

Abstract
Heme-copper oxidases (HCO), nitric oxide reductases (NOR), and sulfite reductases (SiR) catalyze the multi-electron and multi-proton reductions of O2, NO, and SO32−, respectively. Each of these reactions is important to drive cellular energy production through respiratory metabolism and HCO, NOR, and SiR evolved to contain heteronuclear active sites containing heme/copper, heme/nonheme iron, and heme-[4Fe-4S] centers, respectively. The complexity of the structures and reactions of these native enzymes, along with their large sizes and/or membrane associations, make it challenging to fully understand the crucial structural features responsible for the catalytic properties of these active sites. In this review, we summarize progress that has been made to better understand these heteronuclear metalloenzymes at the molecular level though study of the native enzymes along with insights gained from biomimetic models comprising either small molecules or proteins. Further understanding the reaction selectivity of these enzymes is discussed through comparisons of their similar heteronuclear active sites, and we offer outlook for further investigations and areas of ongoing study.
1. Introduction
Respiratory metabolism is the cornerstone of cellular energy processes.1–3 To drive cellular energy production, organisms utilize a wide variety of so-called “terminal electron acceptors”4–8 that are often simple small molecules or ions that are prevalent throughout the organism’s immediate environment and possess thermodynamically favorable reduction potentials. For example, anaerobic respiration using sulfate (SO42−) as the terminal electron acceptor in oxidative phosphorylation can be traced to some of the earliest lineages of bacterial life.9,10 These organisms, and extant sulfate-reducing organisms, release sulfide (H2S) as a byproduct and play a crucial part in the biogeochemical cycle of sulfur.1,10 Similarly, the globally-relevant process of respiratory denitrification uses nitrate (NO3−) for this purpose, ultimately yielding gaseous nitrogen (N2) and water.11,12 Eventually, molecular oxygen (O2), an energetically potent terminal electron acceptor, became sufficiently abundant on the planet to allow for the developing prevalence of cellular aerobic respiration—reducing O2 to H2O.13,14 The greater amounts of energy that cellular respiration can provide to the organism is considered to be an underlying cause for the development of higher organisms.15,16
While these small molecules are good terminal electron acceptors for cellular energy processes, efficiently reducing them can be challenging due to high energetic barriers for their activations. To address this issue, nature has recruited metal ions as cofactors because they possess tuneable thermodynamic reduction potentials and electron configurations to catalyze the activation of these small molecules that cannot be achieved easily with nonmetals. Therefore, metalloproteins are ubiquitous components of cellular respiratory pathways and serve roles both in electron transfer (ET) processes and as catalysts for bond-breaking/bond-forming chemical transformations. These metalloenzymes have evolved to efficiently catalyze small molecule redox transformations, in many cases with high degrees of energy conservation and selectivity while avoiding the release of toxic or reactive intermediates. As a result, many different metal ions and coordination environments have been discovered in these proteins to perform a variety of reactions involved in cellular respiration. Even though the building-blocks for the construction of these metalloproteins are limited to 20 natural amino acids, a handful of naturally-derived organic cofactors (e.g. porphyrin), and Earth abundant metal ions—considerably less diverse than the vast number of building-blocks which can be used to prepare synthetic inorganic and organometallic compounds—metalloenzymes display remarkably high activity and efficiency while operating under relatively mild reaction conditions. The protein structures that produce these remarkable metal catalysts, each specialized for a particular reaction, are an inspiration to chemists interested in the development of effective catalysts relevant to the current needs of society.
A type of reaction central to nearly all cellular respiratory pathways, including sulfate reduction, denitrification, and aerobic respiration, is multi-electron, multi-proton small molecule transformation. Most metalloenzymes that catalyze these reactions use multiple redox-active metal centers in close proximity within the enzyme active site (Figure 1). The precise reasons why these complex heteronuclear catalysts evolved for these reactions and how they carry out these complex reactions efficiently and selectively pose some of the greatest unknowns in biochemistry. Recent advances have begun to fill in many of the blanks through increasingly versatile and inexpensive molecular biology tools available today, along with the development and advancement of multiple computational, spectroscopic and crystallographic techniques, which have allowed more detailed studies of complex native enzymes than ever before. In parallel, numerous bio-inspired synthetic, small molecular model complexes, including functional catalysts, have been created to reconstruct the core features of these metalloenzymes. Their ability to go beyond the relatively confined space of biological coordination chemistry has allowed us to begin to understand the deeper underlying chemical principles at work within these complex metalloenzymes. In a complementary approach to studying native enzymes and their synthetic models, our lab and others have used small and robust proteins as scaffolds to design “biosynthetic” models, or artificial metalloenzymes (ArMs), to recreate the complex catalytic centers of native metalloenzymes from the bottom-up while preserving the advantages of facile design and stability inherent to biomimetic modelling.17–21 Biosynthetic modelling of metalloproteins has also been pursued through new-to-nature de novo designed protein scaffolds, further expanding the structural diversity of protein-based models.17,22–26 Both synthetic and biosynthetic models provide several opportunities to complement the study of native enzymes: targeted construction of minimal catalytic components have led to models that helped identify specific, local factors that lead to efficient bond activation and reactivity and these models have allowed for the isolation and characterization of catalytic intermediates whose presence in the enzyme mechanism is implied but has otherwise not been possible to directly interrogate in the native system. The variety of available functional groups, many of which are non-biological, has led to a number of probing, systematic studies. The approach to model native enzymes by redesigning smaller proteins has further provided experimental support for the catalytic roles of complex protein structures beyond the metal primary coordination sphere.
Figure 1.
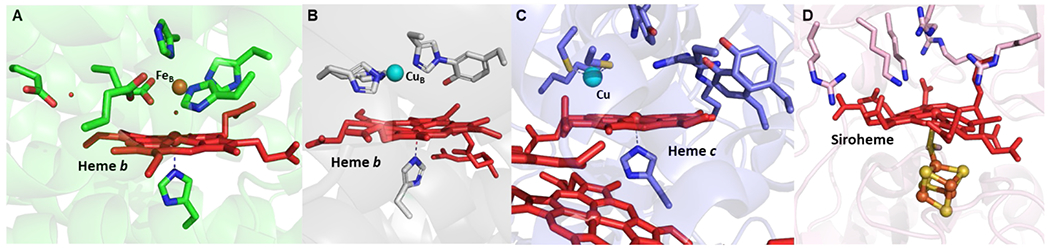
Active site structures of heteronuclear metalloenzymes (A) NOR (PDB ID: 3O0R), (B) HCO (PDB ID: 5B1B), (C) Heme/Cu SiR (SiRA; PDB ID: 4RKM), and (D) siroheme-[4Fe-4S] SiR (PDB ID: 1AOP).
In this review we will focus on recent advances in our molecular understanding of the heteronuclear metallocofactor-containing nitric oxide reductase (NOR), terminal oxidase (heme-copper oxidase, HCO), and sulfite reductase (SiR), along with insights gained from the study of biomimetic transition metal complexes. Each of these enzymes catalyzes a key multi-electron reduction reaction relevant to their respective respiratory pathways in the cell: the 2 e− reduction of nitric oxide (NO) to nitrous oxide (N2O) by NOR, the 4 e− reduction of O2 to water by HCO, and the 6 e− reduction of sulfite (SO32−) to hydrogen sulfide (H2S) in SiR. Notably, these metalloenzymes share a number of structural similarities within their active sites and highlight the way similar building-blocks and catalyst design strategies can accomplish distinct chemical transformations. All three classes of proteins use a heme cofactor to bind and activate the substrate molecule. In the case of NOR and HCO, which are closely related proteins that fall within the same enzyme superfamily, their active sites display a neighboring nonheme metal center that participates in the reduction reaction. A recently determined structure of a multiheme class of SiR has shown a similar heme/nonheme active site structure, although little is currently known about the functional role of this nonheme metal. Regardless, another group of SiRs also utilize a heme cofactor, which is part of a different heteronuclear active site structure, by virtue of a covalent link to a [4Fe-4S] cluster. These native heteronuclear metallocofactor active sites and their biomimetic models have proved to be a challenging but fruitful area of study for structural and computational biologists, biochemists, spectroscopists, and synthetic chemists.
2. Heme-Copper Oxidase (HCO) and Related Biomimetic Models
2.1. The Active Site of Heme-Copper Oxidases.
Aerobic cellular respiration utilizes terminal oxidases, which couple the 4e− reduction of O2 to water with proton translocation, to generate potential energy in the form of a proton gradient that leads to ATP production by ATP synthase (eq 1).14,27
| (1) |
One of the most prevalent groups of terminal oxidases are integral membrane proteins present across all kingdoms of life collectively known as heme-copper oxidases (HCO). They belong to the heme-copper oxidase superfamily, which also include the heme/FeB-containing NOR enzymes (section 3). During O2 reduction, electron and proton transfer steps are highly regulated, which is achieved through precise interactions between the metallocofactors and residues in the proton transfer (PT) channel(s) located across different protein subunits. This synergy helps ensure that harmful, partially reduced oxygen species (PROS)–1e−-, 2e−-, and 3e−-reduced superoxide, peroxide, and hydroxyl radical, respectively–are not released while maintaining energy conservation necessary for proton translocation. HCOs are divided into three main types (A, B, and C), based on their subunit compositions and proton-pumping functions (Figure 2).14,28,29 HCOs are also divided into two distinct classes: one is quinol oxidase (QO) which obtain electrons from quinol, and the other is cytochrome c oxidase (CcO), which obtains reducing equivalents from cytochrome c.14 Due to the wide diversity of their protein structures, electron donors, proton pathways, and metallocofactors, the evolutionary lineage between members in the HCO superfamily remains challenging to fully elucidate.14,30,31
Figure 2.

Sub-classes of HCO protein superfamily. Figure adapted from ref32. Copyright American Chemical Society 2014.
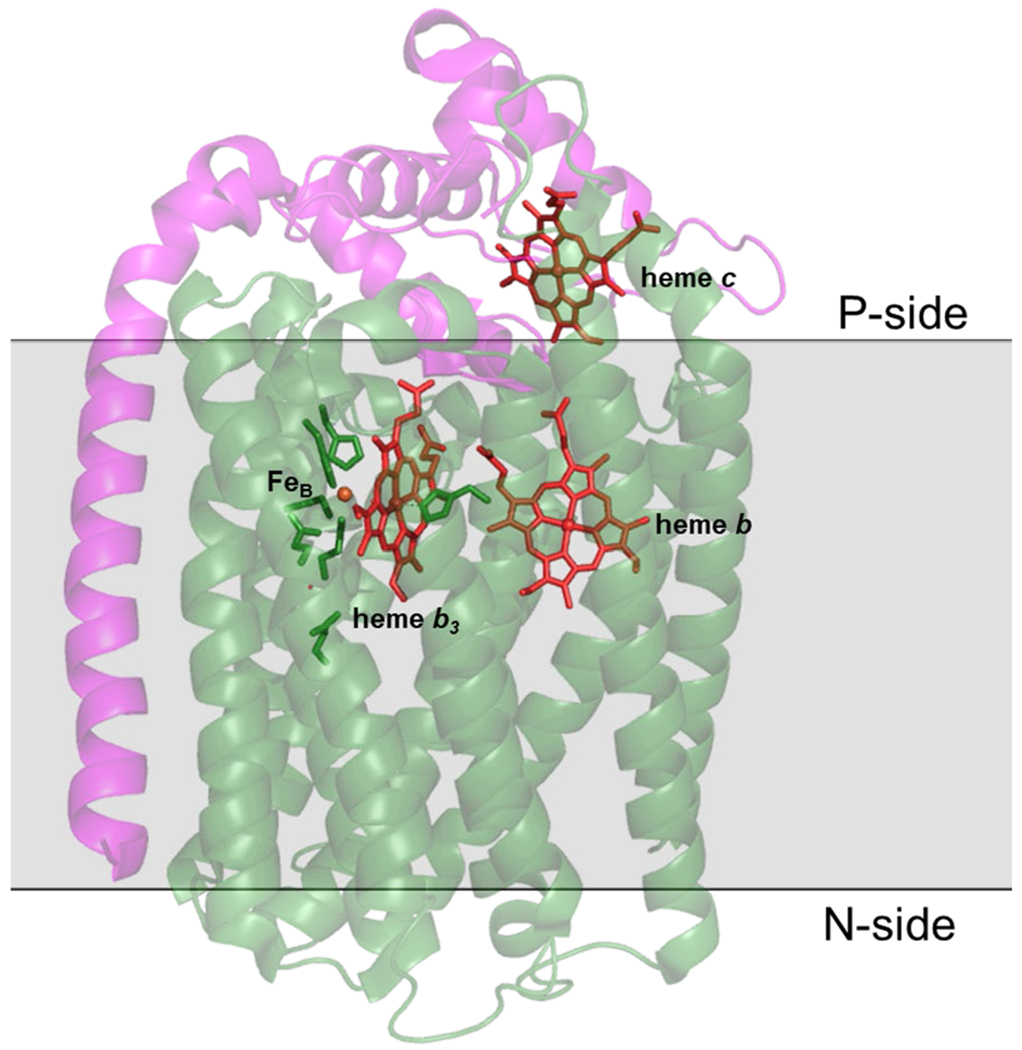
All HCOs share a similar subunit I, which contains the catalytic binuclear center (BNC) that is composed of a high spin heme and copper ion (CuB). Arguably, the most well-studied is the A1 subfamily, particularly aa3-type HCO that can be found in mitochondria, Rhodobacter sphaeroides, and Paracoccus denitrificans.14 HCOs from the B subfamily have been found mainly in archaea but some bacterial B-type HCOs have also been identified and characterized, including ba3 oxidases from Thermus thermophilus and Rhodothermus marinus.33 In A- and B-type CcO, electrons are transferred from cytochrome c to a CuA site in subunit II on the P side of the membrane (Figure 3). These electrons are then passed to a six-coordinate heme a in aa3 oxidase or heme b in ba3 oxidase, which are transferred to the BNC composed of the high-spin heme a3 or b3 and CuB. Other versions of A- and B-type HCOs contain a high-spin heme o3 cofactor in the BNC (Scheme 1).34 The C type HCO is comprised only of cbb3 oxidases, which have been purified from several species, including R. sphaeroides, Vibrio cholerae, Paracoccus denitrificans and Pseudomonas stutzeri.35–37 These enzymes are commonly expressed under conditions of low O2. Among the different types of HCOs, cbb3 oxidase has more in common structurally with bacterial NOR.31 Unlike aa3 and ba3 oxidase, cbb3 oxidase contains heme b3 at the BNC.27 These oxidases contain a subunit N, which is the central subunit containing the BNC, and either an O subunit which has one heme c or both O and P subunits where subunit P contains two heme c cofactors.38
Figure 3.

ET and PT Pathways in subunit I and II of bovine aa3 CcO (PDB ID 5B1B). The schematic diagram depicts chemical protons transferred to the BNC through the D and K pathways while pumped protons are translocated from N- to P-side of the membrane via the H pathway. HCO types without the H pathway are proposed to translocate pumped protons via spatial analogues of the D and K pathways near the BNC, and then to the P-side through a poorly understood PT pathway.
Scheme 1.

Observed metallocofactors in HCO and ET Pathway
Besides differences in their ET pathways, there is also a wide variation between the PT channel(s) among HCO families. Protons that are transferred from the N-side of the membrane to the BNC active site to form water from O2 are called “chemical” protons, while the remainder of protons are separately translocated to the P-side of the membrane to generate the so-called electrochemical gradient for ATP synthase, known as the “pumped” protons.35,36,39 Type A oxidases such as aa3 oxidase can have three PT pathways named D, K, and H pathways (Figure 3). The D and K pathways are named for the residues, Asp and Lys, respectively, which define these channels.28,33 The H pathway only exists in A type CcOs (not QOs), and a standing hypothesis is that this pathway serves as “dielectric well” to help compensate for the thermodynamically unfavorable ET from the P side of the membrane.40 Several articles and reviews have discussed the structure and potential roles of the H pathway.34,40–42 It has been suggested that the H pathway is responsible for translocating all the pumped protons in mammalian A type CcO, while the D and K channels transfer the chemical protons to the BNC.34,43–45 Bacterial A-type CcOs do not rely on the H pathway and it has been established that the K pathway transfers two chemical protons while D pathway is thought to transfer the remaining chemical and pumped protons.43 A recent mutagenesis study has also shown that S. cerevisiae mitochondrial CcO potentially use a similar D pathway for translocation of pumped protons.44 On the other hand, in the B and C type HCOs, including ba3 and cbb3 oxidase, only a single PT pathway is present, which spatially aligns with the A-type K pathway, and is responsible for transferring all of the chemical and pumped protons.28,38,46,47 It has been speculated that the single PT pathway in type B and C oxidases contributes to their lower proton pump stoichiometry, along with differences in ligand binding (O2, NO, CO, etc.) since the D pathway in aa3 oxidase overlaps with hydrophobic residues essential for O2 transfer to the BNC.33,35,48 Regardless of the type of HCO, pumped protons are transferred to a hypothesized “proton loading site” close to the BNC; the exact identity of this loading site is unknown, although it is speculated to be propionate A of heme a3 which receives protons from E242 (Figure 3); therefore, the precise PT pathway for pumped protons to enter the P-side is not currently well understood.49,50 Since this review focuses on insights gained from biomimetic models of native enzyme active sites, which is principally concerned with the efficient and selective O–O activation in HCO (see section 2.4), detailed discussion concerning the proposed mechanisms of the proton pumping activity of HCOs will not be included. Other recent reviews have extensively covered the proton pumping aspect of HCO.49–51
There are many reported structures of type A and B HCO.46,52–54 Currently, there is only one structure of a type C oxidase reported.38 All three types of HCO enzymes display a highly conserved BNC active site structure buried within the transmembrane domain of the catalytic subunit. The heme cofactor is bound by a single His residue, and the distal pocket contains the CuB center ~ 5 Å away, which is coordinated by three conserved His residues. Diffusion of O2 to the BNC has been studied crystallographically with bovine aa3 and Tt ba3 oxidase.55–57 By collecting X-ray diffraction (XRD) data of Xe-pressurized crystals of HCO, these studies demonstrate that O2 migrates through a hydrophobic channel in HCO enzymes (Figure 4) to reach the active site.
Figure 4.

XRD structure of Xe-pressurized cyrstals of HCO, showing the hydrophobic channel for O2 transfer. Figure adapted from ref. 55 Copyright American Chemical Society 2012.
Structures of bovine CcO with various substrate analogues (NO, CO, CN−) suggest there may be functionally significant variation in CuB binding modes (Figure 5).58,59 Largely, the CuB center adopts a trigonal planar geometry in the absence of any ligand, or a pseudo-tetrahedral geometry when a ligand bridging to heme is present. Binding of NO to the heme a3 of bovine CcO has little effect on the Cu geometry; however, in one instance, CN− binding leads to dissociation of a His ligand with CuB and a pseudo trigonal planar geometry by binding to the cyanide nitrogen (PDB ID: 3AG4). Changes to the coordination of CuB have been implicated in modulating the reduction potential of the metal for ET during O–O cleavage or maintaining energy conservation during particular steps of catalytic turnover.58,60
Figure 5.
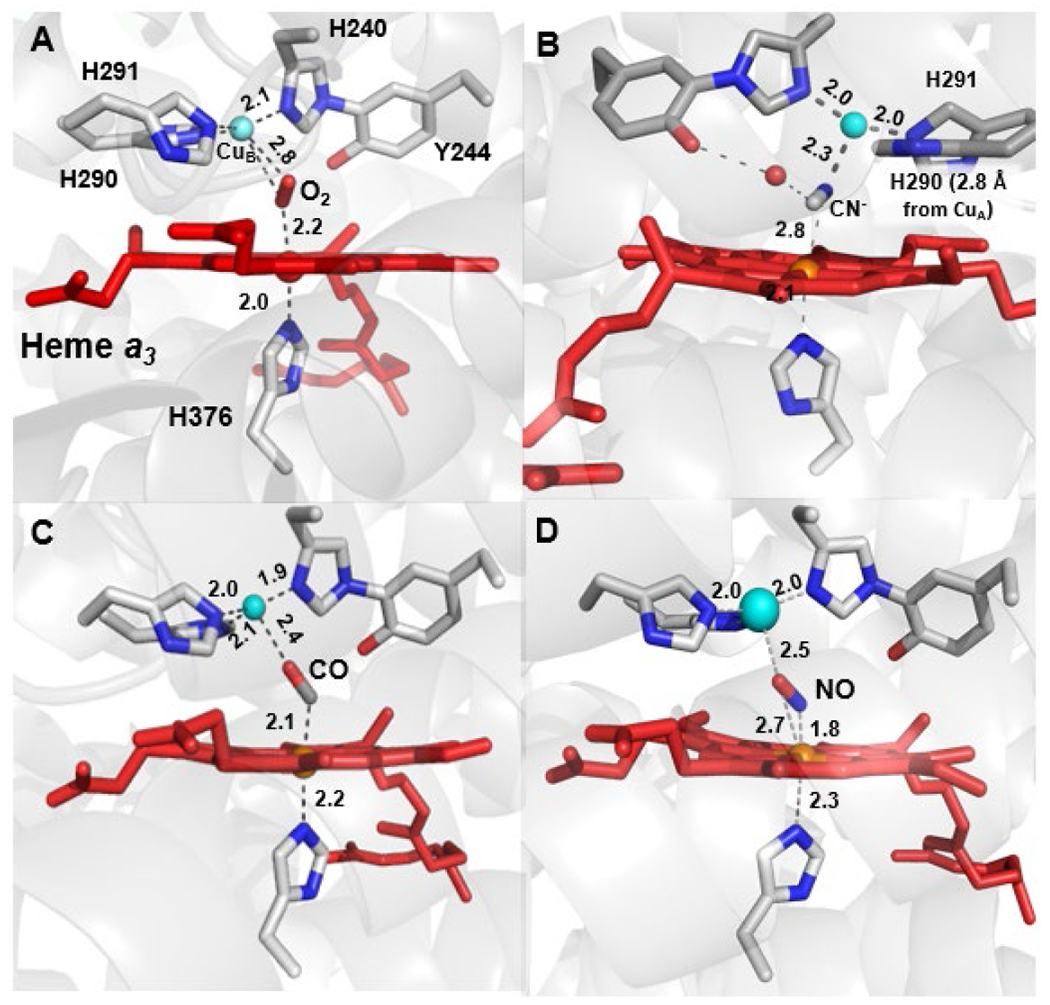
Structures of (A) O2- (PDB ID: 2Y69)-, (B) CN−- (PDB ID: 3AG4), (C) CO- (PDB ID: 5X1F), and (D) NO-bound (PDB ID 3AG3) forms of bovine HCO. The BNC in (A) is in the oxidized state while (B), (C) and (D) are in the fully reduced state.
Another notable feature in HCO is a His-Tyr crosslink–a covalent bond between one of the His residues that binds CuB and a functionally crucial Tyr residue. This Tyr residue serves as a proton and electron donor during catalysis. Small organic models of the phenol-imidazole crosslink suggest it raises the pKa and reduction potential of the Tyr phenol sidechain in HCO.61,62 Implications of the structural and electron donating effect of the cross-linked His to CuB coordination have also been considered.63 This residue is also part of the K PT channel (vide supra), and has H-bonding interactions with a water network in the BNC that interacts with the substrate O2, suggesting it has an important role in PT during turnover.33,58 The position of this Tyr residue in the polypeptide sequence is not conserved, however; C-type cbb3 oxidase displays a His-Tyr crosslink at a different position, where the Tyr is further away in sequence and located on a neighboring helix.38,63 A phylogeny study of HCO sequences has revealed groups of oxidases that lack Tyr at either of these two positions.64 Homology modelling of these sequences reveal two other possible sites near the BNC that can have Tyr residues, which may or may not be covalently linked to one of the CuB His residues. The observation of functionally conserved, but sequentially dissimilar Tyr residues in HCO is interesting from the perspective of understanding the evolution of HCOs.31
2.2. Other Oxidases that Reduce O2 to Water
There are a multitude of metalloenzymes that bind and reduce O2. Along with respiratory enzymes that utilize O2 as an electron acceptor, there are numerous enzymes that couple O2 reduction to the oxidation of organic substrates. Similar to HCOs, the large majority of these metalloproteins use Fe or Cu centers to activate O2. Covering the vast literature concerning these proteins is outside the scope of this review. Instead, we refer the reader to recent reviews that have focused on these other O2 activating metalloenzymes containing heme,65–67 nonheme Fe,68–70 and Cu32,66,71 active sites. This section will focus on metalloenzymes that selectively catalyze the complete 4 e− reduction of O2 to two molecules of water as their native reaction.
2.2.1. bd Oxidase.
These integral membrane proteins exist in a number of prokaryotes and are involved in O2 scavenging and respiratory pathways.72,73 Specifically, all known bd oxidases are quinol oxidases which belong to a family distinct from HCOs.72 These enzymes do not pump protons across the membrane, but are able to generate a proton gradient during O2 reduction by consuming protons from the N-side of the membrane, while oxidizing quinol reducing equivalents near the P-side of the membrane. The bd oxidase contains three heme cofactors, a six-coordinate heme b558 involved in ET, along with a high-spin heme b595 and a heme d (Figure 6A). The heme d cofactor binds to the protein through a weakly-coordinating Glu and displays high O2 affinity.74 Heme d is typically invoked as the site of O2 reduction; however, the high-spin heme b595 appears to be capable of reacting with O2 and other various ligands, leading some to propose it also plays a role in O2 activation.72,75 Flow-flash kinetics experiments of O2 reduction by bd oxidase are consistent with a mechanism similar to HCO (see section 2.3), with a notable difference that there is evidence for an observable FeIII-OOH (hydroperoxide) intermediate.76
Figure 6.

Active site structures of non-HCO oxidase enzymes (A) bd oxidase from Geobacillus thermodenitrificons (PDB ID 5IR6), (B) alternative oxidase (AOX) from Trypanosoma brucei (PDB ID 5ZDP) with ferulenol bound, (C) a flavodiiron oxidase (FDP) from Giardia intestinalis showing its diiron binding site (PDB ID 2Q9U), and (D) laccase, a multi-copper oxidase (MCO) from Trametes versicolor containing a T1 and a trinuclear copper center (T2 and T3 Cu) (PDB ID 1GYC).
2.2.2. Alternative Oxidase (AOX).
These membrane-associated enzymes are found in all domains of life and are closely linked to aerobic respiration, although these oxidoreductases are distinct in being incapable of generating any electrochemical gradient. These enzymes are thought to have various regulatory roles: for example, heat generation in tissue, metabolic homeostasis, defense against oxidative stress, and regulation of cellular signaling pathways.77,78 AOX contain a diiron active site (Figure 6B), which has been unambiguously confirmed recently by the first reported crystal structure.79 The proposed O2-activating mechanism is similar to other carboxylate-bridged diiron proteins, and also shares some features reminiscent to HCO, like a universally conserved Tyr residue near the binuclear active site, which may serve as an electron donor during turnover.80
2.2.3. Flavodiiron Protein (FDP).
FDPs, like the HCO superfamily, are a large family of metalloproteins that catalyze either O2 or NO reduction.81–83 Many FDPs are capable of catalyzing both of these reactions with nearly equivalent activities, while some appear to be selective for one reaction. FDP are globular proteins typically found in anaerobic organisms and are thought to play a role in O2 detoxification. Some FDPs have also been identified in photosynthetic cyanobacteria and are involved in regulating the pool of photosynthetic reducing equivalents under changing light conditions.81,84 Like AOX, the FDP active site comprises a carboxylate bridged diiron center (Figure 6C). Crystal structures of A-type FDPs shows that the diiron metal center is on the opposite end of the flavin mononucleotide cofactor. In order to maintain close proximity for efficient electron transfer, the monomers can form dimer pairs in which the C-terminus of one monomer interacts with the N-terminus of the other.83,85
2.2.4. Multicopper Oxidase (MCO).
MCOs are a large family of enzymes that couple the reduction of dioxygen to water with the oxidation of organic molecules or metal ions.32,86 The site of substrate oxidation is distinct from the O2-reducing site, making MCOs unique from most enzymes that couple O2 reduction to substrate oxidation. MCOs have a mononuclear type 1 (T1) Cu center, which serves as the primary electron acceptor from the substrate molecule, along with the O2-reducing trinuclear copper center (TNC), comprised of a binuclear type 3 (T3) site and neighboring type 2 (T2) Cu (Figure 6D). Complete O2 reduction occurs from the fully reduced form (four CuI), which cleaves the O–O bond in two steps, forming a transient peroxy intermediate (PI), followed by the so-called native intermediate (NI).87 In the presence of substrate, the NI form is re-reduced and H2O is released. One turnover of the O2 reduction catalytic cycle leads to multiple turnovers of substrate oxidation.
2.3. Mechanism of O2 Reduction by HCOs.
Although certain aspects of the O2 reduction mechanism by HCO remain unclear, a general consensus of the basic steps has emerged through extensive structural and spectroscopic studies of bovine aa3 oxidase and other HCOs (Scheme 2). In this section, we will summarize the mechanistic steps of O2 reduction by bovine aa3 oxidase, along with discussion of some remaining mechanistic uncertainties. For more detailed information of the spectroscopic and kinetics data pertaining to our understanding of the mechanism of O2 reduction and proton translocation in HCO, we refer the reader to other recent reviews.34,49,88 In brief, much of the mechanistic insight into O2 reduction at the BNC comes from time-resolved spectroscopies, including resonance Raman (rR) and UV-Vis absorption. One particular method that has been key to our understanding of the mechanism is flow-flash spectroscopy.34,49,89,90 This technique starts with a reduced, or partially reduced, CO-bound HCO in O2 saturated buffer. Time-resolved spectra of O2 reduction are obtained upon photolysis of the CO ligand from the BNC. A drawback of this technique is that it relies on fast dissipation of CO from the active site, which happens to be the case for bovine aa3 oxidase but not all HCOs.91 More recently, analogous experiments have been performed without CO, using a photolabile O2-carrier, which have determined an overall identical O2-reduction process.92
Scheme 2.
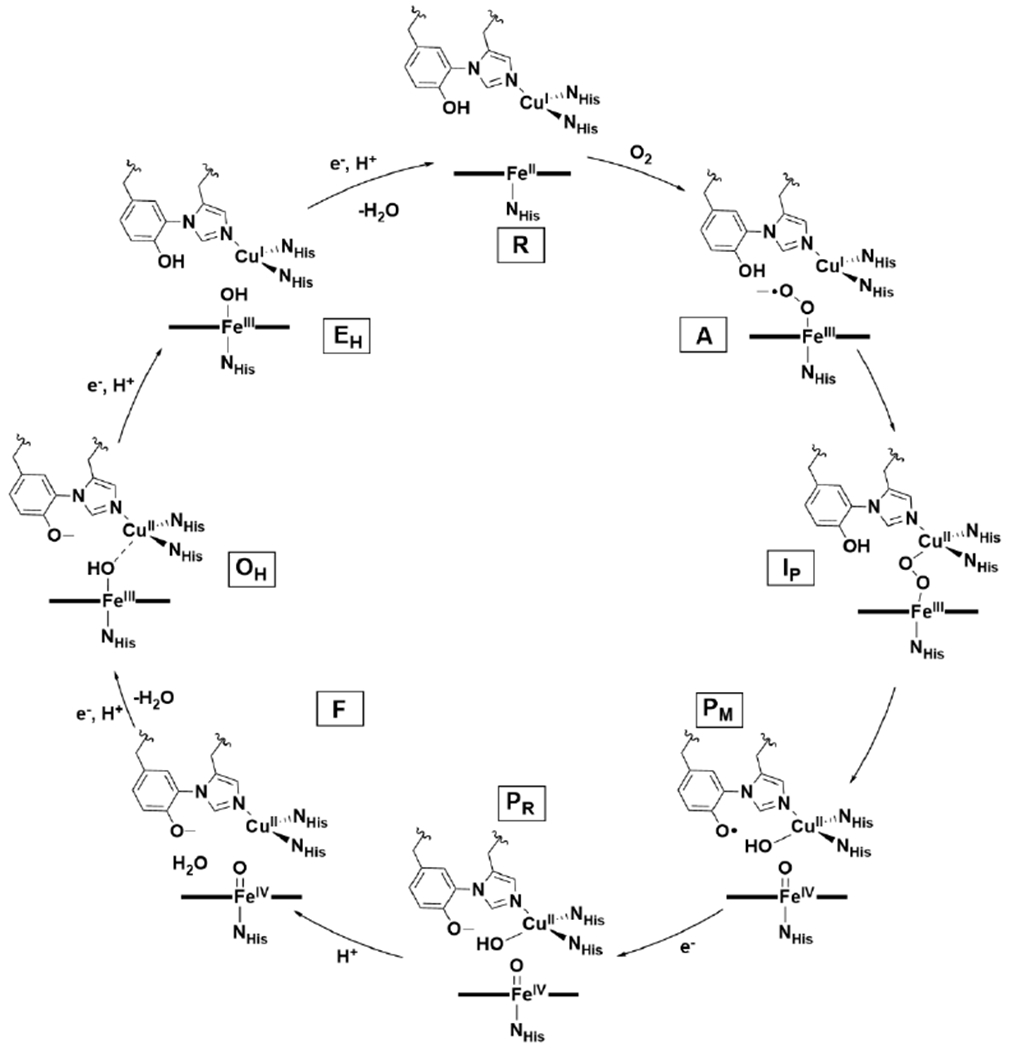
Proposed Mechanism of O2 Reduction by HCO.
2.3.1. O2 binding in HCO.
In flow-flash experiments with fully reduced bovine aa3 oxidase (R), the photolabile CO leaves with an apparent lifetime of ~1 μs, and the process of O2 binding can be observed with a lifetime about an order of magnitude greater, corresponding to a rate constant of ~108 M−1 s−1.90 In the absence of O2, CO still dissociates from the BNC relatively quickly but heme a3 will rebind exogenous CO after tens of milliseconds.93 Time-resolved vibrational spectroscopic studies reveal formation of a transient CuB–CO adduct, concomitant with protein conformational changes that affect the ligand affinity of heme a3, along with solvent access to a nearby water channel.91 Based on these observations, it is proposed that CuB serves as the initial site of O2 binding, which is coupled to protein conformational changes that ensure that there is no back-flow of four proton equivalents, before O2 is transferred and reduced at heme a3 (Figure 7).93
Figure 7.
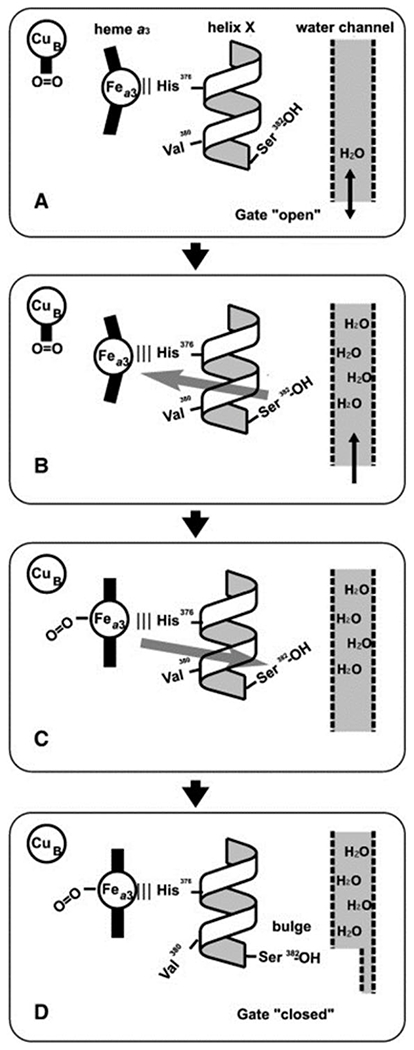
Proposed mechanism of gated O2 binding to BNC with protein conformational changes. (A) O2 binds initially to CuB since the conformation of helix X lowers the O2-affinity of Fea3. The neighboring water channel is based on the helix X conformation. (B) Ser382 is proposed to act as a sensor of H2O and, once it hydrogen bonds with the water channel, it leads to a change in protein structure which increases Fea3 O2-affinity through changes to helix X and the Fe-His376 bond (gray arrow). (C) In this manner, the O2-affinity of Fea3 is highest when enough H2O is present in the water channel. Ligand binding to Fea3 leads to further changes to helix X (gray arrow): a ‘bulge’ conformation on Val380 shifts to Ser382 ‘closing’ the water channel (D).Figure adapted from ref. 93 Copyright Elsevier 2015.
2.3.2. O–O Cleavage Steps (Oxidative Phase‡).
After O2 binding, the next stage of the HCO mechanism is complete O–O bond cleavage, which is known as the oxidative phase of the catalytic cycle.66,94 Coordination of O2 to reduced heme a3 leads to a species with an O2-sensitive rR signal at 571 cm−1, which is characteristic of the so-called A state.34 This rR peak is indicative of the Fe–O stretching vibration (νFe–O) of an end-on Fe–O2 species, but the structure can be Fea3II–O2 (oxy), Fea3III–O2− (superoxo), Fea3III–O–O–CuII (peroxo), or Fea3II–OOH (hydroperoxo) (Scheme 3). The oxy or superoxo assignments for the A state have been favored, due to its similar νFe–O to oxymyoglobin (569 cm−1)—an end-on FeII–O2 or FeIII–O2− hemoprotein;34 however, vibrational studies of heme model complexes are consistent with the peroxo or hydroperoxo forms being equally plausible.66
Scheme 3.
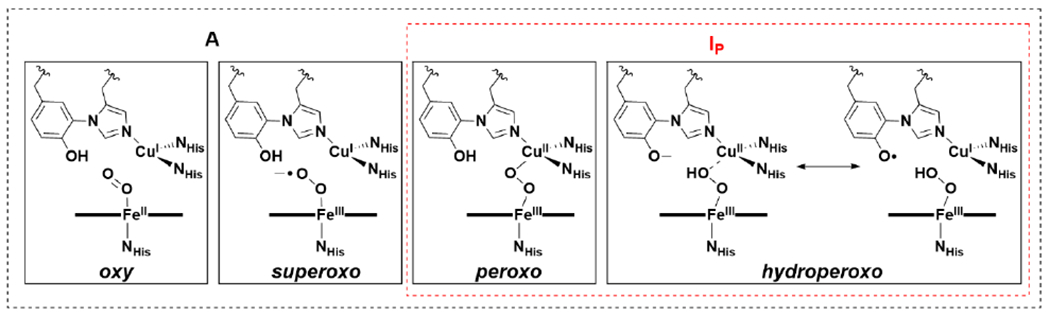
Possible Structures of A and IP Intermediates.
The intermediate of the A state decays forming a so-called P state intermediate with a process lifetime of 32 – 40 μs. It was named the P state because it was initially thought to contain a peroxide; however, rR with isotopic labeling ruled this possibility out. In fact, spectroscopic data is consistent with complete O–O bond cleavage at this stage, to yield an Fea3IV=O/CuII–OH/Tyr-O• species (PM state).34 The exact formulation of the P state depends on whether O2 is introduced at the fully reduced (R) state, or the two-electron reduced state (where CuA and heme a have no reducing equivalents to transfer to the BNC). When heme a is reduced, Tyr-O• is not observed in the P state, instead heme a is oxidized and Tyr is anionic (PR state). Therefore, there is uncertainty whether the PM state is a relevant catalytic intermediate, but the answer likely depends largely on the specific physiological concentrations of O2 and reducing equivalents in vivo.95 Evidence for the relevance of the Tyr-O• in the HCO mechanism has only been indirectly supported through an iodine radical trapping experiment.96 The PM state is EPR silent;49 however, treatment of oxidized bovine aa3 oxidase with H2O2 produces an intermediate that bears weak EPR signals consistent with a delocalized radical on Tyr244, Trp236, and Tyr129, supporting the notion that a Tyr-O• is a feasible intermediate in HCO during turnover.97
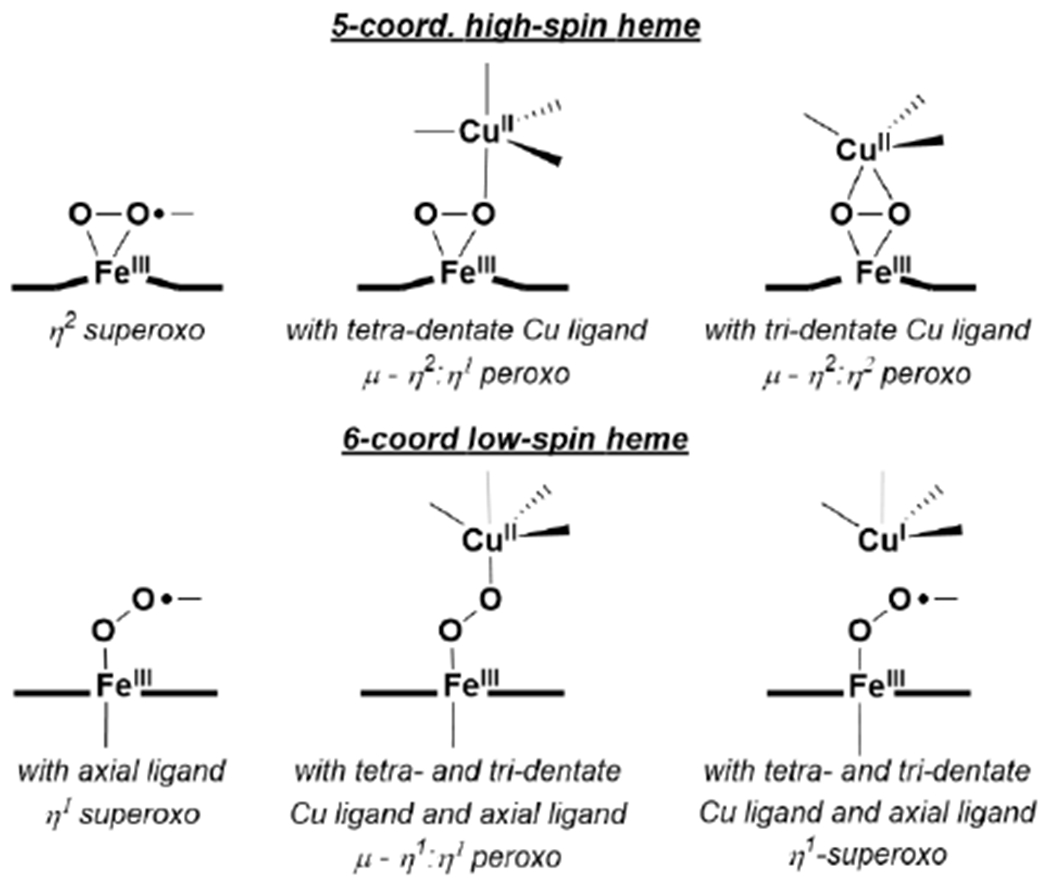
No other intermediates have been observed during the A to P state transition, although often a transient peroxo-containing state, IP, is invoked as a logical species between these intermediates.49,66,98 The exact nature of this peroxo intermediate is debated, specifically whether CuB binds to the distal oxygen, and whether this oxygen is protonated (Scheme 3). Computational studies suggest proton transfer is necessary to lower the barrier to O–O bond cleavage.99,100 Numerous crystallographic studies of HCO enzymes have purportedly isolated peroxo complexes within the BNC, which would correspond to this IP state, initially casting doubt on its catalytic relevance; however, analysis of these structures by Adam et al. have identified inconsistencies of the proposed peroxo assignments from a general coordination chemistry perspective in all but one of these structures, warranting caution in taking these HCO structure assignments at face-value.66 Numerous models of HCO display observable bridging peroxo-intermediates, FeIII–O22−CuII, and their relevance to O–O cleavage processes are further discussed in section 2.4.
2.3.3. Reductive Phase.
The remaining steps of the catalytic cycle involve transfer of protons and electrons to regenerate the R state. Each of these steps in the ‘reductive phase’ are coupled to a proton translocation (for the A type oxidases), which is discussed more extensively elsewhere.34,49–51 Single electron injection experiments have been useful for studying the processes of this phase and these studies conclude that the intermediates in the reductive phase proceed through mostly the same steps: reduction of heme a, followed by proton transfer to BNC, which raises the potential of heme a3 to allow for reduction of the active site.34,101 Studies of the mechanism of proton pumping by type A oxidase suggests it is triggered by the oxidation of heme a, which occurs four times during the reductive cycle, and based on the general principle of Coulombic balance of the electron injection into the BNC at a nearby “proton loading site” (Figure 3).49,50,102 This process is separate from the transfer of protons to the BNC to produce water. The protons that are used to form H2O are denoted “chemical protons”, and, for the remainder of this discussion, only the chemical protons will be considered.
In the PM state, the BNC is fully oxidized, which, in the presence of a reducing equivalent from heme a, will instead rapidly form the previously discussed PR state. Transition from the PR to the F state occurs through transfer of a proton, leading to formation of a BNC structure of Fea33IV=O/CuBII-OH2/TyrO−. Another series of electron and proton transfers produce the fully oxidized (OH) state of HCO. This state is poorly understood due to the lack of direct experimental data for it. It is known that the OH state structurally differs from the as-isolated fully oxidized HCO state that can be structurally and spectroscopically characterized (O) since the O state has a reduction potential too low for the energy conservation function of HCO to be catalytically relevant.103,104 Computational studies of HCO suggest potential structures of OH are Fea3III–OH---CuBII/TyrO−, Fea3III–OH/CuBI/TyrO•, or Fea3III–μ2–O–CuBII/TyrOH (Scheme 4).105,106 It is hypothesized that the OH state readily converts to O under conditions where reducing equivalents are limiting. Theoretical studies suggest either proton transfer105,107 or some other structural rearrangement, like change in metal coordination number,99,106 is responsible for the lower reduction potential in O. For further discussion of the O/OH states, we refer the reader to recent comprehensive reviews of HCO.49,66
Scheme 4.

Possible Structures of the High Potential OH and Lower Potential O States in HCO.
Transition from OH to the partially reduced (E) state has been studied through electron injection experiments with P. denitrificans aa3 oxidase, with reduction and proton translocation occurring on the sub-millisecond timescale.104,108 The exact structure of the E state is unknown, but proposed to be Fea3III–OH/CuI/TyrOH, based on the reduction potentials of each redox active center in the BNC.66 The final E to R state conversion has also been examined with Pd aa3 oxidase, through two-electron reduction of the F state with CO, showing a similar series of steps as the other protonation/reduction/proton translocation steps in the reductive phase.34,109
2.4. Insights Gained from Biomimetic Models of HCO.
HCOs have been studied structurally and mechanistically for several decades and these studies have helped establish an overall view of the H-bonding network at the BNC, the ET pathway that carries electrons to the active site, and the PT channel(s) that transfer protons from the N-side of the membrane towards the BNC, and across the membrane to the P-side. Despite this progress, many aspects of the structural features responsible for such an efficient and selective reduction of O2 to water are still not fully understood (see section 2.3) because it is quite challenging to purify and study these megadalton-sized membrane proteins with multi-domain structures. There are structural features of HCO that are hypothesized to be essential but are difficult to directly interrogate due to the limitations of site-directed mutagenesis using just 20 natural amino acids. In addition to these substitutional limitations, mechanistic studies are also limited to available spectroscopy or crystallographic techniques compatible with these membrane-bound proteins. Furthermore, it can be difficult to capture and study intermediates in HCO due to presence of spectroscopic features of other cofactors that may interfere or even dominate those of BNC where the O2 reduction occurs. These challenges motivate researchers to prepare biomimetic models of HCO as a means to gain deeper insight into the O2 reduction process by this enzyme. These models are much smaller, and can be easier to study because they are free of other cofactors. In addition, it is possible to introduce non-native metal ions or ligands in these models, to probe the roles of each functional group more systematically.
A recent review comprehensively covers studies of synthetic Fe/Cu complexes relevant to HCO and other O2-reducing metalloenzymes.66 The goal of this section is to provide an overview of how these models have helped our understanding of the underlying structural features of native HCO relevant for efficient and selective O2 reduction. Specifically, we will focus on biomimetic studies relevant towards O2 binding, O–O cleavage, and catalytic dioxygen reduction.
Many approaches to mimicking the heme/Cu active site structure of HCO have been employed.19,66,110,111 One is to chemically synthesize heme and Cu complexes separately and then combine them in the presence of O2 or H2O2. This approach has been useful for understanding how the coordination of Fe and Cu affect the structure and stability of reduced O2 intermediates. A notable recent example is the preparation of a “naked” copper-heme peroxo complex, which can serve as a starting point to generate a variety of heme-Cu assemblies.112 A second approach is the synthesis and study of tethered heme/Cu complexes. While most heme/Cu complexes are highly reactive towards O2, and require low temperatures for spectroscopic study, one tethered heme/Cu-peroxo complex has been synthesized which is stable enough to be structurally characterized.113 These systems have also been applied to electrocatalytic O2 reduction reaction (ORR), which has been a useful way to interrogate O2 activation.114–116 Lastly, another approach to model the HCO active site is to use a smaller and robust protein, such as sperm whale myoglobin (swMb), as a scaffold to engineer structural features which mimic those of HCOs. This approach, called biosynthetic modelling, can take advantage of the well-defined protein scaffolds to introduce amino acids in the secondary coordination to probe the roles of weak non-covalent interactions, such as hydrogen bonding networks involving water, more precisely which facilitates systematic study of their structure-property relationships.117–119
2.4.1. O2 Binding of Biomimetic HCO Models.
Studies of simple synthetic Fe porphyrin complexes show that reactions with O2 are commonly encountered in the reduced FeII state.66 The greater propensity of small molecule complexes towards intermolecular reactions often leads to formation of unreactive bridging-oxo complexes with sterically unencumbered porphyrins ((porph)FeIII–μ2–O–FeIII(porph)). Reactivity with O2 at a single Fe site has been examined through the use of sterically demanding porphyrins, to disfavor dimerization, along with the use of cryogenic temperatures to stabilize the early Fe–O2 intermediates. These studies reveal that O2 readily reacts with FeII and typically yields a side-on or end-on FeIII–O2−, depending on whether a trans axial ligand is present. In the presence of CuI, further activation of O2 is often observed, leading to bridging-peroxo intermediates (FeIII-(O22−)-CuII) (see section 2.4.2) (Scheme 5).
Scheme 5.
Characterized Synthetic Fe and Fe/Cu O2 Species Relevant to HCO.
Tethered heme/Cu ligand scaffolds have been useful tools to compare the effect of Cu on O2 binding, since the Fe porphyrin can be prepared with or without the nonheme metal. For example, Collman et al. have reported a study of (L1)FeII and [(L1)FeII/CuI]+, which both form an FeIII–O2− upon reaction with O2 (Scheme 6).120 With CuI, irreversible binding of O2 occurs at room temperature, and the superoxo complex is stable to multiple degassing freeze-pump-thaw cycles. On the other hand, the Fe-only complex only binds O2 once cooled −60 °C. Related model complexes that are able to electrocatalytically reduce O2, [(L2)FeII/CuI]+ and (L2)FeII, display rate limiting O2 binding.121 Further study of the L2 system showed that ZnII has a similar effect as CuI on the rate constant of O2 binding (kon). Interestingly, the corresponding kon for the Fe-only complex was higher, but also highly sensitive to the presence of H2O, which inhibits O2 binding.122 Halime et al. reported a HCO model system, comprised of a tethered high-spin Fe porphyrin and appended Cu, [(L3)FeII/CuI]+, and its Cu-free form, (L3)FeII, are competent for O2 reduction catalysts (Figure 8).123 The rate limiting step of O2 reduction of both complexes is O2 binding at room temperature, and the ORR rate of [(L3)FeII/CuI]+ is higher than (L3)FeII under these conditions. At low temperature, O–O cleavage is the rate limiting step and there is no difference in rate between [(L3)FeII/CuI]+ and (L3)FeII. These results are consistent with Cu principally promoting O2 binding in this system, but having a negligible role in the subsequent O–O bond cleavage.
Scheme 6.

Tethered Heme/Cu Complexes Which Display Effect of Nonheme Metal on O2 Binding Reported by Collman et al. 120,121
Figure 8.
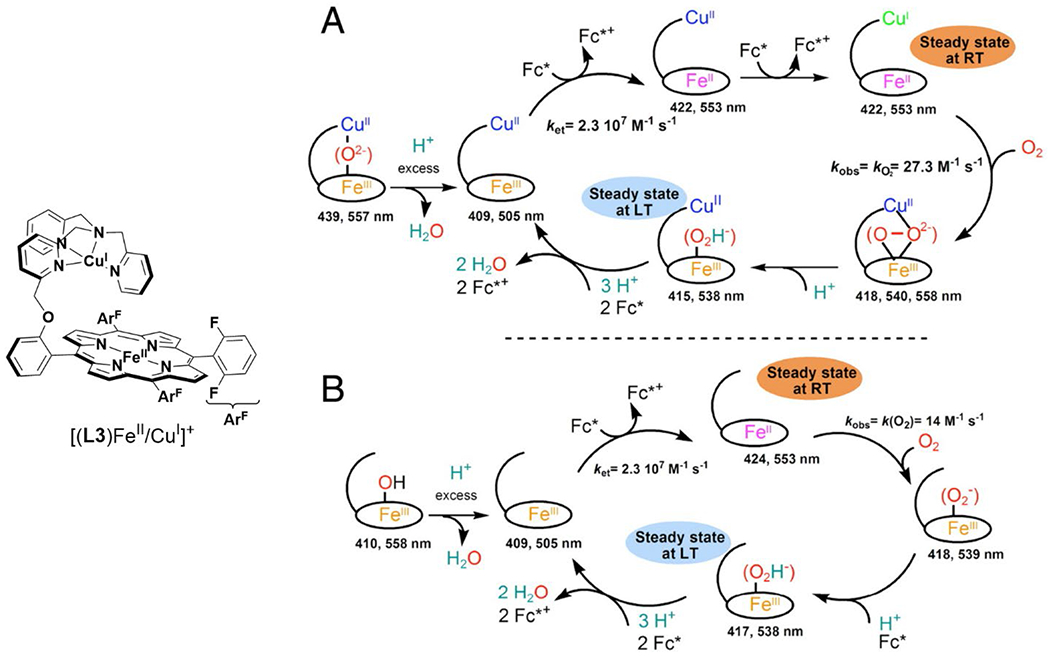
Proposed mechanisms of homogenous catalytic O2 reduction by (A) [(L3)FeII/CuI]+ and (B) (L3)FeII. Mechanism reproduced from ref. 123 Copyright National Academy of Sciences 2011.
The Lu group has used a rationally-designed mutant of sperm-whale myoglobin (Leu29His, Phe43His swMb; CuBMb) to model aspects of the BNC structure in HCO (Figure 9). By incorporating different heme cofactors into CuBMb, the influence of heme reduction potential on O2 binding was investigated.119 Both the kon and koff O2 binding rate constants increase with higher heme reduction potential, but koff displays a steeper dependence on potential, thus making the O2 affinity decrease overall. The presence of a nonheme metal also leads to higher O2 affinity, based on studies using AgI as a redox-inactive analogue of CuI.124 A related biosynthetic protein (Leu29His, Phe43His, Val68Glu swMb; FeBMb), that differs by the presence of a coordinating Glu residue to the nonheme metal, demonstrate that the Lewis acidity of the nonheme metal impacts O2 binding. The rate of formation and stability of oxyheme species was shown to depend on the identity of the nonheme metal (FeII, CuI, CoII, MnII, and ZnII), and could be directly compared when the nonheme metal was not able to be oxidized.117,118
Figure 9.

Overlay of structure of Phe33Tyr CuBMb (magenta) with HCO active site (white).
In native HCO, insight into the effect of CuB on O2 binding are informed by CO photolysis studies (see section 2.3.1), which reveal that when CO dissociates from heme a3 it transiently binds to CuB. This is thought to model, in reverse, the steps of O2 binding to the BNC active site. Similar studies have been carried out with biomimetic model complexes, which generally display this same property as the native enzyme.125–128 Other examples of model substrate binding studies include the [(L4)FeIII/CuII]3+ complex reported by Dallacosta et al., which displays a fivefold increase in azide binding affinity relative to [(L4)FeIII]+ (Scheme 7).129
Scheme 7.
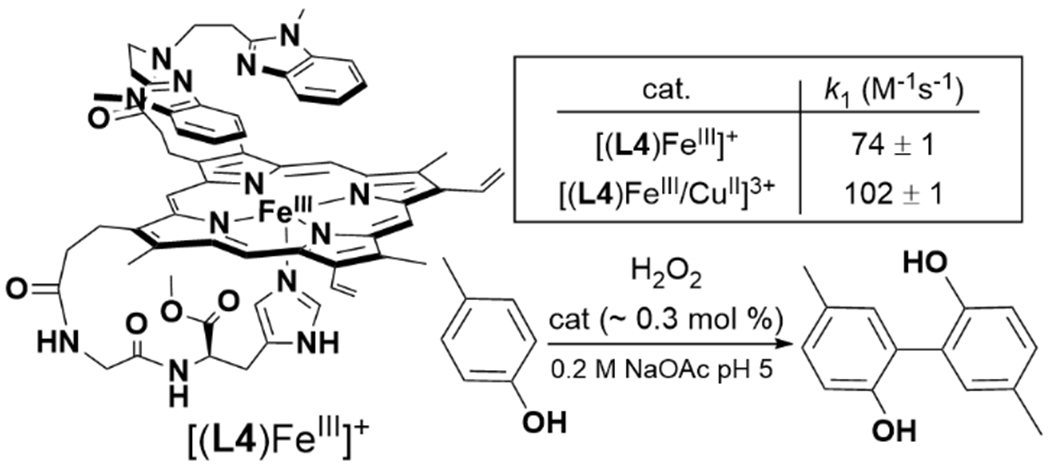
Protoporphyrin IX-Derived Tethered Heme HCO Model Complex and Catalytic H2O2-mediated para-Cresol Oxidation.129
2.4.2. O–O Activation and Cleavage.
Direct studies of the effect of CuI on the properties of the initial heme-O2 intermediates are challenging, due to their propensity for oxidation of CuI and formation of bridging-peroxo complexes. As previously mentioned, examination of redox-inactive metals in place of CuI has been useful for probing the early stages of O2 binding and activation. In FeBMb, increased Lewis acidity of the nonheme metal increases the observed νFe–O of the oxyheme, which indicates a strengthened Fe–O interaction, and subsequent weakening of the O–O bond.118 Only a few stable FeIII-O2−/CuI HCO model complexes have been characterized, although short-lived Fe-superoxo species have been observed in stopped-flow kinetic studies of FeIII–(O22−)–CuII formation.130 The previously discussed picket-fence porphyrin complex L1 with Fe and Cu forms a stable FeIII-O2−.120 Studies of the corresponding Co-porphyrin complex with an empty distal pocket, or with nonheme CuI, CoII, or ZnII, show that these nonheme metals have only a small effect on the νO–O of the CoIII–O2−.131 In an intriguing example of a FeIII-O2−/CuI complex, Liu et al. reported that this species forms in [(L5)FeII/CuI]+ through a transient bridging-peroxo intermediate, seemingly the reverse of the formation pathways of a majority of FeIII–(O22−)–CuII complexes (Scheme 8).132 Two factors were proposed to be crucial for the shift in equilibrium towards FeIII-O2−/CuI with L5: (i) the ‘push’ effect of the axial imidazolyl weakens the binding interactions between Cu and the peroxide and (ii) the relative order of reduction potentials of Fe and Cu in this system thermodynamically favor CuI. This conclusion is corroborated by a report of a stable peroxide in a related system that bears no heme axial ligand, which would both raise the heme potential and remove any ‘push’ effect.133
Scheme 8.

Formation of FeIII-O2− Through a Transient FeIII–(O22−)–CuII. 132
Numerous studies of biomimetic FeIII–(O22−)–CuII intermediates have been performed to gain a deeper understanding of how the coordination environment of the metals influence the binding and reactivity of the peroxide moiety.66,130 For example, the properties of a series of (L)(L6)FeIII–(O22−)–CuII(L’) complexes were investigated, where the presence of an axial ligand (L = DCHIm [1,5-dicyclohexylimidazole] or nothing) and chelating ligand to Cu (L’ = TMPA [tris(2-pyridylmethyl)amine] or AN [bis(3-[dimethylamino]propyl)amine]) had significant impact on the peroxo moiety (Scheme 9).112,134–136 The HS-TMPA (no L, L’ = TMPA) complex displays a side-on binding between Fe and O22−,134,137 which is confirmed by characterization of a tethered complex that has a highly similar ligand architecture and spectroscopic properties (Figure 10).113 The related HS-AN complex displays a greater degree of O–O activation, a consequence of the Cu center shifting from binding end-on to the peroxide in HS-TMPA to side-on in HS-AN due to the change in coordination number about Cu.135,137 Addition of an axial ligand to Fe leads to a change in its spin state, and loss of one Fe–O interaction, resulting in end-on binding in LS-TMPA and LS-AN for both Fe and Cu centers.112,136,138 The consequences of the end-on/end-on binding mode include decreased activation of O22− (based on vibrational spectroscopy) and an increased Fe–Cu distance.
Scheme 9.

Influence of Fe and Cu Coordination Environment on Peroxo-Moiety.
Figure 10.
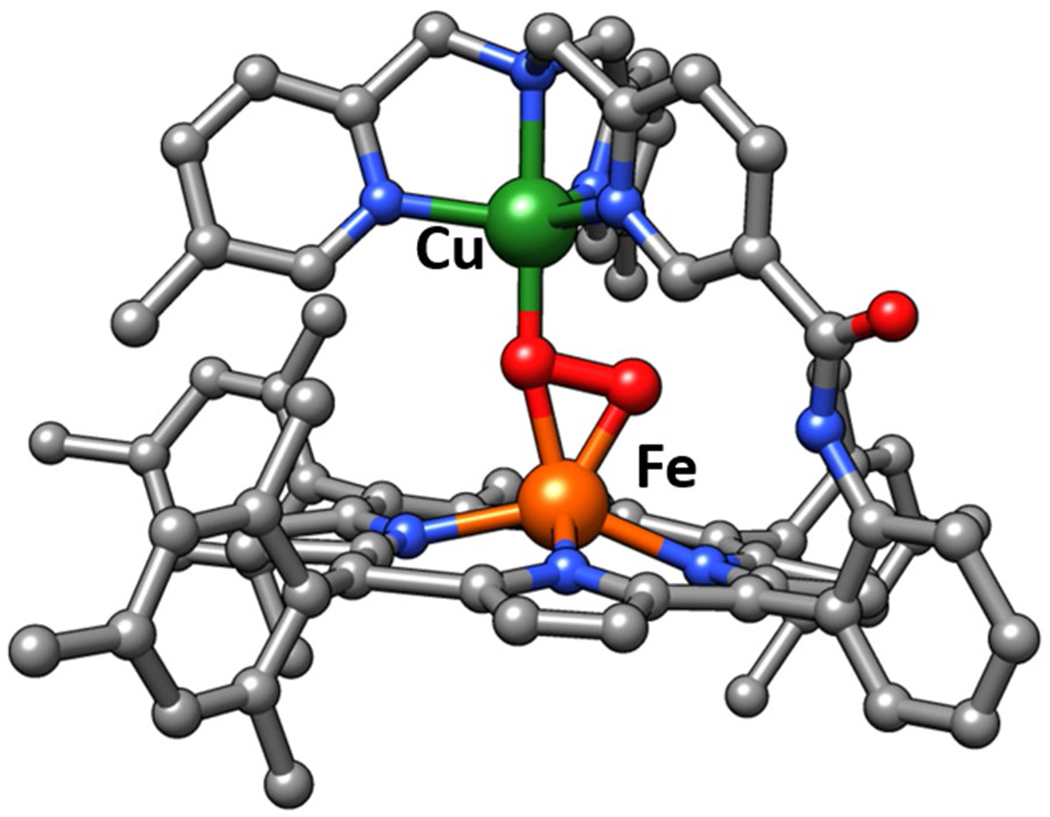
XRD structure of stable Fe/Cu-peroxo complex reported by Chishiro et al.113.
The characterization of heme/Cu-superoxo and -peroxo complexes discussed here have important implications for understanding the cooperativity between Fea3 and CuB in HCO during O2 activation. A major conclusion from these HCO model studies is that the precise role of CuB will be highly dependent on its coordination geometry, distance from the heme, and protonation state of the reduced O2 species. Whether the early intermediates in HCO involve end-on bridging peroxo species, like LS-AN, or if the protein environment shifts the equilibrium towards an FeIII–O2−/CuI, similar to what has been observed for other tethered heme/Cu assemblies, would effect our perspective of a number of aspects of HCO catalysis, such as the role of protons in formation of the P state, and the selectivity the native enzyme displays for complete O2 reduction to water.
We can consider most of the discussed FeIII–O2−/CuI and FeIII–(O22−)–CuII complexes as models of HCO active site in the early A or IP states. Further activation to completely cleave the O–O bond in synthetic biomimetic complexes nearly always requires the addition of some form of reducing equivalent. Interestingly, this observation is despite the fact that there are available electrons from the metal centers, or porphyrin ligand. An exception of complete O–O bond cleavage of H2O2 with an FeIII/CuII complex has been reported by the Casella group: the tethered protoporphyrin IX/Cu complex demonstrates peroxidase-like activity with (L4)FeIII/CuII for the oxidation of para-cresol (Scheme 7).129 Further study of the effect of nonheme metal on this peroxidase-like activity confirm the major role of the metal is to enhance H2O2 binding to heme, and promote heterolytic cleavage to form the compound I-like active oxidant, (porph•+)FeIV=O.139 Although this reaction is not totally relevant to O2 reduction in HCO, it demonstrates that obtaining additional reducing equivalents from the ligand is a viable way to cleave the O–O bond. In contrast, in initial attempts to promote O–O cleavage in HS-AN and HS-TMPA, the addition of strong acid to these FeIII–(O22−)–CuII intermediates simply leads to release of H2O2 (Scheme 10).135 This is a different outcome from related mononuclear FeIII–O22− complexes, which can form analogues to compound I upon addition of acid.66,140 Complete O–O cleavage from HS-AN and HS-TMPA requires addition of at least two equivalents of reductant, yielding the corresponding bridging-oxo product [(L6)FeIII–μ2-O–CuIIL’]+.135
Scheme 10.

Reactivity of HS-TMPA and HS-AN Towards Acid and Reductant to Yield H2O2 and H2O, Respectively.135
There are examples of phenols being used to promote complete O–O cleavage in heme/Cu assemblies, which can support the important role of the active site Tyr as a proton and electron donor in HCO. Collman et al. reported the reaction of [(L1)FeIII–O2−/CuI]+ with sterically-hindered phenols generates a phenoxyl radical along with [(L1)FeIv=O/CuII–OH]+, analogous to the PM state of HCO (Scheme 11).141 In a later study, they showed that a modified tethered ligand (L2), bearing a mimic of the His-Tyr crosslink could similarly achieve complete O–O cleavage of O2 with intramolecular PCET from the appended phenol.142 More recently, a detailed computational and kinetic isotope effect study of the reaction between LS-AN with phenol has provided insight to the mechanism of the phenol-promoted O–O cleavage.143 A mechanism involving hydrogen-bond assisted homolytic cleavage of the bridging peroxo was favored over an alternative that involved initial proton transfer from phenol to generate a transient hydroperoxo (Scheme 12). The potential relevance of this mechanism to HCO was discussed, and it was concluded that proton transfer from the His-Tyr crosslink is an essential prerequisite for its capacity to serve as an electron donor for O–O cleavage.143 On the other hand, many other reported heme/Cu assemblies do not display complete O–O cleavage in the presence of phenols, or appended phenol moieties. It is thought that the low spin ferric heme of [(L1)FeIII–O2−/CuI]+ is crucial for its difference in observed reactivity.137 It is surprising, then, that a similar phenol-appended ferric-superoxo complex reported by Liu et al. is unreactive (Scheme 8).132 Furthermore, Adam et al. have recently demonstrated that two low spin FeIII–(O22−)–CuII complexes (LS-4DCHIm and LS-3DCHIm) display different reactivity towards weak phenolic acid and reductant, based on differences in Cu coordination (Scheme 13).144 There are likely other important structural and electronic features of heme/Cu assemblies which determine their propensity to completely cleave the double-bond of O2 which remain to be elucidated.
Scheme 11.

O–O Cleavage Induced by Inter- and Intramolecular Proton Coupled Electron Transfer from Phenol.141,142
Scheme 12.
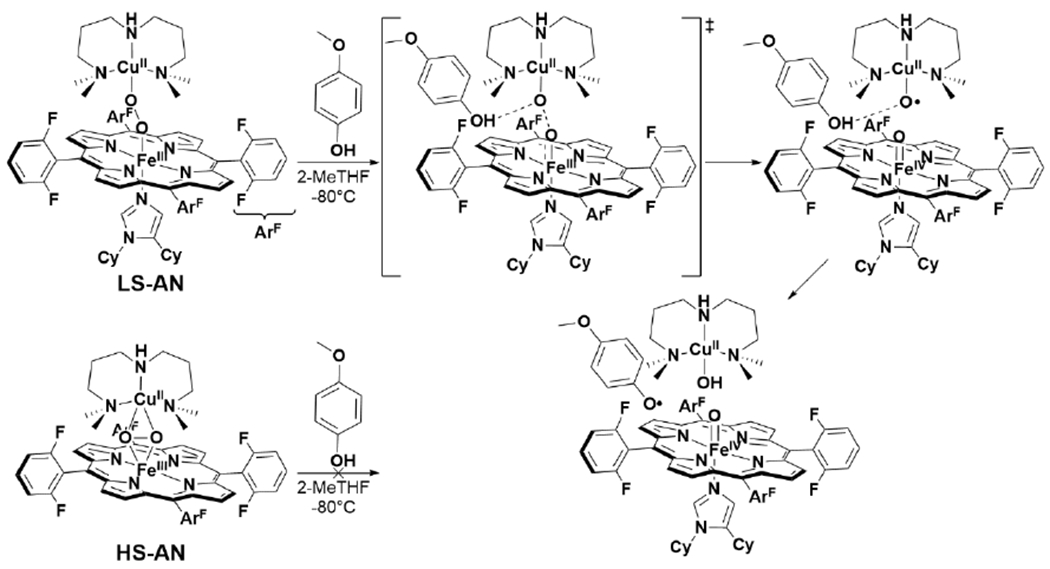
Hydrogen Bond-Assisted Mechanism of O–O Cleavage in LS-AN.143
Scheme 13.
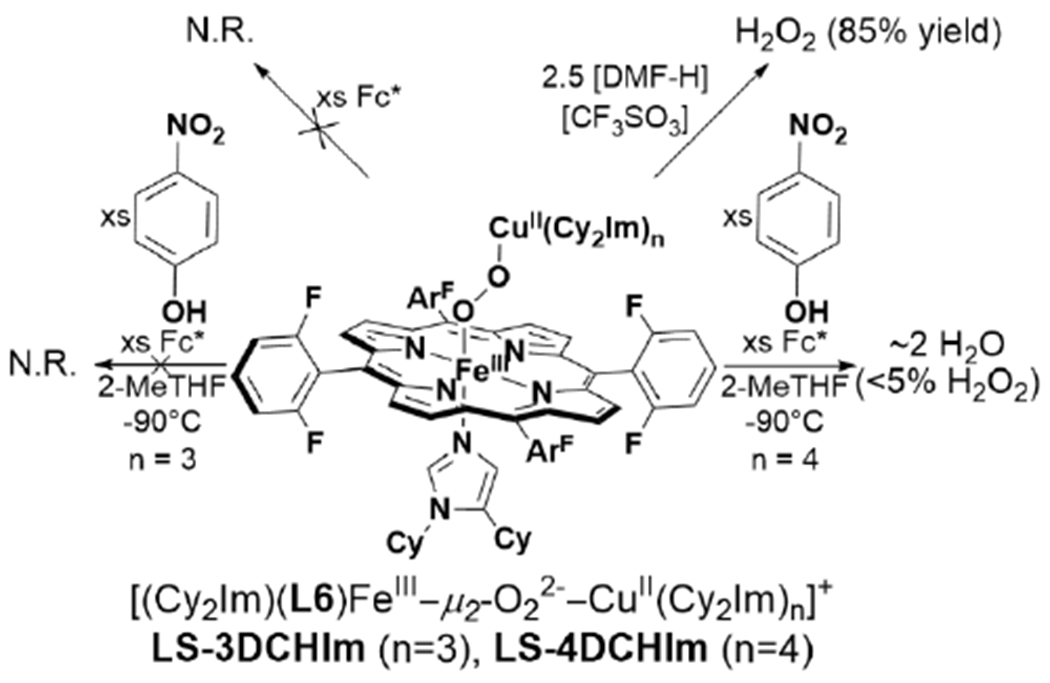
Reactivities of Low-Spin FeIII–(O22−)–CuII Complexes with Different Cu Coordination Number.144
2.4.3. Catalytic Dioxygen Reduction by HCO Models.
Study of the catalytic performance of biomimetic heme/Cu assemblies has been complementary to low-temperature spectroscopic studies of heme/Cu O2 intermediates relevant to HCO, as discussed in the previous sections. In particular, the application of biomimetic heme/Cu complexes to electrocatalysis has been particularly useful in understanding the importance of reduction potentials and electron flux on efficient and selective O2 reduction. More extensive reviews covering the breadth of (electro)catalytic O2 reduction by synthetic complexes related to HCO have been published.66,145–147 Here we will highlight insights gained relevant to our understanding of the mechanism of the native enzyme. Catalytic studies are useful for investigating key structural features that are relevant for high ORR activity, with high selectivity (low PROS formation), and, in some cases, can also be studied using in situ spectroscopic techniques to directly examine the nature of reactive, catalytically-relevant intermediates.
Many relatively complex synthetic porphyrin structures have been examined for catalytic ORR activity. Among these that are relevant to HCO, interesting dependences (or lack thereof) of the Cu center on the extent of PROS formation have been observed. For example, a series of ‘capped’ porphyrin complexes reported by Collman et al. are able to electrocatalytically reduce O2 to form 2 or 4 e− reduced products (Scheme 14).148 The complexes where the Cu is easier to reduce than the Fe porphyrin display mixtures of 2 and 4 e− O2 reduction, where the 2 e− reduced products are attributed to reaction pathways that only involve Cu. Substitution of Fe for Co, which is easier to reduce than the Cu center, leads to exclusive formation of H2O.149 Interestingly, many other heme/Cu ORR catalysts display no increase in selectivity with Cu, likely due to the catalytic conditions that employ a large excess of reducing equivalents (Scheme 15).114,123,150,151 In other examples of electrocatalytic ORR by tethered and supramolecular heme/Cu assemblies, Cu has been shown to slightly increase the average electrons transferred under electrocatalytic conditions, suggesting the Cu is playing a role in storing electrons (Scheme 15; (L11)FeIII/CuII(TerpyCD2) and [(L12)FeII/CuI]+);116,152,153 however, no dramatic rate enhancement comes from the presence of Cu.
Scheme 14.
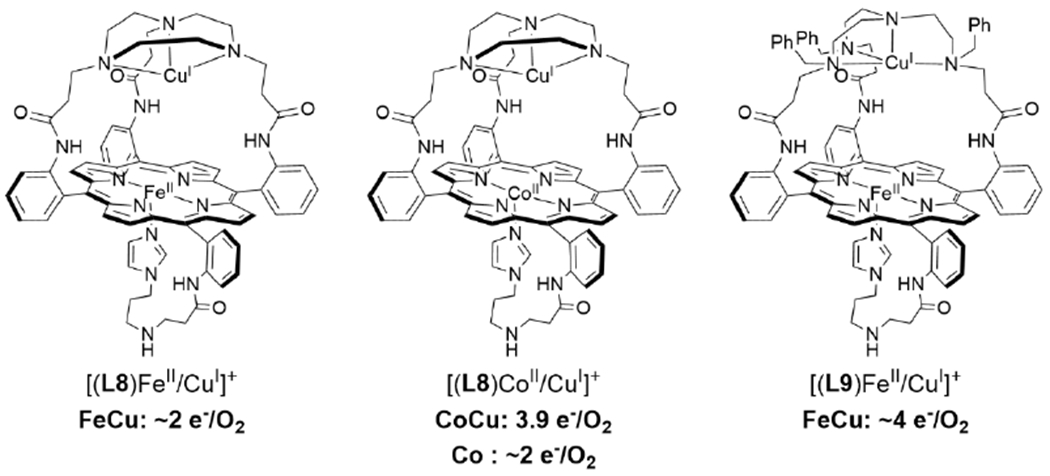
Selectivity of Electrocatalytic ORR By Heme/Cu Complexes.148,149
Scheme 15.
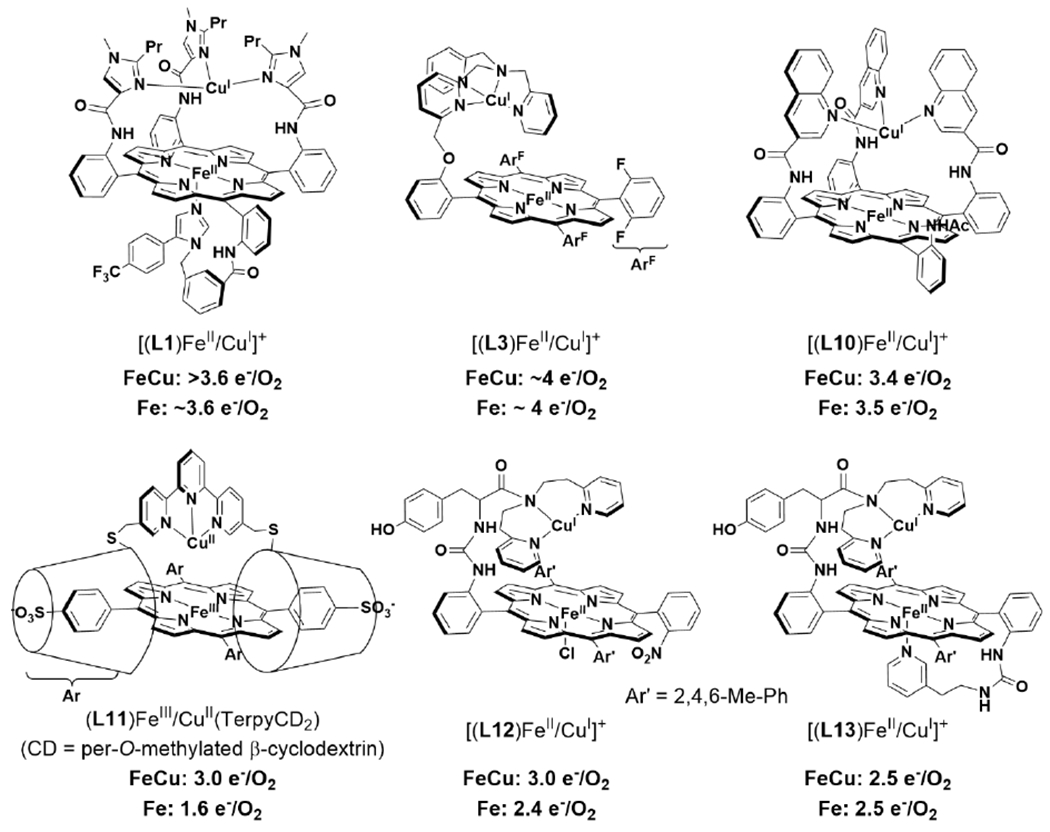
Effect of Cu on Catalytic ORR Selectivity for Various HCO Model Complexes. 114,116,123,150–153
Another structural feature of HCO that has been mimicked in a number of ORR catalysts is the active site Tyr residue. Electrocatalytic ORR studies of synthetic tethered heme/Cu complexes containing a pendant phenol demonstrate that, in some cases, the phenol moiety has no effect on ORR,154 while, in slightly different metal coordination environments under different catalytic conditions, the phenol can help reduce PROS formation.115 In the latter case, the phenol is postulated to reduce PROS through mimicking the role of the His-Tyr in HCO, which is supported through single-turnover studies (Scheme 11).142
The effect of the active site Tyr, and the His-Tyr crosslink, on catalytic ORR has been thoroughly investigated through the biosynthetic CuBMb system. Miner et al. demonstrated that introducing a Tyr residue near the active site (Phe33Tyr or Gly65Tyr) dramatically increases the rate and turnover number (Figure 11).150 The Gly65Tyr CuBMb mutant displays more than double the activity relative to Phe33Tyr CuBMb, demonstrating the importance of this residue’s positioning relative to the heme active site. Furthermore, these Tyr-containing mutants display greater selectivity for complete O2 reduction, which was directly confirmed by measuring H217O formation from 17O2. Remarkably, further engineering of Gly65Tyr CuBMb through introducing positively charged residues on the protein surface, in order to facilitate faster ET, produces an ORR enzyme with activity comparable to that of a native HCO.155 An XRD structure of reduced Phe33Tyr CuBMb with O2 shows that the introduced Tyr residue participates in hydrogen-bonding interactions with a water network leading to the bound O2 (Figure 12).156 This proton-delivery pathway was proposed to be a major reason for the higher activity of Phe33Tyr CuBMb, relative to WT swMb. Further investigation of this structural feature in Phe33Tyr was done through replacing the Tyr with unnatural amino acid analogues of Tyr bearing different electron-withdrawing groups.157 Decrease of the pKa of the phenol sidechain leads to an increase in ORR activity, consistent with this residue being involved in PT during turnover. Evidence for Tyr residue also serving as an electron donor, and forming a Tyr-O•, similar to the mechanistic proposals of HCO, were observed by treating oxidized Phe33Tyr CuBMb (or the unnatural Tyr analogues) with H2O2.157,158 An EPR signal consistent with a phenoxyl radical is observed, which is not present in CuBMb without this Phe33Tyr mutation. Lower amounts of this radical are also observed when the reduced biosynthetic protein is incubated with O2, supporting its relevance to the catalytic mechanism.158 Finally, to understand the potential importance of the covalent link between His and Tyr in native HCO, an unnatural amino acid mimicking this cross-link (imidazolyl-tyrosine; imiTyr) was incorporated into CuBMb (imiTyr CuBMb).159 This biosynthetic protein was unique for displaying higher CuII affinity, and higher ORR activity than Phe33Tyr CuBMb (12 μM O2 min−1 for imiTyr CuBMb+CuII versus ~5 μM O2 min−1 for Phe33Tyr CuBMb, under identical conditions). Furthermore, addition of CuII was shown to significantly decrease PROS formation from 30% to 6%, suggesting this crosslink may have a role in retaining and tuning the Cu center for more selective O2 reduction.
Figure 11.
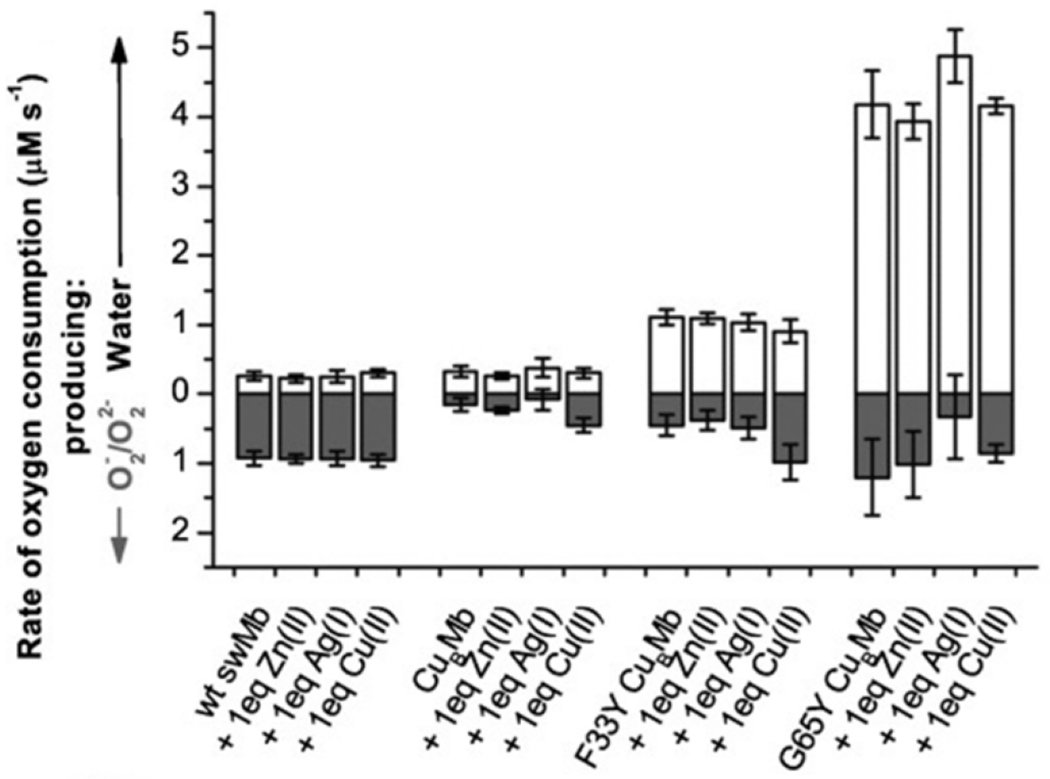
ORR activity of swMb and biosynthetic HCO models. Figure adapted from ref. 150 Copyright Wiley-VHC 2012
Figure 12.

H-bonding network in O2-bound Phe33Tyr CuBMb crystal structure. Figure adapted from ref. 156 Copyright American Chemical Society 2016.
As mentioned previously, the majority of direct electrocatalytic or homogenous ORR studies of heme/Cu systems show little benefit from Cu or, in some cases, display greater propensity for PROS production when Cu is present. Collman et al. hypothesized that to observe HCO-like ORR catalysis, the nature of ET rate and active site isolation should be closer to the conditions of the native enzyme.160 Indeed, depositing the [(L1)FeII/CuI]+ complex into a phosphtidylcholine film on the electrode surface ensures site-isolation of the catalyst and reduces the rate of ET to being diffusion limited.160 Under these conditions, [(L1)FeII/CuI]+ displays selective 4 e-reduction of O2. Further control of the ET rate from electrode to catalyst could be accomplished by covalent attachment of heme/Cu complexes onto self-assembled monolayer (SAM) films on Au electrodes.114,115 These studies use functionalized SAM components that facilitate fast (~103 - 104 e− s−1) or slow (4 - 6 e− s−1) ET to various HCO mimics. Comparison studies of related picket-fence porphyin complexes, (L1)FeII,[(L1)FeII/CuI]+, and [(L2)FeII/CuI]+, demonstrate that the catalyst with all 3 redox active centers, [(L2)FeII/CuI]+ (FeCuArOH), produces the least amount of PROS under fast and slow ET regimes, however the difference between the three catalysts is much more pronounced when ET is slow (Figure 13).115 Analogous studies with (L3)FeII, [(L3)FeII/CuI]+, and [(Im)(L3)FeII/CuI]+ show similar effects: Cu decreases PROS formation, which is further reduced when an addition axial heme ligand is present, but only under low ET rates with C16SH (Figure 14).114 Together, these results demonstrate that, under conditions where reducing equivalents are transferred slowly, structural features that more faithfully model the features of HCO (nonheme CuI, nearby phenol, and low-spin ferric heme) produce a more selective ORR catalyst.
Figure 13.

Effect of Cu and phenol on electrocatalytic ORR selectivity under ‘fast’ and ‘slow’ ET regimes, through covalent tethering of catalyst to different Au SAM electrodes. Figure adapted from ref. 115 Copyright the American Association for the Advancement of Science 2007.
Figure 14.

Effect of Cu and axial ligand on PROS formation on electrocatalytic ORR with (L3)FeIII (6L-Fe) and related complexes. Electron flux is controlled by measuring catalysis with different electrodes: fast ET with edge-plane graphite (EPG), slow ET with 1-octanethiol SAM on Au (C8SH), and very slow ET with 1-1-hexadecanethiol SAM on Au (C16SH). Figure adapted from ref. 114 Copyright American Chemical Society 2015.
An advanced spectroscopic technique, known as surface-enhanced resonance Raman spectroscopy (SERRS) has been used by the Dey group for the study of catalytic intermediates of some of these biomimetic ORR catalysts.147 Using SERRS to examine the mechanism of ORR by (L3)FeII, [(L3)FeII/CuI]+, and [(Im)(L3)FeII/CuI]+ the authors observed [(Im)(L3)FeII/CuI]+ produces a six-coordinate low-spin (l.s.) FeII species upon reduction, with or without O2 present (Scheme 16).114 The sluggish substitution of the sixth ligand (presumably H2O) for O2 was attributed to this complex’s higher PROS at high electron flux, due to side reactions of CuII-O2− species. Furthermore, spectroscopic evidence for significant formation of the (L3)FeIII–O22−–CuII intermediate is observed, with and without an imidazole axial ligand. Build-up of FeIV=O is observed with (L3)FeII, but not the other two complexes, which the authors attributed to a high-potential compound I analogue forming upon heterolytic cleavage of the peroxo intermediates.114,147 A similar mechanistic study of ORR by Gly65TyrCuBMb yielded SERRS spectra of numerous oxidized Fe signals under catalytic turnover, including l.s. FeIII—possibly superoxo or peroxo intermediates—and FeIV=O.161 By attaching Gly65Tyr CuBMb to this electrode, this biosynthetic protein is able to display ORR activity over 100 times faster than other synthetic models, and 10 times faster than electrode-immobilized native HCO.
Scheme 16.

Proposed Mechanism of ORR by [(Im)(L3)FeII/CuI]+ Based on intermediates Observed Via SERRS. Figure Adapted from Ref. 147. Copyright American Chemical Society 2016.
2.5. Summary and Outlook of HCO and Biomimetic Models.
Through decades of studying native HCO and biomimetic models, we have improved our understanding of key structural features responsible for efficient and selective reduction of O2 to H2O through the proposed mechanism that has been illustrated in Scheme 2. Probing the O2 reactivity of heme/Cu assemblies has demonstrated that both the primary coordination sphere (i.e., the heme with axial ligand and tridentate coordination of CuB) and the surrounding secondary coordination sphere (including hydrogen-bonding interactions with water molecules anchored by the phenol of the highly conserved His-Tyr moiety) is crucial for various aspects of catalytic O2 reduction, including O2 binding and O–O cleavage. Specifically, while the precise role of Cu is highly dependent on its coordination geometry, distance from the heme, and protonation state of the reduced O2 species, it has a functional role in promoting O2 binding through its Lewis acidity, along with increasing the binding affinity of the heme to various other ligands. In addition, coordination of an axial ligand to heme can lead to a change in the Fe spin state, affecting the binding properties towards O2; this axial ligand also has an effect on the binding interactions between Cu and O2. Synthetic heme/Cu systems have been observed with a range of O2 binding modes, from close side-on/side-on binding (μ-η2,η2) to negligible Cu-O2 interactions, favoring a FeIII–O2− complex. Furthermore, the active site Tyr residue in HCO is an essential proton and electron donor for O–O cleavage and its covalent crosslink to the Cu-coordinating His residue tunes the reactivity of the phenol and CuB that further promote O2 activate. Interestingly, while many catalytic ORR studies of heme/Cu systems show little benefit from Cu, conditions where reducing equivalents are transferred slowly reveal that models with structural features that more faithfully model the core components of HCO (nonheme Cu, nearby phenol, and low-spin ferric heme) produce a more selective ORR catalyst. The application of biomimetic HCO models towards catalytic ORR has not only improved our understanding of these enzymes, but has shown that, by incorporating these crucial structural features from the native active site, catalytic activity that matches, or even surpasses, native enzymes can be accomplished. One crucial characteristic of the native HCO mechanism that is relatively underdeveloped in biomimetic complexes, however, is that it is capable of ORR with considerable energy conservation, to generate potential energy though proton translocation. This functional feature of HCO is derived from precise control of the reduction potentials and pKa values of the BNC during turnover. Learning how to mimic these aspects in simpler models would further improve our molecular understanding of HCOs.
While significant progress has been made concerning our mechanistic understanding of HCO, there remain a number of key questions:
What is the nature of the (transient) peroxo intermediate, IP? Is this intermediate protonated prior to O–O cleavage? Does Cu bind to this intermediate prior to transition to P state?
What structural features in the OH state lead to its high reduction potential?
How is the His-Tyr cross-link formed?
While it is challenging to trap these intermediates in the native system, synthetic models have been an invaluable means to interrogate these proposed intermediates. Further study of these model systems may inform how the structural aspects of these proposed intermediates impact their reactivity and potential catalytic relevance.
3. Bacterial Nitric Oxide Reductase (NOR) and Related Biomimetic Models
3.1. The Heme/FeB Active Site of Bacterial NORs.
Certain bacteria are able to reduce NO to nitrous oxide (N2O) during a metabolic process known as denitrification, which converts nitrate (NO3−) and nitrite (NO2−) to N2.162,163 Denitrification can be considered an anaerobic version of respiration—the corresponding aerobic respiration process performs 4 e− reduction of O2 to H2O by heme-Cu oxidase enzymes (HCO), which generates chemical potential energy through proton translocation (see section 2). HCO and bacterial NORs belong to the same enzyme superfamily known as the heme-Cu oxidase superfamily, which are a diverse group of integral transmembrane proteins that share a relatively similar catalytic subunit, with a diverse array of secondary subunits involved in proton and electron transfer (Figure 2).164,165 The NOR enzyme class has been sub-divided principally on the initial electron donors (Scheme 17): cytochrome c for cNOR and quinol for qNOR. A third sub-class of NOR has been discovered which contains a CuA cofactor and obtains its electrons from either cytochrome c or quinol, denoted CuANOR. The CuANOR subclass is further subdivided into bNOR, eNOR, and sNOR, based on homology models of predicted genes.30 XRD and cryogenic electron microscopy (cryo-EM) structures of cNORs and qNORs have been obtained from various bacteria.166–172 To date, no structures of CuANORs have been reported, but biochemical studies from a CuANOR from Bacillus azotoformans have been described.173,174
Scheme 17.

Various Cofactors Observed in NORs and ET Pathway to the BNC Active Site.
The study of bacterial NORs have been challenging due to being integral membrane proteins that contain multiple redox active transition metal cofactors. The first in vitro study of NO-reducing activity of membrane fractions of extracts from Alcaligenes faecalis IAM 1015 was reported in 1971.164,175 However, it wasn’t until 1989 that a preparation of purified, active NOR, cNOR from Pseudomonas stutzeri (PsNOR) was established by Heiss et aI., using Triton X-100 detergent as a stabilizing agent.176 Biochemical studies of PsNOR, and later purified NORs from other bacteria, confirmed that these enzymes selectively catalyze the 2 e− reductive coupling of two NO molecules to form N2O (eq 2),176–178 avoiding alternative reactions, including reductive NO disproportionation (eq 3) which is considered a more common reaction that occurs between NO and transition metal complexes.179
| (2) |
| (3) |
Later efforts established that cNOR contains three heme cofactors (heme c, b, and b3, where b3 denotes a high spin heme b cofactor), along with a nonheme Fe (FeB).180,181 In 1994, analysis of the available sequences from bacterial NORs and multiple HCOs established for the first time an evolutionary link between these two enzyme classes.182 A suitably high resolution XRD structure of a bacterial NOR enzyme was not obtained until 2010,166 but by 1998 the overall protein structure of NOR—and the positioning of its metallocofactors—had been inferred via homology modelling to a published HCO crystal structure.183 By this time, it was widely accepted that bacterial NORs had an architecture similar to the catalytic subunit of HCO, including an analogous bimetallic active site, denoted the binuclear center (BNC), where a histidine-coordinated high-spin heme cofactor is positioned next to a nonheme metal that is bound to three conserved His residues.163 A distinguishing feature of NOR sequences, compared to HCO, is the presence of multiple conserved glutamate residues near the BNC, including Glu211 (the numbering from Pseudomonas aeruginosa cNOR; PaNOR) which was proposed to be an additional ligand to FeB, to satisfy a preferred octahedral geometry.184 Biochemical studies by Butland et al. in the early 2000’s demonstrated that some of the conserved Glu residues in P. denitirificans cNOR (PdNOR) are essential for enzymatic activity (Glu198 and Glu125, PdNOR numbering), while not significantly disrupting the assembly and coordination of the metal cofactors (including FeB).185 Glu198 of PdNOR corresponds to Glu211 in PaNOR, the potential FeB binding residue.
When the first XRD structure of a bacterial NOR, PaNOR, was disclosed by Hino et al. in 2010, it confirmed much of the prevailing ideas concerning the general organization of the polypeptide backbone, and the molecular arrangement of the BNC active site (PDB ID: 3O0R).166 PaNOR is composed of two subunits, NorB (56 kDa), which displays 12 transmembrane α-helices that contain the BNC and the heme b cofactor, coordinated by two His residues, and NorC (17 kDa), a single transmembrane α-helix attached to a periplasmic-facing domain that contains the heme c center coordinated by His and Met residues (Figure 15). An ET pathway from the periplasmic surface of PaNOR can be traced from the heme c, b, and b3 centers, with a CaII ion bridging the propionates of hemes b and b3, which is thought to aid in arranging these cofactors for efficient ET. The enzyme was crystallized in its oxidized state, leading the BNC to contain a μ2-O ligand between heme b3 and FeB (Figure 16A). The Fe–Fe distance of the BNC is relatively short, 3.9 Å, compared to the Fe–Cu distance of 4.4 Å in Tt ba3 oxidase (an HCO).166 FeB is 5- or 6-coordinate, with either k1-O or k2-O,O’ binding to the carboxylate side chain of Glu211 (Fe–O distances of 2.04 and 2.47 Å), along with bonds to the μ2-O and the three conserved His residues (His207, 258, and 259). Multiple Glu residues near FeB (Glu 211, 215, and 280) lead to a relatively electronegative distal pocket for heme b3, significantly lowering its reduction potential in PdNOR to ~ 60 mV (vs NHE), compared to the potentials of heme c and heme b, (310 mV and 345 mV, respectively).186 These Glu residues are also thought to be part of a putative PT pathway from the periplasm to the buried active site (Figure 17). The distance between one of the heme b3 propionates and the next hydrogen-bonding residue (Thr330) is 8.0 Å in the crystal structure, suggesting conformational changes in PaNOR are necessary to form a complete PT pathway.166 Molecular dynamics simulations of PaNOFt suggest two possible PT pathways, based on protein structural rearrangements over time.187
Figure 15.
XRD structure of PaNOR (PDB ID: 3O0R), comprised of NorB (green) and NorC (magenta) subunits.
Figure 16.

Different XRD structures of BNC of NORs. (A) Resting state PaNOR (PDB ID 300R) with μ2-O, (B) reduced PaNOR (PDB ID 3WFB) with μ2-Cl−, (C) reduced PaNOR (PDB ID 3WFC) with CO bound, (D) reduced PaNOR (PDB ID 3WFD) with acetaldoxime, (E) GsNOR (PDB ID 3AYF) with ZnII at the FeB site, (F) NmNOR (PDB ID 6LIX) with ZnII inhibitor coordinating 3 Glu residues, (G) cryo-EM structure of NmNOR (PDB ID 6L3H), and (H) cryo-EM structure of AxNOR (PBD ID 6QQ5).
Figure 17.

Structure of PaNOR showing the putative PT channels (1 and 2), along with the hydrophobic channel for NO transfer (yellow). Figure adapted from ref. 166 Copyright the American Association for the Advancement of Science 2010.
PaNOR has also been crystallized in reduced and ligand-bound forms, to gain further insight into the possible BNC structures during turnover.169 The reduced structure (PDB ID: 3WFB) contains a bridging Cl− between heme b3 and FeB and displays a slightly increased distance between the Fe centers (Fe–Fe distance of 4.2 Å, compared to 3.9 Å of oxidized form) (Figure 16B). This greater distance is still significantly shorter than the Fe–Cu distances in structures of most HCO enzymes (see Table 3 in section 5),188 leading to speculation that two diatomic molecules may not be able to bind the metals simultaneously within the BNC. On the other hand, the CO-bound structure of reduced PaNOR contains an ovoid electron density between heme b3 and FeB, which is difficult to model (PDB ID: 3WFC), but detailed analysis by Sato et al. concluded that, while ambiguous, the electron density is only suitably accounted for with four non-hydrogen atoms in the active site (a CO and two H2O, or two CO ligands) (Figure 16C).169 Similarly, acetaldoxime (CH3CHNOH) binds to the BNC in PaNOR, and it can be considered an analogue to hyponitrite (ONNO2−), a key intermediate in all major NOR mechanistic proposals (see section 3.3). The acetaldoxime-bound structure (PDB ID: 3WFD) more clearly demonstrates that four non-hydrogen atoms can fit within the BNC of NOR, although the binding mode displays a relatively short Feb3–N(R)–O(H)–FeB bridging motif, which doesn’t necessarily support an interpretation where each metal can bind NO simultaneously (Figure 16D). It has been proposed that Glu211 may dissociate from FeB to increase the Fe–Fe distance, and facilitate binding of a second NO molecule during catalysis;185,189 despite this proposal, all three of these reduced PaNOR XRD structures show Glu211 bound to FeB, with relatively short Fe–Fe distances of 4.2 – 4.4 Å.169 Interestingly, flash photolysis vibrational spectroscopy of CO-bound CuANOR from B. azotoformans clearly demonstrates that two CO molecules bind to heme b3 and FeB simultaneously (see section 3.3.3);190 although it should be noted that, based on the sequence and homology structure of this CuANOR, there may be no fifth residue to coordinate FeB.174
Table 3.
Representative NOR and HCO Heme-Nonheme Distances.
| Class | PDB Code | Form | Nonheme metal | Distance (Å) |
|---|---|---|---|---|
| cNOR (Pa) | 3O0R | oxidized | Fe | 3.8 |
| cNOR (Pa) | 3WFB | reduced | Fe | 4.2 |
| cNOR (Pa) | 3WFC | reduced, CO-bound | Fe | 4.4 |
| qNOR (Ax) | 6QQE | oxidized | Fe | 4.1 |
| qNOR (Nm) | 6L1X | oxidized, Zn(II)-inhibited | Fe | 3.8 |
| qNOR (Gs) | 3AYF | oxidized, inactive | Zn | 4.6 |
| A-type CcO (Rs) | 1M56 | oxidized | Cu | 4.8 |
| A-type CcO (Rs) | 3FYE | reduced | Cu | 5.3 |
| B-type CcO (Tt) | 1EHK | oxidized | Cu | 4.4 |
| B-type CcO (Tt) | 3EH3 | reduced | Cu | 5.1 |
| C-type CcO (Ps) | 3MK7 | oxidized | Cu | 4.6 |
Variations in the FeB coordination geometry have been observed in crystal structures of qNORs from Geobacillus stearothermophilus (GsNOR), Neisseria meningitidis (NmNOR), and Acaligenes xylosoxidans (AxNOR), that may be relevant to our mechanistic understanding of NOR.167,170–172 qNOR proteins are single subunit (~85 kDa), with 14 transmembrane α-helices arranged in a pattern similar to the NorB/NorC architecture of cNORs. The ZnII-inhibited crystal structure of NmNOR (PDB ID: 6L1X) displays FeB coordinated by only three His residues and a μ2-O to heme b3, where conserved Glu residues near the BNC bind to a ZnII ion instead (Glu494, 498, and 563 with NmNOR numbering) (Figure 16F).172 While this structure is not catalytically relevant due to the presence of ZnII interacting with residues in the PT pathway to the BNC, it demonstrates that FeB remains present in NOR without Glu coordination. A recent cryo-EM structure of active NmNOR without ZnII, although at a very low resolution of 9 Å, appears to display a relatively weak monodentate Glu494–FeB interaction (Fe–O ~ 2.4 Å), while also having a relatively large Fe–Fe distance (~4.5 Å), which supports the hypothesis that ZnII interferes with Glu494 binding to FeB in the previous NmNOR structure (Figure 16G).172 A separate cryo-EM structure of AxNOR (3.2 Å resolution) further demonstrates that Glu coordination is not necessary for FeB binding (Figure 16H).171 The structure of the oxidized enzyme also displays a larger Fe–Fe distance of 4.1 Å, compared to 3.9 Å in PaNOR, supporting the notion that Glu dissociation could play a role in increasing the distance between the Fe centers in the BNC. Importantly, this structure is a catalytically active form of qNOR. It is known that mutation of the nearby Glu490 residue in AxNOR (analogous to Glu211 in PaNOR) greatly diminishes catalytic activity, consistent with a crucial role of this residue during catalysis, either in stabilizing FeB and/or facilitating PT.185,189 while the structural studies of NOR have revealed a range of coordination geometries that FeB can adopt, it is currently unclear whether this possible lability in Glu coordination has any functional significance.
3.2. Other Enzymes that Catalyze NO Reduction.
Bacterial NORs are not the only metalloenzymes that have evolved to achieve the selective reductive coupling of NO to N2O. The active sites of the other NO-reducing enzymes display a diversity of structures, which highlights the versatility within living organisms towards developing transition metal catalysts (Scheme 18).191 The scope of this review, concerning NO and O2 reduction reactions, is focused on the heme/nonheme active sites of the HCO superfamily and the following section will provide only a brief overview of the other NO reducing enzymes that have been described. More extensive reviews detailing their structure and mechanisms have been published.82,165,192–194
Scheme 18.
Metalloenzyme Active Sites of Native and Promiscuous NO Reductases.
Two well-studied examples of NO reducing enzymes are flavodiiron NO reductases (FNORs), which contain a bimetallic nonheme Fe active site,82,83,193 and fungal NOR enzymes called P450nor, which contain a single cysteine-coordinated heme b.165,195 A common feature of these enzymes is that they belong to protein superfamilies that catalyze either NO or O2 reduction (NOR and HCO, FNOR and FDP, P450nor and P450s).82,165,196 This is particularly intriguing since there is no a priori reason to expect the 2 e− bimolecular coupling of NO and 4 e− reduction of O2 would share sets of similar active sites in nature. More recently, NO reductase activity has also been described in a protein bearing a unique tetranuclear FeS cluster, denoted the hybrid cluster protein (Hcp);197 however, biologically-relevant in vivo NOR activity by Hcp is currently inconclusive.198 There are other examples of metalloproteins that appear to have promiscuous NO reductase-like activity in vitro, although the biological relevance of these reactions is unclear. Nevertheless, these metalloenzymes also add to the diversity of biological active site structures that can achieve NO coupling. These enzymes include close relatives to native NOR enzymes (i.e. HCO, or O2-reducing flavodiiron proteins),82,199,200 and certain nitrite reductase hemoproteins, like cytochrome c nitrite reductase.201
3.2.1. Flavodiiron Nitric Oxide Reductase (FNOR).
FNOR enzymes are expressed by certain bacteria under conditions of high concentrations of NO, as a means of detoxification. NO production is part of mammalian immune response to pathogenic organisms; therefore, FNOR are part of the defense strategy of many pathogenic bacteria.193 Instead of a heteronuclear heme-nonheme BNC as in bacterial NOR, the FNOR active site is comprised of two nonheme Fe centers, which are each coordinated by one or two His, a Glu/Asp carboxylate, and a bridging Asp residue, similar to many binuclear nonheme Fe proteins.202 NO is proposed to bind to each Fe prior to N–N bond formation, similar to the trans mechanism in bacterial NORs (see section 3.3),203 although alternative mechanisms have been considered.165,204 It is unknown whether electron donation from the FMN cofactor to the putative Fe–NO species is required prior to N2O formation, or if it regenerates the active diferrous state after N–N bond formation occurs.193 Efforts towards synthetic models of FNOR have demonstrated the feasibility of many of these mechanistic proposals, with diiron dinitrosyl and diiron mononitrosyl complexes producing N2O upon 1 or 2 e− reduction.205–207
3.2.2. Cytochrome P450 Nitric Oxide Reductase (P450nor).
P450nor catalyzes NO reduction in denitrifying fungi using a single heme cofactor without the need for another nonheme Fe.195,208 These enzymes belong to the cytochrome P450 family of enzymes, which are widely known for coupling O2 reduction to the oxidation of organic substrates.65,209 Like other P450 enzymes, the active site of P450nor is a cysteine-coordinated heme b cofactor. P450nor is distinguished from other P450s by a more open distal heme pocket, which facilitates binding of NAD(P)H next to the metal center.210,211 Detailed mechanistic studies of P450nor favor a unique direct hydride transfer mechanism, from the NAD(P)H cofactor to the NO ligand that binds to the initial FeIII heme b.212,165,213 The resulting HNO(H) (nitroxide or hydroxylamide) species is proposed to remain bound to Fe, and produce N2O upon reaction with an incoming NO molecule.214,215 Recent efforts to mimic P540nor with synthetic models by the Lehnert group have demonstrated the importance of axial thiolate ligation on Fe–NO binding, and characterization of an analogue to the putative hydroxylamide intermediate.216,217
3.2.3. (Putative) Nitric Oxide Reductase Activity of Hybrid Cluster Protein (Hcp).
Hcp was first isolated in 1992 from a Desulfovibrio vulgaris substrain.218 Since then, decades of study of Hcp has led to extensive spectroscopic and structural characterization of its metal cofactors; however, determination of the biological role of this protein remains elusive.197 It is known that this protein bears a unique FeS cluster that contains bridging-oxido ligands, best formulated as [Fe4S2O3] (with a unique persulfido cysteine) in its oxidized states, and [Fe4S3] composition in its reduced form.219,220 In vitro catalytic activity towards various small molecules, including NO,221 NH2OH,222 and H2O2,223 have been reported, along with auto-S-nitrosothiol formation.224 It is unclear if any of these reactions are relevant to its biological function; however, studies over the past 5 years support Hcp playing a role in protecting the cell against nitrosative stress under anaerobic conditions.198 A recent study of the molecular mechanism of NO reduction by Hcp confirms it is competent to catalyze reductive NO coupling, and also identified a putative dinitrosyl intermediate by EPR spectroscopy.221
3.3. Mechanism of NO Reduction by Bacterial NORs.
The precise catalytic mechanism of the heme/FeB active site in bacterial NOR enzymes remain elusive. One complication to our mechanistic understanding of NORs is the substrate inhibition it displays, which makes it challenging to interpret which reactions with NO are catalytically relevant.225–227 Extensive spectroscopic and computational studies have focused on three major mechanisms of N2O formation (Scheme 19). These are the so-called (i) cis-FeB, (ii) cis-heme b3, and (iii) trans mechanisms, which are named based on the structure of the putative dinitrosyl intermediate that forms prior to N–N bond formation. The section will describe each mechanism, along with supporting data obtained from studies of native NORs.
Scheme 19.

Proposed Mechanisms of Bacterial NOR.
Common to all mechanistic proposals is the initial 2 e− reduction of the NOR resting state (Feb3III–μ2-O–FeBIII) with the transfer of 2 H+, to arrive at the reduced Feb3II/FeBII state. Binding of NO to the BNC forms an Fe–NO adduct at one or both of the Fe centers. Due to the nature of NO acting as a redox ‘non-innocent’ ligand to transition metal complexes, further discussion of Fe–NO species will use Enemark-Feltham notation, which distinguishes M–NO species based on the sum of electrons occupying the metal d and N–O π* orbitals.228 For example, binding of gaseous NO (which contains a single unpaired electron in the N–O π* orbital) to FeII ion (d6) is described as an {FeNO}7 species, which can be subdivided into the formal metal–ligand oxidation state assignments of FeI–NO+, FeII–NO•, or FeIII–NO−—although in many cases, the precise description will be somewhere between these extremes.229
3.3.1. Cis-FeB Mechanism.
In this mechanism, two NO molecules bind to the nonheme FeB center forming a dinitrosyl species, FeB(NO)2. From this intermediate, formation of a cis-hyponitrite (ONNO2−) occurs after electron transfer from heme b3. Proton transfer to this species results in N–O cleavage and N2O formation. This was an early proposed mechanism of NOR, based on the postulate that heme {FeNO}7 species are unreactive catalytic ‘dead-ends’ and was further supported by existing precedent of NO coupling with synthetic nonheme organometallic complexes,12,230,231 even though later studies have demonstrated that heme {FeNO}7 species can be reactive under certain conditions.232–234 Furthermore, detailed characterization of the bonding and reactivity of a synthetic mononuclear dinitrosyl Fe complex suggest there is a high inherent kinetic barrier to N–N coupling from Fe(NO)2 species, due to the parallel alignment of spin on NO from coupling to the unpaired electrons on Fe.235 Later, a modified cis-FeB mechanism was proposed, which suggest free NO attacks a nonheme {FeNO}7.225 Supporting data for this mechanistic proposal include the observation of a significantly lower reduction potential for heme b3 in PdNOR than FeB or the other heme cofactors.186 This has led some to suggest that semi-reduction of the BNC, and binding of NO at FeBII while heme b3 remains FeIII, is catalyticaIly relevant;236 however, recent studies of electrocatalytic NO reduction by PaNOR shows that heme b3 must be reduced for catalysis.237 Furthermore, electrochemical studies with PdNOR and CO suggest substrate binding can raise the potential of heme b3, and may serve as a way to control its activity.238 Much of the uncertainty about the functionally relevant redox states of the NOR BNC stems from the inconsistent reduction potential values observed for FeB. A number of studies have measured a very low potential for FeB (−0.37 V, below the −0.16 V potential of heme b3) by cyclic voltammetry239 and supported by Mössbauer spectroscopy of Pseudomonas nautica NOR (PnNOR).240 Despite these findings of the low FeB reduction potential, relatively high potentials have been observed with spectroelectrochemical experiments of PdNOR and PaNOR186,237 which happen to be more consistent with potentials observed in biosynthetic and computational model studies.117,241 Further studies are necessary to establish any mechanistic relevance of partially reduced states of the BNC, based on the relative order of reduction potentials of FeB and heme b3. Based on the measured potentials of hemes b and c in the ET pathway, structural changes to the BNC are likely necessary for ET to take place for NO reduction, which will have significant implications for the reduction potentials of the BNC metal centers.
There is spectroscopic evidence that heme b3 remains six-coordinate upon reduction, binding water or hydroxide, and may participate principally as an electron donor to FeB.240 This proposal was based on study of PnNOR, which displays differences in the properties of its heme b3 compared to other NORs, including a low spin resting state,240 and a higher reduction potential than FeBthat is also insensitive to NO.239 It is conceivable that sufficient differences in primary sequence among NOR enzymes can lead to a difference in mechanism, which can possibly explain these disparate observations between PnNOR and other NORs. Recent computational modelling of the BNC active site during turnover determined that the cis-FeB mechanism, specifically binding of NO to FeB and the resulting hyponitrite geometry upon N–N coupling, is energetically less favorable than other mechanisms.242 Differences in the coordination number or spin state of FeB and heme b3 would be expected to alter the potential energy landscape of turnover, possibly in ways that could favor the cis-FeB mechanism.
3.3.2. Cis-Heme b3 Mechanism.
Similar to the modified cis-FeB mechanism, the cis-heme b3 mechanism proposes NO binds only to the heme b3 cofactor, with a subsequent electrophilic attack of incoming NO to the resulting heme {FeNO}7. Experimental data distinctly supporting this mechanism over the trans mechanism (vide infra) is sparse.164 NO reduction by other metalloproteins at a single heme site, such as P450nor or nitrite reductases, certainly provide general precedent for this mechanism, although in some cases hydroxylamine (NH2OH) is formed instead of N2O.165,195,243 Recent characterization of PdNOR isolated in the absence of NorD and NorQ—two proteins necessary for insertion of the FeB cofactor—unambiguously demonstrate the essential role of nonheme Fe for NOR activity.244 Possible roles of FeB in the cis-heme b3 mechanism, then, include: promoting N–O activation at heme {FeNO}7, stabilizing the hyponitrite intermediate, and electron donation at some step after N–N bond formation. Time-resolved spectroscopy of NOR under single turnover conditions support that NO binds preferentially to heme b3.245,246 Lack of NO binding at FeB could be a consequence of the relatively small space between Fe in the BNC that has been observed in structures of NORs. Reduced and ligand-bound crystal structures showed evidence that four atoms may fit in the reduced BNC (see section 3.1), but modeled distances between these four atoms are unreasonably short (~1-2 Å) to be consistent with two different diatomic molecules.169 Notably, nearly every computational study of the NOR catalytic mechanism has found that the cis-heme b3 mechanism is the most energetically favorable.241,242,247–249 Furthermore, calculated vibrational frequencies for a cis-hyponitrite originating from this mechanism better match with an assigned N–N stretching vibration from a spectroscopically observed hyponitrite species in NOR (υN–N: 1332 cm−1);250 the calculated frequency is closer to the experimental value by about 400 cm−1, compared to the calculated trans-hyponitrite in the trans mechanism.242 It should be noted, however, that this assigned hyponitrite signal is in very low abundance and difficult to confirm its position or identity with confidence; similarly, vibrational frequency calculations are highly dependent on the model and basis set used and, while they can reflect relative changes in vibrational frequencies, do not typically predict experimental IR stretches with absolute accuracy.
3.3.3. Trans Mechanism.
The trans mechanism hypothesizes that NO binds to both heme b3 and FeB before N–N coupling. Spectroscopic evidence for this trans dinitrosyl intermediate was reported in 2004: rapid freeze quench EPR of reduced PaNOR in the presence of NO displays new signals at g ~ 4 and g ~ 2.01, which are consistent with nonheme and heme {FeNO}7 EPR signals, respectively.251 These signals disappear upon annealing at higher temperatures, suggesting they are intermediates relevant to N2O formation. However, it was noted in the original report that these signals represent only ~30% of the total BNC, and later re-evaluation of these spectra has cast doubt on their relevance to the NOR catalytic cycle.189 This is due, in part, to synthetic and computational modelling of NOR, which predicts an EPR silent trans dinitrosyl intermediate, because of exchange between the two {FeNO}7 centers. The unambiguous identification of a trans-dicarbonyl species in a CuANOR strongly supports the proposal that a trans-dinitrosyl intermediate is achievable in the BNC.189 Confirmation that vibrational signals in the dicarbonyl of CuANOR arise from the heme and non-heme carbonyl species within a single enzyme, as opposed to a mixture or equilibrium of monocarbonyl species, was obtained by signal integration relative to total enzyme concentration, along with low-temperature identification of a semi-bridging carbonyl form originating from the heme carbonyl, which has a measurable effect on the nonheme Fe carbonyl υC–O.190 Notably, the analogous experiments with PdNOR is more consistent with a mixture of a single CO binding to each metal center,189 possibly indicating substrate binding variation among the NOR classes. As mentioned previously in section 3.1, the CuANOR BNC site may be uniquely more open as a consequence of differences in the coordination of FeB.
3.3.4. Steps After N–N Bond Formation.
After formation of the hyponitrite intermediate, through any of the proposed pathways (cis-FeB, cis-heme b3, or trans), N–O bond cleavage is required to complete the catalytic cycle. Three possibilities for this process include: (i) reformation of the oxidized resting state of NOR (Feb3III-μ2-O-FeBIII), followed by subsequent proton/electron transfer to regenerate the reduced state, (ii) proton transfer steps occurring at the hyponitrite intermediate, and concomitant loss of H2O and N2O to obtain a vacant oxidized state (Feb3III and FeBIII), which is later reduced to turn over the enzyme, or (iii) an intermediate mechanism, where one proton is transferred to the hyponitrite, inducing N–O cleavage and forming an FeIII–OH (bridging or terminal) and N2O, following protonation/reduction in later steps (Scheme 20). Recent, stopped-flow time resolved spectroscopy of NOR supports either proton-dependent pathway ii or iii, based on an observed pH effect of N2O formation.246 A separate study concluded, however, that no protons are transferred from bulk solvent during the first turnover, suggesting pathway i may be operative under certain conditions.252 Freeze-quench EPR of NOR under single turnover shows an increase in g ~ 6 and g ~ 4 signals upon warming to room temperature, which are interpreted as formation of the uncoupled oxidized heme b3 and FeB after NO reduction.251 These signals are also seen in EPR spectra of the NOR resting state in small amounts and have also been attributed to inactive states of the enzyme or impurities.164,251,253 It should be noted that formation of oxidized BNC with a μ2-O ligand is unobservable by EPR. Computational models of NOR show it is energetically feasible to cleave the N–O bond of hyponitrite without any proton transfer. In fact, this mechanism is favored, because the vacant oxidized state of the BNC is very high in energy.241,242,249 Multiple PT pathways have been proposed in NORs, with major differences between nonelectrogenic cNOR and proposed electrogenic PT pathways in qNOR from the cytoplasmic and not periplasmic side, which introduce certain thermodynamic constraints involved in energy conservation.166,170,172 Therefore, it is possible that the precise mechanism(s) of N–O cleavage that occur during turnover may vary between NOR subclasses, or reaction conditions.
Scheme 20.
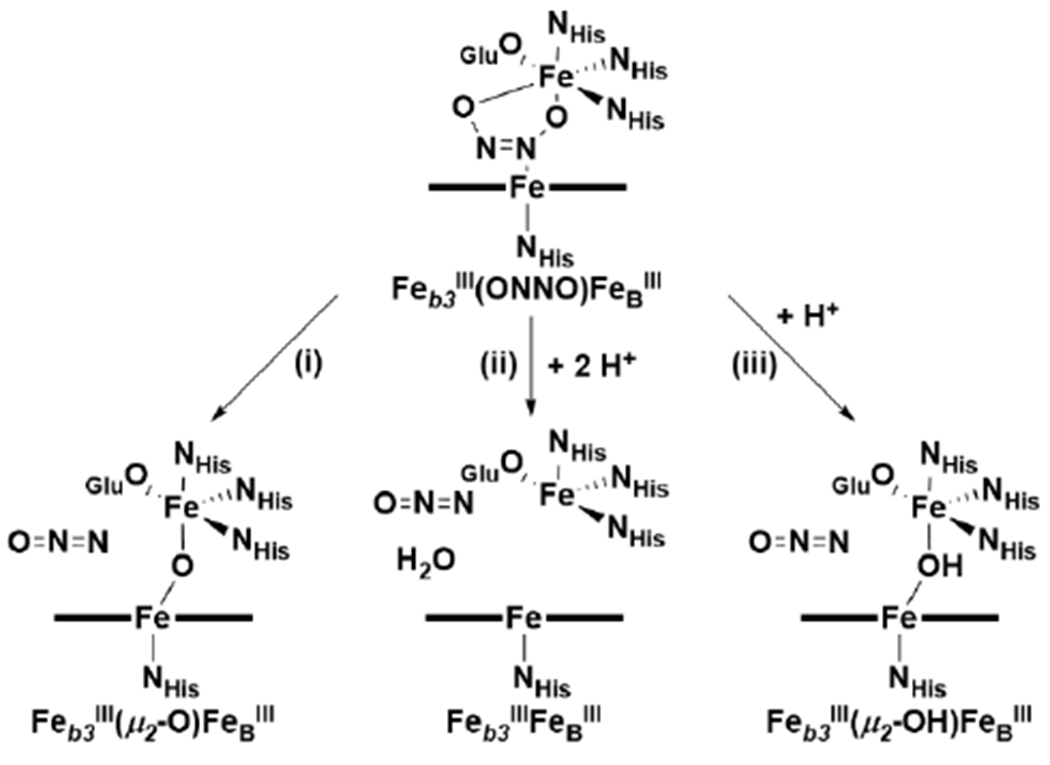
Proposed N–O Bond Cleavage Pathways in Bacterial NORs.
3.4. Insights Gained from Biomimetic Models of Bacterial NORs.
Like HCOs, bacterial NORs are large, membrane bound enzymes containing multiple cofactors, making it difficult to carry out biochemical and biophysical studies due to the challenge in purifying NORs with high homogeneity and yields, along with studying them spectroscopically without interference from other cofactors. Therefore, biomimetic modelling through simpler and smaller compounds or proteins that are free of other cofactors have played key roles in understanding structural features responsible for the activity and mechanism, similarly to the success demonstrated in biometric modelling of HCOs. Since there remain many uncertainties concerning the molecular mechanism of NOR (see section 3.3), it is tempting to look to these simpler models for answers to these mechanistic questions. However, as explained by Kim et al. in their review of synthetic HCO model complexes, models, by their very nature, cannot conclusively prove a mechanism of a native protein, but instead their goal is to “sharpen or focus relevant questions” for further study of the native system.111
In this review, we will provide an overview of models that mimic bacterial NORs both structurally and functionally, because these models provide insight into reactivity and mechanisms concerning NO binding, N–N bond forming, and N–O bond cleaving chemistry relevant to NOR. Literature concerning the properties of heme and nonheme Fe–NO complexes is vast, and many extensive reviews surveying NOR-relevant {FeNO}7 complexes have also been reported.165,179,192,194,229,254–256
3.4.1. Structural and Functional NOR Models.
An early reported synthetic heme/nonheme complex with putative NOR-like activity was disclosed in 2000 by Ju et al (Scheme 21).257 A 5-coordinate Fe-porphyrin complex tethered to a nonheme Fe is reduced in the presence of dithionite to afford a vacant diferrous complex, [(L14)FeII/FeII(Cl)]+. Based on UV-Vis spectroscopy, at low concentrations, this species converts back to the oxidized μ2-O complex upon addition of excess NO. Performing the same experiment at higher concentrations, for identification of gaseous products by GC-MS, led to both N2O and NO2 formation along with a new Fe-containing product with a diferrous oxidation state.257 The change in overall reaction products based on experimental conditions complicates validation that reductive NO coupling occurs with this complex at low concentrations, or whether an alternative mechanism is operative, such as NO disproportionation.
Scheme 21.
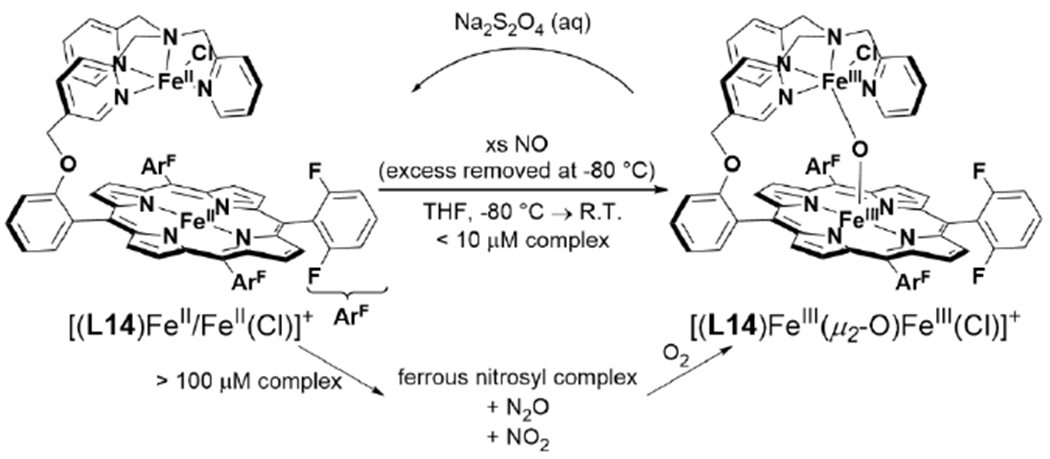
Functional Heme/Fe NOR model reported by Ju et al.257
Mimicking the structure of the BNC was also pursued through ‘biosynthetic’ modelling using small and robust heme proteins, such as myoglobin, as modelling scaffolds based on the hypothesis that introducing a nonheme metal site into such a protein—one that is easier to prepare and study than native NOR—would be able to suitably model the structure and function of the native enzyme. In 2006, Zhao et al. reported that swMb which has been engineered to contain a nonheme binding pocket similar to the CuB binding site of HCO enzymes (CuBMb) could catalytically reduce NO to N2O.258 The production of N2O requires the presence of CuI; the hemoprotein-only or ZnII-bound forms are catalytically inactive. Later in 2009, another engineered mutant of myoglobin (Leu29His, Phe43His, Val68Glu swMb; FeBMb) was reported to be able to bind FeII and subsequently react with NO to form N2O.259 Comparison of the crystal structure of FeBMb (PDB ID: 3K9Z) with the structure of PaNOR reported a year later (PDB ID: 3O0R) showed close agreement between the placement and coordination geometry of the nonheme Fe and the native FeB cofactor (Figure 18).260 It should be recognized that similar to the functional model by Ju et al., it remains unclear whether N2O forms via a 2 e− reductive coupling of NO in FeBMb. One e− oxidation of the heme, based on the shift in its Soret peak, has been confirmed in single-turnover reactions, but no spectroscopic evidence suggesting oxidation of the nonheme Fe has been reported.259,261 Later catalytic studies demonstrate that the absence of nonheme FeII does not halt N2O formation, and FeII can even be substituted for redox-inactive ZnII and still produce similar amounts of N2O.262 These data suggest that FeBMb, is capable of performing 1 e− reductive disproportionation of NO (eq 3), while its potential to follow a 2e− reduction mechanism (eq 2) under other reaction conditions has not been established.
Figure 18.

Overlay of FeBMb structure (cyan; PDB ID: 3K9Z) and PaNOR (yellow; PDB ID: 3O0R). Figure adapted from ref. 260 Copyright American Chemical Society 2015.
A NOR model system which unambiguously performs 2 e− reduction of 2 NO to N2O has been reported by Collman et al.233 It is a synthetic diferrous heme/nonheme complex which forms N2O quantitatively upon addition of excess NO (Scheme 22). The resulting metal-containing species is assigned to a [(L15){FeNO}6/FeIII–OH]2+ complex, based on UV-Vis and EPR spectroscopy, consistent with the oxidation of the initial complex by 2 e−. NO reduction does not occur with the mixed-valent complex ([(L15)FeIII/FeII(Cl)]+), or the complex without nonheme Fe ((L15)FeM), demonstrating the important role of the two redox active metals in this model complex.233
Scheme 22.
Structural and Functional NOR Model Reported by Collman et al. and Mechanism of N2O Formation.233
3.4.2. NO Binding at Biomimetic NOR Models.
Mechanistic studies of NO reduction by structural and functional NOR model systems are aided by the fact that metalation of the porphyrin and/or nonheme binding sites can be controlled in a systematic way. For example, Collman et al. demonstrated that ZnII or FeII can be selectively inserted into the porphyrin of their tethered heme/nonheme ligand L15.233 Further metalation of the nonheme binding site with ZnII or FeII allows for characterization of stable NO adducts of the heme and nonheme Fe, separately. The (L15){FeNO}7 complex displays a υN–O of 1630 cm−1 and a υFe–N at 581 cm−1 consistent with a 6-coordinate heme S = 1/2 {FeNO}7. These IR stretches change little in the presence of ZnII in the nonheme site (1635 cm−1 and 583 cm−1, respectively), suggesting no significant interaction between the {FeNO}7 and the nonheme metal.234 Similarly, it is possible to characterize NO binding in the [(L15)ZnII/{FeNO}7]2+ complex; the υN–O is 1810 cm−1. An EPR signal for the nonheme {FeNO}7 is observed at g ~ 4, consistent with an S = 3/2 complex. Characterization of the separate heme/nonheme {FeNO}7 complexes allowed for a meaningful analysis of the low-temperature intermediates that form when the diferrous complex reacts with NO. At −80 °C in dichloromethane, the first intermediate has EPR and resonance Raman (rR) spectral features nearly identical to the [(L15)ZnII/{FeNO}7]2+ complex. Warming to −40 °C leads to disappearance of the EPR signal, and a rR band at 587 cm−1 which is close to the Fe–N stretch of the heme {FeNO}7. The authors interpret this data as consistent with a trans dinitrosyl intermediate, which is EPR silent due to exchange between the heme and nonheme {FeNO}7 spin systems. Warming this intermediate to room temperature produces the previously mentioned oxidized complex ([(L15){FeNO}6/FeIII–OH]2+) (Scheme 22). These data support a mechanism similar to the proposed trans NOR mechanism (see section 3.3.3) for this functional model.
Similar heme/nonheme substitution studies were conducted on the engineered FeBMb protein by Chakraborty et al.263 Substitution of heme b for Zn(protoporphyrin IX), followed by binding of nonheme FeII and incubation with excess NO, produces an EPR signal at g ~ 4, consistent with a S = 3/2 {FeNO}7. QM/MM studies of the Fe-NO species led the authors to favor a description of the {FeNO}7 as an anti-ferromagnetically coupled high-spin FeII–NO• species; however, it should be noted that the spectral properties (namely, the υN–O, 57Fe Mössbauer, and EPR spectra) of this species conforms to nearly all other characterized nonheme S = 3/2 {FeNO}7 complexes, which have also been described as high-spin FeIII–NO− by DFT.194,255 Careful addition of 1 equivalent NO to FeBMb, or its more reactive mutant FeBMb Ile107Glu,264 leads to NO binding exclusively to the heme cofactor. FeBMb is able to bind a variety of nonheme metals, including FeII, CuI, or ZnII, which have different effects on the degree of N–O bond activation by the heme cofactor.265 With no nonheme metal, the heme {FeNO}7 of FeBMb displays a υN–O of 1601 cm−1, similar to other six-coordinate S = 1/2 {FeNO}7 heme complexes.229,254 The presence of FeII in the nonheme site significantly decreases the υN–O to 1549 cm−1. A similar effect is observed for ZnII in the Ile107Glu mutant (υN–O of 1550/1577 cm−1), but not CuI (υN–O of 1601 cm−1). Recently, Abucayon et al. have demonstrated a similar effect of exogenous Lewis acids on {FeNO}7 heme complexes.232 Addition of BF3•OEt2 to (OEP){FeNO}7 (OEP = octaethylporphyin) leads to weakening of the N–O bond, as evidenced by a dramatic decrease in the υN–O by ~ 200 cm−1 (Scheme 23). Interestingly, this shift in υN–O is roughly equivalent to related anionic {FeNO}8 heme complexes, suggesting a suitable Lewis acid can be as activating as reducing the Fe-NO moiety by one e−.232 The activating effect of this Lewis acid leads to N2O formation via reductive disproportionation, from an initially stable {FeNO}7 complex. A weaker Lewis acid, [K(2.2.2-cryptand)]+, can similarly induce N–N coupling, albeit giving a lower yield of N2O.232,266 These studies suggest that one of the potential roles of FeB in NOR may be to promote the reactivity of the heme nitrosyl by acting as a Lewis acid, which would be relevant to the proposed cis-heme b3 pathway.
Scheme 23.

Lewis acid-assisted NO Reduction by (OEP){FeNO}7.232
Mechanistic study of the NOR-like activity of FeBMb Ile107Glu with excess NO shows that, similar to the [(L15)FeII/FeII]2+, NO binds initially to the nonheme Fe.261 This assignment is based on rapid freeze quench rR spectra of FeBMb Ile107Glu, which show a υN–O band at 1755 cm−1 at the earliest time point (6 ms) upon NO addition. This intermediate leads to the formation of a trans dinitrosyl, with six-coordinate heme {FeNO}7 and nonheme {FeNO}7, identified by growth of the heme υFe–N at 568 cm−1. From the trans dinitrosyl intermediate, FeBMb Ile107Glu either decays to an oxidized heme species with a rate of ~0.7 s−1 at 4 °C, or shifts to a ‘dead-end’ state containing a five-coordinate heme {FeNO}7. The main source of this deactivation appears to be due to attack of NO on the proximal face of the heme.267
Lastly, while reductive NO-coupling could not be unambiguously confirmed from the tethered [(L14)FeII/FeII(Cl)]+ complex, initial attempts to characterize the reaction intermediate at low temperature by UV-Vis were consistent with a mono or dinitrosyl heme species. It is unclear whether the nonheme Fe binds NO (UV-Vis absorbance is not very sensitive to binding of NO by nonheme Fe). Surprisingly, when these experiments were conducted on a compositional isomer of L14 (where the tether attaches to the 6-position of the pendant pyridyl moiety, instead of the 5-position; [(L3)FeII/FeII(Cl)]+), no NOR-like activity was observed.268 Instead, a stable dinitrosyl species forms, based on the presence of two υN–O bands in the FTIR at 1689 and 1798 cm−1 for heme and nonheme {FeNO}7 moieties, respectively (Scheme 24). Characterization of the heme {FeNO}7 in the absence of the nonheme Fe shows almost no difference in the υN–O (1683 cm−1), consistent with little effect of the nonheme Fe on NO activation in this model system. Since the heme {FeNO}7 complexes are 5-coordinate, one possible explanation for its inactivity is that NO binds at the opposite face of the tethered nonheme, preventing N–N bond formation.268 Similarly, Berto et al. have reported that combining a six-coordinate heme {FeNO}7 complex with a nonheme {FeNO}7 model compound of the FeB center displays no intermolecular N2O formation (Scheme 25).269 Overall, these studies suggest that there are certain geometric or electronic requirements that must be met before bimetallic N–N coupling occurs from a putative trans dinitrosyl intermediate in NOR, or its functional models.
Scheme 24.
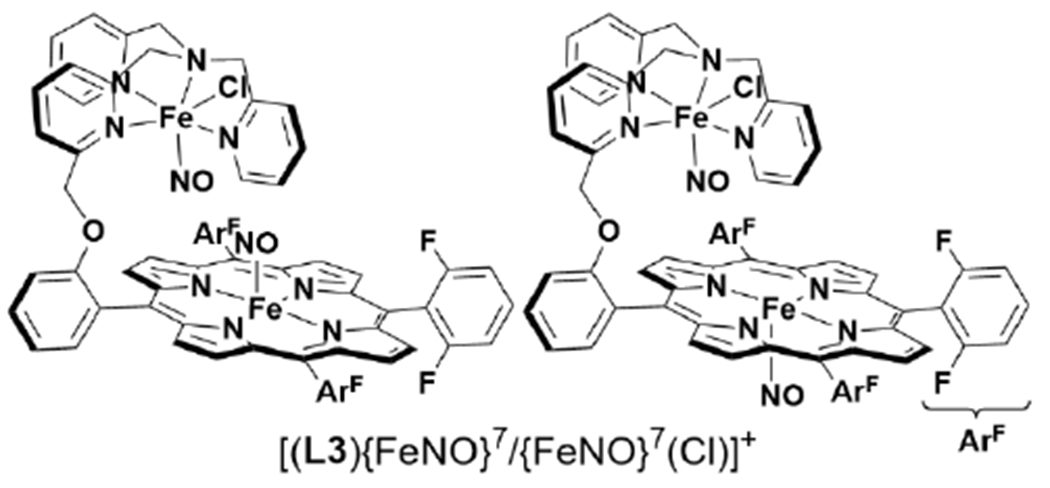
Proposed structures of Stable Dinitrosyl Complex [(L3){FeNO}7/{FeNO}7(CI)]+268
Scheme 25.
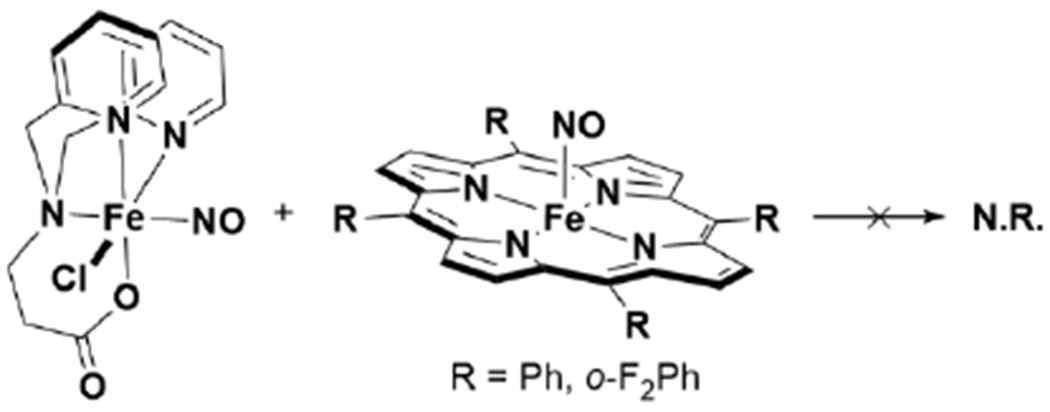
Attempted intermolecular Reactivity of Synthetic FeB Model {FeNO}7 and Heme {FeNO}7.269
3.4.3. N–N Bond Formation.
Direct studies of N–N bond formation in functional NOR models are challenging due to the relatively fast kinetics of the steps after binding of the second NO.234,261 For FeBMb, Bhagi-Damodaran et al. have recently shown that the rate of decay of the heme-nitrosyl intermediate during N2O formation is highly dependent on the electronics of the heme cofactor.270 By substituting different heme cofactors (heme b substituted for mono-formyl, and di-formyl analogues) and introducing a mutation to create a hydrogen bond to the δ1 NH of the proximal His (Leu89Ser), the reduction potential of the heme cofactor could be tuned from −130 mV to +148 mV (vs SHE) (Figure 19A). The rate of heme {FeNO}7 decay varies over 2 orders magnitude based on these differences in heme reduction potential (Figure 19B). The degree of N–O bond activation is also impacted, with the most reducing {FeNO}7 displaying a υN–O of 1527 cm−1, which increases up to 1570 cm−1 in the highest reduction potential cofactor.270 These results demonstrate the crucial role of the heme reduction potential on the nature of the {FeNO}7 and the rate of N–N coupling. For further discussion of the importance of heme reduction potential on NOR activity, see section 5.1.2.
Figure 19.
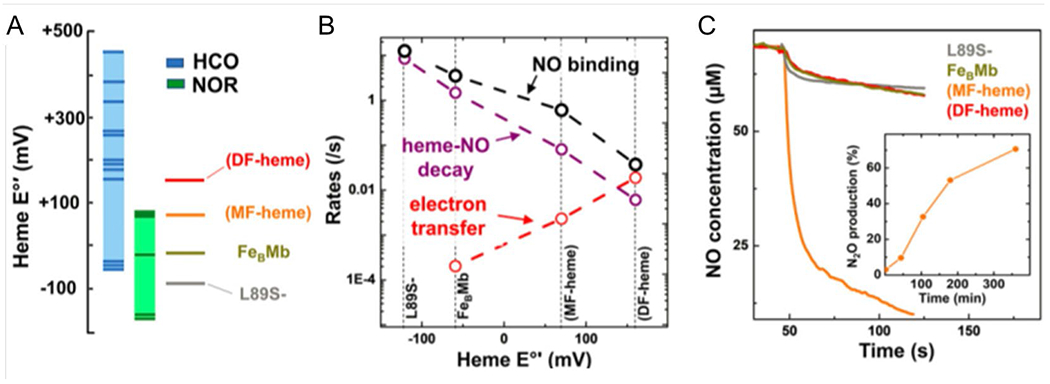
(A) Heme substitution and mutagenesis of FeBMb creates a range of heme reduction potentials that span the range that is observed with the HCO superfamily. (B) Rates of NO binding, heme-NO decay, and electron transfer based on heme reduction potential. (C) NO reduction activity of FeBMb with range of heme reduction potentials. Figure adapted from ref. 270 Copyright the National Academy of Sciences 2018.
More general reviews of N–N bond formation by transition metal-nitrosyl complexes have been published.165,256,271 Among these complexes, a relevant example is the Tp2{Ru2(NO)2}12(μ-Cl)(μ-pz) complex (Tp = trispyrazolylborate; pz = pyrazolate), which can achieve reversible interconversion of dinitrosyl to O=N–N=O complexes upon 2 e− reduction/oxidation (Scheme 26).272 This is a rare example of well-defined radical NO coupling from two {M-NO}7 species, and is related to proposed N–N bond forming steps in the trans mechanism of bacterial NORs, along with some proposed dinitrosyl mechanisms of nonheme diiron FNORs (see section 3.2.2). The bridging O=N–N=O complex, (TpRu)2(μ-Cl)(μ-pz)(μ-N(=O)–N(=O)-k2-N,N′), is capable of N2O formation, and produces a μ2-O dimer upon addition of strong acid.272 it is interesting that protons are required, despite the balanced reaction having no protons being transferred. Theoretical investigations on the role of protons on N2O formation suggest they are important for the second N–N bond forming step, facilitating the conversion of the O=N–N=O species to hyponitrite (−O–N=N–O−), which is a prerequisite to evolve N2O.273
Scheme 26.

Reversible Radical Coupling of {(RuNO)2}12 Upon 2 e− Reduction and Proton-Induced N–O Cleavage.272
3.4.4. N–O Cleavage.
Formation of a hyponitrite intermediate is a ubiquitous step of NO coupling mechanisms. Biomimetic models of NOR display both proton-dependent and -independent mechanisms of cleaving one of the N–O bonds in hyponitrite to release N2O. Both of the tethered heme/nonheme NOR functional models accomplish NO reduction in organic solvent, without the addition of exogenous protons.233,257 By UV-Vis absorbance spectroscopy, Ju et al. observed formation of a [(L14)FeIII–μ2-O–FeIII(Cl)]+ species, a model of the NOR enzyme resting state, after reduction of NO.257 Similarly in the [(L15)FeII/FeII]2+ functional NOR model, Collman et al. observed that one of the oxygen atoms remains bound to the metal complex, forming a nonheme FeIII–OH, which is confirmed by its comparable spectroscopic features to an independently synthesized analogue, (L15){FeNO}6/FeIII–OMe.233 In contrast to these examples, Wang et al. showed that protons are necessary to promote NO reductive coupling, over competing reductive disproportionation, with both synthetic tethered and bimolecular heme/Cu model systems (Scheme 27).274,275 The authors propose that protons are involved in the N–O cleaving step of N2O formation, because a [(L3)FeIII-μ2-O-CuII]+ species is never observed in these reactions, despite multiple examples of such complexes being stable and isolable.
Scheme 27.

Formation of N2O Upon Addition of CuI and H+ to a {Fe(NO}2}8 Complex.274
A number of well-defined binuclear metal-hyponitrite complexes require protons for N–O bond cleavage. Xu et al. have synthesized a stable binuclear heme hyponitrite species by addition of hyponitrous acid (HO–N=N–OH) to ((OEP)FeIII)2(μ2-O) (Scheme 28).276 Addition of strong acid would induce N–O cleavage, with corresponding N2O and H2O formation. Surprisingly, this hyponitrite species has also been observed to perform the microscopic reverse of N–N coupling to form two equivalents of (OEP){FeNO}7 upon slow thermal decomposition.277 Xu et al. more recently reported a modified bridging heme complex (OEP–CH2–CH2–OEP)(FeIII)2(μ2–O) which forms another bridging hyponitrite complex in the presence of hyponitrous acid (as observed by IR spectroscopy).278 This complex, however, is unstable in the absence of excess hyponitrous acid, and releases N2O to re-form the initial μ2-O complex. The authors suggest a consequence of the ethylene bridge is moving the Fe centers closer together to destabilize the hyponitrite intermediate, an observation which may be a relevant to the close Fe–Fe distances observed in NOR structures.
Scheme 28.
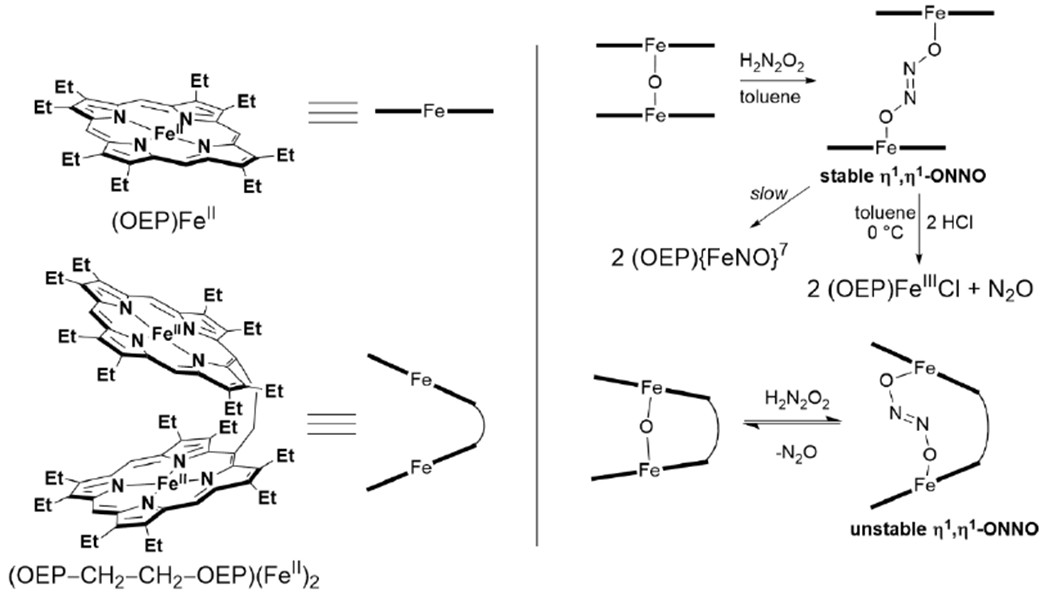
Stable and Reactive Heme Hyponitrite Species Reported by Xu et al.276,278
3.5. Summary and Outlook of NOR and Biomimetic Models.
Native bacterial NOR enzymes have proven to be challenging proteins to study. Like many other integral membrane enzymes, purification of stable, active NOR was an initial difficulty. Despite decades of study, it is only within the last 10 years that a structure of NOR has been reported. Since then, researchers have been able to obtain a handful of other NOR crystal structures, many of which display unique structural variations that may be relevant to catalysis. Another complicating factor includes the presence of multiple auxiliary heme and other redox-active cofactors necessary for electron transfer to the interior BNC, which can convolute spectroscopic study of the heme/nonheme active site. While the detailed molecular mechanism of NOR is not completely understood, much insight has been gained through the combination of spectroscopic, structural, and theoretical studies of the native enzymes. Concurrent study of simpler biomimetic models has allowed for further exploration of the mechanistic possibilities. Specifically, the model studies support the possible role of FeB to be promoting activation of heme-NO intermediate through acting as a Lewis acid, or by binding a second equivalent of NO and forming the N–N bond through a trans dinitrosyl. Further cleavage of the N–O bond can occur through proton dependent or independent pathways.
The following are examples of questions concerning the mechanism of NOR that are crucial to our full understanding of NOR:
What is the role of FeB in the catalytic cycle? Is it necessary for binding of NO and coupling of NO, via the proposed cis-FeB or trans mechanisms, or does it have a role in activating the heme {FeNO}7 for electrophilic attack by free NO, via the cis-heme b3 mechanism? While the heme reduction potential has been shown to play a role in NOR reaction, what about the role of FeB potential?
Is the conserved Glu residue that is observed to coordinate FeB in the BNC always bound during the catalytic cycle? In addition to binding FeB in the resting state, does this Glu and other conserved Glu around the BNC have other roles in the catalytic mechanism, such as gating NO binding, PT, etc.?
Is the resting state of NOR (FeIII–μ2-O–FeIII) relevant to the catalytic cycle? In what ways does the BNC depend on protons to cleave the N–O bond?
Is a single mechanism for N–N formation (cis-FeB, cis-heme b3 or trans) and N–O cleavage operative in the majority of NORs or can the mechanism change between different enzyme sub-classes or catalytic conditions?
Biomimetic models have helped greatly in refining these questions and further studies stand to improve them moving forward.
4. Sulfite Reductases (SiR) and Related Biomimetic Models
4.1. Sulfite Reduction in Cellular Metabolism.
Sulfate-reducing bacteria are widespread among many of Earth’s environments, and more than 250 prokaryotic species with sulfate-reducing genes have been isolated and characterized.10,279 These organisms can couple the reduction of sulfate and sulfite to energy production in the form of a proton motive force, analogous to the respiratory mechanisms of denitrification and aerobic respiration. A notable difference in respiratory sulfate reduction compared to aerobic respiration and denitrification is that the sulfate and sulfite reducing enzymes are globular proteins rather than integral membrane proteins. The mechanism of how these proteins produce a proton gradient for energy production is not completely understood, but the prevalent understanding is that specific respiratory membrane complexes are crucial for generating the proton motive force. These membrane complexes transfer electrons from the periplasm to sulfite reductase, concomitant with the release of protons into the periplasm via oxidation of membrane-associated quinols.280 Multiple respiratory membrane complexes have been observed to be associated with sulfate reducing pathways.279 The cellular pathway for sulfate respiration relies on three enzymes: ATP sulfurylase, 5′-adenylylsulfate reductase (APS reductase), and sulfite reductase (SiR). The initial steps of sulfate uptake require ATP sulfurylase, which generates 5′-adenylylsulfate (adenosine-5′-phosphosulfate, APS) from ATP and sulfate, followed by APS reductase, which transfers 2 e− to APS, generating sulfite and AMP (Figure 20).281 Sulfite reductase then performs the remaining 6 e− and 7 H+ reduction of SO32− to HS− and water in a series of reductive dehydration steps.
Figure 20.
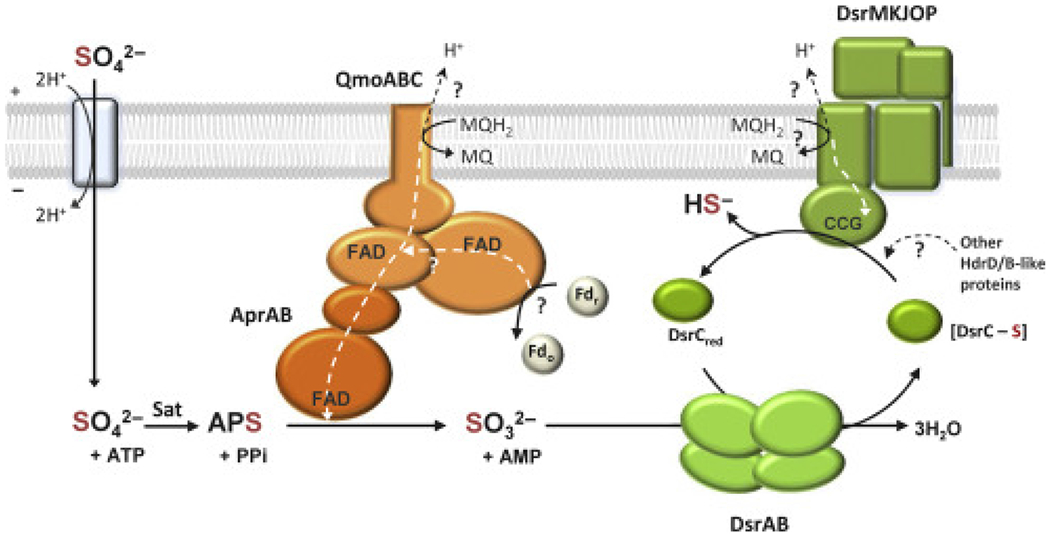
Enzymes involved in the sulfate reduction respiratory pathway. Figure adapted from ref. 279 Copyright Elsevier 2015.
Two different forms of sulfite reductase have been described in sulfate respiratory pathways. The most prevalent is known as dissimilatory sulfite reductase (dSiR), which contains a unique heme-like cofactor (siroheme) and a [4Fe-4S] cluster in its active site (Figure 21).282,283 The proximal ligand to siroheme is also one of four cysteine ligands that coordinate the [4Fe-4S] cluster, creating a covalent bridge between the two iron cofactors. Another dSiR that has been identified is a member of the octaheme nitrite reductase family (ONR) known as multicytochrome c sulfite reductase A (MccA), or simply SiRA, which contains a heme c/Cu active site. The arrangement of heme and Cu is somewhat similar to HCO; however, the coordination of Cu is drastically different, using two Cys residues in a linear binding mode, instead of the His2His-Tyr of HCO.284,285
Figure 21.

Active site structures of siroheme-[4Fe-4S] SiRs (A) Dvir (PDB ID: 2V4J), (B) SiRHP (PDB ID: 1AOP), and (C) dSiR (3MM5).
Sulfite reducing enzymes with siroheme-[4Fe-4S] cofactors are also involved in the production of bio-available sulfur from sulfate/sulfite and are known as assimilatory sulfite reductases (aSiR). Sulfide generated from aSiR is the major sulfur source for biological sulfur accumulation in these organisms by the conversion of L-serine to L-cysteine via O-acetylserine (thiol)-lyase,286 and disruption of this pathway typically substantially inhibits growth. These enzymes are found across all domains of life but have been principally studied in bacteria and plants. The siroheme-[4Fe-4S] active site of aSiR has been studied extensively and is a major source of our spectroscopic and mechanistic understanding of this unique cofactor. aSiRs may be broadly grouped by whether they are NADPH-dependent (prokaryotic) or ferredoxin-dependent (Fd-SiR, eukaryotic). The NADPH-dependent enzymes are oligomeric and comprise electron acceptor subunits with FAD/FMN cofactors and catalytic hemoprotein (HP) subunits that bind the siroheme-[4Fe-4S] cofactors. The most well-studied NADPH-dependent SiR is the aSiR expressed in E. coli, which has an α8ϐ4 composition (a 780 kDa holoenzyme) with either one FAD or FMN in each NADPH-binding flavoprotein α subunit and one siroheme-[4Fe-4S] site per HP ϐ subunit; both subunits are ~60 kDa each.287,288 NADPH binds and delivers electrons to the complex via an FAD flavoprotein subunit which subsequently reduces FMN. Electrons are transferred from the FMN subunit to the HP subunit where the siroheme-[4Fe-4S] cofactor is reduced by electron uptake first through the [4Fe-4S] cluster.288 Fd-SiR isolated from plants are monomeric (~65 kDa) and bear little sequence similarity to the catalytic subunits of the NADPH-dependent aSiR outside of the siroheme-binding domain.289–291 A type of low molecular weight, monomeric aSiR (alSiR) has also been identified, such as the a ISiR from the Hildenborough strain of D. vulgaris. These much smaller enzymes (~27 kDa) are soluble monomers that exhibit spectroscopic features that are similar yet distinct from the oligomeric aSiRs, but they retain the same catalytic site architecture.292,293
4.2. Structures of Sulfite Reductases.
4.2.1. Siroheme-[4Fe-4S] SiRs.
Dissimilatory enzymes (dSiR) like those identified in the sulfate reducing genera Desulfovibrio (desulfoviridin, Dvir),292,294–297 Desulfotomaculum,298,299 and Archeoglobus300,301 are crucial components of the extensive anaerobic sulfate respiratory apparatus of sulfate reducing organisms. These enzymes were initially called bisulfite reductase due to the prevalence of HSO3− at their slightly acidic pH optima (pH 6.0). They were also variably denoted by their siroheme pigmentation as desulfoviridin,292,295,297,302 desulforubidin,303 P-582,298,299 or desulfofuscidin.304–306 The active form of dSiR was first thought to be a tetramer with α2ϐ2 composition (DsrAB),292 and each subunit was found to contain one HP and one ferredoxin-like domain, the latter of which is not present in aSiRs. Quantitative amino acid analysis later suggested that the enzyme was actually a hexamer of α2ϐ2γ2 composition with the DsrAB subunits being ~50 kDa and ~40 kDa, respectively, with a much smaller (~11 kDa) unidentified γ subunit (DsrC).297 Furthermore, metal analyses of dSiRs often gave conflicting degrees of Fe content and siroheme metalation290,300,303,305,307 as well as distinct EPR and Mössbauer spectra that conflicted with the established cofactor structure of aSiR (discussed further in section 4.3). Sulfite reduction assays of purified dSiR in vitro typically resulted in lower activity than aSiR with incomplete sulfite reduction, generating primarily thiosulfate (S2O32−) and trithionate (S3O62−).281,292,302 A thiosulfate reducing pathway has been partially identified,308,309 and it was thought that a possible function of dSiRs was to reduce sulfite by only 2 or 4 e−, unlike the complete 6 e− reduction achieved by all purified aSiR HP subunits, but the function of this pathway in live cells has been disputed, perhaps most effectively by sulfur radiolabeling experiments that demonstrated thiosulfate forms in a process that is inconsistent with a sulfite reduction reaction intermediate.310 Publication of the high resolution X-ray crystal structure of Dvir resolved many of these long-standing issues: the structure elucidated the DsrAB complex with DsrC to form an active α2ϐ2γ2 complex, and each ferredoxin domain was found to bind one [4Fe-4S] cluster;311 homologous structures were also found for other dSiRs.301,312,313 There are four total siroheme binding sites, but only two of these sites are metalated in Dvir, giving rise to its characteristic 628 nm absorption peak of sirohydrochlorin that is present in desulfoviridin dSiRs but not desulforubidin dSiRs, whose absorption spectra are essentially the same as aSiRs.303 The involvement of DsrC in soluble dSiR leads to mechanistic differences that distinguish dSiR from aSiR that will be discussed further in section 4.4. While most dSiR identified have been cytosolic, a membrane-bound DsrABC dSiR (mSiR) was identified in the D. desuifuricans Essex 6 strain that exhibited hydrogenase-coupled activity and accepted electrons from cytochrome c3.314–316 Regardless of the large differences in holoenzyme structure, the siroheme binding domains of all aSiRs, dSiRs, siroheme-binding nitrite reductases (NiRs), and also the so-called “reverse” dSiRs (rSiR) expressed in Thiobacillis, which catalyze the oxidation of sulfane sulfur to sulfite,317 are highly conserved. This conserved structural motif for the active site is termed the sulfite or nitrite reductase repeat (SNiRR), and siroheme has been identified as a cofactor only in sulfite and nitrite reducing enzymes.290
4.2.2. Comparison of aSiR and dSiR.
The major differences between aSiR and dSiR mechanisms are tied to differences of their structures. While siroheme in the aSiR is saddle-shaped and in edge contact with the [4Fe-4S] (Figure 21B), the dSiR siroheme is essentially planar (Figure 21C).301 The saddling of the siroheme in aSiR is induced by the protein scaffold related to sidechains that are not conserved between the different active sites: Asn121 and Thr439 in the hemoprotein subunit of the E. coli enzyme (SiRHP) are replaced by Arg80α in dSiR; dSiR have additional Thr and Tyr residues that contact the siroheme propionates but are absent in aSiR.301,311 Furthermore, the siroheme in NADPH-dependent aSiR has fewer basic residue contacts than in dSiRs.301 While these differences in siroheme-protein interactions are likely responsible for siroheme saddling, the consequences for catalysis are not as clear. It is possible that the saddling in aSiR siroheme may even further enhance the electron “push” effect from siroheme π orbitals while allowing easier π-cation radical formation in siroheme, but despite the frequent observation that it is possible for the siroheme-[4Fe-4S] cofactor to donate up to 3 e− in one step, this process has never been directly implicated in catalysis. Another possible purpose of siroheme ruffling is to tune the reduction potential of both siroheme and the [4Fe-4S] cluster through their strong coupling. Reported reduction potentials vary broadly between SiRs (SiRHP: EH°′ = −340 mV, Ec°′ = −405 mV; alSiR: EH°′ = −21 mV, Ec°′ = −303 mV;318,319 dSiR: EH+C°′ = −298 mV, EH+C°′ = −620 mV),319 where EH°′ and EC°′ denote the midpoint potentials of the siroheme and FeS cluster, respectively. Cofactor reduction in dSiR—for which FeS cluster contact with the siroheme edge is less than in aSiR—is apparently concerted and has not been accomplished in the clean, stepwise manner as in SiRHP; furthermore, the cofactor redox potentials in SiRHP were determined by redox titration rather than the direct electrochemical measurements used for dSiR.290 While differences in redox potential could certainly be related to siroheme coordination, saddling, and siroheme-edge/[4Fe-4S] cluster distance, the relationship is clearly complex.
4.2.3. Heme c/Cu SiR.
A member of the ONR family from W. succinogenes (multiheme cytochrome c sulfite reductase A, SiRA) has been recently characterized that displays high selectivity for sulfite reduction, with catalytic activity (kcat = 200 electrons s−1 heme−1) that far exceeds reported activity for most aSiRs.284,285 SiRA is a soluble homotrimeric enzyme with 7 electron transfer heme c cofactors and one catalytic heme c per monomer (Figure 23). The catalytic site contains a high number of basic residues reminiscent of the conserved Lys/Arg residues conserved in SiRs, along with a unique, linearly coordinated Cys2-CuI site (Figure 24).285 Two Lys and one Arg residue (Lys208, Arg366, and Lys393) are present in the active site and make direct hydrogen bonding contacts with sulfite oxygens, and three Tyr residues (Tyr123, Tyr285, and Tyr301) coordinate multiple ordered waters that may be involved in proton transfer, removal of water from dehydration, or both. The enzyme isolated in a Cu-depleted form (0.2-0.5 Cu per monomer) displayed approximately half the activity of the fully metalated form (0.7-1.0 Cu per monomer), demonstrating the crucial role of CuI in its catalytic activity. Furthermore, the CuI was not observed to undergo spectroscopic changes indicative of redox cycling at any point in the reaction, and exposure to oxygen led to irreversible copper loss, leading to the hypothesis that Cu remains reduced during turnover. Interestingly, reoxidation titration of the fully reduced monomer indicated that the substrate obtained only 4 e− from the 8 total hemes and that the homotrimer (24 total hemes) could fully reduce only 2 sulfite anions at a time, indicating either incomplete sulfite reduction or that the total charge of the homotrimer plays a role in its activation.285
Figure 23.
Overall structure of SiRA with its heme cofactors (PDB ID: 4RKM).
Figure 24.

Active site structure of SiRA (PDB ID: 4RKM).
4.3. Characteristic Spectroscopic Features of SiRs.
Spectroscopy of the siroheme-[4Fe-4S] active site in aSiR and dSiR enzymes is relatively complex, due to the combination of the unique siroheme cofactor and its interactions with the adjacent [4Fe-4S] cluster. Interpreting UV-Vis, EPR, and Mössbauer spectra of these enzymes has been crucial to our growing understanding of their catalytically-relevant structural features. Before an XRD structure had been obtained, enzymes capable of reducing sulfite, nitrite, and hydroxylamine were noted in bacteria, yeast, and plants in the early 1960s, but it was first concluded from studies of the E. coli enzyme that the nitrite and sulfite reducing enzymes were one and the same and that the primary function of this enzyme was sulfite reduction.320 The various SiRs share a common chromophore with absorption peak maxima in both the 380 nm and 580 nm regions, and they could receive electrons from NADPH, reduced methyl viologen (MV+), orferredoxin (Fd). Studies of yeast aSiRs established that there was likely a catalytic subunit common to all of them that contained this unique chromophore.321,322 The chromophore extracted from SiRHP was called “siroheme” and identified as a tetrapyrrole macrocycle of the isobacteriochlorin type (reductively methylated at two adjacent pyrroline rings) with eight carboxylate moieties.323–326 The additional 628 nm absorption peak in Dvir is due to demetalated siroheme (sirohydrochlorin).324 In addition to serving as the catalytic center in SiRs, siroheme is capable of efficiently reducing nitrite and sulfite even outside of the protein matrix.327,328
The property of SiRs that has been the subject of the most study and of the greatest dispute is the interaction of the siroheme and [4Fe-4S] cofactors. The siroheme and [4Fe-4S] cofactors are magnetically exchange coupled in all physiological oxidation states. The oxidized enzyme (SiR0) has a single S = 5/2 spin arising from the high-spin FeIII siroheme and Mossbauer parameters for a [4Fe-4S]2+ cluster (S = 0) but also exhibits field dependence associated with a paramagnetically active iron-sulfur cluster. 329 The 1 e− reduced state (SiR1−) comprises a high-spin FeII siroheme (S = 1 or 2) and oxidized iron-sulfur cluster, meaning the first electron localizes to siroheme, but the total number of electrons for both cofactors becomes a non-Kramer’s system; thus, the two cofactors behave like a single 5-Fe system with a single shared spin. The observed behavior is possible only when considering exchange interactions between the siroheme Fe and individual Fe sites of the [4Fe-4S] cluster and not a dipolar interaction: the two Fe sites of the cluster are strongly exchange-coupled and result in the classical diamagnetic state of the [4Fe-4S]2+ cluster, but they have weaker exchange interactions with the siroheme Fe so that a paramagnetic contribution is always mixed with the cluster ground state.329,330 Novel EPR features arising from exchange interactions between integer spin siroheme and the [4Fe-4S]+ cluster in the 2 e− reduced enzyme (SiR2−) demonstrates that the cofactors remain coupled in the reduced state.331 Some aSiR have been isolated with low-spin siroheme, but the [4Fe-4S]2+ cluster Mössbauer parameters are identical to the high-spin siroheme enzymes.332 The [4Fe-4S] cluster does not contribute significantly to the electronic absorption spectrum, and the S = 1/2 paramagnetic spectrum of the 1 e− reduced [4Fe-4S]+ cluster does not appear in X-band EPR spectra of SiR2−; rather, the classical g = 1.94 ferredoxin-like features predicted by Mössbauer are significantly perturbed into novel features with g = 2.53, 2.29, and 2.07 (“S = 1/2”-like) and characteristic low-field features between g = 4.7-5.4 (“S = 3/2”-like) (Figure 25).331 Reduction of SiRHP in the presence of an inhibitory strong-field ligand such as CN− or CO results in S = 0 siroheme, revealing the “classical” g = 1.94 spectrum of a cubane [4Fe-4S]+ cluster. The perturbed [4Fe-4S] features resulting from exchange coupling with the paramagnetic siroheme could be further modulated with siroheme weak-field ligands or completely silenced by other strong-field ligands such as AsO2−, S2−, and chaotropes (guanidinium, urea, DMSO).318,333–335 Electron-nuclear double resonance (ENDOR) analysis of the 14N hyperfine coupling constants in SiRHP definitively ruled out a histidine proximal ligand to siroheme and noted ϐ-CH proton coupling constants (1.88 MHz) consistent with a cysteine or serine residue as the proximal ligand.336 A low resolution (3.0 Å) X-ray crystal structure of E. coli SiRHP provided evidence that the coupling between the siroheme and [4Fe-4S] cluster prosthetic groups was due to a shared ligand and that the [4Fe-4S] cluster was in near van der Waals contact with the siroheme macrocycle edge,282 and comparison of the sequences of spinach assimilatory nitrite reductase (aNiR) and Salmonella aSiR led to the creation of a model of the siroheme-[4Fe-4S] active site for which one of the Cys residues that coordinate the [4Fe-4S] cluster is the siroheme proximal ligand.337 This model was confirmed by a 1.6 Å X-ray diffraction structure of SiRHP by published by Crane, Siegel, and Getzoff in 1995, which clearly showed the bridging cysteinyl ligand and a saddle-shaped siroheme bent toward the [4Fe-4S] cluster as postulated by the ENDOR results.283 Early characterization of Dvir reported unusually low-field S = 9/2 EPR features that were attributed to one or more cubane [4Fe-4S] clusters, and it was proposed that the siroheme and FeS cluster were not actually coupled and that the signals might even arise from a 6Fe “super cluster.”307,338 However, at the time of these studies, there was significant disagreement regarding protein Fe content, and the presence of these features was subject to purification conditions.290,297,316 The dispute was conclusively resolved with the report of the Dvir structure, which contained two metalated sirohemes coupled to two cubane [4Fe-4S] clusters, two demetalated, uncoupled sirohydrochlorins and corresponding [4Fe-4S] clusters, and two electron transfer [4Fe-4S] clusters near the Fd docking site (Figure 22).311
Figure 25.

EPR Spectra of SiRHP displaying g ~ 2.29 features characteristic of the exchange-coupled FeS cluster. Figure adapted from ref. 331 Copyright American Chemical Society 1982.
Figure 22.

Structure of α2ϐ2γ2 complex of dSiR with DsrA (geen), DsrB (gray), and DsrC (cyan) (PDB ID: 2V4J).
4.4. Other Sulfite Reducing Enzymes.
4.4.1. Siroheme-containing nitrite reductase (NiR).
The conserved SNiRR domain for binding the siroheme-[4Fe-4S] cofactor is present both in SiRs and NiRs—enzymes responsible for the 6 e− reduction of nitrite (NO2) to ammonium (NH4+). Since the active sites are nearly identical, it is not surprising that proteins bearing the SNiRR domain often display both SiR and NiR activity.318,339,340 All SiRs studied to date are capable of nitrite reduction, usually with kcat that is 10-103 fold faster than sulfite as a substrate, but with overall catalytic efficiency several orders of magnitude lower (except alSiR) due to higher nitrite KM.290 NiRs are distinguished by their relatively higher affinity for NO2− over SO32−. Differences in the amino acids forming the distal pocket of the siroheme between NiR and SiR have been implicated as a crucial determinant for the enzyme’s selectivity. Alignment of the siroheme-[4Fe-4S] cofactors from spinach aNiR and SiRHP show that three of the catalytic residues important in SiRHP (Arg83, Arg153, and Lys215) are conserved with analogous residues in aNiR, but two of the residues (His123 and Lys217) are replaced in aNiR by Arg149 and Asn226, respectively. Studies of tobacco NiR have demonstrated that mutating Asn226 to Lys increased sulfite reduction activity by an order of magnitude.341
A ferredoxin-dependent aSiR (called NirA) isolated from Mycobacterium tuberculosis was found to crystallize with an unusual cross-link between the siroheme-contacting residue Tyr69 and Cys161.342 This type of thioether cross-link is present in galactose oxidase where it lowers the reduction potential of the catalytic CuII and facilitates the 2 e− oxidation of alcohols to aldehydes.343 In NADPH- and Fd-dependent aSiR, the Tyr residue is conserved, but Cys is not.342 Mutation of either Tyr or Cys significantly reduced activity in NiRA, and the authors interpreted this to mean that the thioether cross-link is important for NiRA activity. They also noted that the Tyr residue is conserved in Fd-dependent aSiRs but not in Fd-dependent aNiRs, and they proposed that the presence or absence of this Tyr could be used as a genetic marker of substrate preference for sulfite or nitrite.342 These substitutions paint a gradually resolving picture of the structural bases underpinning the selectivity for nitrite over sulfite in aNiRs, but few of the proposed effects of these structural differences have been directly tested to-date.344
4.4.2. Pentaheme Cytochrome c NiR.
An important ancestral structure in bacterial nitrate metabolism is pentaheme cytochrome c nitrite reductase (ccNiR), which phylogenetic analysis has identified to be a likely precursor to hydroxylamine oxidoreductases and octaheme nitrite reductases (ONR).345 ccNiRs vary in sequence but have tightly conserved arrangements of heme c cofactors for which a centrally located Lys-coordinated heme serves as the active site (Figure 26). A ccNiR known as NrfA has been observed to reduce sulfite as well as nitrite.346 Conserved His, Tyr, and Arg residues make hydrogen bonding interactions with bound sulfite. The mutation Tyr218Phe significantly impairs nitrite reduction, but sulfite reduction by NrfA is unaffected, demonstrating a potentially intriguing difference between nitrite and sulfite selectivity.346 However, due to the nature of the NH4+ and S2− quantification assays used, it is unclear whether these mutations truly reduce the rate of nitrite to ammonium reduction or whether they alter the number of electrons transferred per substrate.
Figure 26.
Active site structure of ccNiR (PDB ID: 3BNF).
4.5. Mechanism of Sulfite Reduction in SiRs.
4.5.1. Siroheme-[4Fe-4S] SiR.
The basic mechanistic proposal of sulfite reduction in SiR follows a “push-pull” strategy that is similar to the strategy adopted by O–O bond cleaving enzymes.290,347,348 The electron-rich siroheme-[4Fe-4S] cofactor donates charge to the π-accepting sulfur (or nitrogen) atom of the substrate. The partially saturated siroheme (isobacteriochlorin) is easier to oxidize and a superior π-donor relative to porphyrins (Scheme 29),349–351 and it has been proposed to transfer electrons to the sulfur π-antibonding orbitals to weaken the short, stable S–O bonds; although, this proposal has never been experimentally verified. The high density of conserved positively charged residues in the distal siroheme pocket further promote S–O activation through protonation of the substrate oxygens.283,290. In-depth computational studies of the SiR mechanism have been limited due to the complexities of the coupled siroheme-[4Fe-4S] cofactor;352 therefore, a majority of mechanistic insight has been gained through crystallographic studies of SiRs with their native substrate and substrate analogues.
Scheme 29.

Structural Difference Between Heme and Siroheme Cofactors.
For aSiR, the catalytic cycle for the full reduction to S2− begins with reduction of high-spin FeIII siroheme to high-spin FeII, allowing sulfite to displace a phosphate anion bound in the oxidized resting state (Scheme 30). Substrate binding is accompanied by the rearrangement of a loop region that encloses the anion binding pocket,283 and substrate binding likely affects protein-siroheme contacts, activating the site through redox-gating.318,353 Following substrate binding, electrons are transferred to siroheme through the coupled [4Fe-4S], accompanied by protonation from either ordered waters or from active site Lys/Arg residues for three successive dehydrations (Scheme 30). Typically, these steps are depicted as a transfer of 2 e−/2 H+ to the active site to form S(IV), S(II), S(0), and S(2−) intermediates, but the presence of radical intermediates from single ET steps has not been ruled out. Co-crystallization with inhibitors (CN− and CO) and substrate anions revealed that the active site adopts several conformations to accommodate the substrate as it is deoxygenated (Figure 27). The sidechain of conserved Arg153 rotates from its phosphate-bound state to a sulfite-bound/closed loop state with sulfite (but not nitrite) binding, and Lys215 rotates to interact directly with sulfite. Arg153 then returns to its original conformation after the first dehydration.354 Single mutant experiments with SiRHP revealed that the first two protons likely come from a protonated substrate and an ordered water. Lys215 is essential for anion binding throughout the cycle, but Lys217 and Arg153 are involved in late-stage proton transfer as Ser mutants of either of these residues reduced total electrons transferred to the substrate to 4 e− and 5 e−, respectively, but still allowed efficient 2 e− reduction of hydroxylamine.354,355
Scheme 30.
Proposed Catalytic Mechanisms and the Observed Intermediates of aSiR and dSiR.
Figure 27.
Crystal structures of SiRHP bound to substrate and inhibitors where siroheme Fe is either in its reduced (orange) or oxidized (red) state: (A) PO43− (PDB ID: 1AOP), (B) SO32− (PDB ID: 2GEP), (C) NO2− (PDB ID: 3GEO), (D) NO (PDB ID: 6GEP), (E) CN− (PDB ID: 4GEP), and (F) NO3− (PDB ID: 8GEP).
As mentioned in section 4.2, the heterotrimeric structure of dSiRs and, in particular, the role played by the DsrC subunit suggest that the dSiR mechanism differs substantially from the aSiR mechanism to better facilitate the physiological role of dSiRs. In contrast to aSiR, the siroheme sites of dSiRs are located at the interior interface of the A/B subunits and are more deeply buried. The [4Fe-4S] and proximal face of siroheme are coordinated by residues in DsrB, and strictly conserved positively charged Lys and Arg residues homologous to the distal residues of aSiR are provided by DsrA.311 This active site opens to a substrate channel that is blocked by Tyr-334B in Dvir and Trp-119β in the A. fulgidus dSiR so that only half of the siroheme sites are active.301,311 The C-terminal arm of DsrC reaches into the active siroheme distal pocket and positions a highly conserved cysteine, Cys104, within reach of bound substrate. Recently, Santos et al. have demonstrated that the role of DsrC in the dSiR mechanism is to attack partially reduced sulfur species bound to siroheme, and release from DsrAB as a trisulfide (Cys93–S–Cys104) (Scheme 30).356 Effectively, this means dSiR natively only performs a 2 e− reduction of SO32−, and the remaining 4 e− reduction of the trisulfide DsrC would be accomplished by the DsrK subunit of the associated respiratory membrane complex (DsrMKJOP).311,356 This proposed mechanism essentially reconciles the observation of incomplete sulfite reduction by Dvir purified from the cytosolic fraction while sulfide is obtained as the only product of the enzyme obtained from the membrane fraction315 and finds further support from its co-purification with the sulfite reducing products of the dsr genes and DsrKJO membrane components of sulfur oxidizing A. vinosum.357 However, DsrC is not expressed constitutively with DsrAB, and its association with DsrAB appears to be relatively low;358 therefore, some questions remain concerning the exact cellular role(s) of DsrC in sulfate reducing bacteria. An alternative—though more convoluted—mechanism was proposed from the observation that in some structures of DsrABC the Sγ of Cys104 is covalently linked to a siroheme meso carbon (Figure 21A). By this mechanism, a persulfide forms from reaction with 6 e− reduced S2− before protonation can occur, a second equivalent of SO32− binds to siroheme, and after 2 e− transfer to an incoming substrate anion it reacts with the nearby persulfide to form and then release S2O32− or repeats the process to yield S3O62−.312 However, this mechanism is not as well supported as the transient involvement of DsrC, and there is a greater body of evidence that suggests thiosulfate and trithionate are not produced physiologically.
In nature, siroheme is universally found covalently linked to a [4Fe-4S] cluster, and functional implications for this conserved structural feature have been hypothesized but remain unconfirmed. Structures of the SiRHP subunit in aSiR and the multi-subunit complexes of dSiR show that the [4Fe-4S] cluster likely serves a role in ET between the siroheme active site and the other ET protein subunits.283,311,359 Spectroscopic studies have shown that electronic coupling between siroheme and the [4Fe-4S] cluster cause siroheme to exhibit paramagnetic character in all of its physiological oxidation states (see section 4.3); however, the functional implications of this exchange coupling remain unclear. Reduction of the [4Fe-4S] cluster in CO-bound SiRHP lowers the υC–O from 1920 cm−1 to 1904 cm−1, demonstrating the coupled cluster has some impact on the degree of small molecule activation by siroheme.360 Recent mutation studies of residues near the [4Fe-4S] cluster in SiRHP demonstrate efficient ET to the [4Fe-4S] cluster is an important factor in catalytic activity.361 In these studies, Cepeda et al. modified the bulky surface residues that block solvent access to the [4Fe-4S] to less bulky Ala residues. Phe437Ala and Thr477Ala mutations increase sulfite reduction activity of both the SiRHP subunit and the holo-aSiR complex (dodecamer of SiRHP and flavoprotein subunits), consistent with improved access of reductant to the [4Fe-4S] cluster (Figure 28). Computational analysis of the ET pathways between siroheme and [4Fe-4S] consider the covalent Cys bridge between the Fe in siroheme and Fe in [4Fe-4S] is the most favorable ET pathway.362 This analysis further suggests a functional role of the isobacteriochlorin ring structure of siroheme, which reduces the propensity of the [4Fe-4S] cluster to directly transfer electrons to the π-system. The authors speculate that avoiding radical character on the porphyrin ring may avoid potential side-reactions between the partially reduced sulfur species bound to Fe during turnover.
Figure 28.
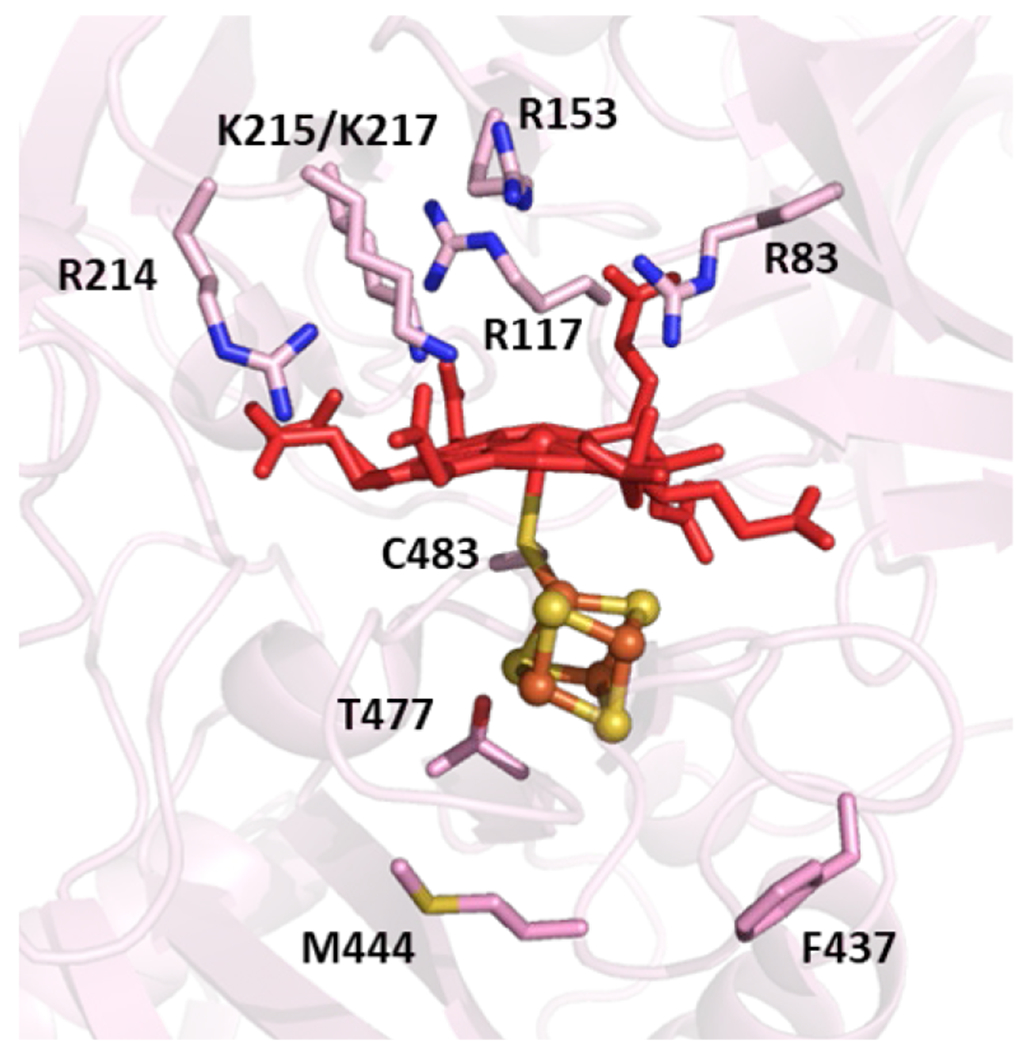
Position of non-coordinating residues in SiRHP (PDB ID: 1AOP) Phe437, Met444, and Thr477 that control ET rate to [4Fe-4S] cluster for SiR activity.
4.5.2. Heme c/Cu (SiRA).
Considerably less is known about the mechanism of sulfite reduction by the heme c/Cu active site of SiRA, which has been studied less than other SiRs.284 The recent XRD structures of different forms of SiRA in the presence of sulfite has led to an initial mechanistic proposal reliant on the nonheme CuI ion that differs from aSiRs and dSiRs (Figure 29D).285 In the Cu-depleted form, sulfite binds to the active site heme in a manner reminiscent of SiRHP (Figure 29C); however, incubation of sulfite with the Cu-containing form leads to a mixture of two ligands observed bound to heme c. The first is SO2, which the authors propose to be the product of dehydration without ET promoted by the [(Cys)2Cu]− center to yield SIVO2 (Figure 29A). A second ligand, SO, was observed bound to heme c, which the authors attribute to a product of partial turnover from photoreduction during data collection (Figure 29B). In both cases, the Cu remains reduced, and its coordination state is unaltered. Thereafter, the SiRA mechanism is proposed to follow three successive 2 e− reductions and two dehydrations, reminiscent of the aSiR mechanism. Further study is necessary to confirm this proposed mechanism and whether the essential Cu center does indeed play no role in ET or the binding of any intermediates.
Figure 29.

XRD structures of SiRA: (A) dehydrated form of SO32− substrate (SO2) (PDB ID: 4RKM), (B) photoreduced structure with SO bound to heme (PDB ID: 4RKM), (C) Cu-depleted form with SO32− bound to heme (PDB ID: 4RKN). (D) Proposed mechanism of sulfite reduction, based on these structures. Figure adapted from ref. 285 Copyright Springer Nature 2015.
4.6. Biomimetic and Biosynthetic Models of SiR.
Comparatively few structural and functional models of SiR have been developed, and those that have been reported focus mainly on the siroheme-[4Fe-4S] cofactor. The Holm group has developed a series of structural SiR models, [(4Fe-4S)(L16)–μ2–S–(L)Fe], using a strategy of cluster sub-site differentiation that could undergo regiospecific substitution in the presence H2S to form a bridge between the FeS and Fe complex (Scheme 31).363,364 The [4Fe-4S] cluster was formed within a tridentate ligand (L16) and was successfully bridged by a variety of routes to either (salen)FeIII or (OEP)FeIII (OEP = octaethylporphyrin). The resulting bridged assemblies demonstrate spin-delocalization from high-spin FeIII to the cluster, and both complexes exhibit two redox transitions in DMF at low potential; though, the order of cofactor reduction could not be determined: −0.71, −0.96 V (salen) and −1.05, −1.32 V (OEP).363 This lab subsequently produced a bridged assembly that was a closer analogue of the SiR catalyst from a [4Fe-4S] cluster and an isobacteriochlorin-type complex (OEiBC) by the same method ([(4Fe-4S)(L16)-μ2-S-FeIII(OEiBC)]2−) that also demonstrated spin delocalization;364 however, this complex was not electrochemically characterized. An alternative strategy to mimic the [4Fe-4S]-heme structure in SiRs has been reported by Gerlach et al., who utilized pyridyl- and imidazoylthiolate linkers between a site-differentiated [4Fe-4S] cluster and Zn(porph) complexes.365 These studies clearly demonstrate an electronic effect of tethering a metal porphyrin to an [4Fe-4S] on the cluster redox potentials, which shift from −440 mV ([4Fe-4S]2+/1+ vs NHE) to −780 mV. None of these structural models have been reported to show any sulfite reductase-like catalytic activity.
Scheme 31.
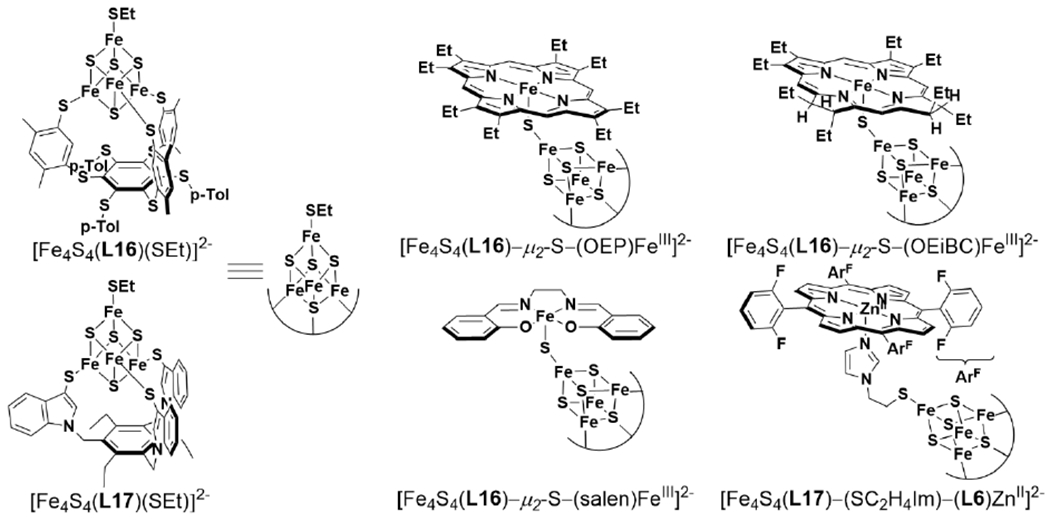
Reported Structural Models of Siroheme-[4Fe-4S] SiRs. 363,365
It was found that siroheme is a competent sulfite reduction catalyst using MV+ as an electron donor without the protein scaffold, though not as efficiently as isolated HP subunits of native SiRs.327 A study of {FeNO}7 complexes ligated by model porphyrin (OEP), chlorin (OEC = 7′,8′-dihydro-octaethylporphyrin), and isobacteriochlorin (OEiBC: 2′,3′,7′,8′-tetrahydro-octaethylrphyrin) ligands found that there was no significant difference between the isobacteriochlorin versus the porphyrin or chlorin for single electron reduction, but it was notable that OEiBC preferentially formed a macrocycle cation radical over oxidation to {FeNO}6 (Scheme 32)366, which typically display much higher reduction potentials in related Fe-porphyrin complexes.367 Examples of electrocatalytic sulfite reduction by other heme complexes have been reported. The six electron reduction of sulfite to sulfide has been reported for the synthetic water-soluble Fe-porphyrin complex, (L11)Fe (L11 = meso-tetrakis(p-sulfanatophenyl)porphyrin).368 Electrocatalytic sulfite reduction has also been described with surfactant-based films of myoglobin.369 These studies determined that the protein likely releases its heme cofactor. Both of these examples observe the catalytic wave at or near the FeII/FeI couple, with potentials much lower than what is relevant for the native enzyme (< −0.4 V and < −0.9 V vs SHE for the model complexes, respectively) (Scheme 33). Nevertheless, these examples demonstrate sulfite reduction can be accomplished by an Fe center with various porphyrin ligands.
Scheme 32.
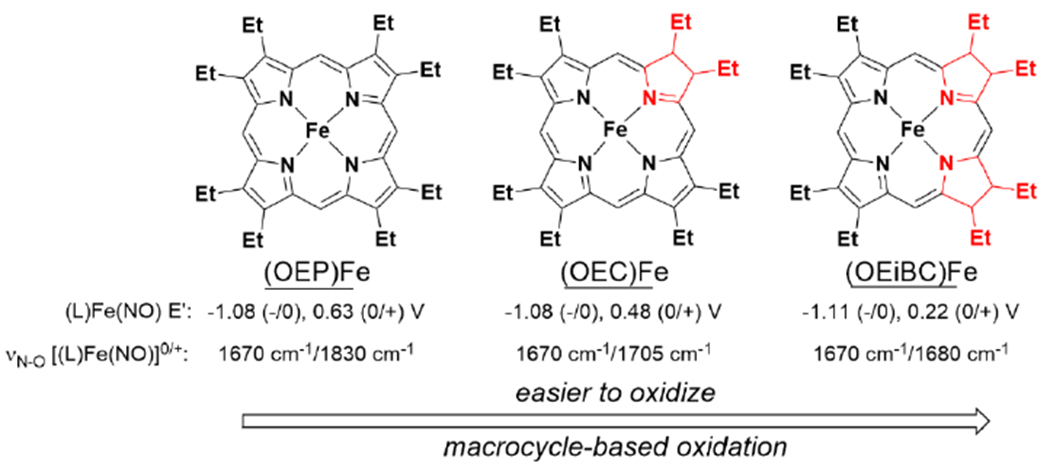
Reduction Potentials and υN–O of Model Porphyrin (OEP), Chlorin (OEC), and isobacterochlorin (OEiBC) Complexes of Fe.366
Scheme 33.
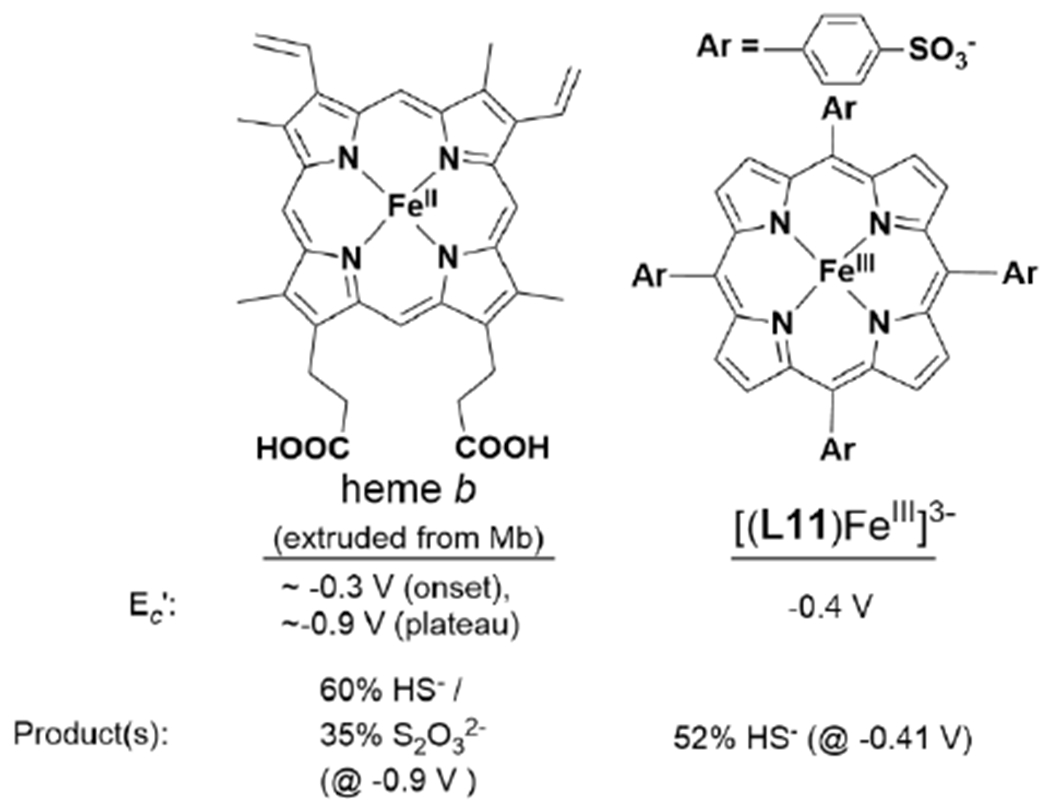
Reported Electrocatalytic SO32− Reducing Heme Complexes and Catalytic Properties.368,369
An early biosynthetic modification of the E. coli aSiR to create a minimal functional holoenzyme with only a single flavoprotein and hemoprotein subunit was achieved by Zeghouf et al.370 They found that they could crystallize the aSiR flavoprotein subunit (SiR-FP, the α subunit of the α8ϐ4 holoenzyme) by truncation of the first 51 amino acids to yield a functional monomeric form of SiR-FP (α′) that they called SiR-FP60.371 The truncated SiR-FP60 formed a functional dimer with purified SiRHP of the form α′ ϐ and was competent to perform NADPH-dependent catalysis at 20% the rate of the native holoenzyme. Given the stability of this complex, they disputed the accepted α8ϐ4 oligomeric state of the holoenzyme, proposing that, based on titration experiments and in the absence of a holoenzyme structure, α8ϐ8 was also plausible.370 The first true structural and functional biosynthetic model of sulfite reductase was developed by Mirts et al., by the redesign of the heme proximal binding site in cytochrome c peroxidase (CcP) to bind a [4Fe-4S] cluster adjacent to the native heme b prosthetic group (Figure 30).372 Like native SiR this biosynthetic model incorporated a bridging Cys residue to act as the heme proximal ligand and a ligand to one of the Fe sites in the [4Fe-4S], building off an earlier attempt to create a cytochrome P450-like Cys-heme active site in CcP.373 The designed enzyme, called SiRCcP, explored several combinations of SCS mutations around the designed [4Fe-4S] cluster site and substrate coordinating distal site. Mutations were introduced that mimicked the conserved Arg153, Lys215, and Lys217 residues in SiRHP, while the native CcP residue Arg48 served an analogous role to SiRHP residue Arg83 as both a (siro)heme and substrate contacting residue. The most active mutant (called KRK-SiRCcP.3) could catalyze the reduction of sulfite to sulfide at ~18% the rate of the M. tuberculosis aSiR.372
Figure 30.
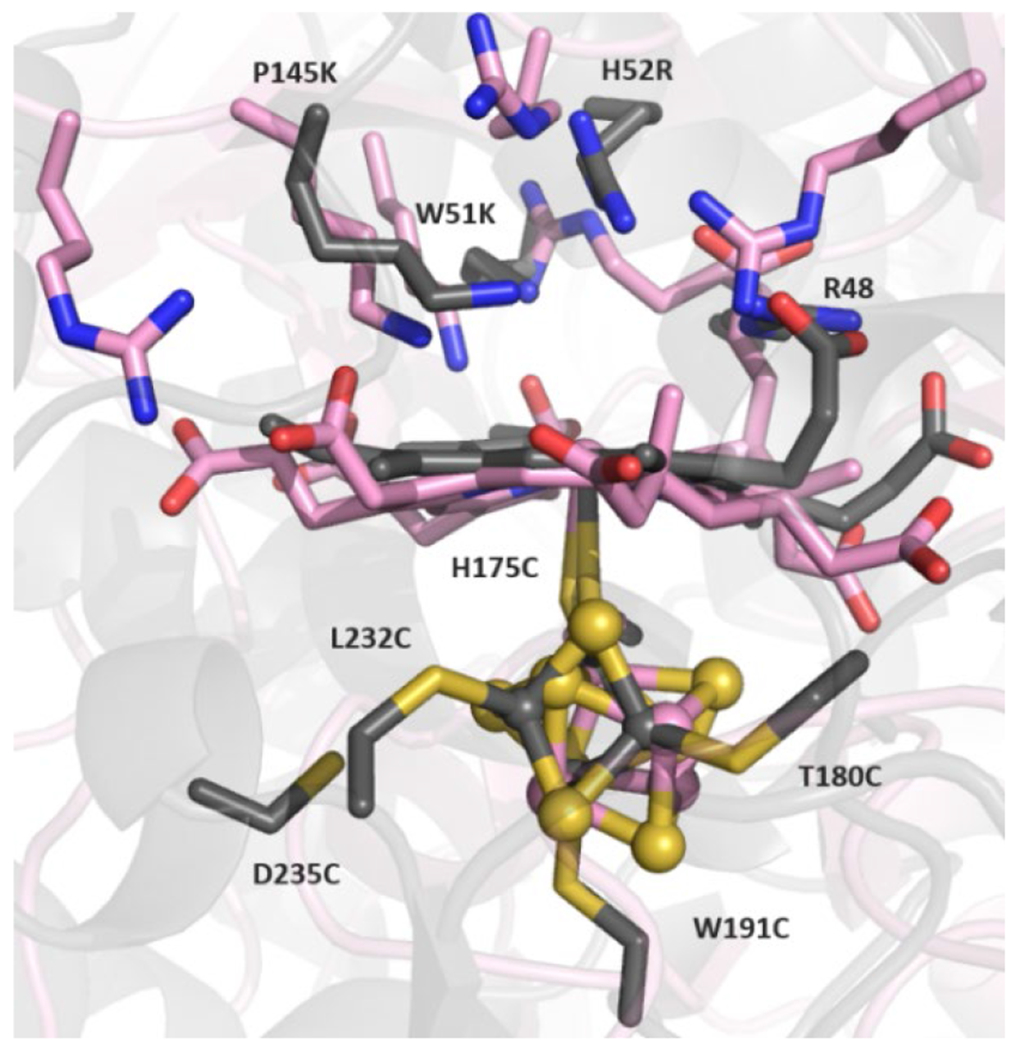
Modelled structure of KRK-SiRCcP.3 (gray) overlaid with SirHP (pink).
4.7. Summary and Outlook of SiR and Biomimetic Models.
In terms of multi-electron chemical transformations, the 6 e− reduction of sulfite to sulfide is one of the most complex small molecule transformations performed by a single enzyme, matched by 6 e− nitrite reduction, or the overall 8 e− N2 reduction process in nitrogenase to form ammonium and dihydrogen.374 Obtaining a clear understanding of the SiR catalytic center proved challenging due to the complex nature of the covalently linked and magnetically exchange-coupled [4Fe-4S] cluster and siroheme, a unique isobacteriochlorin that has been found in no other enzymes in living organisms. Examination of the crystal structures of dissimilatory and assimilatory SiRs combined with electrochemical and computational studies of model heme, chlorin, and (iso)bacteriochlorin compounds have revealed in greater detail how subtle differences in siroheme ruffling induced by protein contacts and coupling to the [4Fe-4S] cluster control the flow of electrons through the siroheme Fe and suppress oxidation of the siroheme ring to prevent the formation and release of partially reduced intermediates. Though few structures of each enzyme class have been obtained so far, they have provided the clearest insights into how the different holoenzyme structures of dSiRs and aSiRs contribute to their functions in respiratory metabolism and the generation of sulfur-containing biomolecules, respectively, as well as the number of successive dehydrations accomplished in each enzyme in its native role. Biomimetic synthetic complexes and proteins have further demonstrated that while many heme molecules can achieve sulfite reduction to a considerable degree, selective reduction to S2− is highly dependent on the inclusion of substrate coordinating and ET-promoting residues provided by the protein scaffold. It is notable also that the two forms of sulfite reductase active sites described so far (siroheme-[4Fe-4S] SiRs and the heme c/Cu multiheme SiRA) utilize unique heteronuclear active sites with heme cofactors. While our understanding of features that promote the reduction of oxyanions such as sulfite and nitrite at an Fe catalyst is growing, many questions remain to be answered for a deeper understanding of the structural bases of the efficient catalytic properties of sulfite reductases. Some of these questions include:
What role does the nonheme cofactor ([4Fe-4S] in SiRs and Cu in SiRA) play in the binding and activation of substrate during turnover? With so little—and generally inconsistent—information regarding the redox potentials and the detailed process of cofactor redox cycling at specific reaction steps, there remain many unknowns of how these auxiliary cofactors may control redox activation, substrate/intermediate stabilization, or modulation of the redox properties of the (siro)heme cofactor at various stages of this multi-step reaction.
For siroheme-containing SiRs, does the siroheme ring ever act as an electron donor during turnover? Are there any functional reasons this ancient cofactor remains one of the most prevalent tools in living organisms for sulfite and nitrite reduction, or is siroheme merely a convenient relic of facile iron macrocycle synthesis in early anaerobes?
What is the nature of the partially-reduced sulfur intermediates (−SOx), and how do their interactions with the enzyme active site influence their reactivity towards sulfide formation versus off-pathway products (S2O32− and S3O62−) from reaction with additional substrate anions?
As has been demonstrated throughout this review, biomimetic models are an invaluable tool in expanding our understanding of the underlying chemical principles in these relatively complex active sites. The complementary study of native sulfite-reducing metalloenzymes and their biomimetic models will hopefully continue to deepen our understanding of the sulfite reduction mechanism down to the molecular level.
5. Comparison of Heme/Nonheme Active Sites of HCO, NOR, and SiR.
5.1. HCO and NOR
5.1.1. Cross-Reactivity of HCO Superfamily.
A handful of bacterial HCOs, representing two of the three main types (B and C; see section 2.1), are able to accomplish catalytic NO reduction.199,200,375,376 Similarly, oxidase activity has been observed in cNOR from P. denitrificans.185,377,378 In cases where an enzyme within this superfamily displays both NO and O2 reducing activity, it exhibits much higher activity for its native reaction (Table 1). A number of the previously discussed synthetic models of BNC active sites display both NO and O2 reduction reactivity.117,258,274,379 Despite their similar roles in biological anaerobic and aerobic respiratory pathways, the molecular mechanisms of O2 reduction to H2O and NO coupling to N2O are quite dissimilar from a chemical perspective. Therefore, it is noteworthy that similar heme/nonheme catalysts for these distinct reactions are found in nature—catalysts similar enough that enzymes in the HCO superfamily can achieve both the 2 e− reduction of NO and 4 e− reduction of O2. On the other hand, if we choose to view the differences between HCO and NOR structure as the result of natural selection which has optimized for efficiency of each of their native reactions, they become a valuable case study in catalyst design for small molecule transformations. What makes the heme/CuB active site better for O2 reduction, and the heme/FeB site for NO coupling? Based on our current knowledge of these enzymes, there is no definitive answer but studies of these native proteins and their biomimetic models provide some possible explanations. In the following sections, we will consider insights gained on the functional implications of differences between the BNC sites of HCO and NOR, in particular the characteristics of heme and nonheme cofactors, along with differences in enzyme PT pathways.
Table 1.
NO and O2 Reduction Activity of HCO Superfamily Enzymes.
5.1.2. Effect of Heme Cofactor.
A range of reduction potentials for the active site heme cofactors in NOR and HCO have been measured, and the general trend is that HCO contains higher potential heme cofactors than the heme b3 center in NOR. The average potential of HCO heme is greater than that of NOR heme by ~200 mV (Table 2).270 The heme b3 center in NOR has a relatively small variation in potential, between −170 mV (vs SHE) and 80 mV.238,382 The range of active site heme potentials in HCO is much greater (between −120 mV and 460 mV), partly due to the occurrence of b3, a3, and o3 forms.383–385 The electron withdrawing formyl groups on heme a are one reason HCO potentials tend to be higher than NOR. Studies of native enzymes and related models suggest the neighboring conserved glutamate residues in NOR, which are absent in HCO, also play a key role in tuning the heme reduction potential.185,264,386 There appears to be functional significance of these heme reduction potentials: HCO enzymes that are competent for NO reduction tend to contain lower heme potentials that are close to those of native NORs, and there is a general correlation of higher NO reductase activity in HCOs as heme potentials decrease (see Tables 1 and 2). One of the highest HCO active site heme potentials reported is the a3 heme of mitochondrial CcO, which is around 460 mV.383 While initially considered to have NO reduction activity,387 later studies ruled out such reactivity and show that it is, instead, inhibited by binding NO.388 The possible implication of heme reduction potential on NO reduction by HCOs was analysed in a computational study.241 This study hypothesized that a conserved Val residue in oxidases leads to a high energy barrier of N2O formation by a mechanism that derives both reducing equivalents from the BNC active site, and therefore NO reduction in HCO relies on an alternative pathway, which involves proton-coupled reduction of the hyponitrite intermediate—the rate of which would be highly dependent on the heme reduction potential.
Table 2.
Measured Reduction Potentials of the Active Site Heme and Nonheme Cofactors of HCO Superfamily Enzymes.
| Enzyme | heme | nonheme | ref |
|---|---|---|---|
| PdNOR | 78-80 mV / 60 mV | 320 mV (FeB) | 185,186,238 |
| PnNOR | −38 ±10 mV / −162 ±9 mV | −369 ± 14 mV (FeB) | 239 |
| PaNOR | −440 mV | −110 mV | 237 |
| M. hydrocarbonclasticus cNOR | −168 ±15 mV | n.r. | 382 |
| Bovine mitochondrial CcO (aa3 ox.) | 460 mV | 400 mV (CuB) | 383 |
| A. ambivalens aa3 oxidase | 390 ±20 mV | n.r. | 389 |
| B. pseudofirmus caa3 ox. | 334 mV | n.r. | 390 |
| B. japonicum cbb3 ox. | 275 mV | n.r. | 391 |
| Bacillus YN-2000 aco ox. | 250 mV | n.r. | 392 |
| E. coli bo3 ox. | 200 mV | n.r. | 385 |
| T. thermophilus caa3 ox. | 133 mV / 248 mV / 378 mV | n.r. | 393 |
| T. thermophilus ba3 ox. | 199 mV | n.r. | 394 |
| R. marinus cbb3 ox. | −50 mV | 120 mV (CuB) | 395 |
| P. stutzeri cbb3 ox. | −54 mV | 372 mV (CuB) | 384 |
| R. sphaeroides cbb3 ox. | −59 mV / −95 mV | 415 mV (CuB) | 384 |
| V. cholera cbb3 ox. | −120 mV | n.r. | 384 |
A recent study of biomimetic heme/nonheme models of NOR supports the hypothesis that heme potential plays an important role on NO and O2 reactivity. FeBMb proteins were prepared with a variety of heme cofactors, leading to a range of observed heme reduction potentials from −130 mV to 148 mV.270 These different heme cofactors would display faster NO binding, greater NO activation (lower υN–O), and faster NO decay with lower reduction potential (Figure 19B). Lower reduction potential would also, however, lead to reduced electron transfer rates, which could possibly slow down NO reduction catalysis. It was determined that the most active NO reduction catalyst of these proteins was one with an intermediate reduction potential of 53 mV (Figure 19C), suggesting efficient catalysis relies on a crucial balance between fast ET and sufficient reducing potential of the active site heme.
5.1.3. Effect of Nonheme Cofactor.
Another prominent difference between NOR and HCO enzymes is the identity and geometry of the nonheme cofactor. Reduction potentials of the nonheme cofactor in the HCO superfamily are much harder to measure, since they are not associated with a distinctive shift in a protein’s UV-Vis absorbance spectrum (unlike a heme Soret band). In the cases where they have been measured, similar to the trend observed with heme cofactor, the reduction potential of the CuB center in HCO is higher than FeB in NOR (Table 2). Based on extensive computational analysis of O2 and NO reduction by the HCO enzyme superfamily, Blomberg proposed that the lower reduction potentials of the metal cofactors in the NOR BNC is advantageous for promoting irreversible NO binding and fast reduction of this toxic substrate, whereas the relatively higher potentials in HCO are important for energy conservation and proton pumping.107,396,397
Another major difference between the FeB and CuB centers is their distance to the heme cofactor. Crystal structures of NORs show a heme/nonheme Fe–Fe distance that ranges between 3.8 and 4.4 Å, whereas HCO structures typically have Fe–Cu distances somewhere between 4.4 and 5.3 Å (Table 3). These differences can be considered the consequence of two major structural features: (i) Glu coordination of FeB in NOR tends to move the nonheme metal closer to heme, and (ii) the His-Tyr crosslink in HCO moves the third His ligand to CuB down (towards the heme), which results in shifting the nonheme metal away from heme, to achieve a stable trigonal planar geometry (Figure 31).54 Functional implications of these structural differences (for promoting O2 over NO reduction and vice versa) are unclear, however, due to an incomplete understanding of potential active site conformational changes that occur during turnovers.
Figure 31.
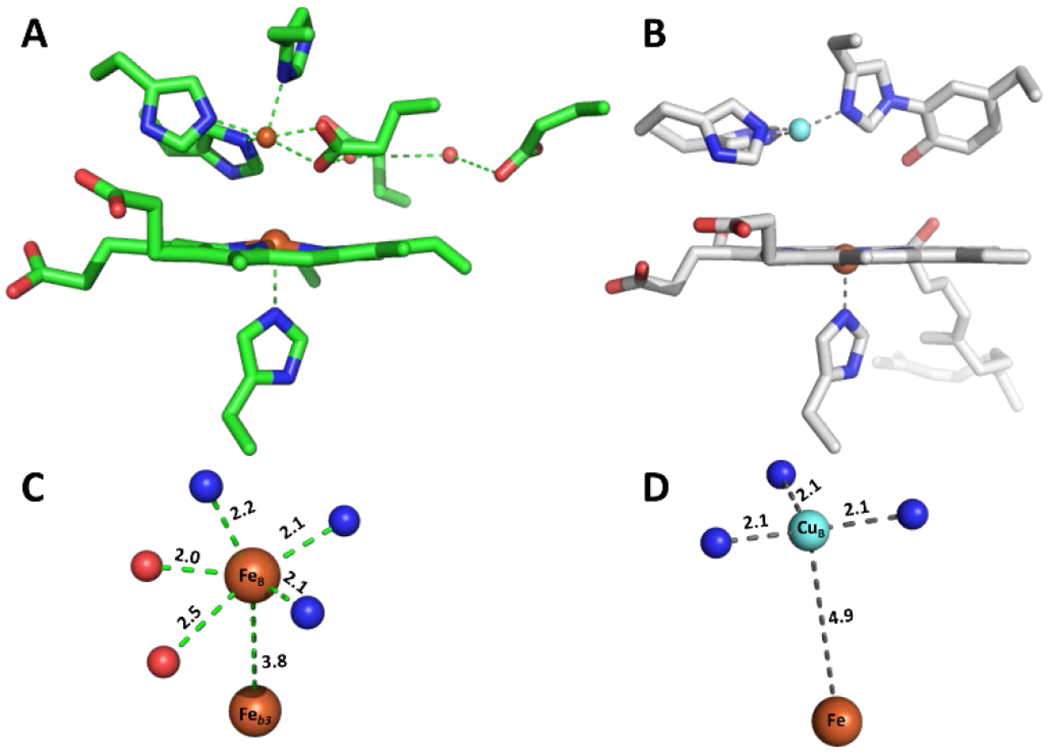
Active site structures of (A) PaNOR, and (B) bovine aa3 oxidase. Coordination geometry and distance to heme of (C) FeB in PaNOR, and (D) CuB in bovine aa3 oxidase.
One way to probe the effect of the nonheme cofactors on the enzymatic activity would be to replace CuB in HCO with FeII, or the FeB center in NOR with CuI. However, there is no report of such a study, probably because of the challenge of introducing a non-native metal ion into the native binding site. In contrast, it is quite easy to prepare biomimetic models of HCO superfamily enzymes with either nonheme Cu or Fe. These models have provided some insight into the role of the nonheme metal in promoting selective O2 or NO reduction. HCO and NOR models reported by Collman et al., [(L14)FeII/CuI]+ and ([(L14)FeIII/FeII(Cl)]+ respectively, have compared the role of the nonheme metal ion in O2 reduction.379 Under slow electron flux (1 electron s−1), the absence of a nonheme metal (L14)FeII) results in PROS production of 20%. Addition of Cu ([(L14)FeII/CuI]+) leads to a decrease of PROS to 6%, while the ([(L14)FeIII/FeII(Cl)]+ complex results in 6% total PROS, which further decreases to 3% at fast electron flux, lower than nearly all similar picket-fence porphyrin complexes tested. This study demonstrates that either nonheme metal (Fe or Cu) can promote complete 4 e− reduction of O2. Similarly, in FeBMb, FeII and CuI forms were both able to catalyze O2 reduction, with rates and selectivity for H2O much higher than the metal-free or ZnII bound forms of the biosynthetic proteins (Figure 32).117 Consistent with the trend observed for NOR and HCO, the reduction potential of the nonheme FeII was found to be lower than CuI (259 and 387 mV, respectively), and it was rationalized that the higher reduction potential of CuI improves catalytic activity by increasing the rate of electron transfer. Furthermore, DFT calculations of the peroxy-bound FeBMb active site with FeIII and CuII showed greater degree of O–O activation with CuII, possibly due to its higher d-electron count.117 These studies suggest CuB is better for O2 reduction by improving energy conservation (by virtue of its higher reduction potential) and leading to higher O2 activation compared to FeB.
Figure 32.
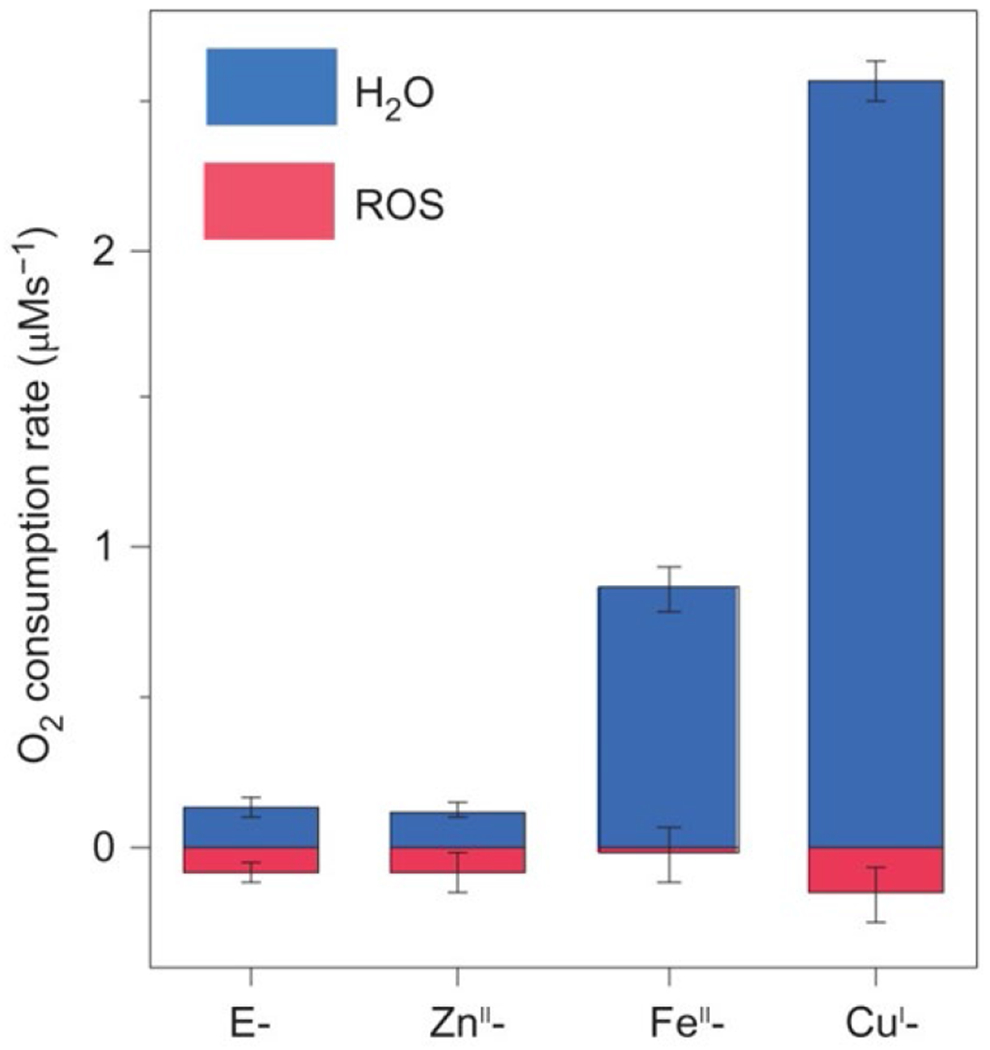
ORR activity data of FeBMb with various nonheme metals. Figure adapted from ref. 117 Copyright Springer Nature 2017.
HCO is also distinguished from NOR by having another redox active cofactor in its binuclear active site. The conserved Tyr residue is understood to be a crucial feature for efficient O2 reduction. Its presence in HCO provides the fully reduced BNC with a total of four reducing equivalents, which allows for complete O–O cleavage without exogenous ET, which provides a simple rationalization for the enzyme’s complete selectivity for H2O over other PROS. Therefore, the absence of a corresponding Tyr in NOR BNC is likely one reason for its lower selectivity for O2 reduction. Computational studies of O2 reduction by NOR suggest a proton and electron are required to form an FeIII–OOH intermediate prior to O–O bond cleavage, while such an intermediate in HCO (Ip) is considered a transient step by virtue of Tyr oxidation and/or PT.396
5.1.4. Effect of PT Pathways.
Unlike HCO, NOR are not able to pump protons; although recently, some quinol- and CuA-dependent NORs were found to be electrogenic by virtue of consuming reducing equivalents from the periplasm and protons from the cytoplasm.170,174 Natively, HCOs produce a proton gradient during O2 reduction through a combination of a similar electrogenic property, along with their unique proton pumping mechanism. Remarkably, catalytic NO reduction by Rs cbb3 oxidase was found to be non-electrogenic, and consumes both protons and electrons from the periplasm, instead of utilizing the cytosolic PT pathways employed for O2 reduction.200 It is proposed that O2 reduction intermediates can be protonated by the cytosolic PT pathway though a residue in the pathway with a relatively high pKa, which cannot protonate NO reduction intermediates that have a lower proton affinity. Kinetics studies of O2 reduction by Rs aa3 oxidase show that a Glu residue near the cytoplasmic surface is important for transferring protons during O2 reduction, and acts as a proton donor with a pKa > 9.398 In contrast, O2 reduction by PdNOR displays pH dependence based on a Glu residue implicated in a periplasmic PT pathway (Glu122) with pKa ~ 6.6.399 Interestingly, O2 reduction is not diminished when this residue is mutated to Asp, which appears to have a pKa similar to the PT pathway in HCO, which is >9. Overall, these studies support the hypothesis that HCO utilize residues in PT pathways with higher pKa values, which are likely important for energy conservation and directional proton pumping, but inefficient for PT to intermediates formed during NO reduction that have lower proton affinity.
5.2. HCO and SiRA.
While significantly less is understood of the sulfite reduction mechanism in the heme c/Cu active site of SiRA than is understood of O2 and NO reduction in the heme/nonheme Cu/Fe centers of HCO and NOR, it is worthwhile to briefly touch on the intriguing similarities and differences of these catalytic centers (Figure 33). While HCO displays promiscuity for reduction of other substrates, such as NO, it is a notable feature of SiRA that it is highly selective for its native reaction. Unlike other SiRs, SiRA displays negligible activity towards nitrite, or other substrates.400 Despite its apparent similarity to the heme/Cu active site of HCO, O2 does not react in a similar way by SiRA; instead, oxidation of the active site leads to loss of Cu and disulfide bond formation.285 The CuI coordination in SiRA ([(Cys)2Cu]−) leaves the Cu ion with a net negative charge, which is distinct from the net positive charge on nonheme Cu or Fe ions in HCO and NOR. This negative charge likely stabilizes the SIV state of the heme-bound sulfur atom prior to electron transfer. This effect would persist through the two subsequent 2 e− reduction steps, but it would be abolished if the CuI ion were to donate an electron, in contrast to the redox-active nonheme Fe/Cu ions. The evidence so far seems to agree that the Cu ion is indeed redox inactive, but this question has only barely been explored. Another common feature between SiRA and HCO active sites is the presence of Tyr that participates in active site water coordination. The crystal structure of SiRA suggests that Tyr285 plays a role in PT with the added role of stabilizing the liberated water molecule once the substrate has been dehydrated.285 The presence of a similarly positioned, conserved Tyr in structurally related ccNiR and the substrate-dependent role it plays in nitrite vs sulfite reduction raises intriguing questions regarding the full functional scope of the active site Tyr residues in SiRA and why SiRA demonstrates such high substrate selectivity for sulfite over nitrite compared to the siroheme-[4Fe-4S] SiRs. Obviously, given the extensive and informative work to understand the similarities and differences between HCO and NOR, it would be of great interest to further develop the functional scope of heme/nonheme Fe and Cu active sites and explore how the specific structural features of SiRA (or a biomimetic model of it) determine its reaction selectivity.
Figure 33.

(A) Active site structures of bovine aa3 oxidase (PDB ID 2Y69) and (B) SiRA (PDB ID 4RKM) and coordination geometry and distance to heme in CuB of (C) aa3 oxidase and (D) SiRA.
6. Outlook.
Multi-electron small-molecule transformations catalyzed by a single enzyme are among the most challenging in biology and are important for many biological processes from cellular respiration to the global cycles of the life-essential elements. To accomplish this difficult task, biology often employs heteronuclear metalloenzymes, such as the HCO, NOR and SiR active sites described in this review. However, most of these enzymes are less well-understood compared to mono- or homonuclear metalloenzymes, in part due to the inherent complexity of different metal ions arranged in close proximity. In this review, we have summarized progress that has been made in studies of each of these enzymes and their biomimetic models, with focus on insights gained in structural features responsible for each step of the reaction mechanisms.
Despite the progress made in the study of the individual enzymes, we still lack a holistic understanding of this class of heteronuclear metalloenzymes. Specifically, a major unresolved issue is what structural features are responsible for the differences in their functions; i.e., why is the heme/nonheme Fe in NOR effective in 2e− reduction of NO to form the N–N bond, while the heme/Cu center in HCO is efficient in 4e− reduction of O2 to break the O–O bond, and why do the heme/Cu in SiRA and the even more complex heme-[4Fe-4S] in SiR perform 6e− reduction of SO32− to break the S–O bond? The answer to these questions will rely on a deep molecular-level understanding of these enzyme mechanisms, which is aided through complementary study of these native active sites and related biomimetic complexes.
Acknowledgements
The Lu group research described in this review has been supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number GM062211 and Department of Energy’s Center for Advanced Bioenergy and Bioproducts Innovation (Office of Science, Office of Biological and Environmental Research, under award DE-SC0018420). C.R. would like to acknowledge support from the National Institutes of Health Postdoctoral Fellowship (GM136177). We thank our group members and collaborators who have contributed to the results and insights gained from our studies.
List of abbreviations:
- ET
electron transfer
- NOR
nitric oxide reductase
- HCO
heme-copper oxidase
- SiR
sulfite reductase
- PT
proton transfer
- PROS
partially reduced oxygen species
- QO
quinol oxidase
- CcO
cytochrome c oxidase
- BNC
binuclear center
- AOX
alternative oxidase
- FDP
flavodiiron protein
- MCO
multicopper oxidase
- PI
peroxy intermediate
- NI
native intermediate
- rR
resonance Raman
- ORR
oxygen reduction reaction
- swMb
sperm whale myoglobin
- CuBMb
Leu29His Phe43His swMb
- FeBMb
Leu29His Phe43His Val68Glu swMb
- DCHIM
1,5-dicyclohexylimidazole
- TMPA
tris(2-pyridylmethyl)amine
- AN
bis(3-[dimethylamino]propyl)amine
- imiTyr
imidazolyl-tyrosine
- SAM
self-assembled monolayer
- SERRS
surface-enhanced resonance Raman spectroscopy
- l.s
low-spin
- XRD
X-ray diffraction
- cryo-EM
cryogenic electron microscopy
- FNOR
flavodiiron NO reductase
- Hcp
hybrid cluster protein
- OEP
octaethylporphyrin
- Tp
trispyrazolylborate
- pz
pyrazolate
- APS
adenosine-5’-phosphosulfate
- ONR
octaheme nitrite reductase
- MccA
multicytochrome c sulfite reductase A
- aSiR
assimilatory sulfite reductase
- Fd
ferredoxin
- HP
hemoprotein
- alSiR
low-molecular weight monomeric aSiR
- dSiR
dissimilatory sulfite reductase
- Dvir
desulfoviridin
- NiR
nitrite reductase
- SNiRR
sulfite or nitrite reductase repeat
- MV
methyl viologen
- aNiR
assimilatory nitrite reductase
- ccNiR
cytochrome c nitrite reductase
- OEC
7′,8′-dihydro-octaethylporphyrin
- OEiBC
2′,3′,7′,8′-tetrahydro-octaethylrphyrin
- SiR-FP
aSiR flavoprotein subunit
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
There is inconsistency in the literature in what steps comprise the ‘oxidative’ and ‘reductive’ phases. Some consider the oxidative phases complete when the BNC is completely oxidized (PM state), others consider it to be when all four reducing equivalents from the fully-reduced enzyme are consumed (OH state). For this review, we will conform the former convention.
references
- 1.Falkowski PG, Fenchel T and Delong EF, Science, 2008, 320, 1034–1039. [DOI] [PubMed] [Google Scholar]
- 2.Zannoni D, Ed., Respiration in archaea and bacteria: diversity of prokaryotic respiratory systems, Springer, Dordrecht, The Netherlands, 2004. [Google Scholar]
- 3.Muras V, Toulouse C, Fritz G and Steuber J, in Bacterial Cell Walls and Membranes, ed. Kuhn A, Springer International Publishing, Cham, 2019, vol. 92, pp. 301–335. [DOI] [PubMed] [Google Scholar]
- 4.Richardson DJ, Microbiology, 2000, 146, 551–571. [DOI] [PubMed] [Google Scholar]
- 5.Linden G and Bongaerts J, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1997, 1320, 217–234. [DOI] [PubMed] [Google Scholar]
- 6.Weiner JH and Cheng VWT, EcoSal Plus, , DOI: 10.1128/ecosalplus.3.2.8. [DOI] [PubMed] [Google Scholar]
- 7.Lecomte SM, Achouak W, Abrouk D, Heulin T, Nesme X and el Z F. Haichar, Front. Environ. Sci, 2018, 6, 139. [Google Scholar]
- 8.Atashgahi S, Häggblom MM and Smidt H, Environ Microbiol, 2018, 20, 934–948. [DOI] [PubMed] [Google Scholar]
- 9.Shen Y and Buick R, Earth-Science Reviews, 2004, 64, 243–272. [Google Scholar]
- 10.Barton LL, Fardeau M-L and Fauque GD, in The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment, eds. Kroneck PMH and Torres MES, Springer Netherlands, Dordrecht, 2014, vol. 14, pp. 237–277. [Google Scholar]
- 11.Moura I, Moura JJG, Pauleta SR and Maia LB, Eds., Metalloenzymes in Denitrification: Applications and Environmental Impacts, Royal Society of Chemistry, Cambridge, 2016. [Google Scholar]
- 12.Ye RW, Averill BA and Tiedje JM, Applied and Environmental Microbiology, 1994, 60, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canfield DE, Annu. Rev. Earth Planet. Sci, 2005, 33, 1–36. [Google Scholar]
- 14.Pereira MM, Santana M and Teixeira M, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2001, 1505, 185–208. [DOI] [PubMed] [Google Scholar]
- 15.Raymond J, Science, 2006, 311, 1764–1767. [DOI] [PubMed] [Google Scholar]
- 16.Dismukes GC, Klimov VV, Baranov SV, Kozlov YN, DasGupta J and Tyryshkin A, Proceedings of the National Academy of Sciences, 2001, 98, 2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H and Pecoraro VL, Chem. Rev, 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Köhler V, Lewis JC and Ward TR, Chem. Rev, 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- 19.Bhagi-Damodaran A, Petrik I and Lu Y, Israel Journal of Chemistry, 2016, 56, 773–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirts EN, Bhagi-Damodaran A and Lu Y, Acc. Chem. Res, 2019, 52, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nastri F, D’Alonzo D, Leone L, Zambrano G, Pavone V and Lombardi A, Trends in Biochemical Sciences, 2019, 44, 1022–1040. [DOI] [PubMed] [Google Scholar]
- 22.Pinter TBJ, Koebke KJ and Pecoraro VL, Angewandte Chemie International Edition, 2020, 59, 7678–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dutton PL and Moser CC, Faraday Discuss, 2011, 148, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korendovych IV and DeGrado WF, Quarterly Reviews of Biophysics, 2020, 53, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy M and Gibney BR, Current Opinion in Structural Biology, 2001, 11, 485–490. [DOI] [PubMed] [Google Scholar]
- 26.Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y and Lombardi A, Chem. Soc. Rev, 2016, 45, 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Horsman JA, Barquera B, Rumbley J, Ma J and Gennis RB, Journal of Bacteriology, 1994, 176, 5587–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Ballmoos C, Smirnova I, Poiana F, Gonska N, Chang H-Y, Gennis RB, Brzezinski P and Ädelroth P, Israel Journal of Chemistry, 2017, 57, 424–436. [Google Scholar]
- 29.Gribaldo S, Talla E and Brochier-Armanet C, Trends in Biochemical Sciences, 2009, 34, 375–381. [DOI] [PubMed] [Google Scholar]
- 30.Hemp J and Gennis RB, in Bioenergetics, eds. Schäfer G and Penefsky HS, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, vol. 45, pp. 1–31. [Google Scholar]
- 31.Sharma V and Wikström M, FEBS Letters, 2014, 588, 3787–3792. [DOI] [PubMed] [Google Scholar]
- 32.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG and Tian L, Chem. Rev, 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HJ, Reimann J, Huang Y and Ädelroth P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2012, 1817, 537–544. [DOI] [PubMed] [Google Scholar]
- 34.Yoshikawa S and Shimada A, Chem. Rev, 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- 35.Lee HJ, Gennis RB and Ädelroth P, PNAS, 2011, 108, 17661–17666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y, Reimann J, Singh LMR and Ädelroth P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2010, 1797, 724–731. [DOI] [PubMed] [Google Scholar]
- 37.Chang H-Y, Ahn Y, Pace LA, Lin MT, Lin Y-H and Gennis RB, Biochemistry, 2010, 49, 7494–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buschmann S, Warkentin E, Xie H, Langer JD, Ermler U and Michel H, Science, 2010, 329, 327–330. [DOI] [PubMed] [Google Scholar]
- 39.von Ballmoos C, Gennis RB, Ädelroth P and Brzezinski P, PNAS, 2011, 108, 11057–11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rich PR and Maréchal A, J. R. Soc. Interface, 2013, 10, 20130183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wikström M, Sharma V, Kaila VRI, Hosler JP and Hummer G, Chem. Rev, 2015, 115, 2196–2221. [DOI] [PubMed] [Google Scholar]
- 42.Yano N, Muramoto K, Shimada A, Takemura S, Baba J, Fujisawa H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Tsukihara T and Yoshikawa S, J. Biol. Chem, 2016, 291, 23882–23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brzezinski P and Gennis RB, J Bioenerg Biomembr, 2008, 40, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Björck ML, Vilhjálmsdóttir J, Hartley AM, Meunier B, Näsvik Äjemyr L, Maréchal A and Brzezinski P, Scientific Reports, 2019, 9, 20207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wikström M, in Photosynthesis and Bioenergetics, WORLD SCIENTIFIC, 2017, pp. 55–63. [Google Scholar]
- 46.Soulimane T, The EMBO Journal, 2000, 19, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang H-Y, Hemp J, Chen Y, Fee JA and Gennis RB, Proceedings of the National Academy of Sciences, 2009, 106, 16169–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rauhamäki V and Wikström M, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2014, 1837, 999–1003. [DOI] [PubMed] [Google Scholar]
- 49.Wikström M, Krab K and Sharma V, Chem. Rev, 2018, 118, 2469–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wikström M and Sharma V, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2018, 1859, 692–698. [DOI] [PubMed] [Google Scholar]
- 51.Ishigami I, Hikita M, Egawa T, Yeh S-R and Rousseau DL, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2015, 1847, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R and Yoshikawa S, Science, 1996, 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- 53.Iwata S, Ostermeier C, Ludwig B and Michel H, Nature, 1995, 376, 660–669. [DOI] [PubMed] [Google Scholar]
- 54.Tosha T and Shiro Y, IUBMB Life, 2013, 65, 217–226. [DOI] [PubMed] [Google Scholar]
- 55.Luna VM, Fee JA, Deniz AA and Stout CD, Biochemistry, 2012, 51, 4669–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luna VM, Chen Y, Fee JA and Stout CD, Biochemistry, 2008, 47, 4657–4665. [DOI] [PubMed] [Google Scholar]
- 57.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P and Iwata S, Journal of Molecular Biology, 2002, 321, 329–339. [DOI] [PubMed] [Google Scholar]
- 58.Yoshikawa S, Muramoto K and Shinzawa-Itoh K, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2011, 1807, 1279–1286. [DOI] [PubMed] [Google Scholar]
- 59.Muramoto K, Ohta K, Shinzawa-Itoh K, Kanda K, Taniguchi M, Nabekura H, Yamashita E, Tsukihara T and Yoshikawa S, Proc Natl Acad Sci USA, 2010, 107, 7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han Du W-G and Noodleman L, Inorg. Chem, 2015, 54, 7272–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCauley KM, Vrtis JM, Dupont J and van der Donk WA, J. Am. Chem. Soc, 2000, 122, 2403–2404. [Google Scholar]
- 62.Pratt DA, Pesavento RP and van der Donk WA, Org. Lett, 2005, 7, 2735–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemp J, Robinson DE, Ganesan KB, Martinez TJ, Kelleher NL and Gennis RB, Biochemistry, 2006, 45, 15405–15410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ducluzeau A-L, van Lis R, Duval S, Schoepp-Cothenet B, Russell MJ and Nitschke W, Trends in Biochemical Sciences, 2009, 34, 9–15. [DOI] [PubMed] [Google Scholar]
- 65.Denisov IG, Makris TM, Sligar SG and Schlichting I, Chem. Rev, 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- 66.Adam SM, Wijeratne GB, Rogler PJ, Diaz DE, Quist DA, Liu JJ and Karlin KD, Chem. Rev, 2018, 118, 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang X and Groves JT, Chem. Rev, 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costas M, Mehn MP, Jensen MP and Que L, Chem. Rev, 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- 69.Komor AJ, Jasniewski AJ, Que L and Lipscomb JD, Nat. Prod. Rep, 2018, 35, 646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kovaleva EG and Lipscomb JD, Nat Chem Biol, 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Solomon EI, Sundaram UM and Machonkin TE, Chem. Rev, 1996, 96, 2563–2606. [DOI] [PubMed] [Google Scholar]
- 72.Borisov VB, Gennis RB, Hemp J and Verkhovsky MI, Biochim Biophys Acta, 2011, 1807, 1398–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Safarian S, Rajendran C, Müller H, Preu J, Langer JD, Ovchinnikov S, Hirose T, Kusumoto T, Sakamoto J and Michel H, Science, 2016, 352, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Belevich I, Borisov VB, Bloch DA, Konstantinov AA and Verkhovsky MI, Biochemistry, 2007, 46, 11177–11184. [DOI] [PubMed] [Google Scholar]
- 75.Theßeling A, Rasmussen T, Burschel S, Wohlwend D, Kägi J, Möller R, Böttcher B and Friedrich T, Nature Communications, 2019, 10, 5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Belevich I, Borisov VB and Verkhovsky MI, J. Biol. Chem, 2007, 282, 28514–28519. [DOI] [PubMed] [Google Scholar]
- 77.Vanlerberghe G, IJMS, 2013, 14, 6805–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore AL, Shiba T, Young L, Harada S, Kita K and Ito K, Annu. Rev. Plant Biol, 2013, 64, 637–663. [DOI] [PubMed] [Google Scholar]
- 79.Shiba T, Kido Y, Sakamoto K, Inaoka DK, Tsuge C, Tatsumi R, Takahashi G, Balogun EO, Nara T, Aoki T, Honma T, Tanaka A, Inoue M, Matsuoka S, Saimoto H, Moore AL, Harada S and Kita K, Proceedings of the National Academy of Sciences, 2013, 110, 4580–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Young L, Shiba T, Harada S, Kita K, Albury MS and Moore AL, Biochemical Society Transactions, 2013, 41, 1305–1311. [DOI] [PubMed] [Google Scholar]
- 81.Vicente JB, Carrondo MA, Teixeira M and Frazão C, in Encyclopedia of Inorganic and Bioinorganic Chemistry, American Cancer Society, 2011. [Google Scholar]
- 82.Kurtz DM Jr., Dalton Trans, 2007, 4115. [Google Scholar]
- 83.Romão CV, Vicente JB, Borges PT, Frazão C and Teixeira M, J Biol Inorg Chem, 2016, 21, 39–52. [DOI] [PubMed] [Google Scholar]
- 84.Santana-Sanchez A, Solymosi D, Mustila H, Bersanini L, Aro E-M and Allahverdiyeva Y, eLife, 2019, 8, e45766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Folgosa F, Martins MC and Teixeira M, FEMS Microbiology Letters, , DOI: 10.1093/femsle/fnx267. [DOI] [PubMed] [Google Scholar]
- 86.Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S and Sannia G, Cell. Mol. Life Sci, 2010, 67, 369–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Solomon EI, Inorg. Chem, 2016, 55, 6364–6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rich PR, Biochemical Society Transactions, 2017, 45, 813–829. [DOI] [PubMed] [Google Scholar]
- 89.Gibson Q and Greenwood C, Biochemical Journal, 1963, 86, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sucheta A, Szundi I and Einarsdóttir Ó, Biochemistry, 1998, 37, 17905–17914. [DOI] [PubMed] [Google Scholar]
- 91.Einarsdóttir Ó, McDonald W, Funatogawa C, Szundi I, Woodruff WH and Dyer RB, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2015, 1847, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Einarsdóttir Ó, Funatogawa C, Soulimane T and Szundi I, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2012, 1817, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nakashima S, Ogura T and Kitagawa T, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2015, 1847, 86–97. [DOI] [PubMed] [Google Scholar]
- 94.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI and Wikstrom M, Proceedings of the National Academy of Sciences, 2004, 101, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wikström M, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2012, 1817, 468–475. [DOI] [PubMed] [Google Scholar]
- 96.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL and Babcock GT, Science, 2000, 290, 1588–1591. [DOI] [PubMed] [Google Scholar]
- 97.Yu MA, Egawa T, Shinzawa-Itoh K, Yoshikawa S, Guallar V, Yeh S-R, Rousseau DL and Gerfen GJ, J. Am. Chem. Soc, 2012, 134, 4753–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schaefer AW, Roveda AC, Jose A and Solomon EI, J. Am. Chem. Soc, 2019, 141, 10068–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han Du W-G, Götz AW, Yang L, Walker RC and Noodleman L, Phys. Chem. Chem. Phys, 2016, 18, 21162–21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blomberg MRA, Siegbahn PEM and Wikström M, Inorg. Chem, 2003, 42, 5231–5243. [DOI] [PubMed] [Google Scholar]
- 101.Siletsky SA and Konstantinov AA, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2012, 1817, 476–488. [DOI] [PubMed] [Google Scholar]
- 102.Brändén G, Gennis RB and Brzezinski P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2006, 1757, 1052–1063. [DOI] [PubMed] [Google Scholar]
- 103.Kaila VRI, Verkhovsky MI and Wikström M, Chem. Rev, 2010, 110, 7062–7081. [DOI] [PubMed] [Google Scholar]
- 104.Belevich I, Bloch DA, Belevich N, Wikstrom M and Verkhovsky MI, Proceedings of the National Academy of Sciences, 2007, 104, 2685–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blomberg MRA, Journal of Inorganic Biochemistry, 2020, 206, 111020. [DOI] [PubMed] [Google Scholar]
- 106.Sharma V, Karlin KD and Wikström M, Proc Natl Acad Sci U S A, 2013, 110, 16844–16849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Blomberg MRA and Siegbahn PEM, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2015, 1847, 1173–1180. [DOI] [PubMed] [Google Scholar]
- 108.Verkhovsky MI, Tuukkanen A, Backgren C, Puustinen A and Wikström M, Biochemistry, 2001, 40, 7077–7083. [DOI] [PubMed] [Google Scholar]
- 109.Ruitenberg M, Kannt A, Bamberg E, Fendler K and Michel H, Nature, 2002, 417, 99–102. [DOI] [PubMed] [Google Scholar]
- 110.Collman JP, Boulatov R and Sunderland CJ, in The Porphyrin Handbook, eds. Kadish KM, Smith KM and Guilard R, Academic Press, Amsterdam, 2003, pp. 1–49. [Google Scholar]
- 111.Kim E, Chufán EE, Kamaraj K and Karlin KD, Chem. Rev, 2004, 104, 1077–1134. [DOI] [PubMed] [Google Scholar]
- 112.Garcia-Bosch I, Adam SM, Schaefer AW, Sharma SK, Peterson RL, Solomon EI and Karlin KD, J. Am. Chem. Soc, 2015, 137, 1032–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chishiro T, Shimazaki Y, Tani F, Tachi Y, Naruta Y, Karasawa S, Hayami S and Maeda Y, Angew. Chem. Int. Ed, 2003, 42, 2788–2791. [DOI] [PubMed] [Google Scholar]
- 114.Chatterjee S, Sengupta K, Hematian S, Karlin KD and Dey A, J. Am. Chem. Soc, 2015, 137, 12897–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Collman JP, Devaraj NK, Decréau RA, Yang Y, Yan Y-L, Ebina W, Eberspacher TA and Chidsey CED, Science, 2007, 315, 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ladomenou K, Charalambidis G and Coutsolelos AG, Inorganica Chimica Acta, 2010, 363, 2201–2208. [Google Scholar]
- 117.Bhagi-Damodaran A, Michael MA, Zhu Q, Reed J, Sandoval BA, Mirts EN, Chakraborty S, Moënne-Loccoz P, Zhang Y and Lu Y, Nature Chem, 2017, 9, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reed JH, Shi Y, Zhu Q, Chakraborty S, Mirts EN, Petrik ID, Bhagi-Damodaran A, Ross M, Moënne-Loccoz P, Zhang Y and Lu Y, Journal of the American Chemical Society, 2017, 139, 12209–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bhagi-Damodaran A, Kahle M, Shi Y, Zhang Y, Ädelroth P and Lu Y, Angewandte Chemie International Edition, 2017, 56, 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Collman JP, Sunderland CJ, Berg KE, Vance MA and Solomon EI, J. Am. Chem. Soc, 2003, 125, 6648–6649. [DOI] [PubMed] [Google Scholar]
- 121.Collman JP, Decréau RA, Lin H, Hosseini A, Yang Y, Dey A and Eberspacher TA, PNAS, 2009, 106, 7320–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Collman JP, Decreau RA, Dey A and Yang Y, Proceedings of the National Academy of Sciences, 2009, 106, 4101–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Halime Z, Kotani H, Li Y, Fukuzumi S and Karlin KD, PNAS, 2011, 108, 13990–13994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sigman JA, Kim HK, Zhao X, Carey JR and Lu Y, Proceedings of the National Academy of Sciences, 2003, 100, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fry HC, Cohen AD, Toscano JP, Meyer GJ and Karlin KD, J. Am. Chem. Soc, 2005, 127, 6225–6230. [DOI] [PubMed] [Google Scholar]
- 126.Lucas HR, Meyer GJ and Karlin KD, J. Am. Chem. Soc, 2009, 131, 13924–13925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nienhaus K, Olson JS and Nienhaus GU, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 2013, 1834, 1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu C, Zhao X, Lu Y, Rousseau DL and Yeh S-R, J. Am. Chem. Soc, 2010, 132, 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dallacosta C, Alves WA, da AM Ferreira C, Monzani E and Casella L, Dalton Trans., 2007, 2197–2206. [DOI] [PubMed] [Google Scholar]
- 130.Chufán EE, Puiu SC and Karlin KD, Acc. Chem. Res, 2007, 40, 563–572. [DOI] [PubMed] [Google Scholar]
- 131.Collman JP, Berg KE, Sunderland CJ, Aukauloo A, Vance MA and Solomon EI, Inorg. Chem, 2002, 41, 6583–6596. [DOI] [PubMed] [Google Scholar]
- 132.Liu J-G, Naruta Y and Tani F, Angew. Chem. Int. Ed, 2005, 44, 1836–1840. [DOI] [PubMed] [Google Scholar]
- 133.Liu J-G, Naruta Y, Tani F, Chishiro T and Tachi Y, Chem. Commun, 2004, 120–121. [DOI] [PubMed] [Google Scholar]
- 134.Ghiladi RA, Hatwell KR, Karlin KD, Huang H, Moënne-Loccoz P, Krebs C, Huynh BH, Marzilli LA, Cotter RJ, Kaderli S and Zuberbühler AD, J. Am. Chem. Soc, 2001, 123, 6183–6184. [DOI] [PubMed] [Google Scholar]
- 135.Chufán EE, Mondal B, Gandhi T, Kim E, Rubie ND, Moënne-Loccoz P and Karlin KD, Inorg. Chem, 2007, 46, 6382–6394. [DOI] [PubMed] [Google Scholar]
- 136.Kieber-Emmons MT, Qayyum MF, Li Y, Halime Z, Hodgson KO, Hedman B, Karlin KD and Solomon EI, Angew. Chem. Int. Ed, 2012, 51, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Halime Z, Kieber-Emmons MT, Qayyum MF, Mondal B, Gandhi T, Puiu SC, Chufán EE, Sarjeant AAN, Hodgson KO, Hedman B, Solomon EI and Karlin KD, Inorg. Chem, 2010, 49, 3629–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kieber-Emmons MT, Li Y, Halime Z, Karlin KD and Solomon EI, Inorg. Chem, 2011, 50, 11777–11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pirota V, Gennarini F, Dondi D, Monzani E, Casella L and Dell’Acqua S, New J. Chem, 2014, 38, 518–528. [Google Scholar]
- 140.Mandon D, Weiss R, Franke M, Bill E and Trautwein AX, Angew. Chem. Int. Ed. Engl, 1989, 28, 1709–1711. [Google Scholar]
- 141.Collman JP, Decréau RA and Sunderland CJ, Chem. Commun, 2006, 3894–3896. [DOI] [PubMed] [Google Scholar]
- 142.Collman JP, Decréau RA, Yan Y, Yoon J and Solomon EI, J. Am. Chem. Soc, 2007, 129, 5794–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schaefer AW, Kieber-Emmons MT, Adam SM, Karlin KD and Solomon EI, J. Am. Chem. Soc, 2017, 139, 7958–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Adam SM, Garcia-Bosch I, Schaefer AW, Sharma SK, Siegler MA, Solomon EI and Karlin KD, J. Am. Chem. Soc, 2017, 139, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Collman JP, Boulatov R, Sunderland CJ and Fu L, Chem. Rev, 2004, 104, 561–588. [DOI] [PubMed] [Google Scholar]
- 146.Collman JP and Decréau RA, Chem. Commun, 2008, 5065. [DOI] [PubMed] [Google Scholar]
- 147.Sengupta K, Chatterjee S and Dey A, ACS Catal, 2016, 6, 6838–6852. [Google Scholar]
- 148.Collman JP, Inorg. Chem, 1997, 36, 5145–5155. [Google Scholar]
- 149.Collman JP, Fu L, Herrmann PC and Zhang X, Science, 1997, 275, 949–951. [DOI] [PubMed] [Google Scholar]
- 150.Miner KD, Mukherjee A, Gao Y-G, Null EL, Petrik ID, Zhao X, Yeung N, Robinson H and Lu Y, Angew. Chem. Int. Ed, 2012, 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ricard D, Didier A, L’Her M and Boitrel B, ChemBioChem, 2001, 2, 144–148. [DOI] [PubMed] [Google Scholar]
- 152.Kitagishi H, Shimoji D, Ohta T, Kamiya R, Kudo Y, Onoda A, Hayashi T, Weiss J, Wytko JA and Kano K, Chem. Sci, 2018, 9, 1989–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Boulatov R, Collman JP, Shiryaeva IM and Sunderland CJ, J. Am. Chem. Soc, 2002, 124, 11923–11935. [DOI] [PubMed] [Google Scholar]
- 154.Charalambidis G, Ladomenou K, Boitrel B and Coutsolelos AG, European Journal of Organic Chemistry, 2009, 2009, 1263–1268. [Google Scholar]
- 155.Yu Y, Cui C, Liu X, Petrik ID, Wang J and Lu Y, J. Am. Chem. Soc, 2015, 137, 11570–11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Petrik ID, Davydov R, Ross M, Zhao X, Hoffman B and Lu Y, J. Am. Chem. Soc, 2016, 138, 1134–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yu Y, Lv X, Li J, Zhou Q, Cui C, Hosseinzadeh P, Mukherjee A, Nilges MJ, Wang J and Lu Y, J. Am. Chem. Soc, 2015, 137, 4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yu Y, Mukherjee A, Nilges MJ, Hosseinzadeh P, Miner KD and Lu Y, J. Am. Chem. Soc, 2014, 136, 1174–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu X, Yu Y, Hu C, Zhang W, Lu Y and Wang J, Angewandte Chemie International Edition, 2012, 51, 4312–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Collman JP and Boulatov R, Angewandte Chemie International Edition, 2002, 41, 3487–3489. [DOI] [PubMed] [Google Scholar]
- 161.Mukherjee S, Mukherjee A, Bhagi-Damodaran A, Mukherjee M, Lu Y and Dey A, Nature Communications, 2015, 6, 8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I and Karlin KD, Chemical Reviews, 2002, 102, 1201–1234. [DOI] [PubMed] [Google Scholar]
- 163.Zumft WG, Microbiol. Mol. Biol. Rev, 1997, 61, 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tosha T and Shiro Y, in Metallobiology, eds. Moura I, Moura JJG, Pauleta SR and Maia LB, Royal Society of Chemistry, Cambridge, 2016, pp. 114–140. [Google Scholar]
- 165.Ferousi C, Majer SH, DiMucci IM and Lancaster KM, Chem. Rev, 2020, 120, 5252–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S and Shiro Y, Science, 2010, 330, 1666–1670. [DOI] [PubMed] [Google Scholar]
- 167.Matsumoto Y, Tosha T, Pisliakov AV, Hino T, Sugimoto H, Nagano S, Sugita Y and Shiro Y, Nat Struct Mol Biol, 2012, 19, 238–245. [DOI] [PubMed] [Google Scholar]
- 168.Crow A, Matsuda Y, Arata H and Oubrie A, Biochemistry, 2016, 55, 3198–3203. [DOI] [PubMed] [Google Scholar]
- 169.Sato N, Ishii S, Sugimoto H, Hino T, Fukumori Y, Sako Y, Shiro Y and Tosha T, Proteins, 2014, 82, 1258–1271. [DOI] [PubMed] [Google Scholar]
- 170.Gonska N, Young D, Yuki R, Okamoto T, Hisano T, Antonyuk S, Hasnain SS, Muramoto K, Shiro Y, Tosha T and Ädelroth P, Sci Rep, 2018, 8, 3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gopalasingam CC, Johnson RM, Chiduza GN, Tosha T, Yamamoto M, Shiro Y, Antonyuk SV, Muench SP and Hasnain SS, Sci. Adv, 2019, 5, eaax1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jamali MAM, Gopalasingam CC, Johnson RM, Tosha T, Muramoto K, Muench SP, Antonyuk SV, Shiro Y and Hasnain SS, IUCrJ, 2020, 7, 404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Suharti MJF Strampraad I. Schröder and de Vries S, Biochemistry, 2001, 40, 2632–2639. [DOI] [PubMed] [Google Scholar]
- 174.Al-Attar S and de Vries S, FEBS Letters, 2015, 589, 2050–2057. [DOI] [PubMed] [Google Scholar]
- 175.Matsubara T and Iwasaki H, The Journal of Biochemistry, 1971, 69, 859–868. [DOI] [PubMed] [Google Scholar]
- 176.Heiss B, Frunzke K and Zumft WG, Journal of Bacteriology, 1989, 171, 3288–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Carr GJ and Ferguson SJ, Biochemical Journal, 1990, 269, 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Turk T and Hollocher TC, Biochemical and Biophysical Research Communications, 1992, 183, 983–988. [DOI] [PubMed] [Google Scholar]
- 179.Lee D-H, Mondal B and Karlin KD, in Activation of Small Molecules, ed. Tolman WB, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, pp. 43–79. [Google Scholar]
- 180.Hendriks J, Warne A, Gohlke U, Haltia T, Lüdovici C, Lübben M and Saraste M, Biochemistry, 1998, 37, 13102–13109. [DOI] [PubMed] [Google Scholar]
- 181.Girsch P and de Vries S, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1997, 1318, 202–216. [DOI] [PubMed] [Google Scholar]
- 182.Saraste M and Castresana J, FEBS Letters, 1994, 341, 1–4. [DOI] [PubMed] [Google Scholar]
- 183.Kannt A, Michel H, Cheesman MR, Thomson AJ, Dreusch AB, Körner H and Zumft WG, in Biological Electron Transfer Chains: Genetics, Composition and Mode of Operation, eds. Canters GW and Vijgenboom E, Springer Netherlands, Dordrecht, 1998, pp. 279–291. [Google Scholar]
- 184.Watmough NJ, Butland G, Cheesman MR, Moir JWB, Richardson DJ and Spiro S, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1999, 1411, 456–474. [DOI] [PubMed] [Google Scholar]
- 185.Butland G, Spiro S, Watmough NJ and Richardson DJ, J. Bacteriol, 2001, 183, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grönberg KLC, Roldán MD, Prior L, Butland G, Cheesman MR, Richardson DJ, Spiro S, Thomson AJ and Watmough NJ, Biochemistry, 1999, 38, 13780–13786. [DOI] [PubMed] [Google Scholar]
- 187.Pisliakov AV, Hino T, Shiro Y and Sugita Y, PLoS Comput Biol, 2012, 8, e1002674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shiro Y, Sugimoto H, Tosha T, Nagano S and Hino T, Philos. Trans. R. Soc., B, 2012, 367, 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Moënne-Loccoz P, Nat. Prod. Rep, 2007, 24, 610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lu S, Suharti S de Vries and Moënne-Loccoz P, J. Am. Chem. Soc, 2004, 126, 15332–15333. [DOI] [PubMed] [Google Scholar]
- 191.Zumft W, Journal of Inorganic Biochemistry, 2005, 99, 194–215. [DOI] [PubMed] [Google Scholar]
- 192.Lehnert N, Berto TC, Galinato MGI and Goodrich LE, in Handbook of Porphyrin Science (Volume 14), World Scientific Publishing Company, 2011, vol. 15, pp. 1–247. [Google Scholar]
- 193.Khatua S and Majumdar A, Journal of Inorganic Biochemistry, 2015, 142, 145–153. [DOI] [PubMed] [Google Scholar]
- 194.Berto TC, Speelman AL, Zheng S and Lehnert N, Coordination Chemistry Reviews, 2013, 257, 244–259. [Google Scholar]
- 195.Shoun H, Fushinobu S, Jiang L, Kim S-W and Wakagi T, Phil. Trans. R. Soc. B, 2012, 367, 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shoun H and Takaya N, International Congress Series, 2002, 1233, 89–97. [Google Scholar]
- 197.Bailey S, Cooper SJ, Hagen W, Arendsen A and Lindley PF, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. Scott RA, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. eibc0569. [Google Scholar]
- 198.Wang J, Vine CE, Balasiny BK, Rizk J, Bradley CL, Tinajero-Trejo M, Poole RK, Bergaust LL, Bakken LR and Cole JA, Molecular Microbiology, 2016, 100, 877–892. [DOI] [PubMed] [Google Scholar]
- 199.Forte E, Urbani A, Saraste M, Sarti P, Brunori M and Giuffrè A, European Journal of Biochemistry, 2001, 268, 6486–6491. [DOI] [PubMed] [Google Scholar]
- 200.Huang Y, Reimann J, Lepp H, Drici N and Adelroth P, Proceedings of the National Academy of Sciences, 2008, 105, 20257–20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Maia LB and Moura JJG, Chem. Rev, 2014, 114, 5273–5357. [DOI] [PubMed] [Google Scholar]
- 202.Kurtz DM, Boice E, Caranto JD, Frederick RE, Masitas CA and Miner KD, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. Scott RA, John Wiley & Sons, Ltd, Chichester, UK, 2004, pp. 1–18. [Google Scholar]
- 203.Weitz AC, Giri N, Frederick RE, Kurtz DM, Bominaar EL and Hendrich MP, ACS Catal, 2018, 8, 11704–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Blomberg LM, Blomberg MRA and Siegbahn PEM, J Biol Inorg Chem, 2006, 12, 79–89. [DOI] [PubMed] [Google Scholar]
- 205.Van Stappen C and Lehnert N, Inorg. Chem, 2018, 57, 4252–4269. [DOI] [PubMed] [Google Scholar]
- 206.Jana M, White CJ, Pal N, Demeshko S, Cordes C (Kupper née), Meyer F, Lehnert N and Majumdar A, J. Am. Chem. Soc, 2020, 142, 6600–6616. [DOI] [PubMed] [Google Scholar]
- 207.Dong HT, White CJ, Zhang B, Krebs C and Lehnert N, J. Am. Chem. Soc, 2018, 140, 13429–13440. [DOI] [PubMed] [Google Scholar]
- 208.Maeda K, Spor A, Edel-Hermann V, Heraud C, Breuil M-C, Bizouard F, Toyoda S, Yoshida N, Steinberg C and Philippot L, Sci Rep, 2015, 5, 9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.McLean KJ, Luciakova D, Belcher J, Tee KL and Munro AW, in Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450, eds. Hrycay EG and Bandiera SM, Springer International Publishing, Cham, 2015, vol. 851, pp. 299–317. [Google Scholar]
- 210.Park S-Y, Shimizu H, Adachi S, Nakagawa A, Tanaka I, Nakahara K, Shoun H, Obayashi E, Nakamura H, lizuka T and Shiro Y, Nat Struct Mol Biol, 1997, 4, 827–832. [DOI] [PubMed] [Google Scholar]
- 211.Oshima R, Fushinobu S, Su F, Zhang L, Takaya N and Shoun H, Journal of Molecular Biology, 2004, 342, 207–217. [DOI] [PubMed] [Google Scholar]
- 212.Lehnert N, Praneeth VKK and Paulat F, J. Comput. Chem, 2006, 27, 1338–1351. [DOI] [PubMed] [Google Scholar]
- 213.Krámos B, Menyhárd DK and Oláh J, J. Phys. Chem. B, 2012, 116, 872–885. [DOI] [PubMed] [Google Scholar]
- 214.Riplinger C, Bill E, Daiber A, Ullrich V, Shoun H and Neese F, Chem. Eur. J, 2014, 20, 1602–1614. [DOI] [PubMed] [Google Scholar]
- 215.Riplinger C and Neese F, ChemPhysChem, 2011, 12, 3192–3203. [DOI] [PubMed] [Google Scholar]
- 216.Hunt AP and Lehnert N, Inorg. Chem, 2019, 58, 11317–11332. [DOI] [PubMed] [Google Scholar]
- 217.McQuarters AB, Blaesi EJ, Kampf JW, Alp EE, Zhao J, Hu M, Krebs C and Lehnert N, Inorg. Chem, 2019, 58, 1398–1413. [DOI] [PubMed] [Google Scholar]
- 218.Pierik AJ, Wolbert RBG, Mutsaers PHA, Hagen WR and Veeger C, Eur J Biochem, 1992, 206, 697–704. [DOI] [PubMed] [Google Scholar]
- 219.Macedo S, Mitchell EP, Romão CV, Cooper SJ, Coelho R, Liu MY, Xavier AV, LeGall J, Bailey S, Garner DC, Hagen WR, Teixeira M, Carrondo MA and Lindley P, J Biol Inorg Chem, 2002, 7, 514–525. [DOI] [PubMed] [Google Scholar]
- 220.Aragão D, Mitchell EP, Frazão CF, Carrondo MA and Lindley PF, Acta Crystallogr D Biol Crystallogr, 2008, 64, 665–674. [DOI] [PubMed] [Google Scholar]
- 221.Hagen WR, FEBS Lett, 2019, 593, 3075–3083. [DOI] [PubMed] [Google Scholar]
- 222.Wolfe MT, Heo J, Garavelli JS and Ludden PW, JB, 2002, 184, 5898–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Almeida CC, Romão CV, Lindley PF, Teixeira M and Saraiva LM, J. Biol. Chem, 2006, 281, 32445–32450. [DOI] [PubMed] [Google Scholar]
- 224.Seth D, Hess DT, Hausladen A, Wang L, Wang Y and Stamler JS, Molecular Cell, 2018, 69, 451–464.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Duarte AG, Cordas CM, Moura JJG and Moura I, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2014, 1837, 375–384. [DOI] [PubMed] [Google Scholar]
- 226.Blomberg MRA and Siegbahn PEM, Biochemistry, 2012, 51, 5173–5186. [DOI] [PubMed] [Google Scholar]
- 227.Koutný M and Kučera I, Biochemical and Biophysical Research Communications, 1999, 262, 562–564. [DOI] [PubMed] [Google Scholar]
- 228.Enemark JH and Feltham RD, Coordination Chemistry Reviews, 1974, 13, 339–406. [Google Scholar]
- 229.Lehnert N, Scheldt WR and Wolf MW, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine II, ed. Mingos DMP, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, vol. 154, pp. 155–223. [Google Scholar]
- 230.Lorković IM and Ford PC, Inorg. Chem, 2000, 39, 632–633. [DOI] [PubMed] [Google Scholar]
- 231.Berto TC, Praneeth VKK, Goodrich LE and Lehnert N, J. Am. Chem. Soc, 2009, 131, 17116–17126. [DOI] [PubMed] [Google Scholar]
- 232.Abucayon EG, Khade RL, Powell DR, Zhang Y and Richter-Addo GB, J. Am. Chem. Soc, 2018, 140, 4204–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Collman JP, Yang Y, Dey A, Decreau RA, Ghosh S, Ohta T and Solomon EI, Proceedings of the National Academy of Sciences, 2008, 105, 15660–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Collman JP, Dey A, Yang Y, Decréau RA, Ohta T and Solomon EI, J. Am. Chem. Soc, 2008, 130, 16498–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Speelman AL, Zhang B, Silakov A, Skodje KM, Alp EE, Zhao J, Hu MY, Kim E, Krebs C and Lehnert N, Inorg. Chem, 2016, 55, 5485–5501. [DOI] [PubMed] [Google Scholar]
- 236.Grönberg KLC, Watmough NJ, Thomson AJ, Richardson DJ and Field SJ,!. Biol. Chem, 2004, 279, 17120–17125. [DOI] [PubMed] [Google Scholar]
- 237.Kato M, Nakagawa S, Tosha T, Shiro Y, Masuda Y, Nakata K and Yagi I, The Journal of Physical Chemistry Letters, 2018, 9, 5196–5200. [DOI] [PubMed] [Google Scholar]
- 238.Field SJ, Roldan MD, Marritt SJ, Butt JN, Richardson DJ and Watmough NJ, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2011, 1807, 451–457. [DOI] [PubMed] [Google Scholar]
- 239.Cordas CM, Duarte AG, Moura JJG and Moura I, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2013, 1827, 233–238. [DOI] [PubMed] [Google Scholar]
- 240.Timóteo CG, Pereira AS, Martins CE, Naik SG, Duarte AG, Moura JJG, Tavares P, Huynh BH and Moura I, Biochemistry, 2011, 50, 4251–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Blomberg MRA and Ädelroth P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2018, 1859, 1223–1234. [DOI] [PubMed] [Google Scholar]
- 242.Blomberg MRA, Biochemistry, 2017, 56, 120–131. [DOI] [PubMed] [Google Scholar]
- 243.Costa C, Macedo A, Moura I, Moura JJG, Le Gall J, Berlier Y, Liu M-Y and Payne WJ, FEBS Letters, 1990, 276, 67–70. [DOI] [PubMed] [Google Scholar]
- 244.Kahle M, Blomberg MRA, Jareck S and Ädelroth P, FEBS Lett, 2019, 593, 1351–1359. [DOI] [PubMed] [Google Scholar]
- 245.Hendriks JHM, Jasaitis A, Saraste M and Verkhovsky MI, Biochemistry, 2002, 41, 2331–2340. [DOI] [PubMed] [Google Scholar]
- 246.Takeda H, Kimura T, Nomura T, Horitani M, Yokota A, Matsubayashi A, Ishii S, Shiro Y, Kubo M and Tosha T, Bull. Chem. Soc. Jpn, 2020, 93, 825–833. [Google Scholar]
- 247.Shoji M, Hanaoka K, Kondo D, Sato A, Umeda H, Katsumasa K and Shiraishi K, Mol. Phys, 2014, 112, 393–397. [Google Scholar]
- 248.Attia AAA and Silaghi-Dumitrescu R, J. Mol. Model, 2015, 21, 130. [DOI] [PubMed] [Google Scholar]
- 249.Blomberg MRA and Siegbahn PEM, J. Comput. Chem, 2016, 37, 1810–1818. [DOI] [PubMed] [Google Scholar]
- 250.Daskalakis V, Ohta T, Kitagawa T and Varotsis C, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2015, 1847, 1240–1244. [DOI] [PubMed] [Google Scholar]
- 251.Kumita H, Matsuura K, Hino T, Takahashi S, Hori H, Fukumori Y, Morishima I and Shiro Y, J. Biol. Chem, 2004, 279, 55247–55254. [DOI] [PubMed] [Google Scholar]
- 252.Lachmann P, Huang Y, Reimann J, Flock U and Ädelroth P, J. Biol. Chem, 2010, 285, 25531–25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sakurai N and Sakurai T, Biochemistry, 1997, 36, 13809–13815. [DOI] [PubMed] [Google Scholar]
- 254.Decréau RA, in Handbook of Porphyrin Science (Volume 22), World Scientific Publishing Company, 2012, vol. 25, pp. 235–305. [Google Scholar]
- 255.Harrop TC, in Advances in Inorganic Chemistry, Elsevier, 2015, vol. 67, pp. 243–263. [Google Scholar]
- 256.Arikawa Y and Onishi M, Coordination Chemistry Reviews, 2012, 256, 468–478. [Google Scholar]
- 257.Ju TD, Woods AS, Cotter RJ, Moënne-Loccoz P and Karlin KD, Inorganica Chimica Acta, 2000, 297, 362–372. [Google Scholar]
- 258.Zhao X, Yeung N, Russell BS, Garner DK and Lu Y, J. Am. Chem. Soc, 2006, 128, 6766–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yeung N, Lin Y-W, Gao Y-G, Zhao X, Russell BS, Lei L, Miner KD, Robinson H and Lu Y, Nature, 2009, 462, 1079–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chakraborty S, Reed J, Sage JT, Branagan NC, Petrik ID, Miner KD, Hu MY, Zhao J, Alp EE and Lu Y, Inorg. Chem, 2015, 54, 9317–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Matsumura H, Hayashi T, Chakraborty S, Lu Y and Moënne-Loccoz P, J. Am. Chem. Soc, 2014, 136, 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sabuncu S, Reed JH, Lu Y and Moënne-Loccoz P, J. Am. Chem. Soc, 2018, 140, 17389–17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Chakraborty S, Reed J, Ross M, Nilges MJ, Petrik ID, Ghosh S, Hammes-Schiffer S, Sage JT, Zhang Y, Schulz CE and Lu Y, Angew. Chem. Int. Ed, 2014, 53, 2417–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lin Y-W, Yeung N, Gao Y-G, Miner KD, Tian S, Robinson H and Lu Y, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 8581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hayashi T, Miner KD, Yeung N, Lin Y-W, Lu Y and Moënne-Loccoz P, Biochemistry, 2011, 50, 5939–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Abucayon EG, Khade RL, Powell DR, Zhang Y and Richter-Addo GB, Angew. Chem. Int. Ed, 2019, 58, 18598–18603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Matsumura H, Chakraborty S, Reed J, Lu Y and Moënne-Loccoz P, Biochemistry, 2016, 55, 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wasser IM, Huang H, Moënne-Loccoz P and Karlin KD, J. Am. Chem. Soc, 2005, 127, 3310–3320. [DOI] [PubMed] [Google Scholar]
- 269.Berto TC, Hoffman MB, Murata Y, Landenberger KB, Alp EE, Zhao J and Lehnert N, J. Am. Chem. Soc, 2011, 133, 16714–16717. [DOI] [PubMed] [Google Scholar]
- 270.Bhagi-Damodaran A, Reed JH, Zhu Q, Shi Y, Hosseinzadeh P, Sandoval BA, Harnden KA, Wang S, Sponholtz MR, Mirts EN, Dwaraknath S, Zhang Y, Moënne-Loccoz P and Lu Y, Proc. Natl. Acad. Sci, 2018, 115, 6195–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wright AM and Hayton TW, Inorg. Chem, 2015, 54, 9330–9341. [DOI] [PubMed] [Google Scholar]
- 272.Arikawa Y, Asayama T, Moriguchi Y, Agari S and Onishi M, J. Am. Chem. Soc, 2007, 129, 14160–14161. [DOI] [PubMed] [Google Scholar]
- 273.Suzuki T, Tanaka H, Shiota Y, Sajith PK, Arikawa Y and Yoshizawa K, Inorg. Chem, 2015, 54, 7181–7191. [DOI] [PubMed] [Google Scholar]
- 274.Wang J, Schopfer MP, Sarjeant AAN and Karlin KD, J. Am. Chem. Soc, 2009, 131, 450–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wang J, Schopfer MP, Puiu SC, Sarjeant AAN and Karlin KD, Inorg. Chem, 2010, 49, 1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Xu N, Campbell ALO, Powell DR, Khandogin J and Richter-Addo GB, J. Am. Chem. Soc, 2009, 131, 2460–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Berto TC, Xu N, Lee SR, McNeil AJ, Alp EE, Zhao J, Richter-Addo GB and Lehnert N, Inorg. Chem, 2014, 53, 6398–6414. [DOI] [PubMed] [Google Scholar]
- 278.Xu N, Abucayon EG, Powell DR and Richter-Addo GB, Nitric Oxide, 2016, 52, 16–20. [DOI] [PubMed] [Google Scholar]
- 279.Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD and Pereira IAC, in Advances in Microbial Physiology, Elsevier, 2015, vol. 66, pp. 55–321. [DOI] [PubMed] [Google Scholar]
- 280.Duarte AG, Catarino T, White GF, Lousa D, Neukirchen S, Soares CM, Sousa FL, Clarke TA and Pereira IAC, Nat Commun, 2018, 9, 5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Barton L, Sulfate-Reducing Bacteria, Springer Science & Business Media, 1995. [Google Scholar]
- 282.McRee DE, Richardson DC, Richardson JS and Siegel LM, J. Biol. Chem, 1986, 261, 10277–10281. [PubMed] [Google Scholar]
- 283.Crane BR, Siegel LM and Getzoff ED, Science, 1995, 270, 59–67. [DOI] [PubMed] [Google Scholar]
- 284.Kern M, Klotz MG and Simon J, Molecular Microbiology, 2011, 82, 1515–1530. [DOI] [PubMed] [Google Scholar]
- 285.Hermann B, Kern M, La Pietra L, Simon J and Einsle O, Nature, 2015, 520, 706–709. [DOI] [PubMed] [Google Scholar]
- 286.Kredich NM, EcoSal Plus, , DOI: 10.1128/ecosalplus.3.6.1.11. [DOI] [PubMed] [Google Scholar]
- 287.Siegel LM, Murphy MJ and Kamin H, J. Biol. Chem, 1973, 248, 251–264. [PubMed] [Google Scholar]
- 288.Siegel LM, Davis PS and Kamin H, J. Biol. Chem, 1974, 249, 1572–1586. [PubMed] [Google Scholar]
- 289.Krueger RJ and Siegel LM, Biochemistry, 1982, 21, 2892–2904. [DOI] [PubMed] [Google Scholar]
- 290.Crane BR and Getzoff ED, Current Opinion in Structural Biology, 1996, 6, 744–756. [DOI] [PubMed] [Google Scholar]
- 291.Nakayama M, Akashi T and Hase T, Journal of Inorganic Biochemistry, 2000, 82, 27–32. [DOI] [PubMed] [Google Scholar]
- 292.Lee J-P, LeGall J and Peck HD, Journal of Bacteriology, 1973, 115, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Tan J and Cowan JA, Biochemistry, 1991, 30, 8910–7. [DOI] [PubMed] [Google Scholar]
- 294.Findley JE and Akagi JM, Biochemical and Biophysical Research Communications, 1969, 36, 266–271. [DOI] [PubMed] [Google Scholar]
- 295.Lee JP and Peck HD, Biochemical and Biophysical Research Communications, 1971, 45, 583–589. [DOI] [PubMed] [Google Scholar]
- 296.Drake HL and Akagi JM, Journal of Bacteriology, 1978, 136, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wolfe BM, Lui SM and Cowan JA, European Journal of Biochemistry, 1994, 223, 79–89. [DOI] [PubMed] [Google Scholar]
- 298.Trudinger PA, Journal of Bacteriology, 1970, 104, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Akagi JM and Adams V, Journal of Bacteriology, 1973, 116, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Dahl C, Kredich NM, Deutzmann R and Trlfper HG, Microbiology, 1993, 139, 1817–1828. [DOI] [PubMed] [Google Scholar]
- 301.Schiffer A, Parey K, Warkentin E, Diederichs K, Huber H, Stetter KO, Kroneck PMH and Ermler U, Journal of Molecular Biology, 2008, 379, 1063–1074. [DOI] [PubMed] [Google Scholar]
- 302.Peck HD, LeGall J, Van Beeumen J, Postgate JR and Kelly DP, Philosophical Transactions of the Royal Society of London. B, Biological Sciences, 1982, 298, 443–466. [DOI] [PubMed] [Google Scholar]
- 303.Moura I, LeGall J, Lino AR, Peck HD, Fauque G, Xavier AV, DerVartanian DV, Moura JJG and Huynh BH, J. Am. Chem. Soc, 1988, 110, 1075–1082. [Google Scholar]
- 304.Hatchikian EC and Zeikus JG, J. Bacteriol, 1983, 153, 1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Fauque G, Lino AR, Czechowski M, Kang L, DerVartanian DV, Moura JJG, LeGall J and Moura I, Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, 1990, 1040, 112–118. [DOI] [PubMed] [Google Scholar]
- 306.Claude Hatchikian E, in Methods in Enzymology, Academic Press, 1994, vol. 243, pp. 276–295. [DOI] [PubMed] [Google Scholar]
- 307.Marritt SJ and Hagen WR, European Journal of Biochemistry, 1996, 238, 724–727. [DOI] [PubMed] [Google Scholar]
- 308.Haschke RH and Campbell LL, Journal of Bacteriology, 1971, 106, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Hatchikian EC, Arch. Microbiol, 1975, 105, 249–256. [DOI] [PubMed] [Google Scholar]
- 310.Chambers LA and Trudinger PA, J Bacteriol, 1975, 123, 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Oliveira TF, Vonrhein C, Matias PM, Venceslau SS, Pereira IAC and Archer M, J. Biol. Chem, 2008, 283, 34141–34149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Hsieh Y-C, Liu M-Y, Wang VC-C, Chiang Y-L, Liu E-H, Wu W, Chan SI and Chen C-J, Molecular Microbiology, 2010, 78, 1101–1116. [DOI] [PubMed] [Google Scholar]
- 313.Oliveira TF, Franklin E, Afonso JP, Khan AR, Oldham NJ, Pereira IAC and Archer M, Front. Microbiol, , DOI: 10.3389/fmicb.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Fitz RM and Cypionka H, Arch. Microbiol, 1990, 154, 400–406. [Google Scholar]
- 315.Steuber J, Cypionka H and Kroneck PMH, Archives of microbiology, 1994, 162, 255–260. [Google Scholar]
- 316.Steuber J, Arendsen AF, Hagen WR and Kroneck PM, European Journal of Biochemistry, 1995, 233, 873–879. [DOI] [PubMed] [Google Scholar]
- 317.Trüper HG, in Methods in Enzymology, Academic Press, 1994, vol. 243, pp. 422–426. [Google Scholar]
- 318.Siegel LM, Rueger DC, Barber MJ, Krueger RJ, Orme-Johnson NR and Orme-Johnson WH, Journal of Biological Chemistry, 1982, 257, 6343–6350. [PubMed] [Google Scholar]
- 319.Lui SM and Cowan JA, J. Am. Chem. Soc, 1994, 116, 11538–11549. [Google Scholar]
- 320.Kemp JD, Atkinson DE, Ehret A and Lazzarini RA, J. Biol. Chem, 1963, 238, 3466–3471. [PubMed] [Google Scholar]
- 321.Naiki N, Plant Cell Physiol, 1965, 6, 179–194. [Google Scholar]
- 322.Yoshimoto A and Sato R, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1968, 153, 576–588. [DOI] [PubMed] [Google Scholar]
- 323.Murphy MJ, Siegel LM, Kamin H and Rosenthal D, J. Biol. Chem, 1973, 248, 2801–2814. [PubMed] [Google Scholar]
- 324.Murphy MJ and Siegel LM, Journal of Biological Chemistry, 1973, 248, 6911–6919. [PubMed] [Google Scholar]
- 325.Murphy MJ, Siegel LM, Tove SR and Kamin H, Proceedings of the National Academy of Sciences, 1974, 71, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Tripathy BC, Sherameti I and Oelmüller R, Plant Signaling & Behavior, 2010, 5, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Yasuhide S, Sogawa N and Ishimoto M, Journal of biochemistry, 1981, 90, 1487–1492. [DOI] [PubMed] [Google Scholar]
- 328.Soriano A and Cowan JA, Journal of the American Chemical Society, 1995, 117, 4724–4725. [Google Scholar]
- 329.Christner JA, Münck E, Janick PA and Siegel LM, J. Biol. Chem, 1981, 256, 2098–2101. [PubMed] [Google Scholar]
- 330.Christner JA, Münck E, Janick PA and Siegel LM, Journal of Biological Chemistry, 1983, 258, 11147–11156. [PubMed] [Google Scholar]
- 331.Janick PA and Siegel LM, Biochemistry, 1982, 21, 3538–3547. [DOI] [PubMed] [Google Scholar]
- 332.Huynh BH, Kang L, DerVartanian DV, Peck HD and LeGall J, Journal of Biological Chemistry, 1984, 259, 15373–15376. [PubMed] [Google Scholar]
- 333.Janick PA and Siegel LM, Biochemistry, 1983, 22, 504–515. [DOI] [PubMed] [Google Scholar]
- 334.Christner JA, Janick PA, Siegel LM and Münck E, Journal of Biological Chemistry, 1983, 258, 11157–11164. [PubMed] [Google Scholar]
- 335.Christner JA, Muenck E, Kent TA, Janick PA, Salerno JC and Siegel LM, Journal of the American Chemical Society, 1984, 106, 6786–6794. [Google Scholar]
- 336.Cline JF, Janick PA, Siegel LM and Hoffman BM, Biochemistry, 1985, 24, 7942–7947. [DOI] [PubMed] [Google Scholar]
- 337.Ostrowski J, Wu J-Y, Rueger DC, Miller BE, Siegel LM and Kredich NM, Journal of Biological Chemistry, 1989, 264, 15726–15737. [PubMed] [Google Scholar]
- 338.Pierik AJ and Hagen WR, European Journal of Biochemistry, 1991, 195, 505–516. [DOI] [PubMed] [Google Scholar]
- 339.Sekine K, Moriyama T, Kim J, Hase T and Sato N, J Biochem, 2017, mvw103. [DOI] [PubMed] [Google Scholar]
- 340.Sekine K, Sakakibara Y, Hase T and Sato N, Biochemical Journal, 2009, 423, 91–98. [DOI] [PubMed] [Google Scholar]
- 341.Nakano S, Takahashi M, Sakamoto A, Morikawa H and Katayanagi K, Chemistry & Biodiversity, 2012, 9, 1989–1999. [DOI] [PubMed] [Google Scholar]
- 342.Schnell R, Sandalova T, Hellman U, Lindqvist Y and Schneider G, J. Biol. Chem, 2005, 280, 27319–27328. [DOI] [PubMed] [Google Scholar]
- 343.Rokhsana D, Howells AE, Dooley DM and Szilagyi RK, Inorg. Chem, 2012, 51, 3513–3524. [DOI] [PubMed] [Google Scholar]
- 344.Swamy U, Wang M, Tripathy JN, Kim S-K, Hirasawa M, Knaff DB and Allen JP, Biochemistry, 2005, 44, 16054–16063. [DOI] [PubMed] [Google Scholar]
- 345.Klotz MG, Schmid MC, Strous M, Camp HJMOD, Jetten MSM and Hooper AB, Environmental Microbiology, 2008, 10, 3150–3163. [DOI] [PubMed] [Google Scholar]
- 346.Lukat P, Rudolf M, Stach P, Messerschmidt A, Kroneck PM, Simon J and Einsle O, Biochemistry, 2008, 47, 2080–2086. [DOI] [PubMed] [Google Scholar]
- 347.Silaghi-Dumitrescu R and Makarov SV, Int. J. Quantum Chem, 2012, 112, 900–908. [Google Scholar]
- 348.Surducan M, Brânzanic AMV and Silaghi-Dumitrescu R, Int J Quantum Chem, 2018, 118, e25697. [Google Scholar]
- 349.Richardson PF, Chang CK, Spaulding LD and Fajer J, Journal of the American Chemical Society, 1979, 101, 7736–7738. [Google Scholar]
- 350.Chang CK and Fajer J, Journal of the American Chemical Society, 1980, 102, 848–851. [Google Scholar]
- 351.Chang CK, Hanson LK, Richardson PF, Young R and Fajer J, Proceedings of the National Academy of Sciences, 1981, 78, 2652–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Brânzanic AMV, Ryde U and Silaghi-Dumitrescu R, Journal of Inorganic Biochemistry, 2020, 203, 110928. [DOI] [PubMed] [Google Scholar]
- 353.Crane BR, Siegel LM and Getzoff ED, Biochemistry, 1997, 36, 12101–12119. [DOI] [PubMed] [Google Scholar]
- 354.Crane BR, Siegel LM and Getzoff ED, Biochemistry, 1997, 36, 12120–12137. [DOI] [PubMed] [Google Scholar]
- 355.Smith KW and Stroupe ME, Biochemistry, 2012, 51, 9857–9868. [DOI] [PubMed] [Google Scholar]
- 356.Santos AA, Venceslau SS, Grein F, Leavitt WD, Dahl C, Johnston DT and Pereira IAC, Science, 2015, 350, 1541–1545. [DOI] [PubMed] [Google Scholar]
- 357.Dahl C, Engels S, Pott-Sperling AS, Schulte A, Sander J, Lübbe Y, Deuster O and Brune DC, Journal of Bacteriology, 2005, 187, 1392–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Venceslau SS, Cort JR, Baker ES, Chu RK, Robinson EW, Dahl C, Saraiva LM and Pereira IAC, Biochemical and Biophysical Research Communications, 2013, 441, 732–736. [DOI] [PubMed] [Google Scholar]
- 359.Parey K, Schiffer A, Steuber J, Fritz G, Ermler U and Kroneck PM, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. Scott RA, John Wiley & Sons, Ltd, Chichester, UK, 2011, p. eibc0678. [Google Scholar]
- 360.Han S, Madden JF, Siegel LM and Spiro TG, Biochemistry, 1989, 28, 5477–5485. [DOI] [PubMed] [Google Scholar]
- 361.Cepeda MR, McGarry L, Pennington JM, Krzystek J and Stroupe ME, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 2018, 1866, 933–940. [DOI] [PubMed] [Google Scholar]
- 362.Brânzanic AMV, Ryde U and Silaghi-Dumitrescu R, Chem. Commun, 2019, 55, 14047–14049. [DOI] [PubMed] [Google Scholar]
- 363.Cai L and Holm RH, Journal of the American Chemical Society, 1994, 116, 7177–7188. [Google Scholar]
- 364.Zhou C, Cai L and Holm RH, Inorganic Chemistry, 1996, 35, 2767–2772. [DOI] [PubMed] [Google Scholar]
- 365.Gerlach DL, Coucouvanis D and Lehnert N, Eur. J. Inorg. Chem, 2013, 2013, 3883–3890. [Google Scholar]
- 366.Fujita E and Fajer J, Journal of the American Chemical Society, 1983, 105, 6743–6745. [Google Scholar]
- 367.McQuarters AB, Kampf JW, Alp EE, Hu M, Zhao J and Lehnert N, Inorg. Chem, 2017, 56, 10513–10528. [DOI] [PubMed] [Google Scholar]
- 368.Kline MA, Barley MH and Meyer TJ, Inorg. Chem, 1987, 26, 2196–2197. [Google Scholar]
- 369.Abdelatty R, Md. Rahman H and Ryan MD, Electroanalysis, 2017, 29, 2437–2443. [Google Scholar]
- 370.Zeghouf M, Fontecave M and Covès J, J. Biol. Chem, 2000, 275, 37651–37656. [DOI] [PubMed] [Google Scholar]
- 371.Gruez A, Pignol D, Zeghouf M, Covès J, Fontecave M, Ferrer J-L and Fontecilla-Camps JC, Journal of Molecular Biology, 2000, 299, 199–212. [DOI] [PubMed] [Google Scholar]
- 372.Mirts EN, Petrik ID, Hosseinzadeh P, Nilges MJ and Lu Y, Science, 2018, 361, 1098–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Sigman JA, Pond AE, Dawson JH and Lu Y, Biochemistry, 1999, 38, 11122–11129. [DOI] [PubMed] [Google Scholar]
- 374.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR and Seefeldt LC, Chem. Rev, 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Giuffre A, Stubauer G, Sarti P, Brunori M, Zumft WG, Buse G and Soulimane T, Proceedings of the National Academy of Sciences, 1999, 96, 14718–14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Butler CS, Forte E, Maria Scandurra F, Arese M, Giuffré A, Greenwood C and Sarti P, Biochemical and Biophysical Research Communications, 2002, 296, 1272–1278. [DOI] [PubMed] [Google Scholar]
- 377.Flock U, Watmough NJ and Ädelroth P, Biochemistry, 2005, 44, 10711–10719. [DOI] [PubMed] [Google Scholar]
- 378.Fujiwara T and Fukumori Y, Journal of bacteriology, 1996, 178, 1866–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Collman JP, Dey A, Yang Y, Ghosh S and Decréau RA, PNAS, 2009, 106, 10528–10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Fee JA, Choc MG, Findling KL, Lorence R and Yoshida T, Proceedings of the National Academy of Sciences, 1980, 77, 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kita K, Konishi K and Anraku Y, J. Biol. Chem, 1984, 259, 3368–3374. [PubMed] [Google Scholar]
- 382.Ramos S, Almeida RM, Cordas CM, Moura JJG, Pauleta SR and Moura I, Journal of Inorganic Biochemistry, 2017, 177, 402–411. [DOI] [PubMed] [Google Scholar]
- 383.Hendler RW, Sidhu GS and Pardhasaradhi K, Biophysical Journal, 1990, 58, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Melin F, Xie H, Meyer T, Ahn YO, Gennis RB, Michel H and Hellwig P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2016, 1857, 1892–1899. [DOI] [PubMed] [Google Scholar]
- 385.Schultz BE and Chan SI, Proceedings of the National Academy of Sciences, 1998, 95, 11643–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Collman JP, Yan Y-L, Lei J and Dinolfo PH, Inorg. Chem, 2006, 45, 7581–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Borutaité V and Brown GC, Biochemical Journal, 1996, 315, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Stubauer G, Giuffrè A, Brunori M and Sarti P, Biochemical and Biophysical Research Communications, 1998, 245, 459–465. [DOI] [PubMed] [Google Scholar]
- 389.Todorovic S, Pereira MM, Bandeiras TM, Teixeira M, Hildebrandt P and Murgida DH, J. Am. Chem. Soc, 2005, 127, 13561–13566. [DOI] [PubMed] [Google Scholar]
- 390.Muntyan MS and Bloch DA, Biochemistry Moscow, 2008, 73, 107–111. [DOI] [PubMed] [Google Scholar]
- 391.Veríssimo AF, Sousa FL, Baptista AM, Teixeira M and Pereira MM, Biochemistry, 2007, 46, 13245–13253. [DOI] [PubMed] [Google Scholar]
- 392.Orii Y, Yumoto I, Fukumori Y and Yamanaka T, Journal of Biological Chemistry, 1991, 266, 14310–14316. [PubMed] [Google Scholar]
- 393.Hellwig P, Soulimane T and Mäntele W, European Journal of Biochemistry, 2002, 269, 4830–4838. [DOI] [PubMed] [Google Scholar]
- 394.Sousa FL, Veríssimo AF, Baptista AM, Soulimane T, Teixeira M and Pereira MM, Biophysical Journal, 2008, 94, 2434–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Pereira MM, Carita JN, Anglin R, Saraste M and Teixeira M, Journal of Bioenergetics and Biomembranes, 2000, 32, 143–152. [DOI] [PubMed] [Google Scholar]
- 396.Blomberg MRA and Ädelroth P, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 2017, 1858, 884–894. [DOI] [PubMed] [Google Scholar]
- 397.Blomberg MRA, Biochemistry, 2016, 55, 489–500. [DOI] [PubMed] [Google Scholar]
- 398.Namslauer A, Aagaard A, Katsonouri A and Brzezinski P, Biochemistry, 2003, 42, 1488–1498. [DOI] [PubMed] [Google Scholar]
- 399.Flock U, Thorndycroft FH, Matorin AD, Richardson DJ, Watmough NJ and Ädelroth P, J. Biol. Chem, 2008, 283, 3839–3845. [DOI] [PubMed] [Google Scholar]
- 400.Hartshorne RS, Kern M, Meyer B, Clarke TA, Karas M, Richardson DJ and Simon J, Mol Microbiol, 2007, 64, 1049–1060. [DOI] [PubMed] [Google Scholar]