

Nicotinamide adenine dinucleotide (NAD) is one of the most important metabolites in mammalian cells (Figure 1A). Its oxidized form (NAD+) and reduced form (NADH) play a role in many reactions within cells, most prominently in the redox reactions that lead to the production of ATP. NAD functions more broadly than that, however, and is considered to be involved in hundreds of different biological events (Lautrup et al., 2019). Production of NAD+ in mammalian cells occurs mainly via the salvage pathway, which utilizes nicotinamide (NAM), a product of NAD+ degradation, to re-synthesize NAD+ (Figure 1B). This occurs in two steps (Garten et al., 2015): first, NAM and phosphoribosyl pyrophosphate are condensed to nicotinamide mononucleotide (NMN) by the enzyme nicotinamide phosphoribosyl transferase (NAMPT); second, NMN and ATP are used by nicotinamide mononucleotide adenylyltransferase 1–3 to produce NAD+. NAMPT functions as the rate-limiting enzyme of the NAD+ salvage pathway (Revollo et al., 2004). More recently, the therapeutic potential of NAD+ and NAD+ biosynthesis has been investigated, with focus on metabolic diseases, cancer, aging and neurodegeneration (Garten et al. 2015; Lautrup et al., 2019). The relationship between NAD+ and neurodegeneration has been actively investigated; however, less is understood concerning the impact of NAD+ and NAD+ biosynthesis on the neuromuscular junction (NMJ), the synapse where motor neurons interact with skeletal muscle, and how muscles and neurons respond to disruptions in NAD homeostasis. Our recent studies have demonstrated that NAMPT deletion in projection neurons significantly impacts the function of NMJs (Wang et al. 2017; Lundt et al. 2020), indicating a non-cell autonomous effect of neuronal NAMPT on NMJs. NMJs are significantly affected following the loss of NAMPT from projection neurons in mice with a phenotype similar to what is observed in motor neuron diseases, especially amyotrophic lateral sclerosis.
Figure 1.
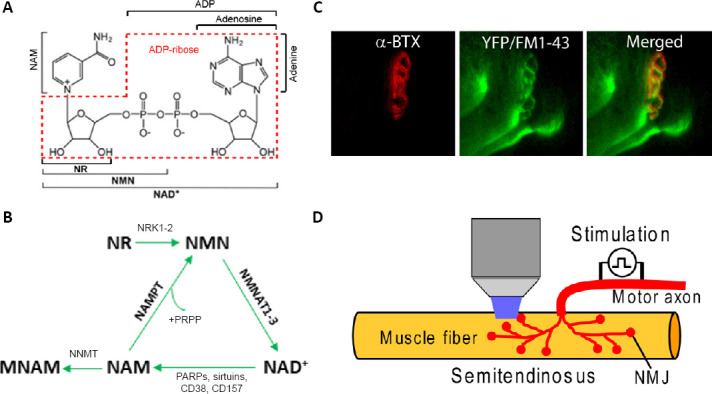
NAD+ salvage pathway and function of NMJs.
(A) Structure of NAD+ and its precursors. (B) Salvage pathway of NAD+ biosynthesis. (C) Motor endplate labeled with α-bungarotoxin (α-BTX-555) and FM1–43. (D) Illustration of FM1–43 imaging of NMJs. Motor axon is stimulated for staining and destaining of FM1–43 to study endocytosis and exocytosis. MNAM: N1-methylnicotinamide; NAD: nicotinamide adenine dinucleotide; NAM: nicotinamide; NMJ: neuromuscular junction; NMN: nicotinamide mononucleotide; NR: nicotinamide riboside; PARP: poly(ADP-ribose) polymerase.
The importance of NAMPT to muscles and neurons has been investigated using gene deletion mouse models. In skeletal muscles, deletion of NAMPT disrupts both normal muscle function and structure. Skeletal muscles are atrophied, are significantly weaker, and seem to be degenerating (Frederick et al., 2016). These mice also have disrupted energy metabolism. The disruption of NAD+ homeostasis can be reversed by administering NMN or nicotinamide riboside (NR), a metabolite that can be converted into NMN by nicotinamide riboside kinases (Figure 1B). The skeletal-muscle NAMPT deleted mice were able to recover the functional deficits by receiving NR in the drinking water (Frederick et al., 2016). Some disease and aging models can also respond to NAD+ supplementation. Ryu et al. (2016) reported that mdx mice, a model for muscular dystrophy, have reduced levels of NAMPT, as well as reduced NAD+ and ATP levels in skeletal muscle. When the mdx mice were fed food pellets containing NR, skeletal muscles had increased levels of NAD+, ATP and NAMPT. The mice also had improved motor performance and skeletal muscle morphology. Unfortunately, neither of these studies investigated how the motor neurons had been impacted before and after NAD+ supplementation. However, the improved motor performance may suggest that motor neurons are positively impacted as a result of the NR treatment. Normal age-related changes to skeletal muscle can also be prevented with NAD+ supplementation. Long-term NMN administration prevented age-associated gene expression changes in skeletal muscle of aged mice and also improved metabolic profiles for glucose and lipids (Mills et al., 2016). Agerholm et al. (2018) examined the effect of NAMPT deletion from intact muscles in mice using Cre recombinase. Results from this study showed the changes in metabolic homeostasis, with reduced levels of Complex I and II of the electron transport chain and elevated glucose uptake. NMN supplementation was able to improve mitochondrial function in aged mice (Mills et al., 2016). Overall, NAMPT dysfunction in skeletal muscles leads to many changes, from gross structure and function to mitochondrial function and metabolic pathways, however, these problems could be alleviated with the treatment of NAD+ precursors. However, these studies were only interested in cell-autonomous effect of skeletal muscle NAMPT.
Recent studies from our laboratory have demonstrated the importance of neuronal NAMPT in NMJ integrity and functions. We demonstrated that inducible deletion of NAMPT from projection neurons causes profound motor deficits, comparable to those observed in the skeletal muscle NAMPT knockout mice, though a significant difference is that deletion of NAMPT from projection neurons was eventually fatal but skeletal muscle deletion has not been shown to be (Wang et al., 2017). Loss of NAMPT in projection neurons produces various NMJ abnormalities including reduced area and increased fragmentation of NMJs, reduced innervation, increased denervation, axonal sprouting and swelling, indicating unhealthy and degenerating axons. Synaptic function at NMJ, which can be studied using FM1-43, a fluorescent styryl dye that can be used to label synaptic vesicles, imaging (Figure 1C and D), is also severely impacted, with evoked endocytosis and exocytosis being significantly impaired after NAMPT deletion in projection neurons (Lundt et al., 2020). These nerves also respond poorly to high frequency stimulation (100 Hz). There is a lot of evidence indicating that the movement and/or the number of synaptic vesicles is reduced following NAMPT deletion. Many of these deficits can be restored through the administration of NMN, with motor tasks and nerve function being improved. However, NMN administration seems only to delay the motor deficits but not prevent them. This differs from the supplemented skeletal muscle NAMPT knockout mice which could fully recover their motor impairments. Complete recovery in projection neuron NAMPT knockout mice may require more than treatment with NAD+ precursors. Also, the beneficial effects of NMN in mouse models is not restricted to motor neurons, Mills et al. (2016) found that NMN can improve eye health and some retinal function.
Deletion of NAMPT in projection neurons also profoundly produces other non-cell autonomous effects, specifically on skeletal muscles (Wang et al., 2017). Following NAMPT deletion in projection neurons, mice had significant muscular atrophy, with significant loss of muscle mass and reduced muscle fiber size in hindlimb muscles. Fiber type switching in skeletal muscles was also observed in these conditional knockout mice. Deletion of NAMPT in projection neurons causes considerable impacts to the motor endplates (Wang et al., 2017; Lundt et al., 2020). Motor endplates were smaller and more fragmented in the conditional knockout mice. Analysis of cross-sections of skeletal muscles indicated similar reductions in motor endplate size. NMN treatments recovered many the changes observed in the cross-sectional parameters and we would expect similar impacts to the entire motor endplate. Mitochondrial morphology in skeletal muscles was altered, with mitochondria becoming misshapen and more elongated. Muscle contraction in skeletal muscles from our conditional knockout mice was impacted, with contractile strength being elevated following loss of NAMPT (Lundt et al., 2020). This was surprising but we also observed that when the mice were given NMN after the deletion, the muscles were contracting with less strength. Overall, the impacts of neuronal deletion of Nampt on skeletal muscles is quite clear. The non-cell autonomous impacts by NAMPT deletion from projection neurons extend beyond skeletal muscle, with heart, lung, liver, and kidneys having reduced mass (Wang et al., 2017). The molecular mechanism by which neuronal NAMPT deletion exerts its non-cell autonomous effect is worth studying further, but the disruption of innervation may be the main reason, as shown in our studies.
The non-cell autonomous influence of NAMPT has also been shown in mice with NAMPT deleted from Schwann cells, which insulate peripheral axons and are important for appropriate nerve function. Sasaki et al. (2018) reported that mice lacking NAMPT in Schwann cells had significantly impaired nerve and NMJ. These mice also developed hindlimb paralysis and had weight loss after 4 weeks of age, similar to what we observed in mice with NAMPT deletion in projection neurons. NAMPT loss in Schwann cells does not seem to induce the degenerative effects observed in mice with NAMPT deleted from skeletal muscles or neurons. These mice did not seem to have increased axonal degeneration, but axonal fragmentation was observed, axonal mitochondrial density was increased, and fewer myelinated axons were present, indicating that axons may attempt to compensate for some metabolic stress. Notably, deletion of NAMPT in Schwann cells was fatal, with mice only surviving for 40 days after birth. Our projection neuron NAMPT knockout mice survived between 3–4 weeks following deletion, so while the loss of Nampt in Schwann’s cells may not induce the same extent of neuron damage, the number of days a mouse survives lacking NAMPT is similar. Comparable to skeletal muscle NAMPT knockout mice, administration of an NAD+ precursor seemed to confer complete recovery.
NAMPT is a critical enzyme for NAD+ biosynthesis in mammalian cells and is essential for survival. It also plays an important role in metabolic homeostasis in cells. Given the strong energetic demands of both skeletal muscles and neurons, loss of NAMPT activity in these cell types produces a broad range of deficits. The NMJs seem particularly susceptible to the NAMPT loss either, through cell-autonomous or non-cell autonomous means. In the absence of NAMPT, treatments with various NAD+ precursors appear to be more effective to muscles than neurons but both cell types respond positively. More investigation is needed to determine the extent that motor neurons are impacted following NAMPT loss in skeletal muscle and how the loss of NAMPT in motor neurons produces the non-cell autonomous changes observed, especially in skeletal muscles.
This work was supported by the National Institute of Health grants R01NS069726 and R01NS094539, and the America Heart Association grants 13GRANT17020004 and 16IRG27780023 to SD.
Additional file: Open peer review report 1 (80.3KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Xusheng Huang, Chinese PLA General Hospital, China.
P-Reviewer: Huang X; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Agerholm M, Dall M, Jensen BAH, Prats C, Madsen S, Basse AL, Graae AS, Risis S, Goldenbaum J, Quistorff B, Larsen S, Vienberg SG, Treebak JT. Perturbations of NAD+ salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. Am J Physiol Endocrinol Metab. 2018;314:E377–395. doi: 10.1152/ajpendo.00213.2017. [DOI] [PubMed] [Google Scholar]
- 2.Frederick DW, Loro E, Liu L, Davila A, Jr, Chellappa K, Silverman IM, Quinn WJ, Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, Redpath P, Migaud ME, Nakamaru-Ogiso E, Rabinowitz JD, Khurana TS, Baur JA. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–546. doi: 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- 4.Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 2019;30:630–655. doi: 10.1016/j.cmet.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundt S, Zhang N, Wang X, Polo-Parada L, Ding S. The effect of NAMPT deletion in projection neurons on the function and structure of neuromuscular junction (NMJ) in mice. Sci Rep. 2020;10:99. doi: 10.1038/s41598-019-57085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016;24:795–806. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 8.Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mázala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Cantó C, Conley KE, Walter L, Lovering RM, Chin ER. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki Y, Hackett AR, Kim S, Strickland A, Milbrandt J. Dysregulation of NAD+ metabolism induces a Schwann cell dedifferentiation program. J Neurosci. 2018;38:6546–6562. doi: 10.1523/JNEUROSCI.3304-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Zhang Q, Bao R, Zhang N, Wang Y, Polo-Parada L, Tarim A, Alemifar A, Han X, Wilkins HM, Swerdlow RH, Wang X, Ding S. Deletion of nampt in projection neurons of adult mice leads to motor dysfunction, neurodegeneration and death. Cell Rep. 2017;20:2184–2200. doi: 10.1016/j.celrep.2017.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.