

Abstract
Background
The vast majority of metastatic cancers cannot be cured. Palliative treatment may relieve disease symptoms by stopping or slowing cancer growth and may prolong patients’ lives, but almost all patients will inevitably develop disease progression after initial response. However, for reasons that are not fully understood, a very few patients will have extraordinary durable responses to standard anticancer treatments.
Materials and Methods
We analyzed exceptional responders treated at Fox Chase Cancer Center between September 2009 and November 2017. An exceptional response was defined as a complete response lasting more than 1 year or a partial response or stable disease for more than 2 years. Tumor samples were analyzed using an Ambry Genetics test kit with a 142‐gene panel. Messenger RNA expression was evaluated using NanoString's nCounter PanCancer Pathways Panel and Immune Profiling Panel and compared with matched controls for gender, age, and cancer type.
Results
Twenty‐six exceptional responders with metastatic bladder, kidney, breast, lung, ovarian, uterine, and colon cancers were enrolled. Mutations were identified in 45 genes. The most common mutation was an EPHA5 nonsynonymous mutation detected in 87.5% of patients. Mutations in DNA damage repair pathway genes were also frequent, suggesting increased genome instability. We also found varying expression of 73 genes in the Pathways panel and 85 genes in the Immune Profiling panel, many of them responsible for improvement in tumor recognition and antitumor immune response.
Conclusions
The genomic instability detected in our exceptional responders, plus treatment with DNA damage compounds combined with favorable anticancer immunity, may have contributed to exceptional responses to standard anticancer therapies in the patients studied.
Implications for Practice
With recent advances in the treatment of cancer, there is increased emphasis on the importance of identifying molecular markers to predict treatment outcomes, thereby allowing precision oncology. In this study, it was hypothesized that there is a “specific biologic signature” in the biology of the cancer in long‐term survivors that allows sensitivity to systemic therapy and durability of response. Results showed that DNA damage repair pathway alterations, combined with favorable anticancer immunity, may have contributed to exceptional responses. It is very likely that an in‐depth examination of outlier responses will become a standard component of drug development in the future.
Keywords: Exceptional responders, Immune system, Precision medicine, Genomic instability, DNA damage repair
Short abstract
Some patients with metastatic cancer will have markedly better responses to treatment than other patients receiving the same treatment. These exceptional responders are not often studied. This article reports the results of a single‐center study that aimed to identify specific molecular signatures among exceptional responders and to unravel molecular patterns that could explain how exceptional responders beat the odds.
Introduction
Some patients with metastatic cancer will have markedly better responses to treatment than other patients receiving the same treatment. The National Cancer Institute (NCI) defines exceptional responders as patients with cancer who have had a complete response (CR) or partial response (PR) for at least 6 months to a treatment in clinical trials in which less than 10% of patients responded overall [1]. Exceptional responders are not often studied because they are quite rare and, within a given trial, form too small a subset for statistical analysis. There is now, however, a growing interest in studying exceptional responders. The NCI's Exceptional Responders pilot clinical trial (NCT02243592) enrolled 100 exceptional responders to analyze tumor tissue and clinical data and to determine whether certain molecular features can predict responses to anticancer drugs. Study accrual goals were met by November 2017, and analysis is ongoing. (The study reported here is separate from NCT02243592).
In the last decade, a number of major advances in molecular biology technology have increased our ability to characterize cancer genes and their structure and expression. There has also been an increasing effort to translate individual genomic information into personalized medicine, with the goal of optimizing survival [2]. A fascinating story of the discovery of somatic gain‐of‐function mutations in hypereosinophilic syndrome came from a group of community practice oncologists who reported a dramatic response to the off‐label use of imatinib [3]. This initial observation generated the hypothesis that hypereosinophilic syndrome is driven by clonal activation of an imatinib‐responsive tyrosine kinase, which has been confirmed later by several trials [4]. Similarly, investigation of a small group of patients with non‐small‐cell lung cancer who had extraordinary responses to gefitinib led to the discovery of a culprit epithelial growth factor receptor (EGFR) mutation [5].
In this single‐center study, we aimed to identify specific molecular signatures among exceptional responders and to unravel molecular patterns that could explain how exceptional responders beat the odds. The hope is that molecular alterations detected in our patients could be explored in others and eventually become a predictor of treatment responses.
Subjects, Materials, and Methods
Study Population
We defined an exceptional responder as a patient with a metastatic solid tumor who had a CR lasting more than 1 year following systemic anticancer therapy or PR or stable disease (SD) lasting more than 2 years at any time during the disease course. Patients with hematologic malignancies (including lymphomas), curable solid tumors (e.g., germ cell tumors), and curable nonmetastatic disease were ineligible for this study. Potential participants were identified and referred by their treating medical oncologists. All patients were aged at least 18 years, had an Eastern Cooperative Oncology Group (ECOG) performance status of ≤1, and had available formalin‐fixed paraffin‐embedded (FFPE) archival tumor tissue. This study was approved by the institutional review board at Fox Chase Cancer Center in September 2014 (IRB 14‐083).
Clinical Data
Each patient gave informed consent, including permission to access their archived tumor samples and collect relevant clinical data from electronic medical records. Information obtained included the patient's age, sex, tumor type and location, date of diagnosis, stage, ECOG performance status, treatments received, duration of best response (CR, PR, SD), and date of recurrence or death if applicable. Response assessment was obtained using the RECIST version 1.1 [6]. Progression‐free survival (PFS) was defined as the time from the first dose of the treatment associated with best response until documented radiological progression or death from any cause. Duration of response was defined as the time from the documented response of the treatment associated with best response until documented progression. The treatment associated with the best response that allowed for inclusion of the patient in the study was determined based on a review of clinical data. Data cutoff date for analysis was November 30, 2017.
Molecular Profiling
Next‐generation sequencing was performed on 16 patients who had a sufficient amount of archived tissue. DNA and RNA were extracted from sections of FFPE tumor specimens from biopsies or surgical resections. If multiple archived tumor specimens were available, the most recent was reviewed. The minimum acceptable tumor cellularity was 10%. Genomic profiling was initially performed to identify molecular aberrations potentially predictive of treatment response and targetable for therapeutics. The samples were analyzed using an Ambry Genetics test kit with a 142‐gene panel for solid tumors. Exons and a small number of select introns from the 142 genes were enriched using a hybridization‐based capture approach and sequenced by next‐generation sequencing on a HiSeq system (Illumina; San Diego, CA). The target average coverage per sample was greater than 250× to allow high‐confidence variant calling [7]. Genomic profiles were compared between different tumor types and treatments associated with the best response. ANNOVAR [8] was used for variant annotation. The functional consequences of nonsynonymous single nucleotide variants and splice variants were predicted using PolyPhen‐2 (Polymorphism Phenotyping v2), SIFT, LRT, MutationTaster, MutationAssessor, and CADD [9]. Variants leading to nonsynonymous changes in exonic regions of encoded proteins or splice variants were selected if they received scores indicating a protein‐damaging function with at least three of six in silico predictors. We also applied a population filter of less than 1% to focus on only rare mutations, and known sequence artifacts were also removed. For indel variants, we only kept those causing frameshifting insertions or deletions. Next, we queried the International Cancer Genome Consortium (ICGC) database with the most commonly observed variants in our analysis (variants present in more than one patient) to look for their distribution in The Cancer Genome Atlas (TCGA) bladder, kidney, breast, and lung cancers, because those are the most common tumors among our data set.
Messenger RNA Expression
Tumor samples from 23 exceptional responders and 23 matched controls were tested for levels of messenger RNA (mRNA) expression using NanoString's nCounter PanCancer panels (Pathways and Immune Profiling). Microdissection of tissue was not performed, and all tissue on unstained slides was used for RNA extraction. Each sample from exceptional responders was paired with a control from a patient matched for age, sex, and diagnosis, and levels of mRNA expression were compared between the two samples. Protected health information related to the control patients was acquired from the Fox Chase Biosample Repository. Twenty 10‐μm slides were prepared for analysis from each FFPE tissue block. RNA extraction was performed using a High Pure FFPET RNA Isolation Kit (Roche; Penzburg, Germany). Each 50‐ng RNA sample was hybridized with the CodeSets of nCounter PanCancer Pathways and Immune Profiling panels. The hybridized samples and data acquisition were processed with the NanoString Prep Station and Digital Analyzer, respectively. For both panels, NanoString expression counts were normalized using NanoString nSolver [10] using default settings (background subtraction based on geometric mean of negative controls, positive control normalization based on geometric mean of positive control probes, and standard CodeSet normalization). Genes with a maximum log2‐scale normalized expression of <6 were excluded as uniformly low expressors. For comparison of gene expression in patient tumor samples and matched controls, the pairwise t test was performed and a gene was defined as significant if the false discovery rate < 0.05 (Benjamini‐Hochberg method [11]). The significant genes were plotted according to the order of gene expression fold‐change between tumor and control samples.
Results
Patients
Twenty‐six exceptional responders (13 male and 13 female) were identified and enrolled in this study. Seven patients had urothelial carcinoma of the bladder, seven had kidney cancer, five had non‐small‐cell lung cancer, four had breast cancer, and one patient had ovarian, uterine, and colon cancer. CR lasting more than 1 year was observed in 15 patients; 11 patients had either PR or SD. The mean age was 71 years (range, 33–93). Median progression‐free survival was 67.5 months (range, 24–142). Baseline characteristics are summarized in Table 1. Table 2 summarizes patients’ gender, age, cancer type, treatment that led to the extraordinary response, duration of treatment, and duration of response. Two of our patients received concurrent therapies that could potentially modulate response. Patient 11 (Table 2) was diagnosed with clear cell renal cell carcinoma (RCC) metastatic to the adrenal gland and experienced a PR with sorafenib for 88 months. After 69 months of initial sorafenib treatment, the patient underwent adrenalectomy. Sorafenib was held for 1 month, and after surgery he continued on therapy for another 18 months, when he experienced progression and changed therapy. Patient 13 had clear cell RCC metastatic to soft tissue in his right thigh. After 47 months of initial therapy with sunitinib, he received radiotherapy in the right thigh mass. After radiotherapy, he continued sunitinib for another 5 months, when he had progression and changed therapy. The rest of our patients did not receive any therapy that could potentially modulate response, such as metastasectomy, radiation, or any other systemic treatment, except therapies listed in Table 2.
Table 1.
Demographic data on exceptional responders
| Characteristic | Exceptional responders (n = 26) |
|---|---|
| Gender, n (%) | |
| Male | 13 (50) |
| Female | 13 (50) |
| Age, years, median (range) | 71 (33–93) |
| Tumor types, n (%) | |
| Urothelial carcinoma of the bladder | 7 (26.9) |
| Kidney | 7 (26.9) |
| Breast | 4 (15.5) |
| Lung | 5 (19.2) |
| Ovarian | 1 (3.8) |
| Uterine | 1 (3.8) |
| Colon | 1 (3.8) |
| Response type, n | |
| Complete response (>1 yr) | 15 |
| Partial response/stable disease (>2 yr) | 11 |
| Duration of response, months, median (range) | |
| Complete response | 88 (25–132) |
| Partial response/stable disease | 45.5 (24–138) |
Table 2.
Exceptional responders’ treatment and response
| Patient number | Gender | Age | Cancer type | Rx | Rx duration, mo | Best response | Duration of response, mo | Status | Sequencing | NanoString |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 79 | Bladder | Cetuximab + paclitaxel | 3 | CR | 88 | Alive | Yes | Yes |
| 2 | M | 76 | Bladder | MVAC | 4 | CR | 98 | Alive | No | Yes |
| 3 | M | 71 | Bladder | Gem | 2 | CR | 72 | Alive | Yes | Yes |
| 4 | M | 75 | Bladder | Gem + cis | 3 | CR | 79 | Alive | No | Yes |
| 5 | F | 78 | Bladder | ddMVAC | 3 | CR | 39 | Died | Yes | No |
| 6 | M | 84 | Bladder | Gem + carbo | 3 | CR | 25 | Died | Yes | Yes |
| 7 | M | 93 | Bladder | Gem + cis | 4 | CR | 90 | Alive | Yes | Yes |
| 8 | M | 86 | Papillary RCC | Tivozanib | 41 | PR | 41 | Alive | No | Yes |
| 9 | F | 50 | Clear cell RCC | Axitinib | 24 | PR | 24 | Died | No | Yes |
| 10 | M | 58 | Anaplastic RCC | Sunitinib | 39 | PR | 39 | Alive | Yes | Yes |
| 11 | M | 68 | Clear cell RCC | Sorafenib | 88 | PR | 88 | Died | No | Yes |
| 12 | M | 57 | Clear cell RCC | Sunitinib | 20 | PR | 50 | Alive | No | Yes |
| 13 | M | 59 | Clear cell RCC | Sunitinib | 52 | PR | 52 | Died | Yes | Yes |
| 14 | M | 53 | Clear cell RCC | Sunitinib | 138 | PR | 138 | Died | Yes | No |
| 15 | F | 77 | TNBC | Paclitaxel + bev | 6 | CR | 99 | Alive | No | Yes |
| 16 | F | 33 | ER/PR–, HER2+ BC | Paclitaxel + trastuzumab | Paclitaxel: 6, trastuzumab: 93 | CR | 93 | Alive | Yes | Yes |
| 17 | F | 48 | TNBC | Paclitaxel | 6 | CR | 142 | Alive | Yes | Yes |
| 18 | F | 48 | ER/PR–, HER2+ BC | Paclitaxel + trastuzumab | Paclitaxel: 6, trastuzumab: 132 | CR | 132 | Alive | Yes | Yes |
| 19 | F | 59 | Lung adenocarcinoma (KRAS mut.) | Carbo + pemetrexed | Carbo: 8, pemetrexed: 50 | PR | 50 | Alive | Yes | Yes |
| 20 | F | 71 | Lung adenocarcinoma (EGFR mut.) | Osimertinib | 24 | PR | 24 | Alive | No | Yes |
| 21 | M | 83 | Lung adenocarcinoma (EGFR mut.) | Erlotinib | 2 | PR | 25 | Alive | Yes | Yes |
| 22 | F | 65 | Lung adenocarcinoma | Cis + gem | 3 | CR | 117 | Alive | No | Yes |
| 23 | F | 79 | Lung adenocarcinoma | Carbo + paclitaxel | 4 | CR | 62 | Died | No | Yes |
| 24 | F | 71 | Uterine carcinosarcoma | Ifosfamide | 4 | SD | 32 | Died | Yes | Yes |
| 25 | F | 84 | High‐grade serous ovarian | Carbo + gem | 5 | CR | 78 | Alive | Yes | Yes |
| 26 | F | 64 | Colon cancer KRAS wt | FOLFIRI + cetuximab | 11 | CR | 63 | Alive | Yes | No |
Abbreviations: BC, breast cancer; bev, bevacizumab; carbo, carboplatin; cis, cisplatin; CR, complete response; dd, dose‐dense; ER, estrogen; F, female; FOLFIRI, folinic acid, fluorouracil, and irinotecan; gem, gemcitabine; M, male; mut, mutation; MVAC, methotrexate, vincristine, doxorubicin, and cisplatin; mut, mutation; PR–, progesterone negative; PR, partial response; RCC, renal cell carcinoma; Rx, prescription; SD, stable disease; TNBC, triple negative breast cancer; wt, wild type
Molecular Profiling and Targeted Gene Sequencing
Targeted gene sequencing using an Ambry Genetics test kit with a 142‐gene panel for solid tumors was performed on 16 patients. After initial analysis, 191 variants were obtained. The variants predicted to be damaging were identified by three methods. Mutations were identified in 45 genes (Fig. 1), with a total of 76 rare damaging variants (supplemental online Table 1). The most common mutation was an EPHA5 nonsynonymous mutation, identified in 14 patients (87.5%). NF1 splicing mutation was observed in 11 patients (68.7%). This mutation had a population frequency of 1.5%, which is slightly above the cutoff value of 1% and seemed to be damaging by MutationTaster and FATHMM methods but not by ClinVar. Because of the fact that it was present in 68.7% of our patients, we felt that this mutation should be included here. Other mutations also occurred in a high percentage of our patient population. A FOXL2 nonsynonymous mutation was identified in nine patients (56.2%), ABL1 mutations (frameshift and nonsynonymous) were identified in seven patients (43.8%), TP53 mutations (frameshift, nonsynonymous, and stop codon) were identified in six patients (37.5%), ATM frameshift and nonsynonymous mutations were identified in four patients (25%), and ABL2 frameshift mutation, BRCA2 (frameshift, nonsynonymous, and stop codon), VHL (nonsynonymous), NOTCH4 (nonsynonymous), and NOTCH 1 (nonsynonymous) mutations were each identified in three patients (18.7%).
Figure 1.

Genomic alterations. Genomic data are from sequencing using Ambry Genetics test kit with a 142‐gene panel for solid tumors. The target average coverage per sample was greater than 250× to allow high‐confidence variant calling.
A total of seven damaging variants were observed in more than one patient in our analysis (supplemental online Table 1). These variants were observed in genes EPHA5, FOXL2, ABL1, ABL2, APC, ATM, and VHL. We queried the ICGC database with these seven variants, and no specific variant level data could be found in TCGA bladder, kidney, breast, and lung data sets, showing that these variants are indeed rare. Looking at gene levels, we also observed that the mutations were rare in the four tumors analyzed from the ICGC database (bladder, kidney, breast, and lung), with the exception of a VHL mutation in kidney cancer (supplemental online Table 2).
NanoString nCounter System Panels
Twenty‐three exceptional responders and 23 control patients matched by age, gender, and disease underwent gene expression analysis using NanoString's PanCancer Immune Profiling Panel and PanCancer Pathways Panel. These panels analyzed 770 genes involved in immune response and cancer‐associated canonical pathways, respectively. Among the genes analyzed, the level of expression significantly differed between case and control in 73 genes in the Pathways Panel and 85 genes in the Immune Profiling Panel.
The Pathways Panel analyzes 770 genes from 13 cancer‐associated canonical pathways involved in the hallmarks of cancer; some genes are involved in more than one pathway. Genes with the most significantly different expression are shown in Figure 2. The three most overexpressed genes were tenascin‐C (TNC) and thrombospondin‐4 (THBS4; PI3K pathway) and LEFTY2 (TGF‐β pathway). We found six overexpressed genes in the MAPK pathway (CD14, TGFB3, RAC2, CDC25B, PRKACA, and GRB2) and six in the RAS pathway (CSF1R, RAC2, LAT, ABL1, PRKACA, and GRB2). Five overexpressed genes are cancer drivers (CSF1R, AMER1, GNAS, ABL1, and DNMT1). Five of the overexpressed genes we found are in the PI3K pathway (TNC, THBS4, IL2RB, CSF1R, and GRB2) and the cell cycle and apoptosis pathway (PRKAR2B, TGFB3, CDC25B, ABL1, and PRKACA). These pathways are involved in deregulating cellular energetics and cell‐death resistance, respectively. Three genes were overexpressed in the WNT pathway (FZD7, RAC2, and PRKACA) and the transcription misregulation pathway (CD14, IL2RB, and CSF1R), which are involved in replicative immortality and genome instability, respectively. Finally, two genes were overexpressed in the Hedgehog pathway (GAS1 and PRKACA), two in the TGF‐β pathway (LEFTY2 and TGFB3), and two in the JAK‐STAT pathway (PAKAR2B and GRB2). These pathways are involved in cell‐death resistance, growth suppressor evasion, and sustained proliferative signaling, respectively. The most underexpressed gene in the Pathways Panel was PTPRR (greater than twofold), followed by PLA2G10, HHIP, and HNF1A (Fig. 2). Three of the underexpressed genes are cancer drivers (BRAF, SF3B1, and HNF1A). Two genes were underexpressed in the Hedgehog pathway (GSK3B and HHIP), two in the DNA damage repair pathway (GTF2H3 and MNAT1), and two in the MAPK pathway (BRAF and PTPRR).
Figure 2.
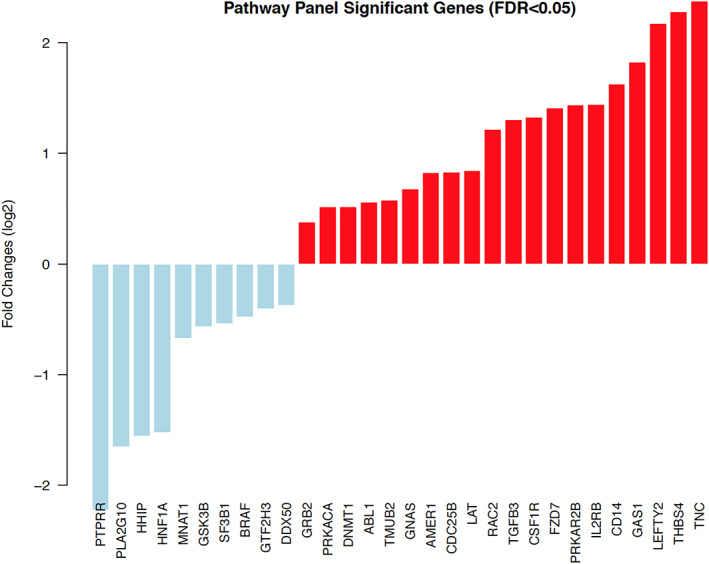
Significant genes from Pathway Panel. Gene expression of 23 paired patient tumor samples and matched control samples were obtained using NanoString nCounter PanCancer Pathway Panel. The pairwise t test was performed for the tumor samples and matched controls, and the cutoff of FDR <0.05 was used to define significantly differentially expressed genes. Red bar, overexpressed; blue bar, underexpressed. Abbreviation: FDR, false discovery rate.
Immune Profiling Panel analyzes genes related to the immune system. The most overexpressed of these was TNFRSF8 (CD30 receptor expressed by activated T and B cells), followed by CYBB, CCL2, SIGLEC1, CD14, FAS, FCER1G, CSF1R, LY96, ITGAM, LAIR2, IL2RB, TNFSF13B, IL21R, SELPLG, CD74, TNFRSF4, ITGB2, and C1QB. The most underexpressed genes were NOS2, ARG2, REPS1, and ATG5 (Fig. 3).
Figure 3.
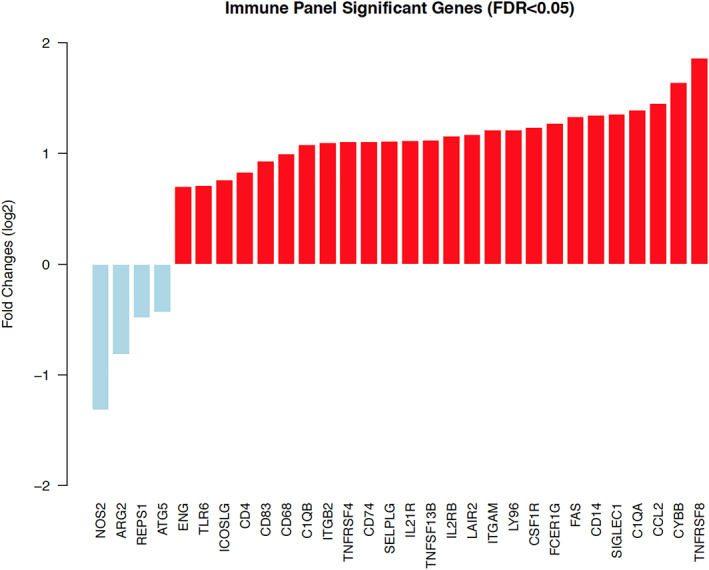
Significant genes from Immune Panel. Gene expression of 23 paired patient tumor samples and matched control samples were obtained using NanoString nCounter PanCancer Immune Panel. The pairwise t test was performed for the tumor samples and matched controls, and the cutoff of FDR <0.05 was used to define significantly differentially expressed genes. Red bar, overexpressed; blue bar, underexpressed. Abbreviation: FDR, false discovery rate.
Discussion
Identifying molecular alterations in exceptional responders’ tumors was a complex process that involved sequencing targeted genes and assessing gene expression on tumors in exceptional responders and matched controls. The most interesting discovery was identifying the same nonsynonymous mutation of the EPHA5 gene in 87.5% of exceptional responders. Mutations in other genes in the EPHA family were also identified: EPHA2, EPHA3, and EPHA7. Another commonly mutated gene was FOXL2, a forkhead family transcriptional factor observed in nine patients (56%). Although its role in cancer is not fully understood, recent evidence has suggested that FOXL2 and its target genes participate in the DNA damage repair (DDR) pathway. Our study also identified additional mutations in DDR pathway such as BRCA2, ATM, RAD50, and FANCA. NanoString analysis also demonstrated change in expression of DDR pathway genes: expression of GTF2H3 (involved in the nucleotide excision repair process) and FANCL was reduced in bladder cancer exceptional responders (supplemental online Figs. 1, 2) in comparison to matched control patients. Other underexpressed DDR genes were SF3B1 [12], GSK3B [13], MNAT1 [14] (Fig. 2), and ATG5 (Fig. 3). ATG5 is a key gene in the autophagy process, playing an important role in the repair and clearance of DNA damage [15].
Eph receptors are the largest family of receptor tyrosine kinases and are known to play an important role in tumor immunity by regulating chemotaxis and migration and enhancement of CD8+ T‐cell activity against tumor cells [16, 17, 18, 19]. A recently study showed that EPHA5 mutations were associated with increased tumor mutation burden, tumor‐infiltrating lymphocytes, and durable responses in patients with lung adenocarcinoma treated with immunotherapy [20]. This study also reported that EPHA5‐mutated tumors were associated with high mutation frequencies of DDR genes and had a tumor microenvironment that was enriched with tumor‐infiltrating CD8+ T cells, CD4+ activated memory T cells, and macrophage M1 cells [20]. Additionally, EPHA5‐mutated tumors had an immune signature characterized by increased expression levels of chemokines and cytolytic activity‐associated gene signatures [20]. Another study indicated that EPHA5 plays a direct role in DDR and interacts with ATM at sites of DNA repair and increases sensitivity to anticancer therapies [21].
Our results revealed increased expression of genes related to antitumor immunity. We identified overexpression of several genes involved in the complex interaction between cancer and the host immune system (Fig. 3). IL21R improves T‐cell responses and activates natural killer (NK) cells. Interleukin (IL)‐21 also improves antigen presentation, and its overexpression is associated with improved outcomes in preclinical models of breast cancer [22]. Similarly, SIGLEC1 (CD169) plays a crucial role in inducing a cytotoxic T‐cell response [23]. Exceptional responders also overexpressed CD4, a membrane glycoprotein of T lymphocytes that interacts with the major histocompatibility complex II antigens [24], and IL2RB, which enriches intratumoral T and NK cells [25] (Fig. 3). TNFRSF4 (OX‐40) promotes the survival and expansion of CD4 and CD8 T cells, enhancing CD8+ T‐cell cytotoxic activity and decreasing T‐cell exhaustion by inhibiting IL‐10 and regulating T‐regulatory cells [26]. OX‐40 agonists are currently being tested in multiple clinical trials [27, 28, 29]. Interestingly, the most overexpressed gene in exceptional responders, analyzed with the Immune panel, was TNFRSF8 (CD30), a member of the TNF receptor superfamily. TNFRSF8 is a positive regulator of apoptosis and is expressed only by activated T and B cells. Other pathways implicated in tumor immunity were also differently expressed in the cohort of exceptional responders. NOTCH1 and NOTCH2 affect the activation of CD8+ T cells [30] and promote activation of the M1 phenotype that has proinflammatory antitumor activity [31]. NOTCH2 was overexpressed in exceptional responders with bladder cancer (supplemental online Figs. 1, 2). Interestingly, among exceptional responders, ARG2 and NOS2 (genes involved in immune escape mechanisms) were significantly underexpressed (Fig. 3).
Huang et al. demonstrated that EPHA5‐mutated lung adenocarcinomas had durable responses to immunotherapy likely related to alterations in the DDR pathway, increased tumor mutational burden, and a favorable antitumor immune signature [20]. We found similar results in our exceptional responders. Some molecular alterations found in our population might help to explain their exceptional responses to chemotherapy. It is well known that tumors with DDR alterations can achieve good responses when treated with DNA damage compounds such as alkylating agents, platinum‐based agents, gemcitabine, paclitaxel, irinotecan, and 5‐fluorouracil, which almost all of our patients received [32]. Additionally, the most overexpressed gene in our cohort, analyzed through NanoString's pathway panel, was TNC, followed by THBS4, LEFTY2, and GAS1 (Fig. 2). TNC is an extracellular matrix molecule that has pleiotropic effects on tumor and stromal cells. It may also affect drug responsiveness and DNA repair through its interaction with fibronectin [33]. THBS4, a tumor‐suppressor gene, is a member of the extracellular calcium‐binding protein family. Overexpression of THBS4 in vitro significantly suppresses cancer‐cell growth [34]. LEFTY2 is a regulator of tumor proliferation whose overexpression is related to sensitivity to cisplatin in preclinical studies [35, 36]. In addition, several studies have shown that GAS1 regulates cell‐growth arrest, apoptosis, and sensitivity to cisplatin [37, 38]. Our study demonstrated an overexpression of the LEFTY2 gene, especially in bladder cancer exceptional responders treated with platinum agents (supplemental online Figs. 1, 2).
Our kidney cancer exceptional responders were treated with antiangiogenic TKIs alone. Antiangiogenic treatment has demonstrated clinical benefit and has been approved for front line treatment of clear cell renal cell carcinoma [39, 40]. Recently, combination trials of TKIs and immunotherapy have shown excellent durable responses [41, 42]. The mechanisms that led our patients to experience these durable responses are not clear but could be related to antitumor immune response. The impact of TKI treatment in the setting of DDR impairment is not known. Our working hypothesis is that TKIs in addition to underlying favorable tumor microenvironment are capable of maintaining durable PRs, similar to the combination of TKIs and immune checkpoint inhibitors. All these patients had EPHA5 mutation, and the RNA analyzes showed overexpression of genes related to chemotaxis and activation of immune response, such as IL6, COLEC12IL1B, CXCL2, and CXCL3; (supplemental online Figs. 3, 4).
Our results demonstrate that outliers have a higher mutational burden in the DDR pathway than is found in an average population of patients with cancer, and mRNA expression analyses confirmed those findings. DDR pathway impairment leads to an accumulation of cytosolic double‐strand DNA in the presence of DNA damage agents. This could lead to a proinflammatory state and a shift in cancer immunoediting toward elimination of tumor cells. The genomic instability detected in our outliers, plus treatment with DNA damage compounds such as alkylating agents, platinum‐based agents, gemcitabine, paclitaxel, irinotecan, and 5‐fluorouracil (in almost all patients), and a favorable immune system could result in durable responses. We also found that outliers overexpressed multiple genes responsible for tumor recognition and tumor‐cell killing by cytotoxic CD8+ T and NK cells.
Because of several limitations of this study, our results should be interpreted cautiously. This single‐center, retrospective pilot study is subject to recall bias because we did not search the entire electronic medical record to identify all exceptional responders. In addition, we had to exclude some patients from the analysis because of a lack of viable archived tumor tissue for next‐generation sequencing. Furthermore, not all patients in this study underwent mRNA expression analysis. Finally, next‐generation sequencing was done using a novel Ambry Genetics platform not certified by the Clinical Laboratory Improvement Amendments at the time of analyses, allowing for the possibility that sequencing data sets may have some artifactual mutations. Another important consideration is that some of our patients who met the eligibility criteria for exceptional response had been treated with targeted therapies that are known to induce durable responses, such as trastuzumab for HER2‐positive breast cancer and osimertinib and erlotinib for lung adenocarcinomas with activating EGFR mutation. Therefore, the exceptional responses could be explained by targeted inhibition of these driver mutations. Nevertheless, these tumors shared similar molecular profiles with the rest of our population, and these patients did better than would be expected for a median duration of response with the same therapies. In addition, two of our patients received metastasis‐directed local therapy during the course of systemic therapy that could have influenced the duration of response. However, the metastasis‐directed therapies were given after a long time of response to systemic therapy (69 and 47 months), which would not exclude these patients based on our exceptional responder criteria.
Conclusion
Cancer development involves alterations in distinct pathways that confer the common cancer characteristics of uncontrolled growth, neoangiogenesis, immune evasion, and the potential to metastasize [43]. Our results may help to elucidate the complex interaction of antineoplastic drugs and molecular characteristics leading to exceptional responses. EPHA5 mutations may contributed to exceptional responses along with other molecular alterations in the DDR pathway and a favorable antitumor immune signature. This is interesting because of the fact that the U.S. Food and Drug Administration approved combination of cytotoxic drugs and immune checkpoint inhibitors, based on their ability to produce more profound and durable responses [44]. This study also highlights the potential for sustained therapeutic response in cancers with a mutation‐disabled DDR pathway with DNA‐damaging therapy when there is an active anticancer immune response. It also demonstrates the value of interrogating RNA expression beyond tumor mutation profiling to elucidate better therapeutic strategies for patients with any cancer. Further exploration of molecular mechanisms that drive exceptional responses is warranted.
Author Contributions
Conception/design: Marijo Bilusic, Elizabeth Plimack
Provision of study material or patients: Marijo Bilusic, Kyungsuk Jung, Katherine Alpaugh, Denise Young, Douglas Flieder, Elizabeth Plimack
Collection and/or assembly of data: Marijo Bilusic, Kyungsuk Jung, Denise Young, Douglas Flieder, Elizabeth Plimack
Molecular profiling: Jianming Pei, Phillip Gray
Data analysis and interpretation: Marijo Bilusic, Daniel Girardi, Kyungsuk Jung, Jianming Pei, Michael Slifker, Qingrong Chen, Phillip Gray Daoud Meerzaman, Elizabeth Plimack Manuscript writing: Marijo Bilusic, Daniel Girardi, Kyungsuk Jung, Elizabeth Plimack
Manuscript review: Marijo Bilusic, Daniel Girardi, Yan Zhou, Kyungsuk Jung, Jianming Pei, Michael Slifker, Qingrong Chen, Daoud Meerzaman, Katherine Alpaugh, Denise Young, Douglas Flieder, Phillip Gray, Elizabeth Plimack
Final approval of manuscript: Marijo Bilusic, Daniel Girardi, Yan Zhou, Kyungsuk Jung, Jianming Pei, Michael Slifker, Qingrong Chen, Daoud Meerzaman, Katherine Alpaugh, Denise Young, Douglas Flieder, Phillip Gray, Elizabeth Plimack
Disclosures
Elizabeth Plimack: Genentech, Merck, Seattle Genetics, Janssen, Flatiron, Bristol‐Myers Squibb (SAB), AstraZeneca, Pfizer, Infinity Pharma (Data Safety Monitoring), AUA, ASCO GU, Clinical Care Options, Fox Chase Cancer Center, Georgetown, Mt. Sinai Ichan School of Medicine, Medscape, NCCN, Omniprex, OncLive, PER, Prime Oncology, Research to Practice, Spire Learning, University of Pennsylvania, Thomas Jefferson Kimmel Cancer Center (H). Astellas, Bristol‐Myers Squibb, Genentech, Merck (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Figures.
Appendix S2. Tables.
Acknowledgments
The authors thank Bonnie L. Casey for editorial assistance in the preparation of this manuscript.
This research was funded by a Fox Chase Cancer Center Pilot Grant for Rare Tumor Subtypes and Exceptional Responders, Fox Chase Cancer Center NCI Core Grant no. P30CA00692.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Footnotes
For Further Reading: Masayuki Takeda, Takayuki Takahama, Kazuko Sakai et al. Clinical Application of the FoundationOne CDx Assay to Therapeutic Decision‐Making for Patients with Advanced Solid Tumors. The Oncologist First published: 16 December 2020.
Implications for Practice: This prospective cohort study was initiated to investigate the feasibility and utility of clinical application of FoundationOne CDx. A total of 181 samples were processed for genomic testing between September 2018 and June 2019, with data being successfully obtained for 175 of these samples, yielding a success rate of 96.7%, and 24 individuals (14%) received matched targeted therapy.
References
- 1. Abrams J, Conley B, Mooney M et al. National Cancer Institute's Precision Medicine Initiatives for the new National Clinical Trials Network. Am Soc Clin Oncol Educ Book 2014:71–76. [DOI] [PubMed] [Google Scholar]
- 2. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015;372:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schaller JL, Burkland GA. Case report: Rapid and complete control of idiopathic hypereosinophilia with imatinib mesylate. MedGenMed 2001;3:9. [PubMed] [Google Scholar]
- 4. Licht JD, Chomienne C, Goy A et al. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 1995;85:1083–1094. [PubMed] [Google Scholar]
- 5. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139. [DOI] [PubMed] [Google Scholar]
- 6. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 7. Gray PN, Vuong H, Tsai P et al. TumorNext: A comprehensive tumor profiling assay that incorporates high resolution copy number analysis and germline status to improve testing accuracy. Oncotarget 2016;7:68206–68228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu X, Wu C, Li C et al. dbNSFP v3.0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Hum Mutat 2016;37:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kudo M, Chung H, Osaki Y. Prognostic staging system for hepatocellular carcinoma (CLIP score): Its value and limitations, and a proposal for a new staging system, the Japan Integrated Staging Score (JIS score). J Gastroenterol 2003;38:207–215. [DOI] [PubMed] [Google Scholar]
- 11. Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Series B Stat Methodol 1995;57:289–300. [Google Scholar]
- 12. Te Raa GD, Derks IA, Navrkalova V et al. The impact of SF3B1 mutations in CLL on the DNA‐damage response. Leukemia 2015;29:1133–1142. [DOI] [PubMed] [Google Scholar]
- 13. Ding L, Madamsetty VS, Kiers S et al. Glycogen synthase kinase‐3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR‐mediated DNA damage response. Clin Cancer Res 2019;25:6452–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kap EJ, Seibold P, Richter S et al. Genetic variants in DNA repair genes as potential predictive markers for oxaliplatin chemotherapy in colorectal cancer. Pharmacogenomics J 2015;15:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eliopoulos AG, Havaki S, Gorgoulis VG. DNA damage response and autophagy: A meaningful partnership. Front Genet 2016;7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shiuan E, Chen J. Eph receptor tyrosine kinases in tumor immunity. Cancer Res 2016;76:6452–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tatsumi T, Herrem CJ, Olson WC et al. Disease stage variation in CD4+ and CD8+ T‐cell reactivity to the receptor tyrosine kinase EphA2 in patients with renal cell carcinoma. Cancer Res 2003;63:4481–4489. [PubMed] [Google Scholar]
- 18. Aasheim HC, Delabie J, Finne EF. Ephrin‐A1 binding to CD4+ T lymphocytes stimulates migration and induces tyrosine phosphorylation of PYK2. Blood 2005;105:2869–2876. [DOI] [PubMed] [Google Scholar]
- 19. Zhang J, Zhang Z, Song W et al. EPHA5 mutation impairs natural killer cell‐mediated cytotoxicity against non‐small lung cancer cells and promotes cancer cell migration and invasion. Mol Cell Probes 2020;52:101566. [DOI] [PubMed] [Google Scholar]
- 20. Huang W, Lin A, Luo P et al. EPHA5 mutation predicts the durable clinical benefit of immune checkpoint inhibitors in patients with lung adenocarcinoma. Cancer Gene Ther 2020. [Epub ahead of print[. [DOI] [PubMed] [Google Scholar]
- 21. Staquicini FI, Qian MD, Salameh A et al. Receptor tyrosine kinase EphA5 is a functional molecular target in human lung cancer. J Biol Chem 2015;290:7345–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mittal D, Caramia F, Michiels S et al. Improved treatment of breast cancer with anti‐HER2 therapy requires interleukin‐21 signaling in CD8+ T Cells. Cancer Res 2016;76:264–274. [DOI] [PubMed] [Google Scholar]
- 23. Fraschilla I, Pillai S. Viewing Siglecs through the lens of tumor immunology. Immunol Rev 2017;276:178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ostroumov D, Fekete‐Drimusz N, Saborowski M et al. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci 2018;75:689–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chifman J, Pullikuth A, Chou JW et al. Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 2016;16:911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol 2015;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ito T, Wang YH, Duramad O et al. OX40 ligand shuts down IL‐10‐producing regulatory T cells. Proc Natl Acad Sci USA 2006;103:13138–13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris A, Vetto JT, Ramstad T et al. Induction of anti‐mammary cancer immunity by engaging the OX‐40 receptor in vivo. Breast Cancer Res Treat 2001;67:71–80. [DOI] [PubMed] [Google Scholar]
- 29. Curti BD, Kovacsovics‐Bankowski M, Morris N et al. OX40 is a potent immune‐stimulating target in late‐stage cancer patients. Cancer Res 2013;73:7189–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell 2018;34:536–548. [DOI] [PubMed] [Google Scholar]
- 31. Hossain F, Majumder S, Ucar DA et al. Notch signaling in myeloid cells as a regulator of tumor immune responses. Front Immunol 2018;9:1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012;12:587–598. [DOI] [PubMed] [Google Scholar]
- 33. Spenlé C, Saupe F, Midwood K et al. Tenascin‐C: Exploitation and collateral damage in cancer management. Cell Adh Migr 2015;9:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Greco SA, Chia J, Inglis KJ et al. Thrombospondin‐4 is a putative tumour‐suppressor gene in colorectal cancer that exhibits age‐related methylation. BMC Cancer 2010;10:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao X, Cai Y, An R. miR‐215 promotes epithelial to mesenchymal transition and proliferation by regulating LEFTY2 in endometrial cancer. Int J Mol Med 2018;42:1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Akiya M, Yamazaki M, Matsumoto T et al. Identification of LEFTY as a molecular marker for ovarian clear cell carcinoma. Oncotarget 2017;8:63646–63664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang YW, Zheng Y, Wang JZ et al. Integrated analysis of DNA methylation and mRNA expression profiling reveals candidate genes associated with cisplatin resistance in non‐small cell lung cancer. Epigenetics 2014;9:896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Q, Qin Y, Wei P et al. Gas1 inhibits metastatic and metabolic phenotypes in colorectal carcinoma. Mol Cancer Res 2016;14:830–840. [DOI] [PubMed] [Google Scholar]
- 39. Motzer RJ, Hutson TE, Tomczak P et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. N Engl J Med 2007;356:115–124. [DOI] [PubMed] [Google Scholar]
- 40. Motzer RJ, Hutson TE, Cella D et al. Pazopanib versus sunitinib in metastatic renal‐cell carcinoma. N Engl J Med 2013;369:722–731. [DOI] [PubMed] [Google Scholar]
- 41. Motzer RJ, Penkov K, Haanen J et al. Avelumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019;380:1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rini BI, Plimack ER, Stus V et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019;380:1116–1127. [DOI] [PubMed] [Google Scholar]
- 43. Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res 2017;7:1016–1036. [PMC free article] [PubMed] [Google Scholar]
- 44. Goldberg SB, Herbst RS. Should chemotherapy plus immune checkpoint inhibition be the standard front‐line therapy for patients with metastatic non‐small cell lung cancer? Cancer 2018;124:4592‐4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1. Figures.
Appendix S2. Tables.