

Abstract
Over the past 50 years, ketamine has solidified its position in both human and veterinary medicine as an important anesthetic with many uses. More recently, ketamine has been studied and used for several new indications, ranging from chronic pain to drug addiction and post-traumatic stress disorder. The discovery of the rapid-acting antidepressant effects of ketamine has resulted in a surge of interest towards understanding the precise mechanisms driving its effects. Indeed, ketamine may have had the largest impact for advancements in the research and treatment of psychiatric disorders in the past few decades. While intense research efforts have been aimed towards uncovering the molecular targets underlying ketamine’s effects in treating depression, the underlying neurobiological mechanisms remain elusive. These efforts are made more difficult by ketamine’s complex dose-dependent effects on molecular mechanisms, multiple pharmacologically active metabolites, and a mechanism of action associated with the facilitation of synaptic plasticity. This review aims to provide a brief overview of the different uses of ketamine, with an emphasis on examining ketamine’s rapid antidepressant effects spanning molecular, cellular, and network levels. Another focus of the review is to offer a perspective on studies related to the different doses of ketamine used in antidepressant research. Finally, the review discusses some of the latest hypotheses concerning ketamine’s action.
Keywords: Ketamine, Subanesthetic, Rapid-acting antidepressant, Depression, Dose, Anesthesia
Introduction
Over the past 50 years, the use of ketamine for anesthesia has become widespread in both human and veterinary medicine. Its safety and short duration and unique mode of action have made ketamine an important drug in emergency medicine and pain management around the world. The study and use of ketamine for the treatment of several new indications, ranging from pain syndromes to drug addiction and psychiatric disorders, is constantly growing. In experimental research, ketamine has unarguably contributed to a better understanding of the glutamatergic system and the development of animal models of schizophrenia [1].
Perhaps the greatest contribution to the legacy of ketamine has been the discovery of its rapid-acting antidepressant effects [2], which has effectively revitalized interest in the development of new antidepressant drugs with a novel mode of action. Indeed, the past decade has seen a rapid increase in the number of research papers associated with ketamine’s antidepressant effects [3]. It can be argued that even beyond its important role in anesthesia, ketamine is a top contender for being the drug with the largest impact for research and treatment of psychiatric disorders in the past several decades.
The ability of ketamine to provide a rapid relief of depressive symptoms, often within hours, has brought it to the forefront of treating severe treatment-resistant depression [4]. Despite its relatively fast pharmacokinetics, the antidepressant effects of ketamine are sustained for up to 1–2 weeks [5]. This rapid onset of ketamine’s action combined with a sustained response lasting far beyond its acute pharmacological effects has puzzled researchers and medical doctors. In contrast, typical antidepressant drugs, such as selective serotonin reuptake inhibitors, often require weeks or even months of continuous medication to elicit their beneficial effects [6]. While the last 2 decades of basic and clinical research have significantly contributed to the understanding of ketamine’s complex profile of effects and its potential for the treatment of psychiatric disorders, the precise neurobiological basis of its antidepressant action remains unclear.
With a vast amount of research aimed at untangling the mysteries of its action, an ever-increasing number of molecular targets and mechanisms have been associated with the antidepressant effects of ketamine [7]. A myriad of novel drug candidates, essentially mimicking certain aspects of ketamine’s action, have risen out of preclinical research. However, to date, no new drug has conformed to the high expectations, with multiple candidates failing in recent clinical trials [8–10]. The shortcomings of animal models of depression are often discussed as a plausible explanation for these failures of translational research, but many other aspects also require further examination. In the rush to understand the molecular complexities of ketamine’s action, both basic and clinical researches have overlooked details related to the administration and dosing time of ketamine [11, 12]. For example, studies directly comparing different dosing parameters and pharmacokinetics between human and rodent studies are still lacking.
With many new ketamine studies published each year, staying up to date becomes a challenge. Thus, the purpose of this review is both to introduce the reader to the various uses of ketamine from anesthesia to pain management and psychiatric use and to provide a thorough overview of the current understanding of ketamine’s effects in treating depression. Herein, the multifaceted profile of ketamine’s effects is reviewed, and studies addressing molecular, cellular, and network mechanisms are introduced. A special emphasis is placed on discussing issues associated with the dosing of ketamine in both basic and clinical research as well as on discussing some of the latest hypotheses related to ketamine’s antidepressant mechanisms.
An overview of ketamine and its many uses
What is ketamine?
Ketamine is an anesthetic drug first synthesized in 1962 at the Parke-Davis pharmaceutical company following the discovery of phencyclidine (PCP), another anesthetic belonging to the arylcyclohexylamine chemical class [13]. At that time, several different drugs sharing a similar structure were screened in search of an optimal candidate for further development—preferably one that lacked the unwanted psychotropic effects of PCP. On the basis of the results of these experiments, ketamine became the lead compound for future development. Edward Domino led the first human experiments, which found ketamine to be a safe and short-acting anesthetic. However, ketamine was not entirely devoid of psychotropic effects and was considered a dissociative anesthetic [14].
Ketamine refers to the mixture of two water-soluble, optical stereoisomers: S( +) and R(−)-ketamine. It is pharmaceutically produced in both racemic and enantiopure preparations. The main mode of ketamine’s pharmacological action is the blockade of N-methyl-d-aspartate receptors (NMDARs), ion channels mainly involved in excitatory glutamatergic neurotransmission. Both enantiomers share the ability to block NMDARs but differ slightly in their potency. S-ketamine is often preferred in clinical anesthesia owing to its stronger ability to block NMDARs, whereas R-ketamine has a lower affinity for NMDARs [15]. Ketamine, PCP, and dizocilpine (also known as MK-801)—the classical NMDAR antagonists—are non-competitive inhibitors of NMDARs [16, 17]. By entering the ion channel and then being captured inside the closing pore, they elicit a trapping block (Fig. 1). In contrast, drugs such as memantine act as partial trapping blockers, which only hinder the channel closure but do not entirely prevent it from functioning [18].
Fig. 1.
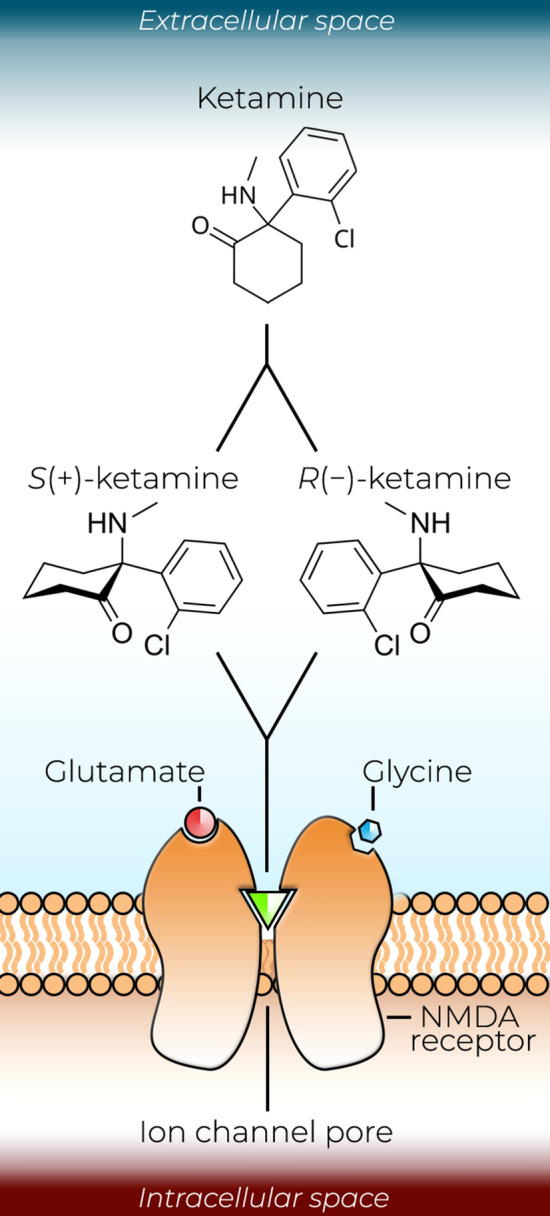
Ketamine acts as a non-competitive blocker N-methyl-d-aspartate receptor (NMDAR) ion channels. Racemic ketamine contains two optical stereoisomers: S( +) and R(−)-ketamine. Both isomers act as non-competitive blockers, sharing the ability to enter NMDAR ion channels and to exhibit a trapping block. S-ketamine is often preferred in clinical anesthesia because it has a higher affinity for NMDARs than R-ketamine
Although ketamine exerts its most pronounced effects through the blockade of NMDARs, it has been proposed to also affect many other targets, including dopaminergic, serotonergic, adrenergic, opioidergic, cholinergic, and sigma receptors [1, 15, 19]. Ketamine also acts on serotonin, noradrenaline, and dopamine reuptake transporters (SERT, NET, and DAT respectively) and various ion channels, such as voltage-gated sodium channels (VGSCs) [20] and hyperpolarization-activated cyclic nucleotide (HCN)-gated channels [21].
After administration, ketamine is rapidly distributed in the body and has low plasma protein binding and a short elimination half-life of approximately 2–4 h in humans [22–25]. The rate of metabolism and elimination of ketamine in mice is much faster than that in humans, with a serum half-life of approximately 13 min [26]. The initial metabolite is (R,S)-norketamine (NK), but (2R,6R;2S,6S)-hydroxynorketamine (HNK) and (R,S)-dehydronorketamine (DHNK) are the major circulating metabolites in human plasma [27, 28]. The peak plasma concentration for NK is reached in approximately 1.3 h, and that for DHNK and HNK is reached in 3.8 h [28]. Plasma levels of HNK and DHNK can be measured 24 h after the infusion and may remain detectable for up to 48 h. The initial metabolic reaction—N-demethylation to NK—is mainly catalyzed by liver cytochrome P450 enzymes CYP2B6 and CYP3A4. These steps are followed by hydroxylation to generate HNK and DHNK [29]. In addition, several minor metabolic pathways also exist. For a more thorough take on ketamine’s basic pharmacology, pharmacokinetics, and metabolism, see the review by Zanos et al. [15].
Dissociation and anesthesia
What makes ketamine unique from most other sedatives and anesthetics is the state of dissociative anesthesia produced by the blockade of NMDARs at high doses. Ketamine does not primarily act through gamma-aminobutyric acid (GABA) receptors like most other anesthetics that possess sedative or hypnotic properties [19]. The dissociative effects of ketamine can be described by the experience of being conscious while being drawn away from sensory perceptions. Higher doses produce a dose-dependent deepening of the dissociative state towards dream-like states of open- and closed-eye visuals and strong perturbations of thought and bodily sensation [30], which may be explained by the unique shifting of intracortical dynamics under the effects of ketamine [31, 32]. A high dose of ketamine results in a state of deep dissociation accompanied by amnesia and loss of consciousness because NMDARs are important components of excitatory neurotransmission, long-term potentiation (LTP), and memory formation. Notably, the psychoactive qualities of ketamine are also sought by recreational users, as evidenced by the surge in using ketamine as a drug of abuse from the 1970s to this day [23].
The electrophysiological effects of ketamine can be measured using electroencephalography (EEG), where ketamine demonstrates different dynamics than many other anesthetics [33]. In nonhuman primates, upon ketamine anesthesia, high beta-gamma electroencephalogram (EEG) oscillations emerge at first and deepen towards slow-delta oscillations [31]. In human volunteers, the loss of behavioral responsiveness has been associated with the onset of EEG slow-wave activity (SWA) [33]. Moreover, high-density EEG studies in human volunteers demonstrate that during ketamine anesthesia, theta, gamma, and delta power increased in frontal and posterior channel clusters, whereas posterior alpha power decreased both under anesthetic and subanesthetic doses [34].
Owing to its non-GABAergic mechanism of action, ketamine is a safe and effective choice for emergency anesthesia in a prehospital setting. Ketamine offers a relatively wide dosing range, produces a sympathomimetic effect that supports cardiovascular stability along with maintenance of respiratory function, and provides a good level of analgesia comparable to that produced by morphine [35]. During dissociative anesthesia, the basic reflexes are well preserved [36]. For the induction of anesthesia, doses of racemic ketamine are typically in the range of 1–2 mg/kg administered as an intravenous bolus (Fig. 2), which produce a state of dissociative anesthesia within 1–2 min of injection [35]. After induction, a continuous dose of 1–6 mg/kg per h is required to maintain the effect. In contrast, doses of S-ketamine required for the induction of general anesthesia are 0.5–1 mg/kg, followed by a continuous infusion of 0.5–3 mg/kg per h [19]. In veterinary medicine and animal research, ketamine is very commonly used in combination with xylazine, a sedative α2 adrenergic receptor agonist [37].
Fig. 2.

Some of the current and emerging clinical uses of ketamine along with examples of commonly used doses. Clinical applications not yet commonly adopted are followed by an asterix
Ketamine can be efficiently administered via multiple routes, including oral, sublingual, intranasal, intramuscular, intraosseous, rectal, and subcutaneous routes, but the highest bioavailability and fastest onset are achieved by intravenous administration [35, 38]. At present, ketamine is used in emergency units for anesthesia and procedural sedation in various patient populations ranging from children to adults. In this context, the wide dosing range, the ability to administer intramuscular doses, and the analgesic properties are particularly useful and make it possible to administer ketamine in field conditions where no anesthesiologist or monitoring equipment is available [39]. However, ketamine is not widely preferred as a sole agent for general anesthesia performed in hospitals owing to its psychotomimetic effects and the potential to produce emergence phenomena, manifesting as profound confusion or hyperexcitation upon waking up from the dissociative state in up to 20% of adults [35].
Pain management
Ketamine’s analgesic effects, which emerge even at subanesthetic doses, and its good safety profile have prompted its use in the treatment of both acute and chronic pain and in procedural sedation of pediatric patients [40]. The analgesic effects of racemic ketamine become apparent at intravenous doses of 0.25–0.5 mg/kg, which corresponds to 0.125–0.25 mg/kg of S-ketamine [19]. In a prehospital setting, ketamine may produce analgesia comparable to that produced by morphine or fentanyl without as much respiratory depression [41, 42]. For short-term pain relief, low intravenous doses of ketamine provide analgesic effectiveness and safety equivalent to those of intravenous morphine for short-term pain relief [43]. However, ketamine is often used together with other pain medications. For example, a low dose of ketamine combined with a reduced dose of hydromorphone promotes rapid and effective pain relief in emergency department patients suffering from acute pain [44].
A large Cochrane review found that perioperative intravenous ketamine reduces the need for postoperative analgesics and decreases perceived pain intensity [45]. Ketamine alone, however, may not prevent chronic postoperative pain [46–49]. In addition to its use in treating acute pain, ketamine is also used in chronic pain syndromes, where it exerts beneficial effects through the inhibition of NMDARs [50]. Ketamine has been suggested to be effective in treating many different types of chronic pain, such as neuropathic pain [51] and phantom limb pain [52, 53], cancer pain [54], and migraine with prolonged aura [55]. Ketamine may be particularly useful in instances where opioids no longer provide sufficient pain relief because it has been suggested to reduce opioid tolerance and pain hypersensitivity [56, 57]. Moreover, the antidepressant effects of ketamine may be beneficial when treating patients with chronic pain because depression and pain syndromes often co-exist [58].
Psychiatric use
In the 1970s, the psychoactive side effects of ketamine were desirable in psychiatric investigations where the drug was used as an abreactive agent. A variety of doses more than capable of producing psychotropic experiences were employed in early trials [59, 60] with relatively positive, although anecdotal, outcomes. Moreover, early observations of NMDAR antagonist-induced transient psychotomimetic effects paved the way for the advancement of the idea that glutamatergic neurotransmission was a key component in schizophrenia, and led to the development of ketamine-, PCP-, and MK-801-induced animal models of schizophrenia [61]. More recently, the use of ketamine in treating various psychiatric disorders has regained momentum. Apart from ketamine’s use as a rapid-acting antidepressant drug—which will be more thoroughly discussed in the subsequent section—its use as a novel pharmacotherapeutic intervention for the treatment of post-traumatic stress disorder (PTSD) [62] and substance use disorders [63] has also been explored.
Post-traumatic stress disorder is a condition triggered by the experience of going through a traumatic event [64, 65]. Symptoms may include mental re-experience of the traumatic event and uncontrollable thoughts related to it as well as severe anxiety, arousal, and reactivity. Post-traumatic stress disorder may manifest as negative cognition and mood, and it is often comorbid with depression. Moreover, current pharmacotherapies are often insufficiently effective [66]. Because ketamine is commonly used in emergency anesthesia and analgesia, studies have examined the effects of ketamine administered acutely after physical trauma on the subsequent emergence of PTSD symptoms. On the one hand, two observational studies suggest that ketamine worsens the risk of developing PTSD symptoms [67, 68]; however, questions have been raised about their methodology [62]. On the other hand, a retrospective review of medical records of burn victims suggested reduced prevalence of PTSD among those receiving intraoperative ketamine [69]. However, a subsequent study found no differences [70]. Animal studies of PTSD-like behavior have also shown no changes [71] or slightly detrimental [72] outcomes.
Clinical studies of ketamine in patients suffering from chronic PTSD have been positive. The first randomized controlled study in this context demonstrated that a single subanesthetic dose of iv ketamine (0.5 mg/kg) was superior to midazolam (0.045 mg/kg) in rapidly reducing PTSD (and depressive) symptoms within 24 h [73]; however, PTSD symptoms often started to recur 48 h post-infusion, and no significant difference was detected at 1 week post-infusion. Another open-label trial studied repeated iv ketamine infusions (0.5 mg/kg; six infusions over 12 days) and found rapid improvement in both PTSD and depressive symptoms, with the median time to relapse in PTSD remitters being 41 days [74]. Most recently, the first randomized controlled trial of repeated ketamine in PTSD found ketamine to elicit better improvement than the active control group receiving midazolam [75]. Overall, ketamine seems to hold promise in treating PTSD, but more studies are warranted. For a more thorough take on ketamine in the treatment of PTSD, see the review by Feder et al. [62].
Ketamine has been suggested to have beneficial effects in treating drug addiction. Rodent studies have shown that ketamine reduces ethanol consumption in alcohol-preferring rats [76, 77] and reduces morphine-induced place preference [78]. Several clinical studies provide support for the potential of ketamine in the treatment of alcoholism as well as opioid and cocaine addiction and cannabis use disorder [79–85]. In this context, mystical-type experiences mediated by the psychoactive profile of the drug have been suggested to be important [86]. At present, evidence for the use of ketamine in this context is only beginning to emerge, and further research is needed to properly evaluate its effectiveness and safety in treating substance use disorders [63].
Ketamine as an antidepressant drug
Rapid onset of action
The seminal study by Berman et al. [2] was the first to demonstrate the rapid-acting antidepressant effects of an intravenous subanesthetic infusion of ketamine in patients suffering from major depressive disorder (MDD). Since then, a number of clinical trials have replicated these results [87–89] and extended the findings to treatment-resistant patients, which has rapidly increased the use of ketamine for the treatment of depression. Other administration routes and doses have also been successfully used, with several trials indicating the antidepressant action of intranasally [90, 91] and orally [92, 93] administered ketamine. However, despite 2 decades of active research, there is still no clear consensus on the dose dependency of ketamine’s effects or the superiority of different administration routes in treating depression.
The factors that separate ketamine from traditional antidepressant drugs are its rapid onset of action and effectiveness in treatment-resistant patients. Ketamine’s antidepressant effects develop within hours of drug administration and may last from a couple of days to approximately 2 weeks following a single dose [5]. The antidepressant effects often peak at 24 h after the infusion [94]. Similar antidepressant effects have also been observed in patients suffering from bipolar depression [95, 96]. These effects of ketamine on depression are remarkably more rapid than those of traditional antidepressants, which may take several weeks or months to develop. Ketamine’s rapid onset of action is particularly useful in treating depressed patients who exhibit suicidality because ketamine also rapidly ameliorates suicidal ideation [97, 98]. Because the antidepressant effects of ketamine are relatively transient, repeated doses are often required to maintain them. However, repeated doses may not produce markedly enhanced antidepressant effects than a single ketamine treatment [99].
Intravenous, intramuscular, and subcutaneous administration
For the treatment of depression, ketamine is typically infused intravenously over 40 min at a subanesthetic dose of 0.5 mg/kg; the intramuscular and subcutaneous routes are generally less used [38]. The low subanesthetic doses combined with a slow iv infusion rate produce only minor psychoactive effects and mitigate the occurrence of dissociative/psychedelic experiences—generally considered unwanted—that become more common towards the higher end of the subanesthetic spectrum. Bolus doses are rarely used owing to poor tolerability. However, an open longitudinal study suggested that a rapid bolus injection of 0.5 mg/kg ketamine also has rapid antidepressant effects with relatively good tolerability [100], whereas a study of S-ketamine (0.25 mg/kg; iv over 10 min) reported that 11.1% of the patients described their experience as very disturbing.
Several small studies have examined the dose-dependent effects of ketamine in depressed patients while also comparing different administration routes. For example, a placebo-controlled cohort-based pilot trial investigated the effects of ketamine on depressive patients using dose titration from 0.1 to 0.5 mg/kg with intravenous, intramuscular, and subcutaneous routes of delivery [101]. The experiment included 15 patients who received ascending doses. Different administration routes produced comparable antidepressant effects, whereas the dose required for an antidepressant response varied between individuals, suggesting that dose titration should be done on an individual basis. In this study, higher doses resulted in greater antidepressant effects and, as expected, more pronounced psychotomimetic effects. Notably, as discussed by the authors, subcutaneous delivery is a promising method for ketamine administration because the plasma levels of ketamine are similar to those observed in intravenous administration and because it is convenient to administer.
Another small dose–response trial compared four different ketamine doses (0.1, 0.2, 0.3, and 0.4 mg/kg) administered to four patients as an iv infusion over 2–5 min in a placebo-controlled double-blind crossover design [102]. The results of this study neatly demonstrate the dose dependency of the psychoactive effects and suggest that antidepressant efficacy of ketamine increases with increasing subanesthetic dose. The dose dependency of ketamine’s antidepressant effects has been further addressed in a systematic review and meta-analysis of nine ketamine trials for the treatment of depression [103]. The authors categorized ketamine doses used in the different studies into low (0.5 mg/kg iv) or very low doses (50 mg intranasal spray, 0.1–0.4 mg/kg iv, and 0.1–0.5 mg/kg iv or sc). Six of these trials used a low dose of ketamine, whereas three used a very low dose. They reported that a low dose of ketamine appeared superior to a very low dose, but a substantial heterogeneity was observed in the clinical response; one-fifth of patients showed remission at 1 week, whereas most others experienced short-lasting benefits. More recently, a double-blind active placebo-controlled trial compared several subanesthetic ketamine doses, but only the higher doses (0.5 mg/kg and 1.0 mg/kg) were found to have clinically meaningful effects [104].
S-ketamine has been studied considerably less in terms of the iv dosage range, but 0.25 mg/kg of S-ketamine has been shown to be non-inferior to the standard dose (0.5 mg/kg) of racemic ketamine [105]. One trial used 0.2 and 0.4 mg/kg of S-ketamine administered intravenously over 40 min and demonstrated robust efficacy [106]. The authors note that the lower dose may allow for higher tolerability while maintaining efficacy.
Taken together, these studies suggest that there is a range of therapeutically active subanesthetic doses of ketamine. For racemic ketamine, doses towards the higher end of the range may be more effective; however, existing studies do not provide conclusive evidence. Large-scale studies comparing the various routes of administration are also lacking, as are studies comparing the effectiveness of different infusion rates. Moreover, it also remains unclear whether there are minimum and maximum effective doses for achieving antidepressant responses and whether patients who do not respond to lower doses would benefit from higher doses—and vice versa.
Intranasal administration
The majority of studies concerning the antidepressant effects of ketamine have focused on investigating intravenous administration, in which doses are determined by the subject’s weight. In contrast, trials for intranasal ketamine typically use predetermined bolus doses. These intranasal doses of ketamine (and S-ketamine) commonly range between 20 and 100 mg [91, 107–110]. While the bioavailability of intranasal ketamine is estimated to be approximately 25–50% [19, 111], this route of administration is faster to use and more convenient than intravenous infusion. With S-ketamine being approved by the U.S. Food and Drug Administration (FDA) for alleviating symptoms of treatment-resistant depression in 2019, the use of intranasal S-ketamine is likely to increase in the coming years. However, concerns have been expressed over the cost-effectiveness of this intranasal formulation [112].
Intranasal administration of 50 mg of ketamine has shown relatively similar treatment outcomes to those of standard ketamine infusion [91]. Similarly, intranasal S-ketamine doses ranging 28–84 mg have been compared by the pharmaceutical company Janssen in a phase II trial [90]. Remarkably, this study also found that higher doses produced more sustained remission. Similar to racemic ketamine administered via infusion, intranasally administered S-ketamine (84 mg) also reduces suicidality in patients at imminent risk of suicide [113]. The safety and efficacy of intranasal S-ketamine have been studied in several phase III trials; Popova et al. [114] demonstrated a statistically significant effect of intranasal S-ketamine combined with a new oral antidepressant in treatment-resistant depression in comparison with antidepressant/placebo, whereas two other studies failed to find statistically significant differences [108, 109]. In addition, one study addressed intranasal S-ketamine combined with an oral antidepressant for relapse prevention [107]. This study found that the continuation of S-ketamine treatment with an oral antidepressant was superior in delaying relapse than the antidepressant plus placebo.
While intranasal S-ketamine clearly has some beneficial effects in the treatment of depression, the magnitude of these effects, among other things, remains a matter of debate [115–117]. Indeed, a re-analysis of the four phase III studies submitted to the FDA suggests that intranasal S-ketamine may only improve depressive symptoms by reducing 4 points on the Montgomery-Åsberg Depression Rating Scale (MADRS), a scale that ranges from 0 to 60 [118]. At present, it remains unclear how well S-ketamine, or intranasal administration in general, performs when compared with racemic ketamine administered as an intravenous infusion. A recent non-inferiority study comparing intravenous S-ketamine versus racemic ketamine suggests that their antidepressant effects may not be markedly different [105]. Indeed, off-label administration of racemic ketamine remains an attractive option for clinicians for intravenous use because it is readily available, low cost, and easily adaptable to nasal administration via sinus nebulizers.
Psychoactive effects and the therapeutic context
Most clinical research on ketamine’s antidepressant action has focused on using low subanesthetic doses that minimize the emergence of psychotomimetic side effects. Moreover, the majority of studies have addressed the effectiveness of ketamine from a purely pharmacotherapeutic rather than psychotherapeutic perspective. With the resurgence of psychedelic research and therapy, the issue of the importance of subjective experience and the set and setting has been brought up also in association with ketamine treatment [119]. The underlying idea is that psychedelic therapy can be considered to be a drug-assisted form of psychotherapy rather than pure pharmacotherapy [120]. In this context, psychological factors such as the preceding preparation as well as subsequent integration of the experience are thought to be important for influencing therapeutic outcomes by some researchers. While some preliminary studies of psychedelics support the notion that psychoactive effects and therapeutic context are associated with therapeutic effects [120–122], further research is required to thoroughly address their significance.
Regarding ketamine treatment, these questions have not been studied to any significant extent and remain largely unanswered. However, some clinical studies have suggested that the psychoactive or dissociative effects of ketamine during administration could be associated with producing therapeutic responses. For example, Sos et al. [123] performed a double-blind, crossover, placebo-controlled trial and found a substantial relationship between the antidepressant effects and psychotomimetic effects of ketamine. In this study, the more intense psychotomimetic effects, assessed by the Brief Psychiatric Rating Scale (BPRS), were positively correlated with improved mood ratings on the MADRS 7 days after ketamine infusion. A study by Luckenbaugh et al. [124] also investigated whether psychoactive effects were important for the emergence of antidepressant responses by analyzing 108 treatment-resistant depressive patients from three studies that used a single subanesthetic ketamine dose (0.5 mg/kg iv over 40 min). Two of these were double blind, and the third had an open-label design. The authors found a significant correlation between increased Clinician-Administered Dissociative States Scale (CADSS) score at 40 min and improved Hamilton Depression Rating Scale (HDRS) scores at 230 min and on day 7. Moreover, a more recent study by Niciu et al. [125] reported that dissociative symptoms (also measured by CADSS) were associated with the antidepressant responses after 0.5 mg/kg (iv) ketamine. In contrast, another study found no correlation between CADSS and HDRS responses at any time following the ketamine infusion [126].
Notably, some recent studies have focused on examining the subjective properties of ketamine. A small study—not related to depression—investigating eight cocaine-dependent individuals found that ketamine produced significantly greater acute mystical-type effects (measured by Hood’s Mysticism Scale) than the active control lorazepam [127]. The HMS score, but not the CADSS score, was found to mediate the effect of ketamine on the motivation to quit cocaine on the following day. In another interesting approach, Stocker et al. [128] screened 62 YouTube videos of depressed patients narrating their subjective experience of receiving ketamine and found that 27.4% of the individuals reported an experience of floating that they associated with the amelioration of their depressive symptoms. The connection between treatment response and the floating sensation was subsequently investigated in a double-blind, crossover, placebo-controlled clinical trial, which concluded that the two were not associated [129]. However, identifying clinically usable markers of treatment response and new means to improve the psychological effectiveness of psychoactive pharmacotherapies remains an important avenue for further research.
Towards a neurobiological understanding of ketamine’s antidepressant effects
Discovery of the role of NMDARs
Among the first studies to promote the idea of the antidepressant-like action of NMDAR antagonists was the work by Trullas and Skolnick [130] in mice. They demonstrated that the NMDAR antagonists AP-7 and MK-801 reduced the behavioral immobility of animals subjected to the forced swimming test (FST), a classical test to assess antidepressant-like properties. In a subsequent study, Skolnick et al. [131] demonstrated that chronic treatments with traditional antidepressants alter radioligand binding to NMDARs in the cerebral cortex. These results suggested NMDARs as a common target for the action of traditional antidepressants and prompted further investigations into the modulation of glutamatergic neurotransmission in the treatment of depression [132]. Remarkably, the first clinical note of the putative antidepressant effects of d-cycloserine, an NMDAR modulator, was published in 1959 [133], but it took several decades before NMDAR antagonists were considered as potential antidepressant therapeutics.
Since the discovery of ketamine’s antidepressant potential in the late 1990s [2], basic research has focused on identifying the neuronal, molecular, and metabolic targets important for ketamine’s effects. A large number of studies have examined the effects of ketamine in various animal models of depression, with most studies providing strong support for the antidepressant-like action of ketamine in rodents [134–139]. An important research line in the study of ketamine’s effects emerged from the study of chronic stress-induced synaptic alterations and dendritic atrophy [140], also associated with dysfunctional neurotrophic support [141], processes that were found to be positively affected by ketamine [142]. Most importantly, the relative similarity of ketamine’s effects in animal models of depression and in clinical studies provides some basis for the translational significance of these findings.
Cortical excitation and glutamate
The main mechanisms believed to underlie ketamine’s antidepressant effects converge on the increase in cortical excitation and glutamate release and burst, which are thought to trigger subsequent molecular and physiological changes leading to the remediation of depressive symptoms [138]. Because NMDARs are important components of excitatory glutamatergic neurotransmission, one could expect that by blocking these receptors, ketamine treatment would cause cortical inhibition rather than excitation. These paradoxical effects may be explained by the disinhibition hypothesis, which proposes that subanesthetic doses of ketamine preferentially inhibit NMDARs present in GABAergic interneurons (Fig. 3a). This decreases the inhibition of excitatory pyramidal neurons and increases glutamate release and burst [132, 143–145], which continue to activate neurotrophic signaling mechanisms leading to the amelioration of chronic stress-induced synaptic deficits [146]. Two recently published animal studies strongly support the disinhibition hypothesis [147, 148] as the mechanism for subanesthetic ketamine-induced neuronal activation.
Fig. 3.

An overview of some of the proposed molecular mechanisms underlying ketamine’s rapid antidepressant action. a The disinhibition hypothesis. Ketamine preferentially blocks N-methyl-d-aspartate receptors (NMDARs) on gamma-aminobutyric acid (GABA)-ergic inhibitory interneurons, leading to a decrease in the inhibitory tone exerted on excitatory pyramidal neurons. Increased glutamate release acts on postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs) and induces cellular effects such as the release of brain-derived neurotrophic factor (BDNF) and activation of its receptor tropomyosin receptor kinase B (TrkB) and the regulation of downstream pathways important for synaptic plasticity and protein synthesis. Downstream effects include activation of mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) and regulation of AMPAR dynamics and scaffolding proteins such as postsynaptic density protein 95 (PSD95). b Hydroxynorketamine metabolites may modulate postsynaptic AMPAR signaling, leading to various downstream effects. c Inhibition of glycogen synthase kinase 3 β (GSK3β) by ketamine reduces phosphorylation of PSD95, which augments AMPAR signaling by reducing the internalization of AMPAR subunits, among other effects. d Ketamine may also block extrasynaptic NMDARs, normally tonically activated by glutamate, and induce mTOR activity. e The blockade of spontaneous NMDAR-mediated neurotransmission can have effects that lead to the disinhibition of BDNF translation via eukaryotic elongation factor 2 (eEF2)-dependent mechanisms
Other proposed molecular hypotheses of ketamine’s action include the direct antagonism of extrasynaptic NMDARs on pyramidal neurons, which disrupts the tonic activation of NMDARs by ambient glutamate and results in homeostatic synaptic plasticity and a compensatory increase in excitatory drive in the prefrontal cortex (Fig. 3d) [145]. These changes are thought to be mediated by the blockade of extrasynaptic NMDARs containing GluN2B subunits [149]. Furthermore, ketamine has been suggested to inhibit NMDAR-mediated spontaneous neurotransmission (Fig. 3e) [150] and to trigger an increase in the translation of proteins such as the brain-derived neurotrophic factor (BDNF), a crucial mediator of synaptic plasticity, by reducing the phosphorylation of eukaryotic elongation factor 2 (eEF2), thus leading to antidepressant-like effects [150–152]. In contrast, several recent studies suggest that ketamine’s effects are mediated through its HNK metabolites independently of NMDAR inhibition (Fig. 3b) [134]. Altogether, these hypotheses are not mutually exclusive and may together explain the molecular changes observed after ketamine administration.
Regardless of the chosen hypothetical framework, studies have demonstrated that ketamine possesses excitatory effects that are highly dose dependent. In rats, anesthetic doses of 200 mg/kg (ip) decreased acute glutamate activity measured by microdialysis, whereas lower doses (10, 20, and 30 mg/kg) increased glutamate outflow in the prefrontal cortex [144]. In another study, subanesthetic ketamine provoked transient changes in glutamate cycling in the medial prefrontal cortex of rats [153]. The glutamate surge following ketamine administration may produce its rapid-acting antidepressant effects through the modulation of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs) because the blockade of these receptors abolishes ketamine’s antidepressant-like behavioral responses in mice and rats [154–156]. Moreover, positive allosteric AMPAR modulators produce antidepressant-like behavioral effects in rodents (Knapp et al., 2002; Li et al., 2001) and upregulate BDNF synthesis [159, 160].
Intracellular signaling pathways
Based on rodent studies, ketamine-induced activation of AMPARs is thought to result in the modulation of a subset of molecular pathways involved in synaptic plasticity, which lead to antidepressant-like effects. For example, ketamine induces the rapid translation and release of BDNF [150, 161], resulting in the activation and phosphorylation of the BDNF receptor, tropomyosin receptor kinase B (TrkB) [150, 162]. BDNF-TrkB signaling constitutes a major component of synaptic plasticity regulation [163, 164], which is crucial for counteracting the negative synaptic effects of stress, restoring altered network activity and producing antidepressant-like effects [142, 146, 165–168]. Indeed, the antidepressant-like effects of subanesthetic ketamine and its HNK metabolites are diminished in BDNFmet66met knock-in mice that exhibit compromised activity-dependent BDNF release [169, 170]. The rapid onset of ketamine’s antidepressant effects is hypothesized to be associated with the rapid activity-dependent release of BDNF. However, somewhat unexpectedly, our recent studies demonstrate that the activation of TrkB and associated signaling pathways is more prominent with high, sedative-anesthetic doses of ketamine than with subanesthetic doses of ketamine [171] and sedative doses of non-excitatory drugs such as medetomidine [172].
Other downstream signaling pathways and targets considered to be important in ketamine’s effects are glycogen synthase kinase 3 beta (GSK3β) [135, 150], extracellular signal-regulated kinase 1/2 [ERK1/2, also known as p44/42-mitogen-activated protein kinase (p44/42-MAPK)] [173], and mammalian target of rapamycin (mTOR) [136] along with its effector p70-S6 kinase (p70-S6K) [174]. In mice, the inhibition of GSK3β by ketamine through the phosphorylation of the serine 9 residue has been suggested to be necessary for the rapid antidepressant-like effects observed [135], which may lead to the augmentation of AMPAR-mediated signaling by diminished internalization of GluA1 subunits (Fig. 3c) [175]. Studies investigating ketamine together with co-administered lithium—an unspecific GSK3 inhibitor—found additive antidepressant-like effects in rodents [176]; however, lithium did not facilitate or prolong the effects of ketamine in depressed patients [177]. Moreover, it is important to remember that GSK3β is a promiscuous kinase involved in various cellular processes [178] and the serine 9 phosphorylation is also regulated by several anesthetic drugs other than ketamine [171, 172, 179, 180].
Increased glutamate activity under the effects of subanesthetic ketamine leads to upregulation of p44/42-MAPK activation, whereas higher anesthetic doses reduce MAPK phosphorylation [136, 171]. Increased MAPK signaling is correlated with the antidepressant effects of ketamine in rodents [173], which could be associated with its role in regulating AMPAR trafficking and synaptic potentiation [181]. Indeed, studies have demonstrated that the acute blockade of MAPK signaling results in a depressive phenotype in rodents, and that BDNF ± mice are more sensitive to develop a depressive phenotype when administered a low dose of MAPK kinase inhibitor [182]. Moreover, increased expression of mitogen-activated protein kinase phosphatase 1 (MKP1), a negative regulator of MAPK, results in the emergence of depressive behavior in rodents, whereas mice lacking MKP1 are resilient to stress [183].
Ultimately, activity-induced signaling cascades such as MAPK mediate their effects to the nucleus, where the activity of transcription factors is regulated [184]. Subsequent changes in the expression of immediate early genes (IEGs), particularly those encoding proteins involved in synaptic transmission and function, could be involved in the emergence of antidepressant effects. Here, ketamine-induced expression of IEGs such as Homer1 [185–189] could be particularly important. This notion is supported by a recent study, where the administration of cell-permeable TAT-Homer1a essentially recapitulated the behavioral responses induced by ketamine in rodents [190].
Finally, the molecular determinants of ketamine’s action are thought to converge on the mTOR complex, which is involved in cellular protein synthesis and metabolism. As demonstrated by Li et al. (2010), subanesthetic doses of ketamine increase levels of synaptic protein and formation of dendritic spines in the prefrontal cortex of rats. Ketamine acutely induces the phosphorylation of mTOR, p70-S6K, eukaryotic translation initiation factor 4E-binding protein 1, p44/42-MAPK, and Akt, which are believed to influence the subsequently upregulated expression of synaptic proteins such as Arc, synapsin I, postsynaptic density protein 95 (PSD95), and GluR1 [136]. Notably, the effects of ketamine on synaptogenesis and behavior are reversed with the blockade of mTOR activity by the intracerebroventricular administration of rapamycin [136]. Following these pivotal findings, several other animal studies have investigated the role of mTOR in the therapeutic effects of ketamine, with promising results [137, 191, 192]. In particular, mTOR has been suggested to be involved in the sustained, rather than acute, antidepressant effects following ketamine administration [137]. However, understanding the function of the mTOR complex in ketamine’s antidepressant effects is complicated by a recent clinical trial, which found that mTOR inhibitor rapamycin increases the duration of ketamine’s antidepressant effects rather than diminishing them [193].
Cellular-level findings in animal models
Several studies have identified the actions of ketamine on the infralimbic prefrontal cortex (IL-PFC). The inactivation of this region using muscimol blocked the antidepressant effects of systemic ketamine, whereas microinfusion of ketamine into the IL-PFC essentially recapitulated them [194]. Optogenetic targeting and stimulation of the IL-PFC was also found to produce both rapid and sustained antidepressant effects and to increase the number and function of dendritic spines. Recent rodent studies have further elaborated the cellular targets underlying these effects by examining the role of pyramidal neurons expressing dopamine receptor D1 and D2 (Drd1 and Drd2, respectively) [195]. Optogenetic activation of Drd1 pyramidal cells in the medial PFC resulted in rapid and sustained antidepressant effects, whereas stimulation of Drd2 neurons was found to be ineffective.
In rodents, the ketamine-induced amelioration of depressive-like behavior is particularly associated with acute changes in prefrontal circuit function, which is followed by increased dendritic spine formation [137]. The increase in or restoration of dendritic spines is not required for exerting ketamine’s immediate effects but are instead critical for sustaining the antidepressant effect over time. Moreover, interference with ketamine-induced prefrontal spine formation blocked effects on motivated escape behavior but had no influence on sucrose preference, indicating that spinogenesis in other brain areas may support other behaviors [137].
Ketamine also decreases the activation and burst firing of neurons in the lateral habenula (LHb), effects that were associated with acute antidepressant effects in congenitally helpless rats [196]. The local blockade of either NMDARs or low-voltage-sensitive T-type calcium channels in the LHb was found to be sufficient for the induction of rapid antidepressant effects. Essentially, these findings suggest that ketamine’s mood elevating effects result from the blockade of NMDAR-dependent LHb bursts, which disinhibits downstream monoaminergic reward centers [196].
Electrophysiological and metabolic measures
In terms of more general mechanisms, subanesthetic doses of ketamine increase metabolic activity in the prefrontal cortex in healthy volunteers, as measured by [18F]-fluorodeoxyglucose positron emission tomography (PET) [197]. These findings correspond well to other findings suggesting increased glutamate neurotransmission in the prefrontal cortex of healthy and depressed subjects receiving ketamine [198, 199]. The disinhibition hypothesis, which entails a decrease in the activity of GABAergic interneurons and disinhibition of excitatory pyramidal neurons, also provides a putative mechanism for the commonly observed increase in high-frequency gamma oscillations on cortical EEG following ketamine administration [172, 200–202]. Notably, Nugent et al. [202] found that large increases in gamma power were associated with better antidepressant outcomes in subjects with MDD with lower baseline gamma, whereas this relationship was inverse in subjects with MDD with higher baseline gamma. Ketamine also enhanced gamma responses to somatosensory stimuli at 230 min and day 1 in ketamine responders when compared with non-responders, suggesting that increases in synaptic strength coincide with antidepressant effects [203, 204]. A 7 T 1H-magnetic resonance spectroscopy study showed no difference in glutamate levels of the pregenual cingulate cortex 24 h post ketamine administration, suggesting that glutamate levels are not altered in the long term [205]. These studies, coupled with abundant molecular evidence, support the notion that subanesthetic ketamine acutely increases cortical excitation and glutamate activity, which drive subsequent synaptic alterations.
In rats, lower doses (60 mg/kg) of ketamine promote behavioral arousal along with theta-range EEG activity and induction of Fos immunoreactivity in the arousal system [206]. In contrast, rats treated with higher doses (150 mg/kg) of ketamine are briefly sedated, but signs of hyperarousal emerge once the acute effects of ketamine subside. Notably, the arousal-promoting effects of subanesthetic doses of ketamine are followed by the increase of delta frequency power during subsequent sleep [207]. Similar intriguing findings were also observed in human studies, in which increased sleep SWA the night following subanesthetic ketamine administration was associated with the therapeutic efficacy of the treatment [208]. The facilitation of SWA is also evident after sleep deprivation, electroconvulsive therapy, repetitive transcranial magnetic stimulation (rTMS), and nitrous oxide administration in humans—all treatments with rapid antidepressant potential [172, 209–211]. These observations suggest that the facilitation of SWA is a homeostatic response to the preceding cortical activation elicited by ketamine and other treatments potentially sharing rapid antidepressant effects [12, 209].
Clinical neuroimaging
The neuroimaging literature related to ketamine’s antidepressant effects has identified convergent brain regions of interest in the prefrontal cortex, including specific cortical areas such as the subgenual anterior cingulate cortex (sgACC) and the posterior cingulate cortex (PCC) as well as the hippocampus (for a thorough review, see Ionescu et al. [212]). The sgACC, in particular, has been a focus of numerous studies because earlier studies have found depressed patients to exhibit overactivity of the sgACC, which is suggested to normalize upon recovery after treatment [213–216]. A recent study in nonhuman primates demonstrated that ketamine is indeed able to reverse the depressive-like impairments and metabolic changes produced by the overactivation of the sgACC [217]. The hyperactivity of the sgACC has also been observed during task performance in depressed patients, where ketamine may act to normalize sgACC hyperactivity to positive incentives [218]. However, many findings related to the activity of the sgACC remain inconclusive. For example, PET studies have reported no change [219] or increased metabolism in the sgACC in response to ketamine in depressed patients [220]. One study compared ketamine and lanicemine, a promising putative rapid-acting antidepressant at the time, and found them to increase sgACC activation in depressed patients [221]. Since then, lanicemine development has been abandoned after a failure to meet trial endpoints [10]. Nevertheless, another trial found ketamine, but not lanicemine, to increase prefrontal global connectivity [222], hinting at a possible difference between effective and non-effective NMDAR antagonists. This effect of ketamine on connectivity has also been observed in other trials [223, 224] and hypothesized to represent the effects of ketamine on the induction of glutamate neurotransmission.
One focus of functional imaging studies is related to the activity and connectivity of the default-mode network (DMN), which correlates with the severity of depression and rumination [225–227]. A recent double-blind, placebo-controlled, crossover study demonstrated that the connectivity between insula and the DMN was normalized when measured 2 days after ketamine treatment [228]. This change was reversed after 10 days, which corresponds well with the duration of ketamine’s antidepressant effects. Moreover, a study examining ketamine’s effects on fMRI activity during an emotional processing task showed that participants with MDD exhibited greater brain activity than healthy controls after placebo infusion, whereas ketamine reduced brain activity to a level similar to that in placebo-treated healthy controls [229]. Reduction in activity was observed in large regions throughout the brain, including areas related to the DMN. Several studies by the same group support the notion of ketamine normalizing patterns of brain activity in MDD patients [202, 228, 230].
Emerging hypotheses of ketamine’s action
Understanding the precise mechanistic basis of ketamine’s antidepressant effects has been the target of hundreds of studies in the past decades. These studies have verified the ability of subanesthetic ketamine doses to boost glutamatergic firing and to increase AMPAR function, which continue to be among key areas of interest [7, 138]. For many researchers, the prevailing view is that ketamine has almost a unique ability to regulate a chain of molecular events connected with the facilitation of synaptic plasticity, ultimately steering functional activity towards the amelioration of depressive symptoms. This view has prompted the targeting of individual effectors within complex signaling pathways with the deterministic aim of isolating the root of antidepressant action. These efforts have not been entirely in vain because several studies have demonstrated the antidepressant-like behavioral actions of BDNF [231] as well as the direct activation of TrkB [232] and mTOR [233] in rodents. However, these perspectives may fail to appreciate the full complexity of the molecular signaling events associated with neuronal plasticity, where single effectors are part of a much larger web of activity-dependent molecular interactions. Most importantly, clinical evidence strongly connecting any of the proposed molecular targets to the amelioration of depressive symptoms is currently lacking.
The remarkably rapid yet relatively sustained antidepressant effects of ketamine—often emerging within hours and lasting for up to a week—are a challenging problem to understand. Notably, the recent discovery of the antidepressant-like effects of ketamine’s HNK metabolites have led to a hypothesis in which the gradual buildup of these metabolites, (2R,6R)-hydroxynorketamine in particular, has been suggested to explain ketamine’s antidepressant potency [134] (Fig. 4a). The HNK metabolites differ from the pharmacological effects of ketamine and challenge the notion of ketamine’s effects being dependent on NMDAR inhibition. Antidepressant-relevant concentrations of (2R,6R)-hydroxynorketamine do not block NMDAR function but still result in antidepressant-like responses in rodents [234]. This hypothesis, however, remains controversial in light of more recent preclinical [171, 235, 236] and clinical data [237, 238]. More specifically, recent studies evaluating the plasma levels of these metabolites after ketamine treatment reported contradictory observations—higher levels of 2R,6R-HNK were associated with less clinical improvement [237, 238]. These metabolites remain the topic of ongoing research and debate, while patient studies are still underway.
Fig. 4.

Emerging hypotheses of ketamine’s action. a The metabolism of ketamine to its hydroxynorketamine metabolites, and particularly (2R,6R)-hydroxynorketamine, is responsible for mediating the rapid antidepressant effects. b Ketamine disrupts neuronal homeostasis, which triggers self-correcting mechanisms. c Ketamine regulates both circadian and homeostatic processes of sleep to elicit antidepressant effects. d The hypothesis of encoding, consolidation and renormalization in depression (ENCORE-D) proposes that ketamine alters neural encoding of information, the consolidation of synaptic change, and the renormalization of synaptic strength during sleep to elicit acute and sustained antidepressant effects
Several new hypotheses emerged in the recent years explore the neurobiological phenomena underlying antidepressant effects instead of solely crediting ketamine, its metabolites, or any other specific molecular entity for the amelioration of depressive symptoms. It can be argued that because ketamine is rapidly metabolized, its lasting antidepressant effects must be the result of neurobiological adaptations triggered during the brief period of its acute pharmacological action. On the basis of the noteworthy findings of rapid antidepressant-like effects produced by NMDAR enhancers in rodents and those showing the antidepressant-like effects of the negative allosteric GABA-A receptor modulator MRK-015, Workman et al. [239] proposed that rapid-acting antidepressants engage mechanisms of homeostatic plasticity to treat depression (Fig. 4b). In particular, they highlight the ability of these treatments to either strongly increase or decrease neuronal activity, which causes neurons and neuronal circuits to homeostatically self-correct. In this context, ketamine and other putative treatments may act as disruptors of the homeostatic balance, triggering the activation of self-correcting homeostatic mechanisms that are otherwise insufficiently activated in MDD. Importantly, their work also discusses the idea that drug–target interactions should be considered as a means of inducing homeostatic responses within neurons, rather than solely focusing on the target (i.e., receptor–ligand) activity.
A substantially large number of molecular and cellular changes associated with ketamine are also associated with the physiological processes of sleep and wake [12]. While a general lack of consideration of sleep exists in both basic and clinical research involving ketamine’s antidepressant effects [11], some recent studies and hypotheses have begun to investigate the effects of ketamine from the perspective of the neurobiology of sleep and circadian rhythms. For example, a model by Duncan et al. [240] aims to uncover the effects of ketamine through the homeostatic and circadian components of sleep. In this context, treatment with ketamine leads not only to increased neuronal plasticity, slow-wave sleep (SWS), and enhanced sleep quality but also to modulation of circadian timing and output (Fig. 4c). These effects culminate in the reduction in the circadian mood component. Indeed, clinical studies have demonstrated an association between ketamine’s antidepressant effects and circadian rhythms [240, 241] as well as a correlation between ketamine-induced changes in the levels of BDNF, amount of SWS, quality of sleep, and subsequent mood changes in MDD [208, 242]. Moreover, it remains plausible that the antidepressant effects of ketamine are modulated by homeostatic sleep pressure and/or circadian time at the time of administration since cortical excitability in humans [243–245], and the anesthetic effects of ketamine in animal studies, are regulated by these factors [246, 247]. In any case, a significant translational gap exists due to the fact that most rodent species are nocturnal and sleep predominantly during the day when most drug treatments are given, whereas patients receive ketamine almost exclusively during the waking hours [11]. Understanding how sleep and circadian rhythms may influence treatment outcomes remains a very important theme for future studies in both basic and clinical research.
Building upon the themes of synaptic homeostasis and sleep, the hypothesis of encoding, consolidation, and renormalization in depression (ENCORE-D) proposes that ketamine’s rapid and sustained effects are produced and consolidated throughout wake and sleep in several phases [209] (Fig. 4d). It suggests that ketamine’s ability to increase cortical excitation triggers intrinsic mechanisms of synaptic plasticity both acutely and upon drug withdrawal, leading to the facilitated encoding of information and increase in synaptic strength. These changes are further consolidated during subsequent steps involving changes in transcription and protein synthesis. The final stage of ketamine-induced change is suggested to be reached on the night following treatment, during a period of deep sleep dominated by SWA. Here, ENCORE-D suggests that synapses in previously activated networks undergo renormalization of synaptic strength, a concept hypothesized within the framework of the the synaptic homeostasis hypothesis (SHY) [248, 249], ultimately giving rise to sustained alterations in network function. While remaining highly speculative and in need of experimental proof, the ENCORE-D framework provides several novel conceptual ideas about how the effects of rapid-acting antidepressants may be associated with the physiological mechanisms of wake and sleep in producing antidepressant outcomes [209].
Conclusions
The past 50 years have transformed ketamine from a powerful battlefield anesthetic to the wonder drug of modern psychiatry, with new indications still being uncovered. Intensive research efforts aimed at understanding the precise mechanisms underlying ketamine’s effects have resulted in important advances in our understanding of depression and stimulated new concepts of molecular and cellular neuropharmacology. However, as with anything new, the glimmer of ketamine may have distracted both basic and clinical researchers from addressing fundamental issues. Certain questions, such as whether increasing doses in patients that respond poorly to lower doses is beneficial and whether anesthetic doses lack antidepressant effects, remain unanswered. There are also no comprehensive comparisons of the effectiveness of different administration routes or the rate and time administration. Moreover, the circadian time, dose, and method of administration in animal studies often differ significantly from those in clinical practice and lack proper translational validation. Future studies aimed at addressing these and other basic questions will hopefully advance our understanding of the pharmacological and neurobiological mechanisms of ketamine in the treatment of psychiatric disorders.
Acknowledgements
I wish to thank Dr. Tomi Rantamäki, Dr. Piia Kohtala, and Dr. Niko Lindholm for their valuable comments on the manuscript.
Abbreviations
- AMPAR
α-Amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor
- BDNF
Brain-derived neurotrophic factor
- BPRS
Brief Psychiatric Rating Scale
- CADSS
Clinician-Administered Dissociative States Scale
- DAT
Dopamine reuptake transporter
- DHNK
Dehydronorketamine
- DMN
Default-mode network
- Drd1
Dopamine receptor D1
- eEF2
Eukaryotic elongation factor 2
- ECT
Electroconvulsive therapy
- EEG
Electroencephalogram
- ENCORE-D
Encoding, consolidation, and renormalization in depression
- ERK
Extracellular signal-regulated kinase
- FST
Forced swimming test
- GABA
Gamma-aminobutyric acid
- GSK3β
Glycogen synthase kinase 3 beta
- HCN
Hyperpolarization-activated cyclic nucleotide
- HDRS
Hamilton Depression Rating Scale
- HMS
Hood’s Mysticism Scale
- HNK
Hydroxynorketamine
- IEG
Immediate early gene
- IL-PFC
Infralimbic prefrontal cortex
- im
Intramuscular
- ip
Intraperitoneal
- iv
Intravenous
- LHb
Lateral habenula
- LTP
Long-term potentiation
- MADRS
Montgomery-Åsberg Rating Scale
- MAPK
Mitogen-activated protein kinase
- MDD
Major depressive disorder
- MKP1
Mitogen-activated protein kinase phosphatase 1
- mTOR
Mammalian target of rapamycin
- NET
Noradrenaline reuptake transporter
- NK
Norketamine
- NMDAR
N-Methyl-d-aspartate receptor
- PCC
Posterior cingulate cortex
- PCP
Phencyclidine
- PET
Positron emission tomography
- PSD95
Postsynaptic density protein 95
- p70-S6K
P70-S6 kinase
- rTMS
Repetitive transcranial magnetic stimulation
- sc
Subcutaneous
- SERT
Serotonin reuptake transporter
- sgACC
Subgenual anterior cingulate cortex
- SWA
Slow-wave activity
- SWS
Slow-wave sleep
- TrkB
Tropomyosin receptor kinase B
- VGSC
Voltage-gated sodium channel
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital.. This work has been supported by the Orion Research Foundation, the Sigrid Jusélius Foundation, and the Emil Aaltonen Foundation. The funding sources were not involved in the writing process or the decision to submit the work.
Compliance with ethical standards
Conflict of interest
S.K. is listed as a co-inventor on a patent application wherein new tools enabling the development of rapid-acting antidepressants and the efficacy monitors thereof are disclosed based on the basic principles of ENCORE-D. S.K. has assigned his patent rights to the University of Helsinki but will share a percentage of any royalties that may be received by the University of Helsinki.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Frohlich J, Van Horn JD. Reviewing the ketamine model for schizophrenia. J Psychopharmacol. 2014;28:287–302. doi: 10.1177/0269881113512909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 3.Rantamäki T. Searching for ketamine’s antidepressant mechanisms: from synaptic plasticity to dentate gyrus cell proliferation. Acta Physiol. 2019;225:e13252. doi: 10.1111/apha.13252. [DOI] [PubMed] [Google Scholar]
- 4.Sanacora G, Frye MA, McDonald W, Mathew SJ, Turner MS, Schatzberg AF, et al. A consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry. 2017;74:399–405. doi: 10.1001/jamapsychiatry.2017.0080. [DOI] [PubMed] [Google Scholar]
- 5.Corriger A, Pickering G. Ketamine and depression: a narrative review. Drug Des Devel Ther. 2019;13:3051–3067. doi: 10.2147/DDDT.S221437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53:649–659. doi: 10.1016/S0006-3223(03)00231-2. [DOI] [PubMed] [Google Scholar]
- 7.Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23:801–811. doi: 10.1038/mp.2017.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quiroz JA, Tamburri P, Deptula D, Banken L, Beyer U, Rabbia M, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry. 2016;73:675–684. doi: 10.1001/jamapsychiatry.2016.0838. [DOI] [PubMed] [Google Scholar]
- 9.Ibrahim L, DiazGranados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, et al. A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32:551–557. doi: 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, et al. Adjunctive lanicemine (AZD6765) in patients with major depressive disorder and history of inadequate response to antidepressants: a randomized placebo-controlled study. Neuropsychopharmacology. 2017;42:844–853. doi: 10.1038/npp.2016.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alitalo O, Saarreharju R, Zarate CA, Kohtala S, Rantamaki T. A wake-up call—revealing the oversight of sleep physiology and related translational discrepancies in studies of rapid-acting antidepressants. MedRxiv. 2020 doi: 10.1101/2020.09.29.20204008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohtala S, Alitalo O, Rosenholm M, Rozov S, Rantamäki T. Time is of the essence: coupling sleep-wake and circadian neurobiology to the antidepressant effects of ketamine. Pharmacol Ther. 2020 doi: 10.1016/j.pharmthera.2020.107741. [DOI] [PubMed] [Google Scholar]
- 13.Denomme N. The Domino effect: Ed Domino’s early studies of psychoactive drugs. J Psychoactive Drugs. 2018;50:298–305. doi: 10.1080/02791072.2018.1506599. [DOI] [PubMed] [Google Scholar]
- 14.Domino EF, Chodoff P, Corssen G. Pharmacologic effects of CI-581, a new dissociative anesthetic, in man. Clin Pharmacol Ther. 1965;6:279–291. doi: 10.1002/cpt196563279. [DOI] [PubMed] [Google Scholar]
- 15.Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. 2018;70:621–660. doi: 10.1124/pr.117.015198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolshakov KV, Gmiro VE, Tikhonov DB, Magazanik LG. Determinants of trapping block of N-methyl-D-aspartate receptor channels. J Neurochem. 2003;87:56–65. doi: 10.1046/j.1471-4159.2003.01956.x. [DOI] [PubMed] [Google Scholar]
- 17.MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, et al. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J Physiol. 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 19.Sinner B, Graf BM. Modern anesthetics. Berlin: Springer Berlin Heidelberg; 2011. Ketamine; pp. 313–333. [Google Scholar]
- 20.Haeseler G, Tetzlaff D, Bufler J, Dengler R, Münte S, Hecker H, et al. Blockade of voltage-operated neuronal and skeletal muscle sodium channels by S(+)- and R(-)-ketamine. Anesth Analg. 2003;96:1019–1026. doi: 10.1213/01.ANE.0000052513.91900.D5. [DOI] [PubMed] [Google Scholar]
- 21.Zhou C, Douglas JE, Kumar NN, Shu S, Bayliss DA, Chen X. Forebrain HCN1 channels contribute to hypnotic actions of ketamine. Anesthesiology. 2013;118:785–795. doi: 10.1097/ALN.0b013e318287b7c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clements JA, Nimmo WS. Pharmacokinetics and analgesic effect of ketamine in man. Br J Anaesth. 1981;53:27–30. doi: 10.1093/bja/53.1.27. [DOI] [PubMed] [Google Scholar]
- 23.Mathew SJ, Zarate CA. Ketamine for treatment-resistant depression. Cham: Springer International Publishing; 2016. [Google Scholar]
- 24.Adams JD, Baillie TA, Trevor AJ, Castagnoli N. Studies on the biotransformation of ketamine 1—identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spectrom. 1981;8:527–538. doi: 10.1002/bms.1200081103. [DOI] [PubMed] [Google Scholar]
- 25.Woolf TF, Adams JD. Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica. 1987;17:839–847. doi: 10.3109/00498258709043993. [DOI] [PubMed] [Google Scholar]
- 26.Maxwell CR, Ehrlichman RS, Liang Y, Trief D, Kanes SJ, Karp J, et al. Ketamine produces lasting disruptions in encoding of sensory stimuli. J Pharmacol Exp Ther. 2006;316:315–324. doi: 10.1124/jpet.105.091199. [DOI] [PubMed] [Google Scholar]
- 27.Zarate CA, Brutsche N, Laje G, Luckenbaugh DA, Venkata SLV, Ramamoorthy A, et al. Relationship of Ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72:331–338. doi: 10.1016/j.biopsych.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Venkata SLV, Moaddel R, Luckenbaugh DA, Brutsche NE, Ibrahim L, et al. Simultaneous population pharmacokinetic modelling of ketamine and three major metabolites in patients with treatment-resistant bipolar depression. Br J Clin Pharmacol. 2012;74:304–314. doi: 10.1111/j.1365-2125.2012.04198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portmann S, Kwan HY, Theurillat R, Schmitz A, Mevissen M, Thormann W. Enantioselective capillary electrophoresis for identification and characterization of human cytochrome P450 enzymes which metabolize ketamine and norketamine in vitro. J Chromatogr A. 2010;1217:7942–7948. doi: 10.1016/j.chroma.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 30.Garfield JM, Garfield FB, Stone JG, Hopkins D, Johns LA. A comparison of psychologic responses to ketamine and thiopental–nitrous oxide–halothane anesthesia. Anesthesiology. 1972;36:329–338. doi: 10.1097/00000542-198003000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Ballesteros JJ, Huang P, Patel SR, Eskandar EN, Ishizawa Y. Dynamics of Ketamine-induced loss and return of consciousness across primate neocortex. Anesthesiology. 2020;132:750–762. doi: 10.1097/ALN.0000000000003159. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder KE, Irwin ZT, Gaidica M, Bentley JN, Patil PG, Mashour GA, et al. Disruption of corticocortical information transfer during ketamine anesthesia in the primate brain. Neuroimage. 2016;134:459–465. doi: 10.1016/j.neuroimage.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sleigh J, Pullon RM, Vlisides PE, Warnaby CE. Electroencephalographic slow wave dynamics and loss of behavioural responsiveness induced by ketamine in human volunteers. Br J Anaesth. 2019;123:592–600. doi: 10.1016/j.bja.2019.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vlisides PE, Bel-Bahar T, Lee U, Li D, Kim H, Janke E, et al. Neurophysiologic correlates of ketamine sedation and anesthesia. Anesthesiology. 2017;127:58–69. doi: 10.1097/ALN.0000000000001671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marland S, Ellerton J, Andolfatto G, Strapazzon G, Thomassen O, Brandner B, et al. Ketamine: use in anesthesia. CNS Neurosci Ther. 2013;19:381–389. doi: 10.1111/cns.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White PF, Way WL, Trevor AJ. Ketamine—its pharmacology and therapeutic uses. Anesthesiology. 1982;56:119–136. doi: 10.1097/00000542-198202000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Mees L, Fidler J, Kreuzer M, Fu J, Pardue MT, García PS. Faster emergence behavior from ketamine/xylazine anesthesia with atipamezole versus yohimbine. PLoS ONE. 2018;13:e0199087. doi: 10.1371/journal.pone.0199087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrade C. Ketamine for depression, 4: In what dose, at what rate, by what route, for how long, and at what frequency? J Clin Psychiatry. 2017;78:e852–e857. doi: 10.4088/JCP.17f11738. [DOI] [PubMed] [Google Scholar]
- 39.Guldner GT, Petinaux B, Clemens P, Foster S, Antoine S. Ketamine for procedural sedation and analgesia by nonanesthesiologists in the field: a review for military health care providers. Mil Med. 2006;171:484–490. doi: 10.7205/MILMED.171.6.484. [DOI] [PubMed] [Google Scholar]
- 40.Gao M, Rejaei D, Liu H. Ketamine use in current clinical practice. Acta Pharmacol Sin. 2016;37:865–872. doi: 10.1038/aps.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonanno FG. Ketamine in war/tropical surgery (a final tribute to the racemic mixture) Injury. 2002;33:323–327. doi: 10.1016/S0020-1383(01)00209-1. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy RM, Porter FL, Miller JP, Jaffe DM. Comparison of fentanyl/midazolam with ketamine/midazolam for pediatric orthopedic emergencies. Pediatrics. 1998;102:956–963. doi: 10.1542/peds.102.4.956. [DOI] [PubMed] [Google Scholar]
- 43.Motov S, Rockoff B, Cohen V, Pushkar I, Likourezos A, McKay C, et al. Intravenous subdissociative-dose ketamine versus morphine for analgesia in the emergency department: a randomized controlled trial. Ann Emerg Med. 2015;66:222–229. doi: 10.1016/j.annemergmed.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Ahern TL, Herring AA, Stone MB, Frazee BW. Effective analgesia with low-dose ketamine and reduced dose hydromorphone in ED patients with severe pain. Am J Emerg Med. 2013;31:847–51. https://doi.org/10.1016/j.ajem.2013.02.008. [DOI] [PubMed]
- 45.Brinck E, Tiippana E, Heesen M, Bell RF, Straube S, Moore RA, et al. Perioperative intravenous ketamine for acute postoperative pain in adults. Cochrane Database Syst Rev. 2018 doi: 10.1002/14651858.CD012033.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaparro LE, Smith SA, Moore RA, Wiffen PJ, Gilron I. Pharmacotherapy for the prevention of chronic pain after surgery in adults. Cochrane Database Syst Rev. 2013 doi: 10.1002/14651858.CD008307.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dualé C, Sibaud F, Guastella V, Vallet L, Gimbert YA, Taheri H, et al. Perioperative ketamine does not prevent chronic pain after thoracotomy. Eur J Pain. 2009;13:497–505. doi: 10.1016/j.ejpain.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 48.Ryu HG, Lee CJ, Kim YT, Bahk JH. Preemptive low-dose epidural ketamine for preventing chronic postthoracotomy pain: a prospective, double-blinded, randomized, clinical trial. Clin J Pain. 2011;27:304–308. doi: 10.1097/AJP.0b013e3181fd5187. [DOI] [PubMed] [Google Scholar]
- 49.McNicol ED, Schumann R, Haroutounian S. A systematic review and meta-analysis of ketamine for the prevention of persistent post-surgical pain. Acta Anaesthesiol Scand. 2014;58:1199–1213. doi: 10.1111/aas.12377. [DOI] [PubMed] [Google Scholar]
- 50.Niesters M, Martini C, Dahan A. Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol. 2014;77:357–367. doi: 10.1111/bcp.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eide PK, Stubhaug A, Øye I, Breivik H. Continuous subcutaneous administration of the N-methyl-d-aspartic acid (NMDA) receptor antagonist ketamine in the treatment of post-herpetic neuralgia. Pain. 1995;61:221–228. doi: 10.1016/0304-3959(94)00182-E. [DOI] [PubMed] [Google Scholar]
- 52.Eichenberger U, Neff F, Sveticic G, Björgo S, Petersen-Felix S, Arendt-Nielsen L, et al. Chronic phantom limb pain: the effects of calcitonin, ketamine, and their combination on pain and sensory thresholds. Anesth Analg. 2008;106:1265–1273. doi: 10.1213/ane.0b013e3181685014. [DOI] [PubMed] [Google Scholar]
- 53.Mitra S, Kazal S. Oral ketamine for phantom limb pain: an option for challenging cases. Indian J Anaesth. 2015;59:446. doi: 10.4103/0019-5049.160963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mercadante S, Arcuri E, Tirelli W, Casuccio A. Analgesic effect of intravenous ketamine in cancer patients on morphine therapy. J Pain Symptom Manag. 2000;20:246–252. doi: 10.1016/S0885-3924(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 55.Afridi SK, Giffin NJ, Kaube H, Goadsby PJ. A randomized controlled trial of intranasal ketamine in migraine with prolonged aura. Neurology. 2013;80:642–647. doi: 10.1212/WNL.0b013e3182824e66. [DOI] [PubMed] [Google Scholar]
- 56.Kissin I, Bright CA, Bradley EL. The effect of ketamine on opioid-induced acute tolerance: can it explain reduction of opioid consumption with ketamine-opioid analgesic combinations? Anesth Analg. 2000;91:1483–1488. doi: 10.1097/00000539-200012000-00035. [DOI] [PubMed] [Google Scholar]
- 57.Chazan S, Ekstein MP, Marouani N, Weinbroum AA. Ketamine for acute and subacute pain in opioid-tolerant patients. J Opioid Manag. 2008;4:173–180. doi: 10.5055/jom.2008.0023. [DOI] [PubMed] [Google Scholar]
- 58.Robinson MJ, Edwards SE, Iyengar S, Bymaster F, Clark M, Katon W. Depression and pain. Front Biosci. 2009;14:5031–5051. doi: 10.2741/3585. [DOI] [PubMed] [Google Scholar]
- 59.Wolfson PE. Psychedelic experiential pharmacology pioneering clinical explorations with salvador roquet (how I came to all of this: ketamine, admixtures and adjuvants, don juan and carlos castaneda too): an interview with Richard Yensen. Int J Transpers Stud. 2014;33:160–174. doi: 10.24972/ijts.2014.33.2.160. [DOI] [Google Scholar]
- 60.Khorramzadeh E, Lotfy AO. The use of ketamine in psychiatry. Psychosomatics. 1973;14:344–346. doi: 10.1016/S0033-3182(73)71306-2. [DOI] [PubMed] [Google Scholar]
- 61.Winship IR, Dursun SM, Baker GB, Balista PA, Kandratavicius L, Maia-de-Oliveira JP, et al. An overview of animal models related to schizophrenia. Can J Psychiatry. 2019;64:5–17. doi: 10.1177/0706743718773728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feder A, Rutter SB, Schiller D, Charney DS. The emergence of ketamine as a novel treatment for posttraumatic stress disorder. Adv Pharmacol. 2020;89:261–286. doi: 10.1016/bs.apha.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 63.Jones JL, Mateus CF, Malcolm RJ, Brady KT, Back SE. Efficacy of ketamine in the treatment of substance use disorders: a systematic review. Front Psychiatry. 2018 doi: 10.3389/fpsyt.2018.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kirkpatrick H, Heller G. Post-traumatic stress disorder: theory and treatment update. Int J Psychiatry Med. 2014;47:337–346. doi: 10.2190/PM.47.4.h. [DOI] [PubMed] [Google Scholar]
- 66.Hoskins M, Pearce J, Bethell A, Dankova L, Barbui C, Tol WA, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206:93–100. doi: 10.1192/bjp.bp.114.148551. [DOI] [PubMed] [Google Scholar]
- 67.Schönenberg M, Reichwald U, Domes G, Badke A, Hautzinger M. Ketamine aggravates symptoms of acute stress disorder in a naturalistic sample of accident victims. J Psychopharmacol. 2008;22:493–497. doi: 10.1177/0269881107082481. [DOI] [PubMed] [Google Scholar]
- 68.Schönenberg M, Reichwald U, Domes G, Badke A, Hautzinger M. Effects of peritraumatic ketamine medication on early and sustained posttraumatic stress symptoms in moderately injured accident victims. Psychopharmacology. 2005;182:420–425. doi: 10.1007/s00213-005-0094-4. [DOI] [PubMed] [Google Scholar]
- 69.McGhee LL, Maani CV, Garza TH, Gaylord KM, Black IH. The correlation between ketamine and posttraumatic stress disorder in burned service members. J Trauma Inj Infect Crit Care. 2008;64:S195–S199. doi: 10.1097/TA.0b013e318160ba1d. [DOI] [PubMed] [Google Scholar]
- 70.Highland KB, Soumoff AA, Spinks EA, Kemezis PA, Buckenmaier CC. Ketamine administration during hospitalization is not associated with posttraumatic stress disorder outcomes in military combat casualties. Anesth Analg. 2020;130:402–408. doi: 10.1213/ANE.0000000000004327. [DOI] [PubMed] [Google Scholar]
- 71.McGowan JC, LaGamma CT, Lim SC, Tsitsiklis M, Neria Y, Brachman RA, et al. Prophylactic ketamine attenuates learned fear. Neuropsychopharmacology. 2017;42:1577–1589. doi: 10.1038/npp.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juven-Wetzler A, Cohen H, Kaplan Z, Kohen A, Porat O, Zohar J. Immediate ketamine treatment does not prevent posttraumatic stress responses in an animal model for PTSD. Eur Neuropsychopharmacol. 2014;24:469–479. doi: 10.1016/j.euroneuro.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 73.Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. 2014;71:681–688. doi: 10.1001/jamapsychiatry.2014.62. [DOI] [PubMed] [Google Scholar]
- 74.Albott CS, Lim KO, Forbes MK, Erbes C, Tye SJ, Grabowski JG, et al. Efficacy, safety, and durability of repeated ketamine infusions for comorbid posttraumatic stress disorder and treatment-resistant depression. J Clin Psychiatry. 2018 doi: 10.4088/JCP.17m11634. [DOI] [PubMed] [Google Scholar]
- 75.Feder A, Costi S, Rutter SB, Collins AB, Govindarajulu U, Jha MK, et al. A randomized controlled trial of repeated ketamine administration for chronic posttraumatic stress disorder. Am J Psychiatry. 2021;178:193–202. doi: 10.1176/appi.ajp.2020.20050596. [DOI] [PubMed] [Google Scholar]
- 76.Rezvani AH, Levin ED, Cauley M, Getachew B, Tizabi Y. Ketamine differentially attenuates alcohol intake in male versus female alcohol preferring (P) rats. J Drug Alcohol Res. 2017;6:236030. doi: 10.4303/jdar/236030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruda-Kucerova J, Babinska Z, Luptak M, Getachew B, Tizabi Y. Both ketamine and NBQX attenuate alcohol drinking in male Wistar rats. Neurosci Lett. 2018;666:175–180. doi: 10.1016/j.neulet.2017.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKendrick G, Garrett H, Jones HE, McDevitt DS, Sharma S, Silberman Y, et al. Ketamine blocks morphine-induced conditioned place preference and anxiety-like behaviors in mice. Front Behav Neurosci. 2020;14:1–13. doi: 10.3389/fnbeh.2020.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krupitsky EM, Grinenko AY. Ketamine psychedelic therapy (KPT): a review of the results of ten years of research. J Psychoactive Drugs. 1997;29:165–183. doi: 10.1080/02791072.1997.10400185. [DOI] [PubMed] [Google Scholar]
- 80.Krupitsky E, Burakov A, Romanova T, Dunaevsky I, Strassman R, Grinenko A. Ketamine psychotherapy for heroin addiction: immediate effects and two-year follow-up. J Subst Abuse Treat. 2002;23:273–283. doi: 10.1016/S0740-5472(02)00275-1. [DOI] [PubMed] [Google Scholar]
- 81.Krupitsky EM, Burakov AM, Dunaevsky IV, Romanova TN, Slavina TY, Grinenko AY. Single versus repeated sessions of ketamine-assisted psychotherapy for people with heroin dependence. J Psychoactive Drugs. 2007;39:13–19. doi: 10.1080/02791072.2007.10399860. [DOI] [PubMed] [Google Scholar]
- 82.Dakwar E, Levin F, Foltin RW, Nunes EV, Hart CL. The effects of subanesthetic ketamine infusions on motivation to quit and cue-induced craving in cocaine-dependent research volunteers. Biol Psychiatry. 2014;76:40–46. doi: 10.1016/j.biopsych.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dakwar E, Levin F, Hart CL, Basaraba C, Choi J, Pavlicova M, et al. A single ketamine infusion combined with motivational enhancement therapy for alcohol use disorder: a randomized midazolam-controlled pilot trial. Am J Psychiatry. 2020;177:125–133. doi: 10.1176/appi.ajp.2019.19070684. [DOI] [PubMed] [Google Scholar]
- 84.Dakwar E, Nunes EV, Hart CL, Foltin RW, Mathew SJ, Carpenter KM, et al. A single ketamine infusion combined with mindfulness-based behavioral modification to treat cocaine dependence: a randomized clinical trial. Am J Psychiatry. 2019;176:923–930. doi: 10.1176/appi.ajp.2019.18101123. [DOI] [PubMed] [Google Scholar]
- 85.Azhari N, Hu H, O’Malley KY, Blocker ME, Levin FR, Dakwar E. Ketamine-facilitated behavioral treatment for cannabis use disorder: a proof of concept study. Am J Drug Alcohol Abuse. 2020 doi: 10.1080/00952990.2020.1808982. [DOI] [PubMed] [Google Scholar]
- 86.Rothberg RL, Azhari N, Haug NA, Dakwar E. Mystical-type experiences occasioned by ketamine mediate its impact on at-risk drinking: results from a randomized, controlled trial. J Psychopharmacol. 2020 doi: 10.1177/0269881120970879. [DOI] [PubMed] [Google Scholar]
- 87.Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, Aanhet Rot M, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74:250–256. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 89.Chen M-H, Lin W-C, Tu P-C, Li C-T, Bai Y-M, Tsai S-J, et al. Antidepressant and antisuicidal effects of ketamine on the functional connectivity of prefrontal cortex-related circuits in treatment-resistant depression: a double-blind, placebo-controlled, randomized, longitudinal resting fMRI study. J Affect Disord. 2019;259:15–20. doi: 10.1016/j.jad.2019.08.022. [DOI] [PubMed] [Google Scholar]
- 90.Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression. JAMA Psychiatry. 2018;75:139. doi: 10.1001/jamapsychiatry.2017.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lapidus KAB, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76:970–976. doi: 10.1016/j.biopsych.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arabzadeh S, Hakkikazazi E, Shahmansouri N, Tafakhori A, Ghajar A, Jafarinia M, et al. Does oral administration of ketamine accelerate response to treatment in major depressive disorder? Results of a double-blind controlled trial. J Affect Disord. 2018;235:236–241. doi: 10.1016/j.jad.2018.02.056. [DOI] [PubMed] [Google Scholar]
- 93.Domany Y, Bleich-Cohen M, Tarrasch R, Meidan R, Litvak-Lazar O, Stoppleman N, et al. Repeated oral ketamine for out-patient treatment of resistant depression: randomised, double-blind, placebo-controlled, proof-of-concept study. Br J Psychiatry. 2019;214:20–26. doi: 10.1192/bjp.2018.196. [DOI] [PubMed] [Google Scholar]
- 94.Walter M, Li S, Demenescu LR. Multistage drug effects of ketamine in the treatment of major depression. Eur Arch Psychiatry Clin Neurosci. 2014;264:55–65. doi: 10.1007/s00406-014-0535-3. [DOI] [PubMed] [Google Scholar]
- 95.Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A Randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zarate CA, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71:939–946. doi: 10.1016/j.biopsych.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ballard ED, Ionescu DF, Vande Voort JL, Niciu MJ, Richards EM, Luckenbaugh DA, et al. Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J Psychiatr Res. 2014;58:161–166. doi: 10.1016/j.jpsychires.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shiroma PR, Thuras P, Wels J, Albott CS, Erbes C, Tye S, et al. A randomized, double-blind, active placebo-controlled study of efficacy, safety, and durability of repeated vs single subanesthetic ketamine for treatment-resistant depression. Transl Psychiatry. 2020;10:206. doi: 10.1038/s41398-020-00897-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vidal S, Gex-Fabry M, Bancila V, Michalopoulos G, Warrot D, Jermann F, et al. Efficacy and safety of a rapid intravenous injection of ketamine 0.5 mg/kg in treatment-resistant major depression: an open 4-week longitudinal study. J Clin Psychopharmacol. 2018;38:590–597. doi: 10.1097/JCP.0000000000000960. [DOI] [PubMed] [Google Scholar]
- 101.Loo CK, Gálvez V, O’Keefe E, Mitchell PB, Hadzi-Pavlovic D, Leyden J, et al. Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatr Scand. 2016;134:48–56. doi: 10.1111/acps.12572. [DOI] [PubMed] [Google Scholar]
- 102.Lai R, Katalinic N, Glue P, Somogyi AA, Mitchell PB, Leyden J, et al. Pilot dose-response trial of I.V. ketamine in treatment-resistant depression. World J Biol Psychiatry. 2014;15:579–584. doi: 10.3109/15622975.2014.922697. [DOI] [PubMed] [Google Scholar]
- 103.Xu Y, Hackett M, Carter G, Loo C, Gálvez V, Glozier N, et al. Effects of low-dose and very low-dose ketamine among patients with major depression: a systematic review and meta-analysis. Int J Neuropsychopharmacol. 2016;19:1–15. doi: 10.1093/ijnp/pyv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD) Mol Psychiatry. 2020;25:1592–1603. doi: 10.1038/s41380-018-0256-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Correia-Melo FS, Leal GC, Vieira F, Jesus-Nunes AP, Mello RP, Magnavita G, et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: a randomized, double-blind, non-inferiority study. J Affect Disord. 2020;264:527–534. doi: 10.1016/j.jad.2019.11.086. [DOI] [PubMed] [Google Scholar]
- 106.Singh JB, Fedgchin M, Daly E, Xi L, Melman C, De Bruecker G, et al. Intravenous esketamine in adult treatment-resistant depression: a double-blind, double-randomization, placebo-controlled study. Biol Psychiatry. 2016;80:424–431. doi: 10.1016/j.biopsych.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 107.Daly EJ, Trivedi MH, Janik A, Li H, Zhang Y, Li X, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76:893–903. doi: 10.1001/jamapsychiatry.2019.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ochs-Ross R, Daly EJ, Zhang Y, Lane R, Lim P, Morrison RL, et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment-resistant depression—TRANSFORM-3. Am J Geriatr Psychiatry. 2020;28:121–141. doi: 10.1016/j.jagp.2019.10.008. [DOI] [PubMed] [Google Scholar]
- 109.Fedgchin M, Trivedi M, Daly EJ, Melkote R, Lane R, Lim P, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1) Int J Neuropsychopharmacol. 2019;22:616–630. doi: 10.1093/ijnp/pyz039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gálvez V, Li A, Huggins C, Glue P, Martin D, Somogyi AA, et al. Repeated intranasal ketamine for treatment-resistant depression—the way to go? Results from a pilot randomised controlled trial. J Psychopharmacol. 2018 doi: 10.1177/0269881118760660. [DOI] [PubMed] [Google Scholar]
- 111.Malinovsky JM, Servin F, Cozian A, Lepage JY, Pinaud M. Ketamine and norketamine plasma concentrations after iv, nasal and rectal administration in children. Br J Anaesth. 1996;77:203–207. doi: 10.1093/bja/77.2.203. [DOI] [PubMed] [Google Scholar]
- 112.Dadiomov D. Dissociating the clinical role and economic value of intranasal esketamine. J Manag Care Spec Pharm. 2020;26:20–22. doi: 10.18553/jmcp.2020.26.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Canuso CM, Singh JB, Fedgchin M, Alphs L, Lane R, Lim P, et al. Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am J Psychiatry. 2018;175:620–630. doi: 10.1176/appi.ajp.2018.17060720. [DOI] [PubMed] [Google Scholar]
- 114.Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry. 2019;176:428–438. doi: 10.1176/appi.ajp.2019.19020172. [DOI] [PubMed] [Google Scholar]
- 115.Schatzberg AF. A word to the wise about intranasal esketamine. Am J Psychiatry. 2019;176:422–424. doi: 10.1176/appi.ajp.2019.19040423. [DOI] [PubMed] [Google Scholar]
- 116.Drevets WC, Singh JB, Hough D, Daly EJ, Popova V, Manji H. Comment on a word to the wise about intranasal esketamine. Am J Psychiatry. 2019;176:856–857. doi: 10.1176/appi.ajp.2019.19070688. [DOI] [PubMed] [Google Scholar]
- 117.Schatzberg AF. More thoughts on intranasal esketamine: response to Drevets et al. Am J Psychiatry. 2019;176:858–859. doi: 10.1176/appi.ajp.2019.19070688r. [DOI] [PubMed] [Google Scholar]
- 118.Gastaldon C, Papola D, Ostuzzi G, Barbui C. Esketamine for treatment resistant depression: a trick of smoke and mirrors? Epidemiol Psychiatr Sci. 2020;29:e79. doi: 10.1017/S2045796019000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dore J, Turnipseed B, Dwyer S, Turnipseed A, Andries J, Ascani G, et al. Ketamine assisted psychotherapy (KAP): patient demographics, clinical data and outcomes in three large practices administering ketamine with psychotherapy. J Psychoactive Drugs. 2019;51:189–198. doi: 10.1080/02791072.2019.1587556. [DOI] [PubMed] [Google Scholar]
- 120.Roseman L, Nutt DJ, Carhart-Harris RL. Quality of acute psychedelic experience predicts therapeutic efficacy of psilocybin for treatment-resistant depression. Front Pharmacol. 2018;8:974. doi: 10.3389/fphar.2017.00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kaelen M, Giribaldi B, Raine J, Evans L, Timmerman C, Rodriguez N, et al. The hidden therapist: evidence for a central role of music in psychedelic therapy. Psychopharmacology. 2018;235:505–519. doi: 10.1007/s00213-017-4820-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carhart-Harris RL, Roseman L, Haijen E, Erritzoe D, Watts R, Branchi I, et al. Psychedelics and the essential importance of context. J Psychopharmacol. 2018;32:725–731. doi: 10.1177/0269881118754710. [DOI] [PubMed] [Google Scholar]
- 123.Sos P, Klirova M, Novak T, Kohutova B, Horacek J, Palenice T. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuroendocrinol Lett. 2013;34:287–293. [PubMed] [Google Scholar]
- 124.Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche NE, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? J Affect Disord. 2014;159:56–61. doi: 10.1016/j.jad.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Niciu MJ, Shovestul BJ, Jaso BA, Farmer C, Luckenbaugh DA, Brutsche NE, et al. Features of dissociation differentially predict antidepressant response to ketamine in treatment-resistant depression. J Affect Disord. 2018;232:310–315. doi: 10.1016/j.jad.2018.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [1H]-MRS. Psychiatry Res Neuroimaging. 2011;191:122–127. doi: 10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dakwar E, Anerella C, Hart CL, Levin FR, Mathew SJ, Nunes EV. Therapeutic infusions of ketamine: do the psychoactive effects matter? Drug Alcohol Depend. 2014;136:153–157. doi: 10.1016/j.drugalcdep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stocker K, Hasler G, Hartmann M. The altered-state-of-consciousness aspect of a feeling of lightness is reported to be associated with antidepressant benefits by depressed individuals receiving ketamine infusions: a systematic analysis of internet video testimonials. Psychother Psychosom. 2019;88:182–183. doi: 10.1159/000497441. [DOI] [PubMed] [Google Scholar]
- 129.Acevedo-Diaz EE, Cavanaugh GW, Greenstein D, Kraus C, Kadriu B, Park L, et al. Can ‘floating’ predict treatment response to ketamine? Data from three randomized trials of individuals with treatment-resistant depression. J Psychiatr Res. 2020;130:280–285. doi: 10.1016/j.jpsychires.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185:1–10. doi: 10.1016/0014-2999(90)90204-J. [DOI] [PubMed] [Google Scholar]
- 131.Skolnick P, Layer RT, Popik P, Nowak G, Paul IA, Trullas R. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry. 1996;29:23–26. doi: 10.1055/s-2007-979537. [DOI] [PubMed] [Google Scholar]
- 132.Abdallah CG, Sanacora G, Duman RS, Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: is it glutamate inhibition or activation? Pharmacol Ther. 2018;190:148–158. doi: 10.1016/j.pharmthera.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Crane GE. Cycloserine as an antidepressant agent. Am J Psychiatry. 1959;115:1025–1026. doi: 10.1176/ajp.115.11.1025. [DOI] [PubMed] [Google Scholar]
- 134.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–486. doi: 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beurel E, Song L, Jope R. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol Psychiatry. 2011;16:1068–1070. doi: 10.1038/mp.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science (80-) 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science (80-). 2019;364:8078. doi: 10.1126/science.aat8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zanos P, Thompson SM, Duman RS, Zarate CA, Gould TD. Convergent mechanisms underlying rapid antidepressant action. CNS Drugs. 2018;32:197–227. doi: 10.1007/s40263-018-0492-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, et al. Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology. 2015;232:4325–4335. doi: 10.1007/s00213-015-4062-3. [DOI] [PubMed] [Google Scholar]
- 140.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 141.Duman RS. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 142.Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22:238–249. doi: 10.1038/nm.4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Miller OH, Moran JT, Hall BJ. Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: direct inhibition and disinhibition. Neuropharmacology. 2016;100:17–26. doi: 10.1016/j.neuropharm.2015.07.028. [DOI] [PubMed] [Google Scholar]
- 146.Duman RS, Aghajanian GK. synaptic dysfunction in depression: potential therapeutic targets. Science (80-) 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ali F, Gerhard DM, Sweasy K, Pothula S, Pittenger C, Duman RS, et al. Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nat Commun. 2020;11:72. doi: 10.1038/s41467-019-13809-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Widman AJ, McMahon LL. Disinhibition of CA1 pyramidal cells by low-dose ketamine and other antagonists with rapid antidepressant efficacy. Proc Natl Acad Sci. 2018;115:E3007–E3016. doi: 10.1073/pnas.1718883115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Miller OH, Yang L, Wang C-C, Hargroder EA, Zhang Y, Delpire E, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;3:1–22. doi: 10.7554/eLife.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 152.Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci. 2013;33:6990–7002. doi: 10.1523/JNEUROSCI.4998-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chowdhury GMI, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. 2017;22:120–126. doi: 10.1038/mp.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Koike H, Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav Brain Res. 2014;271:111–115. doi: 10.1016/j.bbr.2014.05.065. [DOI] [PubMed] [Google Scholar]
- 155.Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–111. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 156.Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 157.Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098) Neuropharmacology. 2001;40:1028–1033. doi: 10.1016/S0028-3908(00)00194-5. [DOI] [PubMed] [Google Scholar]
- 158.Knapp RJ, Goldenberg R, Shuck C, Cecil A, Watkins J, Miller C, et al. Antidepressant activity of memory-enhancing drugs in the reduction of submissive behavior model. Eur J Pharmacol. 2002;440:27–35. doi: 10.1016/S0014-2999(02)01338-9. [DOI] [PubMed] [Google Scholar]
- 159.Mackowiak M, O’Neill MJ, Hicks CA, Bleakman D, Skolnick P. An AMPA receptor potentiator modulates hippocampal expression of BDNF: an in vivo study. Neuropharmacology. 2002;43:1–10. doi: 10.1016/S0028-3908(02)00066-7. [DOI] [PubMed] [Google Scholar]
- 160.Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci. 2000;20:8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lepack AE, Bang E, Lee B, Dwyer JM, Duman RS. Fast-acting antidepressants rapidly stimulate ERK signaling and BDNF release in primary neuronal cultures. Neuropharmacology. 2016;111:242–252. doi: 10.1016/j.neuropharm.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun H-L, Zhou Z-Q, Zhang G-F, Yang C, Wang X-M, Shen J-C, et al. Role of hippocampal p11 in the sustained antidepressant effect of ketamine in the chronic unpredictable mild stress model. Transl Psychiatry. 2016;6:e741. doi: 10.1038/tp.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 164.Song M, Martinowich K, Lee FS. BDNF at the synapse: why location matters. Mol Psychiatry. 2017;22:1370–1375. doi: 10.1038/mp.2017.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, et al. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- 166.Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- 167.Rantamäki T, Vesa L, Antila H, Di Lieto A, Tammela P, Schmitt A, et al. Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. PLoS ONE. 2011;6:e20567. doi: 10.1371/journal.pone.0020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rantamäki T. TrkB neurotrophin receptor at the core of antidepressant effects, but how? Cell Tissue Res. 2019;377:115–124. doi: 10.1007/s00441-018-02985-6. [DOI] [PubMed] [Google Scholar]
- 169.Fukumoto K, Fogaça MV, Liu R-J, Duman C, Kato T, Li X-Y, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2 R,6 R )-hydroxynorketamine. Proc Natl Acad Sci. 2019;116:297–302. doi: 10.1073/pnas.1814709116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71:996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kohtala S, Theilmann W, Rosenholm M, Müller HK, Kiuru P, Wegener G, et al. Ketamine-induced regulation of TrkB-GSK3β signaling is accompanied by slow EEG oscillations and sedation but is independent of hydroxynorketamine metabolites. Neuropharmacology. 2019;157:107684. doi: 10.1016/j.neuropharm.2019.107684. [DOI] [PubMed] [Google Scholar]
- 172.Kohtala S, Theilmann W, Rosenholm M, Penna L, Karabulut G, Uusitalo S, et al. Cortical excitability and activation of TrkB signaling during rebound slow oscillations are critical for rapid antidepressant responses. Mol Neurobiol. 2019;56:4163–4174. doi: 10.1007/s12035-018-1364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Réus GZ, Vieira FG, Abelaira HM, Michels M, Tomaz DB, dos Santos MAB, et al. MAPK signaling correlates with the antidepressant effects of ketamine. J Psychiatr Res. 2014;55:15–21. doi: 10.1016/j.jpsychires.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 174.Dwyer JM, Maldonado-Avilés JG, Lepack AE, DiLeone RJ, Duman RS. Ribosomal protein S6 kinase 1 signaling in prefrontal cortex controls depressive behavior. Proc Natl Acad Sci U S A. 2015;112:6188–6193. doi: 10.1073/pnas.1505289112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Beurel E, Grieco SF, Amadei C, Downey K, Jope RS. Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord. 2016;18:473–480. doi: 10.1111/bdi.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Costi S, Soleimani L, Glasgow A, Brallier J, Spivack J, Schwartz J, et al. Lithium continuation therapy following ketamine in patients with treatment resistant unipolar depression: a randomized controlled trial. Neuropsychopharmacology. 2019;44:1812–1819. doi: 10.1038/s41386-019-0365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation. Neuropsychopharmacology. 2010;35:2143–2154. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Leikas JV, Kohtala S, Theilmann W, Jalkanen AJ, Forsberg MM, Rantamäki T. Brief isoflurane anesthesia regulates striatal AKT-GSK3β signaling and ameliorates motor deficits in a rat model of early-stage Parkinson′s disease. J Neurochem. 2017;142:456–463. doi: 10.1111/jnc.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kohtala S, Theilmann W, Suomi T, Wigren HK, Porkka-Heiskanen T, Elo LL, et al. Brief isoflurane anesthesia produces prominent phosphoproteomic changes in the adult mouse hippocampus. ACS Chem Neurosci. 2016;7:749–756. doi: 10.1021/acschemneuro.6b00002. [DOI] [PubMed] [Google Scholar]
- 181.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/S0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- 182.Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 183.Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of MAP kinase causes depressive behavior. Nat Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/S0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 185.Blüthgen N, van Bentum M, Merz B, Kuhl D, Hermey G. Profiling the MAPK/ERK dependent and independent activity regulated transcriptional programs in the murine hippocampus in vivo. Sci Rep. 2017;7:45101. doi: 10.1038/srep45101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.De Bartolomeis A, Sarappa C, Buonaguro EF, Marmo F, Eramo A, Tomasetti C, et al. Different effects of the NMDA receptor antagonists ketamine, MK-801, and memantine on postsynaptic density transcripts and their topography: role of Homer signaling, and implications for novel antipsychotic and pro-cognitive targets in psychosis. Prog Neuro-Psychopharmacology Biol Psychiatry. 2013;46:1–12. doi: 10.1016/j.pnpbp.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 187.Ficek J, Zygmunt M, Piechota M, Hoinkis D, Rodriguez Parkitna J, Przewlocki R, et al. Molecular profile of dissociative drug ketamine in relation to its rapid antidepressant action. BMC Genomics. 2016;17:362. doi: 10.1186/s12864-016-2713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Conti B, Maier R, Barr AM, Morale MC, Lu X, Sanna PP, et al. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol Psychiatry. 2007;12:167–189. doi: 10.1038/sj.mp.4001897. [DOI] [PubMed] [Google Scholar]
- 189.Serchov T, Heumann R, van Calker D, Biber K. Signaling pathways regulating Homer1a expression: implications for antidepressant therapy. Biol Chem. 2016;397:207–214. doi: 10.1515/hsz-2015-0267. [DOI] [PubMed] [Google Scholar]
- 190.Holz A, Mülsch F, Schwarz MK, Hollmann M, Döbrössy MD, Coenen VA, et al. Enhanced mGlu5 signaling in excitatory neurons promotes rapid antidepressant effects via AMPA receptor activation. Neuron. 2019;104(338–352):e7. doi: 10.1016/j.neuron.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 191.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Girgenti MJ, Ghosal S, LoPresto D, Taylor JR, Duman RS. Ketamine accelerates fear extinction via mTORC1 signaling. Neurobiol Dis. 2017;100:1–8. doi: 10.1016/j.nbd.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology. 2020;45:990–997. doi: 10.1038/s41386-020-0644-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fuchikami M, Thomas A, Liu R, Wohleb ES, Land BB, DiLeone RJ, et al. Optogenetic stimulation of infralimbic PFC reproduces ketamine’s rapid and sustained antidepressant actions. Proc Natl Acad Sci. 2015;112:8106–8111. doi: 10.1073/pnas.1414728112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hare BD, Shinohara R, Liu RJ, Pothula S, DiLeone RJ, Duman RS. Optogenetic stimulation of medial prefrontal cortex Drd1 neurons produces rapid and long-lasting antidepressant effects. Nat Commun. 2019;10:223. doi: 10.1038/s41467-018-08168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yang Y, Cui Y, Sang K, Dong Y, Ni Z, Ma S, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature. 2018;554:317–322. doi: 10.1038/nature25509. [DOI] [PubMed] [Google Scholar]
- 197.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 198.Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, et al. The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology. 2018;43:2154–2160. doi: 10.1038/s41386-018-0136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li C-T, Chen M-H, Lin W-C, Hong C-J, Yang B-H, Liu R-S, et al. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: a randomized controlled study. Hum Brain Mapp. 2016;37:1080–1090. doi: 10.1002/hbm.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Maksimow A, Särkelä M, Långsjö JW, Salmi E, Kaisti KK, Yli-Hankala A, et al. Increase in high frequency EEG activity explains the poor performance of EEG spectral entropy monitor during S-ketamine anesthesia. Clin Neurophysiol. 2006;117:1660–1668. doi: 10.1016/j.clinph.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 201.Carlén M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nugent AC, Ballard ED, Gould TD, Park LT, Moaddel R, Brutsche NE, et al. Ketamine has distinct electrophysiological and behavioral effects in depressed and healthy subjects. Mol Psychiatry. 2019;24:1040–1052. doi: 10.1038/s41380-018-0028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nugent AC, Wills KE, Gilbert JR, Zarate CA. Synaptic potentiation and rapid antidepressant response to ketamine in treatment-resistant major depression: a replication study. Psychiatry Res Neuroimaging. 2019;283:64–66. doi: 10.1016/j.pscychresns.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cornwell BR, Salvadore G, Furey M, Marquardt CA, Brutsche NE, Grillon C, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–561. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Evans JW, Lally N, An L, Li N, Nugent AC, Banerjee D, et al. 7T 1 H-MRS in major depressive disorder: a ketamine treatment study. Neuropsychopharmacology. 2018;43:1908–1914. doi: 10.1038/s41386-018-0057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–662. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Feinberg I, Campbell IG. Stimulation of NREM Delta EEG by ketamine administration during waking: demonstration of dose dependence. Neuropsychopharmacology. 1995;12:89–90. doi: 10.1016/0893-133X(94)00072-8. [DOI] [PubMed] [Google Scholar]
- 208.Duncan WC, Sarasso S, Ferrarelli F, Selter J, Riedner BA, Hejazi NS, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2013;16:301–311. doi: 10.1017/S1461145712000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rantamäki T, Kohtala S. Encoding, consolidation, and renormalization in depression : synaptic homeostasis, plasticity, and sleep integrate rapid antidepressant effects. Pharmacol Rev. 2020;72:439–465. doi: 10.1124/pr.119.018697. [DOI] [PubMed] [Google Scholar]
- 210.Huber R, Esser SK, Ferrarelli F, Massimini M, Peterson MJ, Tononi G. TMS-induced cortical potentiation during wakefulness locally increases slow wave activity during sleep. PLoS ONE. 2007;2:e276. doi: 10.1371/journal.pone.0000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sackeim HA, Luber B, Katzman GP, Moeller JR, Prudic J, Devanand D, et al. The effects of electroconvulsive therapy on quantitative electroencephalograms. Relationship to clinical outcome. Arch Gen Psychiatry. 1996;53:814–824. doi: 10.1017/CBO9781107415324.004. [DOI] [PubMed] [Google Scholar]
- 212.Ionescu DF, Felicione JM, Gosai A, Cusin C, Shin P, Shapero BG, et al. Ketamine-associated brain changes: a review of the neuroimaging literature. Harv Rev Psychiatry. 2018;26:320–339. doi: 10.1097/HRP.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 214.Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- 215.Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S, et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000;48:830–843. doi: 10.1016/S0006-3223(00)01036-2. [DOI] [PubMed] [Google Scholar]
- 216.Drevets WC, Savitz J, Trimble M. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 2008;13:663–681. doi: 10.1017/S1092852900013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Alexander L, Gaskin PLR, Sawiak SJ, Fryer TD, Hong YT, Cockcroft GJ, et al. Fractionating blunted reward processing characteristic of anhedonia by over-activating primate subgenual anterior cingulate cortex. Neuron. 2019;101(307–320):e6. doi: 10.1016/j.neuron.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Morris LS, Costi S, Tan A, Stern ER, Charney DS, Murrough JW. Ketamine normalizes subgenual cingulate cortex hyper-activity in depression. Neuropsychopharmacology. 2020;45:975–981. doi: 10.1038/s41386-019-0591-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Carlson PJ, Diazgranados N, Nugent AC, Ibrahim L, Luckenbaugh DA, Brutsche N, et al. Neural correlates of rapid antidepressant response to ketamine in treatment-resistant unipolar depression: a preliminary positron emission tomography study. Biol Psychiatry. 2013;73:1213–1221. doi: 10.1016/j.biopsych.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Nugent AC, Diazgranados N, Carlson PJ, Ibrahim L, Luckenbaugh DA, Brutsche N, et al. Neural correlates of rapid antidepressant response to ketamine in bipolar disorder. Bipolar Disord. 2014;16:119–128. doi: 10.1111/bdi.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Downey D, Dutta A, McKie S, Dawson GR, Dourish CT, Craig K, et al. Comparing the actions of lanicemine and ketamine in depression: key role of the anterior cingulate. Eur Neuropsychopharmacol. 2016;26:994–1003. doi: 10.1016/j.euroneuro.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 222.Abdallah CG, Dutta A, Averill CL, McKie S, Akiki TJ, Averill LA, et al. Ketamine, but not the NMDAR antagonist lanicemine, increases prefrontal global connectivity in depressed patients. Chronic Stress. 2018;2:247054701879610. doi: 10.1177/2470547018796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. 2017;42:1210–1219. doi: 10.1038/npp.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Abdallah CG, Averill CL, Salas R, Averill LA, Baldwin PR, Krystal JH, et al. Prefrontal connectivity and glutamate transmission: relevance to depression pathophysiology and ketamine treatment. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:566–574. doi: 10.1016/j.bpsc.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Berman MG, Peltier S, Nee DE, Kross E, Deldin PJ, Jonides J. Depression, rumination and the default network. Soc Cogn Affect Neurosci. 2011;6:548–555. doi: 10.1093/scan/nsq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Greicius MD, Flores BH, Menon V, Glover GH, Solvason HB, Kenna H, et al. Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry. 2007;62:429–437. doi: 10.1016/j.biopsych.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sheline YI, Price JL, Yan Z, Mintun MA. Resting-state functional MRI in depression unmasks increased connectivity between networks via the dorsal nexus. Proc Natl Acad Sci. 2010;107:11020–11025. doi: 10.1073/pnas.1000446107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Evans JW, Szczepanik J, Brutsché N, Park LT, Nugent AC, Zarate CA. Default mode connectivity in major depressive disorder measured up to 10 days after ketamine administration. Biol Psychiatry. 2018;84:582–590. doi: 10.1016/j.biopsych.2018.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Reed JL, Nugent AC, Furey ML, Szczepanik JE, Evans JW, Zarate CA. Effects of ketamine on brain activity during emotional processing: differential findings in depressed versus healthy control participants. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4:610–618. doi: 10.1016/j.bpsc.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Reed JL, Nugent AC, Furey ML, Szczepanik JE, Evans JW, Zarate CA. Ketamine normalizes brain activity during emotionally valenced attentional processing in depression. NeuroImage Clin. 2018;20:92–101. doi: 10.1016/j.nicl.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shirayama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fukuda M, Takatori A, Nakamura Y, Suganami A, Hoshino T, Tamura Y, et al. Effects of novel small compounds targeting TrkB on neuronal cell survival and depression-like behavior. Neurochem Int. 2016;97:42–48. doi: 10.1016/j.neuint.2016.04.017. [DOI] [PubMed] [Google Scholar]
- 233.Hasegawa Y, Zhu X, Kamiya A. NV-5138 as a fast-acting antidepressant via direct activation of mTORC1 signaling. J Clin Invest. 2019;129:2207–2209. doi: 10.1172/JCI129702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci U S A. 2019;116:5160–5169. doi: 10.1073/pnas.1816071116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yamaguchi J, Toki H, Qu Y, Yang C, Koike H, Hashimoto K, et al. (2R,6R)-Hydroxynorketamine is not essential for the antidepressant actions of (R)-ketamine in mice. Neuropsychopharmacology. 2018;43:1900–1907. doi: 10.1038/s41386-018-0084-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yang C, Qu Y, Abe M, Nozawa D, Chaki S, Hashimoto K. (R)-Ketamine shows greater potency and longer lasting antidepressant effects than its metabolite (2R,6R)-hydroxynorketamine. Biol Psychiatry. 2017;82:e43–e44. doi: 10.1016/j.biopsych.2016.12.020. [DOI] [PubMed] [Google Scholar]
- 237.Farmer CA, Gilbert JR, Moaddel R, George J, Adeojo L, Lovett J, et al. Ketamine metabolites, clinical response, and gamma power in a randomized, placebo-controlled, crossover trial for treatment-resistant major depression. Neuropsychopharmacology. 2020;45:1398–1404. doi: 10.1038/s41386-020-0663-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Grunebaum MF, Galfalvy HC, Choo T-H, Parris MS, Burke AK, Suckow RF, et al. Ketamine metabolite pilot study in a suicidal depression trial. J Psychiatr Res. 2019;117:129–134. doi: 10.1016/j.jpsychires.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Workman ER, Niere F, Raab-Graham KF. Engaging homeostatic plasticity to treat depression. Mol Psychiatry. 2018;23:26–35. doi: 10.1038/mp.2017.225. [DOI] [PubMed] [Google Scholar]
- 240.Duncan WC, Slonena E, Hejazi NS, Brutsche N, Yu KC, Park L, et al. Motor-activity markers of circadian timekeeping are related to ketamine’s rapid antidepressant properties. Biol Psychiatry. 2017;82:361–369. doi: 10.1016/j.biopsych.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Duncan WC, Slonena EE, Hejazi NS, Brutsche N, Park LT, Henter ID, et al. Are 24-hour motor activity patterns associated with continued rapid response to ketamine? Neuropsychiatr Dis Treat. 2018;14:2739–2748. doi: 10.2147/NDT.S172089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Duncan WC, Zarate CA. Ketamine, sleep, and depression: current status and new questions. Curr Psychiatry Rep. 2013;15:394. doi: 10.1007/s11920-013-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Huber R, Mäki H, Rosanova M, Casarotto S, Canali P, Casali AG, et al. Human cortical excitability increases with time awake. Cereb Cortex. 2013;23:332–338. doi: 10.1093/cercor/bhs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ly JQM, Gaggioni G, Chellappa SL, Papachilleos S, Brzozowski A, Borsu C, et al. Circadian regulation of human cortical excitability. Nat Commun. 2016;7:11828. doi: 10.1038/ncomms11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Kuhn M, Wolf E, Maier JG, Mainberger F, Feige B, Schmid H, et al. Sleep recalibrates homeostatic and associative synaptic plasticity in the human cortex. Nat Commun. 2016;7:12455. doi: 10.1038/ncomms12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Giedt WR, Lakin ML, Winters WD. Diurnal response to ketamine and gamma-hydroxybutyrate and its possible relationship to pineal indoleamines. Neuropharmacology. 1978;17:221–228. doi: 10.1016/0028-3908(78)90105-3. [DOI] [PubMed] [Google Scholar]
- 247.Rebuelto M, Ambros L, Montoya L, Bonafine R. Treatment-time-dependent difference of ketamine pharmacological response and toxicity in rats. Chronobiol Int. 2002;19:937–945. doi: 10.1081/CBI-120014572. [DOI] [PubMed] [Google Scholar]
- 248.Tononi G, Cirelli C. Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull. 2003;62:143–150. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 249.Tononi G, Cirelli C. Sleep and synaptic down-selection. Eur J Neurosci. 2020;51:413–421. doi: 10.1111/ejn.14335. [DOI] [PMC free article] [PubMed] [Google Scholar]