

Abstract
Background
Prostate cancer (PC) tissue contains all‐trans retinoic acid (ATRA) at a very low level (10−9 M), at least an order of magnitude lower than in adjacent normal healthy prostate cells or benign prostate hyperplasia. When this is coupled with deregulated expression of the intracellular lipid‐binding proteins FABP5 and CRABP2 that is frequently found in PC, this is likely to result in the preferential delivery of ATRA to oncogenic PPARβ/δ rather than retinoic acid receptors (RARs). There are three isotypes of RARs (RARα, RARβ, and RARγ) and recent studies have revealed discrete physiological roles. For example, RARα and RARγ promote differentiation and self‐renewal, respectively, which are critical for proper hematopoiesis.
Aims
We have previously shown that ATRA stimulates transactivation of RARγ at sub‐nanomolar concentrations (EC50 0.24 nM), whereas an 80‐fold higher concentration was required for RARα‐mediated transactivation (EC50 19.3 nM). Additionally, we have shown that RAR pan‐antagonists inhibit the growth of PC cells (at 16‐34 nM). These findings, together with the low level of ATRA in PC, led us to hypothesize that RARγ plays a role in PC pathogenesis and that RARγ‐selective antagonism may be an effective treatment.
Methods and results
We found that concentrations of 10−9 M and below of ATRA promoted survival/proliferation and opposed adipogenic differentiation of human PC cell lines by a mechanism that involves RARγ. We also found that a RARγ‐selective antagonist (AGN205728) potently induced mitochondria‐dependent, but caspase‐independent, cell death in PC cell lines. Furthermore, AGN205728 demonstrated synergism in killing PC cells in combination with cytotoxic chemotherapeutic agents.
Conclusion
We suggest that the use of RARγ‐selective antagonists may be effective in PC (and potentially other cancers), either as a single agent or in combination with cytotoxic chemotherapy.
Keywords: prostate cancer, RARγ antagonists, retinoids
1. INTRODUCTION
Prostate cancer (PC) is one of the most commonly diagnosed non‐cutaneous malignancies in elderly males living in the industrialized countries of Western Europe and North America and current estimates suggest that approximately 1 in 10 men over the age 65 years will go on to develop an aggressive metastatic form of the disease. 1 Thus due to an increasingly aging population, the incidence of PC is predicted to greatly increase over the next 30 years. 2 Currently, chemo‐ or radiotherapy, often coupled with androgen deprivation therapy (ADT), is the gold‐standard treatment for most PC patients. 3 , 4 However, a highly aggressive metastatic hormone‐independent form of the disease (castration or hormone resistant prostate cancer (CRPC) develops in a significant number of patients and the treatment options for these patients remain limited. Even with the recent introduction of improved therapeutics, the average survival time of CRPC patients is still often less than 36 months, 3 , 4 necessitating the development of new and better treatment approaches.
All‐trans retinoic acid (ATRA) plays crucial roles in early development, organogenesis and tissue homeostasis by regulating the balance between cell proliferation, differentiation and apoptosis 5 . ATRA serves as an activating ligand for a host of obligate dimers between one of the retinoic acid receptor (RARα, RARβ, RARγ), and one of the retinoid X receptor (RXRα, RXRβ, RXRγ) family members. In the absence of ligand, gene expression is repressed, whereas ATRA binding leads to coregulator exchange and gene activation. 6 Individual RAR isotypes have different patterns of temporal and spatial expression during prostate development, suggesting that they play discrete roles during prostate development. 7 ATRA plays a role in the development of early prostate precursor cells derived from the murine fetal urogenital sinus, 8 with signaling by RARα or RARγ‐implicated in the promotion of differentiation vs survival/proliferation, respectively. 9 In adults, an adequate supply of ATRA, possibly acting via RARγ, is required for the proliferation and survival of prostate stem cells, 9 as well as the maintenance of proper prostate functionality. 7 , 8 , 10 , 11 Moreover, both ATRA‐deprivation and genetic ablation of RARγ expression block development of the murine prostate, providing further evidence of an important role for RARγ. 10 PC tissue has been observed to contain ATRA at very low levels (in the region of 10−9 M), which is at least an order of magnitude lower than in adjacent normal healthy prostate cells or benign prostate hyperplasia. 12 Any effects arising from low concentrations of ATRA are predicted to be compounded by over‐ and under‐expression, respectively, of the intracellular lipid‐binding proteins FABP5 and CRABP2, which occurs frequently in PC 13 , 14 , 15 . In addition to functioning as a pan‐RAR agonist, ATRA also activates the “orphan” nuclear receptor PPARβ/δ, which promotes proliferation and survival. Whether ATRA is diverted to RARs or oncogenic PPARβ/δ is dependent on the FABP5:CRABP2 ratio, with FABP5 delivering ATRA to PPARβ/δ and CRABP2 delivering ATRA to RARs. It is therefore likely that this is likely to divert available ATRA to oncogenic PPARβ/δ at the expense of retinoic acid receptors (RARs). It is therefore predicted that PC cells become adapted to survive and proliferate in a low‐concentration ATRA environment, with ATRA diverted to oncogenic PPARβ/δ at the expense of retinoic acid receptors (RARs).
Pharmacological concentrations (10−6 M) of ATRA have demonstrated clinical efficacy in a sub‐type of acute myeloid leukemia (AML) referred to as acute promyelocytic leukemia (APL), that is generally associated with the t(15;17)(q24;q21) reciprocal translocation between the PML and RARA genes. 16 This is primarily due to the fact that APL is associated with few cytogenetic defects apart from t(15;17) and the success of ATRA (in combination with arsenic trioxide) in this case is due to its direct targeting of the PML‐RARα fusion oncoprotein. 16 , 17 This idea is supported by the failure of ATRA to yield positive clinical outcomes in other cancers. 18 , 19 , 20 , 21 This may also be due, at least in part, to the fact that ATRA elicits pleiotropic effects—most likely as a result of multiple RAR isotype‐specific actions. 22 , 23 Thus, ATRA may be of limited therapeutic usefulness in complex tumors and approaches that do not incorporate pan‐agonism of RARs, ideally utilizing isotype‐specific agonists and/or antagonists may prove useful. Consistent with this notion, we have previously shown that, in contrast to ATRA, pan‐RAR antagonists induced growth arrest and apoptosis in PC cell lines and primary patient samples, while normal prostate epithelial cells displayed reduced sensitivity. 24 , 25 In this study, we therefore sought to investigate how ATRA concentration affects the growth and survival characteristics of PC cell lines and whether this would afford the opportunity to develop new therapeutic approaches.
2. METHODS
2.1. Compounds
The synthetic retinoids AGN195183, AGN205327, AGN194310, AGN196996, and AGN205728 (Figure 1 and Table 1) were synthesized at the Shanghai Institute of Materia Medica. Their synthesis, development, and specificities have been described previously in Hughes et al and references within. 26 All‐trans‐retinoic acid (Cat. # 0695); the pan‐RAR agonist TTNPB (4‐[(E)‐2‐(5,6,7,8‐Tetrahydro‐5,5,8,8‐tetramethyl‐2‐naphthalenyl)‐1‐propenyl]benzoic acid, Cat. # 0761) 27 ; the RARα‐selective agonist AM580 (4‐[(5,6,7,8‐Tetrahydro‐5,5,8,8‐tetramethyl‐2‐naphthalenyl)carboxamido]benzoic acid, Cat. # 0760) 28 ; the PPARγ‐selective agonist ciglitazone (Cat. # 1307) 29 ; the PPARγ‐selective irreversible antagonist GW9662 (Cat. # 1508) 30 ; the microtubule stabilizer docetaxel (Cat. # 4056); the thymidylate synthetase inhibitor 5‐fluorouracil (Cat. # 3257); and the topoisomerase II inhibitor etoposide (Cat. # 1226) were purchased from Tocris Biosciences. Retinoids were dissolved in 50% methanol/50% dimethylsulphoxide (DMSO) at a concentration of 10 mM (stored at −20°C), and this stock was diluted using culture medium to the required concentration.
FIGURE 1.
Structures of ATRA and retinoid analogs used in this study. Transactivation properties are detailed in Table 1
TABLE 1.
Binding affinities of synthetic retinoids at different retinoic acid receptor isoforms
| ED50 (nM) | ||||
|---|---|---|---|---|
| Retinoids | RARα | RARβ | RARγ | Classification |
| Retinoic acid receptor agonists | ||||
| AGN195183 a | 20.1 | >5000 | >5000 | RARα‐selective agonist |
| AGN205327 b | 3766.0 | 734 | 32 | RARγ‐selective agonist |
| Retinoic acid receptor antagonists | ||||
| AGN194310 c | 4.3 | 5 | 2 | RARαβγ antagonist |
| AGN196996 d | 3.9 | 4036 | >10 000 | RARα‐selective antagonist |
| AGN205728 e | 2400.0 | 4248 | 3 | RARγ‐selective antagonist |
4‐(4‐Chloro‐3‐hydroxy‐5,5,8,8‐tetramethyl‐5,6,7,8‐tetrahydronaphthalene‐2‐carboxamido)‐2,6‐difluorobenzoic acid.
2‐[(E)‐N‐Hydroxy‐C‐(5,5,8,8‐tetramethyl‐6,7‐dihydronaphthalen‐2‐yl)carbonimidoyl]‐1H‐indole‐5‐carboxylic acid.
4‐[[4‐(4‐Ethylphenyl)‐2,2‐dimethyl‐(2H)‐thiochromen‐6‐yl]‐ethynyl]‐benzoic acid.
4‐(3‐Bromo‐4‐ethoxy‐5‐(4‐methylbenzoyl)benzamido)benzoic acid.
4‐[(E,3E)‐3‐[8,8‐Dimethyl‐5‐(4‐methylphenyl)‐7H‐naphthalen‐2‐yl]‐3‐hydroxyiminoprop‐1‐enyl]benzoic acid.
2.2. Cell lines and cell culture
RWPE‐1 (ATCC# CRL‐11609, RRID:CVCL_3791); LNCaP (ATCC# CRL‐1740, RRID:CVCL_1379); DU 145 (ATCC HTB‐81, RRID:CVCL_0105) and PC‐3 (ATCC# CRL‐1435, RRID:CVCL_0035) cells were purchased specifically for this study from the American Type Culture Collection (ATCC, Manassas, VA). The LNCaP, 31 DU 145 32 and PC‐3 33 cell lines were derived from metastatic prostate carcinoma. The poorly invasive, androgen‐dependent LNCaP cell line expresses RARA and RARG mRNA, and RARα and RARγ, but not RARβ protein. 34 , 35 There are conflicting data regarding expression of RARB mRNA in LNCaP cells. 34 , 35 , 36 , 37 DU145 and PC3 cells exhibit androgen‐independent growth and express RARA and RARG, but RARG mRNA, and also RARα and RARγ, but not RARβ. 34 , 35 Human prostate epithelial non‐neoplastic RWPE‐1 cells 38 express RARα and RARγ, but not RARβ. 37 Cell cultures were maintained under serum‐free conditions using the Prostate Epithelial Cell Growth Medium (PrEGM) BulletKit (Cat. # CC‐3166, Lonza) minus retinoic acid in the presence of antibiotics (Lonza). For experiments, cells were seeded in RPMI‐1640 supplemented with ×1 ITS+1 liquid media (containing insulin, transferrin, selenium dioxide, linoleic acid, and bovine serum albumin, Merck) in the presence of antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin Lonza).
2.3. Analysis of cell growth
Cell proliferation was determined either by direct cell count or indirectly using the ViaLight Plus Cell Proliferation and Cytotoxicity BioAssay Kit (Lonza) using a Berthold LB953 luminometer as previously described. 25 Briefly, cells were seeded (400 cells per well) in RPMI‐1640 + ITS+1 culture medium in 96‐well microtitre plates and treated with retinoids both immediately and at day 2 by replacing the medium. The number of viable cells was assessed at day 5 by measuring cellular ATP levels.
2.4. Analysis of colony forming potential
Single cell suspensions of serum‐free adapted PC cells were prepared by trypsinization of 80% confluent plates using 0.25% (v/v) trypsin in phosphate‐buffered saline for 5 minutes at 37°C. Single cell suspensions containing <90% viable cells as measured by trypan blue exclusion were discarded. Cells were seeded at a density of 1000 cells per 65‐mm diameter plastic cell culture dish. Cells were incubated for 21 days at 37°C in 5% CO2 in 4 mL of RPMI 1640 plus ITS+1. Culture medium was replaced every 5 days, and after 21 days cells were fixed using 1% (v/v) formaldehyde in 0.9% (v/v) NaCl and stained with 1% (v/v) methylene blue in 0.9% (v/v) NaCl. Colony‐forming efficiency was determined by counting the number of colonies that formed: (number of colonies formed/number of cells inoculated) × 100. Determination of clone morphology (holoclone, meroclone, or paraclone) was performed as previously described. 39
2.5. Analysis of transactivation
Transactivation assays using CV‐1 kidney fibroblast cells have been described previously. 40 Briefly, CV‐1 kidney fibroblast cells were first transiently transfected with a mammalian plasmid vector (ERE‐Tk‐Luc) 41 that expresses Firefly luciferase under the transcriptional regulation of the Xenopus vitellogenin estrogen response element (ERE), together with the pGL4.70[hRluc] Renilla luciferase control vector (Promega). Cells were also co‐transfected with expression vectors encoding a fusion protein comprising the estrogen receptor DNA‐binding domain and the ligand‐binding domains of either RARα, RARβ, or RARγ. Cells were transfected using Lipofectamine (Thermo) according to the manufacturer's instructions. Twenty‐four hours post‐transfection, cells were treated with increasing concentrations of ATRA (10‐10‐10‐6) for 16 hours in culture medium‐containing 2.5% charcoal‐stripped FBS (Merck). Transactivation was measured using the Dual‐Luciferase Reporter Assay System (Promega). Firefly luciferase reporter activity was normalized against Renilla luciferase activity, and transactivation data are presented as the percentage of the maximal response produced by 1 μM ATRA.
2.6. Analysis of adipogenic differentiation
Adipocyte differentiation was induced in serum‐free adapted LNCaP, PC‐3 or DU145 as described by Zhau et al. 42 Briefly, cells were plated at a density of 3 × 105 cells/dish in 60 mm cell culture dishes in RPMI 1640 plus ITS+1. In order to induce adipogenic differentiation, this growth medium was supplemented with dexamethasone (10−7 M), indomethacin (5 × 10−5 M), and the phosphodiesterase inhibitor 3‐isobutyl‐l‐methyl‐xanthine (IBMX, 5 × 10−5 M). Adipogenic differentiation medium was refreshed every 3 days and adipogenesis was assessed by Oil red O staining on day 14 and day 21. Cells were washed with ice cold PBS and fixed in 5% formaldehyde (v/v in PBS) for 1 hour. Cells were washed and incubated for 15 minutes with 1 mL of Oil red O (0.6% [w/v] in 60% isopropanol/ PBS 1:1 v/v). After washing, cell‐bound Oil red O was extracted in 1 mL isopropanol and absorbance was measured at 510 nm. Values were normalized using the ViaLight Plus Cell Proliferation and Cytotoxicity BioAssay Kit (Lonza).
2.7. Antibody transfection
Serum‐free grown LNCaP cells were transfected with antibodies directed against either RARα or RARγ (Chariot, Active Motif) 43 according to the manufacturer's recommendations. In brief, 50 μg of antibody and 1 μg of β‐galactosidase were resuspended in 100 μL ×2 sterile phosphate‐buffered saline (PBS) and mixed with freshly sonicated Chariot transfection reagent (6 μL of reagent re‐constituted in 100 μL sterile H2O). This mixture was incubated at room temperature for 30 minutes to form the transfection complex reagent. LNCaP cells (5 × 105) were washed twice with pre‐warmed serum‐free RPMI 1640 and the final cell pellet was mixed with the antibody/Chariot mixture and incubated at 37°C in a humidified atmosphere containing 5% CO2. After 30 minutes a further 400 μL of serum‐free RPMI 1640 were added to the cells to achieve the final transfection concentration. The LNCaP cells were incubated at 37°C for a further 5 hours to allow internalization of the antibody. Control cells were treated with the Chariot reagent alone (6 μg in 200 μL of sterile PBS). After antibody transfection, the cells were washed twice in RPMI 1640 + ITS+1 and re‐suspended in a volume of 1 mL. A 100‐μL aliquot of cells was removed, fixed and stained for β‐galactosidase expression to assess transfection efficiency. Cells were grown for a further 12 days at which time colonies were fixed with 2% buffered formaldehyde, washed and stained with 1.25% (w/v) crystal violet stain. Polyclonal antiserum to RARα was a generous gift from Dr P. Chambon (Institute for Genetics and Cellular and Molecular Biology, Strasbourg, France), and the antibody to RARγ (Cat. # sc‐550) was purchased from Santa Cruz Biotechnology Inc.
2.8. Cell cycle analysis
Cell cycle status was measured by staining harvested cells with propidium iodide (PI, Molecular Probes) according to the manufacturer's instructions. The distribution of cells between phases of the cell cycle was determined using a FACS Calibur flow cytometer and the CellFIT cell‐cycle analysis software (Becton‐Dickinson). The cell cycle distribution is shown as the percentage of cells containing 2n (G1 phase), 4n (G2 and M phases) and 4n > 3 > 2n DNA amount (S phase) judged by propidium iodide staining. The apoptotic population is defined by the percentage of cells with DNA content lower than 2n (sub‐G1 phase).
2.9. Analysis of apoptosis
Apoptosis‐associated DNA fragmentation was measured using the TACS 2 TdT Fluorescein in situ apoptosis detection kit (Trevigen). Briefly, serum‐free adapted PC cells were seeded at a density of 5 × 105 cells per 75 cm2 flask and treated with retinoids of vehicle control. Treated cells (both adherent cells and those in suspension) were trypsinized, cytospun onto microscope slides and analyzed according to the manufacturer's instructions. Induction of apoptosis was assessed using fluorescence microscopy and quantified by counting five separate fields of view (minimum of 500 cells).
Activation of caspase was measured essentially as described by Keedwell et al. 25 The pan‐caspase inhibitor Z‐VAD‐FMK (R&D Systems) was used to investigate the involvement of caspases in AGN205728‐induced apoptosis. A 20 mM stock solution was prepared in DMSO. Z‐VAD‐FMK was added at 50 μM to bulk cultures of cells 1 hour before adding AGN205728, and cells were monitored using the TUNEL assay. Fluorometric assays for caspase activity were conducted in 96‐well microtiter plates. In total, 50 μL of assay buffer (20 mM HEPES, pH 7.5, 10% glycerol, 2 mM dithiothreitol) containing 50 μM of peptide substrates for caspase‐3 (DEVD‐AFC), caspase‐8 (IETD‐AFC), or caspase‐9 (LEHD‐AFC) (all from Biovision Inc.) were added to each well. In total, 50 μL of cell lysate were added to initiate the reactions. Backgrounds were measured in wells that contained assay buffer, substrate, and lysis buffer. Fluorescence was measured on a CytoFluor 4000 fluorescence plate reader (Applied Biosystems) set at 400 nm excitation and 508 nm emission. Caspase activities were calculated as fold increases relative to control wells.
2.10. Software, data manipulation and statistical analyses
At least three experiments of each type were performed with triplicate replicates. Results are expressed as means ± SD (± SD). Statistical significance between groups of data was analyzed by unpaired t‐test or 2‐way ANOVA as appropriate using GraphPad Prism (GraphPad Software, La Jolla, CA) or SigmaStat (Systat Software, San Jose, CA). Statistical significance is represented as follows: ns (not significant), P ≥ .05; *, P < .05; **, P < .01; ***, P < .001; ****, P ≤ .0001. Dose‐response curves for compounds were plotted using the kinetics module of SigmaPlot (Systat Software, San Jose, CA). Marvin was used for drawing and displaying chemical structures of ATRA and synthetic retinoids (Marvin 19.12, 2019, ChemAxon).
3. RESULTS
3.1.1. Low concentrations of all‐trans retinoic acid stimulate proliferation and colony forming ability of prostate cancer cells
In light of our hypothesis that PC cells may be adapted to survive and proliferate in a low ATRA concentration environment, we initially sought to titrate ATRA against three model PC cell lines derived from metastatic prostate carcinoma: LNCaP, PC‐3, and DU145. We found that in all three PC cell lines, ATRA treatment for 5 days within the range of 10−11‐10−9 M resulted in an increase in cell numbers of up to 1.5‐fold (Figure 2A). By contrast, treatment ATRA at concentrations of 10−8 M and above progressively diminished cell growth. In order to assess the long‐term effects of low vs high concentrations of ATRA on cell growth and survival, we performed single‐cell clonogenic assays on LNCaP, PC‐3 and DU145 cells and RWPE‐1 cells, which are derived from HPV‐18‐transformed epithelial cells from the peripheral zone of normal prostate. Consistent with our 5‐day ATRA treatment results, we found that for all three PC cell lines treatment with 10−10 M ATRA increased the colony forming efficiency (CFE) while treatment with 10−6 M ATRA decreased CFE (Figure 2B). By contrast, non‐tumorigenic RWPE‐1 cells, which untreated had a low CFE of 0.43, failed to respond to either high or low concentrations of ATRA. These results indicated that treatment of malignant PC cell lines with low nanomolar concentrations of ATRA favors survival/proliferation of the clonogenic fraction, whereas higher pharmacological concentrations promote growth arrest and/or differentiation.
FIGURE 2.
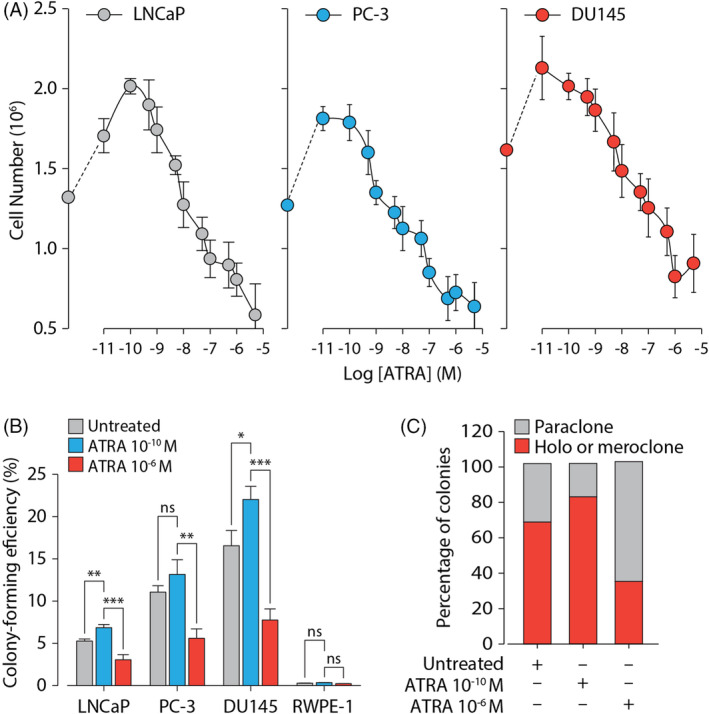
Low doses of all‐trans retinoic acid stimulate growth of prostate cancer cell lines. (A) Serum‐free adapted LNCaP, PC‐3 and DU145 cells were treated with ATRA at the indicated concentrations. Results are presented as the mean number of cells recovered (±SD). (B) The ability of serum‐free adapted LNCaP, PC‐3, or DU145 cells to form colonies in response to 10−10 M and 10−6 M ATRA as indicated was analyzed by methylene blue staining. Results are presented as mean colony‐forming efficiencies (±SD). (C) Serum‐free adapted LNCaP cells were treated with 10−10 M and 10−6 M ATRA as indicated and assessed for ability to form holoclone/meroclone, or paraclone colonies. Results are presented as the percentage of colony types as indicated. Statistical significance is represented as described in Section 2
When subjected to single cell clonogenic assays, malignant PC cells produce colonies of three different types: holocolone (comprising large cells with homogeneous morphology), meroclone (comprising cells with intermediate size and heterogeneous morphology), and paraclone (comprising smaller cells with homogeneous morphology). These morphologically distinct types of colonies are considered to contain cells that possess stem cell‐like (holocolone), early progenitor (meroclone), or late progenitor/differentiation‐committed (paraclone) characteristics. In the absence of ATRA, around 70% of colonies formed by serum‐free adapted LNCaP displayed typical 39 large holoclone/meroclone morphologies with the remaining 30% containing cells with the smaller, differentiated paraclone morphology (Figure 2C). Treatment with 10−10 M ATRA increased the proportion of colonies displaying morphologies consistent with stem cell‐like or early progenitor status to around 85%. Treatment with 10−6 M ATRA, on the other hand, increased the proportion of colonies displaying late progenitor/differentiation‐committed (paraclone) characteristics to more than 60%. Taken together, these results suggested that the low physiological concentrations of ATRA found in PC serve to either increase the clonogenic (stem cell) population by providing a stem‐cell specific survival stimulus or block entry of this cell population into a spontaneous differentiation/apoptosis programme.
3.1.2. Agonism of RARγ promotes growth and inhibits adipogenic differentiation in prostate cancer cells
ATRA is the natural endogenous ligand for all three RAR isotypes and they display similar binding affinities for this retinoid (RARα, 0.37 × 10−9 M; RARβ, 0.37 × 10−9 M; RARγ, 0.22 × 10−9 M). 44 However, we and others have previously shown that while ATRA stimulates transactivation in CV‐1 cells via RARβ and RARγ at sub‐nanomolar concentrations, much higher concentrations are required for RARα‐mediated transactivation. 44 , 45 We also observed this phenomenon in LNCaP prostate cancer cells (that do not express RARβ), where RARγ displayed a half‐maximal response (EC50) value of 0.24 × 10−9 M while RARα displayed an EC50 value of 19 × 10−9 M, which was 79‐fold higher. This prompted us to investigate whether individual RAR isotypes played a role in the differential response of PC cells to low vs high concentrations of ATRA. Consistent with our prior data, we found that RARβ and RARγ displayed EC50 values of 0.39 × 10−9 M and 0.36 × 10−9 M, respectively, while ATRA had a minimal effect on transactivation via RARα at concentrations below 10−9 M (EC50 = 12.4 × 10−9 M) (Figure 3A‐C). This represented a 30‐fold difference and maximal transactivation by RARα did not occur until 10−6 M. We then examined the growth response of LNCaP cells of RAR isotype‐selective agonists in comparison to the pan‐RAR agonist ATRA (Figure 3D). In line with results suggesting opposing roles for RARα and RARγ in prostate growth and differentiation (and potentially in PC), the RARγ‐selective agonist AGN205327 elicited an effect on LNCaP cells that mirrored ATRA while the RARα‐selective agonists AM580 and AGN195183 failed to promote cell growth at low concentrations. It is also important to note that the modest inhibition in the growth of LNCaP cells and concentrations greater than 10−6 M can be explained by a loss of receptor specificity and subsequent agonism of RARα at high concentrations (Table 1). Further supporting the notion that, in contrast to RARγ, RARα has anti‐growth and pro‐differentiative role PC, co‐treatment of AGN195183 with an RARα‐selective antagonist (AGN196996) abolished AGN195183‐mediated growth inhibition in LNCaP cells (Figure 3E). Moreover, co‐treatment of ATRA with AGN196996 promoted proliferation of LNCaP cells, likely due to activity of ATRA on RARγ.
FIGURE 3.
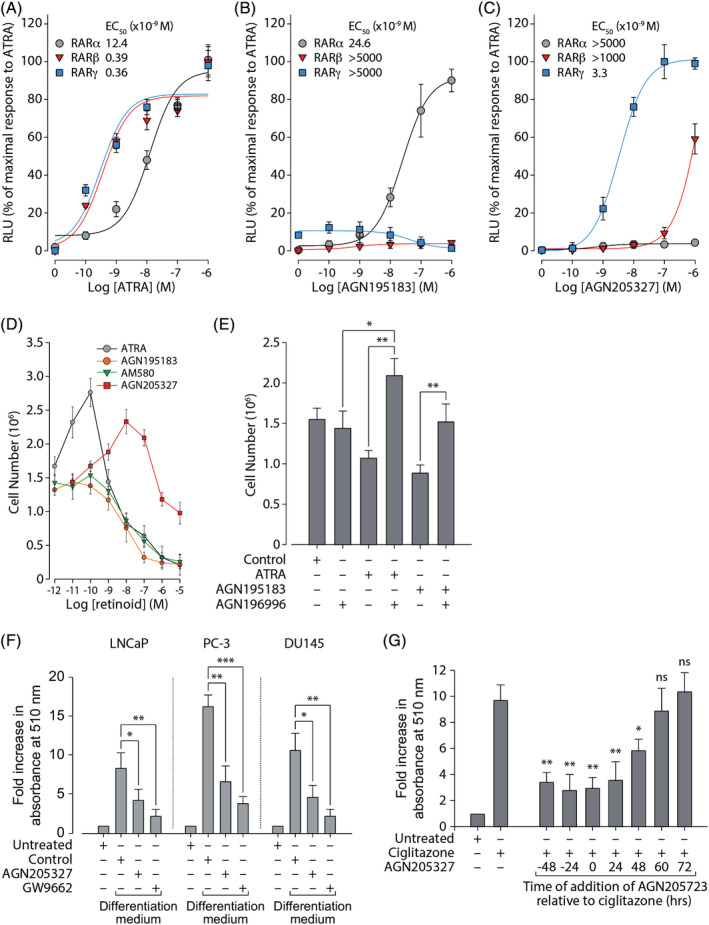
RARγ promotes growth and inhibits differentiation of prostate cancer cell lines. (A‐C) CV‐1 cells were transiently transfected with expression vectors encoding a fusion protein containing the estrogen receptor (ER) DNA binding domain (DBD) and the ligand binding domain (LBD) of the indicated RAR isotype, together with a firefly luciferase reporter under the control of an estrogen response element (ERE‐Tk‐Luc). Differences in transfection efficiency between samples were normalized using a control vector constitutively expressing Renilla luciferase. Transfected cells were treated with (A) ATRA, (B) AGN195183, or (C) AGN205327 at the indicated concentrations for 24 hours analyzed for luciferase activity. Results shown are from a typical experiment performed in triplicate and are expressed as relative light units (RLU, arbitrary units) ± SD that have been normalized to the maximal response to ATRA (10−6 M). The compound concentrations at which half‐maximal effect is observed (EC50) for RARα, RARβ, or RARγ are indicated. (D) Serum‐free adapted LNCaP cells were treated with the indicated concentrations of pan‐RAR agonist ATRA, RARγ‐selective agonist AGN205327, RARα‐selective agonists AGN195183 and AM580. Results are presented as the mean number of cells recovered (±SD). (E) Serum‐free adapted LNCaP cells were treated for 4 days with ATRA (10−8 M), AGN195183 (10−8 M) and the RARα‐selective antagonist AGN196996 (10−8 M) as indicated. Results are presented as the mean number of cells recovered (±SD). (F) Serum‐free adapted LNCaP, PC‐3, or DU145 cells were grown in differentiation medium without compound (Control), containing AGN205327 (10−7 M) or PPARγ‐selective antagonist GW9662 (10−5 M) as indicated. 14 days post‐induction of differentiation, cells were tested for lipid droplet accumulation using the Oil red O assay. Results are presented as mean fold increase in absorbance at 510 nm (±SD) relative to undifferentiated serum‐free adapted cells (Untreated). (G) Adipogenic differentiation of serum‐free adapted LNCaP cells was induced by treatment with PPARγ‐selective agonist ciglitazone (10−6 M). AGN205327 (10−7 M) was added at the indicted times pre‐ or post‐treatment with ciglitazone. Cells were analyzed and results presented as described in (F). Statistical significance is represented as described in Section 2
PC cell lines contain a sub‐population of cells that possess characteristics similar to mesenchymal stem cells that, upon appropriate stimulation, can undergo neuroendocrine, adipogenic, or osteoblastic differentiation. 42 When grown in a pro‐differentiative culture medium containing dexamethasone, indomethacin, and the phosphodiesterase inhibitor IBMX, lipid droplets began to form within cells and 10‐12 days post‐induction of differentiation, lipid accumulation may be quantitatively analyzed. In agreement with our results suggesting that, via either RARα or RARγ, ATRA can elicit either pro‐ or anti‐growth and differentiation responses in PC, it has also been reported that ATRA can block the differentiation of pre‐adipocyte‐like cells into lipid‐loaded adipocytes in several model systems. 46 , 47 , 48 , 49 , 50 We therefore examined whether RARγ agonism could inhibit adipogenic differentiation in PC cell lines. Oil red O staining showed that treatment with AGN205327 (10−7 M) was able to inhibit the accumulation of lipid droplets by around 50% (Figure 3F). These results were of a similar order to those obtained with the PPARγ antagonist GW9662 (10−5 M). We also examined the ability of AGN205327 to diminish adipogenic differentiation arising from PPARγ agonism (Figure 3G). We found that induction of adipogenic differentiation in LNCaP cells by the PPARγ agonist ciglitazone (10−7 M) was inhibited by up 70% when AGN205327 (10−7 M) was given at time points ranging between 48 hours pre‐ and post‐addition of ciglitazone. Addition of AGN205327 later than 48 hours post‐induction of adipogenesis failed to have any effect on ciglitazone‐induced adipogenesis, which is consistent with studies showing that retinoids can only block adipogenesis when added during the early commitment phase. 47 , 50 , 51
3.1.3. Antagonism of RARγ inhibits growth and colony formation in prostate cancer cells
Our results showing that RARγ agonism could promote growth and clonogenicity, and inhibit differentiation strongly suggested that RARγ‐selective antagonism could have the opposite effects. This is in line with our previous data showing that treatment of LNCaP cells with pan‐RAR antagonist AGN194310 (Figure 1, S1A, S1D and Table 1) inhibited colony formation. 25 Consistent with these observations, the RARγ‐selective antagonist AGN205728 (Figure 1, S1B, S1E and Table 1) diminished the CFE of all three PC cell lines in a fashion similar to AGN194310 (Figure 4A). Treatment with neither ATRA, nor the RARα‐selective antagonist AGN196996 (Figure 1, S1C, S1F and Table 1) proved as effective AGN205728 or AGN194310. These data indicate that antagonism of RARγ alone is sufficient to reduce colony formation in PC cell lines. The growth inhibitory effect of treatment AGN205728 in PC cell lines was observed also in short‐term assays, with concentrations of 10−6 M and above abolishing metabolic activity (Figure 4B). By contrast, non‐tumorigenic RWPE‐1 cells showed significantly diminished response to AGN205728. Consistent with this, AGN194310 and AGN205728 but not AGN196996 were able to diminish RAR‐mediated transactivation stimulated by ATRA (Figure 4C).
FIGURE 4.
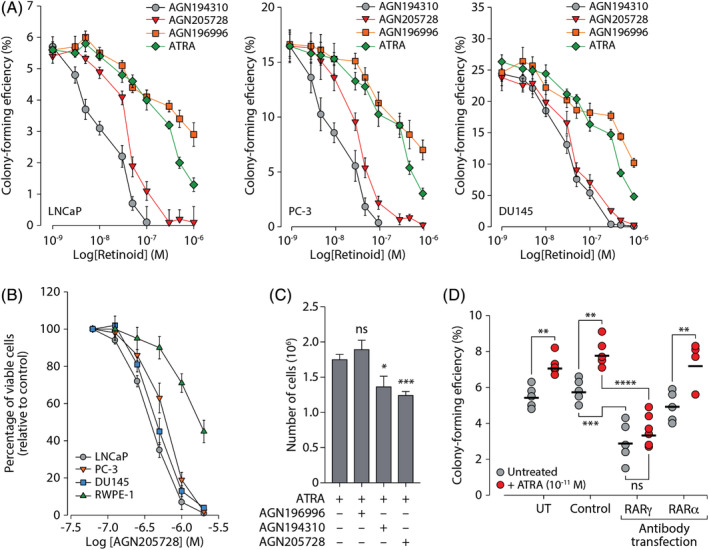
Antagonism of RARγ inhibits growth of prostate cancer cell lines. (A) The ability of serum‐free adapted LNCaP, PC‐3, or DU145 cells to form colonies was analyzed in response to treatment with ATRA, AGN196996, pan‐RAR antagonist AGN194310, or RARγ‐selective antagonist AGN205728 at the indicated concentrations. Results are presented as mean colony‐forming efficiencies (±SD). (B) Serum‐free adapted LNCaP, PC‐3, DU145, or RWPE‐1 cells were treated with AGN205728 as indicated. Metabolic activity (intracellular ATP concentration) was measured using the Vialight HS High Sensitivity Cell Proliferation/Cytotoxicity assay. Results are presented as percentage of viable cells relative to control (±SD). (C) Serum‐free grown LNCaP cells were treated 5 × 10−8 M AGN196996, AGN194310, or AGN205728 or vehicle control as indicated for 12 hours prior to stimulation with 10−10 M ATRA. After 96 hours cells, cells were harvested by trypsinization and counted. The data are presented as the mean number of cells recovered from three separate flasks (±SD). (D) Serum‐free grown LNCaP cells were transfected with polyclonal antibodies directed against RARα or RARγ as indicated. Cells were grown for 12 days post‐transfection ± ATRA as indicated and analyzed for ability to form colonies. Results are presented as mean colony‐forming efficiencies (±SD). Statistical significance is represented as described in Section 2
While the EC50 data indicated a high degree of selectivity AGN205728 towards RARγ (Table 1), strongly supporting a role for this RAR isotype in the pathology of PC, we nevertheless wanted to demonstrate the role of RARγ by other means. To this end, we took a completely different approach to abrogating activation of RARγ and used an anti‐RAR antibody to interfere with its function (Figure 4D). Transfection with this “neutralizing” antibody phenocopied treatment with AGN205728 and diminished CFE, both with and without low dose (10−11 M) ATRA. This result validated RARγ as a bona fide therapeutic target, at least as far as in vitro studies permit. By contrast, and consistent with results from RARα‐selective antagonism, treatment with a neutralizing antibody raised against RARα failed to block increases in CFE arising from low dose ATRA treatment. These data reveal that antagonism of RARγ is sufficient to inhibit the growth PC cell lines in vitro and non‐malignant prostate cells display reduced sensitivity to this treatment.
3.1.4. Antagonism of RARγ in LNCaP cells induces growth arrest followed by caspase‐independent apoptosis
In order to understand the mechanisms underlying the inhibition of growth and clonogenic potential we observed upon abrogation of proper RARγ function, we first analyzed the effects of treatment with AGN194310 (10−6 M) or AGN205728 (10−6 M) on the cell cycle (Figure 5A). The proportion of cells in G1 phase of the cell cycle gradually increased following treatment with AGN194310 or AGN205728, which was paralleled by a fall in the proportion of cells found in S‐phase. After 12 hours the proportion of cells identified as sub‐G0/G1 (indicative of apoptosis) started to rise until at the 48‐hour timepoint almost 60% of cells were sub‐G0/G1 following AGN194310 treatment and almost 40% of cells were sub‐G0/G1 following AGN205728 treatment. The accumulation propidium iodide‐stained DNA in sub‐G0/G1 compartment indicated that cellular DNA had begun to fragment, a biomarker of apoptotic or necrotic cell death. Time‐course agarose gel electrophoresis analysis of DNA from AGN205728‐treated LNCaP cells revealed a discrete laddering pattern that is typical of cells undergoing apoptosis rather the more generalized smeared pattern that is associated with necrosis (Figure 5B). A quantitative readout of the percentage of cells undergoing apoptosis following treatment with AGN194310 (10−6 M) or AGN205728 (10−6 M) was obtained using the terminal deoxynucleotide transferase end‐labeling (TUNEL) assay (Figure 5C). After 24 hours, the percentage of cells undergoing apoptosis gradually increased until 60 hours post‐treatment the proportion of apoptotic cells reached 35% and 40% for AGN194310 and AGN205728, respectively. Again, among the untreated cells and the AGN196996‐treated sample there was little evidence of induction of apoptosis. We then assessed the contribution of caspases to AGN205728‐associated induction of apoptosis. Here, we found that pre‐treatment with the pan‐caspase inhibitor Z‐VAD‐FMK (5 × 10−5 m) failed to diminish induction of apoptosis by AGN205728 (Figure 5D) and, furthermore, AGN205728 failed to activate caspases‐3, −8 or −9 (Figure 5E).
FIGURE 5.
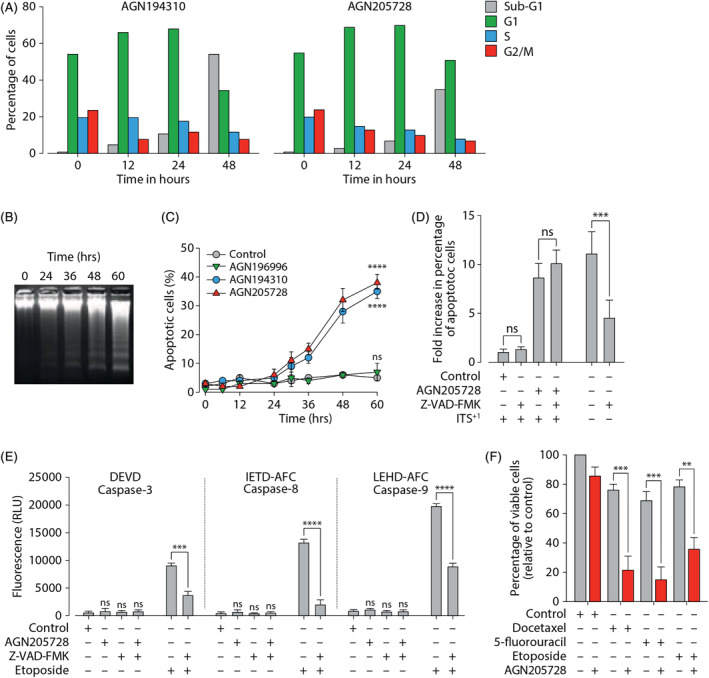
Antagonism of RARγ leads to caspase‐independent apoptosis. (A) Serum‐free adapted LNCaP cells were treated with AGN194310 or AGN205728 as indicated, stained with propidium iodide and analyzed by flow cytometry. (B) Agarose gel electrophoresis analysis of DNA from AGN205728‐treated LNCaP cells. (C) Serum‐free adapted LNCaP cells were treated with 10−6 M AGN194310, AGN205728,or AGN196996 as indicated. Time course of the appearance of DNA‐strand breaks was assessed by TUNEL assay (TACS‐2 TdT Fluorescein in situ apoptosis detection kit). TUNEL‐positive cells were counted for every 500 cells from five fields from each slide, and three different slides were analyzed for each sample. Results are presented as percentage of apoptotic cells (±SD). (D) Serum‐free adapted LNCaP cells were treated with 10−6 M AGN205728 and the pan‐caspase inhibitor Z‐VAD‐FMK as indicated and assayed as described above. (E) Serum‐free adapted LNCaP cells were treated with 10−6 M AGN205728, Z‐VAD‐FMK, or etoposide (positive control) as indicated. Cleavage of caspase 3‐, −8 or −9 was assayed using the caspase‐3 substrate DECD‐AFC, the caspase‐8 substrate IEHD‐AFC or the caspase‐9 substrate LEHD‐AFC, respectively. (F) Serum‐free adapted LNCaP cells were treated with docetaxel (10−10 M), 5‐fluorouracil (5 × 10−6 M) or etoposide (5 × 10−6 M) alone or in combination with AGN205728 (10−7 M) as indicated. Metabolic activity (intracellular ATP concentration) was measured using the Vialight HS High Sensitivity Cell Proliferation/Cytotoxicity assay. Statistical significance is represented as described in Section 2
Chemotherapy has been one of the mainstays in the treatment of castration‐resistant PC, including the taxanes, carbazitaxel, and docetaxel. 52 These agents induce caspase‐dependent apoptosis (similar to RARα agonism) in numerous PC cell lines, which prompted us to test docetaxel in combination with low‐dose AGN205728 (10−7 M). We found that the combination of AGN205728 and docetaxel led to a significant reduction in cell viability compared to their use as single agents (Figure 5F). Similar results were obtained with the pyrimidine analog 5‐fluorouracil (5‐FU) and the topoisomerase 2 inhibitor etoposide.
4. DISCUSSION
Retinoic acid signaling in prostate carcinoma cells is complex and our data strongly suggest that is at least in part because RARα and RARγ play opposing roles in the promotion of differentiation and proliferation/survival, respectively. This notion is in agreement with studies in breast carcinoma cell lines showing that RAR isoforms have opposing and, perhaps, mutually antagonistic roles in controlling proliferation. 14 , 53 , 54 , 55 , 56 While RARγ supports the proliferation and survival of malignant breast epithelial cells RARα, on the other hand, inhibits proliferation and promotes differentiation. Our results suggest that this complexity is due, at least in part, to concentration‐dependent differential responses of RAR isotypes to ATRA. At low nanomolar levels, RARγ is activated preferentially, while higher concentrations of ATRA (including pharmacological levels) activate both. These findings are in keeping with studies that have reported that ATRA exerts concentration‐related biphasic effects on the proliferation of PC cell lines when these cells are grown under serum free conditions. 11 , 35 , 57 , 58 , 59 , 60 , 61 , 62 , 63 Furthermore, we have shown that low levels of ATRA, probably acting via RARγ, increase colony forming efficiency, promote proliferation, and survival and oppose differentiation, suggesting that stem‐cells or early progenitors may be the target cell populations.
It is well‐established that PC cell populations contain fractions possessing mesenchymal stem cell‐like properties and can be differentiated into adipocytes, 42 , 64 and we demonstrate that this can be blocked by activation of RARγ. The differentiation cocktail used in this study for the induction of adipogenesis in LNCaP cells included the glucocorticoid agonist dexamethasone and the phosphodiesterase inhibitor IBMX, the combination of which down‐regulates expression of RARγ in pre‐adipocyte models. 49 The down‐regulation of RARγ expression prior the onset of adipogenesis is an essential molecular signature of several in vitro pre‐adipocyte differentiation models 48 , 49 , 50 while in vivo, an inverse correlation between RARγ expression and adiposity has been demonstrated. 48 , 65 Moreover, RARγ agonism inhibits the expression of the pro‐adipogenic nuclear receptor PPARγ and thereby blocks the adipogenic signals emanating from this receptor. 46 , 51 , 66 , 67 Our observation that the addition of an RARγ agonist prior to adipogenesis induction blocks PPARγ‐stimulated adipogenesis of LNCaP cells is consistent with these findings.
The deregulation of retinoic acid signaling in malignant cells also has implications for the use of retinoids as chemotherapeutic agents. It has long been known that pharmacological concentrations of ATRA (> 10−7 M), acting via RARα, can drive malignant prostate cells into growth arrest and apoptosis both in vitro and in vivo. 68 However, high dose retinoid treatment for PC, as well as most other solid tumors, has had limited success in the clinic. 18 , 19 , 20 , 69 Several issues contribute to this failure, including acquisition of retinoid‐resistance due to promoter hypermethylation‐mediated loss of RARβ expression in patients with advanced PC. 70 , 71 , 72 RARβ is known to play a role in driving growth arrest in malignant prostate cells at physiological ATRA concentrations, so its absence may provide a growth/survival advantage to the malignant cells over their normal counterparts. Second, the altered biology of cancer cells may negatively impact uptake, metabolism, storage of retinoids, thus impeding the ability to achieve a high enough local concentrations of ATRA to activate RARα. Indeed, pharmacodynamic studies have shown that the plasma concentrations of ATRA (in the region of 2.2‐0.4 μM) that have to be achieved before marginal clinical benefits are observed are often associated with therapy‐limiting toxicities. 69
The pan‐RAR agonistic activities of ATRA may well preferentially activate RARγ over RARα in the tumor microenvironment 53 , 55 , 73 and our results indicate that antagonism RARγ may represent a better therapeutic strategy. This notion is in agreement with our previous findings that pan‐RAR antagonists can induce caspase‐independent apoptosis more effectively that ATRA in PC cell lines. 25 Our results also suggest that RARγ antagonists may be effective in combination with cytotoxic chemotherapy. We showed that a sub‐maximally effective concentration of AGN205728 (10−7 M) significantly increased the degree of cell death induced in serum free adapted‐LNCaP cells produced by sub‐maximal concentrations of the taxane docetaxel, as well as sub‐optimal concentrations of the commonly used chemotherapeutic agents 5‐fluorouracil and etoposide. Docetaxel is used for the treatment of castration‐resistant PC but this is limited due to high levels of toxicity (particularly neutropenia) and either innate or acquired resistance in a significant percentage of patients. If the sensitivity of PC towards taxanes and other therapeutic agents could be increased, both the incidence of toxicity and the acquisition of resistance may be reduced. Finally, while we performed experiments in defined media in order to prevent ATRA and, potentially, androgens or other factors found in fetal bovine serum from interfering with our data, the androgen‐deprived environment created mimics the clinical scenario. Recent research indicates that androgen depravation induces a neuroendocrine phenotype in LNCaP cells that is followed by the acquisition of a “stem cell‐like” state characterized by expression of markers including markers CD133, ALDH1A1, and ABCB1A. 74 Many PC patients who are not cured with surgery or radiation and subsequently undergo androgen deprivation therapy (ADT) develop resistance to ADT and relapse with castration resistant prostate cancer (CRPC) that is associated with very poor outcome. Our data indicate RARγ antagonism to be an effective approach in this context and thus merits further investigation.
In conclusion, we have demonstrated that lower than normal concentrations of ATRA oppose differentiation, promote the survival and proliferation of several PC cell lines by activating RARγ. We also showed that RARγ antagonists alone can block proliferation of both androgen‐dependent and independent PC cell lines and eventual induce caspase‐independent apoptosis. Hence, RARγ antagonism alone, or in combination with cytotoxic chemotherapy, may represent a novel approach to the treatment of advanced prostate that targets both androgen‐dependent and androgen‐independent PC cells.
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
AUTHOR CONTRIBUTIONS
Kevin Petrie: Formal analysis; investigation; methodology; supervision; writing‐original draft; writing‐review and editing. Zuzanna Urban‐Wojciuk: Formal analysis; investigation; methodology; writing‐original draft; writing‐review and editing. Yordan Sbirkov: Formal analysis; investigation; methodology; writing‐original draft; writing‐review and editing. Amy Graham: Formal analysis; investigation; writing‐review and editing. Annika Hamann: Formal analysis; investigation; writing‐original draft; writing‐review and editing. Geoffrey Brown: Conceptualization; formal analysis; investigation; methodology; project administration; supervision; writing‐original draft; writing‐review and editing.
ETHICAL STATEMENT
Not Applicable.
Supporting information
Figure S1 Supporting information
ACKNOWLEDGEMENTS
Zuzanna Urban‐Wojciuk was supported by the project “International Centre for Cancer Vaccine Science” carried out within the International Research Agendas Programme of the Foundation for Polish Science co‐financed by the European Union under the European Regional Development Fund.
Petrie K, Urban‐Wójciuk Z, Sbirkov Y, Graham A, Hamann A, Brown G. Retinoic acid receptor γ is a therapeutically targetable driver of growth and survival in prostate cancer. Cancer Reports. 2020;3:e1284. 10.1002/cnr2.1284
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Duncan ME, Goldacre MJ. Mortality trends for benign prostatic hyperplasia and prostate cancer in English populations 1979‐2006. BJU Int. 2011;107(1):40‐45. [DOI] [PubMed] [Google Scholar]
- 2. Maddams J, Utley M, Moller H. Projections of cancer prevalence in the United Kingdom, 2010‐2040. Br J Cancer. 2012;107(7):1195‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sartor O. Androgen deprivation—continuous, intermittent, or none at all? N Engl J Med. 2012;367(10):945‐946. [DOI] [PubMed] [Google Scholar]
- 4. Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14(4):401‐407. [DOI] [PubMed] [Google Scholar]
- 5. Gutierrez‐Mazariegos J, Theodosiou M, Campo‐Paysaa F, Schubert M. Vitamin a: a multifunctional tool for development. Semin Cell Dev Biol. 2011;22(6):603‐610. [DOI] [PubMed] [Google Scholar]
- 6. Samarut E, Rochette‐Egly C. Nuclear retinoic acid receptors: conductors of the retinoic acid symphony during development. Mol Cell Endocrinol. 2012;348(2):348‐360. [DOI] [PubMed] [Google Scholar]
- 7. Prins GS, Putz O. Molecular signaling pathways that regulate prostate gland development. Differentiation. 2008;76(6):641‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vezina CM, Allgeier SH, Fritz WA, et al. Retinoic acid induces prostatic bud formation. Dev Dyn. 2008;237(5):1321‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blum R, Gupta R, Burger PE, et al. Molecular signatures of prostate stem cells reveal novel signaling pathways and provide insights into prostate cancer. PLoS One. 2009;4(5):e5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, Chambon P. Function of retinoic acid receptor gamma in the mouse. Cell. 1993;73(4):643‐658. [DOI] [PubMed] [Google Scholar]
- 11. Peehl DM, Wong ST, Stamey TA. Vitamin a regulates proliferation and differentiation of human prostatic epithelial cells. Prostate. 1993;23(1):69‐78. [DOI] [PubMed] [Google Scholar]
- 12. Pasquali D, Thaller C, Eichele G. Abnormal level of retinoic acid in prostate cancer tissues. J Clin Endocrinol Metab. 1996;81(6):2186‐2191. [DOI] [PubMed] [Google Scholar]
- 13. Morgan E, Kannan‐Thulasiraman P, Noy N. Involvement of fatty acid binding protein 5 and PPARbeta/delta in prostate cancer cell growth. PPAR Res. 2010;2010:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129(4):723‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Napoli JL. Cellular retinoid binding‐proteins, CRBP, CRABP, FABP5: effects on retinoid metabolism, function and related diseases. Pharmacol Ther. 2017;173:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Thé H, Pandolfi PP, Chen Z. Acute promyelocytic leukemia: A paradigm for oncoprotein‐targeted cure. Cancer Cell. 2017;32(5):552‐560. [DOI] [PubMed] [Google Scholar]
- 17. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505‐2515. [DOI] [PubMed] [Google Scholar]
- 18. Bhutani T, Koo J. A review of the chemopreventative effects of oral retinoids for internal neoplasms. J Drugs Dermatol. 2011;10(11):1292‐1298. [PubMed] [Google Scholar]
- 19. Kelly WK, Osman I, Reuter VE, et al. The development of biologic end points in patients treated with differentiation agents: an experience of retinoids in prostate cancer. Clin Cancer Res. 2000;6(3):838‐846. [PubMed] [Google Scholar]
- 20. Pili R, Salumbides B, Zhao M, et al. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13‐cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106(1):77‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cruz FD, Matushansky I. Solid tumor differentiation therapy – is it possible? Oncotarget. 2012;3(5):559‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer therapy. Br J Cancer. 2014;111(11):2039‐2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrie K, Zelent A, Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol. 2009;16(2):84‐91. [DOI] [PubMed] [Google Scholar]
- 24. Hammond LA, Van Krinks CH, Durham J, et al. Antagonists of retinoic acid receptors (RARs) are potent growth inhibitors of prostate carcinoma cells. Br J Cancer. 2001;85(3):453‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keedwell RG, Zhao Y, Hammond LA, et al. An antagonist of retinoic acid receptors more effectively inhibits growth of human prostate cancer cells than normal prostate epithelium. Br J Cancer. 2004;91(3):580‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughes PJ, Zhao Y, Chandraratna RA, Brown G. Retinoid‐mediated stimulation of steroid sulfatase activity in myeloid leukemic cell lines requires RARalpha and RXR and involves the phosphoinositide 3‐kinase and ERK‐MAP kinase pathways. J Cell Biochem. 2006;97(2):327‐350. [DOI] [PubMed] [Google Scholar]
- 27. Loeliger P, Bollag W, Mayer H. Arotinoids, a new class of highly active retinoids. Eur J Med Chem. 1980;15(1):9‐15. [Google Scholar]
- 28. Kagechika H, Kawachi E, Hashimoto Y, Himi T, Shudo K. Retinobenzoic acids. 1. Structure‐activity relationships of aromatic amides with retinoidal activity. J Med Chem. 1988;31(11):2182‐2192. [DOI] [PubMed] [Google Scholar]
- 29. Willson TM, Cobb JE, Cowan DJ, et al. The structure‐activity relationship between peroxisome proliferator‐activated receptor gamma agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem. 1996;39(3):665‐668. [DOI] [PubMed] [Google Scholar]
- 30. Bendixen AC, Shevde NK, Dienger KM, Willson TM, Funk CD, Pike JW. IL‐4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator‐activated receptor gamma 1. Proc Natl Acad Sci U S A. 2001;98(5):2443‐2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horoszewicz JS, Leong SS, Kawinski E, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43(4):1809‐1818. [PubMed] [Google Scholar]
- 32. Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF. Isolation of a human prostate carcinoma cell line (DU 145). Int J Cancer. 1978;21(3):274‐281. [DOI] [PubMed] [Google Scholar]
- 33. Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC‐3). Invest Urol. 1979;17(1):16‐23. [PubMed] [Google Scholar]
- 34. Campbell MJ, Park S, Uskokovic MR, Dawson MI, Koeffler HP. Expression of retinoic acid receptor‐beta sensitizes prostate cancer cells to growth inhibition mediated by combinations of retinoids and a 19‐nor hexafluoride vitamin D3 analog. Endocrinology. 1998;139(4):1972‐1980. [DOI] [PubMed] [Google Scholar]
- 35. Jones HE, Eaton CL, Barrow D, Dutkowski C, Griffiths K. Response of cell growth and retinoic acid receptor expression to retinoic acid in neoplastic and non‐neoplastic prostate cell lines. Prostate. 1997;30(3):174‐182. [DOI] [PubMed] [Google Scholar]
- 36. Moison C, Senamaud‐Beaufort C, Fourriere L, et al. DNA methylation associated with polycomb repression in retinoic acid receptor beta silencing. FASEB J. 2013;27(4):1468‐1478. [DOI] [PubMed] [Google Scholar]
- 37. Long MD, Singh PK, Russell JR, et al. The miR‐96 and RARgamma signaling axis governs androgen signaling and prostate cancer progression. Oncogene. 2019;38(3):421‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis. 1997;18(6):1215‐1223. [DOI] [PubMed] [Google Scholar]
- 39. Beaver CM, Ahmed A, Masters JR. Clonogenicity: holoclones and meroclones contain stem cells. PLoS One. 2014;9(2):e89834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagpal S, Athanikar J, Chandraratna RA. Separation of transactivation and AP1 antagonism functions of retinoic acid receptor. Alpha J Biol Chem. 1995;270(2):923‐927. [DOI] [PubMed] [Google Scholar]
- 41. Klock G, Strahle U, Schutz G. Oestrogen and glucocorticoid responsive elements are closely related but distinct. Nature. 1987;329(6141):734‐736. [DOI] [PubMed] [Google Scholar]
- 42. Zhau HE, He H, Wang CY, et al. Human prostate cancer harbors the stem cell properties of bone marrow mesenchymal stem cells. Clin Cancer Res. 2011;17(8):2159‐2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19(12):1173‐1176. [DOI] [PubMed] [Google Scholar]
- 44. Allegretto EA, McClurg MR, Lazarchik SB, et al. Transactivation properties of retinoic acid and retinoid X receptors in mammalian cells and yeast. Correlation with hormone binding and effects of metabolism. J Biol Chem. 1993;268(35):26625‐26633. [PubMed] [Google Scholar]
- 45. Brown G, Marchwicka A, Cunningham A, Toellner KM, Marcinkowska E. Antagonizing retinoic acid receptors increases myeloid cell production by cultured human hematopoietic stem cells. Arch Immunol Ther Exp (Warsz). 2017;65(1):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kawada T, Kamei Y, Fujita A, et al. Carotenoids and retinoids as suppressors on adipocyte differentiation via nuclear receptors. Biofactors. 2000;13(1–4):103‐109. [DOI] [PubMed] [Google Scholar]
- 47. Berry DC, DeSantis D, Soltanian H, Croniger CM, Noy N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet‐induced obesity. Diabetes. 2012;61(5):1112‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berry DC, Noy N. All‐trans‐retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation‐activated receptor beta/delta and retinoic acid receptor. Mol Cell Biol. 2009;29(12):3286‐3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lahnalampi M, Heinaniemi M, Sinkkonen L, Wabitsch M, Carlberg C. Time‐resolved expression profiling of the nuclear receptor superfamily in human adipogenesis. PLoS One. 2010;5(9):e12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xue JC, Schwarz EJ, Chawla A, Lazar MA. Distinct stages in adipogenesis revealed by retinoid inhibition of differentiation after induction of PPARgamma. Mol Cell Biol. 1996;16(4):1567‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hisada K, Hata K, Ichida F, et al. Retinoic acid regulates commitment of undifferentiated mesenchymal stem cells into osteoblasts and adipocytes. J Bone Miner Metab. 2013;31(1):53‐63. [DOI] [PubMed] [Google Scholar]
- 52. Sumanasuriya S, De Bono J. Treatment of advanced prostate cancer‐a review of current therapies and future promise. Cold Spring Harb Perspect Med. 2018;8(6):a030635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bosch A, Bertran SP, Lu Y, et al. Reversal by RARalpha agonist Am580 of c‐Myc‐induced imbalance in RARalpha/RARgamma expression during MMTV‐Myc tumorigenesis. Breast Cancer Res. 2012;14(4):R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garattini E, Paroni G, Terao M. Retinoids and breast cancer: new clues to increase their activity and selectivity. Breast Cancer Res. 2012;14(5):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu Y, Bertran S, Samuels TA, Mira‐y‐Lopez R, Farias EF. Mechanism of inhibition of MMTV‐neu and MMTV‐wnt1 induced mammary oncogenesis by RARalpha agonist AM580. Oncogene. 2010;29(25):3665‐3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schneider SM, Offterdinger M, Huber H, Grunt TW. Activation of retinoic acid receptor alpha is sufficient for full induction of retinoid responses in SK‐BR‐3 and T47D human breast cancer cells. Cancer Res. 2000;60(19):5479‐5487. [PubMed] [Google Scholar]
- 57. Dulinska J, Gil D, Zagajewski J, et al. Different effect of beta‐carotene on proliferation of prostate cancer cells. Biochim Biophys Acta. 2005;1740(2):189‐201. [DOI] [PubMed] [Google Scholar]
- 58. Esquenet M, Swinnen JV, Heyns W, Verhoeven G. Control of LNCaP proliferation and differentiation: actions and interactions of androgens, 1alpha,25‐dihydroxycholecalciferol, all‐trans retinoic acid, 9‐cis retinoic acid, and phenylacetate. Prostate. 1996;28(3):182‐194. [DOI] [PubMed] [Google Scholar]
- 59. Esquenet M, Swinnen JV, Heyns W, Verhoeven G. LNCaP prostatic adenocarcinoma cells derived from low and high passage numbers display divergent responses not only to androgens but also to retinoids. J Steroid Biochem Mol Biol. 1997;62(5–6):391‐399. [DOI] [PubMed] [Google Scholar]
- 60. Fong CJ, Sutkowski DM, Braun EJ, et al. Effect of retinoic acid on the proliferation and secretory activity of androgen‐responsive prostatic carcinoma cells. J Urol. 1993;149(5):1190‐1194. [DOI] [PubMed] [Google Scholar]
- 61. Li MT, Richter F, Chang C, Irwin RJ, Huang H. Androgen and retinoic acid interaction in LNCaP cells, effects on cell proliferation and expression of retinoic acid receptors and epidermal growth factor receptor. BMC Cancer. 2002;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu Z, Ren G, Shangguan C, et al. ATRA inhibits the proliferation of DU145 prostate cancer cells through reducing the methylation level of HOXB13 gene. PLoS One. 2012;7(7):e40943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Richter F, Huang HF, Li MT, Danielpour D, Wang SL, Irwin RJ Jr. Retinoid and androgen regulation of cell growth, epidermal growth factor and retinoic acid receptors in normal and carcinoma rat prostate cells. Mol Cell Endocrinol. 1999;153(1–2):29‐38. [DOI] [PubMed] [Google Scholar]
- 64. Ding G, Shao J, Ding Q, et al. Comparison of the characteristics of mesenchymal stem cells obtained from prostate tumors and from bone marrow cultured in conditioned medium. Exp Ther Med. 2012;4(4):711‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Perez‐Echarri N, Noel‐Suberville C, Redonnet A, Higueret P, Martinez JA, Moreno‐Aliaga MJ. Role of adipogenic and thermogenic genes in susceptibility or resistance to develop diet‐induced obesity in rats. J Physiol Biochem. 2007;63(4):317‐327. [DOI] [PubMed] [Google Scholar]
- 66. Sakuta T, Uchiyama T, Kanayama T. Topical ER36009, a RARgamma‐selective retinoid, decreases abdominal white adipose tissue and elicits changes in expression of genes related to adiposity and thermogenesis. Endocrine. 2006;30(1):113‐119. [DOI] [PubMed] [Google Scholar]
- 67. Shirakura Y, Takayanagi K, Mukai K, Tanabe H, Inoue M. Beta‐cryptoxanthin suppresses the adipogenesis of 3T3‐L1 cells via RAR activation. J Nutr Sci Vitaminol (Tokyo). 2011;57(6):426‐431. [DOI] [PubMed] [Google Scholar]
- 68. Hammond LA, Brown G, Keedwell RG, Durham J, Chandraratna RA. The prospects of retinoids in the treatment of prostate cancer. Anticancer Drugs. 2002;13(8):781‐790. [DOI] [PubMed] [Google Scholar]
- 69. Trump DL, Smith DC, Stiff D, et al. A phase II trial of all‐trans‐retinoic acid in hormone‐refractory prostate cancer: a clinical trial with detailed pharmacokinetic analysis. Cancer Chemother Pharmacol. 1997;39(4):349‐356. [DOI] [PubMed] [Google Scholar]
- 70. Lotan R, Lotan Y. Retinoic acid receptor beta2 hypermethylation: implications for prostate cancer detection, prevention, and therapy. Clin Cancer Res. 2004;10(12 Pt 1):3935‐3936. [DOI] [PubMed] [Google Scholar]
- 71. Lotan Y, Xu XC, Shalev M, et al. Differential expression of nuclear retinoid receptors in normal and malignant prostates. J Clin Oncol. 2000;18(1):116‐121. [DOI] [PubMed] [Google Scholar]
- 72. Richter F, Joyce A, Fromowitz F, et al. Immunohistochemical localization of the retinoic acid receptors in human prostate. J Androl. 2002;23(6):830‐838. [PubMed] [Google Scholar]
- 73. Yan TD, Wu H, Zhang HP, et al. Oncogenic potential of retinoic acid receptor‐gamma in hepatocellular carcinoma. Cancer Res. 2010;70(6):2285‐2295. [DOI] [PubMed] [Google Scholar]
- 74. Sanchez BG, Bort A, Vara‐Ciruelos D, Diaz‐Laviada I. Androgen deprivation induces reprogramming of prostate cancer cells to stem‐like cells. Cell. 2020;9(6):1441‐1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.