

Abstract
Endogenous biomarkers are emerging to advance clinical drug‐drug interaction (DDI) risk assessment in drug development. Twelve healthy subjects received a multidrug and toxin exclusion protein (MATE) inhibitor (pyrimethamine, 10, 25, and 75 mg) in a crossover fashion to identify an appropriate endogenous biomarker to assess MATE1/2‐K‐mediated DDI in the kidneys. Metformin (500 mg) was also given as reference probe drug for MATE1/2‐K. In addition to the previously reported endogenous biomarker candidates (creatinine and N 1‐methylnicotinamide (1‐NMN)), N 1‐methyladenosine (m1A) was included as novel biomarkers. 1‐NMN and m1A presented as superior MATE1/2‐K biomarkers since changes in their renal clearance (CLr) along with pyrimethamine dose were well‐correlated with metformin CLr changes. The CLr of creatinine was reduced by pyrimethamine, however, its changes poorly correlated with metformin CLr changes. Nonlinear regression analysis (CLr vs. mean total concentration of pyrimethamine in plasma) yielded an estimate of the inhibition constant (Ki) of pyrimethamine and the fraction of the clearance pathway sensitive to pyrimethamine. The in vivo Ki value thus obtained was further converted to unbound Ki using plasma unbound fraction of pyrimethamine, which was comparable to the in vitro Ki for MATE1 (1‐NMN) and MATE2‐K (1‐NMN and m1A). It is concluded that 1‐NMN and m1A CLr can be leveraged as quantitative MATE1/2‐K biomarkers for DDI risk assessment in healthy volunteers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Drug‐drug interactions (DDIs) involving renal MATE1/2‐K inhibition, impacting metformin pharmacokinetics, are important. Therefore, regulators mandate clinical metformin DDI studies when investigational drugs trigger conservative agency DDI risk thresholds (e.g., projected peak plasma concentration (Cmax),u/in vitro MATE1/2‐K half‐maximal inhibitory concentration ratio > 0.1).
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Could an endogenous MATE1/2‐K substrate serve as a clinical biomarker to support inhibition assessment, de‐risk metformin DDI, and obviate the need for a formal DDI study?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Beyond creatinine, two endogenous MATE1/2‐K substrates (N 1‐methyladenosine (m1A) and N 1‐methylnicotinamide (1‐NMN)) present as superior urinary biomarkers. Both support MATE1/2‐K inhibition assessment and the generation of in vivo inhibition constant (Ki) values.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Perpetrator dose‐dependent inhibition of renal MATE1/2‐K can be assessed using m1A and/or 1‐NMN. Once generated, in vivo Ki values could be input for physiologically‐based pharmacokinetic models describing metformin DDI. The output of such models can drive the data translation (e.g., in vitro to in vivo for MATE1/2‐K inhibition) and enable decision making (e.g., the need for/design of a metformin DDI study).
To ensure safety in drug therapy upon concomitant use of drugs, drug‐drug interaction (DDI) risks of new chemical entities with major drug‐metabolizing enzymes and transporters are routinely assessed in drug development by mechanism‐based approaches. Typically, such approaches involve the prediction of DDI based on in vitro data and clinical assessment using recommended probe drugs. 1 , 2 , 3 However, it is generally accepted that the use of in vitro data can render both significant false‐positive and, to a lesser degree, false‐negative predictions. 4 Recently, endogenous biomarkers, generally physiological substrates of drug‐metabolizing enzymes and transporters, are emerging as useful tools to advance DDI risk assessment of investigational drugs in early phases of clinical development before conducting the clinical DDI study using the recommended probe drugs. 2 , 4 , 5 Application of endogenous biomarkers has been expanded to encompass complex DDIs involving multiple potential interaction sites with investigational drug. 6 Although useful, biomarkers require validation and characterization in terms of their ease of analysis, sample requirements, sensitivity, dynamic range, significant correlation with pharmacokinetic parameters of probe drugs, as well as variability (e.g., diurnal, interindividual and intra‐individual differences). 4
The kidneys are essential organs for eliminating drugs and metabolites into the urine from the blood circulation. In addition to glomerular filtration, drugs are eliminated into the urine by drug transport systems in the proximal tubules, which consist of OCT2, and MATE1 and MATE2‐K mediating the uptake and subsequent efflux of water‐soluble cationic compounds, respectively. 7 , 8 , 9 An oral antidiabetic, metformin, is a well‐characterized substrate of these transporters, and serves as a typical probe for assessing potential OCT2 and/or MATE1/2‐K associated DDIs. 10 In addition, endogenous metabolites, such as creatinine and N 1‐methylnicotinamide (1‐NMN), have been also identified as substrates of OCT2 and MATEs. 11 , 12 , 13 , 14 , 15 Creatinine clearance serves as an index of kidney function. 16 On the other hand, drugs known to be inhibitors of OCT2 and MATEs have been shown to cause reversible elevation of serum creatinine without kidney injury, 17 , 18 such as cimetidine, trimethoprim, and pyrimethamine that also reduce the renal clearance of metformin at their therapeutic doses. 19 , 20 , 21 , 22 The 1‐NMN, an endogenous metabolite of nicotinamide (also known as vitamin B3 or niacin) formed by N‐mehtylation, 23 is also known to be secreted into the urine by renal organic cation transporters. 15 , 24 Additionally, based on the genomewide association study of serum metabolite profiles, 25 we recently identified N 1‐methyladenosine (m1A), which is considered derived from transfer RNA, 26 as a novel endogenous substrate of OCT2 and MATE1/2‐K that undergoes significant tubular secretion in the kidneys of humans. 27 Administration of DX‐619, an inhibitor of OCT2 and MATE1/2‐K, 28 delayed the elimination of m1A in monkeys, 27 supporting that it can serve as a DDI biomarker.
Correlation of the pharmacokinetic parameters, such as clearance or area under the plasma concentration time curve (AUC), between endogenous substrates and the typical probe drug when transporters are inhibited in the presence of a perpetrator in a dose‐dependent manner is an important criterion to convince investigators of the appropriateness of a particular biomarker. 29 , 30 To date, however, no such data are available for biomarkers of renal OCT2 and MATE1/2‐K. Therefore, we designed a clinical study to examine the dose‐dependent effect of pyrimethamine on the pharmacokinetic parameters of MATE1/2‐K substrates in a crossover fashion. Pyrimethamine is a potent MATE1/2‐K inhibitor characterized by its long elimination half‐life (ca. 100 hours) in humans, which provides constant inhibition of MATE1/2‐K during a metformin DDI study. 19 In earlier clinical studies with healthy subjects, the pyrimethamine dose was 50 mg (single dose), which caused a slight reduction in renal clearance (CLr) of metformin (at most, 35% inhibition), 19 but a reduction of CLr by 70% that represented an almost complete inhibition of 1‐NMN renal secretory clearance. 15 In a later study by Oh et al., 20 two doses of pyrimethamine 50 mg administered 12 hours apart resulted in stronger impact on metformin AUC and CLr. Based on these clinical studies, pyrimethamine was given orally at 3 different doses, 10, 25, and 75 mg to explore the dose‐dependent inhibition of renal organic cation transporters. In this regard, the use of biomarkers to assess the dose‐dependent inhibition of liver (OATP1B by rifampicin) and renal (OAT by probenecid) transporters has already been described. 29 , 30 , 31 In addition to MATE1/2‐K substrates, the AUC and CLr of pyridoxic acid (PDA) was also determined to examine the effect of pyrimethamine on renal organic anion transporters.
METHODS
Chemicals
All authentic compounds, reagents, and organic solvents were of a commercially available analytical grade. Purchase sources of authentic compounds are listed in Supplementary Methods .
Clinical study design
This study was conducted following the Clinical Trials Act, Japan. The study protocol was reviewed by Certified Review Board, the Graduate School of Medicine, The University of Tokyo (CRB3180024). The study is registered as the specified clinical trial in Japan Registry of Clinical Trials (https://jrct.niph.go.jp/en‐latest‐detail/jRCTs031180125). Written informed consent was provided by all participants prior to their inclusion in the study. The study was an open‐label, dual‐sequence, four‐phase crossover study in 12 healthy Japanese male participants. The schedules of dosing and sampling are both described in Supplementary Methods .
Quantification of test compounds by liquid chromatography with tandem mass spectrometry
Plasma and urine samples analyzed by liquid chromatography with tandem mass spectrometry were prepared by protein precipitation as described previously. 27 All compounds were separated and detected using the QTRAP5500 system (AB SCIEX, Toronto, Canada) equipped with the Nexera X2 LC system (Shimadzu, Kyoto, Japan), operated in electrospray ionization mode. Analyte quantification was performed using Analyst version 1.7 (Sciex, Toronto, ON, Canada). Creatinine and 1‐NMN were analyzed using creatinine‐d3 and 1‐NMN‐d3 as internal standards, respectively. The conditions of liquid chromatography, multiple reaction monitoring precursor/product ion transitions are summarized in Supplementary Methods .
Pharmacokinetic analysis in the human clinical study
The area under the plasma concentration–time curve from zero to 24 hours (AUC0–24) was determined with the linear trapezoidal rule. However, the AUC of 1‐NMN was calculated from time zero to 12 hours postdose given that plasma concentrations of 1‐NMN showed significant intraday variation, and AUC during 12–24 hours was determined only at 2 timepoints (12 and 24 hours), thus potentially impacting interpretation of CLr.
The CLr regarding plasma concentration of test compounds was determined by dividing the amount excreted into the urine from 0 to 24 hours (X urine) by the AUC value. For 1‐NMN, AUC and the amount excreted into the urine from 0 to 12 hours were used.
Ki ,app (apparent inhibition constant of pyrimethamine as the total concentration) and f PYR (the fraction sensitive to inhibition by pyrimethamine) were obtained by iterative nonlinear least squares method 30 (Prism8 version 8.4.1; GraphPad Software, La Jolla, CA) using the following equation under constraint of 0 < fP YR ≤ 1 and Ki ,app> 0, where C av represents the mean plasma concentrations of pyrimethamine in each subject:
C av was obtained by dividing AUC pyrimethamine with the duration time (12 hours for 1‐NMN, otherwise 24 hours).
Statistical analysis
Natural log‐transformed of all parameters were analyzed by cohort using a mixed effect model with treatment as a fixed effect and subject as a random effect. Estimates of the adjusted mean differences (test‐reference) and corresponding 90% confidence intervals (CIs) for AUC and CLr were obtained from the model. The adjusted mean differences and 90% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test/reference) and 90% CI for the ratios. The control phase (metformin alone) was the reference treatment for all analyses except for the assessment of metformin’s impact on biomarkers, in which the baseline phase (placebo) was the reference. The values of plasma concentration and CLr are presented as the mean ± SEM.
RESULTS
Participant demographics
Twelve healthy male Japanese participants were enrolled into the study. The range for age, body mass index, height, and weight was 23–38 years, 20.9–24.1 kg/m2, 165–183 cm, and 59.5–75.8 kg, respectively. The participants did not manifest any abnormality by medical examination or blood or biochemical tests at baseline.
Pyrimethamine plasma concentrations
Time profiles of the plasma concentrations of pyrimethamine are shown in Figure S1 . The mean plasma concentrations (C av) were 0.287 ± 0.024, 0.781 ± 0.031, and 2.47 ± 0.18 μM (mean ± SEM) when the volunteers received 10, 25, and 75 mg single doses of pyrimethamine, respectively. The maximum plasma concentrations (Cmax) were 0.390 ± 0.047, 1.06 ± 0.14, and 3.15 ± 0.072 μM (mean ± SEM), respectively.
Effect of pyrimethamine on the pharmacokinetics of metformin
The effect of pyrimethamine on the AUC geometric mean ratio (± 90% CI) showed clear separation for metformin (Table 1 ). Pyrimethamine 25 mg and 75 mg were shown to increase metformin AUC by 24% and 69%, respectively, although no impact was observed with pyrimethamine 10 mg (Table 1 ). However, pyrimethamine 10 mg, 25 mg, and 75 mg decreased metformin CLr (Figure 1b ) by 12%, 34%, and 55%, respectively (Table 1 ). The greatest impact on the urinary excretion of metformin occurred over 0–4 hours postdose (Figure 1a ). Effect of pyrimethamine on other pharmacokinetic parameters are summarized in Table S1 .
Table 1.
Adjusted geometric means and GMR (90% CI) for metformin and the endogenous biomarkers
| Metformin | 1‐NMN a | m1A | Creatinine | PDA | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC (μM × h) | CLr (ml/min) | AUC (nM × h) | CLr (ml/min) | AUC (nM × h) | CLr (ml/min) | AUC (μM × h) | CLr (ml/min) | AUC (nM × h) | CLr (ml/min) | |
| Baseline, placebo | N/A | N/A | 386.8 | 413.2 | 2,673.1 | 174.5 | 2,370.8 | 131.1 | N/D | N/D |
| Control, metformin | 72.5 | 468.3 | 652.6 | 388.8 | 2,489.9 | 161.6 | 2,119.6 | 126.3 | 291.2 | 545.1 |
| PYR 10 mg, + Met | 74.2 | 411.6 | 402.6 | 360.0 | 2,576.0 | 145.0 | 2,494.9 | 103.8 | 214.2 | 584.6 |
| PYR 25 mg, + Met | 89.8 | 308.9 | 361.4 | 247.2 | 2,694.6 | 128.3 | 2,545.3 | 95.3 | 177.2 | 603.1 |
| PYR 75 mg, + Met | 122.9 | 210.2 | 451.2 | 161.3 | 2,654.5 | 84.9 | 2,303.1 | 95.2 | 164.7 | 423.7 |
| Control vs. baseline | ||||||||||
| GMR | N/A | N/A | 1.69 | 0.94 | 0.93 | 0.93 | 0.89 | 0.96 | N/A | N/A |
| 90% CI | N/A | N/A | 1.39, 2.05 | 0.83, 1.07 | 0.89, 0.98 | 0.84, 1.02 | 0.86, 0.93 | 0.88, 1.05 | N/A | N/A |
| PYR 10 mg vs. control | ||||||||||
| GMR | 1.02 | 0.88 | 0.62 | 0.93 | 1.03 | 0.90 | 1.18 | 0.82 | 0.74 | 1.07 |
| 90% CI | 0.91, 1.15 | 0.80, 0.96 | 0.51, 0.75 | 0.82, 1.05 | 0.99, 1.08 | 0.82, 0.99 | 1.13, 1.23 | 0.75, 0.90 | 0.52, 1.05 | 0.91, 1.26 |
| PYR 25 mg vs. control | ||||||||||
| GMR | 1.24 | 0.66 | 0.55 | 0.64 | 1.08 | 0.79 | 1.20 | 0.75 | 0.61 | 1.11 |
| 90% CI | 1.10, 1.39 | 0.60, 0.72 | 0.49, 0.67 | 0.56, 0.72 | 1.03, 1.13 | 0.72, 0.87 | 1.15, 1.25 | 0.69, 0.83 | 0.43, 0.87 | 0.94, 1.30 |
| PYR 75 mg vs. control | ||||||||||
| GMR | 1.69 | 0.45 | 0.69 | 0.41 | 1.07 | 0.53 | 1.09 | 0.75 | 0.57 | 0.78 |
| 90% CI | 1.51, 1.90 | 0.41, 0.49 | 0.57, 0.84 | 0.37, 0.47 | 1.02, 1.12 | 0.48, 0.58 | 1.04, 1.13 | 0.69, 0.82 | 0.40, 0.81 | 0.66, 0.92 |
1‐NMN, N1‐methylnicotinamide; AUC, area under the plasma concentration–time curve; CI, confidence interval; CLr, renal clearance; GMR, geometric mean ratio; m1A, N 1‐methyladenosine; Met, metformin; N/A, not applicable; N/D, not determined; PDA, pyridoxic acid; PYR, pyrimethamine.
Based on 12‐hour data.
Figure 1.
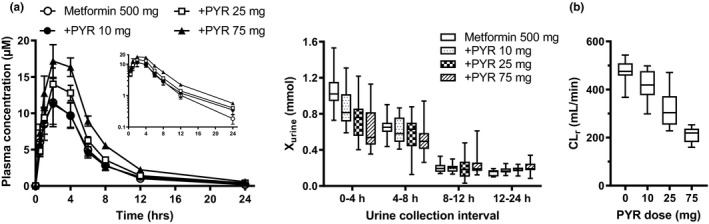
Effect of pyrimethamine on the plasma concentrations, and amount excretion of metformin (a), and renal clearance (b). Plasma concentrations and urinary excretion of metformin were determined in the subjects treated with or without pyrimethamine (10, 25, and 75 mg). Each symbol represents the mean and SEM (n = 12). Urinary excretion during the intervals designated was shown in box‐and‐whisker plot. (b) Renal clearance determined in healthy volunteers treated with or without an oral dose of pyrimethamine (10, 25, and 75 mg) were shown in box‐and‐whisker plots. +PYR, pyrimethamine administrated group.
Effect of pyrimethamine on the pharmacokinetics of endogenous compounds
Plasma concentration profiles and urinary excretion of m1A, creatinine, and 1‐NMN following each dose of pyrimethamine are summarized in Figure 2 . Under control conditions (metformin alone), plasma concentrations of m1A and creatinine were almost identical throughout the day, whereas those of 1‐NMN exhibited marked intraday variation. Differences in m1A, creatinine, and 1‐NMN concentrations were also observed between the baseline and control phases of the study (Table 1 ; Figure 2 ). The AUC ratio was unchanged or slightly higher in the pyrimethamine‐treated groups for m1A and creatinine, respectively, whereas the AUC ratio of 1‐NMN was rather lower in the pyrimethamine‐treated groups (Table 1 ; Figure 3a ). The CLr of m1A, 1‐NMN, and creatinine were decreased in a dose‐dependent manner, although creatinine CLr showed no further decrease beyond the 25 mg dose level (Table 1 ; Figure 3a ).
Figure 2.
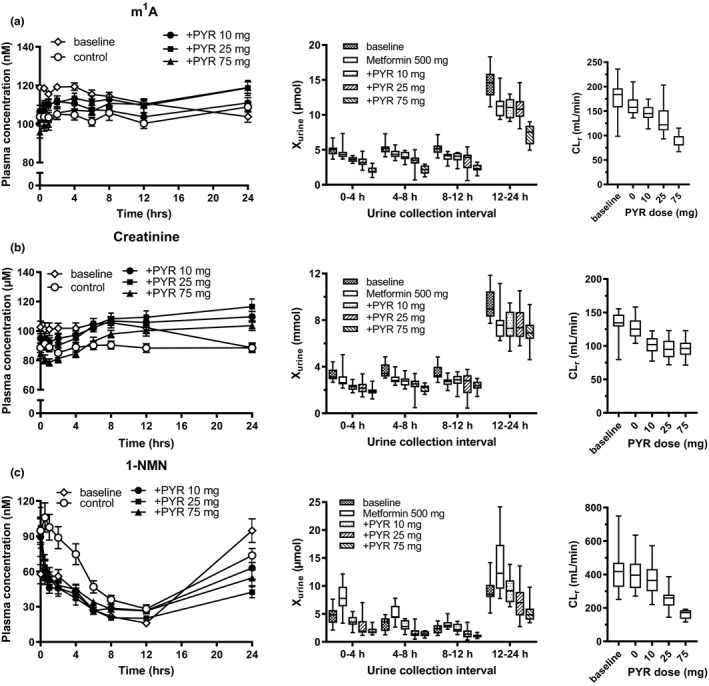
Effect of pyrimethamine on the plasma concentration‐time profiles and amount excreted into the urine and CLr of endogenous OCT2/MATE substrates. Plasma concentrations of m1A (a), creatinine (b), and 1‐NMN (c) were determined at designated times in healthy volunteers in the baseline phase, and drug treatment phases (metformin with or without an oral dose of pyrimethamine 10, 25, and 75 mg). Each symbol represents the mean and SEM (n = 12). Amounts excreted into the urine during the intervals designated and CLr were shown in box‐and‐whisker plot. m1A, N 1‐methyladenosine; 1‐NMN, N 1‐methylnicotinamide; +PYR, pyrimethamine administration; CLr, renal clearance.
Figure 3.
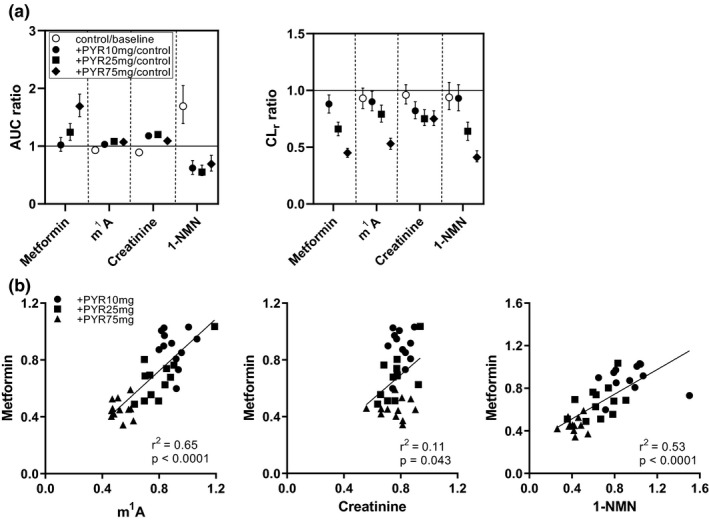
Effect of pyrimethamine on AUC ratio and CLr ratio, and correlation between CLr ratio of endogenous OCT2/MATE substrates against that of metformin. (a) AUC ratio and CLr ratio with 90% confidence intervals were shown. Data were taken from Table 1 . (b) The correlation of the CLr ratio among the test compounds is shown with correlation coefficient and P value. m1A, N 1‐methyladenosine; 1‐NMN, N 1‐methylnicotinamide; AUC, area under the curve; CLr, renal clearance; PYR, pyrimethamine.
The CLr of 1‐NMN was calculated using the plasma and urine date from time 0 to 12 hours because of sparse blood sampling between 12 and 24 hours to obtain reliable AUC. Correlation of CLr calculated from time 0 to 12 or 24 hours are shown in Figure S2 .
Correlation on the pharmacokinetics of metformin and endogenous compounds
The CLr ratio (+PYR/control) of endogenous compounds was compared with that of metformin and pairwise Pearson correlation coefficients (r) were calculated (Figure 3b ). The m1A and 1‐NMN showed positive and good correlations with metformin (r 2 > 0.5), whereas correlation between creatinine and metformin was not observed (r 2 = 0.11).
Nonlinear regression analysis of CLr ratio and pyrimethamine C av
The relationship between CLr ratio and pyrimethamine C av (total concentration in plasma) was analyzed to obtain estimates of apparent inhibition constant (Ki ,app) and fraction sensitive to inhibition by pyrimethamine (f PYR; Figure 4 ). Curve fitting‐based estimates of Ki ,app and f PYR are shown in Table 2 . The CLr of metformin, m1A, and 1‐NMN appeared to be decreased according to pyrimethamine C av. The f PYR of metformin, m1A, and 1‐NMN was almost identical (~ 0.8), whereas that of creatinine was much smaller (0.26).
Figure 4.
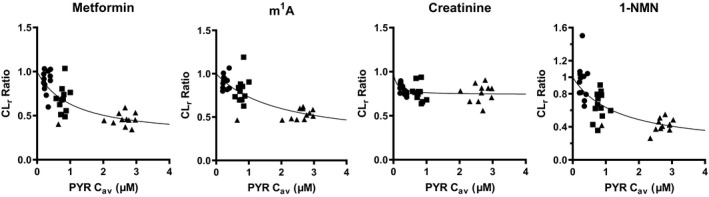
Nonlinear regression analysis CLr ratio of the test compounds and pyrimethamine C av. Ki ,app of pyrimethamine with regard to C av, and f PYR were determined by iterative nonlinear regression analysis, as described in Methods. Rigid lines represent the fitted lines. The fitted parameters were summarized in Table 2 . 1‐NMN, N 1‐methylnicotinamide; Cav, mean plasma concentration; CLr, renal clearance; f PYR, fraction sensitive to inhibition by pyrimethamine; K i,app, apparent inhibition constant of pyrimethamine as the total concentration; PYR, pyrimethamine.
Table 2.
Ki ,app with regard to pyrimethamine C av, f PYR, and in vitro Ki of test compounds
| Compound | Clinical data | In vitro Ki (μM) | |||
|---|---|---|---|---|---|
| Ki ,app, μM | f PYR | OCT2 | MATE1 | MATE2‐K | |
| Metformin | 1.05 ± 0.39 (0.137) | 0.766 ± 0.103 | 37.9 ± 5.6 | 0.154 ± 0.029 | 0.0941 ± 0.016 |
| N 1‐methyladenosine | 1.94 ± 0.96 (0.252) | 0.792 ± 0.188 | 0.466 ± 0.043 | ND | 0.0680 ± 0.015 |
| Creatinine | 0.0914 ± 0.0661 (0.0119) | 0.260 ± 0.026 | 1.57 ± 0.24 | ND | ND |
| N 1‐methylnicotinamide a | 1.37 ± 0.63 (0.178) | 0.863 ± 0.157 | 41.2 ± 9.1 | 0.125 ± 0.026 | 0.0821 ± 0.018 |
Ki ,app and f PYR were determined by iterative nonlinear regression analysis using renal clerance ratio, and average pyrimethamine concentrations, as described in the Methods. In vitro Ki were determined by iterative nonlinear regression analysis using the data shown in Figure 5 . Number in the parenthesis represents the Ki ,app corrected by the unbound fraction of pyrimethamine in the plasma (0.13). Each parameter represents the fitted parameter with computer calculated SD. K i was calculated from IC50, assuming IC50 approximates K i when substrate concentration is lower than the Km.
Cav, mean plasma concentration; f PYR, fraction sensitive to inhibition by pyrimethamine; Ki, inhibition constant; K i,app, apparent inhibition constant of pyrimethamine as the total concentration; ND, not determined.
CLr calculated from time zero to 12 hours was used in the analysis.
Determination of in vitro Ki values of pyrimethamine
For the comparison with in vivo Ki , app values, the in vitro Ki values of pyrimethamine for OCT2 and MATE1/2‐K‐mediated substrate uptake were determined ( Table 2 ; Figure 5 ). MATE1‐mediated m1A uptake and MATE1/2‐K‐mediated creatinine uptake could not be observed in our expression system. The absolute values of the uptake are summarized in Table S3 . After correction of the unbound fraction of pyrimethamine in the plasma (0.1320), the Ki ,app was similar to the in vitro Ki values of metformin, m1A, and 1‐NMN for MATE1 and MATE2‐K (Table 2 ).
Figure 5.
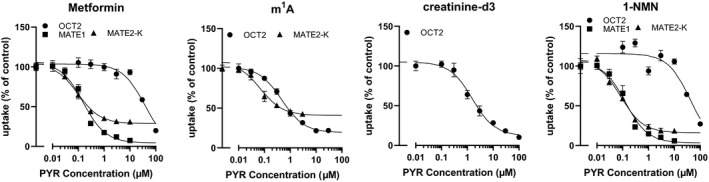
Effect of pyrimethamine on OCT2, MATE1, and MATE2‐K. Uptake of metformin (10 μM), m1A (100 μM), creatinine‐d3 (100 μM), and 1‐NMN (10 μM) was determined in the presence of pyrimethamine at various concentration in the stable expression system of OCT2, MATE1, and MATE2‐K using HEK293 cells. Incubation time was 2 minutes except for m1A (30 minutes) to determine the uptake. Absolute values of the uptake are summarized in Table S3 . Each symbol represents the mean and SEM (n = 3). Rigid lines represent the fitted line. Nonlinear regression analysis was conducted as described in Supplementary Methods . 1‐NMN, N 1‐methylnicotinamide; m1A, N 1‐methyladenosine; PYR, pyrimethamine.
Inhibition of OAT1 and OAT3 by pyrimethamine
As a negative control, we determined the CLr of PDA, a pyridoxine metabolite. Renal OATs, OAT1 and OAT3, are considered to mediate tubular secretion of PDA. 32 , 33 Pyrimethamine is a weak OAT1 and OAT3 inhibitor at its therapeutic dose with half‐maximal inhibitory concentration of 241 ± 55 µM and 9.69 ± 2.27 µM, respectively (Figure S3 ). Low inhibition potency to OAT3 was consistent with our previous report. 34 Pyrimethamine decreased the AUC of PDA in a dose‐dependent manner, but was shown to weakly inhibit the PDA CLr (22% decrease) at the highest dose (Table 1 and Figure S4 ).
Effect of transporter genotype on the plasma concentrations of the probe drugs and endogenous substrates
Genetic polymorphisms have been reported to alter the kinetics of OCT2 and MATE substrate drugs. As such, genetic polymorphisms were assessed and pharmacokinetic profiles were segregated in terms of subject genotype. None of the tested polymorphisms, OCT2 rs316019, MATE1 rs2289669, rs2453579, rs2252281, and MATE2‐K rs12943590, provided significant separation (Figure S5 ).
DISCUSSION
OCT2 and MATE1/2‐K play a pivotal role in the renal elimination of water‐soluble cationic compounds. This study aimed to identify endogenous biomarkers appropriate for assessing MATE1/2‐K inhibition by examining the effect of increasing doses of pyrimethamine on their CLr in comparison to metformin, a frequently used clinical probe to examine in vivo inhibition of either or both OCT2 and MATE1/2‐K.
The decreases in metformin CLr supported the dose‐dependent inhibition of MATE1/2‐K by pyrimethamine (Table 1 ; Figure 1 ). In addition, pyrimethamine may weakly inhibit the oral absorption of metformin because the amount recovered in the urine over 24 hours divided by the dose (fe) and bioavailability (F) were lower at the highest pyrimethamine dose compared in this phase (Table S1 ). The effect of pyrimethamine on the AUC and CLr differed across the endogenous compounds (Table 1 ). A minimal increase in AUC was observed for creatinine and m1A despite significant dose‐dependent reductions in CLr (Figure 3 ). Therefore, consideration should be given to these biomarkers for further analysis. Metformin administration negligibly impacted the CLr of NMN, m1A, and creatinine (< 7%; Figure 3a ). It was evident that there was a notable difference in 1‐NMN AUC, but not m1A and creatinine AUC, between the baseline and control phases (Figure 3a ). Without mass balance data, it is not practical to address the underlying mechanism. Because the plasma concentrations of 1‐NMN in the baseline phases were almost similar to those in pyrimethamine‐treated phases during the duration to calculate AUC (Figure 2c ), metformin administration may accompany with increments of 1‐NMN AUC by unknown mechanism, which was diminished by pyrimethamine administration. It is also possible that there happened to be interday variation in the supply of 1‐NMN as well as its precursor nicotinamide. The plasma concentrations of 1‐NMN did not show a continuous increase for hours in this study, which was reported by Mueller et al., 35 although the subjects were fasted in both studies when they received drug administration. Of note, the same author’s value for metformin CLr was like the result in this study after normalization of the body weights of the participants (9%). In contrast, the CLr of 1‐NMN reported previously 35 was a 38% smaller value by unknown reason. Further studies are necessary to elucidate if ethnicity of participants can be critical in selection of appropriate MATE1/2‐K biomarkers.
Unlike m1A and 1‐NMN, a reduction in CLr of creatinine appeared to reach a plateau at 25 mg pyrimethamine dose (Figure 2 ; Figure 3a ). Consequently, the CLr ratio of m1A and 1‐NMN was reasonably correlated with that of metformin according to the degree of transporter inhibition, whereas correlation of creatinine CLr was poor (Figure 3b ). Furthermore, nonlinear regression analysis provided estimates of Ki ,app values of pyrimethamine, which were similar between metformin and the endogenous compounds (m1A and 1‐NMN), and also to the in vitro Ki values for MATE1/2‐K after correction of Ki ,app for the plasma unbound fraction of pyrimethamine (0.13; Table 2 ). These results convincingly demonstrate that m1A and 1‐NMN are quantitative biomarkers for MATE1/2‐K‐mediated DDI. Considering the long half‐life of pyrimethamine in the plasma (Figure S1 ), mean plasma concentrations (C av) were used in this analysis. Because we have used Cmax for the analysis of other transporters, such as OATP 1B1/3‐mediated DDI caused by a single dose of rifampicin, 36 the analysis using the maximum pyrimethamine concentrations were also conducted. In this instance, it was determined that the fitted parameters were not greatly affected (Figure S6 ).
The reason for the substrate dependence in the dose‐dependent effect of pyrimethamine on CLr between creatinine and other substrates remains unknown. Previously, the Ki values of pyrimethamine for MATE1 and MATE2‐K‐mediated creatinine uptake were reported as 0.17 and 0.22 μM, respectively. 37 Such results are not too different from the corresponding values determined in this study using other test substrates (Table 2 ). OAT2 has also been reported as a creatinine transporter in the kidneys, however, the inhibition potency of pyrimethamine for OAT2 was very weak (half‐maximal inhibitory concentration > 100 μM) to account for this substrate dependence. 37 , 38 Perhaps most importantly, the low contribution of the tubular secretion to the urinary excretion (f PYR, 26%) may limit application of creatinine to such quantitative analysis, when compared with other compounds (Table 2 ).
The absolute value of m1A CLr under control conditions was only 1.3‐fold greater than the corresponding creatinine clearance, and 1.7‐fold greater than the creatinine clearance at 75 mg, which could approximate glomerular filtration rate. Assuming this represents contribution of the tubular secretion (41%), it could not account for f PYR (Table 2 ). Because CLr of m1A at the highest pyrimethamine dose was below the CLr of creatinine (Table 1 ; Figure 2 ), m1A may undergo reabsorption from the urine. Considering that m1A is an adenosine metabolite, a nucleoside transport system might account for such reabsorption. Indeed, it has been suggested that m1A is a substrate of SLC29A1/equilibrative nucleoside transporter 1 in HEK293 cells. 27 However, it has been confirmed that the inhibitory effect of pyrimethamine on this transporter is minimal (Figure S3 ).
Given that the AUC of m1A and 1‐NMN were not increased along with CLr reduction (Figure 2 ), their major elimination pathway is unlikely to be urinary excretion, as unchanged form through pyrimethamine may also affect the synthesis of 1‐NMN. Even though urinary excretion is not major for 1‐NMN and m1A, it is still possible to use changes in their CLr as an index of MATE inhibition. High metabolic activity of 1‐NMN in human hepatocytes was previously reported. 39 Urinary excretion of 1‐NMN was decreased along with an increase in the replacement index of human hepatocytes in human hepatocytes transplanted chimeric mice, suggesting the hepatic metabolism is the major elimination pathway of 1‐NMN. As observed for OCT2, pyrimethamine unlikely inhibits OCT1‐mediated hepatic uptake of 1‐NMN at the doses in this study, considering its Ki for OCT1. 40 The major elimination pathway of m1A remains undefined in humans although its concentration in serum was associated with OCT2 single‐nucleotide polymorphism. 27
We also determined CLr for PDA, a recently identified OAT1 and OAT3 endogenous biomarker, 32 , 33 in the same subjects to exclude the possibility of nonspecific inhibition of the kidney function by pyrimethamine (Table 1 ; Figure S4 ). Based on in vitro inhibition data, pyrimethamine is a weak inhibitor of OAT1 and OAT3 (Figure S3 ). Consistently, pyrimethamine showed only a weak inhibitory effect on CLr of PDA at the highest dose, supporting the specificity of pyrimethamine for MATE1/2‐K in the kidneys (Table 1 ; Figure S4 ). This approach expands the application of endogenous biomarkers in assessing DDIs. Dosing several probe drugs in a cocktail is a way to assess the DDI risk of multiple transporters simultaneously in the same subject. 41 , 42 However, equivalence of the pharmacokinetic parameters of probe drugs between single dose and simultaneous dosing as a cocktail would need to be validated prior to its use. 42 Leveraging of multiplexed endogenous biomarkers is one possible way to facilitate simultaneous DDI assessment across multiple drug transporters early in clinical development without the potential need for DDI studies with individual probes or probe drug cocktails.
As described, this study highlighted the difference in pyrimethamine dose response across different endogenous MATE1/2‐K substrates to identify the appropriate endogenous biomarker for these transporters, and presented integrative analysis of the dose dependence by nonlinear regression analysis. The CLr of 1‐NMN and m1A showed similar performance as DDI biomarkers for MATE1/2‐K, however, because neither 1‐NMN nor m1A present as plasma biomarkers, it is essential to collect urine with appropriate intervals considering the elimination half‐life of the investigational drug in question, which is a limiting factor for their application to drug development. In addition, because diurnal change in the plasma concentration of 1‐NMN was more evident than that of m1A, plasma sampling at multiple timepoints between 12 and 24 hours postdose is warranted to calculate reliable CLr values if a 24‐hour assessment is needed. In this regard, as previously described for OATP1B, it is envisioned that physiologically‐based pharmacokinetic (PBPK) model‐based analysis will aid the design of clinical studies. 41 A PBPK model for metformin has already been constructed and it is able to describe the metformin‐cimetidine DDI involving MATE1 inhibition. 43 Likewise, the construction of PBPK models for 1‐NMN and m1A will advance the prediction of OCT2 and MATE1/2‐K‐mediated DDI in drug development.
In conclusion, the present study assessed the performance of creatinine, 1‐NMN, and m1A as candidate OCT2 and MATE1/2‐K biomarkers, and strongly supports the rationale to leverage the CLr of 1‐NMN and m1A as quantitative MATE1/2‐K biomarkers for the DDI risk assessment in healthy volunteers. Further studies are required to assess the performance of both 1‐NMN and m1A clinical biomarkers for perpetrator drugs that present different OCT2, MATE1, MATE2‐K inhibition signatures vs. pyrimethamine.
Funding
This study was funded and supported by Pfizer Inc.
Conflict of Interest
E.K., L.L., S.M., L.M.H., M.V., V.L., and A.D.R. are employees of Pfizer Inc., USA. R.R. was employed by Pfizer Inc at the time of the study. C.M. is an employee of Pfizer Inc., Japan. All other authors declared no competing interests for this work.
Author Contributions
T.M., E.K., A.D.R., Y.S., and H.K. wrote the manuscript. M.V., A.D.R., E.K., C.M., K.F., Y.S., and H.K. designed the research. T.M., L.L., L.M.H., S.M., R.R., L.S.W., and J.G.J. performed the research. T.M., A.D.R., Y.S., H.K., V.H.L., and M.V. analyzed the data.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1
Table S2
Table S3
Supplementary Material
Acknowledgments
The authors would like to thank Yusuke Kondo (Laboratory of Molecular Pharmacokinetics, Graduate School of Pharmaceutical Sciences, the University of Tokyo) for his bioanalytical technical support, and Kazuaki Ogoe (clerical work), Yasuko Kamiya (CRC), and Issei Kazume (Pharmacist; P‐One Clinic, Japan) for their assistance.
References
- 1. Yoshida, K. et al In vitro‐in vivo extrapolation of metabolism‐ and transporter‐mediated drug‐drug interactions‐overview of basic prediction methods. J. Pharm. Sci. 106, 2209–2213 (2017). [DOI] [PubMed] [Google Scholar]
- 2. Chu, X. et al Clinical probes and endogenous biomarkers as substrates for transporter drug‐drug interaction evaluation: perspectives from the international transporter consortium. Clin. Pharmacol. Ther. 104, 836–864 (2018). [DOI] [PubMed] [Google Scholar]
- 3. Yoshida, K. , Maeda, K. & Sugiyama, Y. Transporter‐mediated drug‐drug interactions involving OATP substrates: predictions based on in vitro inhibition studies. Clin. Pharmacol. Ther. 91, 1053–1064 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Rodrigues, A.D. , Taskar, K.S. , Kusuhara, H. & Sugiyama, Y. Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther. 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Maeda, K. Recent progress in in vivo phenotyping technologies for better prediction of transporter‐mediated drug‐drug interactions. Drug Metab. Pharmacokinet. 35, 76–88 (2020). [DOI] [PubMed] [Google Scholar]
- 6. Jones, N.S. et al Complex DDI by fenebrutinib and the use of transporter endogenous biomarkers to elucidate the mechanism of DDI. Clin. Pharmacol. Ther. 107, 269–277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 72, 253–319 (2020). [DOI] [PubMed] [Google Scholar]
- 8. Wagner, D.J. , Hu, T. & Wang, J. Polyspecific organic cation transporters and their impact on drug intracellular levels and pharmacodynamics. Pharmacol. Res. 111, 237–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motohashi, H. & Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 15, 581–588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gessner, A. , König, J. & Fromm, M.F. Clinical aspects of transporter‐mediated drug‐drug interactions. Clin. Pharmacol. Ther. 105, 1386–1394 (2019). [DOI] [PubMed] [Google Scholar]
- 11. Masuda, S. et al Identification and functional characterization of a new human kidney‐specific H+/organic cation antiporter, kidney‐specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 17, 2127–2135 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Gorboulev, V. et al Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 16, 871–881 (1997). [DOI] [PubMed] [Google Scholar]
- 13. Tanihara, Y. et al Substrate specificity of MATE1 and MATE2‐K, human multidrug and toxin extrusions/H(+)‐organic cation antiporters. Biochem. Pharmacol. 74, 359–371 (2007). [DOI] [PubMed] [Google Scholar]
- 14. Urakami, Y. , Kimura, N. , Okuda, M. & Inui, K. Creatinine transport by basolateral organic cation transporter hOCT2 in the human kidney. Pharm. Res. 21, 976–981 (2004). [DOI] [PubMed] [Google Scholar]
- 15. Ito, S. et al N‐methylnicotinamide is an endogenous probe for evaluation of drugdrug interactions involving multidrug and toxin extrusions (MATE1 and MATE2‐K). Clin. Pharmacol. Ther. 92, 635–641 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Hudson, J.Q. & Nolin, T.D. Pragmatic use of kidney function estimates for drug dosing: the tide is turning. Adv. Chronic Kidney Dis. 25, 14–20 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Chu, X. , Bleasby, K. , Chan, G.H. , Nunes, I. & Evers, R. Transporters affecting biochemical test results: creatinine‐drug interactions. Clin. Pharmacol. Ther. 100, 437–440 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Nakada, T. , Kudo, T. , Kume, T. , Kusuhara, H. & Ito, K. Quantitative analysis of elevation of serum creatinine via renal transporter inhibition by trimethoprim in healthy subjects using physiologically‐based pharmacokinetic model. Drug Metab. Pharmacokinet. 33, 103–110 (2018). [DOI] [PubMed] [Google Scholar]
- 19. Kusuhara, H. et al Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 89, 837–844 (2011). [DOI] [PubMed] [Google Scholar]
- 20. Oh, J. et al Inhibition of the multidrug and toxin extrusion (MATE) transporter by pyrimethamine increases the plasma concentration of metformin but does not increase antihyperglycaemic activity in humans. Diabetes. Obes. Metab. 18, 104–108 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Somogyi, A. , Stockley, C. , Keal, J. , Rolan, P. & Bochner, F. Reduction of metformin renal tubular secretion by cimetidine in man. Br. J. Clin. Pharmacol. 23, 545–551 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grün, B. et al Trimethoprim‐metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br. J. Clin. Pharmacol. 76, 787–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aksoy, S. , Szumlanski, C.L. & Weinshilboum, R.M. Human liver nicotinamide N‐methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 269, 14835–1480 (1994). [PubMed] [Google Scholar]
- 24. Müller, F. et al N(1)‐methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: studies on the metformin‐trimethoprim interaction. Eur. J. Clin. Pharmacol. 71, 85–94 (2015). [DOI] [PubMed] [Google Scholar]
- 25. Shin, S.‐Y. et al An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mishima, E. et al Conformational change in transfer RNA is an early indicator of acute cellular damage. J. Am. Soc. Nephrol. 25, 2316–2326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyake, T. et al Elucidation of N1‐methyladenosine as a potential surrogate biomarker for drug interaction studies involving renal organic cation transporters. Drug Metab. Dispos. 47, 1270–1280 (2019). [DOI] [PubMed] [Google Scholar]
- 28. Imamura, Y. et al Effect of the fluoroquinolone antibacterial agent DX‐619 on the apparent formation and renal clearances of 6β‐hydroxycortisol, an endogenous probe for CYP3A4 inhibition, in healthy subjects. Pharm. Res. 30, 447–457 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Tsuruya, Y. et al Investigation of endogenous compounds applicable to drug‐drug interaction studies involving the renal organic anion transporters, OAT1 and OAT3, in humans. Drug Metab. Dispos. 44, 1825–1933 (2016). [DOI] [PubMed] [Google Scholar]
- 30. Mori, D. et al Dose‐dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin. Pharmacol. Ther. 107, 1004–1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takehara, I. et al Comparative study of the dose‐dependence of OATP1B inhibition by rifampicin using probe drugs and endogenous substrates in healthy volunteers. Pharm. Res. 35, 138 (2018). [DOI] [PubMed] [Google Scholar]
- 32. Shen, H. et al Discovery and validation of pyridoxic acid and homovanillic acid as novel endogenous plasma biomarkers of organic anion transporter (OAT) 1 and OAT3 in cynomolgus monkeys. Drug Metab. Dispos. 46, 178–188 (2018). [DOI] [PubMed] [Google Scholar]
- 33. Shen, H. et al Evidence for the validity of pyridoxic acid (PDA) as a plasma‐based endogenous probe for OAT1 and OAT3 function in healthy subjects. J. Pharmacol. Exp. Ther. 368, 136–145 (2019). [DOI] [PubMed] [Google Scholar]
- 34. Imamura, Y. et al 6β‐hydroxycortisol is an endogenous probe for evaluation of drug‐drug interactions involving a multispecific renal organic anion transporter, OAT3/SLC22A8, in healthy subjects. Drug Metab. Dispos. 42, 685–694 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Müller, F. , König, J. , Hoier, E. , Mandery, K. & Fromm, M.F. Role of organic cation transporter OCT2 and multidrug and toxin extrusion proteins MATE1 and MATE2‐K for transport and drug interactions of the antiviral lamivudine. Biochem. Pharmacol. 86, 808–815 (2013). [DOI] [PubMed] [Google Scholar]
- 36. Mori, D. et al Dose‐dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin. Pharmacol. Ther. 107, 1004–1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mathialagan, S. , Rodrigues, A.D. & Feng, B. Evaluation of renal transporter inhibition using creatinine as a substrate in vitro to assess the clinical risk of elevated serum creatinine. J. Pharm. Sci. 106, 2535–2541 (2017). [DOI] [PubMed] [Google Scholar]
- 38. Shen, H. et al Characterization of organic anion transporter 2 (SLC22A7): a highly efficient transporter for creatinine and species‐dependent renal tubular expression. Drug Metab. Dispos. 43, 984–993 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Kitamura, S. et al Aldehyde oxidase‐catalyzed metabolism of N 1 ‐methylnicotinamide in vivo and in vitro in chimeric mice with humanized liver. Drug Metab. Dispos. 36, 1202–1205 (2008). [DOI] [PubMed] [Google Scholar]
- 40. Kikuchi, R. , Peterkin, V.C. , Chiou, W.J. , de Morais, S.M. & Bow, D.A.J. Validation of a total IC 50 method which enables in vitro assessment of transporter inhibition under semi‐physiological conditions. Xenobiotica 47, 825–832 (2017). [DOI] [PubMed] [Google Scholar]
- 41. Trueck, C. et al A clinical drug‐drug interaction study assessing a novel drug transporter phenotyping cocktail with adefovir, sitagliptin, metformin, pitavastatin, and digoxin. Clin. Pharmacol. Ther. 106, 1398–1407 (2019). [DOI] [PubMed] [Google Scholar]
- 42. Stopfer, P. et al Optimization of a drug transporter probe cocktail: potential screening tool for transporter‐mediated drug‐drug interactions. Br. J. Clin. Pharmacol. 84, 1941–1949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishiyama, K. et al Physiologically‐based pharmacokinetic modeling analysis for quantitative prediction of renal transporter‐mediated interactions between metformin and cimetidine. CPT Pharmacometrics Syst. Pharmacol. 8, 396–406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Table S1
Table S2
Table S3
Supplementary Material