

Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) outbreak initiated the global coronavirus disease 2019 (COVID‐19) pandemic resulting in 42.9 million confirmed infections and > 1.1 million deaths worldwide as of October 26, 2020. Remdesivir is a broad‐spectrum nucleotide prodrug shown to be effective against enzootic coronaviruses. The pharmacokinetics (PKs) of remdesivir in plasma have recently been described. However, the distribution of its active metabolite nucleoside triphosphate (NTP) to the site of pulmonary infection is unknown in humans. Our objective was to use existing in vivo mouse PK data for remdesivir and its metabolites to develop a mechanism‐based model to allometrically scale and simulate the human PK of remdesivir in plasma and NTP in lung homogenate. Remdesivir and GS‐441524 concentrations in plasma and total phosphorylated nucleoside concentrations in lung homogenate from Ces1c −/− mice administered 25 or 50 mg/kg of remdesivir subcutaneously were simultaneously fit to estimate PK parameters. The mouse PK model was allometrically scaled to predict human PK parameters to simulate the clinically recommended 200 mg loading dose followed by 100 mg daily maintenance doses administered as 30‐minute intravenous infusions. Simulations of unbound remdesivir concentrations in human plasma were below 2.48 μM, the 90% maximal inhibitory concentration for SARS‐CoV‐2 inhibition in vitro. Simulations of NTP in the lungs were below high efficacy in vitro thresholds. We have identified a need for alternative dosing strategies to achieve more efficacious concentrations of NTP in human lungs, perhaps by reformulating remdesivir for direct pulmonary delivery.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Remdesivir therapy has shown clinical benefit in patients with coronavirus disease 2019 (COVID‐19) caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Remdesivir is converted intracellularly to its active nucleoside triphosphate metabolite, which terminates RNA transcription in intracellular SARS‐CoV‐2. Target concentrations necessary for remdesivir to inhibit SARS‐CoV‐2 have been determined in vitro.
WHAT QUESTION DID THIS STUDY ADDRESS?
What are the pharmacokinetic (PK) profiles of remdesivir and its metabolites under current clinical treatment regimens and are these treatment regimens able to achieve effective concentrations at target sites of infection to optimally treat COVID‐19 caused by SARS‐CoV‐2?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study leverages existing preclinical in vivo data of remdesivir and its metabolites to examine the PKs of the current clinical treatment regimens for remdesivir and the ability of these regimens to achieve concentrations at target sites of infection necessary for effective treatment of COVID‐19 caused by SARS‐CoV‐2.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
A novel mechanism‐based model of remdesivir and its metabolites describing the active metabolite at the site of action was developed. This study provides an explanation for the need for alternative dosing strategies and the need for optimization of remdesivir treatment for it to be effective to treat COVID‐19 caused by SARS‐CoV‐2.
INTRODUCTION
The severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) outbreak in Wuhan, China, in December 2019 initiated a global pandemic, known as coronavirus disease 2019 (COVID‐19), 1 resulting in over 42.9 million confirmed infections and > 1.1 million deaths worldwide as of October 26, 2020. In order to infiltrate the host cell, SARS‐CoV‐2 binds to angiotensin‐converting enzyme 2 (ACE‐2) receptors, located in the nose with decreasing expression throughout the lower respiratory tract. 2 , 3 , 4 Symptomatic patients with COVID‐19 experience typical symptoms of viral infection, such as fever, cough, shortness of breath, and fatigue. 5 Although many infected individuals experience mild symptoms, including dry cough and sore throat, a significant proportion subsequently develop peripheral lung symptoms, the most serious of which is viral interstitial pneumonia. 6 The progression from initial upper airways disease to serious life‐threatening illness, if not death, occurs rapidly and requires urgent respiratory support. 7
Remdesivir (GS‐5734), originally developed for treatment of Ebola virus, 8 , 9 is a broad‐spectrum nucleotide prodrug effective against many endemic, emerging, and enzootic coronaviruses (CoV). 10 , 11 , 12 , 13 Remdesivir is highly potent with a poor solubility profile (aqueous solubility 0.339 mg/mL, 6 g of solubilizer, sulfobutylether β‐cyclodextrin sodium, SBECD for 5 mg/mL remdesivir, logP 2.0‐2.2, pKa 10.23) and is primarily renally eliminated (74%). 14 Currently approved remdesivir dosing regimen is an i.v. administered 200 mg loading dose on day 1 followed by daily maintenance dose of 100 mg as a 30‐minute infusion for up to 10 days. 15
Remdesivir is a monophosphoramidate prodrug of the C‐adenosine analog (GS‐441524), both of which are metabolized into an active nucleoside triphosphate (NTP) within host cells. Remdesivir is hydrolyzed within host cells into an alanine metabolite (GS‐704277), which is further metabolized into monophosphate and diphosphate derivatives before undergoing triphosphorylation. 8 , 16 The triphosphate form of nucleotide analogue remdesivir acts as a substrate for viral RNA‐dependent RNA polymerase (RdRp) that determines the replication of SARS‐CoV‐2. 17 NTP competes with adenosine triphosphate (ATP) for incorporation as a substitute into the nascent RNA strand resulting in premature termination of RNA synthesis.
Recently, remdesivir received US Food and Drug Administration (FDA) approval for the treatment of adult and pediatric patients 12 years of age and older and weighing at least 40 kilograms for the treatment of COVID‐19 requiring hospitalization. 18 , 19 , 20 Grein et al. observed that 68% of patients with COVID‐19 (N = 53) treated with remdesivir on a compassionate‐use basis showed improvements in oxygen support status compared with baseline. 10 Adaptive COVID‐19 Treatment Trial (ACTT‐1), a placebo‐controlled clinical trial of intravenous remdesivir in adult patients hospitalized with COVID‐19 with lower respiratory involvement (N = 1062) found remdesivir to be superior to placebo in shortening the median time to recovery, defined as hospital discharge or hospitalization for infection‐control purposes only, from 15 (95% confidence interval [CI]: 13–18) to 10 (95% CI: 9–11) days. 19 Another randomized open label clinical trial compared treatment with i.v. remdesivir as a 5‐day (N = 197) and 10‐day (N = 200) treatment in patients with severe COVID‐19 based on the evaluation of clinical status on day 14. Overall, there was no significant difference in clinical status of patients on day 14 and there were no statistically significant differences in recovery rates or mortality rates between the 2 groups. 20 Another clinical study found patients with COVID‐19 symptoms randomly assigned to remdesivir (N = 158) had a numerically faster time to clinical improvement, although not statistically significant compared with those receiving placebo (N = 79) over a duration of 10 days or less. 21
Although the plasma pharmacokinetics (PKs) of remdesivir have been recently described in humans, 22 the distribution of its active metabolite to the site of infection has not. Allometric scaling of PK models developed using available preclinical in vivo data enables the simulation of target site PK in humans, with the assumption that physiological factors are related to body weight. 23 Additionally, allometric scaling is applicable to drugs that are primarily excreted renally (~75%), 24 , 25 such as remdesivir. We can use existing in vivo mouse PK data for remdesivir and its metabolites to develop a mechanism‐based model and simulate human PKs of remdesivir and its active metabolite, NTP, at target sites of infection. In vitro efficacy experiments of SARS‐CoV‐2 infectious viral load have shown the half‐maximal inhibitory concentration (IC50) and 90% inhibitory concentration (IC90) for inhibition in Calu3 2B4 cells by remdesivir to be 0.28 and 2.48 μM, respectively, and by GS‐441524 to be 0.62 and 1.34 μM, respectively. 26 Our objective was to use a mechanistically informed modeling approach leveraging published in vitro efficacy targets to determine the adequacy of current dosing regimens. This approach will develop a more granular understanding of treatment efficacy in patients infected with SARS‐CoV‐2 and aid in identifying alternative dosing strategies.
METHODS
Pharmacokinetic data
PK analyses were performed using in vivo plasma PK data based on a study previously conducted in female Ces1c ‐/‐ mice (from C57BL/6J strain) administered 25 mg/kg of remdesivir. 13 Briefly, plasma samples were collected from mice (3 per timepoint) at eight timepoints (0.25, 0.5, 1, 2, 4, 6, 8, and 12 hours) following s.c. injection. Samples were stored at − 80°C until they were assayed for total remdesivir (molecular weight [MW] = 602.6 g/mol) and GS‐441524 metabolite (MW = 291.26 g/mol) concentrations by liquid chromatography (LC) and dual mass spectrometry (MS‐MS).
In a separate experiment, lung PKs was evaluated following administration of 2 remdesivir doses of 25 mg/kg twice daily or 50 mg/kg once daily s.c. to female Ces1c ‐/‐ mice. 13 Terminal lung samples obtained at 1, 2, 6, 12, and 24 hours after administration were snap frozen, pulverized, weighed, homogenized, and dried. Total nucleosides consisting of nucleoside monophosphate, diphosphate, and triphosphate isolated from lung homogenate were analyzed by LC‐MS/MS. Published and publicly available PK studies were designed by Gilead Sciences and conducted at CRO Jackson.
Model development
Remdesivir and GS‐441524 (referred to as Nuc) concentrations in plasma and total phosphorylated nucleoside (referred to as TNuc) concentrations in the lungs were simultaneously fit to estimate model parameters using the naïve pooled approach in Phoenix WinNonlin 8.2 (Certara L.P., Princeton, NJ). Model discrimination was performed with regard to model structure and error model based on significant change in log‐likelihood ratio, minimization of Akaike information criterion (AIC), parameter estimates, and visual assessment of diagnostic plots.
Remdesivir plasma PK was described by a one‐compartment model with first‐order absorption and elimination via irreversible metabolism to Nuc by hydrolases. The absorption and metabolism of remdesivir were described by:
| (1) |
| (2) |
where is the first‐order absorption rate constant and is the first‐order elimination rate constant for remdesivir via irreversible metabolism to Nuc in plasma. The concentration of remdesivir in plasma was determined by the ratio of the amount of remdesivir in plasma, , and the volume of distribution of remdesivir in plasma, .
Nuc PK was described by a two‐compartment model, where Nuc formation from remdesivir and its elimination from plasma were both described by first‐order rate constants. Equations 3 and 4 describe Nuc PK in plasma and tissue, respectively:
| (3) |
| (4) |
where and describe Nuc volume in plasma and tissue, respectively. is the intercompartmental clearance of Nuc between plasma and tissue. The Nuc plasma concentration was obtained by dividing the amount of Nuc in the plasma, by
The total phosphorylated nucleoside PK consisting of monophosphate, diphosphate, and triphosphate nucleoside metabolites in the lungs (TNuc) were described by a single compartment. Distribution of Nuc in the lungs and subsequent intracellular conversion of Nuc into TNuc and intracellular clearance of the formed TNuc were described using first‐order rates:
| (5) |
where is the combined first‐order rate constant of Nuc distribution and conversion to TNuc within the lungs, is the intracellular clearance of TNuc, and is the cumulative volume of distribution of TNuc. The TNuc concentration in the lungs was described by the ratio of the amount, , and the volume of distribution of TNuc in the lungs, .
Error models assumed pooled estimates of interindividual and intra‐individual mouse variability at each sampling timepoint to describe the residual unexplained variability. Additive, proportional, and proportional error models were evaluated to describe residual variability separately for remdesivir, and its metabolites Nuc and TNuc.
Allometric scaling
Allometric scaling was used to predict human PK parameters for remdesivir and Nuc in plasma and TNuc in the lungs based on PK parameters estimated in mice. Allometric scaling was performed using fixed body weights (BWs) for humans () and mice () of 70 kg and 0.025 kg, respectively, to scale PK parameters from mice () to humans ():
| (6) |
where the values for exponent b for volume, clearance, and first‐order rate constant parameters were 1, 0.75, and −0.25, respectively. 27 Human PK parameters calculated based on simulations performed using allometrically scaled human PK parameters were compared with human PK parameters obtained by modeling digitized data from a single ascending dose remdesivir PK study in healthy volunteers (see Supplementary Methods).
Simulations
Allometrically scaled human PK parameters were used to simulate clinical dosing regimens of remdesivir. 10 Simulated PK profiles included remdesivir and Nuc plasma concentrations and TNuc lung concentrations. Individual simulations without output error were performed in ADAPT5. 28 Remdesivir and Nuc simulated plasma concentrations were compared with PKs in healthy human volunteers. Briefly, based on the European Medicines Agency (EMA) compassionate use of remdesivir, healthy volunteers received a 30‐minute i.v. infusion of 200 mg remdesivir on day 1 and 100 mg daily for 4 days. 29 Mean maximum concentration (Cmax), area under the curve over the dosing interval (AUCτ), and half‐life data for remdesivir and Nuc were recorded on day 1 (N = 8) and day 5 (N = 7). Simulations of TNuc in the lungs were compared with previously described in vitro data 13 and clinical data in healthy human volunteers. 29 Using published protein binding values 29 to determine the free drug concentrations, the simulated free drug concentrations were compared with the in vitro‐determined average IC50 and IC90 effective remdesivir concentrations for inhibiting SARS‐CoV‐2. 30 , 31
RESULTS
PK model
The model simultaneously described the time course of remdesivir and Nuc concentrations in plasma and TNuc in the lungs (Figure 1). The model described the PK data for two different remdesivir doses well based on model fits shown in Figure 2 and parameter estimates reported in Table 1 (percent coefficient of variation [%CV] < 50%).
Figure 1.
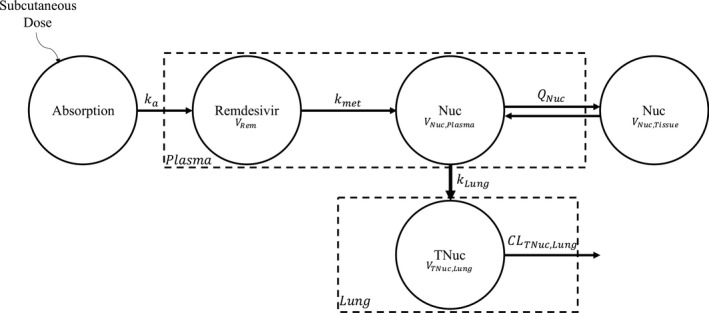
Schematic of the pharmacokinetic mouse model for remdesivir and metabolites. Subcutaneous dosing of remdesivir is described by a one compartment model with first‐order absorption, ka. Remdesivir is irreversibly metabolized to GS‐441524 metabolite (Nuc) via a first‐order rate constant, kmet. This metabolite is characterized by a two‐compartment model with linear distribution, QNuc, between plasma, VNuc,Plasma, and tissue, VNuc,Tissue. Nuc is distributed to lung and phosphorylated to monophosphate, diphosphate, and triphosphate nucleoside, TNuc, via a first‐order rate constant kLung. TNuc is characterized by a one compartment model, only residing in the lungs, VTNuc,Lung. TNuc is eliminated via a linear, intracellular process, CLTNuc,Lung
Figure 2.
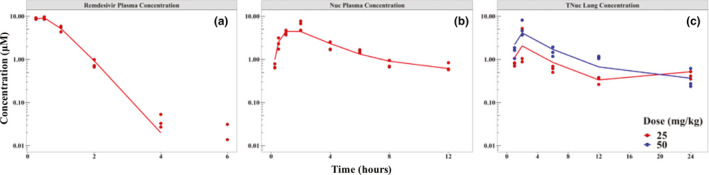
Pharmacokinetic model fits. Semi‐log plot of the concentration of remdesivir and its metabolites versus time in mice following administration of 25 mg/kg (red) or 50 mg/kg (blue) s.c. injection of remdesivir. Model fits (lines) of observed data (points) are shown for (a) total plasma concentration of remdesivir after a single dose, (b) total plasma concentration of nucleoside after a single dose, and (c) total phosphorylated nucleoside (TNuc) concentration in lung after twice‐daily dosing of 25 mg/kg (red) and a single dose of 50 mg/kg (blue)
Table 1.
Final parameter estimates for remdesivir and its metabolites in mice
| Parameter (unit) | Definition | Estimate | %CV |
|---|---|---|---|
| Remdesivir | |||
| ka (h−1) | First‐order absorption rate | 3.31 | 29 |
| VRem (L/kg) | Volume of distribution, plasma | 2.05 | 23 |
| kmet (h−1) a | First‐order metabolic rate constant | 1.95 | 26 |
| ErrorRem,Plasma (μM) | Additive error | 0.412 | 18 |
| Nuc | |||
| VNuc,Plasma (L/kg) | Volume of distribution, plasma | 5.35 | 10 |
| VNuc,Tissue (L/kg) | Volume of distribution, tissue | 11.7 | 49 |
| QNuc (L/hr/kg) | Intercompartmental clearance | 1.15 | 20 |
| kLung (h‐1)b | First‐order metabolic rate constant | 0.217 | 21 |
| ErrorNuc,Plasma (%) | Proportional error | 27.8 | 16 |
| TNuc | |||
| VNTP,Lung (L/kg) | Volume of distribution | 1.69 | 33 |
| CLNTP,Lung (L/hr/kg) | Clearance from lung | 2.26 | 17 |
| ErrorNTP,Lung (%) | Proportional error | 48.9 | 16 |
Abbreviations: %CV, percent coefficient of variation; NTP, nucleoside triphosphate; TNuc, total phosphorylated nucleoside.
kmet affects remdesivir and Nuc.
kLung affects Nuc and TNuc.
Remdesivir and Nuc in plasma were modeled as a one‐compartment and two‐compartment model, respectively (AIC = 119). Alternatively, two compartments were used for both remdesivir and Nuc in plasma (AIC = 123) but this model had parameter identifiability issues related to the remdesivir tissue compartment. Finally, remdesivir and Nuc PK described using a two‐compartment one‐compartment model, respectively, resulted in poor parameter precision compared to the final model (AIC = 124). Models that described bidirectional formation of both Nuc and TNuc and models with additional nonmetabolic routes of elimination for remdesivir and Nuc were assessed. However, parameters describing these models were unidentifiable. Final volume and clearance parameters were conditioned on bioavailability, as remdesivir was administered s.c.
Scaling and human PK parameters
Allometric scaling was performed to predict human PK parameters for remdesivir and Nuc in plasma and TNuc in lungs. 4 Murine PK parameters were scaled to a 70 kg human using published mouse BW of 0.025 kg and established scaling constants of 1, 0.75, and −0.25 for volume, clearance, and rate constants, respectively. 27 , 32 The predicted human PK parameters are reported in Table 2. Predicted human volume and clearance parameters were assumed to be conditioned on bioavailability as these parameters were scaled based on murine volume and clearance PK parameters following s.c. administration.
Table 2.
Predicted human PK parameter estimates based on allometric scaling
| Parameter (unit) | Definition | Estimate |
|---|---|---|
| Remdesivir | ||
| VRem (L/kg) | Volume of distribution, plasma | 2.05 |
| kmet (h‐1)a | First‐order metabolic rate constant | 0.268 |
| Nuc | ||
| VNuc,Plasma (L/kg) | Volume of distribution, plasma | 5.35 |
| VNuc,Tissue (L/kg) | Volume of distribution, tissue | 11.7 |
| QNuc (L/hr/kg) | Intercompartmental clearance | 0.158 |
| kLung (h‐1)b | First‐order metabolic rate constant | 0.030 |
| TNuc | ||
| VNTP,Lung (L/kg) | Volume of distribution, lung | 1.69 |
| CLNTP,Lung (L/hr/kg) | Clearance from lung | 0.311 |
Abbreviations: Nuc, GS‐441524; PK, pharmacokinetic; TNuc, total phosphorylated nucleoside.
kmet affects remdesivir and Nuc.
kLung affects Nuc and TNuc.
Simulations
Model‐predicted human PK parameters were used to simulate currently approved remdesivir treatment regimen for 4 days administered to a 70 kg adult. Unbound concentrations were calculated using a free fraction of 12.1% 29 for remdesivir, 100% for Nuc (range 85–127%), 29 and intracellular TNuc. PK parameters (Cmax, AUCτ, and half‐life) estimated based on the EMA data (observed) and the same PK parameters calculated from simulations performed using allometrically scaled human PK parameters (simulated; Figure 3) are reported in Table 3. Simulated unbound remdesivir plasma concentrations are shown in Figure 3a. Unbound remdesivir Cmax based on simulations was ~4‐fold lower than the observed Cmax on days 1 and 5; whereas the predicted plasma AUCτ and half‐life were within twofold and threefold of the observed values, respectively. Simulations of unbound remdesivir plasma concentrations using current approved dosing regimens were below the in vitro IC50 and IC90 for inhibition of SARS‐CoV‐2 of 0.28 (black dashed line in Figure 3a) and 2.48 μM 26 (black dotted line), respectively. PK parameters (Cmax, AUCτ, and half‐life) calculated based on simulated unbound Nuc plasma PK profiles on days 1 and 5 were within 1.5‐fold of the observed PK parameters based on EMA data. Unbound Nuc PK profiles were within the 95% CI of the predicted human PK (blue shaded region) simulated using PK parameters obtained by modeling digitized human PK data (see Supplementary Results). Simulations of unbound Nuc using allometrically scaled PK parameters were below in vitro IC50 and IC90 for inhibition of SARS‐CoV‐2 of 0.62 (black dashed line in Figure 3b) and 1.34 μM 26 (black dotted line), respectively. Simulated Cmax of TNuc in the lungs (Figure 3c) for days 1 and 5 was 0.027 μM. The simulated drug exposures (AUCτ) of TNuc for days 1 and 5 were 0.523 and 0.679 μM·hours, respectively. The slow formation rate of TNuc based on model predictions identified a formation‐rate limited elimination of TNuc. 33 The simulated half‐life of TNuc in the lungs was 23.1 hours, within 1.1‐fold of the NTP‐predicted half‐life of 22 hours based on in vitro cell culture studies in normal human bronchiolar epithelial cells (Lonza, #CC‐2540, Donor 29132). 13
Figure 3.
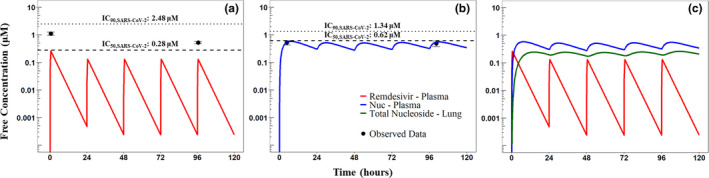
Simulated human pharmacokinetics (PKs). The clinical dosing regimen of a 200 mg loading dose followed by 100 mg daily maintenance dose administered as 30‐minute I.V. infusion was simulated. Simulated PK profiles for (a) remdesivir (red) where the black dashed line indicate the remdesivir IC50 (0.28 uM) and the black dotted line indicates remdesivir IC90 (2.48 uM) for inhibiting SARS‐CoV‐2; (b) Nuc (blue) where the black dashed line indicates the Nuc IC50 (0.62 uM) and the black dotted line indicates Nuc IC90 (1.34 uM) for inhibiting SARS‐CoV‐2; and (c) TNuc (green) metabolites over a 5‐day period is shown. Free concentrations for parent and metabolites in uM are shown on a semi‐log scale. The mean observed peak concentration (Cmax) measured in healthy volunteers is shown as black points. Error bars represent 95% confidence interval (CI) of observed data. The blue shaded region represents the 95% CI of the predicted Nuc plasma PK based on digitized human PK data described in the Supplementary Methods. IC50, half‐maximal inhibitory concentration; IC90, 90% inhibitory concentration; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Table 3.
Observed remdesivir and Nuc PK parameters in plasma (Cmax, drug exposure, AUC, and half‐life) were calculated using EMA compassionate use of remdesivir PK data
| PK Parameter | Observed a | Simulated | Fold change b |
|---|---|---|---|
| Remdesivir day 1 (N = 8) | |||
| Cmax (ng/mL) | 5440 (20.3) | 1300 | 0.24 |
| AUC c (h∙ng/mL) | 2920 (20.6) | 3064 | 1.1 |
| t1/2 (h) | 0.98 (0.82, 1.03) | 2.6 | 2.6 |
| Remdesivir day 5 (N = 7) | |||
| Cmax (ng/mL) | 2610 (12.7) | 653 | 0.25 |
| AUC c (h∙ng/mL) | 1560 (13.9) | 2600 | 1.7 |
| t1/2 (h) | 0.89 (0.82, 1.09) | 2.6 | 2.9 |
| Nuc day 1 (N = 8) | |||
| Cmax (ng/mL) | 152 (25.9) | 169 | 1.1 |
| AUC c (h∙ng/mL) | 2240 (29.1) | 3060 | 1.4 |
| t1/2 (h) | NA | 23.1 | NA |
| Nuc day 5 (N = 7) | |||
| Cmax (ng/mL) | 142 (30.3) | 162 | 1.1 |
| AUC c (h∙ng/mL) | 2230 (30.0) | 3230 | 1.4 |
| t1/2 (h) | 25.3 (24.10, 30.32) | 23.1 | 0.9 |
Simulated remdesivir and Nuc PK parameters in plasma were calculated based on PK profiles simulated using allometrically scaled human PK parameters. Currently approved treatment simulated was 30‐minute i.v. infusion of 200 mg of remdesivir on day 1 followed by 100 mg daily for 4 days in healthy subjects. The difference between the observed and simulated parameters (Fold Change) is reported here.
Abbreviations: %CV, percent coefficient of variation; AUC, area under the curve; Cmax, peak concentration; EMA, European Medicines Agency; Nuc, GS‐441524; PK, pharmacokinetic; t1/2, half‐life.
Observed PK parameters are presented as mean (%CV), whereas the t1/2 as median (Q1, Q3).
Fold‐change was calculated as the ratio of the simulated parameter to the observed parameter.
Area under the curve for day 1 (AUC0–24) and for day 5 (AUCτ).
DISCUSSION
SARS‐CoV‐2 infects the upper and lower airways to cause diffuse alveolar damage. 3 , 34 Serious pulmonary manifestations of COVID‐19 make the lungs an ideal drug target to prevent the onset of local inflammation leading to pneumonia and tissue damage. Remdesivir is a broad‐spectrum antiviral agent with demonstrated in vitro activity against SARS‐CoV‐2, 13 which shortens length of hospital stay in patients with COVID‐19. 10 , 19 , 20 , 21 As of October 26, 2020, clinicaltrials.gov reported two active trials for i.v. remdesivir in patients with moderate to severe COVID‐19: ACTT‐2 (N = 1,034) and WHO‐SOLIDARITY‐GERMANY (N = 400).
In this study, a mechanism‐based PK model describing remdesivir and Nuc in plasma and the active metabolite (NTP) in the lungs was developed using mouse data. Although Nuc (aqueous solubility 13.1 mg/mL, logP −1.9 to −0.58, pKa 12.13) is less lipophilic than remdesivir, a higher percentage of unbound Nuc circulates given its lower protein binding; hence, a larger fraction of free metabolite is available to penetrate tissue, 35 , 36 explaining the use of a tissue compartment to describe Nuc PKs. Leveraging the developed preclinical mechanism‐based PK model, allometric scaling was performed to predict human PK parameters. These PK parameters were used to simulate remdesivir PK in humans to better understand target concentration attainment with the current approved regimen. The simulated PK parameters (Cmax, AUCτ, and half‐life) calculated based on allometrically scaled human PK parameters were reasonably predictive of the 11 observed human PK parameters (Cmax, AUCτ, and half‐life), 29 shown in Table 3 and NTP in vitro‐predicted half‐life, 13 with 67% (8/12) and 83% (10/12) of the simulated parameters within 2‐fold and 3‐fold of observed parameters, respectively. 25 Simulated human PK parameters are in agreement with the currently available observed PK parameters for remdesivir and Nuc in humans and experimentally determined half‐life of NTP in human lung cells. Furthermore, simulated Nuc plasma PK profiles based on the currently approved dosing regimen were within the 95% CI of the Nuc PK profiles simulated using model estimated parameters by modeling digitized human data. The mechanism‐based model was able to accurately simulate remdesivir and Nuc human PK profiles in plasma. 29 Hence, incorporating in vivo murine experimental data of NTP concentrations at the target site (lung) into this model 13 enabled us to simulate predicted NTP concentrations in the human lung. The average NTP exposure (AUCinf) in human peripheral blood mononuclear cells (PBMCs) following a single, 150 mg, 2‐hour i.v. infusion of remdesivir was 0.555 µM·hour (%CV 28.3) in healthy human volunteers (N = 10). 29 Although the observed value is not specific to the human lungs and the loading dose simulated is 33.3% higher (i.e., 200 mg infused over 30 minutes compared with to 150 mg infused over 2 hours), the exposure observed is comparable to the simulated exposures in human lungs predicted using the mechanism‐based PK model estimates for day 1 and day 5. This model can be used to simulate NTP concentrations in human lungs, the site of action, to optimize and assess novel remdesivir treatment regimens that can achieve exposures adequate for SARS‐CoV‐2 inhibition.
Current clinical dosing may be able to achieve unbound concentrations at or above the remdesivir IC50 of 0.28 μM for SARS‐CoV‐2, however, the simulations demonstrate that the Cmax attained with this dosing 29 is below the remdesivir IC90 threshold of 2.48 μM and both IC50 and IC90 thresholds for Nuc of 0.62 μM and 1.34 μM, respectively. Because the IC90 threshold is suggested to be a more stringent threshold of viral inhibition, 37 , 38 maintaining free drug concentrations above the IC90 are likely to lead to more effective COVID‐19 treatment. Thus, current i.v. dosing may not be optimal for achieving the IC90 required to inhibit SARS‐CoV‐2 in the lungs to effectively treat COVID‐19 compared with a targeted, inhaled formulation. Concentration thresholds evaluated using Vero E6 cells were also considered (remdesivir: IC50: 1.65 μM, IC90: 2.40 μM and Nuc: IC50: 0.47 μM, IC90: 0.71 μM) 26 as a less conservative measure of efficacy compared with the previous thresholds reported here using Calu 3B4 cells. Another in vitro study demonstrated that 10 µM of remdesivir resulted in a 9‐log and 10.2‐log reduction in SARS‐CoV‐2 viral nucleocapsid gene expression in alveolar epithelium when tested in organoid and air‐liquid interface cultures, respectively. 39 A target concentration of 10 µM is 10‐fold higher than the observed free drug concentrations in humans, and it is uncertain if this concentration can be achieved in the lungs via the i.v. route or if similar levels of viral inhibition would be possible at lower concentrations. Furthermore, these target concentrations necessary for the inhibition of viral replication are reflective of remdesivir concentrations measured in vitro, not the active NTP metabolite needed in vivo.
Mice infected with 104 or 103 plaque forming unit (pfu)/50 µL of SARS‐CoV were given 25 mg/kg s.c. remdesivir twice daily as prophylaxis (N = 10) or as standard therapy (N = 11), respectively. Lung viral load on day 4 in untreated mice (1.7–3.1 × 106 pfu/lobe) were similar to viral production examined in human bronchial epithelial cells on day 4 in culture (3 × 106 pfu/culture). 40 Treated mice showed significant reductions in day 4 lung viral load and significant improvements in lung function (reduced PenH score) compared with placebo. 29 Further examination of this dose is warranted to determine if the drug‐virus interaction is conserved in SARS‐CoV‐2 infection to assess the contribution of the host immune response to treatment success and to assess therapeutic impact on clinical lung function. The average observed AUCτ of total phosphorylated nucleosides in the mouse lung given this regimen is 10.76 µM·hours (%CV = 61), 13 far above the simulated and observed exposure for NTP in human lungs and plasma under the approved dosing regimen. It is uncertain if this exposure is achievable via i.v. administration or to what extent exposures below this threshold result in significant clinical improvements in lung function. Higher NTP concentrations in the lungs are needed to inhibit viral replication and improve the clinical benefit. 16 Given remdesivir’s poor solubility, reformulating remdesivir to an inhaled formulation may help to achieve higher target NTP concentrations in the lungs. Given the speed of onset of the disease and the role of lung infection in initiating the rapid clinical decline, direct pulmonary delivery of remdesivir as an aerosol formulation is a rational approach to impeding the progress of the disease and the consequent lung damage. Currently, remdesivir as an inhaled nanoparticle formulation is being evaluated for outpatient treatment of COVID‐19 (NCT04480333).
The conclusions of this work should be placed in the context of the underlying assumptions. This model assumed that the total measured concentration of phosphorylated nucleoside metabolites, TNuc, in the lungs (NMP, NDP, and NTP) were equivalent to NTP in the lungs. This assumption is reasonable based on the observation that, on average, NTP constitutes ~80% of the total nucleoside concentration in lungs. 13 In addition, our model assumed complete conversion of remdesivir to Nuc and Nuc to NTP. Although ~10% of remdesivir is eliminated unchanged in urine, 14 there are currently no data to effectively differentiate renal and metabolic elimination of remdesivir, and the current model was not able to differentiate routes of elimination. Additional studies to measure the amounts of remdesivir and its metabolites recovered in urine would make the next generation of this model more robust.
The effective use of allometric scaling of preclinical PKs of an antiviral therapy to describe human PKs has been described. 41 Allometric scaling is an empirical approach that does not take into account differences between species with regard to protein binding, hepatic metabolism, or drug‐receptor interactions. 24 However, these species differences were negligible or accounted for in our approach. The free fraction of remdesivir has a narrow range across species (8.0–14.2%). 29 Remdesivir is primarily metabolized by hydrolase activity and not by hepatic enzymes. Additionally, because carboxylesterase is not found in human plasma, 42 carboxylesterase knockout mice were used in this study to increase the similarity of remdesivir metabolism between mice and humans. Last, receptor interactions of remdesivir are not species‐dependent, indicating allometric scaling is an appropriate tool to predict remdesivir human PK parameters.
The simulated remdesivir Cmax in humans was 0.25‐fold of the average observed Cmax. This parameter is described by the volume of distribution, with the higher predicted volume of distribution value attributable to the inability of the preclinical data to describe the distribution phase of remdesivir because this phase is masked by the absorption phase during s.c. administration. Thus, the available s.c. preclinical data do not fully describe the volume of distribution of remdesivir based on i.v. administration leading to a loss of predictive precision of remdesivir Cmax in humans. Additionally, because mice were administered remdesivir s.c., PK parameters scaled to humans assume 100% bioavailability. Although high s.c. bioavailability is not uncommon for small molecule antivirals, 43 , 44 PK studies with i.v. administration of remdesivir in mice would help determine the true s.c. bioavailability resulting in better characterization of remdesivir PKs in humans. Despite this limitation, accurate conversion from remdesivir to Nuc and from Nuc to NTP was conserved in the scaling process across species, demonstrating the ability of the model to effectively simulate Nuc concentrations in plasma and NTP concentrations in lungs for the assessment of target attainment in a clinical setting.
Additional published PK data have shown biphasic dispositions for remdesivir in rhesus macaques 8 and marmosets, 13 which may have better described the distribution phase of remdesivir PKs. As previously stated, the distribution phase was not apparent in the mouse PK data, likely because it was masked by the absorption phase from the s.c. injection. However, because the rhesus macaque and marmoset data did not include lung NTP concentrations, the data were insufficient to scale and describe NTP concentrations in human lungs. Although total NTP concentration in the lungs was well‐described, NTP concentrations in differentiated cells or regions within the lungs have not been described. Preclinical experiments that differentiate NTP concentrations in lower or upper respiratory tracts could help describe target NTP concentrations at specific sites of infection within the lung. Last, efficacy targets available in the literature 13 , 30 do not describe concentrations of NTP, the active metabolite, necessary to inhibit viral replication. Although a model was created to simulate and effectively describe NTP in human lungs, additional in vitro experiments need to be performed to determine the efficacy target for extracellular and intracellular NTP to better characterize the target concentration of NTP for effective remdesivir dosing. 31 Moreover, the model can assist in the design of preclinical efficacy studies to assess remdesivir and NTP concentrations necessary for SARS‐CoV‐2 inhibition in vivo.
Through this analysis, we have identified a need for alternative dosing strategies to achieve the desired concentrations of NTP in human lungs. Reformulating the poorly soluble remdesivir for direct pulmonary delivery may achieve the desired concentration at the site of infection while also being an appropriate route of administration to mitigate pulmonary progression in critically ill patients with COVID‐19.
Funding
No funding was received for this work.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
P.O.H., B.J., A.J.H., A.V.K., A.D.K., T.P.S., and G.G.R. wrote the manuscript. G.G.R. designed the research. P.O.H. performed the research. P.O.H and G.G.R. analyzed the data.
Supporting information
Fig S1
Fig S2
Table S1
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgments
Pharmacokinetic studies were performed by CRO Jackson laboratories and the study was designed and paid for by Gilead Sciences. The PK data were publicly available and were re‐analyzed herein. Neither entity played a role in the preparation or interpretation of the data in this paper.
REFERENCES
- 1. Lai C‐C, Shih T‐P, Ko W‐C, et al. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and coronavirus disease‐2019 (COVID‐19): The epidemic and the challenges. Int J Antimicrob Agents. 2020;55:105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li W, Moore MJ, Vasilieva N, et al. Angiotensin‐converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis GV, van Goor H . Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hou YJ, Okuda K, Edwards CE, et al. SARS‐CoV‐2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell. 2020;182:429‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo YR, Cao QD, Hong ZS, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID‐19) outbreak – an update on the status. Mil Med Res. 2020;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi H, Han X, Jiang N, et al. Radiological findings from 81 patients with COVID‐19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis. 2020;20:425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ñamendys‐Silva SA. Respiratory support for patients with COVID‐19 infection. Lancet Respir Med. 2020;8:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS‐5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mulangu S, Dodd LE, Davey RT, et al. A randomized, controlled trial of Ebola virus disease therapeutics. N Engl J Med. 2019;381:2293‐2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe Covid‐19. N Engl J Med. 2020;382:2327‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral remdesivir (GS‐5734) is mediated by the viral polymerase and the proofreading exoribonuclease. MBio. 2018;9:e00221‐e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown AJ, Won JJ, Graham RL, et al. Broad spectrum antiviral remdesivir inhibits human endemic and zoonotic deltacoronaviruses with a highly divergent RNA dependent RNA polymerase. Antiviral Res. 2019;169:104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheahan TP, Sims AC, Graham RL, et al. Broad‐spectrum antiviral GS‐5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017;9:eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Veklury (remdesivir) (2020).
- 15. Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID‐19: a randomized clinical trial. JAMA. 2020;324:1048‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun D. Remdesivir for treatment of COVID‐19: combination of pulmonary and IV administration may offer additional benefit. AAPS J. 2020;22:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gordon CJ, Tchesnokov EP, Woolner E, et al. Remdesivir is a direct‐acting antiviral that inhibits RNA‐dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem. 2020;295:6785‐6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Office of the Commissioner . FDA Approves First Treatment for COVID‐19. FDA <https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐first‐treatment‐covid‐19> (2020).
- 19. Beigel JH, Tomashek KM, Dodd LE. Remdesivir for the treatment of Covid‐19 — final report. N Engl J Med. 2020;383:1813‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goldman JD, Lye DCB, Hui DS, et al. Remdesivir for 5 or 10 days in patients with severe Covid‐19. N Engl J Med. 2020;383:1827–1837. 10.1056/NEJMoa2015301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID‐19: a randomised, double‐blind, placebo‐controlled, multicentre trial. Lancet. 2020;395:1569‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Humeniuk R, Mathias A, Cao H, et al. Safety, tolerability, and pharmacokinetics of remdesivir, an antiviral for treatment of COVID‐19, in healthy subjects. Clin Transl Sci. 10.1111/cts.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adolph EF. Quantitative relations in the physiological constitutions of mammals. Science. 1949;109:579‐585. [DOI] [PubMed] [Google Scholar]
- 24. Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 2009;157:907‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poulin P, Jones HM, Do Jones R, et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 1: goals, properties of the phrma dataset, and comparison with literature datasets. J Pharm Sci. 2011;100:4050‐4073. [DOI] [PubMed] [Google Scholar]
- 26. Pruijssers AJ, George AS, Schäfer A, et al. Remdesivir inhibits SARS‐CoV‐2 in human lung cells and chimeric SARS‐CoV expressing the SARS‐CoV‐2 RNA polymerase in mice. Cell Rep. 2020;32:107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alskär O, Karlsson MO, Kjellsson MC. Model‐based interspecies scaling of glucose homeostasis: model‐based interspecies scaling of glucose homeostasis. CPT Pharmacomet Syst Pharmacol. 2017;6:778‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. D’Argenio D, Schumitzky A, Wang X. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. (Biomedical Simulations Resource, Los Angeles, CA, 2009).
- 29. Summary on compassionate use: Remdesivir Gilead <https://www.ema.europa.eu/en/documents/other/summary‐compassionate‐use‐remdesivir‐gilead_en.pdf> (2020).
- 30. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019‐nCoV) in vitro. Cell Res. 2020;30:269‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan J, Zhang X, Liu J, et al. Connecting hydroxychloroquine in vitro antiviral activity to in vivo concentration for prediction of antiviral effect: a critical step in treating COVID‐19 patients. Clin Infect Dis. 10.1093/cid/ciaa623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Houston JB, Taylor G. Drug metabolite concentration‐time profiles: influence of route of drug administration. Br J Clin Pharmacol. 1984;17:385‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martines RB, Ritter JM, Matkovic E, Gary J, Bollweg BC, Bullock H. Pathology and pathogenesis of fatal COVID‐19 cases associated with SARS‐CoV‐2 pandemic. Emerg Infect Dis. 2020;26:2005‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korzekwa K, Nagar S. Drug distribution Part 2. Predicting volume of distribution from plasma protein binding and membrane partitioning. Pharm Res. 2017;34:544‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woodnutt G, Berry V, Mizen L. Effect of protein binding on penetration of beta‐lactams into rabbit peripheral lymph. Antimicrob Agents Chemother. 1995;39:2678‐2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Swinney DC. Molecular Mechanism of Action (MMoA) in drug discovery In Annual Reports in Medicinal Chemistry vol. 46, 301–317 (Elsevier, New York, NY, 2011). [Google Scholar]
- 38. Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin North Am. 2010;24:413‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mulay, A. , Konda, B. , Garcia, G. , et al. SARS‐CoV‐2 infection of primary human lung epithelium for COVID‐19 modeling and drug discovery. bioRxiv. 2020 10.1101/2020.06.29.174623 [DOI] [PMC free article] [PubMed]
- 40. Ehre C. SARS‐CoV‐2 infection of airway cells. N Engl J Med. 2020;383:969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amantana A, Chen Y, Tyavanagimatt SR, et al. Pharmacokinetics and interspecies allometric scaling of ST‐246, an oral antiviral therapeutic for treatment of orthopoxvirus infection. PLoS One. 2013;8:e61514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B, Sedlacek M., Manoharan I. et al. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol. 2005;70:1673‐1684. [DOI] [PubMed] [Google Scholar]
- 43. Cundy KC, Barditch‐Crovo P, Walker RE, et al. Clinical pharmacokinetics of adefovir in human immunodeficiency virus type 1‐infected patients. Antimicrob Agents Chemother. 1995;39:2401‐2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wachsman M, Petty BG, Cundy KC, et al. Pharmacokinetics, safety and bioavailability of HPMPC (cidofovir) in human immunodeficiency virus‐infected subjects. Antiviral Res. 1996;29:153‐161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material