

Abstract
It has been reported that hyperexcitability occurs in a subset of patients with Alzheimer’s disease (AD) and hyperexcitability could contribute to the disease. Several studies have suggested that the hippocampal dentate gyrus (DG) may be an important area where hyperexcitability occurs. Therefore, we tested the hypothesis that the principal DG cell type, granule cells (GCs), would exhibit changes at the single-cell level which would be consistent with hyperexcitability and might help explain it. We used the Tg2576 mouse, where it has been shown that hyperexcitability is robust at 2–3 months of age. GCs from 2–3-month-old Tg2576 mice were compared to age-matched wild type (WT) mice. Effects of muscarinic cholinergic antagonism were tested because previously we found that Tg2576 mice exhibited hyperexcitability in vivo that was reduced by the muscarinic cholinergic antagonist atropine, counter to the dogma that in AD one needs to boost cholinergic function. The results showed that GCs from Tg2576 mice exhibited increased frequency of spontaneous excitatory postsynaptic potentials/currents (sEPSP/Cs) and reduced frequency of spontaneous inhibitory synaptic events (sIPSCs) relative to WT, increasing the excitation:inhibition (E:I) ratio. There was an inward NMDA receptor-dependent current that we defined here as a novel synaptic current (nsC) in Tg2576 mice because it was very weak in WT mice. Intrinsic properties were distinct in Tg2576 GCs relative to WT. In summary, GCs of the Tg2576 mouse exhibit early electrophysiological alterations that are consistent with increased synaptic excitation, reduced inhibition, and muscarinic cholinergic dysregulation. The data support previous suggestions that the DG contributes to hyperexcitability and there is cholinergic dysfunction early in life in AD mouse models.
Keywords: Hyperexcitability, synaptic potentials, synaptic currents, intrinsic properties, muscarinic receptors, acetylcholine
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative and progressive disorder characterized by increasing impairment in learning and memory, and by two major types of neuropathology: amyloid-β (Aβ) plaques and neurofibrillary tangles (Alzheimer et al., 1995; Braak and Braak, 1991; Perl, 2010). The disease is common, debilitating, and there is no cure. As a result, it is increasingly important to understand the contributing factors so that new treatments can be developed.
Several lines of evidence suggest that hyperexcitability is a contributing factor, primarily in familial AD (Chin and Scharfman, 2013; Friedman et al., 2012; Ghatak et al., 2019; Lam et al., 2017; Noebels, 2011; Palop and Mucke, 2009; Palop and Mucke, 2010a; Palop and Mucke, 2010b; Scarmeas et al., 2009; Styr and Slutsky, 2018; Vossel et al., 2013; Vossel et al., 2016; Vossel et al., 2017). Hyperexcitability is manifested by intermittent seizures and/or epileptiform activity in an electroencephalogram (EEG), similar to patients with epilepsy. Seizures may not be accompanied by convulsive behavior, and in these cases, they can easily be missed. An EEG is necessary, but not typically conducted, or recordings are made far from the site of abnormal activity (Lam et al., 2019). For these reasons and others, the extent to which hyperexcitability occurs in AD is unclear (Friedman et al., 2012; Leonard and McNamara, 2007; Scarmeas et al., 2009; Scharfman, 2019; Vossel et al., 2017).
Hyperexcitability also occurs in mouse models of AD (Bearer et al., 2018; Bezzina et al., 2015; Born et al., 2014; Ciccone et al., 2019; Duffy et al., 2015; Fontana et al., 2017; Fu et al., 2019; Kam et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007; Petrache et al., 2019; Reyes-Marin and Nunez, 2017; Sanchez et al., 2012; Siskova et al., 2014; Verret et al., 2012; Xu et al., 2015; You et al., 2017; Ziyatdinova et al., 2016). One of the reasons the mice may show hyperexcitability is that they have mutations that are found in familial AD, and familial AD is often associated with seizures (Duff, 2001; Duyckaerts et al., 2008; Gotz et al., 2018; Gotz and Ittner, 2008; Scharfman, 2012). Mutations in these mouse models are typically in the precursor to Aβ, amyloid precursor protein (APP). Other mouse models of AD that exhibit increased excitability have mutations in molecules that are important to APP metabolism, such as presenilin (Beckman et al., 2020). There are also mouse models with altered ApoE or mutations in tau that show hyperexcitability (Duan et al., 2006; Hunter et al., 2012; Perry et al., 2008; Sanchez et al., 2018). Mouse models often have more than one mutation and these mice also show hyperexcitability (APP/PS1: (Fontana et al., 2017; Minkeviciene et al., 2009; Reyes-Marin and Nunez, 2017). The mouse models have led to many insights into the nature of hyperexcitability and its potential contribution to AD pathophysiology (Gheyara et al., 2014; Palop et al., 2007; Roberson et al., 2007; Sanchez et al., 2012; Verret et al., 2012; Vossel et al., 2010).
One question we addressed was whether hyperexcitability occurs early in life, before cognitive impairment or Aβ plaque and neurofibrillary tangle pathology. To address this question, the Tg2576 mouse model was used because its neuropathology has a slow development of neuropathology (Hsiao et al., 1996). The Tg2576 mice overexpress the Swedish mutation (APPSwe) of human amyloid precursor protein (hAPP), and Aβ plaques are detected starting after 6 months of age (Citron et al., 1992; Hsiao et al., 1996; Jacobsen et al., 2006; Kawarabayashi et al., 2001).
Using Tg2576 mice with implanted electrodes, we found evidence of hyperexcitability by 5 weeks of age, manifested by spontaneous generalized spikes that were large in amplitude and brief (<0.5 ms; Kam et al. (2016)). Although seizures were not detected at 1–2 months of age, they did sporadically afterwards (Kam et al., 2016). The spikes were similar to those occurring in between seizures in epilepsy, called interictal spikes (IIS). Another group also found that IIS occur as early as 6 weeks of age in Tg2576 mice (Bezzina et al., 2015), and it has been known for many years that mice with hAPP mutations exhibit IIS, although early ages were not examined (Born et al., 2014; Palop et al., 2007; Reyes-Marin and Nunez, 2017; Sanchez et al., 2012).
These recordings suggested that hyperexcitability was an early step in the pathophysiology of the Tg2576 mice, so we investigated mechanisms. IIS appeared to be due to excessive activity in the cholinergic system in the very young mice. For example, IIS were reduced by atropine, a muscarinic cholinergic receptor antagonist (Kam et al., 2016). Also, there was increased expression of the rate-limiting enzyme for acetylcholine synthesis, choline acetyltransferase (Kam et al., 2016). The data suggested it was timely to reconsider the idea that the medial septal cholinergic neurons play a critical role in AD (Craig et al., 2011; Davies and Maloney, 1976; Ferreira-Vieira et al., 2016; Francis et al., 1999; Hampel et al., 2018; Hampel et al., 2019; Mufson et al., 2008). Our data suggested that early in life there was increased excitability due to an abnormality of the medial septal cholinergic system and later in life, cholinergic function declines due to the adverse effects of overactivity (Kam et al., 2016). This view is consistent with the idea that cholinergic changes are more complex in AD (Mufson et al., 2008) than the original cholinergic hypothesis which suggested that the cholinergic system declines in AD (Davies and Maloney, 1976). Our hypothesis was also consistent with other studies of Tg2576 mice showing abnormalities in cholinergic markers at 5–8 months of age, and more abnormalities after 17 months (Apelt et al., 2002; Klingner et al., 2003).
To define mechanisms of hyperexcitability and the role of the cholinergic neurons further, several studies suggested it would be useful to focus on the DG. For example, patients with mild cognitive impairment show hyperactivity in the DG/CA3 (Bakker et al., 2012). In mice, several neuroanatomical changes occur in the DG in hAPP models of AD (Bearer et al., 2018; Fontana et al., 2017; Jacobsen et al., 2006; Krezymon et al., 2013; Ohm, 2007; Palop et al., 2005; Palop et al., 2007; Palop et al., 2003; Roberson et al., 2011; You et al., 2017) which also occur in the human DG in AD (Palop et al., 2003; Scharfman, 2012). In addition, the DG is a region that is pivotal in animal models of epilepsy (Dengler et al., 2017; Heinemann et al., 1992; Kobayashi and Buckmaster, 2003; Krook-Magnuson et al., 2015; Lothman et al., 1992; Pun et al., 2012; Scharfman, 2019; Sun et al., 2007).
The hAPP mouse model of AD, the J20 mouse, has been particularly informative. These mice share the APP mutation (KM670/671NL; APPSwe) with Tg2576 mice, as well as a second mutation (V717F; APPInd) and a different promoter (platelet-derived growth factor; PDGF). This model exhibited increased expression of GC c-Fos and ΔFosB, which are upregulated in young GCs by neuronal activity (You et al., 2017). Furthermore, experimental manipulations which affected the DG led to improvement in excitability, behavior, and other measurements of dysfunction (Verret et al., 2012; You et al., 2017).
Regarding the cholinergic system, the DG is of interest because the septocholinergic neurons innervate all cell types in a robust manner (Aznavour et al., 2005; Clarke, 1985; Deller et al., 1999; Dougherty and Milner, 1999; Frotscher, 1991; Frotscher and Leranth, 1985; Frotscher and Leranth, 1986; Leranth and Frotscher, 1987; Milner and Veznedaroglu, 1993; Nyakas et al., 1987; Takacs et al., 2018; Wainer et al., 1985).
For these reasons, we focused on the DG to obtain insights into why hyperexcitability occurs in young Tg2576 mice in vivo, and the role of cholinergic input. We recorded from the principal cells of the DG (granule cells; GCs) in hippocampal slices with whole-cell recordings to understand cellular changes with high resolution. Early ages (less than 3 months) were used because at these ages, hyperexcitability occurs relatively selectively, whereas at older ages there also are changes in sleep and other contributing factors in AD pathophysiology. We studied synaptic events by recording spontaneous excitatory postsynaptic potentials (sEPSPs) / currents (sEPSCs) and spontaneous inhibitory postsynaptic currents (sIPSCs). To obtain an understanding of nonsynaptic effects, intrinsic properties (e.g., resting membrane potential or RMP, input resistance or Rin, time constant or τ, etc.) were also analyzed. Finally, we used the muscarinic antagonist atropine to determine if atropine affected GCs of WT and Tg2576 mice differently.
The results suggest that there are alterations in GCs in Tg2576 mice that could explain hyperexcitability in vivo. The data suggest that young (< 3 months) Tg2576 mice have GCs with increased synaptic excitation and decreased inhibition. There also were several abnormalities in intrinsic properties that may contribute to hyperexcitability. However, some of the alterations in intrinsic properties would be predicted to decrease excitability, and whether the latter is an attempt to compensate for hyperexcitability is discussed. Regarding the cholinergic modulation of GCs, we found several abnormal effects of atropine in Tg2576 mice, some of which are novel.
Together the results suggest explanations for hyperexcitability in the DG of the Tg2576 mouse model of AD. The results suggest that there are very early increases in excitability in the DG mediated by increased synaptic excitation and decreased inhibition. Moreover, there are altered intrinsic properties and abnormal muscarinic cholinergic modulation of GCs
METHODS
I. Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at The Nathan Kline Institute and experiments were carried out in accordance with the National Institutes of Health (NIH) guidelines.
Mice expressing human APP695 with the Swedish (Lys670Arg, Met671Leu) mutations driven by the hamster prion protein promoter (Hsiao et al, 1996) were bred from male heterozygous Tg2576 and female non-transgenic mice (C57BL6/SJL F1 hybrid, Stock# 100012, Jackson Labs), and fed a chow that is commonly used during breeding (Purina 5008, W.F. Fisher).
Mice were housed with same-sex siblings and fed a chow that is common after weaning (Purina 5001, W.F. Fisher). Mice were provided food and water ad libitum, and housed using a 12 hr light-dark cycle. Before an animal was used genotypes were determined using an in-house protocol for detecting the APP695 gene. After use, genotypes were confirmed.
Both male and female mice were used. For electrophysiology, WT mice were 63.16 ± 3.73 days old (n=19) and Tg2576 mice were 68.24 ± 4.39 days old (n=17). The ages were not significantly different (unpaired t-test, p= 0.381).
II. Slice electrophysiology
A. Slice preparation
Mice were deeply anesthetized by isoflurane (1–2%; Aerrane, Piramal Enterprises) inhalation, followed by a subsequent intraperitoneal injection of urethane (2.5 g/kg; i.p.). Mice were perfused transcardially with a cold (4°C) sucrose-based artificial cerebrospinal fluid (sucrose ACSF) containing (in mM) 90 sucrose, 2.5 KCl, 1.25 NaH2PO4, 4.5 MgSO4, 25.0 NaHCO3, 10.0 D-glucose, 80.0 NaCl, and 0.5 CaCl2; pH 7.4. Then mice were decapitated and the brain was removed and dissected in the same sucrose ACSF at 4°C. All ACSF solutions were aerated with carbogen (95% O2, 5% CO2, All-Weld Products). One cerebral hemisphere was mounted on a vibratome stage and slices were cut horizontally in cold (4°C) sucrose ACSF with a vibratome (350 μm thick; Model# HMV450, Micron Instruments). Slices were immediately placed in a custom-made holding chamber containing sucrose ACSF at 30°C for 30 min and aerated with carbogen (95% O2, 5% CO2). Afterwards, slices were stored in the same holding chamber at room temperature for at least 60 min before recording.
B. Whole-cell patch clamp recordings
1). Recording conditions
Slices were transferred to a recording chamber (RC-27LD, Warner) and perfused with ACSF containing NaCl instead of sucrose (NaCl ACSF) which contained (in mM: 130 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 25.0 NaHCO3, 10.0 D-glucose, and 2.4 CaCl2 (pH 7.4). NaCl ACSF was perfused at 6 mL/min with a peristaltic pump (Masterflex C/L, Cole-Parmer) and maintained at 32°C with a temperature controller (TC-324B, Warner) and in-line heater (SH-27B, Warner).
2). Recording electrodes and acquisition
For whole cell current-clamp experiments, borosilicate glass capillaries (1.5 mm outside diameter; 0.86 inner diameter, Sutter Instruments) were pulled horizontally (P-97, Sutter) so that the resistance was 4–9 MΩ. Seal resistances were > 1 GΩ before breaking into whole cell configuration. All data were digitized (Digidata 1440A, Molecular Devices), amplified by a MultiClamp 700B amplifier (Molecular Devices), and low pass filtered using a single-pole RC filter at 10 kHz. Analysis used pClamp software (v11.1, Molecular Devices) and is described further below.
3). Synaptic potentials and synaptic currents
a. Recording
For the evaluation of the synaptic activity, spontaneous events were evaluated using a continuous recording for 3–5 min.
Current-clamp recordings of spontaneous excitatory postsynaptic potentials (sEPSPs) were obtained from GCs using an intracellular solution of the following composition (in mM): K-gluconate 130, NaCl 2, HEPES 10, EGTA 0.2, Mg-ATP 4, Na-GTP 0.3, Na2-phosphocreatine 14, and 0.5% biocytin; adjusted to pH 7.3 and 304 mOsm. Cells were patched-clamped using the whole-cell recording configuration and sEPSPs were evaluated at the resting membrane potential of the cell.
Voltage-clamp recordings of spontaneous excitatory and inhibitory postsynaptic currents (sEPSCs and sIPSCs, respectively) were performed in a different set of neurons, using an intracellular solution of the following composition (in mM): Cesium methanesulfonate 125, NaCl 4, HEPES 10, EGTA 1, MgATP 4, Tris-GTP 0.3, diTris-phosphocreatine 10, QX-314Cl 5, and 0.2% biocytin adjusted to pH 7.3 and 290 mOsm. To evaluate sEPSCs and sIPSCs, cells were voltage-clamped at a holding potential (HP) of −70 mV and 0 mV, respectively, in order to isolate glutamatergic and GABAergic receptor-mediated currents.
b. Analysis
Detection of spontaneous events (EPSP, EPSC and IPSC) was performed off-line using MiniAnalysis 6.0 (Synaptosoft, Inc.). sEPSPs were identified as events having a fast rise time and a peak that was >2–3 standard deviations (SD) from the root mean square (RMS) of the baseline noise (random, background electrical fluctuations) (Hong and Rebec, 2012; Serletis et al., 2011). sEPSCs and sIPSCs were included if the maximum (peak) change in current (negative or positive, respectively) relative to baseline noise had a peak amplitude of >3 SD from the RMS of the noise. This determination was chosen because pilot work showed that discrimination between synaptic events and noise was poor with 2 SD and good with >3 SD. The mean frequency and amplitude were calculated for all events over the entire recording period (each period lasted 3–5 min) and are presented as mean ± SEM.
Kinetics were evaluated off-line by measuring the rise and decay times using MiniAnalysis 6.0 (Synaptodsoft, Inc.). The total rise time was calculated from the onset to peak of a PSC. The decay was the time from the peak to the point during the decay when the peak amplitude had declined to 37%.
The excitation:inhibition ratio was estimated from the mean PSC amplitudes or frequencies. For amplitude, the mean EPSC amplitude was divided by the mean IPSC amplitude. For frequency, the mean EPSC frequency was divided by the mean IPSC frequency. For EPSCs, the data included both AMPA and NMDA receptor-dependent EPSCs. In other words, EPSCs at −70 mV (AMPA receptor-dependent EPSCs) and nsCs at 0 mV (NMDA receptor-dependent EPSCs) were included. IPSCs were calculated from recordings at 0 mV. All recordings used the same internal solution.
Total charge transfer was defined as the sum of integrated areas of all PSCs in a 3 min recording period. The integration began at the onset of a PSC and ended when the event had returned to baseline. In some cases, an event did not return to baseline before another PSC began. In these cases, the integrated area of the first event was truncated at the onset of the second PSC. Therefore, charge transfer for the first event was underestimated. We do not think the underestimation was large because most PSCs occurred without truncation by a second PSC.
4). Intrinsic properties
Current clamp was used so recording electrodes had the same K-gluconate internal solution described above. Supplemental Figure 1 shows how measurements were made. Resting membrane potential (RMP) was defined as the difference between the potential while intracellular and that recorded after withdrawing the microelectrode from the cell. Sub-threshold depolarizing and hyperpolarizing pulses (−30 pA to +30 pA; 5–10 pA steps; 1 sec duration) were delivered to assess input resistance (Rin). The steady-state voltage responses were plotted against the amplitude of current injection and the slope of the linear fit between 0 pA and −5 pA was used to define Rin (Clampfit v. 11.1; Molecular Devices). Time constant (tau; τ) was determined from hyperpolarizing pulses (−20 pA). τ was defined as the time to reach 63% of the steady-state response. To determine characteristics of action potential (AP) discharge, an AP was used close to its threshold. Specifically, depolarizing current pulses of +40 to +100 pA (10 pA steps; 1 sec duration) were delivered to elicit an AP in approximately 50% of trials. We then used a combination of the AP analysis tool and statistics using Clampfit (pClamp software v11.1, Molecular Devices) to determine the characteristics of the APs. Threshold was defined as the membrane potential at which the AP was initiated. This was determined in Clampfit by finding the intersection of the rising phase of the AP and the slope leading up to its initiation. Then a three-point tangent slope vector was used to find the position in the initial region where the slope was ≥ 10 V/s. The mean AP peak amplitude was determined from measuring RMP to the AP peak. The time to the AP peak amplitude was the time from the start of the current step to the AP peak. The time to peak from threshold involved only the period from the point of initiation of the AP to the AP peak. Half-width was defined as the time from the point of initiation to the point when the AP reached half of its peak amplitude. AP rising and decay slopes were defined by the maximum dv/dt of each AP phase, and the dv/dt ratio was defined as the ratio of rising/decay slopes. Afterhyperpolarization (AHP) amplitude was measured from the membrane potential where the AP returned to baseline to the peak of the AHP.
To quantify spike frequency adaptation, we evaluated the time from one AP peak to the next for all the spike pairs in trains of 4 APs. A train with 4 APs was selected because in previous studies, we found that it is relatively easy to study adaptation in GCs with 4 APs, and this can distinguish GCs from other cell types (Scharfman et al., 2000). A train of 4 APs is also useful because one does not elicit extensive firing in granule cells. Extensive firing is good to avoid because even a single granule cell can have strong excitatory effects on mossy cells and hilar GABAergic neurons (Scharfman et al., 1990). Once the hilar cells are activated, they feedback and influence the GC. This makes repeated presentations of the same current command elicit different responses in GCs even in control mice. In Tg2576 mice it could elicit hyperexcitability which we wanted to avoid. With a 4 AP train, we analyzed each interval between APs (interspike interval). We also compared the first and the last interval because the largest differences in the interval are between these two intervals.
In the course of studying trains of 4 APs in GCs, we found limited differences between WT and Tg2576 mice, and no evidence of possible confounding factors such as feedback from mossy cells or hyperexcitability. This led us to ask whether differences in spike frequency adaptation would be more evident if we prolonged the trains. Therefore, we studied trains with 6–7 APs. For these data, the first 6 APs were analyzed so that the number of APs was consistent from cell to cell.
III. Pharmacology
All chemicals used for making the different slicing, recording and internal solutions were American chemical society (ACS) reagent grade. Atropine sulfate salt monohydrate (Catalog #A0257), (+)-Bicuculline (#14340) and DL-2-amino-5-phosphonopentanoinc acid (APV; #A5282) were obtained from Sigma-Aldrich; 6,7-dinitroquinoxaline-2,3(1H,4H)-dione disodium salt (DNQX; #2312) was obtained from Tocris.
Atropine was dissolved in double distilled water and a stock solution at a concentration of 10 mM was prepared. Stock solution was stored at 4°C and protected from light. For bicuculine, APV and DNQX, drugs were dissolved in double distilled water and stock solutions of every drug were prepared at the following concentrations: bicuculine 10 mM, APV 10 mM and DNQX 50 mM. Then, solutions were stored at 4°C and protected from light.
To test the effects of drugs, all recordings were performed initially at resting potential for 3–5 min in voltage- or current-clamp configurations (choice of voltage- or current-clamp are shown in the timelines for the Figures). After a drug was added to the ACSF an additional period of 3–5 min was recorded. The reason for this is that the recordings showed a rapid effect of drugs which plateaued by the end of the 3–5 min recording period. To compare the effects of drugs, data were selected before the drug was added and at the end of the 3–5 min recording period after drug was applied.
IV. Anatomy
A. Granule cell identification
After the completion of recordings, slices were immediately placed in 4% paraformaldehyde (PFA; Sigma) in Tris buffer (TB; 0.1 M, pH 7.4) and kept in fixative at 4°C. For visualization, approximately two weeks after the experiments, slices were permeabilized with 0.7% Triton X-100 in TB with gentle but continuous shaking on a rotator at room temperature for 1 hr. Then slices were incubated in a 0.1% hydrogen peroxide (H2O2) solution in TB for 30 min in order to reduce endogenous peroxidase activity. After a series of washes in 0.25% Triton X-100 in TB at room temperature, the slices were incubated in Avidin Biotin Complex (ABC elite Kit, # PK-6100, Vector Laboratories) for 2 hr at room temperature and then pre-incubated in a solution containing 0.5 mg/mL 3,3’-diaminobenzidine (DAB; Invitrogen) and NiCl2 (50 mM) dissolved in TB. This solution was pre-incubated for 30 min at room temperature and followed by a solution of DAB and 30% H2O2 in TB for 1–2 min. Slices were washed in TB and then passed through a graded series of glycerol solutions (25, 40, 55 %) diluted in TB for 15 min each. A subsequent incubation in 60% 2,2’-thiodiethanol (TDE; Sigma) for 30 min was performed. Then the slices were immediately mounted and coverslipped with 60% TDE. Photomicrographs were made on a brightfield microscope (Model BX61; Olympus of America) equipped with a digital camera (Retiga 2000R, Teledyne QImaging) using Image-Pro Plus software (v.7; Media Cybernetics, Inc).
B. Identification of Aβ pathology
1). Perfusion-fixation and sectioning
Mice were deeply anesthetized by isoflurane inhalation (Patterson Veterinary) followed by an intraperitoneal injection of urethane (2.5 g/kg, Sigma-Aldrich). After opening the heart cavity, a 26-gauge needle was inserted into the left ventricle of the heart, followed by perfusion with 10 mL of cold saline (0.9% NaCl in ddH2O, 4°C) using a peristaltic pump (Minipuls 1, Gilson), and 30 mL of cold 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4, 4°C). The brains were removed immediately and postfixed in 4% PFA at 4°C. After post-fixation, the brains were hemisected and cut in the horizontal plane (50 μm-thick sections) using a vibratome (Model# VT1000p, Leica). Sequentially collected serial sections from similar dorso-ventral levels across the animals were processed.
2). Immunohistochemistry
For immunostaining, we used an antibody to human Aβ (McSA1) which was raised against the N-terminal fragment (residues 1–12; (Grant et al., 2000)). Prior studies showed that the antibody was specific for Aβ and not APP (Billings et al., 2005; Grant et al., 2000). We chose this antibody because it can detect the soluble and insoluble forms of Aβ. The soluble form of Aβ was of interest because the early ages we studied, Tg2576 mice have not been shown to exhibit Aβ plaques.
We adapted a protocol from Kobro-Flatmoen et al. (2016) and used free-floating sections. Both WT and Tg2576 mouse sections were processed together. Initially, sections were treated with an antigen retrieval procedure (3 hrs in 0.1 M PB, pH 7.4, at 60°C) to unmask the epitope of interest. All the following washes and dilutions were performed using 0.1 M PB (pH 7.4). First, sections were incubated for 20 min in 0.5% Triton X-100. Then sections were incubated for 2 hrs in 5% normal goat serum (Vector Laboratories) to block non-specific binding. Subsequently, sections were incubated overnight at 4°C in primary antiserum in 3% normal goat serum and 0.5% Triton X-100 (1:1000, mouse monoclonal antibody to McSA1, Medimabs,). Followed by 2 hrs of incubation with secondary antibody (1:350, goat anti-mouse IgG Alexa Fluor 488, Invitrogen), sections were mounted on 0.1% gelatin-coated slides, allowed to dry, and then coverslipped with a mounting medium that resists fading (Vectashield® HardSetTM, H-1400, Vector Laboratories). Then slides were examined using a fluorescence microscope (Model# BX61, Olympus of America) and photographed using a digital camera (Model# Infinity 3–6UR, Lumenera) and Infinity 3 software (v. 6.5.6, Lumenara). Sections from WT and Tg2576 mice were photographed together using the same microscope and software settings.
3). Thioflavin-S staining
For Thioflavin-S labeling, we followed the protocol from Roberson et al. (2007). Briefly, sections were mounted on 0.1% gelatin-coated slides and incubated in 1% Thioflavin-S (Sigma-Aldrich) in double-distilled (dd) H2O for 10 min at room temperature. Then sections were, dehydrated in a graded series of alcohols (80%, 95% and 100% in ddH20, 4 min each), xylene (4 min; Sigma Aldrich), and coverslipped with Permount (Fisher Chemical Co.). Sections were examined using the fluorescence attachment of the microscope described above and photographed using the same camera. As for immunostained sections, Thoiflavin-S stained sections from WT and Tg2576 mice were processed and photographed together, using the same microscope and software settings.
VI. Statistics
All results are presented as the mean ± standard error of the mean (SEM), and statistical significance was achieved if the p value was <0.05 (denoted on all graphs by an asterisk).
Statistical analyses were performed using Prism (v. 8.3, GraphPad). For parametric data, statistical significance for comparisons between two different groups was determined using an unpaired Student’s t-test, and a Student’s paired t-test was performed to compare the same cells before and after a treatment, or for data at two time points (e.g. during a train of APs). For non-parametric data, Mann-Whitney U test was used for unpaired analysis, and the Wilcoxon test was used for paired analysis. Cumulative distributions were analyzed using the Kolmogorov-Smirnov (KS) test. To compare the differences in binary characteristics of GC localization, Fisher’s exact test was used.
For comparisons between more than two groups, parametric data were analyzed by two-way ANOVA followed by Sidak’s post-hoc test. For example, spike frequency adaptation was analyzed using a two-way ANOVA with AP pairs and genotype as main factors. For ANOVAs, interactions between factors are not reported when they were not significant.
For comparisons of more than two groups, nonparametric data were analyzed by a Kruskal-Wallis test with Dunn’s multiple comparisons test.
RESULTS
A total of 41 cells from 17 Tg2576 mice, and 38 cells from 19 WT mice were used for the electrophysiology in this study. No more than three GCs per slice, from no more than two slices per animal were used.
I. Differences in synaptic activity, intrinsic properties and firing behavior in young WT and Tg2576 mice.
A. Synaptic activity
1). sEPSPs in Tg2576 mice are increased relative to WT mice
GCs were recorded in current clamp to assess sEPSPs (Figure 1A). GCs from Tg2576 mice showed a significantly higher mean frequency of sEPSPs than WT mice (Tg2576: 2.28 ± 0.19 events/sec, WT: 1.59 ± 0.09; unpaired t-test, t=3.250, df=32; p=0.001; Figure 1B1a–b, 1B2a). The representative traces in Figure 1B1 show that there was primarily an increase in small events, which lowered the mean amplitude (Figure 1B2b), whereas the differences in mean amplitudes of sEPSPs were not significantly different (Tg2576: 0.39 ± 0.03 mV, WT: 0.45 ± 0.03; unpaired t-test, t=1.407, df=32; p=0.085; Figure 1B1a–b, 1B2b). The frequency distribution of sEPSPs amplitudes (Figure 1C1) showed a large number of small events in Tg2576 GCs, like the representative examples in Figure 1B1. However, the cumulative distributions of the amplitudes showed no statistical differences (Komologorov-Smirnov test, D=0.187; p=0.567; Figure 1C2), consistent with the emphasis of the cumulative distribution on amplitude rather than frequency.
Figure 1. sEPSPs in GCs from Tg2576 mice are increased relative to WT mice.
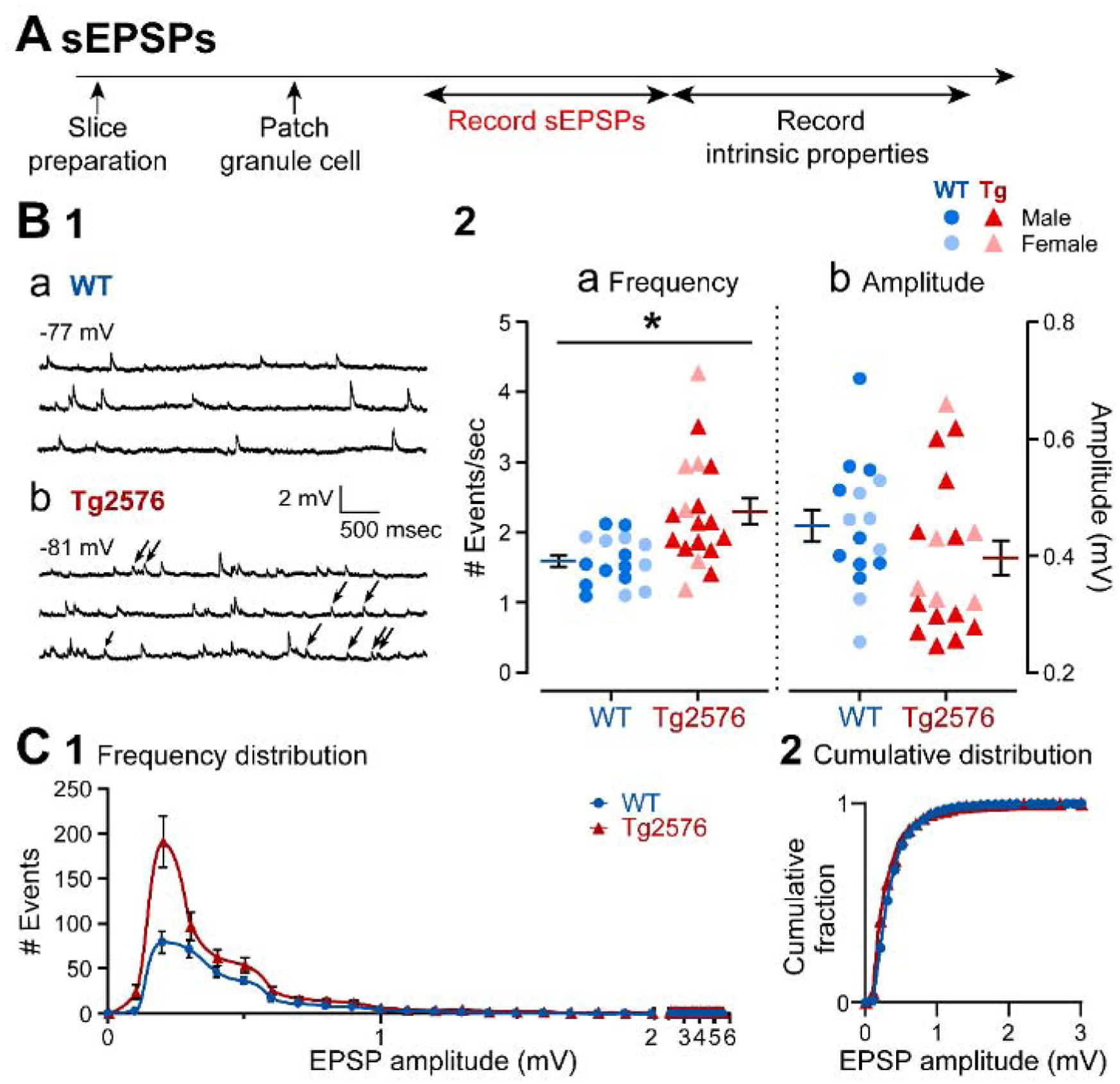
(A) The timeline of the electrophysiological recordings for the determination of the sEPSPs and intrinsic properties from GCs.
(B1) Representative traces show typical sEPSPs obtained in (a) WT and (b) Tg2576 mice.
(B2) Quantification of sEPSP frequency and amplitude in WT and Tg2576 GCs. SEPSP frequency was significantly greater in Tg2576 mice, but not amplitude.
(C1) A histogram shows the frequency distribution of sEPSP amplitudes.
(C2) The cumulative distribution is shown. Differences between WT and Tg2576 GCs were not significant.
Data are represented as mean ± SEM. *p<0.05. Specific p values and statistics are in the text.
2). Like EPSPs, sEPSCs are increased inTg2576 mice relative to WT mice
Next, sEPSCs were compared in WT and Tg2576 GCs in voltage clamp (Figure 2A). The experimental timelines for voltage clamp and current clamp experiments were similar, but different GCs were sampled. Representative traces in Figure 2B1 show that sEPSCs were fast inward currents at −70 mV holding potential. Like sEPSPs, Tg2576 GCs sEPSC frequency was greater in Tg2576 mice compared to WT GCs (Tg2576 mice: 4.07 ± 0.28 events/sec, WT: 3.16 ± 0.33; unpaired t-test, t=1.998, df=18; p=0.031; Figure 2B1a–b, 2B2a).
Figure 2. sEPSCs in GCs from Tg2576 mice are increased relative to WT mice.
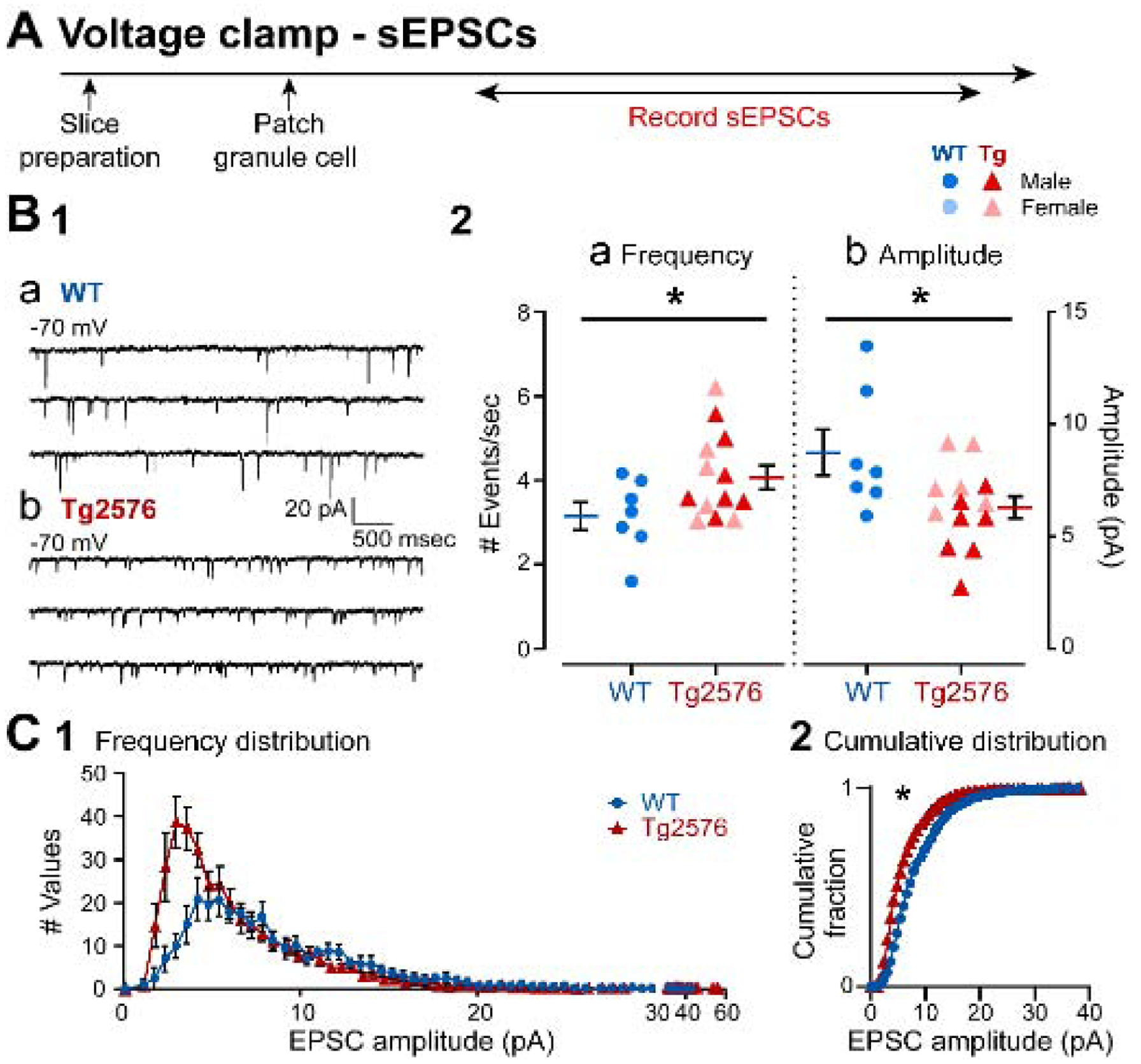
(A) The timeline of the electrophysiological recordings for the determination of sEPSCs from GCs.
(B1) Representative traces show the typical sEPSCs obtained in (a) WT and (b) Tg2576 mice from −70 mV holding potential.
(B2) Quantification of sEPSC frequency and amplitude in WT and Tg2576 GCs. Tg2576 GCs had a significantly greater sEPSC frequency and a reduction in amplitude compared to WT GCs.
(C1) A histogram shows the frequency distribution of sEPSCs amplitudes.
(C2) The cumulative distribution is shown. The distributions of Tg2576 GCs were significantly different from WT GCs.
Also, like sEPSPs, there was a lower mean amplitude of sEPSCs in Tg2576 mice. However, unlike sEPSPs, the difference was significant (Tg2576: 6.27 ± 0.54 pA; WT: 8.72 ± 1.03; unpaired t-test, t=2.432, df=18; p=0.013; Figure 2B2b). The reason why voltage clamp data were significant and current clamp data were not may be that sometimes there is a greater sensitivity in assessing synaptic events in voltage clamp compared to current clamp.
The frequency distribution of sEPSC amplitudes shown in Figure 2C1 illustrates a greater frequency of small sEPSCs, like data from sEPSPs in Figure 1. There was a significant difference in the cumulative distributions (Kolmogorov-Smirnov test, D=0.261, p=0.028; Figure 2C2), consistent with the data showing a significant reduction in sEPSC mean amplitudes (Figure 2B2b).
Taken together, the data from current and voltage clamp in Figures 1 and 2 suggest that Tg2576 mice have a greater frequency of excitatory input to GCs than WT mice. Tg2576 GCs showed this effect both for sEPSPs and sEPSCs. Regarding amplitude, there was a reduction in sEPSC amplitude, and this may have been due to more small events in the Tg2576 mouse. However, the differences in frequency were more robust than differences in amplitude, because reduced sEPSC amplitude was significant but reduced sEPSP amplitude was not. Below we return to the idea that there are additional small EPSCs in Tg2576 mice that are rare in WT mice, which are apparent by another type of experiment (see section about ‘nsCs’ below).
3). sIPSCs in Tg2576 mice are reduced in frequency relative to WT mice
The next experiments were conducted to determine if sIPSCs were different in WT and Tg2576 mice. The timeline (Figure 3A) and GCs that were sampled were the same as those used to record sEPSCs (Figure 2). The data in Figure 3B1 show that sIPSCs were recorded as fast outward currents at 0 mV holding potential. In Tg2576 mice, sIPSC frequency was significantly lower than WT mice (Tg2576: 8.19 ± 0.63 events/sec, WT: 10.38 ± 1.01; unpaired t-test, t=1.862, df=25; p=0.037, Figure 3B1a–b, 3B2a). There were no significant differences in sIPSC mean amplitude (Tg2576: 10.57 ± 0.88 pA; WT: 10.90 ± 1.11; unpaired t-test, t=0.238, df=25; p=0.407; Figure 3B1a–b, 3B2b). The frequency distribution of sIPSCs amplitudes in Tg2576 mice is consistent with a reduced number of events in Tg2576 mice, particularly large (>7 pA) events (Figure 3C1). There was a significant difference in the cumulative distributions (Komologorov-Smirnov test; D=0.267; p=0.002; Figure 3C2), despite the lack of change in sIPSC mean amplitudes (Figure 3B2b). The reason why the WT and Tg2576 GCs were not different in mean amplitude but cumulative distributions were different may be due to the fact that mean and cumulative distribution are not the same: cumulative distributions are more sensitive to the numbers of events of all amplitudes whereas mean amplitude pools all data.
Figure 3. sIPSCs are decreased in GCs from Tg2576 mice relative to WT mice.
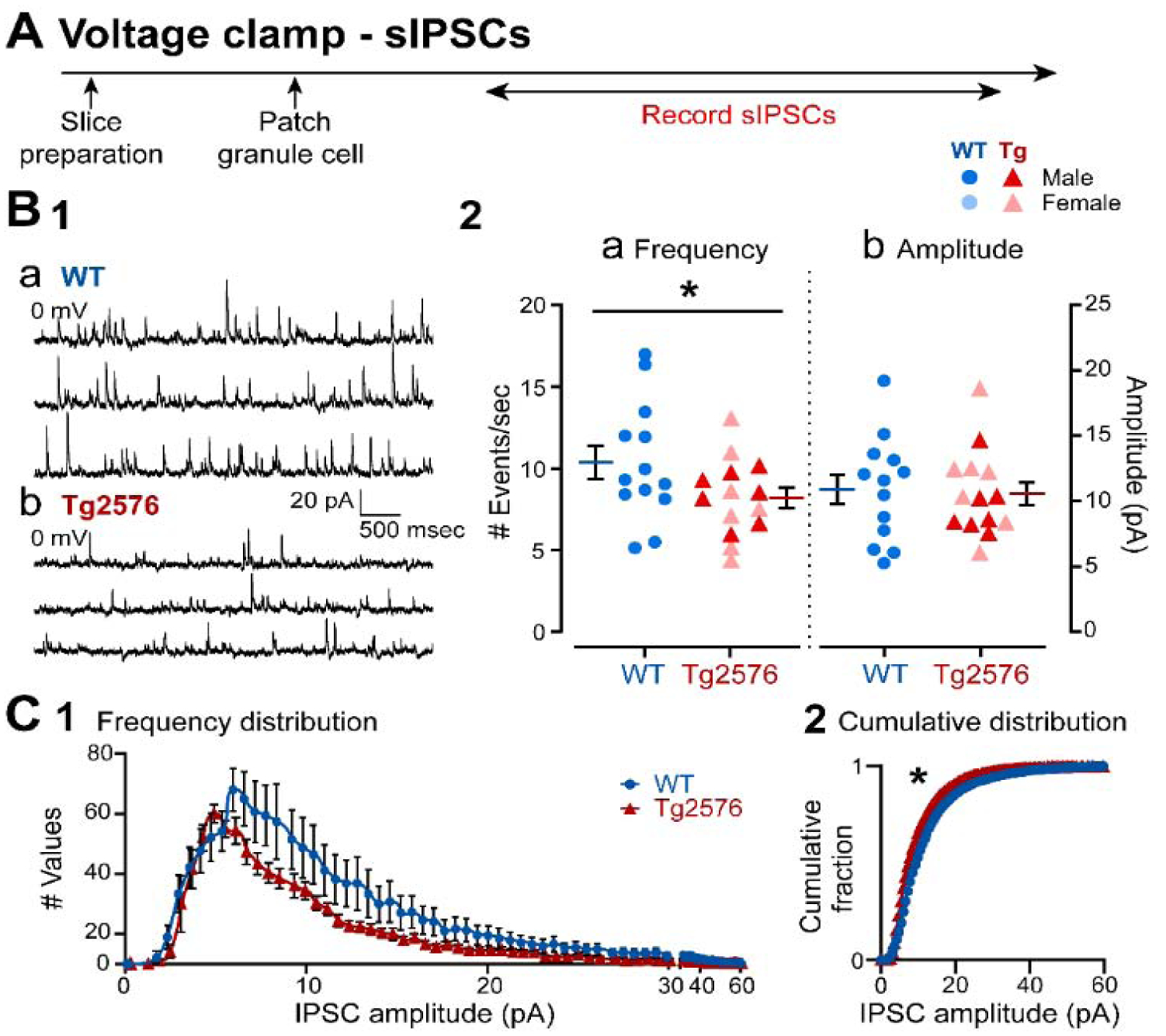
(A) The timeline of the electrophysiological recordings for the determination of the sIPSCs from GCs.
(B1) Representative traces show typical sIPSCs obtained in (a) WT and (b) Tg2576 mice at 0 mV holding potential.
(B2) Quantification of sIPSC frequency and amplitude in WT and Tg2576 GCs. Tg2576 GCs had a significantly reduced sIPSC frequency, but not in amplitude compared to WT GCs.
(C1) A histogram shows the frequency distribution of sIPSC amplitudes.
(C2) The cumulative distributions for WT and Tg2576 GCs were significantly different.
Taken together, results in Figures 1–3 suggest increased excitatory and decreased inhibitory synaptic input to GCs of Tg2576 mice. These data suggest an altered excitatory:inhibitory (E:I) balance. The implications are important because the alterations in E:I balance in Tg2576 GCs could contribute to the increased excitability in Tg2576 mice. Below we discuss E:I ratio further after addressing an additional small EPSC called ‘nsCs.’
B. GC intrinsic properties in Tg2576 mice are different in many ways from WT mice
Next, we examined intrinsic properties to determine if they showed genotypic differences, and they could potentially contribute to the differences in the synaptic events of WT and Tg2576 mice (Figure 4A). The intrinsic properties that were measured were RMP, input resistance (Rin), time constant (τ) and characteristics of APs. The characteristics of APs included amplitude, measurements of duration (time to peak, half-width), threshold, maximum rate of rise, maximum rate of decay, and dv/dt ratio (maximum rate of rise/maximum rate of decay), and the AHP amplitude (see Methods and Supplemental Figure 1).
Figure 4. Differences in intrinsic properties of WT and Tg2576 mice.
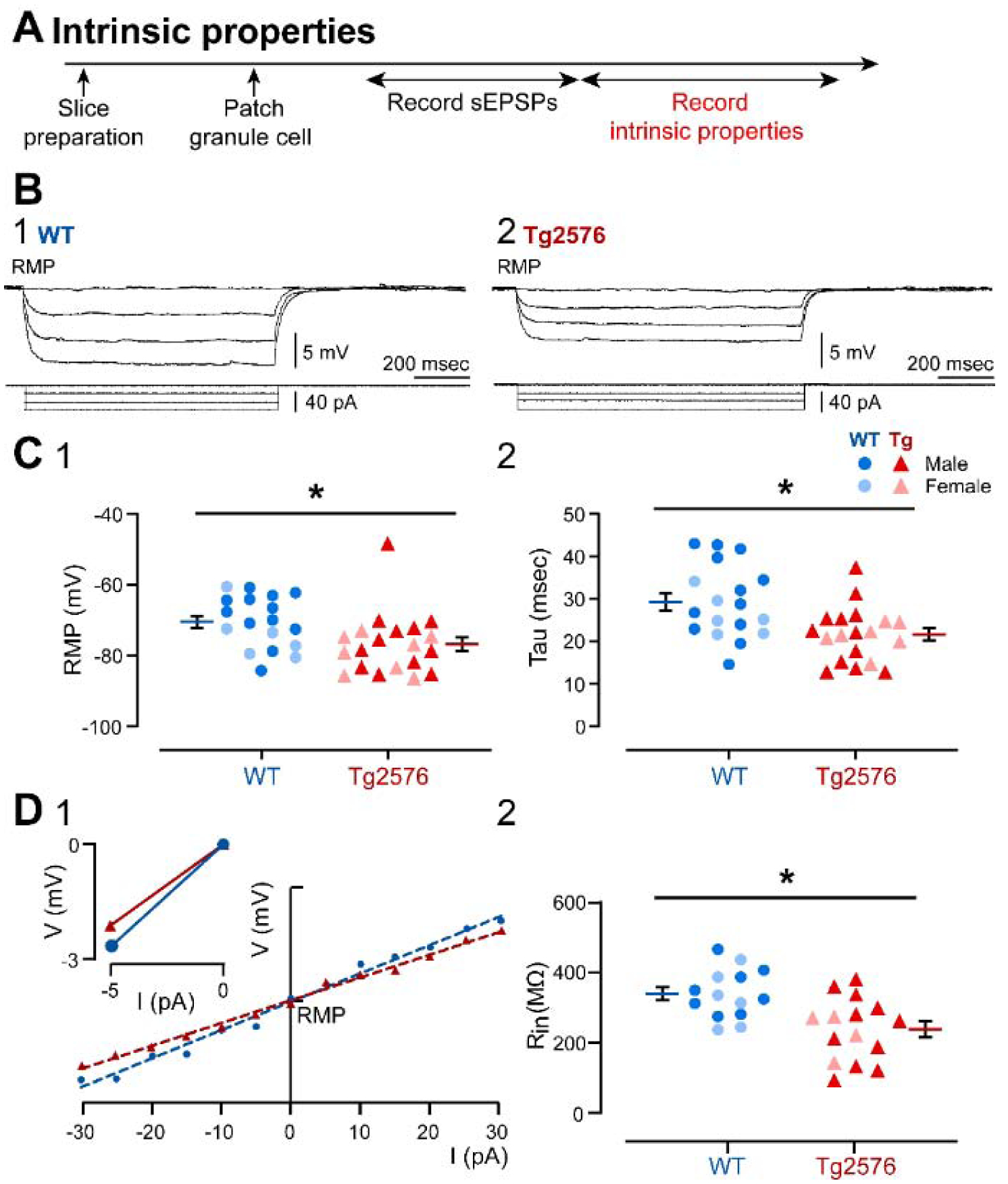
(A) The timeline of the electrophysiological recordings for the determination of ntrinsic properties from GCs.
(B) Membrane potential responses of representative (1) WT and (2) Tg2576 GCs to consecutive negative current steps from −5 pA to −15 pA using a 5 pA increment.
(C) (1) Resting membrane potential (RMP, in mV) and (2) tau (msec) values show that Tg2576 GCs had significantly more hyperpolarized RMPs and shorter tau than WT GCs.
(D1) I-V curves and linear regressions used to calculate (input resistance (Rin). I-V curves were based on responses to positive and negative current pulses (−30 to 30 pA) in WT and Tg2576 GCs. The inset shows a representative response to a −5 pA current step from RMP, from which slope was calculated to provide a second estimation of Rin.
The RMP of GCs from Tg2576 mice was more hyperpolarized (−77.09 ± 1.94 mV) relative to WT mice (−70.74 ± 1.75 mV; unpaired t-test, t=2.414, df=36; p=0.021, Figure 4C1). Tau was smaller in Tg2576 mice (21.46 ± 1.48 msec) than WT mice (29.17 ± 2.02 msec; unpaired t-test, t=3.108, df=35; p=0.004; Figure 4C2, 4B2). Tg2576 GCs showed a significantly smaller Rin (239.1 ± 23.07 MΩ) than WT mice (340.1 ± 18.70 MΩ; unpaired t-test, t=3.373, df=27; p=0.002; Figure 4D1, 4D2).
Representative APs and their corresponding phase plots are shown in Figure 5. Although AP amplitude and threshold were not significantly different between genotypes (Supplemental Table 2), AP duration differed. The time to peak from threshold was greater in Tg2576 (1.28 ± 0.08 msec) than WT mice (1.02 ± 0.09 msec; Mann-Whitney test, U=101; p=0.032; Figure 5A and 5B1). Half-width was also greater in Tg2576 (0.34 ± 0.02 msec) compared to WT mice (0.27 ± 0.02 msec; Mann-Whitney test, U=95; p=0.017; Figure 5B2). Tg2576 GCs did not differ from WT mice in the maximum rate of rise, maximum rate of decay, dv/dt ratio or AHP amplitudes (Supplemental Table 2).
Figure 5. Differences in the properties of APs in WT and Tg2576 mice.
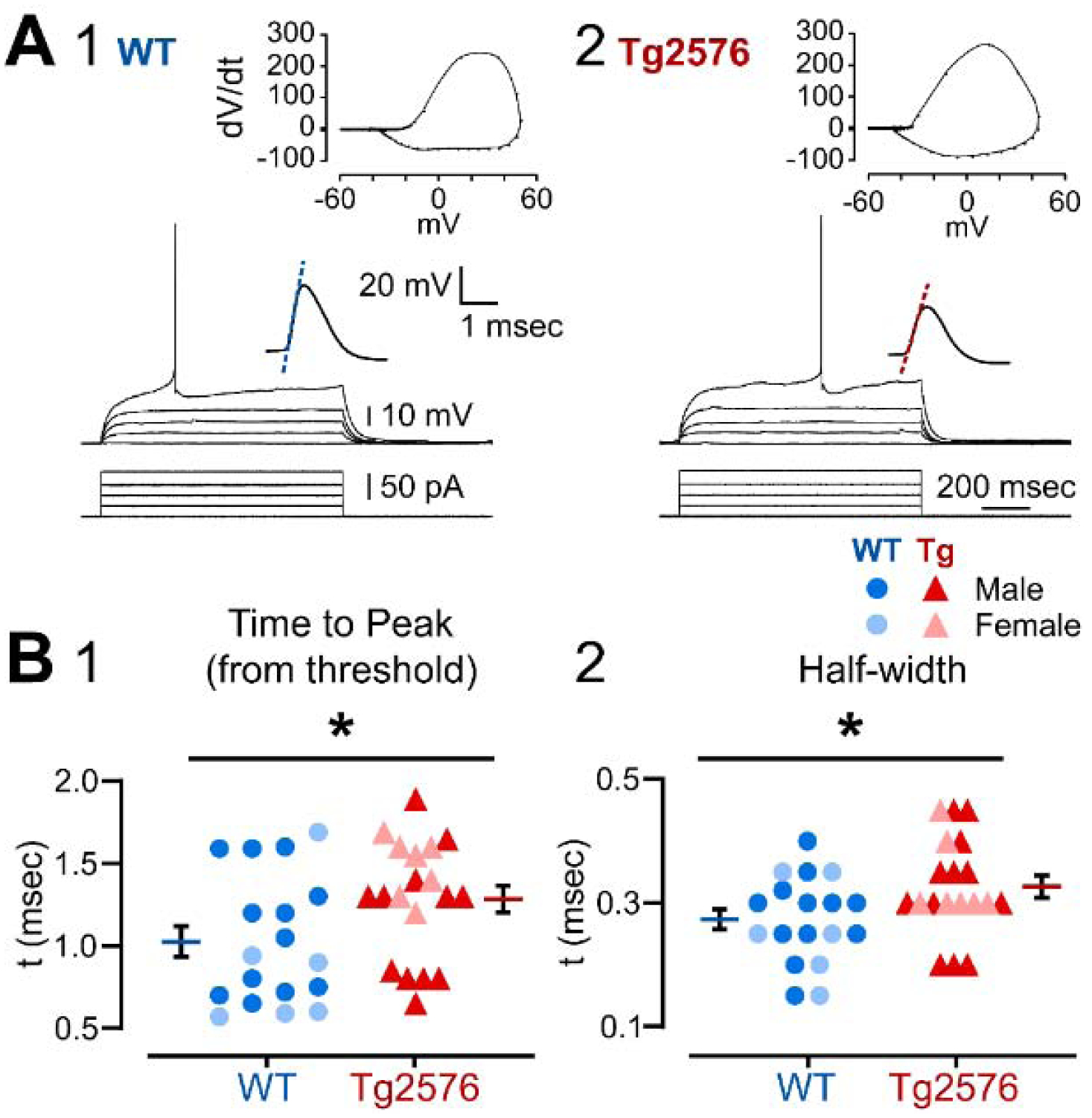
(A) Representative traces from (1) WT and (2) Tg2576 GCs of responses to increasingly larger current steps (+5 pA increment) from RMP until an AP was generated. The AP is shown at higher gain in the inset above the traces. The phase plots corresponding to the traces are shown at the top.
(B) The mean (1) time to peak and (2) half width are shown for an AP at threshold. The APs from Tg2576 GCs had a significantly longer time to peak and half-width compared to WT GCs.
C. Spike frequency adaptation is reduced in GCs from Tg2576 mice compared to WT mice
To address firing behavior, spike frequency adaptation was chosen as a measurement (Figure 6) because the firing behavior of GCs is primarily in trains with strong spike frequency adaptation, as previously reported (Scharfman, 1992; Staley et al., 1992; Williamson and Patrylo, 2007). As shown by the examples in Figure 6Aa–b, Tg2576 GCs showed weaker adaptation than WT mice. To quantify adaptation, the interspike intervals (ISI) were compared either from trains of 4 APs (Figure 6A–B) or trains that were longer (Figure 6C–D). For the comparison of 4 APs, Tg2576 GCs appeared to have less spike frequency adaptation than WT mice (Figure 6Aa–b). However, when all ISIs were compared sequentially, there was only a trend (two-way RMANOVA; F (1, 29) = 3.742; p=0.063; Figure 6B1). When Sidak’s post-hoc comparisons were done, they showed that the first ISI was significantly longer in Tg2576 mice (mean difference −46.98 msec; t=2.842; df=27.37; p=0.025) and other ISIs were not (all p values > 0.05; Figure 6B1).
Figure 6. Spike frequency adaptation is reduced in GCs from Tg2576 mice compared to WT.
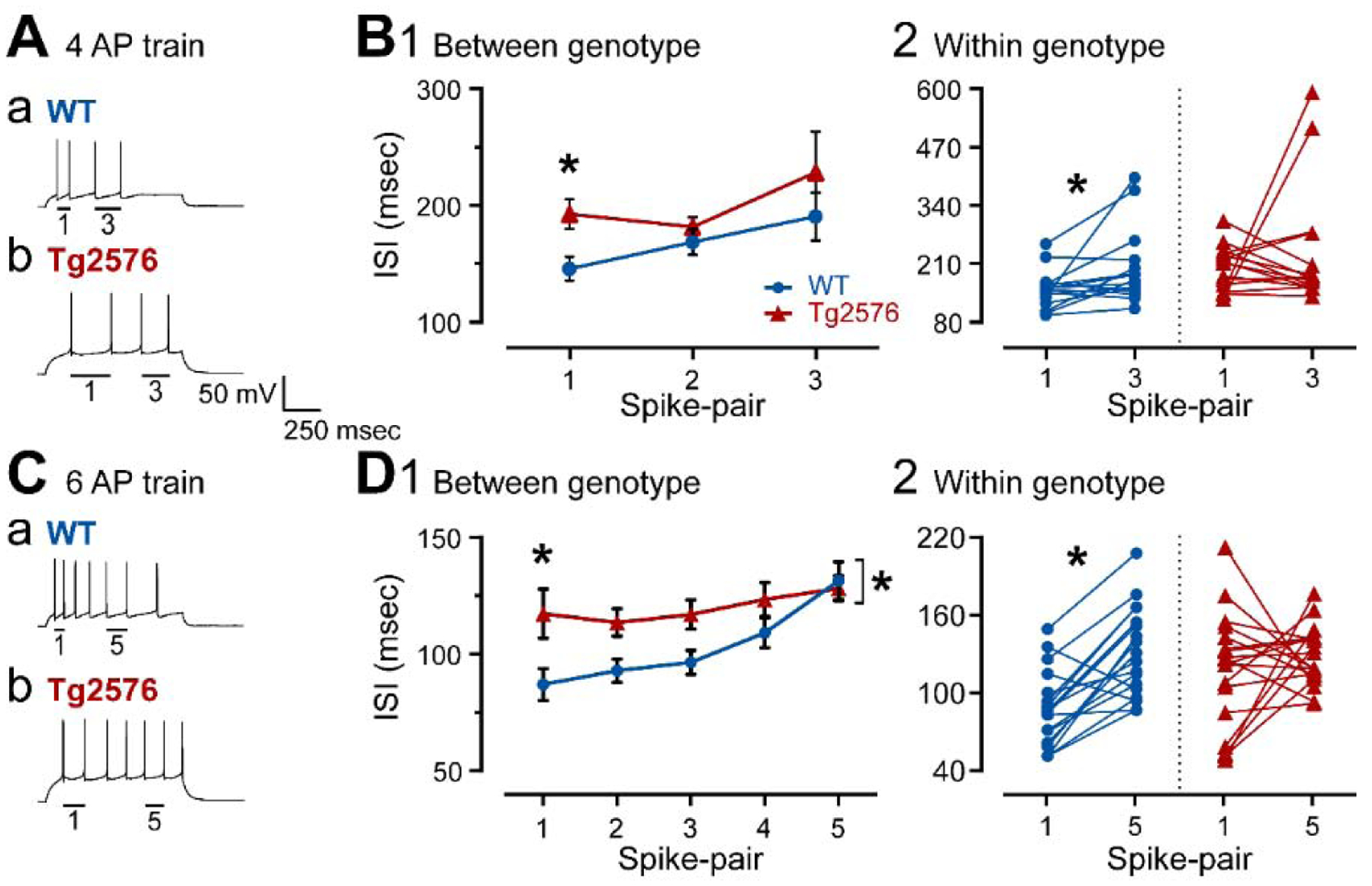
(A) Representative traces of 4 AP trains induced by positive current steps to GCs from (a) WT and (b) Tg2576 mice. The first (1) and the third (3) AP pairs are labeled by the bars below them.
(B1) Spike frequency adaptation was quantified in WT and Tg2576 mice by measuring the ISI for each AP pair and plotting them sequentially. The first ISI was significantly longer ISI in Tg2576 mice.
(B2) Comparisons of the first and third AP pairs showed that the first ISI was significantly shorter in WT mice, indicating adaptation occurred, but not in Tg2576 mice, suggesting Tg2576 mice have weak adaptation.
(C) Representative traces of 7 AP trains induced by positive current steps to GCs from (a) WT and (b) Tg2576 mice. The first (1) and 5th (5) AP pairs are labeled by the bars below the traces.
(D1) Spike frequency adaptation was quantified in WT and Tg2576 mice by measuring the ISI for each AP pair and plotting them sequentially. The first ISI was significantly longer in Tg2576 mice. The sequence of ISIs were also significantly different, with Tg2576 mice exhibiting longer ISIs.
(D2) Comparisons of the first and fifth AP pairs showed that the first ISI was significantly shorter in WT mice, indicating adaptation occurred, but not Tg2576 mice, suggesting Tg2576 mice have weak adaptation.
When the first and third AP pairs were examined specifically, the WT mice showed adaptation (first AP pair: 141.3 ± 10.39 msec; third AP pair: 186.1 ± 20.56; Wilcoxon’s test, W=92, p=0.016; Figure 6B2). In contrast, Tg2576 mice did not. In other words, Tg2576 mice did not exhibit significant differences between the first and third pair of APs (first AP pair: 188.3 ± 12.85 msec; third AP pair: 223.7 ± 35.09; Wilcoxon test, W=−12, p=0.762, Figure 6B2).
These data suggested weak adaptation using a 4 AP train, especially in Tg2576 mice. Adaptation using longer trains (6 APs) was studied to confirm these results (Figure 6Ca–b). The sequence of APs was significantly different in WT and Tg2576 mice (two-way RMANOVA; F(1, 170)= 14.24; p=0.0002; Figure 6D1) and Sidak’s post-hoc test showed a significant difference for the first AP pair (mean difference −30.20 msec; t=3.091; df=170; p=0.012) but not the others (all p values > 0.05; Figure 6D1), similar to the 4 AP train. In Figure 6D2, comparisons were made of WT and Tg2576 GCs for the first and fifth AP pairs only. The data showed that there was adaptation in the WT GCs (first AP pair: 87.08 ± 6.91 msec; fifth AP pair: 131.5 ± 8.08; paired t-test, t=5.458, df=17; p<0.0001; Figure 6D2) but not the Tg2576 GCs (first AP pair: 117.3 ± 10.56 msec; fifth AP pair: 128.1 ± 5.40; paired t-test, t=0.814, df=17; p=0.427; Figure 6D2). These data confirm the results from 4 AP trains.
Taken together, Tg2576 GCs had different intrinsic properties from WT GCs and the differences suggested that Tg2576 mice had reduced excitability. However, firing behavior of Tg2576 mice showed reduced adaptation which would increase excitability. These differences between WT and Tg2576 mice, sometimes decreasing and sometimes increasing excitability, make it difficult to predict whether the differences in intrinsic properties would affect excitability in vivo.
The relatively weak spike frequency adaptation in Tg2576 GCs is interesting because the muscarinic cholinergic “M” current is a major contributing factor to spike frequency adaptation in prior studies of pyramidal cells (Rogawski, 2000; Storm, 1990). To understand cholinergic regulation of GCs in WT and Tg2576 mice further, we examined the role of the muscarinic cholinergic receptor on sPSCs, intrinsic properties, and firing behavior.
II. Muscarinic cholinergic modulation of synaptic activity, intrinsic properties and firing behavior is different in young Tg2576 and WT mice
A. Muscarinic cholinergic modulation differentially affects excitatory and inhibitory synaptic activity differently in WT and Tg2576 mice
1). The muscarinic antagonist atropine produces a small increase in sEPSP frequency in WT GCs but not in Tg2576 GCs
To address how muscarinic cholinergic receptors might contribute to the differences between sEPSPs in WT and Tg2576 mice, the effects of the muscarinic antagonist atropine was tested by adding it to the ACSF (10 μM; Figure 7A).
Figure 7. Atropine produces a small increase in sEPSP frequency in WT GCs but not in Tg2576 GCs.
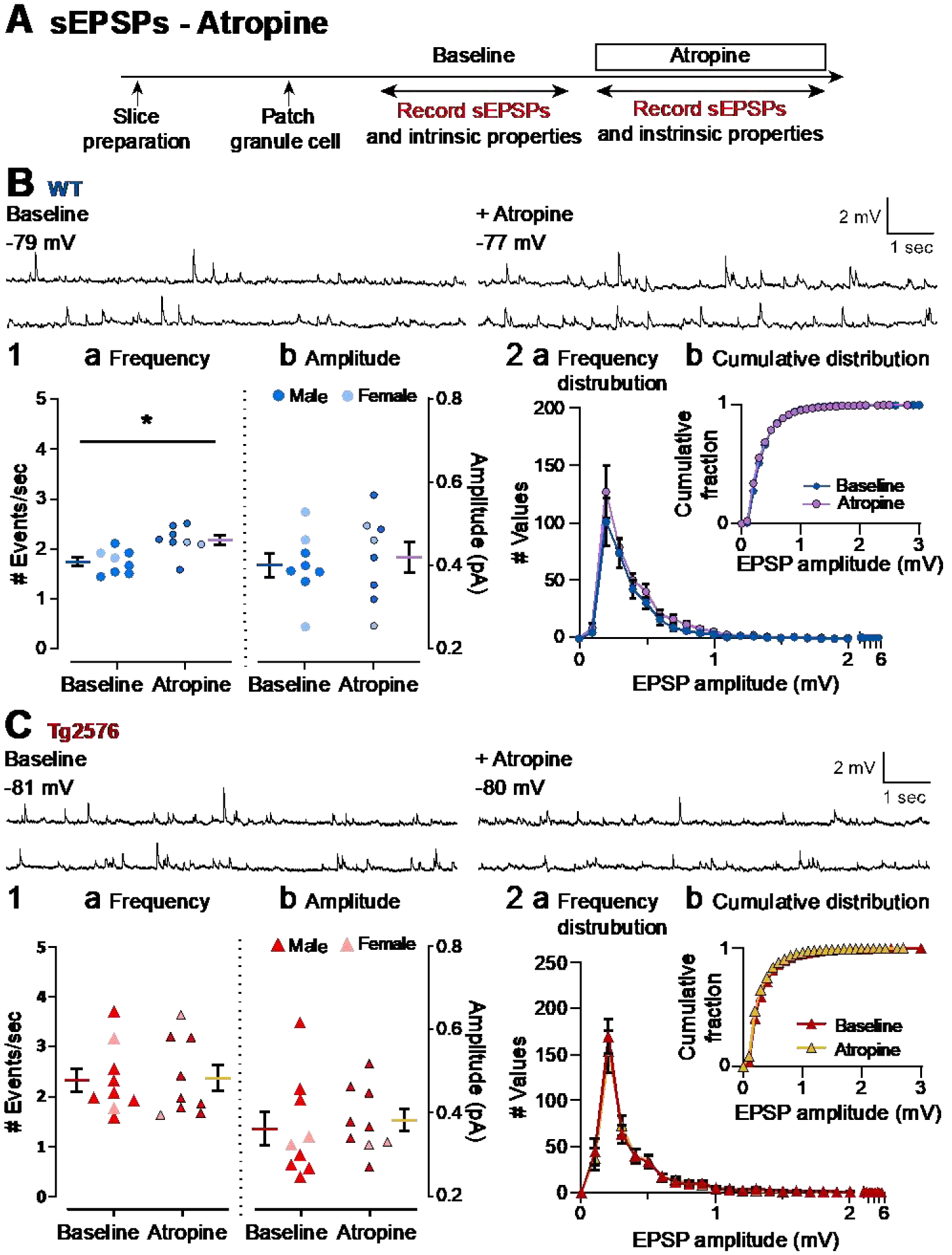
(A) The timeline of the electrophysiological recordings for the evaluation of the effects of atropine on sEPSPs. Atropine (10 μM) was added to the ACSF for the time period indicated by the box.
(B) Representative traces for sEPSPs for WT GCs obtained in baseline conditions (left) and in presence of atropine (right).
(B1) Quantification of the (a) frequency and (b) amplitude of sEPSPs for WT GCs are presented. The only effects of atropine that were significant was that atropine increased the mean sEPSP frequency. The magnitude of this effect was small.
(B2) Histograms showing the (a) frequency distribution of sEPSPs amplitudes for WT GCs. Insets (b) show that there were no significant effects of atropine on cumulative distributions.
(C) Representative traces for sEPSPs for Tg2576 GCs obtained in baseline conditions (left) and in presence of atropine (right).
(C1) Quantification of the (a) frequency and (b) amplitude of sEPSPs for Tg2576 GCs are presented. There was no effect in Tg2576 mice.
(C2) Histograms showing the (a) frequency distribution of sEPSPs amplitudes for Tg2576 GCs. Insets (b) show that there were no significant effects of atropine on cumulative distributions.
Current clamp experiments showed that WT sEPSPs increased in response to atropine, but this was not true for Tg2576 sEPSPs (Figure 7B–C). Thus, the mean frequency of sEPSPs in WT mice was 1.75 ± 0.09 events/sec before atropine and after atropine it was 2.18 ± 0.10 (paired t-test, t=3.309, df=7, p=0.013; Figure 7B1a). In Tg2576 mice, the frequencies before and after atropine were 2.15 ± 0.23 and 2.19 ± 0.26 events/sec, respectively, which was not significantly different (paired t-test, t=0.247, df=8, p=0.811; Figure 7C1a). One reason why the Tg2576 mice did not show an effect of atropine might be that the baseline frequency of Tg2576 mice was already high (2.15 ± 0.23 events/sec) relative to WT mice (1.75 ± 0.09 events/sec) before atropine. Thus, the baseline sEPSP frequency of Tg2576 GCs (2.15 ± 0.23 events/sec) was similar to the post-atropine sEPSP frequency of WT GCs (2.18 ± 0.10 events/sec; unpaired t-test, t=0.127, df=10.89, p=0.901; Figure 7B1a vs. 7C1a).
In contrast to frequency, there was no effect of atropine on sEPSP mean amplitude in WT mice (baseline: 0.40 ± 0.03 mV; atropine: 0.42 ± 0.04; paired t-test, t=0.604, df=7; Figure 7B1b) or Tg2576 mice (baseline: 0.35 ± 0.04 mV; atropine: 0.37 ± 0.03; paired t-test, t=0.985, df=7, p=0.358; Figure 7C1b).
Consistent with a lack of significant effects of atropine on sEPSP amplitude, WT and Tg2576 mice did not show any differences in their frequency distributions (Figure 7B2, 7C2), or cumulative distributions (Kolmogorov-Smirnov tests in WT: D=0.166, p=0.826; Figure 7B2b; Tg2576: D=0.095, p=0.999; Figure 7C2b).
2). Atropine has no significant effect on sEPSCs in WT and Tg2576 mice
Next, atropine was tested using voltage clamp and sEPSCs were analyzed (Figure 8A). Atropine did not have a significant effect on sEPSC frequency in WT mice (baseline: 3.16 ± 0.33 events/sec; atropine: 3.44 ±0.33; paired t-test, t=1.221, df=6, p=0.268; Figure 8B1a) or sEPSC amplitude (baseline 8.72 ± 1.03 pA; atropine: 7.84 ± 0.88; paired t-test, t=1.656, df=6, p=0.149; Figure 8B1b). Consistent with the lack of changes in frequency and amplitude, the frequency distributions showed no differences between recordings before vs. after atropine for WT mice, and there were no significant differences in the cumulative distributions (Kolmogorov-Smirnov test, D=0.129; p=0.734; Figure 8B2b).
Figure 8. Atropine did not affect sEPSCs in GCs of WT and Tg2576 mice.
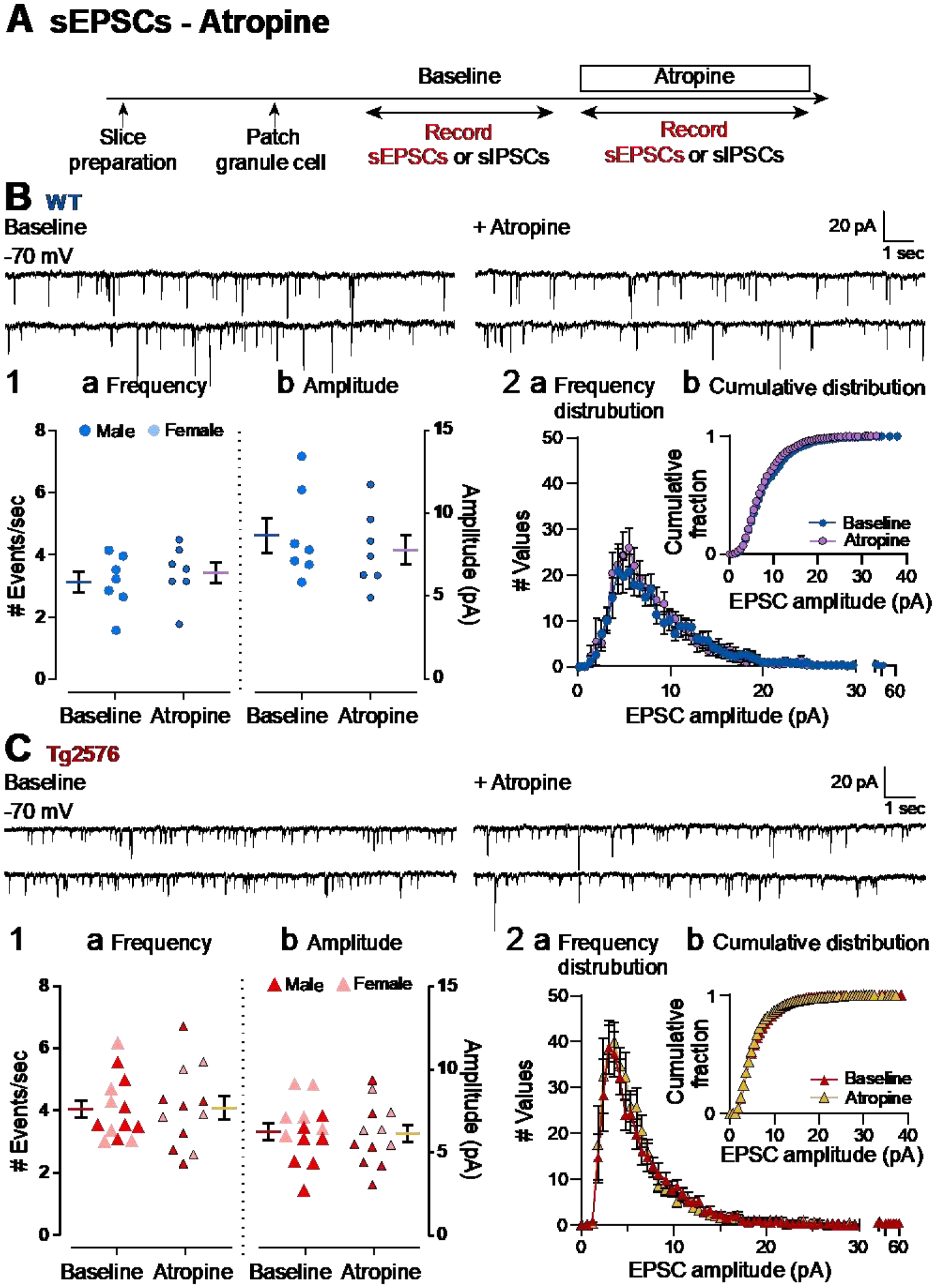
(A) The timeline of the electrophysiological recordings to determine effects of atropine on sEPSCs of WT and Tg2576 GCs.
(B) Representative traces for sEPSCs for WT GCs obtained in baseline conditions (left) and in presence of atropine (right).
(B1) Quantification of the (a) frequency and (b) amplitude of sEPSCs for WT GCs showed no significant effects of atropine.
(B2) Histograms showing the (a) frequency distribution of sEPSCs amplitudes for WT GCs. Insets (b) show that there were no significant effects of atropine on cumulative distributions.
(C) Representative traces for sEPSCs for Tg2576 GCs obtained in baseline conditions (left) and in presence of atropine (right).
(C1) Quantification of the (a) frequency and (b) amplitude of sEPSCs for Tg2576 GCs showed no significant effects of atropine.
(C2) Histograms showing the (a) frequency distribution of sEPSCs amplitudes for Tg2576 GCs. Insets (b) show that there were no significant effects of atropine on cumulative distributions.
Next, Tg2576 sEPSCs were evaluated (Figure 8C). Like WT mice, atropine did not have a significant effect on sEPSC frequency (baseline: 4.05 ± 0.31 events/sec; atropine: 4.10 ± 0.28; paired t-test, t=0.169, df=11; p=0.870; Figure 8C1a) or sEPSC amplitude (baseline: 6.26 ± 0.54 pA; atropine: 6.15 ± 0.54; paired t-test, t=0.311, df=11, p=0.762; Figure 8C1b). Similar to WT mice, there was not a significant effect of atropine on cumulative distributions of Tg2576 mice (Kolmogorov-Smirnov test, D=0.091; p=0.963; Figure 8C2b).
3). Atropine reduces sIPSC frequency and amplitude in WT and Tg2576 mice
Next, the effect of atropine on sIPSCs was addressed using the same experimental timeline to the one used for sEPSCs (Figure 9A). There was a significant reduction in sIPSC frequency in WT GCs (baseline: 10.24 ± 1.09 events/sec; atropine: 7.96 ± 0.86; paired t-test, t=2.456, df=11, p=0.032; Figure 9B1a) and Tg2576 GCs (baseline: 8.19 ± 0.63 events/sec; atropine: 6.42 ± 0.53; paired t-test, t=4.477, df=13, p=0.0006; Figure 9C1a). Atropine also reduced sIPSC mean amplitude in WT GCs (baseline: 10.93 ± 1.21 pA; atropine: 8.74 ± 0.76 pA; paired t-test, t=3.790, df=11, p=0.003; Figure 9B1b) and Tg2576 GCs (baseline: 10.57 ± 0.88 pA; atropine: 9.18 ± 0.74; Wilcoxon test, W=−77.0, p=0.013; Figure 9C1b).
Figure 9. Atropine reduces sIPSC frequency and amplitude in both WT and Tg2576 mice.
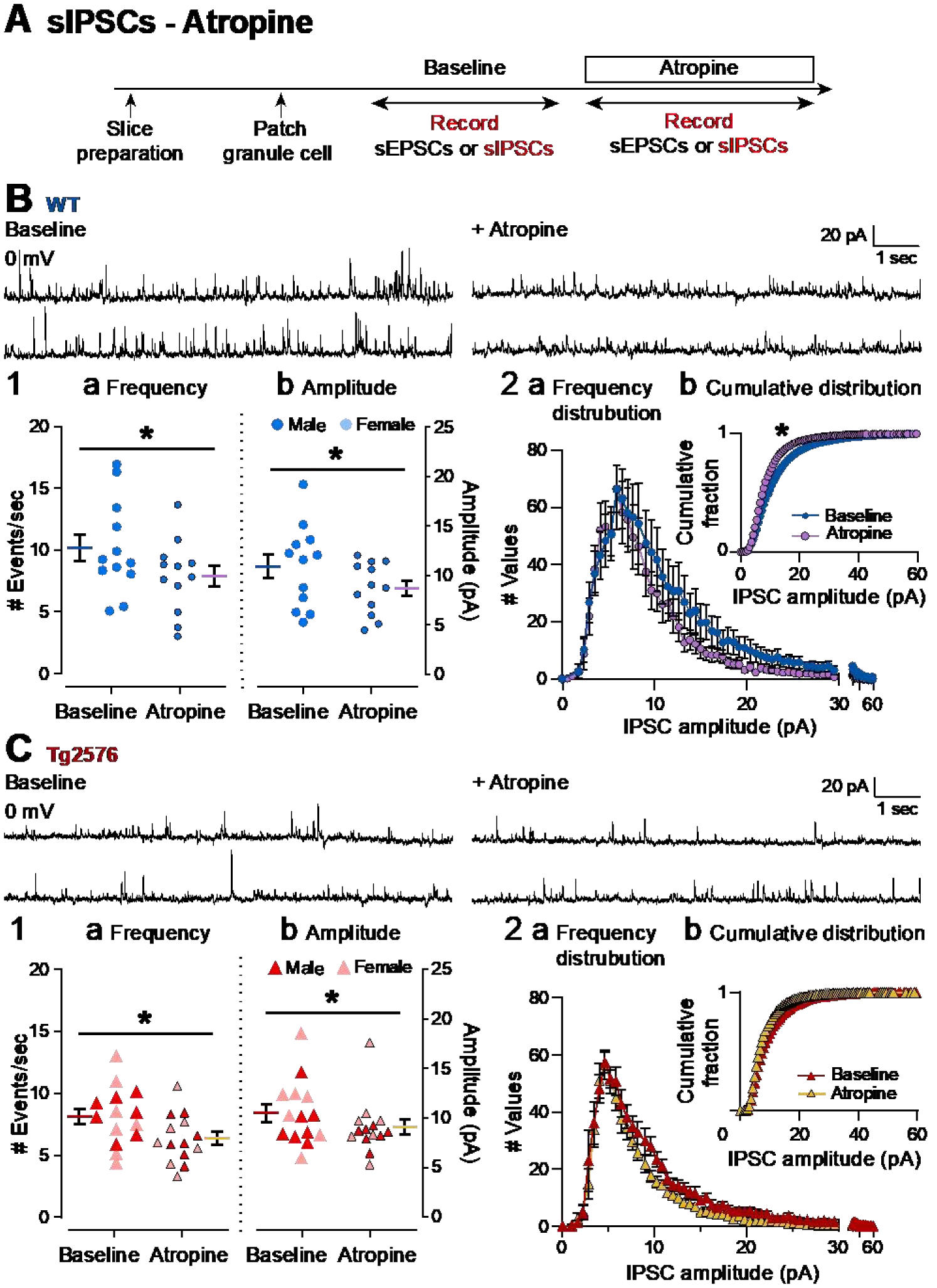
(A) The timeline of electrophysiological recordings used to determine effects of atropine on sIPSCs of GCs from WT and Tg2576 mice.
(B) Representative traces of sIPSCs for WT GCs obtained in baseline conditions (left) and in presence of atropine (right).
(B1) Quantification of the (a) frequency and (b) amplitude of sIPSCs for WT GCs show that atropine reduced frequency and amplitude in WT mice.
(B2) Histograms showing the (a) frequency distribution of sIPSPs amplitudes for WT GCs. Insets (b) show the cumulative distributions where there is a significant effect of atropine in WT mice.
(C) Representative traces of sIPSCs for Tg2576 GCs obtained in baseline conditions (left) and in presence of atropine (right).
(C1) Quantification of the (a) frequency and (b) amplitude of sIPSCs for Tg2576 GCs show that atropine reduced frequency and amplitude in Tg2576 mice.
(C2) Histograms showing the (a) frequency distribution of sIPSPs amplitudes for Tg2576 GCs. Insets (b) show the cumulative distributions. There was not a significant effect of atropine in Tg2576 mice.
The frequency distributions for WT and Tg2576 mice showed different effects of atropine (Figure 9B2, 9C2). The cumulative distributions of sIPSCs showed a decline in sIPSCs after atropine that was significant for WT mice (Kolmogorov-Smirnov test, D=0.250, p=0.006; Figure 9B2b), but not for Tg2576 mice (Kolmogorov-Smirnov test, D=0.104, p=0.740 Figure 9C2b). These data suggest that muscarinic cholinergic receptors decrease sIPSCs in WT and Tg2576 GCs.
4). A novel synaptic current (nsC) that is normally greater in Tg2576 mice compared to WT mice
In the course of the voltage clamp experiments, recordings at 0 mV revealed additional notable findings (Figure 10A, Figure 11A1, 11A2a). There were small inward glutamatergic synaptic currents that we defined as novel (novel spontaneous currents, nsCs) because they were prominent during the baseline in Tg2576 GCs but rare in WT GCs (Figure 10A–D). Atropine increased nsCs in WT mice but had less of an effect in Tg2576 mice (Figure 10A–D), perhaps because there already were many nsCs present.
Figure 10. A novel spontaneous inward current (nsC) that is greater in Tg2576 GCs than WT GCs and the effects of atropine.
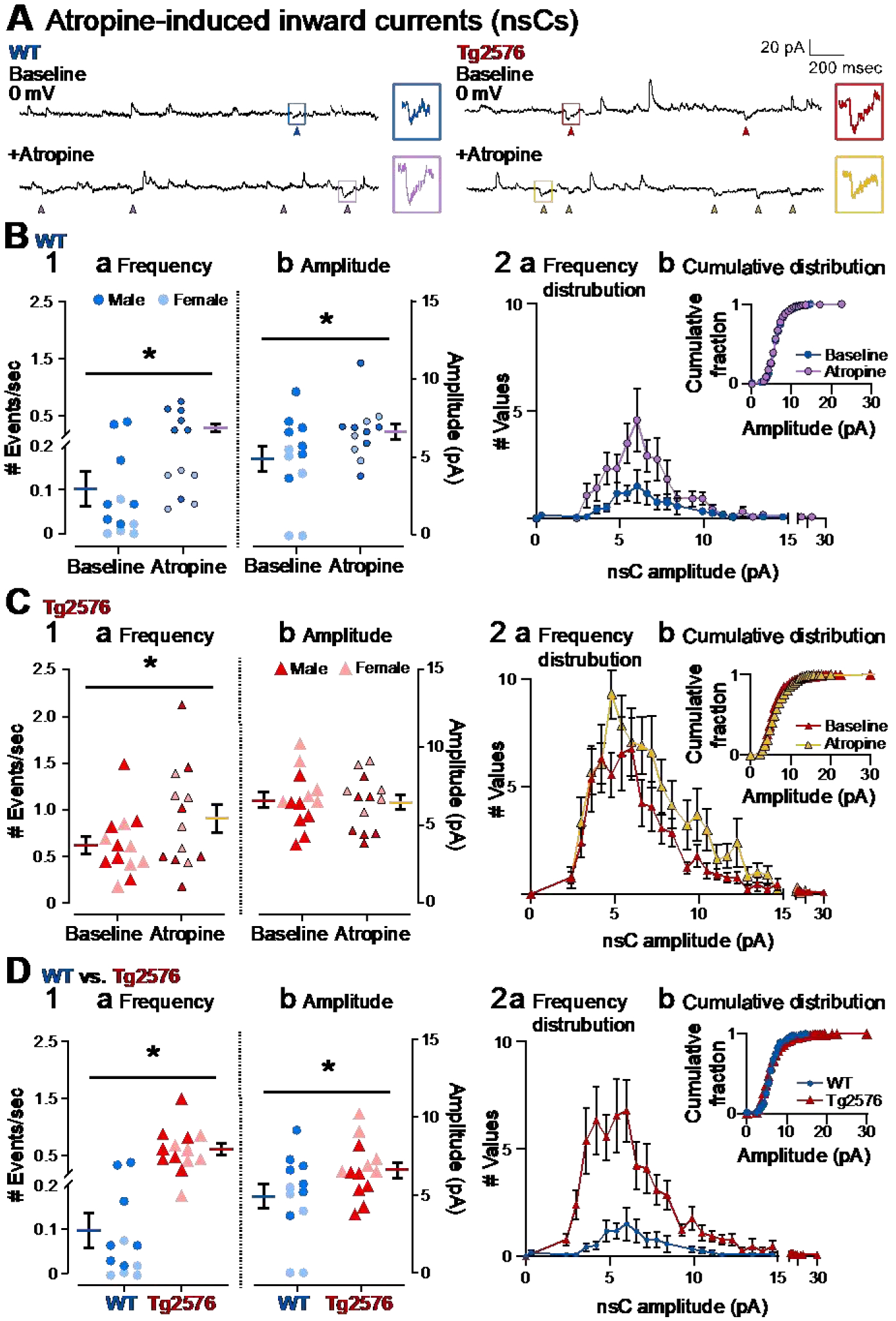
(A) Representative traces show spontaneous inward currents (pointed with arrows) obtained in WT (left) and Tg2576 mice (right) in baseline conditions and in the presence of atropine (10 μM). Holding potential was 0 mV. The small squares mark examples of the spontaneous inward currents, which we refer to as nsCs. The examples of nsCs are expanded on the right.
(B1) Mean frequency (a) and amplitude (b) of nsCs are shown for WT mice during the baseline and after atropine was added. Atropine significantly increased the frequency and amplitude of nsCs.
(B2) WT nsC frequency distributions (a) and cumulative distributions (b) are shown. Atropine did not cause significant changes to the cumulative distributions relative to baseline conditions.
(C1) Mean frequency (a) and amplitude (b) of nsCs are shown for Tg2576 mice. Atropine significantly increased the frequency of nsCs but not the amplitude.
(C2) Tg2576 nsC frequency distributions (a) and cumulative distributions (b) are shown. Atropine did not cause significant changes to the cumulative distributions, like WT mice.
(D1) Comparisons of WT and Tg2576 GCs for nsC frequency (a) and amplitude (b) during baseline conditions. Tg2576 GCs had significantly more frequent and larger nsCs than WT mice.
(D2) NsC frequency distributions (a) and cumulative distributions (b) for WT and Tg2576 GCs during baseline conditions are shown. The cumulative distribution of nsCs did not show a significant difference.
Figure 11. Differences in properties of nsCs from other EPSCs in WT and Tg2576 mice.
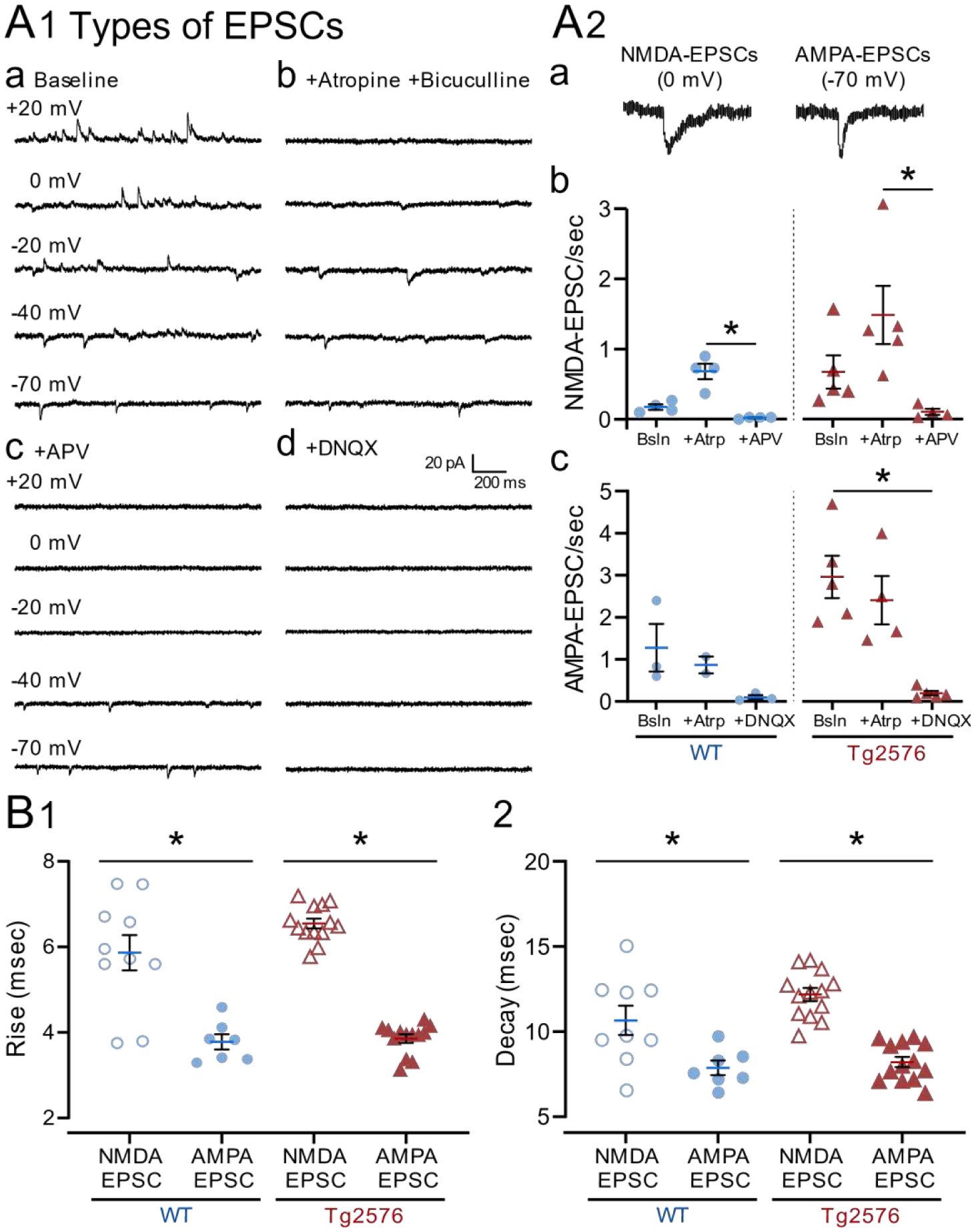
(A1) Representative traces show spontaneous outward and inward currents (a) before and after sequential addition of (b) atropine and bicuculline, (c) APV and (d) DNQX. All concentrations were 10 μM except APV which was 50 μM. Recordings were made for 2 min at each holding potential.
(A2) Representative examples of (a) nsCs at 0 mV (NMDA-EPSCs) and AMPA-EPSCs at −70 mV. (b) Mean frequencies of nsCs (NMDA-EPSCs) are shown for WT (left) and Tg2576 mice (right). (c) Mean frequency of AMPA-EPSCs at −70 mV. Note nsCs (NMDA-EPSCs) were dramatically reduced by APV and AMPA-EPSCs by DNQX.
(B) Mean value for the (1) rise and (2) decay times of nsCs at 0 mV (NMDA-EPSCs) and AMPA-EPSCs at −70 mV for Tg2576 and WT mice. The kinetics of NMDA-EPSCs were significantly slower than AMPA-EPSCs.
We confirmed that nsCs were glutamatergic, like other inward currents at 0 mV (Supplemental Figure 2) and at the same time, confirmed that the sEPSCs described in the previous sections were glutamatergic, and sIPSCs were GABAergic (Supplemental Figure 2B and 2C). In these experiments, after atropine was added, the GABAA receptor antagonist bicuculline was added also (Supplemental Figure 2A), and sIPSCs were blocked (Supplemental Figure 2B3 and 2C2). Afterwards, CNQX and APV were added, and all inward currents were blocked (Supplemental Figure 2B4 and 2C3).
Because of their small size, we discriminated nsCs from the baseline noise by their kinetics (fast rate of rise relative to decay, like synaptic currents) and amplitude >1 standard deviation (SD) greater than the baseline noise (Figure 10A, Figure 11A1, 11A2a, Supplemental Figure 2C1, arrows). Baseline noise was defined as the average peak-to-peak amplitude for a randomly-selected period of the baseline lasting 1 sec.
The presence of nsCs was rare in WT GCs under baseline conditions, as mentioned above. This may be why they have not been previously reported. Also mentioned above, Tg2576 GCs showed more frequent nsCs during the baseline than WT GCs. This and other direct WT vs. Tg2576 comparisons are explained further below (Figure 10D).
Effects of atropine (10 μM) on nsCs showed some similarities and some differences between WT and Tg2576 mice (Figure 10A–C). For WT nsCs, atropine increased frequency (baseline: 0.10 ± 0.04 events/sec; atropine: 0.29 ± 0.07; Wilcoxon test, W=78; p<0.001, Figure 10B1a) as well as amplitude (baseline: 4.91 ± 0.79 pA; atropine: 6.32 ± 0.51; paired t-test, t=2.302, df=11; p=0.021; Figure 10B1b).
Atropine also increased the frequency of nsCs in Tg2576 GCs (baseline: 0.63 ± 0.09 events/sec; atropine: 0.92 ± 0.15; paired t-test, t=2.758, df=12; p=0.017, Figure 10C1a) However, nsC amplitude did not change significantly in response to atropine (baseline: 6.59 ± 0.50 pA; atropine: 6.46 ± 0.48; paired t-test, t=0.523, df=12; p=0.610; Figure 10C1b), probably because Tg2576 nsCs were already large in amplitude before atropine. Thus, WT nsC mean amplitude after atropine was approximately the same as the Tg2576 nsC mean amplitude before atropine.
When comparing the frequency distributions, the data reflected the larger changes after atropine in WT mice (Figure 10B2a), compared with Tg2576 mice (Figure 10C2a). Cumulative distributions did not show any significant difference before and after atropine in WT (Kolmogorov-Smirnov test, D=0.349; p=0.188; Figure 10B2b) and Tg2576 mice (Kolmogorov-Smirnov test, D=0.241; p=0.351; Figure 10C2b).
When WT and Tg2576 mice were compared directly (Figure 10D) several interesting findings were revealed. Under baseline conditions, nsCs were approximately 6-fold greater in frequency in Tg2576 mice compared to WT mice (Tg2576: 0.63 ± 0.09, events/sec; WT: 0.10 ± 0.04; Mann-Whitney test, U=4, p<0.001, Figure 10A and 10D1a). During the baseline, nsCs were also slightly greater in amplitude in Tg2576 mice relative to WT mice (Tg2576: 6.59 ± 0.50 pA, and WT: 4.91 ± 0.79; unpaired t-test, t=1.833, df=23; p=0.040; Figure 10A,10D1b). The frequency distributions reflecting the differences between WT and Tg2576 nsCs are shown in Figure 10D2a. The cumulative distributions were not significantly different (Kolmogorov-Smirnov test, D=0.344, p=0.131; Figure 10D2b).
Taken together, nsCs reflect a glutamatergic synaptic current that was more prevalent in Tg2576 GCs relative to WT GCs. While atropine induced more nsCs in both WT and Tg2576 mice, there were greater effects of atropine in WT mice. These data may explain the more frequent small sEPSPs in Tg2576 compared to WT mice (Figure 1B2a). The data may also reflect an alteration in the actions of muscarinic receptors in young Tg2576 mice so that effects of atropine were diminished compared to effects of atropine in WT mice.
5). nsCs are mediated by NMDA receptors
In a subset of 9 cells (4 WT and 5 Tg2576) from 4 WT and 3 Tg2576 mice, two different approaches were used to identify which type of glutamatergic receptor mediated the nsCs. We found that nsCs were reduced by APV, suggesting that they were mediated by NMDA receptors (WT: baseline 0.18 ± 0.04 nsCs/sec, atropine 0.68 ± 0.11, APV 0.02 ± 0.01; Kruskal-Wallis test, K-W statistic=10.02, p<0.001; Tg2576: baseline 0.67 ± 0.23 nsCs/sec, atropine 1.49 ± 0.42, APV 0.11 ± 0.04; Kruskal-Wallis test, K-W statistic=9.286, p=0.002; Figure 11A1 and 11A2b). In contrast, EPSCs recorded at −70 mV were reduced by DNQX suggesting they were mediated by AMPA receptors (WT: baseline 1.28 ± 0.57 EPSCs/sec, atropine 0.87 ± 0.20, DNQX 0.10 ± 0.05; Kruskal-Wallis test, K-W statistic=5.0, p=0.075; Tg2576: baseline 2.97 ± 0.50 EPSCs/sec, atropine 2.41 ± 0.58, DNQX 0.20 ± 0.05; Kruskal-Wallis test, K-W statistic=9.411, p=0.001; Figure 11A1 and 11A2c). There were no significant effects of APV on EPSCs recorded at −70 mV or DNQX on nsCs (Supplemental Figure 3A). Based on this pharmacology, we often refer to nsCs as NMDA-EPSCs below. For the same reason, we often refer to EPSCs recorded at −70 mV as AMPA-EPSCs.
Analysis of the kinetics was also performed because nsCs often had a slow time course, and NMDA receptor-mediated currents typically possess slower kinetics compared with AMPA receptor-mediated currents (Traynelis et al., 2010). We found that the kinetics of nSCs recorded at 0 mV (NMDA-EPSCs) were significantly slower than the kinetics for EPSCs recorded at −70 mV (AMPA-EPSCs).
Kinetics was measured by the rise time and decay values. For the rise times, WT mice nsCs (5.87 ± 0.41 msec) were slower than AMPA-EPSCs (3.78 ± 0.18 msec; unpaired t-test with Welch’s correction; t=4.647, df=12.02, p=0.0006; Figure 11B1). Tg2576 values for nsCs (6.55 ± 0.12 msec) were also slower than AMPA-EPSCs (3.86 ± 0.10 msec; Mann-Whitney test, U=0, p<0.0001). For the decay values, nsCs of WT mice (10.66 ± 0.86 msec) were slower than AMPA-EPSCs (7.86 ± 0.41 msec; unpaired t-test with Welch’s correction; t=2.933, df=11.26, p=0.013; Figure 11B2). The same was true for Tg2576 mice (nsCs: 12.18 ± 0.38 msec; AMPA-EPSCs: 8.20 ± 0.31 msec; unpaired t-test, t=8.088, df=24, p<0.0001).
There were no significant differences in kinetics of EPSCs between WT and Tg2576 mice (Supplemental Figure 3B3a, 3B3b). Although we used means for the statistical comparisons above, we also analyzed individual values for PSCs and found the same result (Supplemental Figure 3B2a, 3B2b).
III. GCs in Tg2576 mice have an increased E:I ratio
The E:I ratio for the frequency of events used data from both AMPA-EPSCs and nsCs (NMDA-EPSCs). The ratio was significantly increased in Tg2576 mice (WT: 0.21 ± 0.06; Tg2576: 0.60 ± 0.08, Mann-Whitney test, U=12, p=0.0001; Figure 12A1). Similarly, E:I ratio for amplitude showed a significant increase in Tg2576 mice (WT: 1.05 ± 0.20; Tg2576: 1.60 ± 0.15; unpaired t-test, t=2.215, p=0.037; Figure 12A2). Next, we analyzed charge transfer of EPSCs, including both AMPA-EPSCs and nsCs together. The total charge transfer was greater in Tg2576 mice compared to WT mice (WT: 7305 ± 2142; Tg2576: 13626 ± 1885 pA*msec, Mann-Whitney test; U=119, p=0.021; Figure 12B1). However, the charge transfer was not significantly different when Tg2576 and WT IPSCs were compared (WT: 124669 ± 20339; Tg2576: 110209 ± 13221 pA*msec; unpaired t-test, t=0.605, df=25, p=0.551; Figure 12B2). These data show significantly increased E:I ratio in Tg2576 mice, potentially contributing to their increased excitability. Charge transfer suggests most of this change in E:I ratio was due to increased excitation rather than inhibition.
Figure 12. Increased E:I ratio and total charge transfer in Tg2576 compared to WT mice.
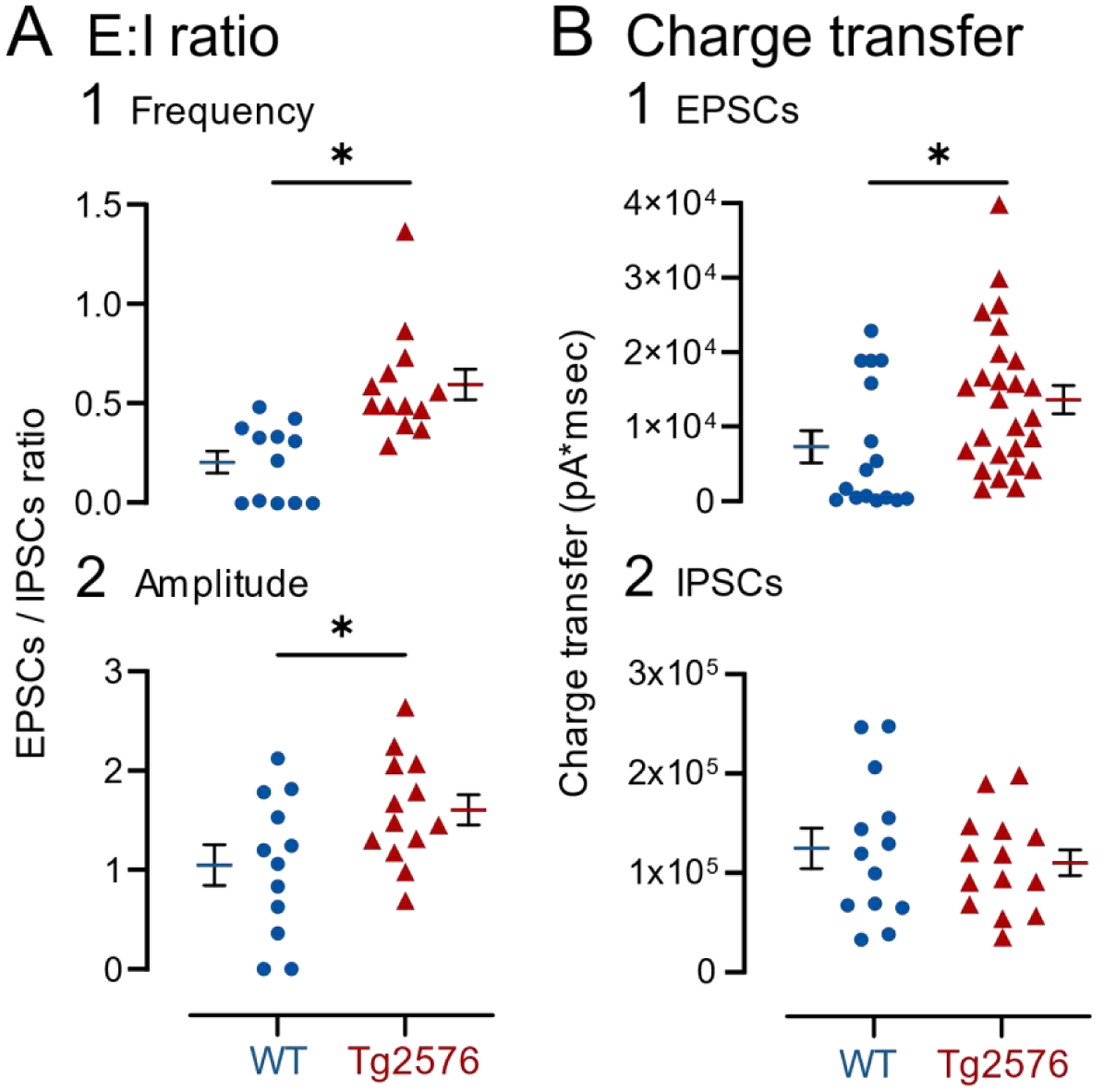
(A) Changes in the ratio of EPSCs (NMDA- and AMPA-EPSCs) to IPSCs based on (1) mean frequency or (2) amplitude showed an increased ratio in Tg2576 mice relative to WT mice.
(B) Changes in the charge transfer of (1) excitatory currents (NMDA- and AMPA-EPSCs) and (2) inhibitory currents (IPSCs). Tg2576 GCs showed increased charge transfer for EPSCs but not IPSCs.
B. Atropine has distinct effects on intrinsic properties of WT and Tg2576 mice
To determine if atropine altered intrinsic properties, GCs used to record sEPSPs were recorded before and after atropine (Figure 7A). In WT and Tg2576 mice, most intrinsic properties were not significantly different when pre- and post-atropine values were compared (Table 1). However, Rin was higher in Tg2576 GCs after atropine (baseline: 262.70 ± 35.64 MΩ; atropine: 451.90 ± 63.68; paired t-test, t=2.767, df=5; p=0.040; Table 1). In WT mice, atropine also increased mean Rin but the difference from the baseline mean was not significant (baseline: 295.30 ± 32.21 MΩ; atropine: 372.80 ± 67.72; paired t-test, t=0.891, df=3; p=0.439; Table 1). Another difference between WT and Tg2576 mice was threshold. The mean threshold was more depolarized after atropine in Tg2576 GCs, although the difference from the baseline mean was only 2 mV (baseline: −33.07 ± 2.23 mV; atropine: −31.37 ± 2.49; paired t-test, t=2.819, df=8; p=0.023; Table 1). Threshold was not significantly changed by atropine in WT mice (baseline: −39.76 ± 2.89 mV; atropine: −39.23 ± 2.95, paired t-test, t=0.951, df=7, p=0.373; Table 1). These data shed light on potential reasons for the increases in nsCs in Tg2576 mice after atropine because the increase in Rin in Tg2576 mice could explain an increased frequency of small synaptic events.
Table 1. Effects of atropine on intrinsic properties were different for WT and Tg2576 mice.
Intrinsic properties of WT and Tg2576 GCs during baseline and after atropine was added. Parametric data were compared using a paired Student’s t-test (1). Non-parametric data were compared by a Wilcoxon matched pairs test (2). Red font indicates significant differences (p<0.05).
| Intrinsic properties Baseline vs. Atropine | ||||||||
|---|---|---|---|---|---|---|---|---|
| RMP (mV) | Rin (−5 pA) (MΩ) | Tau | AHP | |||||
| WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | |
| Baseline | −68.98 ± 2.59 | −73.24 ± 3.44 | 295.30 ± 32.21 | 262.70 ± 35.64 | 28.04 ± 2.82 | 22.02 ± 1.95 | −15.50 ± 1.73 | −18.29 ± 1.94 |
| Atropine | −68.21 ± 2.14 | −72.36 ± 3.62 | 372.80 ± 67.72 | 451.90 ± 63.68 | 30.20 ± 3.31 | 23.61 ± 2.43 | −14.34 ± 2.22 | −19.22 ± 2.23 |
| p value | 0.535 | 0.426 | 0.439 | 0.040 | 0.246 | 0.434 | 0.461 | 0.357 |
| Statistical Test | (1) | (2) | (1) | (1) | (1) | (1) | (2) | (1) |
| t, df | t=0.653, df=7 | W = 15 | t=0.891, df=3 | t=2.767, df=5 | t=1.266, df=7 | t=0.823, df=8 | W=−28.00 | t=0.977, df=8 |
| Nmale | 8 | 9 | 4 | 6 | 8 | 9 | 8 | 9 |
| Nfemale | 8 | 9 | 4 | 6 | 8 | 9 | 8 | 9 |
| Threshold (mV) | Peak Amplitude (mV) | Time to Peak | Time to Peak from threshold | |||||
| WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | |
| Baseline | −39.76 ± 2.89 | −33.07 ± 2.23 | 107.80 ± 3.88 | 106.80 ± 4.62 | 468.70 ± 131.0 | 641.30 ± 92.49 | 1.27 ± 0.14 | 1.19 ± 0.16 |
| Atropine | −39.23 ± 2.95 | −31.37 ± 2.49 | 103.90 ± 4.01 | 105.40 ± 6.00 | 437.20 ± 81.91 | 595.50 ± 107.7 | 1.22 ± 0.13 | 1.12 ± 0.15 |
| p value | 0.373 | 0.023 | 0.147 | 0.457 | >0.999 | 0.820 | 0.666 | 0.906 |
| Statistical Test | (1) | (1) | (1) | (1) | (2) | (2) | (1) | (2) |
| t, df | t=0.951, df=7 | t=2.819, df=8 | t=1.630, df=7 | t=0.782, df=8 | W=0.000 | W=−5.000 | t=0.451, df=7 | W=−2.000 |
| Nmale | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
| Nfemale | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
| Half-width | Max Rise Slope | Max Decay Slope | dv/dt ratio | |||||
| WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | |
| Baseline | 0.27 ± 0.03 | 0.38 ± 0.02 | 295.80 ± 36.31 | 213.90 ± 23.34 | −74.98 ± 6.55 | −68.15 ± 3.58 | 3.88 ± 0.23 | 3.09 ± 0.24 |
| Atropine | 0.29 ± 0.02 | 0.30 ± 0.04 | 255.90 ± 41.52 | 200.90 ± 26.80 | −68.56 ± 6.48 | −63.13 ± 4.76 | 3.58 ± 0.33 | 3.08 ± 0.24 |
| p value | 0.537 | 0.106 | 0.001 | 0.309 | 0.003 | 0.060 | 0.164 | 0.892 |
| Statistical Test | (1) | (2) | (1) | (1) | (1) | (1) | (1) | (1) |
| t, df | t=0.649, df=7 | W=−28.00 | t=5.733, df=7 | t=1.086, df=8 | t=4.466, df=7 | t=2.194, df=8 | t=1.556, df=7 | t=0.140, df=8 |
| Nmale | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
| Nfemale | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
In WT mice, the maximum rate of rise and rate of decay of APs were decreased by atropine (maximum rate of rise, baseline: 295.80 ± 36.31 mV/msec; atropine: 255.90 ± 41.52; paired t-test, t=5.733, df=7, p=0.001; maximum rate of decay, baseline: −74.98 ± 6.55 mV/msec; atropine: −68.56 ± 6.48, paired t-test, t=4.466, df=7, p=0.003; Table 1). However, atropine did not have a significant effect in Tg2576 mice, possibly because the rate of rise and decay were already low.
C. Atropine had little effect on spike frequency adaptation
Next, spike frequency adaptation was examined. When trains of 4 APs were analyzed, there were no differences between WT and Tg2576 mice in the sequence of APs compared by two-way RMANOVA (WT: F(1,21)=0.1724, p=0.682; Tg2576: F(1,20)=0.3530, p=0.559; Supplemental Figure 4A–B1). When the first and third AP pairs were compared, WT mice showed adaptation during the baseline, but there was no significant effect of atropine (paired t-test, t=1.423, df=6, p=0.102; Supplemental Figure 4B2). Tg2576 mice did not show adaptation and there was no significant effect of atropine (paired t-tests, t=0.671, df=6, p=0.264; Supplemental Figure 4B2).
When trains of 6 APs were analyzed (Supplemental Figure 4C), there was adaptation in WT mice and atropine had no detectable effect, like the results from the 4 AP trains (two-way RMANOVA, F(1,143)=1.608, p=0.207; Supplemental Figure 4D1). In Tg2576 mice, there was no significant effect of atropine either (two-way RMANOVA, F(1,138)=0.534, p=0.466; Supplemental Figure 4D1). When we compared the first and fifth AP pairs in WT mice, there was adaptation before as well as post-atropine (paired t-test, t=3.478, df=7, p=0.010; Supplemental Figure 4D2), suggesting that atropine did not have an effect. In Tg2576 mice, there was no significant effect of atropine when the first and fifth AP pairs were analyzed (paired t-test, t=0.623, df=7, p=0.553; Supplemental Figure 4D2). Thus, atropine had little effect on spike frequency adaptation. The small effects of atropine on adaptation in WT mice can be explained by a role of K+ currents other than M-current in spike frequency adaptation (e.g., IA, ID, IC; (Aiken et al., 1995; Storm, 1990). The lack of effect of atropine in Tg2576 mice is probably due to the very weak adaptation in the baseline conditions.
Taken together, the effects of atropine revealed different muscarinic cholinergic regulation of GC synaptic events and intrinsic properties in WT and Tg2576 mice. Regarding EPSCs, NMDA-EPSCs increased after atropine, suggesting that normally muscarinic receptors keep NMDA-EPSCs depressed. However, NMDA-EPSCs were already high in Tg2576 mice before atropine was added, suggesting a reduction in the normal inhibitory effects of muscarinic receptors in Tg2576 mice. This idea is consistent with cholinergic dysfunction in Tg2576 mice. Regarding IPSCs, atropine reduced IPSCs in both WT and Tg2576 mice, suggesting dysfunction of muscarinic receptors primarily affected excitation.
For intrinsic properties, atropine increased Rin and threshold for Tg2576 but not WT mice. Atropine decreased adaptation in WT mice but not Tg2576 mice. The effect on Rin may have only been evident in Tg2576 mice because Rin was lower than WT before atropine was added. Regarding firing behavior, atropine reduced adaptation in WT GCs but not Tg2576 GCs One reason may be that adaptation was already weak in Tg2576 mice before atropine was added. Thus, preexisting dysfunction of muscarinic receptors may have been a reason why some effects of atropine were different in WT and Tg2576 mice. This idea is elaborated upon further in the Discussion.
IV. The morphology and locations of recorded cells were similar in WT and Tg2576 mice
A. Morphology of recorded cells were similar in WT and Tg2576 mice
Confirmation that recorded cells were GCs was important because several types of GABAergic neurons are present in the GC layer (GCL;(Hosp et al., 2014; Houser, 2007; Scharfman, 1995). Recorded cells were filled with biocytin by including it in the internal solution (see Methods). A randomly-selected group of recorded cells were evaluated by processing them to visualize biocytin (n=39; see Methods). An example of a filled GC is shown in Supplemental Figure 5A3, and its morphology is consistent with a normal adult GC. GC somata were round or oval, and size (8–10 μm long) corresponded to past reports of rodent GCs (Amaral et al., 2007; Pierce et al., 2011; Rahimi and Claiborne, 2007). The dendrites extended throughout the molecular layer and there were numerous spines, similar to mature GCs (Amaral et al., 2007; Frotscher et al., 2000; Rahimi and Claiborne, 2007). The intrinsic properties also suggested that the recorded cells were GCs because intrinsic properties were consistent with previous studies of GCs rather than GABAergic neurons (Supplemental Table 2;(Scharfman, 1992).
B. Positions of recorded GCs along the septotemporal axis were similar in WT and Tg2576 mice
To determine if there was a preference for the WT GCs to be in a particular part of the DG that was distinct from Tg2576 mice, slices were examined to determine if they were from the dorsal, mid, or ventral DG. For the purposes of this comparison, the dorsal DG was defined as horizontal sections approximately 3.0 to 3.7 mm below the cortical surface, ventral DG as horizontal sections between approximately 4.3 to 5.0 mm, and the middle of the DG was defined as the area between the dorsal and ventral sites. As shown in Supplemental Figure 5B1, there was no difference in the proportions of WT GCs recorded from dorsal, mid or ventral DG when they were compared to GCs from Tg2576 mice by Fisher’s exact test (all p>0.206).
C. Position of recorded GCs within the GCL were similar in WT and Tg2576 mice
We also compared the location of GCs in different subdivisions of the GCL (Supplemental Figure 5B2). This was done to address whether GCs might be adult-born GCs, which are usually located near the hilus (Gould and Cameron, 1996), or semilunar GCs which are located near or in the IML (Williams et al., 2007). Therefore, we compared three subdivisions of the GCL: the GCL border with the IML, center of the GCL, and GCL border with the hilus (Supplemental Figure 5B2). We found that similar proportions of GCs in WT and Tg2576 mice were located in these three parts of the GCL (Fisher’s exact test, all p>0.183; Supplemental Figure 5B2).
D. Position of recorded GCs within the DG blades were similar in WT and Tg2576 mice
Next, we analyzed whether the GCs were preferentially localized to the upper blade, crest or lower blade, defining these areas as before (Bernstein et al., 2020). Supplemental Figure 5B3 shows that the GCs from WT and Tg2576 mice did not appear to differ in the frequency they were recorded in the upper blade, crest, or lower blade. For statistical comparisons, WT was compared to Tg2576 for each location (Fisher’s exact test, all p>0.721).
V. Sex differences in synaptic properties suggest increased excitation in female Tg2576 mice
Female and male mice were compared for most measurements described above. A caveat in the comparisons of sex differences is that females were studied without knowing the stage of the estrous cycle. Therefore, females at different stages of the estrous cycle were likely to be pooled.
Regarding excitatory and inhibitory synaptic activity, WT males and females showed no statistical differences (Table 2). However, there were differences in male and female Tg2576 mice, where the females showed greater EPSC and nsC amplitudes than males (EPSCs: males: 5.25 ± 0.58 pA; females: 7.46 ± 0.54, unpaired t-test, t=2.761, df=11; p=0.019. NsC: males: 5.65 ± 0.57 pA; females: 7.69 ± 0.63, unpaired t-test, t=2.414, df=11; p=0.034; Table 2).
Table 2. Sex differences in GC synaptic properties.
GC synaptic properties from male and female mice. WT females were not studied where they are not listed. Statistical comparisons are shown for female WT vs. male WT and female Tg2576 vs. male Tg2576 GCs. (1) Unpaired student’s t-test for parametric data, and (2) Mann-Whitney test for non-parametric data. Two-way ANOVA was also conducted, where female WT data were available (sEPSPs). This analysis confirmed the lack of effect of sex on EPSP frequency and amplitude by t-test (Supplemental Table 2).
| Synaptic properties | ||||||||
|---|---|---|---|---|---|---|---|---|
| Frequency | ||||||||
| EPSPs | IPSCs | EPSCs | nsCs | |||||
| WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | |
| Male | 1.56 ± 0.12 | 2.12 ± 0.16 | 10.38 ± 1.01 | 8.31 ± 0.59 | 3.16 ± 0.33 | 4.04 ± 0.34 | 0.10 ± 0.04 | 0.71 ± 0.15 |
| Female | 1.62 ± 0.14 | 2.54 ± 0.46 | 8.08 ± 1.17 | 4.10 ± 0.50 | 0.53 ± 0.10 | |||
| p value | 0.768 | 0.288 | 0.860 | 0.926 | 0.358 | |||
| Statistical Test | (1) | (1) | (1) | (1) | (1) | |||
| t, df | t=0.301, df=14 | t=1.097, df=17 | t=0.181, df=12 | t=0.095, df=11 | t=0.960, df=11 | |||
| Nmale | 9 | 13 | 13 | 7 | 7 | 7 | 12 | 7 |
| Nfemale | 7 | 6 | 7 | 6 | 6 | |||
| Amplitude | ||||||||
| EPSPs | IPSCs | EPSCs | nsCs | |||||
| WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | WT | Tg2576 | |
| Male | 0.48 ± 0.04 | 0.39 ± 0.04 | 10.90 ± 1.11 | 9.67 ± 0.91 | 8.72 ± 1.03 | 5.25 ± 0.58 | 4.91 ± 0.79 | 5.65 ± 0.57 |
| Female | 0.42 ± 0.04 | 0.42 ± 0.05 | 11.46 ± 1.50 | 7.46 ± 0.54 | 7.69 ± 0.63 | |||
| p value | 0.333 | 0.630 | 0.326 | 0.019 | 0.034 | |||
| Statistical Test | (1) | (1) | (1) | (1) | (1) | |||
| t, df | t=1.002, df=14 | t=0.491, df=17 | t=1.024, df=12 | t=2.761, df=11 | t=2.414, df=11 | |||
| Nmale | 9 | 13 | 13 | 7 | 7 | 7 | 12 | 7 |
| Nfemale | 7 | 6 | 7 | 6 | 6 | |||
These data were also analyzed using a two-way ANOVA with sex and genotype as main factors. We did not find any significant sex differences, but genotype was a significant factor (Supplemental Table 3). Intrinsic properties did not appear to differ between sexes (Supplemental Table 4). It is recognized that comparison of more mice could have revealed more evidence of sex differences.
VI. Limited Aβ pathology in the DG at early ages in Tg2576 mice
It has been reported that Aβ plaques are present after 6 months in Tg2576 mice (Hsiao et al., 1996). To confirm this in the mice used in the present study, and study the DG specifically, additional WT and Tg2576 were studied at 2–3 months of age mice (n=6 per genotype, 3 males and 3 females). In addition, 12–15 month-old Tg2576 mice were used (n=6, 3 males and 3 females). We used Thioflavin-S to identify plaques (Roberson et al., 2007) and an antibody to Aβ, McSA1, to study plaques as well as intracellular (soluble) Aβ (Kobro-Flatmoen et al., 2016).
Representative micrographs of the Thioflavin-S and McSA1-stained sections are presented in Supplemental Figure 6. At 2–3 months of age, there was no evidence of Thioflavin-S in WT or Tg2576 mice. However, it was present at older ages. For McSA1, there was no immunofluorescence at 2–3 months in the WT mice and a low level of fluorescence in the hippocampus in Tg2576 mice. At older ages, there was strong fluorescence in Tg2576 mice, similar to the staining for Thioflavin-S.
These data suggest that Aβ plaques are not present at the young ages of Tg2576 mice used in this study. The McSA1 fluorescence in young Tg2576 mice showed that intracellular Aβ was present, but outside the GCL. These data are consistent with previous findings which suggested that soluble Aβ levels were elevated at 3 months of age in Tg2576 mouse hippocampus although plaques were not detected (Duffy et al., 2015).
Because soluble Aβ was not evident in GCs at the ages we studied, the differences in synaptic events and intrinsic properties of WT and Tg2576 GCs may be due to altered input in Tg2576 GCs rather than effects of soluble Aβ from within the GC. The inputs that might be altered at 2–3 months of age could be from the cells that are highly vulnerable such as entorhinal cortical and cholinergic neurons. This idea is supported by numerous studies of soluble (oligomeric) Aβ which have shown that it can lead to altered cellular and synaptic function (Chen, 2005; Chen et al., 2002; Ciccone et al., 2019; Eslamizade et al., 2015; Hou et al., 2009; Pena et al., 2010; Rovira et al., 2002; Tamagnini et al., 2015; Wu et al., 1995).
DISCUSSION
I. Synaptic activity in WT and Tg2576 mice
The results from our studies of synaptic activity suggest that sEPSPs and sEPSCs were greater and sIPSPs were reduced in Tg2576 mice compared to WT. For sEPSPs and sEPSCs, the frequency increased, particularly of small events. The mean amplitude decreased, although it was significant only for sEPSCs. For sIPSPs, frequency was reduced, not amplitude. Together these changes suggest a potential reason why the Tg2576 mouse was found previously to exhibit increased excitability at early ages (Bezzina et al., 2015; Kam et al., 2016): GCs showed increased excitation and decreased inhibition. Having said that, it is recognized that the DG might be only one of several areas contributing to increased excitability in hAPP mice (Bezzina et al., 2015; Kam et al., 2016; Palop et al., 2007; Verret et al., 2012; You et al., 2017). However, a focus on the DG is appropriate given the DG has been implicated in the pathophysiology of mouse models of AD neuropathology (Noebels, 2011; Palop et al., 2007; Palop and Mucke, 2009; Scharfman, 2012; You et al., 2017). Moreover, DG and CA3 hyperactivity have been implicated in early stages of AD (Bakker et al., 2012). In addition, the DG is considered to be a critical regulator of hippocampal hyperexcitability in epilepsy research (Heinemann et al., 1992; Krook-Magnuson et al., 2015; Lothman et al., 1992; Scharfman, 2019). Notably, our data are consistent with the enhanced expression of markers of neuronal activity observed in GCs in another mouse model of human APP mutations (J20 mice) (You et al., 2017).
There could be many reasons why sEPSP and sEPSC frequency was high in Tg2576 GCs relative to WT GCs. One possibility is that there was increased activity in glutamatergic afferents to the GCs. An attractive candidate is the perforant path input from the entorhinal cortex because it is considered to be the major excitatory input to the DG (Amaral et al., 2007). Also, the entorhinal cortex is highly vulnerable in AD, exhibiting early pathology (Braak and Braak, 1991; Kordower et al., 2001) especially in the perforant path connections to the DG (de Leon et al., 2007; de Toledo-Morrell et al., 2000). In the Tg2576 mouse, we found that there also are early signs of degenerative changes in the entorhinal cortex, and there was increased excitability in slices of the entorhinal cortex at 2–4 months of age (Duffy et al., 2015). Another study also found increased excitability at early ages in Tg2576 mice using in vivo recordings of entorhinal cortical units (Xu et al., 2015). It would be logical that increased entorhinal cortical excitability would contribute to an increase in perforant path release of glutamate, leading to increased sEPSP/Cs in GCs.
Notably, a previous study showed that at 4 months of age, GCs of Tg2576 mice exhibited reduced spines (Jacobsen et al., 2006). This result might be a response to overactivity at early ages because spine loss is observed in GCs in response to hyperexcitability (Isokawa, 2000) and in cultures after increased excitation due to GABA receptor antagonism (Drakew et al., 1996). Interestingly, long-term potentiation of the perforant path was impaired at 5 months of age in the Tg2576 mice in the study where GCs had reduced spines (Jacobsen et al., 2006), consistent with a reduction in spines in other experiments when LTP is impaired (D’Agostino et al., 2013). These studies are important to consider together because they suggest that early hyperexcitability precedes and possibly contributes to impairments later in life, which has been hypothesized before (Kam et al., 2016).
There are other possible contributing factors that explain why sEPSPs and sEPSCs increased in GCs in Tg2576 mice. For example, a previously undetectable input might become detectable. It seems unlikely that altered intrinsic properties would be the cause, because Rin decreased in Tg2576 mice, and decreased Rin usually reduces the sensitivity of a neuron to its afferent input. Instead, one possible cause of increased sensitivity is that the normal role of muscarinic receptors to suppress glutamate release was reduced in Tg2576 mice This suppression of normal muscarinic receptor function could also explain why there were increased nsCs in Tg2576 mice relative to WT mice under basal conditions (discussed further below).
This possible role of muscarinic receptors in regulating excitatory input to GCs is consistent with prior studies. Septohippocampal neurons are the major cholinergic input to the DG and hippocampus (Aznavour et al., 2005; Clarke, 1985; Deller et al., 1999; Dougherty and Milner, 1999; Frotscher, 1991; Frotscher and Leranth, 1985; Frotscher and Leranth, 1986; Leranth and Frotscher, 1987; Milner and Veznedaroglu, 1993; Nyakas et al., 1987; Takacs et al., 2018; Wainer et al., 1985) and normally one of the roles of acetylcholine is to suppress release of transmitter from external inputs to the DG and hippocampus, a suppression that occurs during sleep and allows more emphasis on circuits intrinsic to the DG/hippocampus and memory consolidation (Hasselmo et al., 1996). However, in the Tg2576 mouse there appears to be early abnormalities of the septohippocampal input (Kam et al., 2016). Specifically, septal neurons appear to be overactive in that they express c-Fos (Kam et al., 2016). In addition, young Tg2576 mice have increased hippocampal expression of the synthetic enzyme for acetylcholine, choline acetyl transferase (ChAT), which is present in cholinergic fibers from cholinergic neurons extrinsic to the DG (Kam et al., 2016). ChAT also rises early in AD patients (DeKosky et al., 2002; Ikonomovic et al., 2007). One possibility is that muscarinic receptors were downregulated in young Tg2576 mice in response to increased release of acetylcholine from septohippocampal nerve terminals. As a result, Tg2576 mice would have less cholinergic suppression of glutamate release. Indeed, atropine had less of an effect on Tg2576 nsCs than WT nsCs. Atropine also had little effect on spike frequency adaptation in Tg2576 mice when WT mice showed an effect of atropine.
The finding in the present study, that sIPSCs were reduced in Tg2576 mice, could also contribute to the increased frequency of sEPSPs and sEPSCs. Reduced sIPSCs is consistent with prior studies which suggest that the GABAergic neurons in mice with human APP mutations develop deficient sodium channels (Nav1.1) which make them less able to release GABA (Martinez-Losa et al., 2018; Verret et al., 2012). Another possibility is that the deficient sodium channels occur later in life, and the first defect in sIPSCs is due to another mechanism. One early mechanism would be alteration of the normal effects of septohippocampal terminals on GABAergic neurons. Normally acetylcholine has diverse effects on GABAergic neurons which depend in part on behavioral state and in part on the type of GABAergic neurons (Behrends and ten Bruggencate, 1993; Chiang et al., 2010; Dannenberg et al., 2017; Dougherty and Milner, 1999; Frazier et al., 1998; Frazier et al., 2003; Leranth and Frotscher, 1987; McQuiston and Madison, 1999; Pabst et al., 2016; Pitler and Alger, 1992; Raza et al., 2017; Yi et al., 2014), making a clear mechanism hard to propose at the present time.
II. Intrinsic properties and firing behavior of WT and Tg2576 mice
The results showed that intrinsic properties and firing behavior were altered in Tg2576 mice relative to WT. RMP was more hyperpolarized, and both Rin and τ were reduced in Tg2576 mice. Also, APs in Tg2576 GCs had a longer time to peak and a longer half-width. Firing behavior showed that adaptation in Tg2576 GCs was weak. Together these differences suggest complex changes in GCs in young Tg2576 mice. While weaker adaptation would make a cell more likely to fire APs, a more hyperpolarized RMP and reduced Rin would potentially decrease the ability of afferent input to influence the cell. The predicted net effect would be to reduce the ability of GCs to respond to afferent input while increasing the ability of GCs to fire APs.
If this is true, it would be likely to cause substantial network defects because APs would fire outside of the times they would normally be triggered by afferent input. The DG would become unlikely to play its normal role in cognition and behavior because it would fire at inappropriate times. In addition, the longer time to peak and half-width of Tg2576 GC APs might also render GCs less effective in their normal roles because APs would be slower to peak. The slow time-to-peak could lead to delays in the timing of AP discharge and transmitter release.
Together the results suggest possible cellular mechanisms for decreased cognitive function in Tg2576 mice. The idea that cognitive impairments develop early in life (2–4 months-old) is supported by a prior study showing worse performance of approximately 3 month-old Tg2576 mice on tasks that test spatial memory (e.g. novel object location; Duffy et al. (2015)). In the CRND8 mouse, which has two human APP mutations (APPSwe, APPInd), the novel object location task has also been found to be deficient, and the animals were just 2 months of age (Francis et al., 2012). In the J20 mouse, the novel object location task is impaired early in life too (You et al., 2017). Notably, task performance was improved by specific experimental manipulations of GCs (You et al., 2017), implicating the DG in cognitive impairments.
It is remarkable that the changes in synaptic events and intrinsic properties in Tg2576 mice occurred so early in life. It begs the question how Aβ could trigger effects so early and how there could be so many effects. One potential answer is that effects of Aβ are not the only cause, but APP and APP metabolites also contribute. This idea has support from an earlier study of Tg2576 mice (Xu et al., 2015) as well as a wealth of evidence that APP metabolites other than Aβ cause numerous adverse effects (Nixon, 2017). Regarding the diversity of effects, some effects may be primary whereas others are compensatory. For example, there may first be an increase in excitability due to greater activity of excitatory input with reduced inhibitory input, and then a hyperpolarization of the RMP and reduction in Rin to compensate.
III. Atropine in WT and Tg2576 mice
A. Synaptic activity
1). sEPSPs, sEPSCs and sIPSCs
Atropine had diverse effects in Tg2576 mice compared to WT mice, sometimes being similar and sometimes quite different. WT and Tg2576 mice were similar in the effect of atropine to reduce sIPSC frequency and amplitude. WT and Tg2576 mice were different in the effects of atropine on excitatory events. For excitatory activity, frequency increased in WT mice and it was not evident in Tg2576 mice. Why atropine would fail to show an effect in Tg2576 mice could be due to a deficiency in receptors as described above. Also noted above, one mechanism would be that muscarinic receptors are downregulated by hyperactivity of cholinergic inputs early in life. In other words, high activity of cholinergic inputs would lead to high concentrations of released acetylcholine. In response to a high concentration of agonist, there could be a compensatory internalization of muscarinic receptors.
2). nsCs (NMDA-EPSCs)
Atropine also had a very interesting effect when GC holding potential was 0 mV. Small inward currents were present, especially in Tg2576 mice. Atropine increased these inward currents in WT mice and had a small effect in Tg2576 mice, possibly because the frequency was already high before atropine was applied. In addition, muscarinic receptor internalization (discussed above) could have played a role in preventing effects of atropine. The inward currents, which we call nsCs (or NMDA-EPSCs since they were blocked by APV and not DNQX), may have contributed to the increase in sEPSP and sEPSC frequency in Tg2576 mice at −70 mV (Figures 1, 2) but were simply much clearer using voltage clamp at 0 mV. They are potentially important because they suggest another way the cholinergic system is abnormal in young Tg2576 mice and could contribute to increased excitability of GCs.
The results showing that nsCs are elevated in Tg2576 mice and are mediated by NMDA receptors are interesting. Based on the idea that NMDA receptors normally mediate a small component of glutamatergic transmission, because NMDA receptors are extrasynaptic, the results suggest that glutamatergic inputs increase in the Tg2576 mice, and the higher concentration of glutamate leads to spillover at NMDA receptors.
There are two likely candidate inputs to explain these nsCs (NMDA-EPSCs) because GCs primarily have two glutamatergic inputs, the perforant path and mossy cells. Regarding mossy cells, they could be responsible for the nsCs because they have small excitatory effects on GCs relative to their strong indirect inhibitory effects (Bernstein et al., 2020). The small effects would be consistent with the small size of nsCs. However, whether muscarinic receptors regulate glutamate release from mossy cells is unclear (Hofmann and Frazier, 2010).
It seems likely that the perforant path would contribute to nsCs for reasons described above. Importantly, the entorhinal cortex receives extensive innervation by septal cholinergic afferents (Desikan et al., 2018; Heys et al., 2012), making it likely that the perforant path could be altered by atropine. These findings support the view that the cholinergic system in Tg2576 mice is altered at very early ages in multiple ways.
3). Intrinsic properties and firing behavior
Atropine also had effects on intrinsic properties that differed between WT and Tg2576 mice. Thus, atropine reduced Rin but only in Tg2576 mice. Atropine made threshold more depolarized in Tg2576 mice, but did not in WT. The difference between WT and Tg2576 threshold would make the Tg2576 GCs less able to fire APs. Together these effects help explain the ability of atropine to decrease aberrant interictal spikes in vivo in the Tg2576 mice (Kam et al., 2016). On the other hand, if atropine induced nsCs and decreased sIPSCs, atropine would be likely to increase excitability.
Thus, a remaining question is how the results explain the effect of atropine to decrease IIS in vivo (Kam et al., 2016). In the GCs of the present study, atropine increased excitability by increasing nsCs, and by depressing IPSCs. It also had effects on intrinsic properties and firing behavior that might decrease excitability. An important point is that many of the animals tested with atropine in the in vivo study by Kam et al. (2016) were older than 2–3 months. At that time, atropine might have less effect on afferent input because of the gradual reduction in entorhinal, cholinergic and GABAergic function caused by Aβ pathology. That would leave the effects of atropine on intrinsic properties and those effects reduced excitability. Thus, at early ages atropine might have a limited effect on IIS in vivo but at older ages atropine might reduce IIS but reducing excitability.
This discussion is potentially relevant to cholinergic treatment in AD where acetylcholine levels are increased by a cholinesterase inhibitor. Our data suggest that individuals with AD that are treated in this way may exert different effects depending on the age and stage of pathology. This is consistent with clinical data showing that some individuals are helped by these drugs, and others are not (Hampel et al., 2018; Richter et al., 2018).
IV. Sex differences
Sex differences were important to address because of widespread observations that female mice with hAPP mutations are more affected than males (Bangasser et al., 2017; Clinton et al., 2007; Jiao et al., 2016; Roy et al., 2018; Yang et al., 2018). In the present study, females had larger sEPSCs (AMPA-EPSCs) and larger nsCs (NMDA-EPSCs) in Tg2576 mice. These effects might make Tg2576 females more likely to exhibit hyperexcitability. Hyperexcitability could lead to a worse phenotype in females relative to males because hyperexcitability in glutamatergic neurons can lead to overexcitation of downstream targets. Hyperactivity of the DG and CA3 also has been shown to cause impaired cognition (Bakker et al., 2012). In addition, hyperexcitability of GCs can lead to excitotoxicity of hilar cells and CA3 pyramidal cells because the GC boutons are unusually large and can release high concentrations of glutamate (Acsady et al., 1998; Chicurel and Harris, 1992; Rama et al., 2019; Rollenhagen and Lubke, 2010; Scharfman, 2016; Scharfman and Bernstein, 2015). Hilar and CA3 cells might die, but even if they did not die, they would be likely to become impaired by the metabolic demand to support normal function after exposure to high concentrations of glutamate (Choi, 1994; Olney et al., 1986; Sattler and Tymianski, 2001; Schwarcz et al., 1984).
V. Role of Aβ
Aβ in its oligomeric (“soluble”) form may have contributed to the changes reported in this study by an indirect mechanism, because at the ages studied, there was soluble Aβ in neurons of the hippocampus and hilus, near the GCs. Extrinsic inputs may also have had soluble Aβ. In addition, the precursor APP or APP metabolites could have contributed. APP is likely to have played a role because it is overexpressed from birth. However, to date none of the synaptic changes or intrinsic properties we identified have been shown to occur in response to APP.
In prior studies, there are diverse effects of adding APP, its metabolites, or Aβ to hippocampal slices. Among these, multiple studies have documented synaptic effects on hippocampal neurons and a few on GCs (Chen, 2005; Chen et al., 2002; Ciccone et al., 2019; Eslamizade et al., 2015; Hou et al., 2009; Pena et al., 2010; Rovira et al., 2002; Tamagnini et al., 2015; Wu et al., 1995). One of the problems in relating these papers to the results presented here is that it is hard to equate superfusion of synthetic peptides to endogenous production of Aβ from mutated APP. Nevertheless, the published data using exogenous application of Aβ peptides are consistent with what we have shown, because diverse changes in synaptic activity have been documented, both increases and decreases (Gulisano et al., 2019; Kelly et al., 1996; Parameshwaran et al., 2007; Ripoli et al., 2014; Yao et al., 2013).
VI. Conclusions
This study provided insight into the early changes in GCs of the Tg2576 mouse model of AD neuropathology. These data help address the hypothesis that the DG is an area that is affected at early ages and participates in the progressive pathophysiology of the mouse model. By examining ages that were younger than those examined previously, and by using whole cell recording, detailed insight into early synaptic and intrinsic properties were obtained. The results suggest diverse synaptic changes develop in GCs that could contribute to increased excitability at young ages, and ultimately cognitive impairment. The results are the first to show that intrinsic characteristics are affected at extremely young ages. Finally, by elaborating the effects of atropine, the results support the hypothesis that the cholinergic system is altered in diverse ways at early ages. The results support the view that AD pathology begins long before the presence of Aβ plaques.
Supplementary Material
HIGHLIGHTS.
Granule cells (GCs) in young Tg2576 mice had increased spontaneous excitatory synaptic input.
Tg2576 GCs had reduced spontaneous inhibitory synaptic input.
Resting potential and other intrinsic properties were abnormal in Tg2576 GCs.
Muscarinic cholinergic regulation of GCs was altered in Tg2576 mice.
A spontaneous NMDA receptor-mediated current was robust in Tg2576 GCs relative to WT.
ACKNOWLEDGEMENTS
We thank Dr. Aine Duffy and Yi-Ling Lu for their contributions to the initiation of this project. We also thank Drs. Justin Botterill, Chiara Criscuolo, Christos Lisgaras and Yi-Ling Lu for discussion during the preparation of this paper. This project was supported by NIH R01 AG-055328 to H.E.S. and the New York State Office of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
REFERENCES
- Acsady L, et al. , 1998. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 18, 3386–403. 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken SP, et al. , 1995. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 115, 1163–8. 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A, et al. , 1995. An english translation of Alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin Anat. 8, 429–31. 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Amaral DG, et al. , 2007. The dentate gyrus: Fundamental neuroanatomical organization (dentate gyrus for dummies). Prog Brain Res. 163, 3–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apelt J, et al. , 2002. Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res. 953, 17–30. 10.1016/s0006-8993(02)03262-6. [DOI] [PubMed] [Google Scholar]
- Aznavour N, et al. , 2005. Postnatal development of the cholinergic innervation in the dorsal hippocampus of rat: Quantitative light and electron microscopic immunocytochemical study. J Comp Neurol. 486, 61–75. 10.1002/cne.20501. [DOI] [PubMed] [Google Scholar]
- Bakker A, et al. , 2012. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 74, 467–74. 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangasser DA, et al. , 2017. Corticotropin-releasing factor overexpression gives rise to sex differences in Alzheimer’s disease-related signaling. Mol Psychiatry. 22, 1126–1133. 10.1038/mp.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearer EL, et al. , 2018. Alterations of functional circuitry in aging brain and the impact of mutated APP expression. Neurobiol Aging. 70, 276–290. 10.1016/j.neurobiolaging.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman M, et al. , 2020. Loss of presenilin 2 age-dependently alters susceptibility to acute seizures and kindling acquisition. Neurobiol Dis. 136, 104719. 10.1016/j.nbd.2019.104719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends JC, ten Bruggencate G, 1993. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: Excitation of GABAergic interneurons and inhibition of GABA-release. J Neurophysiol. 69, 626–9. 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- Bernstein HL, et al. , 2020. Excitatory effects of dentate gyrus mossy cells and their ability to influence granule cell firing: An optogenetic study in adult mouse hippocampal slices. bioRxiv. 2020.06.06.137844.10.1101/2020.06.06.137844. [Google Scholar]
- Bezzina C, et al. , 2015. Early onset of hypersynchronous network activity and expression of a marker of chronic seizures in the Tg2576 mouse model of Alzheimer’s disease. PLoS One. 10, e0119910. 10.1371/journal.pone.0119910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, et al. , 2005. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 45, 675–88. 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Born HA, et al. , 2014. Genetic suppression of transgenic APP rescues hypersynchronous network activity in a mouse model of Alzeimer’s disease. J Neurosci. 34, 3826–40. 10.1523/JNEUROSCI.5171-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–59. 10.1007/bf00308809. [DOI] [PubMed] [Google Scholar]
- Chen C, 2005. Beta-amyloid increases dendritic Ca2+ influx by inhibiting the A-type K+ current in hippocampal CA1 pyramidal neurons. Biochem Biophys Res Commun. 338, 1913–9. 10.1016/j.bbrc.2005.10.169. [DOI] [PubMed] [Google Scholar]
- Chen QS, et al. , 2002. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 77, 354–71. 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Chiang PH, et al. , 2010. M(1)-like muscarinic acetylcholine receptors regulate fast-spiking interneuron excitability in rat dentate gyrus. Neuroscience. 169, 39–51. 10.1016/j.neuroscience.2010.04.051. [DOI] [PubMed] [Google Scholar]
- Chicurel ME, Harris KM, 1992. Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J Comp Neurol. 325, 169–82. 10.1002/cne.903250204. [DOI] [PubMed] [Google Scholar]
- Chin J, Scharfman HE, 2013. Shared cognitive and behavioral impairments in epilepsy and Alzheimer’s disease and potential underlying mechanisms. Epilepsy Behav. 26, 343–51. 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, 1994. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 100, 47–51. 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Ciccone R, et al. , 2019. Amyloid beta-induced upregulation of Nav1.6 underlies neuronal hyperactivity in Tg2576 Alzheimer’s disease mouse model. Sci Rep. 9, 13592. 10.1038/s41598-019-50018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, et al. , 1992. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 360, 672–4. 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Clarke DJ, 1985. Cholinergic innervation of the rat dentate gyrus: An immunocytochemical and electron microscopical study. Brain Res. 360, 349–54. 10.1016/0006-8993(85)91253-3. [DOI] [PubMed] [Google Scholar]
- Clinton LK, et al. , 2007. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 28, 76–82. 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig LA, et al. , 2011. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci Biobehav Rev. 35, 1397–409. 10.1016/j.neubiorev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- D’Agostino G, et al. , 2013. Prolyl endopeptidase-deficient mice have reduced synaptic spine density in the CA1 region of the hippocampus, impaired LTP, and spatial learning and memory. Cereb Cortex. 23, 2007–14. 10.1093/cercor/bhs199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg H, et al. , 2017. Modulation of hippocampal circuits by muscarinic and nicotinic receptors. Front Neural Circuits. 11, 102. 10.3389/fncir.2017.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies P, Maloney AJ, 1976. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 2, 1403. 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- de Leon MJ, et al. , 2007. Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann N Y Acad Sci. 1097, 114–45. 10.1196/annals.1379.012. [DOI] [PubMed] [Google Scholar]
- de Toledo-Morrell L, et al. , 2000. From healthy aging to early Alzheimer’s disease: In vivo detection of entorhinal cortex atrophy. Ann N Y Acad Sci. 911, 240–53. 10.1111/j.1749-6632.2000.tb06730.x. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, et al. , 2002. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 51, 145–55. 10.1002/ana.10069. [DOI] [PubMed] [Google Scholar]
- Deller T, et al. , 1999. Cholinergic innervation of mossy cells in the rat fascia dentata. Hippocampus. 9, 314–20.. [DOI] [PubMed] [Google Scholar]
- Dengler CG, et al. , 2017. Massively augmented hippocampal dentate granule cell activation accompanies epilepsy development. Sci Rep. 7, 42090. 10.1038/srep42090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan S, et al. , 2018. Target selectivity of septal cholinergic neurons in the medial and lateral entorhinal cortex. Proc Natl Acad Sci U S A. 115, E2644–E2652. 10.1073/pnas.1716531115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KD, Milner TA, 1999. Cholinergic septal afferent terminals preferentially contact neuropeptide Y-containing interneurons compared to parvalbumin-containing interneurons in the rat dentate gyrus. J Neurosci. 19, 10140–52. 10.1523/JNEUROSCI.19-22-10140.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakew A, et al. , 1996. Spine loss in experimental epilepsy: Quantitative light and electron microscopic analysis of intracellularly stained CA3 pyramidal cells in hippocampal slice cultures. Neuroscience. 70, 31–45. 10.1016/0306-4522(95)00379-w. [DOI] [PubMed] [Google Scholar]
- Duan RS, et al. , 2006. Apolipoprotein E deficiency increased microglial activation/CCR3 expression and hippocampal damage in kainic acid exposed mice. Exp Neurol. 202, 373–80. 10.1016/j.expneurol.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Duff K, 2001. Transgenic mouse models of Alzheimer’s disease: Phenotype and mechanisms of pathogenesis. Biochem Soc Symp. 195–202. 10.1042/bss0670195. [DOI] [PubMed] [Google Scholar]
- Duffy AM, et al. , 2015. Entorhinal cortical defects in Tg2576 mice are present as early as 2–4 months of age. Neurobiol Aging. 36, 134–48. 10.1016/j.neurobiolaging.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, et al. , 2008. Alzheimer disease models and human neuropathology: Similarities and differences. Acta Neuropathol. 115, 5–38. 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eslamizade MJ, et al. , 2015. Alterations in CA1 pyramidal neuronal intrinsic excitability mediated by Ih channel currents in a rat model of amyloid beta pathology. Neuroscience. 305, 279–92. 10.1016/j.neuroscience.2015.07.087. [DOI] [PubMed] [Google Scholar]
- Ferreira-Vieira TH, et al. , 2016. Alzheimer’s disease: Targeting the cholinergic system. Curr Neuropharmacol. 14, 101–15. 10.2174/1570159x13666150716165726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana R, et al. , 2017. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol Aging. 50, 64–76. 10.1016/j.neurobiolaging.2016.10.027. [DOI] [PubMed] [Google Scholar]
- Francis BM, et al. , 2012. Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging. 33, 555–63. 10.1016/j.neurobiolaging.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT, et al. , 1999. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J Neurol Neurosurg Psychiatry. 66, 137–47. 10.1136/jnnp.66.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, et al. , 1998. Acetylcholine activates an α-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 18, 1187–95. 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, et al. , 2003. Nicotinic receptors on local circuit neurons in dentate gyrus: A potential role in regulation of granule cell excitability. J Neurophysiol. 89, 3018–28. 10.1152/jn.01036.2002. [DOI] [PubMed] [Google Scholar]
- Friedman D, et al. , 2012. Seizures and epilepsy in Alzheimer’s disease. CNS Neurosci Ther. 18, 285–94. 10.1111/j.1755-5949.2011.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, 1991. Target cell specificity of synaptic connections in the hippocampus. Hippocampus. 1, 123–30. 10.1002/hipo.450010202. [DOI] [PubMed] [Google Scholar]
- Frotscher M, et al. , 2000. Role of afferent innervation and neuronal activity in dendritic development and spine maturation of fascia dentata granule cells. Cereb Cortex. 10, 946–51. 10.1093/cercor/10.10.946. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Leranth C, 1985. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: A combined light and electron microscopic study. J Comp Neurol. 239, 237–46. 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Leranth C, 1986. The cholinergic innervation of the rat fascia dentata: Identification of target structures on granule cells by combining choline acetyltransferase immunocytochemistry and Golgi impregnation. J Comp Neurol. 243, 58–70. 10.1002/cne.902430106. [DOI] [PubMed] [Google Scholar]
- Fu CH, et al. , 2019. Early seizure activity accelerates depletion of hippocampal neural stem cells and impairs spatial discrimination in an Alzheimer’s disease model. Cell Rep. 27, 3741–3751 e4. 10.1016/j.celrep.2019.05.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatak S, et al. , 2019. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife. 8. 10.7554/eLife.50333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheyara AL, et al. , 2014. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 76, 443–56. 10.1002/ana.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, et al. , 2018. Rodent models for Alzheimer disease. Nat Rev Neurosci. 19, 583–598. 10.1038/s41583-018-0054-8. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner LM, 2008. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci. 9, 532–44. 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Gould E, Cameron HA, 1996. Regulation of neuronal birth, migration and death in the rat dentate gyrus. Dev Neurosci. 18, 22–35. 10.1159/000111392. [DOI] [PubMed] [Google Scholar]
- Grant SM, et al. , 2000. Abeta immunoreactive material is present in several intracellular compartments in transfected, neuronally differentiated, p19 cells expressing the human amyloid beta-protein precursor. J Alzheimers Dis. 2, 207–22. 10.3233/jad-2000-23-403. [DOI] [PubMed] [Google Scholar]
- Gulisano W, et al. , 2019. Neuromodulatory action of picomolar extracellular Abeta42 oligomers on presynaptic and postsynaptic mechanisms underlying synaptic function and memory. J Neurosci. 39, 5986–6000. 10.1523/JNEUROSCI.0163-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, et al. , 2018. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 141, 1917–1933. 10.1093/brain/awy132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, et al. , 2019. Revisiting the cholinergic hypothesis in Alzheimer’s disease: Emerging evidence from translational and clinical research. J Prev Alzheimers Dis. 6, 2–15. 10.14283/jpad.2018.43. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, et al. , 1996. Encoding and retrieval of episodic memories: Role of cholinergic and GABAergic modulation in the hippocampus. Hippocampus. 6, 693–708.. [DOI] [PubMed] [Google Scholar]
- Heinemann U, et al. , 1992. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 7, 273–80 [PubMed] [Google Scholar]
- Heys JG, et al. , 2012. Effects of acetylcholine on neuronal properties in entorhinal cortex. Front Behav Neurosci. 6, 32. 10.3389/fnbeh.2012.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann ME, Frazier CJ, 2010. Muscarinic receptor activation modulates the excitability of hilar mossy cells through the induction of an afterdepolarization. Brain Res. 1318, 42–51. 10.1016/j.brainres.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SL, Rebec GV, 2012. A new perspective on behavioral inconsistency and neural noise in aging: Compensatory speeding of neural communication. Front Aging Neurosci. 4, 27. 10.3389/fnagi.2012.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp JA, et al. , 2014. Morpho-physiological criteria divide dentate gyrus interneurons into classes. Hippocampus. 24, 189–203. 10.1002/hipo.22214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou JF, et al. , 2009. Intracellular amyloid induces impairments on electrophysiological properties of cultured human neurons. Neurosci Lett. 462, 294–9. 10.1016/j.neulet.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Houser CR, 2007. Interneurons of the dentate gyrus: An overview of cell types, terminal fields and neurochemical identity. Prog Brain Res. 163, 217–32. 10.1016/S0079-6123(07)63013-1. [DOI] [PubMed] [Google Scholar]
- Hsiao K, et al. , 1996. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 274, 99–102. 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hunter JM, et al. , 2012. Emergence of a seizure phenotype in aged apolipoprotein epsilon 4 targeted replacement mice. Brain Res. 1467, 120–32. 10.1016/j.brainres.2012.05.048. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, et al. , 2007. Superior frontal cortex cholinergic axon density in mild cognitive impairment and early Alzheimer disease. Arch Neurol. 64, 1312–7. 10.1001/archneur.64.9.1312. [DOI] [PubMed] [Google Scholar]
- Isokawa M, 2000. Remodeling dendritic spines of dentate granule cells in temporal lobe epilepsy patients and the rat pilocarpine model. Epilepsia. 41 Suppl 6, S14–7. 10.1111/j.1528-1157.2000.tb01550.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, et al. , 2006. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 103, 5161–6. 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao SS, et al. , 2016. Sex dimorphism profile of Alzheimer’s disease-type pathologies in an APP/PS1 mouse model. Neurotox Res. 29, 256–66. 10.1007/s12640-015-9589-x. [DOI] [PubMed] [Google Scholar]
- Kam K, et al. , 2016. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of beta-amyloid neuropathology. Sci Rep. 6, 20119. 10.1038/srep20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, et al. , 2001. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 21, 372–81. 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JF, et al. , 1996. Amyloid beta-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci U S A. 93, 6753–8. 10.1073/pnas.93.13.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingner M, et al. , 2003. Alterations in cholinergic and non-cholinergic neurotransmitter receptor densities in transgenic Tg2576 mouse brain with beta-amyloid plaque pathology. Int J Dev Neurosci. 21, 357–69. 10.1016/j.ijdevneu.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS, 2003. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci. 23, 2440–52. 10.1523/JNEUROSCI.23-06-02440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobro-Flatmoen A, et al. , 2016. Reelin-immunoreactive neurons in entorhinal cortex layer II selectively express intracellular amyloid in early Alzheimer’s disease. Neurobiol Dis. 93, 172–83. 10.1016/j.nbd.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Kordower JH, et al. , 2001. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 49, 202–13.. [DOI] [PubMed] [Google Scholar]
- Krezymon A, et al. , 2013. Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer’s disease. PLoS One. 8, e76497. 10.1371/journal.pone.0076497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, et al. , 2015. In vivo evaluation of the dentate gate theory in epilepsy. J Physiol. 593, 2379–88. 10.1113/JP270056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, et al. , 2019. New approaches to studying silent mesial temporal lobe seizures in Alzheimer’s disease. Front Neurol. 10, 959. 10.3389/fneur.2019.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, et al. , 2017. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat Med. 23, 678–680. 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, McNamara JO, 2007. Does epileptiform activity contribute to cognitive impairment in Alzheimer’s disease? Neuron. 55, 677–8. 10.1016/j.neuron.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Leranth C, Frotscher M, 1987. Cholinergic innervation of hippocampal GAD- and somatostatin-immunoreactive commissural neurons. J Comp Neurol. 261, 33–47. 10.1002/cne.902610104. [DOI] [PubMed] [Google Scholar]
- Lothman EW, et al. , 1992. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 7, 301–13 [PubMed] [Google Scholar]
- Martinez-Losa M, et al. , 2018. Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron. 98, 75–89 e5. 10.1016/j.neuron.2018.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV, 1999. Muscarinic receptor activity has multiple effects on the resting membrane potentials of CA1 hippocampal interneurons. J Neurosci. 19, 5693–702. 10.1523/JNEUROSCI.19-14-05693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Veznedaroglu E, 1993. Septal efferent axon terminals identified by anterograde degeneration show multiple sites for modulation of neuropeptide Y-containing neurons in the rat dentate gyrus. Synapse. 14, 101–12. 10.1002/syn.890140202. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, et al. , 2009. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 29, 3453–62. 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, et al. , 2008. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev Neurother. 8, 1703–18. 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, 2017. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 31, 2729–2743. 10.1096/fj.201700359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels J, 2011. A perfect storm: Converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia. 52 Suppl 1, 39–46. 10.1111/j.1528-1167.2010.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyakas C, et al. , 1987. Detailed projection patterns of septal and diagonal band efferents to the hippocampus in the rat with emphasis on innervation of CA1 and dentate gyrus. Brain Res Bull. 18, 533–45. 10.1016/0361-9230(87)90117-1. [DOI] [PubMed] [Google Scholar]
- Ohm TG, 2007. The dentate gyrus in Alzheimer’s disease. Prog Brain Res. 163, 723–40. 10.1016/S0079-6123(07)63039-8. [DOI] [PubMed] [Google Scholar]
- Olney JW, et al. , 1986. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 44, 857–77 [PubMed] [Google Scholar]
- Pabst M, et al. , 2016. Astrocyte intermediaries of septal cholinergic modulation in the hippocampus. Neuron. 90, 853–65. 10.1016/j.neuron.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Palop JJ, et al. , 2005. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 25, 9686–93. 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, et al. , 2007. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 55, 697–711. 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, et al. , 2003. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A. 100, 9572–7. 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, 2009. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 66, 435–40. 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, 2010a. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci. 13, 812–8. 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, 2010b. Synaptic depression and aberrant excitatory network activity in Alzheimer’s disease: Two faces of the same coin? Neuromolecular Med. 12, 48–55. 10.1007/s12017-009-8097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameshwaran K, et al. , 2007. Amyloid beta-peptide Abeta(1–42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse. 61, 367–74. 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- Pena F, et al. , 2010. Beta-amyloid protein (25–35) disrupts hippocampal network activity: Role of Fyn-kinase. Hippocampus. 20, 78–96. 10.1002/hipo.20592. [DOI] [PubMed] [Google Scholar]
- Perl DP, 2010. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 77, 32–42. 10.1002/msj.20157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S, et al. , 2008. Dietary supplementation with s-adenosyl methionine was associated with protracted reduction of seizures in a line of transgenic mice. Comp Med. 58, 604–6 [PMC free article] [PubMed] [Google Scholar]
- Petrache AL, et al. , 2019. Aberrant excitatory-inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s disease. Cereb Cortex. 29, 1834–1850. 10.1093/cercor/bhz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JP, et al. , 2011. Morphometry of hilar ectopic granule cells in the rat. J Comp Neurol. 519, 1196–218. 10.1002/cne.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE, 1992. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J Physiol. 450, 127–42. 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun RY, et al. , 2012. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 75, 1022–34. 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi O, Claiborne BJ, 2007. Morphological development and maturation of granule neuron dendrites in the rat dentate gyrus. Prog Brain Res. 163, 167–81. 10.1016/S0079-6123(07)63010-6. [DOI] [PubMed] [Google Scholar]
- Rama S, et al. , 2019. Glutamate imaging reveals multiple sites of stochastic release in the CA3 giant mossy fiber boutons. Front Cell Neurosci. 13, 243. 10.3389/fncel.2019.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza SA, et al. , 2017. HIPP neurons in the dentate gyrus mediate the cholinergic modulation of background context memory salience. Nat Commun. 8, 189. 10.1038/s41467-017-00205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Marin KE, Nunez A, 2017. Seizure susceptibility in the APP/PS1 mouse model of Alzheimer’s disease and relationship with amyloid beta plaques. Brain Res. 1677, 93–100. 10.1016/j.brainres.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Richter N, et al. , 2018. Effect of cholinergic treatment depends on cholinergic integrity in early Alzheimer’s disease. Brain. 141, 903–915. 10.1093/brain/awx356. [DOI] [PubMed] [Google Scholar]
- Ripoli C, et al. , 2014. Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci. 34, 12893–903. 10.1523/JNEUROSCI.1201-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, et al. , 2011. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 31, 700–11. 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, et al. , 2007. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 316, 750–4. 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, 2000. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: Implications for therapy. Trends Neurosci. 23, 393–8. 10.1016/s0166-2236(00)01629-5. [DOI] [PubMed] [Google Scholar]
- Rollenhagen A, Lubke JH, 2010. The mossy fiber bouton: The “common” or the “unique” synapse? Front Synaptic Neurosci. 2, 2. 10.3389/fnsyn.2010.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira C, et al. , 2002. Abeta(25–35) and Abeta(1–40) act on different calcium channels in CA1 hippocampal neurons. Biochem Biophys Res Commun. 296, 1317–21. 10.1016/s0006-291x(02)02072-7. [DOI] [PubMed] [Google Scholar]
- Roy U, et al. , 2018. Sex- and age-specific modulation of brain GABA levels in a mouse model of Alzheimer’s disease. Neurobiol Aging. 62, 168–179. 10.1016/j.neurobiolaging.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, et al. , 2018. Tau-induced pathology in epilepsy and dementia: Notions from patients and animal models. Int J Mol Sci. 19. 10.3390/ijms19041092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, et al. , 2012. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 109, E2895–903. 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Tymianski M, 2001. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 24, 107–29. 10.1385/MN:24:1-3:107. [DOI] [PubMed] [Google Scholar]
- Scarmeas N, et al. , 2009. Seizures in Alzheimer disease: Who, when, and how common? Arch Neurol. 66, 992–7. 10.1001/archneurol.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, 1992. Differentiation of rat dentate neurons by morphology and electrophysiology in hippocampal slices: Granule cells, spiny hilar cells and aspiny ‘fast-spiking’ cells. Epilepsy Res Suppl. 7, 93–109 [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, 1995. Electrophysiological diversity of pyramidal-shaped neurons at the granule cell layer/hilus border of the rat dentate gyrus recorded in vitro. Hippocampus. 5, 287–305. 10.1002/hipo.450050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, 2012. Alzheimer’s disease and epilepsy: Insight from animal models. Future Neurol. 7, 177–192. 10.2217/fnl.12.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, 2016. The enigmatic mossy cell of the dentate gyrus. Nat Rev Neurosci. 17, 562–75. 10.1038/nrn.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, 2019. The dentate gyrus and temporal lobe epilepsy: An “exciting” era. Epilepsy Curr. 19, 249–255. 10.1177/1535759719855952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Bernstein HL, 2015. Potential implications of a monosynaptic pathway from mossy cells to adult-born granule cells of the dentate gyrus. Front Syst Neurosci. 9, 112. 10.3389/fnsys.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, et al. , 2000. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: Functional implications of seizure-induced neurogenesis. J Neurosci. 20, 6144–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, et al. , 1990. Synaptic connections of dentate granule cells and hilar neurons: Results of paired intracellular recordings and intracellular horseradish peroxidase injections. Neuroscience. 37, 693–707. 10.1016/0306-4522(90)90100-i. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, et al. , 1984. Excitotoxic models for neurodegenerative disorders. Life Sci. 35, 19–32. 10.1016/0024-3205(84)90148-6. [DOI] [PubMed] [Google Scholar]
- Serletis D, et al. , 2011. Complexity in neuronal noise depends on network interconnectivity. Ann Biomed Eng. 39, 1768–78. 10.1007/s10439-011-0281-x. [DOI] [PubMed] [Google Scholar]
- Siskova Z, et al. , 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron. 84, 1023–33. 10.1016/j.neuron.2014.10.024. [DOI] [PubMed] [Google Scholar]
- Staley KJ, et al. , 1992. Membrane properties of dentate gyrus granule cells: Comparison of sharp microelectrode and whole-cell recordings. J Neurophysiol. 67, 1346–58. 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- Storm JF, 1990. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 83, 161–87. 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Styr B, Slutsky I, 2018. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat Neurosci. 21, 463–473. 10.1038/s41593-018-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, et al. , 2007. Selective loss of dentate hilar interneurons contributes to reduced synaptic inhibition of granule cells in an electrical stimulation-based animal model of temporal lobe epilepsy. J Comp Neurol. 500, 876–93. 10.1002/cne.21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs VT, et al. , 2018. Co-transmission of acetylcholine and GABA regulates hippocampal states. Nat Commun. 9, 2848. 10.1038/s41467-018-05136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagnini F, et al. , 2015. Intrinsic excitability changes induced by acute treatment of hippocampal CA1 pyramidal neurons with exogenous amyloid beta peptide. Hippocampus. 25, 786–97. 10.1002/hipo.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, et al. , 2010. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev. 62, 405–96. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, et al. , 2012. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 149, 708–21. 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. , 2013. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–66. 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. , 2016. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol. 80, 858–870. 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. , 2017. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 16, 311–322. 10.1016/S1474-4422(17)30044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. , 2010. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 330, 198. 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainer BH, et al. , 1985. Cholinergic and non-cholinergic septohippocampal pathways. Neurosci Lett. 54, 45–52. 10.1016/s0304-3940(85)80116-6. [DOI] [PubMed] [Google Scholar]
- Williams PA, et al. , 2007. Semilunar granule cells: Glutamatergic neurons in the rat dentate gyrus with axon collaterals in the inner molecular layer. J Neurosci. 27, 13756–61. 10.1523/JNEUROSCI.4053-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, 2007. Physiological studies of human dentate granule cells. Prog Brain Res. 163, 183–98. 10.1016/S0079-6123(07)63011-8. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. , 1995. Beta-amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 6, 2409–13. 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- Xu W, et al. , 2015. Early hyperactivity in lateral entorhinal cortex is associated with elevated levels of abetapp metabolites in the Tg2576 mouse model of Alzheimer’s disease. Exp Neurol. 264, 82–91. 10.1016/j.expneurol.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, et al. , 2018. Sex differences in neuropathology and cognitive behavior in APP/PS1/tau triple-transgenic mouse model of Alzheimer’s disease. Neurosci Bull. 34, 736–746. 10.1007/s12264-018-0268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W, et al. , 2013. Abeta induces acute depression of excitatory glutamatergic synaptic transmission through distinct phosphatase-dependent mechanisms in rat CA1 pyramidal neurons. Brain Res. 1515, 88–97. 10.1016/j.brainres.2013.03.049. [DOI] [PubMed] [Google Scholar]
- Yi F, et al. , 2014. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: Roles in cellular excitability, inhibitory transmission and cognition. J Physiol. 592, 3463–94. 10.1113/jphysiol.2014.275453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JC, et al. , 2017. Epigenetic suppression of hippocampal calbindin-D28k by ΔFosB drives seizure-related cognitive deficits. Nat Med. 23, 1377–1383. 10.1038/nm.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyatdinova S, et al. , 2016. Increased epileptiform EEG activity and decreased seizure threshold in arctic APP transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res. 13, 817–30. 10.2174/1567205013666160129095508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.