Two of these repeats include the core RLM (three parallel β-strands, flanked by two α-helices) and a single additional α-helix. Glycoside hydrolase belongs to the GxGYxYP family that appeared sparsely in eukaryotes through multiple occasions of lateral gene transfer from gut microbiota [76]. These proteins are involved in carbohydrate metabolism and their genes are responsive to α-mannans (mannose polysaccharides) that are used by bacteria as nutrients [76]. Two additional proteins with known 3D structure have a similar trimer tandem duplication formed by three RLM domains: cell wall binding protein 8 (Cwp8, PDB: 5j6q) and cell wall binding protein 6 (Cwp6, PDB: 5j72). RLM domain trimers of these closely related proteins define the cell wall binding 2 (CWB2) family that is required for non-covalent anchoring to the cell wall components of the surface layers [77], and exhibits 148 unique domain architectures over 48 species [78]. Overall, the CWB2 RLM trimer structure is very similar to the B.thetaiotaomicron glycoside hydrolase N-terminal domains, with a minor difference – the middle layer of each CWB2 RLM domain consists of four parallel β-strands (insertions relative to B.thetaiotaomicron glycoside hydrolase N-terminal domains shown in magenta) (Fig 9A–B).
Fig 9. Trimer tandem duplication of RLM-containing domains in distant homologs.

Individual RLMs from (A) B.thetaiotaomicron glycoside hydrolase N-terminal domains (PDB: 3sgg) and (B) C.difficile Cwp8 RLM trimer (PDB: 5j6q) are colored blue, cyan and orange from the N- to the C-terminus. Conserved residues are shown in red stick. Insertions in Cwp8 are colored magenta.
CWB2 RLM trimers bind PS-II – a six-member ring polysaccharide [77,78] using conserved amino acids (Fig 9B, red sticks). The GxGYxYP protein has similar negatively charged residues at corresponding positions (Fig 9A, red sticks). Taken together the similar hydrolase activity, polysaccharide binding, and similarity in conserved positions and structure indicate that RLM trimer domains of GxGYxYP and Cwp proteins are homologs.
Taxonomic distribution of RLM homology groups reveals universal nature of the folds
Rossmann folds are found in ancient, frequently diverged domains that adopt a large variety of functions and perform many types of extant enzymatic reactions. They are known to utilize iron-sulfur clusters, which have been used as electron carriers even when the atmosphere had no oxygen [79]. RLM-containing enzymes take part in the ancient Wood-Ljungdahl metabolic pathway thought to be used by the LUCA [5]. Unsurprisingly, these proteins are found in all kingdoms of life and play roles in various crucial processes.
We determined the overall taxonomic distribution across RLM family and homology groups (Fig 10) using the Pfam database [36] (see Materials and Methods). RLM homology groups with families that are universal, containing proteins from all three major taxonomic lineages (archaeal, bacterial and eukaryotic), constitute 59 out of 156 (37.8%) groups. The proportion of homology groups containing universal protein families is higher when viral proteins are considered (68 out of 156, 43.6%) and constitute the majority of H-groups. These universal H-groups include all of the largest groups (e.g. Rossmann-like, P-loop like, Flavodoxin like, etc.) as well as some smaller ones that consist of a single family (e.g., GT-D type glycosyltransferase, PF08759). The remaining H-groups (29 out of 156, 18.6%) are represented by small, varying fractions of differing combinations of taxonomic groups. Notably, we observed homologous RLM groups with domains from families that belong solely to a single taxonomic group in bacteria, eukaryotes and viruses, but not in archaea. There are only 7 exclusively archaeal RLM-containing F-groups at the family level. Three of these F-groups have unknown function and contain only one known 3D protein structure (2003.1.1.430, 2004.1.1.84, 2005.1.1.78). Domains from DUF1890 F-group (2003.1.1.430, PF09001) belong to the largest RLM-containing Rossmann-related ECOD H-group (2003.1) and contain conserved Gly residues at the location of Gly-rich loop, which is common for members of the Rossmann-related H-group. F-groups 2004.1.1.84 (PF03192) and 2005.1.1.78 (PF19025) do not contain any conserved motifs, however they revealed structure similarity to domains of their homology groups – P-loop domains-related (2004.1) and HUP domains (2005.1) respectively. Enzymes from the F420-dependent methylenetetrahydromethanopterin dehydrogenase (MTD) F-group (2007.1.18.1, PF01993) participate in methane metabolism, which is unique to bacteria and archaea [80]. Members of the Pyrrolysine synthase (PylD) F-group (2007.1.1.39) are responsible for pyrrolysine biosynthesis. Pyrrolysine is an amino acid, which is unique to small groups of archaea and bacteria [81]. Two final F-groups represent functions that are unique to archaea. tRNA (cytidine(56)-2’-O)-methyltransferase (2488.1.1.9, PF01994) is responsible for methylation of cytidine at position 56 in archaea tRNAs. In contrast to archaea, bacteria and eukaryotes contain an unmodified cytidine residue at position 56 [82]. This protein contains a signature alpha/beta knot structural motif and belongs to the diverse alpha/beta knot ECOD H-group (2488.1), which includes all major taxonomical groups. Alpha/beta knots were hypothesized to have evolved from Rossmanoidal precursor, however nothing is known about the origin of the knot in this fold. It was suggested that exclusively archaeal alpha/beta knot family groups were acquired by horizontal gene transfer from bacteria living in similar environmental conditions [83]. Finally, DUF1246 F-group (2003.1.10.6, PF06849) includes 5-formaminoimidazole-4-carboxamide-1-(beta)-D-ribofuranosyl 5’-monophosphate synthetase (FAICAR), which catalyze steps 9 and 10 of archaea purine biosynthesis and mechanism of these steps has differences with bacteria and eukaryote mechanisms [84]. FAICAR belongs to the largest H-group in our dataset – Rossmann-like (2003.1). Thus, these two families (2007.1.18.1 and 2007.1.1.39) represent metabolic functions that are specific to archaea and bacteria. Another two families (2488.1.1.9 and 2003.1.10.6) represent functions that unique to archaea, however members of these F-groups belong to large and diverse H-groups that include all major taxonomical groups. There are 4 exclusively eukaryotic homologous groups in our dataset. All of them are involved in processes that are unique to eukaryotes. For example, “Peroxisome assembly protein 22” H-group (ECOD id: 7593.1), includes of one family: Peroxin-22 (PF12827), which is exclusive to eukaryotes. Peroxin-22 (Pex22p) is membrane protein, which forms the complex with Pex4p protein and catalyze the ubiquitination of other proteins, as well as takes part in peroxisome biogenesis [85,86]. Viruses contain RLM proteins that form three exclusively viral homologous groups. However, due to the fast evolution of viral proteins, most are difficult to classify. For example, the cystovirus bacteriophage phi12 encodes a unique P7 protein with an RLM (PDB: 2q82). We classified Phi12 P7 as its own unique possible homology group (X-group), implying a lack of evidence for homology to existing folds. P7 serves as a putative virion assembly cofactor thought to bind the unique three-segmented double-stranded cystovirus RNA genome [33,87]. While the Phi12 P7 protein probably evolved to bind its unusual genome from a DNA/RNA-binding Rossmann-like protein, the identity of the ancestor remains elusive. Bacteria can also include fast evolving components of the genome that function in host immune response evasion or the bacterial arms race [88]. There are 3 homologous groups with exclusively bacterial families in our dataset. For example, H-group “a/b domain in family 98 glycoside hydrolases”. Family 98 glycoside hydrolases is known to play an important role in bacterial virulence and is specific for distinct host carbohydrate antigens [89,90].
Fig 10. Taxonomic distribution of RLM protein across family and homology groups.

A - Archaea, B - Bacteria, E – Eukaryotes, V – Viruses. Percentage shows the ratio of particular taxonomic groups combination from total number of family or homologous groups respectively. The thickness of lines represents the number of RLM F-groups.
Disease-associated mutations within human RLM proteins
By now, genomes of more than 25,000 species have been sequenced, and more than 160,000 protein structures have been determined. Sequencing of human genomes facilitated the determination of disease-causing mutations (DCMs) in the human proteome. Using bioinformatic methods we identified RLM-containing protein sequences in human (see Materials and Methods) and found that RLM-containing proteins constitute approximately 15% of the human proteome. Such a high proportion can be explained by the adaptability of RLM proteins to multiple functions. Amino acid mutations may alter local structural features of proteins, thereby causing protein dysfunction and disease. RLM enzymes contribute to 38% of all known metabolic reactions [5] and are crucial for human cellular function. We mapped all known DCMs to RLM-containing proteins with determined (from Protein Data Bank) or modeled (from SWISS-MODEL) 3D structures using a single-amino acid variations database recently developed in our lab [91]. We defined 613 human RLM-containing proteins (UniProt IDs) that contain DCMs within characterized residues of determined or modeled structures. A majority of these proteins (361 out of 613, 59%) contain DCMs limited to the RLM domains, 70 (11%) contain DCMs only within the non-RLM domains, and 184 (30%) contain DCMs within both RLM and non-RLM domains. Figure 11 shows the distribution of DCMs among the 613 defined human RLM-containing proteins within their RLM and non-RLM domains. The overall fraction of DCMs within RLM domains is higher than within non-RLM domains, although the difference is not significant according to the Fisher exact test.
Fig 11. Distribution of DCM fraction within RLM and non-RLM domains.

DCM fraction is defined as ratio of number of residues linked with DCM to the total number of residues in the chain.
Changes in RLM domains classification with DCMs might reveal new insights in the significance of a particular mutation in the light of newly identified homologous relationship. Overall 114 RLM proteins with DCMs underwent classification changes in ECOD (Supplementary Table 4). Most of these changes are linked to the reclassification of homology groups to new X-groups during the elimination of “Other Rossmann-like” X-group, discussed above. However, there are some exceptions. Human ornithine transcarbamylase (PDB: 1ep9), previously formed an independent H-group, was found to have significant structure similarity with domains from Rossmann-related H-group (Dali Z-score=10.8 with formate dehydrogenase, PDB: 4xye) and was moved to this H-group (2003.1). Ornithine transcarbamylase also contains conserved Gly residue in the loop between β1 and α1 of RLM, which is the reduced Gly-rich loop, common for Rossmann-related H-group domains. Mutation of the conserved G197 (as well as most of known DCMs in this protein) is linked with ornithine carbamoyltransferase deficiency (OTCD) – a disorder of the urea cycle which causes a form of hyperammonemia [92,93]. This protein has one of the largest fraction values of DCMs in RLM domain in our dataset – 29% (Supplementary Table 4).
We classified each of the 613 defined human RLM-containing proteins with DCMs based on disease class using the Genetic Association Database (GAD) [94]. Table 1 shows top three GAD disease classes in which human RLM proteins with DCMs are significantly overrepresented based on Fisher exact test (P<0.05).
Table 1. Top three Genetic Association Database (GAD) disease classes with significantly overrepresented RLM proteins.
Percentage was calculated based on number of RLM proteins (UniProt IDs) found in GAD (613 out of 615).
| GAD disease class | Number of proteins | Percentage | P-value | Fisher exact test |
|---|---|---|---|---|
| Metabolic | 236 | 38.5 | 2.8E-2 | 2.5E-2 |
| Cancer | 176 | 28.7 | 3.4E-6 | 2.5E-6 |
| Neurological | 156 | 25.4 | 5.4E-5 | 3.9E-5 |
Almost 40% of all RLM proteins with DCMs are linked to metabolic diseases (236 out of 613). This result agrees with our recent finding that RLM enzymes comprise 38% of reference metabolic pathways and are overrepresented in nucleotide metabolism, energy metabolism, and metabolism of amino acids [5]. For example, aromatic-L-amino-acid decarboxylase (AAD) catalyzes the decarboxylation of L-3,4-dihydroxyphenylalanine (DOPA) to dopamine and L-5-hydroxytryptophan to serotonin, utilizing pyridoxal 5’-phosphate (PLP) as a cofactor [95]. This enzyme adopts a fold with a seven-stranded β-sheet (order 3245671, with 324 forming RLM, and with antiparallel β7), flanked by nine α-helices (Fig 12A) and belongs to the PLP-dependent transferases ECOD homology group. Mutations within AAD cause aromatic L-amino-acid decarboxylase deficiency (AADCD), which is a disorder of monoamine neurotransmitter metabolism, characterized by significant developmental and psychomotor delay and autonomic dysfunction [96]. Mutations within AAD lead to decreased binding affinity of the enzyme for substrate (DOPA), which results in decreasing dopamine levels [97].
Fig 12. Disease-causing mutations within RLM-containing domains.

(A) Aromatic-L-amino-acid decarboxylase (PDB: 3rbf). Positions of the mutations associated with aromatic L-amino-acid decarboxylase deficiency are colored in magenta. (B) BRCT domains of Breast cancer type 1 (BRCA1) susceptibility protein (PDB: 4y2g). Positions of mutations associated with ovarian and breast cancer are colored in magenta. (C) Calcium-sensing receptor (CaSR) domains of G-protein-coupled receptor (GPCR) (PDB: 5k5s). Positions of mutations associated with hypocalciuric hypercalcemia, hyperparathyroidism, hypocalcemia and epilepsy are colored by magenta. (D) Nucleotide-binding domain of Ras protein (PDB: 4dsn). Positions of mutations associated with Noonan syndrome are colored by magenta. (A-D) RLMs are colored by rainbow.
About 30% of all RLM proteins with DCMs are linked to cancer (28.7%). RLM proteins are also involved in different processes related to cancer, for example Breast cancer type 1 susceptibility protein (BRCA1). BRCA1 is a large protein of 1863 amino acids, which consists of an N-terminal RING domain and two C-terminal Rossmann-like tandem BRCT domains (Fig 12B). BRCA1 interacts with DNA damage response sensors, coordinating the recognition of DNA damage sites and DNA repair [16]. BRCA1 is a tumor suppressor, and various mutations within it are associated with breast and ovarian cancer [30]. BRCT domains adopt a four-stranded parallel β-sheet (order 2134) flanked by four helices. These domains can recognize phosphorylated proteins [98], and this ability is essential for BRCA1’s tumor suppression function [99]. There were 254 identified BRCA1 mutations linked to cancer development, and at least 19 positions with DCMs are in BRCT domains (Fig 12B, colored in magenta), which can cause the malfunction of this protein and increased breast cancer risks [100–102].
Calcium-sensing receptor (CaSR) domains are extracellular domains of G-protein-coupled receptor (GPCR) that maintain extracellular Ca2+ homeostasis in blood [103]. CaSR domains belong to the Flavodoxin-like ECOD X-group and usually form a dimer (Fig 12C). Mutations within these domains are linked to hypocalciuric hypercalcemia [104], hyperparathyroidism [105], hypocalcemia [106] and epilepsy [31]. Epilepsy belongs to the neurological class of diseases and 25.4% of all RLM proteins with DCM are linked to neurological diseases. CaSR domains play crucial roles in GPCR activation, which usually proceeds in two steps. During the first step, an amino acid agonist binds at the interface between two CaSR domains (Fig 12C, shown by sticks).
The second step of activation requires binding of a Ca2+ ion [105]. Similar to other GPCRs [107], CaSR exists in a conformational equilibrium between inactive and active states. Some DCMs within the CaSR domain can be described as loss-of-function mutations. For example, mutation R465Q within RLM-containing domain is characterized by a blunted response to calcium stimulation, thereby causing hypocalciuric hypercalcemia [108].
Additionally, about 13% of all RLM proteins with DCMs are linked to developmental diseases (80 out of 613). Ras is a GTP-binding protein that regulates cellular responses to different extracellular stimuli and is oncogenic; cancer cells often express mutant Ras proteins [109]. The nucleotide-binding domain of Ras proteins adopts a six-stranded β-sheet with antiparallel β3 (order 321456) flanked by five helices (Fig 12D) and belongs to the P-loop homology group, since it contains a classical Walker A motif GxxxxGKTS [110]. Although DCMs in Ras proteins are primarily associated with cancer [111], they can also be linked to several developmental disorders, such as Noonan, Costello, and cardio-facio-cutaneous syndromes [32,109]. Noonan syndrome is an autosomal dominant dysmorphic syndrome characterized by short stature, facial dysmorphism, skeletal abnormalities, cardiac defects, and learning disabilities [109]. Ras proteins bind GDP/GTP and possess intrinsic GTPase activity [112], cycling between active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound conformations [113]. Mutations within RLM-containing GDP/GTP-binding domain of Ras proteins reduce their activity and impair their responsiveness to GTPase activating proteins [113]. Ras proteins are also an important part of the Ras–Raf–MEK–ERK pathway, malfunction of which may cause developmental anomalies and carcinogenesis [109].
Conclusions
In this study, we defined the minimal Rossmann-like structural motif (RLM) and identified RLM-containing domains among known 3D structures from the Protein Data Bank. These domains were found in 38,685 PDB structures, more than 20% of known structures. Classification of these RLM-containing domains into 123 possible homology groups (X-groups) and 156 homology groups (H-groups) was non-trivial, requiring detailed manual analysis of about 70% of H-groups. Our new classification revised the largest RLM-containing ECOD X-group (“Other Rossmann-like domains”) by creating independent X-groups for most of its previously assigned H-groups, as well as by merging some of its H-groups with other large X-groups such as Rossmann-like (ECOD id: 2003), P-loop domains-like (ECOD id: 2004) and Flavodoxin-like (ECOD id: 2007). These new classifications were incorporated into the ECOD database, beginning with version v275 (20200517). In spite of the minimal Rossmann-like motif being the structural core of the majority of RLM-containing domains, manual consideration of homology revealed deterioration of RLM structure elements in some homologs, for example pyruvate-ferredoxin oxidoreductase domain III and Lipase_3 family. These are evolutionary exceptions, nevertheless they should be considered as Rossmann-like domains because they are homologous to other F-groups that contain RLMs.
Our analysis showed that, confined in the diversity of RLM domains, there are homologous domains that belong solely to a single taxonomic group (bacteria, eukaryotes or viruses). However, we did not find exclusively archaeal RLM-containing H-groups. We showed that RLM-containing proteins constitute approximately 15% of the human proteome. Analyzing disease-causing mutations (DCMs) in RLM-containing proteins, we found that the overall fraction of DCMs within RLM domains is higher than within non-RLM domains. The top Genetic Association Database disease classes with significantly overrepresented RLM proteins with DCMs are metabolic, cancer and neurological.
RLM proteins are ubiquitous in nature. As we recently showed, one of the reasons for their wide distribution is their ability to bind various types of ligands, which is provided by the incorporation of an RLM into a broad array of structural contexts [5]. Moreover, protein promiscuity was shown to be an important component of protein evolution [114,115]. Another possible reason for the success of RLM proteins might be their repetitive topology type, which makes them more likely to emerge during evolution in comparison to non-repetitive topologies. A comparison of contact order distributions for RLM-containing domains and immunoglobulin-like domains (a non-repetitive superfold) revealed significant differences between these two distributions. This observation agrees with the likely multiple independent origins of RLM domains in comparison to the non-repetitive topology of IG-like domains and might explain the large number of possible homology groups (X-groups) in the evolutionary classification of RLM domains. Taken together, our data reveal that RLM-containing proteins represent an example of highly successful evolutionary structural unit, which arose multiple times in evolution and adopted a large variety of functions among all domains of life.
Materials and Methods
Identifying RLMs in ECOD domains using ProSMoS
The minimal RLM was defined as a three-layer α/β/α sandwich with the central β-sheet containing a minimum of three parallel β-strands (β1, β2, and β3 in Fig 1). We require the second element to be α-helical to maintain the α/β doubly-wound characteristic of Rossmann-like folds and to maintain the known ligand binding site. To accurately represent all known Rossmann-like crossover connections between β-strands β2 and β3, element IV includes three variations: α-helix, β-strand or linker (Fig 1B).
We used the minimal RLMs described above as queries to search against all known protein structures in the Protein Data Bank [116] using the ProSMoS program developed in our lab [34]. We used PALSSE [117] to generate a database of secondary-structure interaction matrices derived from ECOD domains. Each matrix describes the interactions (parallel or antiparallel) and hydrogen-bonding of the PDB structure. This minimal structural consensus of RLM domains was represented as three ProSMoS query matrices. Query matrices specified the number and types of secondary structure elements in the motif under consideration, the hydrogen-bonding and parallel or anti-parallel relationships between its elements, and minimum and maximum length of the three component β-strands. All β-strands were required to be at least three amino acids in length. Out of more than 117,000 domains, which contain the RLM, only 840 domains were not identified by ProSMoS. There were multiple reasons for this: deteriorated or missing RLM β-strands (e.g., e5da1A2) or an unusual element IV (e.g., e3i12C4). The flowchart of data collecting is shown in Fig S2.
Domains were considered to belong to a Rossmann-like fold when the RLM overlapped with the evolutionarily conserved structural core. Domains from each PDB structure from the ProSMoS search results were annotated using ECOD. ECOD domains are identified by an identifier (e.g. “e1l5jB5”), which incorporates a) the PDB identifier, b) a chain identifier (sometimes multicharacter), and c) a domain number. In the current work we used the following hierarchy of structural definitions: structures/depositions from the PDB contain multiple proteins/chains which can contain one to many ECOD domains. We define “fold” as synonymous to ECOD topology groups. All RLMs identified must overlap completely with an ECOD domain to be considered. Consequently, each identified RLM is fully contained as part of an ECOD domain and cannot belong to two different domains at the same time.
Clustering of RLM domains into homologous groups
For each identified RLM domain, we built a sequence profile using HHblits [118] with default settings against the NCBI non-redundant protein sequence database (National Center for Biotechnology Information, NIH, Bethesda, MD). Sequence profiles of RLM domains were integrated into an RLM sequence library using the HHpred package [49]. All-against-all searches querying domain profiles against the RLM sequence library were performed using the HHsearch method from the HHpred package [49].
Clustering was performed in several steps. First, we clustered all RLM domains using the BLASTClust program [119] with a 40% identity threshold. At this step we obtained 6518 clusters. Then, from each BLASTClust cluster we randomly picked one representative domain. To determine sequence similarity between cluster representatives, we ran HHsearch using sequence profiles of BLASTClust cluster representative domains against the RLM sequence library. All hits with a HHsearch probability higher or equal 99% for particular representative domain were considered as members of this cluster. Thereby we obtained 6518 redundant clusters (one domain may belong to more than one cluster). Next, we merged all clusters that contain at least one common domain. At this step we obtained 594 clusters. For new clusters we again randomly picked 594 representative domains and calculated structure similarity between them using Dali all-against-all [59]. All cases with Dali Z-score ≥ 8 [120] were manually investigated. For these cases we considered additional functional justifications for homology defined by conservation of residues in the active site, as well as the position and type of ligands found there (Fig 4). Cluster representatives with similarity in binding site, ligands type, and position were considered homologs and all members of both clusters were placed in a single homology group. In total we obtained 156 RLM homology groups, which are represented in ECOD as H-groups. The flowchart of data processing is shown in Fig S3.
Supplementary Fig S1 illustrates the conserved core of RLM-containing ECOD topology groups (T-groups). The total number of RLM family (F-group) is 1260. For each ECOD RLM F-group we choose one representative domain. To determine the conserved core of a particular T-group we picked the longest F-group representative domain and calculated its structural alignment to the remainder of representatives within this T-group using TMalign [50]. Using this TMalign structural alignment we also obtained “master-slave” sequence alignments for the longest representative. Then we added sequence profiles of each representative to the “master-slave” alignment and calculated frequency of gaps in each position. All positions with gaps frequency ≤ 20% were included into core and colored in rainbow in Fig S1.
Fig 3 displays distributions of HH/TM average scores within and between homology groups. HH/TM average score was calculated as the average between HHsearch probability (scaled between 0 to 1) and TMalign score. These distributions were calculated for each F-group representative domain. For each representative domain we chose values of HHsearch probability, TMalign score, and average score against all other representative domains within the homology group and against all RLM domains not within the representative’s homology group. Aggregation of these values for all representative domains is illustrated by the distributions in Fig 3. These distribution plots were created using heatmaply R package [121].
Taxonomic distribution of RLM protein families
To determine the taxonomic distribution of ECOD RLM homology groups, we first checked the taxonomy of each family group from the Pfam database [36] associated with a particular homology group. For this analysis we considered all domain sequences (not only proteins with known 3D structure) that belong to a particular family and their distribution between the four major taxonomic groups: archaea, bacteria, eukaryotes and viruses. Combinations of taxonomical groups (see Fig 10) for domain families associated with a particular homology group, revealed overall combination of taxonomical groups for this H-group. Each H-group belongs to one combination of taxonomical groups. One H-group (ECOD id: 7602.1) from our data set has no Pfam database assignment and is therefore excluded from taxonomical analysis. Our BLAST sequence search for a representative domain revealed that Bacteria and Archaea proteins are linked to this H-group.
Mapping of disease-causing mutations to human RLM protein structures
To map disease-causing mutations to human RLM proteins we first identified all RLM proteins in human proteome. We used the reference human proteome from UniProt KB [122], proteome ID: UP000005640. For this analysis we used only reviewed proteins. Identification of RLM proteins within the human proteome was performed in three steps. At the first step we collected all human UniProt IDs with identified 3D structure which contained an identified RLM. At the second step we identified all human UniProt IDs without known 3D structure that belong to RLM Pfam database v32.0 [36] families. The mapping of RLM ECOD families to Pfam families was derived from ECOD database [25,26]. At the third step we use BLAST [119] on the remaining UniProt sequences in the human proteome against sequences of all RLM domains. If the query hit an RLM domain with E-value cutoff 0.05, the query protein was considered to be RLM-containing. We mapped disease-causing mutations to human RLM protein structures using a single-amino acid variations database developed in our lab (http://prodata.swmed.edu/DBSAV/) [91]. For mapping, we used only RLM proteins with known 3D structure from Protein Data Bank [116] or with predicted 3D structure from SWISS-MODEL [123]. Disease classes were collected from the Genetic Association Database (GAD) [94]. RLM proteins were assigned to disease classes using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 [124].
Contact order calculation
For contact order (CO) calculation we used a program developed by Plaxco et al. [51], which calculated relative CO as average sequence separation divided to the protein length. We used default contact cutoff (6 Angstroms). We calculated contact orders for all F-group representative RLM domains (1260 representatives) and compared them with the contact order of F-group representative domains from Immunoglobulin-like beta-sandwich (ECOD id: 11) ECOD X-group (693 representatives) and OB-fold (ECOD id: 2) ECOD X-group (236 representatives).
Supplementary Material
The Rossmann-like domains are the most populated among known structures of α/β-folds in PDB.
Their functional features and evolutionary relationship might suggest targets for drug design
Classification revealed 123 possible homology groups and 156 homology groups
Fraction of disease-causing mutations within RLM domains is higher than within non-RLM domains
Acknowledgements
This work was supported by the National Institutes of Health [GM127390 to N.V.G.] and the Welch Foundation [I-1505 to N.V.G.].
Footnotes
Competing interests
The authors have declared that no competing interests exist.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aravind L, Anantharaman V, & Koonin EV (2002). Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP‐ATPase nucleotide‐binding domains: implications for protein evolution in the RNA world. Proteins. 48(1), 1–14. [DOI] [PubMed] [Google Scholar]
- 2.Aravind L, de Souza RF, & Iyer LM (2010). Predicted class-I aminoacyl tRNA synthetase-like proteins in non-ribosomal peptide synthesis. Biol. direct 5(1), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orengo CA, Jones DT, & Thornton JM (1994). Protein superfamilles and domain superfolds. Nature. 372(6507), 631–634. [DOI] [PubMed] [Google Scholar]
- 4.Rossmann MG, Moras D, & Olsen KW (1974). Chemical and biological evolution of a nucleotide-binding protein. Nature. 250(5463), 194–199. [DOI] [PubMed] [Google Scholar]
- 5.Medvedev KE, Kinch LN, Schaeffer RD, & Grishin NV (2019). Functional analysis of Rossmann-like domains reveals convergent evolution of topology and reaction pathways. PLoS Comput. Biol 15(12), e1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aravind L, Mazumder R, Vasudevan S, & Koonin EV (2002). Trends in protein evolution inferred from sequence and structure analysis. Curr. Opin. Struct. Biol 12(3), 392–399. [DOI] [PubMed] [Google Scholar]
- 7.Bashton M, & Chothia C. (2002). The geometry of domain combination in proteins. J. Mol. Biol 315(4), 927–939. [DOI] [PubMed] [Google Scholar]
- 8.Makarova KS, Anantharaman V, Grishin NV, Koonin EV, & Aravind L. (2014). CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front. Genet 5, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koonin EV, & Makarova KS (2018). Discovery of oligonucleotide signaling mediated by CRISPR-associated polymerases solves two puzzles but leaves an enigma. ACS. Chem. Biol 13(2), 309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kopec KO, & Lupas AN (2013). β-Propeller blades as ancestral peptides in protein evolution. PLoS One, 8(10), e77074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galperin MY, & Koonin EV (2012). Divergence and convergence in enzyme evolution. J. Biol. Chem 287(1), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards H, Abeln S, & Deane CM (2013). Exploring fold space preferences of new-born and ancient protein superfamilies. PLoS Comput. Biol 9(11), e1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todd AE, Orengo CA, & Thornton JM (2002). Plasticity of enzyme active sites. Trends Biochem. Sci 27(8), 419–426. [DOI] [PubMed] [Google Scholar]
- 14.Kuriyan J, Krishna TSR, Wong L, Guenther B, Pahler A, Williams CH, & Model P. (1991). Convergent evolution of similar function in two structurally divergent enzymes. Nature. 352(6331), 172–174. [DOI] [PubMed] [Google Scholar]
- 15.Apic G, Gough J, & Teichmann SA (2001). Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mol. Biol 310(2), 311–325. [DOI] [PubMed] [Google Scholar]
- 16.Wu Q, Paul A, Su D, Mehmood S, Foo TK, Ochi T, … & Blundell TL (2016). Structure of BRCA1-BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol. Cell 61(3), 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desir GV, Wang L, & Peixoto AJ (2012). Human renalase: a review of its biology, function, and implications for hypertension. J. Am. Soc. Hypertens 6(6), 417–426. [DOI] [PubMed] [Google Scholar]
- 18.Buraczynska M, Zukowski P, Buraczynska K, Mozul S, & Ksiazek A. (2011). Renalase gene polymorphisms in patients with type 2 diabetes, hypertension and stroke. Neuromolecular Med. 13(4), 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beauharnois JM, Bolívar BE, & Welch JT (2013). Sirtuin 6: a review of biological effects and potential therapeutic properties. Mol. Biosyst 9(7), 1789–1806. [DOI] [PubMed] [Google Scholar]
- 20.Tennen RI, & Chua KF (2011). Chromatin regulation and genome maintenance by mammalian SIRT6. Trends. Biochem. Sci 36(1), 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan S, Garg A, Sharma A, Camacho N, Picchioni D, Saint-Léger A, de Pouplana L, Yogavel M, & Sharma A. (2013). An appended domain results in an unusual architecture for malaria parasite tryptophanyl-tRNA synthetase. PLoS One. 8(6), e66224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatt TK, Khan S, Dwivedi VP, Banday MM, Sharma A, Chandele A, Camacho N, De Pouplana LR, Wu Y, Craig AG and Mikkonen AT (2011). Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat. Commun 2(1), 1–11. [DOI] [PubMed] [Google Scholar]
- 23.Dwivedi GR, Srikanth KD, Anand P, Naikoo J, Srilatha NS, & Rao DN (2015). Insights into the functional roles of N-terminal and C-terminal domains of Helicobacter pylori DprA. PLoS One. 10(7), e0131116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honbou K, Suzuki NN, Horiuchi M, Niki T, Taira T, Ariga H, & Inagaki F. (2003). The crystal structure of DJ-1, a protein related to male fertility and Parkinson’s disease. J. Biol. Chem 278(33), 31380–31384. [DOI] [PubMed] [Google Scholar]
- 25.Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim BH and Grishin NV (2014). ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol 10(12), e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer RD, Kinch L, Medvedev KE, Pei J, Cheng H, & Grishin N. (2019). ECOD: identification of distant homology among multidomain and transmembrane domain proteins. BMC Mol. Cell. Biol 20(1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murzin AG, Brenner SE, Hubbard T, & Chothia C. (1995). SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol 247(4), 536–540. [DOI] [PubMed] [Google Scholar]
- 28.Dawson NL, Lewis TE, Das S, Lees JG, Lee D, Ashford P, Orengo CA and Sillitoe I. (2017). CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 45(D1), D289–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreeva A, Kulesha E, Gough J. and Murzin AG (2020). The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 48(D1), D376–D382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J. and Miki Y. (1994). BRCA1 mutations in primary breast and ovarian carcinomas. Science. 266(5182), 120–122. [DOI] [PubMed] [Google Scholar]
- 31.Kapoor A, Satishchandra P, Ratnapriya R, Reddy R, Kadandale J, Shankar SK, & Anand A. (2008). An idiopathic epilepsy syndrome linked to 3q13. 3‐q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann. Neurol 64(2), 158–167. [DOI] [PubMed] [Google Scholar]
- 32.Kratz CP, Zampino G, Kriek M, Kant SG, Leoni C, Pantaleoni F, Oudesluys‐Murphy AM, Di Rocco C, Kloska SP, Tartaglia M. and Zenker M. (2009). Craniosynostosis in patients with Noonan syndrome caused by germline KRAS mutations. Am. J. Med. Genet. A 149(5), 1036–1040. [DOI] [PubMed] [Google Scholar]
- 33.Medvedev KE, Kinch LN, & Grishin NV (2018). Functional and evolutionary analysis of viral proteins containing a Rossmann‐like fold. Protein Sci. 27(8), 1450–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi S, Zhong Y, Majumdar I, Sri Krishna S, & Grishin NV (2007). Searching for three-dimensional secondary structural patterns in proteins with ProSMoS. Bioinformatics. 23(11), 1331–1338. [DOI] [PubMed] [Google Scholar]
- 35.Kinch LN, & Grishin NV (2002). Evolution of protein structures and functions. Curr. Opin. Struct. Biol 12(3), 400–408. [DOI] [PubMed] [Google Scholar]
- 36.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A. and Sonnhammer ELL, 2019. The Pfam protein families database in 2019. Nucleic Acids Res. 47(D1), D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wierenga RK, Terpstra P, & Hol WG (1986). Prediction of the occurrence of the ADP-binding βαβ-fold in proteins, using an amino acid sequence fingerprint. J. Mol. Biol 187(1), 101–107. [DOI] [PubMed] [Google Scholar]
- 38.Pai CH, Chiang BY, Ko TP, Chou CC, Chong CM, Yen FJ, Chen S, Coward K, Wang AHJ and Lin CH (2006). Dual binding sites for translocation catalysis by Escherichia coli glutathionylspermidine synthetase. EMBO J. 25(24), 5970–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laurino P, Tóth-Petróczy Á, Meana-Pañeda R, Lin W, Truhlar DG, & Tawfik DS (2016). An ancient fingerprint indicates the common ancestry of Rossmann-fold enzymes utilizing different ribose-based cofactors. PLoS Biol. 14(3), e1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neuwald AF, Aravind L, Spouge JL, & Koonin EV (1999). AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9(1), 27–43. [PubMed] [Google Scholar]
- 41.Aubourg S, Kreis M, & Lecharny A. (1999). The DEAD box RNA helicase family in Arabidopsis thaliana. Nucleic Acids Res. 27(2), 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, & Tong L. (2006). Polyadenylation factor CPSF-73 is the pre-mRNA 3’-end-processing endonuclease. Nature. 444(7121), 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorynia S, Lorenz TC, Costaguta G, Daboussi L, Cascio D, & Payne GS (2012). Yeast Irc6p is a novel type of conserved clathrin coat accessory factor related to small G proteins. Mol. Biol. Cell 23(22), 4416–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen PYT, Aman H, Can M, Ragsdale SW, & Drennan CL (2018). Binding site for coenzyme A revealed in the structure of pyruvate: ferredoxin oxidoreductase from Moorella thermoacetica. Proc. Natl. Acad. Sci. U S A 115(15), 3846–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Håkansson K, Wang AHJ, & Miller CG (2000). The structure of aspartyl dipeptidase reveals a unique fold with a Ser-His-Glu catalytic triad. Proc. Natl. Acad. Sci. U S A 97(26), 14097–14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lecerof D, Fodje MN, Leon RA, Olsson U, Hansson A, Sigfridsson E, Ryde U, Hansson M. and Al-Karadaghi S. (2003). Metal binding to Bacillus subtilis ferrochelatase and interaction between metal sites. J. Biol. Inorg. Chem 8(4), 452–458. [DOI] [PubMed] [Google Scholar]
- 47.Lewis M, Chang G, Horton NC, Kercher MA, Pace HC, Schumacher MA, Brennan RG and Lu P. (1996). Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 271(5253), 1247–1254. [DOI] [PubMed] [Google Scholar]
- 48.Galperin MY (2006). Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J. Bacteriol 188(12), 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Söding J. (2005). Protein homology detection by HMM–HMM comparison. Bioinformatics. 21(7), 951–960. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, & Skolnick J. (2005). TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33(7), 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Plaxco KW, Simons KT, & Baker D. (1998). Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol 277(4), 985–994. [DOI] [PubMed] [Google Scholar]
- 52.Chiti F, Taddei N, White PM, Bucciantini M, Magherini F, Stefani M, & Dobson CM (1999). Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struc. Biol 6(11), 1005–1009. [DOI] [PubMed] [Google Scholar]
- 53.Fersht AR (2000). Transition-state structure as a unifying basis in protein-folding mechanisms: contact order, chain topology, stability, and the extended nucleus mechanism. Proc. Natl. Acad. Sci. U S A 97(4), 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Houwman JA, & van Mierlo CP (2017). Folding of proteins with a flavodoxin‐like architecture. FEBS J. 284(19), 3145–3167. [DOI] [PubMed] [Google Scholar]
- 55.Toofanny RD, Calhoun S, Jonsson AL, & Daggett V. (2019). Shared unfolding pathways of unrelated immunoglobulin-like β-sandwich proteins. Protein Eng. Des. Sel 32(7), 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Theobald DL, Mitton-Fry RM and Wuttke DS (2003). Nucleic acid recognition by OB-fold proteins. Annu Rev Biophys Biomol Struct. 32(1), 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zimmermann L, Stephens A, Nam SZ, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN and Alva V. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol 430(15), 2237–2243. [DOI] [PubMed] [Google Scholar]
- 58.Martin JL, & McMillan FM (2002). SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struc. Biol 12(6), 783–793. [DOI] [PubMed] [Google Scholar]
- 59.Holm L. (2019). Benchmarking fold detection by DaliLite v. 5. Bioinformatics. 35(24), 5326–5327. [DOI] [PubMed] [Google Scholar]
- 60.Chouhan BPS, Maimaiti S, Gade M, & Laurino P. (2018). Rossmann-fold methyltransferases: taking a “β-Turn” around their cofactor, S-adenosylmethionine. Biochemistry. 58(3), 166–170. [DOI] [PubMed] [Google Scholar]
- 61.Ferrer JL, Zubieta C, Dixon RA, & Noel JP (2005). Crystal structures of alfalfa caffeoyl coenzyme A 3-O-methyltransferase. Plant Physiol. 137(3), 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hori T, Yokomizo T, Ago H, Sugahara M, Ueno G, Yamamoto M, Kumasaka T, Shimizu T. and Miyano M. (2004). Structural basis of leukotriene B4 12-hydroxydehydrogenase/15-Oxo-prostaglandin 13-reductase catalytic mechanism and a possible Src homology 3 domain binding loop. J. Biol. Chem 279(21), 22615–22623. [DOI] [PubMed] [Google Scholar]
- 63.Horton JR, Liebert K, Hattman S, Jeltsch A, & Cheng X. (2005). Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of dam methyltransferase. Cell. 121(3), 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pieulle L, Guigliarelli B, Asso M, Dole F, Bernadac A, & Hatchikian EC (1995). Isolation and characterization of the pyruvate-ferredoxin oxidoreductase from the sulfate-reducing bacterium Desulfovibrio africanus. Biochim. Biophys. Acta 1250(1), 49–59. [DOI] [PubMed] [Google Scholar]
- 65.Schulz GE (1992). Binding of nucleotides by proteins. Curr. Opin. Struc. Biol 2(1), 61–67. [Google Scholar]
- 66.Leipe DD, Wolf YI, Koonin EV, & Aravind L. (2002). Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol 317(1), 41–72. [DOI] [PubMed] [Google Scholar]
- 67.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J. and Sussman JL (1992). The α/β hydrolase fold. Protein Eng. 5(3), 197–211.1409539 [Google Scholar]
- 68.Xu T, Liu L, Hou S, Xu J, Yang B, Wang Y, & Liu J. (2012). Crystal structure of a mono-and diacylglycerol lipase from Malassezia globosa reveals a novel lid conformation and insights into the substrate specificity. J. Struct. Biol 178(3), 363–369. [DOI] [PubMed] [Google Scholar]
- 69.Brzozowski AM, Savage H, Verma CS, Turkenburg JP, Lawson DM, Svendsen A, & Patkar S. (2000). Structural origins of the interfacial activation in Thermomyces (Humicola) lanuginosa lipase. Biochemistry. 39(49), 15071–15082. [DOI] [PubMed] [Google Scholar]
- 70.van Tilbeurgh H, Egloff MP, Martinez C, Rugani N, Verger R, & Cambillau C. (1993). Interfacial activation of the lipase–procolipase complex by mixed micelles revealed by X-ray crystallography. Nature. 362(6423), 814–820. [DOI] [PubMed] [Google Scholar]
- 71.Fritsche E, Paschos A, Beisel HG, Böck A, & Huber R. (1999). Crystal structure of the hydrogenase maturating endopeptidase HYBD from Escherichia coli. J. Mol. Biol 288(5), 989–998. [DOI] [PubMed] [Google Scholar]
- 72.Kwon S, Nishitani Y, Watanabe S, Hirao Y, Imanaka T, Kanai T, Atomi H. and Miki, (2016). Crystal structure of a [NiFe] hydrogenase maturation protease HybD from Thermococcus kodakarensis KOD1. Proteins. 84(9), 1321–1327. [DOI] [PubMed] [Google Scholar]
- 73.Kumoi K, Satoh T, Murata K, Hiromoto T, Mizushima T, Kamiya Y, Noda M, Uchiyama S, Yagi H. and Kato K. (2013). An archaeal homolog of proteasome assembly factor functions as a proteasome activator. PLoS One. 8(3), e60294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ferri-Fioni ML, Fromant M, Bouin AP, Aubard C, Lazennec C, Plateau P, & Blanquet S. (2006). Identification in archaea of a novel D-Tyr-tRNATyr deacylase. J. Biol. Chem 281(37), 27575–27585. [DOI] [PubMed] [Google Scholar]
- 75.Štefanić Z, Narczyk M, Mikleušević G, Kazazić S, Bzowska A, & Luić M. (2018). Crystallographic snapshots of ligand binding to hexameric purine nucleoside phosphorylase and kinetic studies give insight into the mechanism of catalysis. Sci. Rep 8(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rigden DJ, Eberhardt RY, Gilbert HJ, Xu Q, Chang Y, & Godzik A. (2014). Structure-and context-based analysis of the GxGYxYP family reveals a new putative class of glycoside hydrolase. BMC Bioinformatics. 15(1), 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Willing SE, Candela T, Shaw HA, Seager Z, Mesnage S, Fagan RP, & Fairweather NF (2015). C lostridium difficile surface proteins are anchored to the cell wall using CWB 2 motifs that recognise the anionic polymer PSII. Mol. Microbiol 96(3), 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Usenik A, Renko M, Mihelič M, Lindič N, Borišek J, Perdih A, Pretnar G, Müller U. and Turk D. (2017). The CWB2 cell wall-anchoring module is revealed by the crystal structures of the Clostridium difficile cell wall proteins Cwp8 and Cwp6. Structure. 25(3), 514–521. [DOI] [PubMed] [Google Scholar]
- 79.Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A. and Lu Y. (2014). Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev 114(8), 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ceh K, Demmer U, Warkentin E, Moll J, Thauer RK, Shima S. and Ermler U. (2009). Structural basis of the hydride transfer mechanism in F420-dependent methylenetetrahydromethanopterin dehydrogenase. Biochemistry. 48(42), 10098–10105. [DOI] [PubMed] [Google Scholar]
- 81.Quitterer F, Beck P, Bacher A. and Groll M. (2013). Structure and reaction mechanism of pyrrolysine synthase (PylD). Angew Chem Int Ed Engl. 52(27), 7033–7037. [DOI] [PubMed] [Google Scholar]
- 82.Kuratani M, Bessho Y, Nishimoto M, Grosjean H. and Yokoyama S. (2008). Crystal structure and mutational study of a unique SpoU family archaeal methylase that forms 2′-O-methylcytidine at position 56 of tRNA. J Mol Biol. 375(4), 1064–1075. [DOI] [PubMed] [Google Scholar]
- 83.Tkaczuk KL, Dunin-Horkawicz S, Purta E. and Bujnicki JM. (2007). Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC bioinformatics. 8(1), p.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y, White RH and Ealick SE (2008). Crystal structure and function of 5-formaminoimidazole-4-carboxamide ribonucleotide synthetase from Methanocaldococcus jannaschii. Biochemistry. 47(1), 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Williams C, Van Den Berg M, Panjikar S, Stanley WA, Distel B. and Wilmanns M. (2012). Insights into ubiquitin‐conjugating enzyme/co‐activator interactions from the structure of the Pex4p: Pex22p complex. EMBO J. 31(2), 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Williams C, Van Den Berg M, Stanley WA, Wilmanns M. and Distel B. (2013). A disulphide bond in the E2 enzyme Pex4p modulates ubiquitin-conjugating activity. Sci Rep. 3, 2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eryilmaz E, Benach J, Su M, Seetharaman J, Dutta K, Wei H, Gottlieb P, Hunt JF and Ghose R. (2008). Structure and dynamics of the P7 protein from the bacteriophage ϕ12. J. Mol. Biol 382(2), 402–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng Y, Roberts RJ, & Kasif S. (2004). Identification of genes with fast-evolving regions in microbial genomes. Nucleic Acids Res. 32(21), 6347–6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Higgins MA, Whitworth GE, El Warry N, Randriantsoa M, Samain E, Burke RD, Vocadlo DJ and Boraston AB (2009). Differential recognition and hydrolysis of host carbohydrate antigens by Streptococcus pneumoniae family 98 glycoside hydrolases.J Biol Chem. 284(38), 26161–26173.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kwan DH, Constantinescu I, Chapanian R, Higgins MA, Kötzler MP, Samain E, Boraston AB, Kizhakkedathu JN and Withers SG (2015). Toward efficient enzymes for the generation of universal blood through structure-guided directed evolution. J Am Chem Soc. 137(17), 5695–5705. [DOI] [PubMed] [Google Scholar]
- 91.Pei J, Kinch LN, Otwinowski Z, & Grishin NV (2020). Mutation severity spectrum of rare alleles in the human genome is predictive of disease type. PLoS Comput. Biol 16(5), e1007775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tuchman M, Plante RJ, McCann MT and Qureshi AA (1994). Seven new mutations in the human ornithine transcarbamylase gene. Hum. Mutat 4(1), 57–60. [DOI] [PubMed] [Google Scholar]
- 93.Climent C. and Rubio V. (2002). Identification of seven novel missense mutations, two splice‐site mutations, two microdeletions and a polymorphic amino acid substitution in the gene for ornithine transcarbamylase (OTC) in patients with OTC deficiency. Hum. Mutat 19(2), 185–186. [DOI] [PubMed] [Google Scholar]
- 94.Becker KG, Barnes KC, Bright TJ, & Wang SA (2004). The genetic association database. Nat. Genet 36(5), 431–432. [DOI] [PubMed] [Google Scholar]
- 95.Giardina G, Montioli R, Gianni S, Cellini B, Paiardini A, Voltattorni CB, & Cutruzzolà F. (2011). Open conformation of human DOPA decarboxylase reveals the mechanism of PLP addition to Group II decarboxylases. Proc. Natl. Acad. Sci. U S A 108(51), 20514–20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pons R, Ford B, Chiriboga CA, Clayton PT, Hinton V, Hyland K, Sharma R. and De Vivo DC (2004). Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology. 62(7), 1058–1065. [DOI] [PubMed] [Google Scholar]
- 97.Chang YT, Sharma R, Marsh JL, McPherson JD, Bedell JA, Knust A, Bräutigam C, Hoffmann GF and Hyland K, 2004. Levodopa‐responsive aromatic L–amino acid decarboxylase deficiency. Ann. Neurol 55(3), 435–438. [DOI] [PubMed] [Google Scholar]
- 98.Yu X, Chini CCS, He M, Mer G, & Chen J. (2003). The BRCT domain is a phosphor-protein binding domain. Science. 302(5645), 639–642. [DOI] [PubMed] [Google Scholar]
- 99.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, DeRooij DG, Hirsch S, Ravi K, Hicks JB and Szabolcs M. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 334(6055), 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Couch FJ, & Weber BL (1996). Mutations and polymorphisms in the familial early‐onset breast cancer (BRCA1) gene. Human Mutat. 8(1), 8–18. [DOI] [PubMed] [Google Scholar]
- 101.Williams RS, Lee MS, Hau DD, & Glover JM (2004). Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat. Struct. Mol. Biol 11(6), 519–525. [DOI] [PubMed] [Google Scholar]
- 102.Antoniou AC, Sinilnikova OM, McGuffog L, Healey S, Nevanlinna H, Heikkinen T, Simard J, Spurdle AB, Beesley J, Chen X. and Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer. (2009). Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum. Mol. Genet 18(22), 4442–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hofer AM, & Brown EM (2003). Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol 4(7), 530–538. [DOI] [PubMed] [Google Scholar]
- 104.Cole DE, Yun FH, Wong BY, Shuen AY, Booth RA, Scillitani A, Pidasheva S, Zhou X, Canaff L. and Hendy GN (2009). Calcium-sensing receptor mutations and denaturing high performance liquid chromatography. J. Mol. Endocrinol 42(4), 331–339. [DOI] [PubMed] [Google Scholar]
- 105.Geng Y, Mosyak L, Kurinov I, Zuo H, Sturchler E, Cheng TC, Subramanyam P, Brown AP, Brennan SC, Mun HC and Bush M. (2016). Structural mechanism of ligand activation in human calcium-sensing receptor. Elife. 5, e13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Okazaki R, Chikatsu N, Nakatsu M, Takeuchi Y, Ajima M, Miki J, Fujita T, Arai M, Totsuka Y, Tanaka K. and Fukumoto S. (1999). A novel activating mutation in calcium-sensing receptor gene associated with a family of autosomal dominant hypocalcemia. J. Clin. Endocrinol. Metab 84(1), 363–366. [DOI] [PubMed] [Google Scholar]
- 107.Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, DeVree BT, Sunahara RK and Chae PS (2011). Structure and function of an irreversible agonist-β 2 adrenoceptor complex. Nature. 469(7329), 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leech C, Lohse P, Stanojevic V, Lechner A, Göke B, & Spitzweg C. (2006). Identification of a novel inactivating R465Q mutation of the calcium-sensing receptor. Biochem. Biophys. Res. Commun 342(3), 996–1002. [DOI] [PubMed] [Google Scholar]
- 109.Schubbert S, Shannon K, & Bollag G. (2007). Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7(4), 295–308. [DOI] [PubMed] [Google Scholar]
- 110.Stenmark H, & Olkkonen VM (2001). The rab gtpase family. Genome Biol. 2(5), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D. and Ludlam MJ (2012). Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U S A 109(14), 5299–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang MH, Nickerson S, Kim ET, Liot C, Laurent G, Spang R, Philips MR, Shan Y, Shaw DE, Bar-Sagi D. and Haigis MC (2012). Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. U S A 109(27), 10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE and Nguyen H. (2006). Germline KRAS mutations cause Noonan syndrome. Nat. Genet 38(3), 331–336. [DOI] [PubMed] [Google Scholar]
- 114.Nobeli I, Favia AD, & Thornton JM (2009). Protein promiscuity and its implications for biotechnology. Nat. Biotechnol 27(2), 157–167. [DOI] [PubMed] [Google Scholar]
- 115.Alhindi T, Zhang Z, Ruelens P, Coenen H, Degroote H, Iraci N, & Geuten K. (2017). Protein interaction evolution from promiscuity to specificity with reduced flexibility in an increasingly complex network. Sci. Rep 7, 44948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE, 2000. The protein data bank. Nucleic Acids Res. 28(1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Majumdar I, Krishna SS, & Grishin NV (2005). PALSSE: A program to delineate linear secondary structural elements from protein structures. BMC Bioinformatics. 6(1), 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Remmert M, Biegert A, Hauser A, & Söding J. (2012). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9(2), 173–175. [DOI] [PubMed] [Google Scholar]
- 119.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, & Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17), 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Holm L, & Sander C. (1998). Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 26(1), 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Galili T, O’Callaghan A, Sidi J, & Sievert C. (2018). heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics. 34(9), 1600–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.UniProt Consortium. (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47(D1), D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L. and Lepore R. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sherman BT, & Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4(1), 44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.