

Abstract
Motivation
The binding of T-cell receptors (TCRs) to their target peptide MHC (pMHC) ligands initializes the cell-mediated immune response. In autoimmune diseases such as multiple sclerosis, the TCR erroneously recognizes self-peptides as foreign and activates an immune response against healthy cells. Such responses can be triggered by cross-recognition of the autoreactive TCR with foreign peptides. Hence, it would be desirable to identify such foreign-antigen triggers to provide a mechanistic understanding of autoimmune diseases. However, the large sequence space of foreign antigens presents an obstacle in the identification of cross-reactive peptides.
Results
Here, we present an in silico modeling and scoring method which exploits the structural properties of TCR-pMHC complexes to predict the binding of cross-reactive peptides. We analyzed three mouse TCRs and one human TCR isolated from a patient with multiple sclerosis. Cross-reactive peptides for these TCRs were previously identified via yeast display coupled with deep sequencing, providing a robust dataset for evaluating our method. Modeling query peptides in their associated TCR-pMHC crystal structures, our method accurately selected the top binding peptides from sets containing more than a hundred thousand unique peptides.
Availability and implementation
Analyses were performed using custom Python and R scripts available at https://github.com/weng-lab/antigen-predict.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
As a surveillance mechanism against pathogens and cancer, T cells of the host immune system use their αβ T-cell receptors (TCRs) to inspect other cells. Targets recognized by TCRs are peptides bound and presented by the host major histocompatibility complex (MHC) proteins on the outer surface of the cellular membrane, and the peptide epitope may derive for instance from a viral protein. TCR recognition triggers complex signaling pathways that lead to a variety of outcomes, such as the destruction of infected or diseased cells, T-cell proliferation and release of proinflammatory cytokines.
Determining peptide epitopes that can be recognized by TCRs is of considerable interest, impacting fields ranging from virology to cancer immunotherapy. Peptide immunogenicity involves three steps, each of which have been addressed via various predictive algorithms: peptide processing (Bhasin and Raghava, 2004; Nielsen et al., 2005), peptide binding to an MHC (Andreatta and Nielsen, 2016; Jurtz et al., 2017; O’Donnell et al., 2018) and TCR recognition of the peptide-MHC (pMHC) complex (Lanzarotti et al., 2019; Ogishi and Yotsuyanagi, 2019; Pierce and Weng, 2013; Riley et al., 2019; Schneidman-Duhovny et al., 2018; Tung et al., 2011). While progress has been made in predicting the outcome of each step, the fixed size of the TCR repertoire relative to the much larger number of possible peptide epitopes that T cells may encounter presents a particularly significant challenge.
Even with a TCR repertoire estimated to lie in the tens of millions, estimates are that any particular TCR would need to recognize at least one million different pMHC complexes in order to provide sufficient immune coverage (Mason, 1998; Sewell, 2012). This high level of cross-reactivity has been verified using combinatorial peptide libraries (Maynard et al., 2005; Wooldridge et al., 2012). Thus, although specificity is considered a hallmark of immunity, TCRs display significant cross-reactivity. Even if such cross-reactivity can be rationalized at a high level from structural and biophysical principles (Singh et al., 2017), determining the range of peptides recognized by a specific TCR remains a major goal in immunology. Demonstrating the biological significance of the problem, TCR cross-recognition of self-peptides is believed to underlie various autoimmune disorders (Gravano and Hoyer, 2013), and patient deaths have occurred due to unanticipated ‘off-target’ recognition of TCRs used in clinical trials for cancer immunotherapy (Linette et al., 2013; Morgan et al., 2013).
Given the availability of TCR-pMHC structural information, together with advances in protein design and prediction methodologies, in principle, the peptide specificity profile of a TCR should be predictable using in silico methods. One challenge, however, is the availability of detailed experimental datasets against which such prediction methods could be benchmarked. In addition to combinatorial peptide libraries, Garcia and colleagues have used yeast display of pMHC libraries coupled with TCR staining and deep sequencing to assess the specificity profiles of TCRs (Adams et al., 2016; Birnbaum et al., 2014; Gee et al., 2018). With each yeast cell expressing a unique random peptide, these libraries allow for affinity-based interrogation of over one hundred million peptides against a query TCR. Affinity based selection proceeds through multiple rounds where yeast libraries are enriched for yeast that bind bead-multimerized TCR. Subsequent deep sequencing of yeast DNA from final selection rounds produces enrichment counts for peptides selected by the query TCR. Thereby peptides with the highest read counts in the last round of selection are indicative of cross-reactive peptides capable of binding the TCR. Such experiments provide rich datasets for developing and benchmarking in silico approaches to evaluate TCR specificity.
Here, we used structure-based in silico methods to predict the specificity profiles for four TCRs assessed using yeast display and deep sequencing: 2B4, 226, 5cc7 and Ob.1A12 (Birnbaum et al., 2014). Three of these TCRs recognize a peptide derived from moth cytochrome C presented by the murine class II MHC protein I-Ek (Newell et al., 2011). The fourth (Ob.1A12) was isolated from a patient with relapsing-remitting multiple sclerosis and recognizes a peptide derived from the myelin basic protein presented by the human class II MHC protein HLA-DR2 (Wucherpfennig et al., 1994). The deep sequencing data provided more than 100 000 peptides for each TCR, including both binders and non-binders, ideal for benchmarking structure-based in silico methods.
Using the crystal structures for the four TCR-pMHC complexes (Birnbaum et al., 2014; Hahn et al., 2005; Newell et al., 2011), we modeled all of the query peptides within the TCR-pMHC complexes and scored the structural models to predict cross-reactive peptides for each of the four TCRs. Our modeling and scoring approach was capable of recovering cross-reactive peptides from large pools of primarily non-binding peptides for each TCR tested. We compared our method with an approach of selecting peptides with similar sequences to each TCR’s cognate peptide epitope (i.e. the target peptide found in the crystallographic structure). Our structure-modeling approach out-performed the sequence-similarity approach for one of the four TCRs investigated. Furthermore, combining the two approaches yields the best performance for two other TCRs while maintaining the performance for the last TCR, underscoring the value of including structural information in epitope prediction.
2 Materials and methods
2.1 Peptide sequence extraction
Sequencing data were downloaded from the Sequence Read Archive (SRA) under project accession SRP040021 and converted to FASTQ files using the SRA Toolkit. Each sequenced read contains the nucleotide sequence encoding one of the random peptides of the combinatorial yeast display library. Prior to sequencing, barcodes were appended to distinguish the selection round and the TCR used for selection in the pooled sequencing results (Birnbaum et al., 2014). As a first step in processing, we split reads by barcode into their individual selection rounds along with the TCR used for selection. The randomized peptide in each read was generated by mutagenic primers allowing all 20 amino acids via NNK codons, with the exception of some restricted positions for anchoring to the MHC (Birnbaum et al., 2014). We identified the start of the nucleotide sequence encoding the peptide via its specific position given in the primer sequence and thereby extracted the nucleotide sequence encoding the full 13-mer or 14-mer peptide recognized by the mouse or human TCR, respectively. Next, the nucleotide sequences encoding the peptides were translated into amino acid sequences and peptide sequences containing stop codons or unknown amino acids were discarded. The resulting read counts for each unique peptide were recorded for each round of selection for each TCR.
To validate our procedure of extracting peptide sequences with that of Birnbaum et al. (2014), we compared the read counts of the top 25 most abundant peptides given in Birnbaum et al. in the fourth round of selection for the 2B4 TCR with our extracted read counts for the same peptides in the same round. The Spearman correlation of our read counts versus read counts from Birnbaum et al. was 0.9998. Birnbaum et al. performed surface plasmon resonance (SPR) experiments on two of these top recovered peptides and reported binding of the 2B4 TCR for both, validating the use of yeast display libraries in recovery of cross-reactive peptides. For further experimental details regarding library creation, list of primers used for randomization and for deep sequencing, we defer the reader to the original publication from Birnbaum et al. (2014).
2.2 Peptide structure modeling
Template TCR-pMHC complex structures were downloaded from the protein data bank (PDB) with the following PDB IDs: 3QIB (2B4-MCC-I-Ek), 3QIU (226-MCC-I-Ek), 4P2R (5cc7-5c1-I-Ek) and 1YMM (Ob.1A12-MBP-HLA-DR2). To reduce computation time the structures were truncated to contain only the binding interface (up to residue 83 for the class II MHC α chain and residue 93 for the β chain). Each TCR was truncated to just contain the variable domains, excluding the constant domains that are distal from the binding interface for pMHC. Water molecules were also removed to simplify scoring and for consistency across TCR-pMHC structures with different resolutions.
Prior to modeling peptides onto TCR-pMHC structure templates, we prepared structures using the refinement application, relax, of the Rosetta suite of programs (Version 3.5) (Leaver-Fay et al., 2011; Nivon et al., 2013). The goal of relaxing structures prior to modeling and scoring is to resolve clashes and other errors that may negatively impact performance of Rosetta energy functions. We followed a relax protocol that was previously tested on a benchmark set of 51 proteins which increased sequence recovery in enzyme design while keeping the RMSD between relaxed and original input structures minimal (Nivon et al., 2013). An example of the cleaned and relaxed 2B4-MCC-I-Ek complex is provided as a PDB file in Supplementary Data (3QIB_relax.trunc.pdb). The following is an example Rosetta command used for relaxing our initial TCR-pMHC complex templates prior to modeling:
rosetta_source/bin/relax.linuxgccrelease –database rosetta_database/ -relax:constrain_relax_to_start_coords -relax:coord_constrain_sidechains -relax:ramp_constraints false -s my_pdb.pdb -ex1 -ex2 -use_input_sc -flip_HNQ -no_optH false
To model peptides onto the template TCR-pMHC structures, we utilized the fixed backbone application, fixbb, of the Rosetta suite of programs (Leaver-Fay et al., 2008, 2011), with parameters ‘extrachi_cutoff 1 –ex1 –ex2 –ex3’ to increase χ angle rotamer sampling for side-chain placement of peptide residues. All side chains of the TCR-pMHC aside from those modeled on the peptide were left in their original poses. An example resfile specifying amino acid mutations to model the 2A peptide in the 2B4-MCC-I-Ek complex as in W Figure 1B is given in Supplementary Data (resfile_2A.txt). The following is an example Rosetta command used for peptide structural modeling:
Fig. 1.
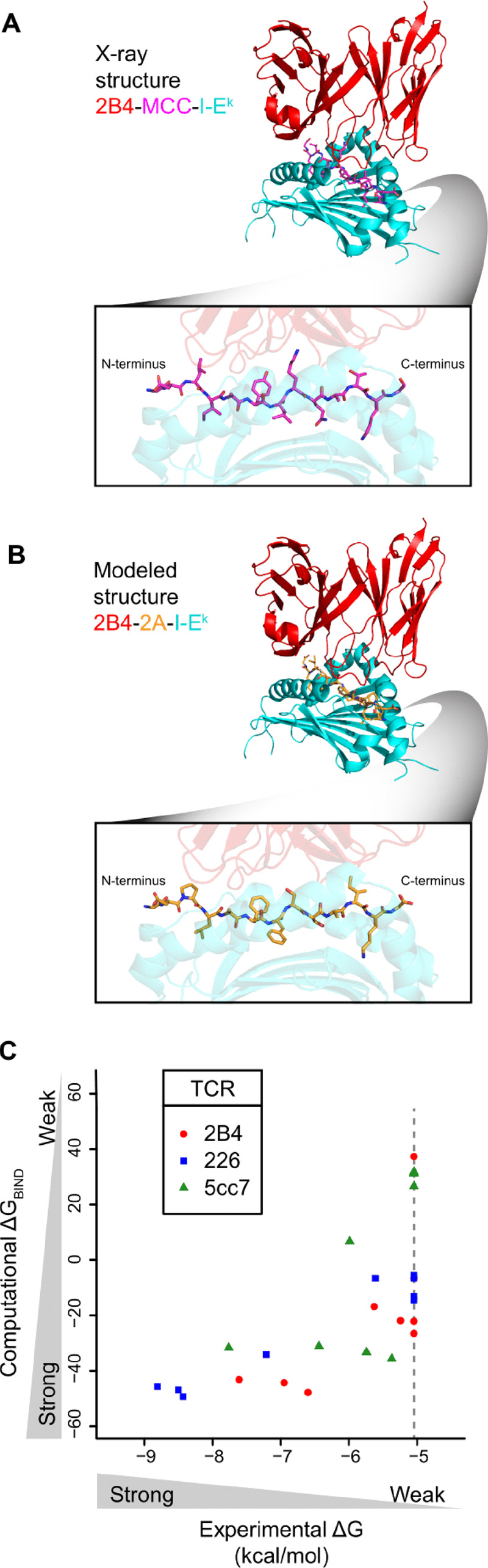
Prediction of TCR-pMHC binding free energies. (A) Crystal structure of TCR-pMHC interface for the 2B4 TCR (red) interacting with the MCC peptide (magenta) displayed by the I-Ek MHC (cyan). Bottom box: profile of the MCC peptide. (B) Fixed backbone model structure of TCR-pMHC interface for the 2B4 TCR (red) interacting with the 2A peptide (orange) displayed by the I-Ek MHC (cyan). 2A peptide was modeled onto the backbone of the MCC peptide in (A), TCR and MHC protein structures remain identical to (A). Bottom box: profile of the 2A peptide. (C) Scatter plot of ΔGBIND from computational modeling and scoring versus ΔG from experimental binding energies. Each point represents a peptide in Supplementary Table S1, where experimental ΔG is determined by binding affinity of pMHC with the indicated TCR (red, blue, green) via surface plasmon resonance from Birnbaum et al., and computational ΔGBIND is determined from modeling and scoring of peptide in TCR-pMHC complex. Pearson correlations are 0.67, 0.97 and 0.67 for TCRs 2B4, 226 and 5cc7, respectively. Correlation across the entire set of peptides is 0.69. Peptides with unreliable KDs due to weak or non-binding interactions were assigned a KD of 200 μM (ΔG = -5.05 kcal/mol, gray dotted line). If we assigned a ΔG of 0 kcal/mol for these weak/non-binding peptides the correlation across the entire set of peptides was similar (r = 0.74). (Color version of this figure is available at Bioinformatics online.)
rosetta_source/bin/fixbb.linuxgccrelease –database rosetta_database/ -s my_pdb.pdb -resfile my_resfile -suffix my_label -extrachi_cutoff 1 -ex1 -ex2 -ex3
2.3 Prediction of peptide-MHC/TCR binding free energy
We investigated three scoring approaches, GCOMPLEX, ΔGBIND and ΔGBIND.2, where GCOMPLEX is the Rosetta score for the entire TCR-pMHC complex, and the latter two scoring methods are defined using the following formulas:
| (1) |
| (2) |
such that GTCR/MHC is the Rosetta score for the TCR and MHC chains bound without the peptide, GPEPTIDE is the score for the isolated peptide in its bound conformation, GTCR is the score for the isolated TCR in its bound conformation and GpMHC is the score for the peptide and MHC chains bound without the TCR. To score each component, we used Rosetta’s scoring application, score (Leaver-Fay et al., 2011). This scoring function is a linear combination of 19 energy terms, including van der Waals, solvation, electrostatics and hydrogen bonding interactions along with other statistical potentials. The weights for the energy terms were left in their default settings (those specified in the standard.wts file along with the score12.wts_patch file found in Rosetta’s weights directory). The following is an example Rosetta command used for scoring modeled TCR-pMHC structures (i.e. scoring commands for GCOMPLEX, GTCR/MHC, GPEPTIDE, GTCR and GpMHC):
rosetta_source/bin/score.linuxgccrelease –database rosetta_database/ -s my_pdb.pdb -out:file:scorefile outputfile.sc
2.4 Heatmaps of amino acid frequency
Heatmaps representing the cross-reactivity for individual TCRs were generated using either the top 50 most enriched peptides in the fourth round of TCR selection (top 50 experimentally selected) or the top 50 peptides with the most favorable ΔGBIND (top 50 computationally selected) (Fig. 3A and B). Each cell of the heatmap represents the amino acid frequency in the top 50 peptides for the specific peptide residue position, such that, for example, a value of 1 for Ala at residue position -3 indicates all of the top 50 peptides carry an Ala at this position of the peptide, while a value of 0 indicates none of the top 50 peptides carry an Ala at this position. Hence, the sum of each column in the heatmap is 1. Peptide residue positions are in order from N- to C-terminus with residue labels (-3…13) used to be consistent with the nomenclature in Birnbaum et al.
Fig. 3.

Amino acid frequencies for top peptides selected by yeast display or by computation for mouse and human TCRs. (A) Heatmaps represent the amino acid frequencies at peptide positions for the 50 peptides with the most abundant reads in the fourth round of selection for four TCRs (2B4, 226, 5cc7 and Ob.1A12). (B) Amino acid frequencies at peptide positions for the 50 peptides with the most favorable ΔGBIND. The peptide pool for ΔGBIND computation was the union of positive and negative binding sets (>105 peptides). The peptide residues from the template TCR-pMHC structures used for modeling, MCC (for the 2B4 and 226 TCRs), 5c1 (for the 5cc7 TCR) and MBP (for the Ob.1A12 TCR) are outlined in black. Peptide positions restricted in the yeast display libraries to maintain MHC binding are marked in red beneath the heatmap. Shared amino acid substitutions between experimental and computational heatmaps with frequencies two-fold higher than expected are marked by displaying the amino acid frequency at the substitution position. Correlations of frequencies between the experimental and computational heatmap for 2B4, 226, 5cc7 and Ob.1A12 TCRs are 0.91, 0.86, 0.84 and 0.53, respectively (excluding restricted positions). (C) Precision-recall curves assessing binding prediction performance of three methods: (1) computational ΔGBIND, (2) BLOSUM62 sequence similarity to cognate peptide and (3) the average rank of the two methods. Values for the area under the curve (AUC) are displayed in the upper right corner. (Color version of this figure is available at Bioinformatics online.)
2.5 BLOSUM62 sequence similarity
To measure sequence similarity between query peptides in our positive and negative binding sets against the cognate peptide found in the crystal structure of the TCR-pMHC complex, we used the BLOSUM62 substitution matrix designed for sequence alignment of proteins (Henikoff and Henikoff, 1992). The BLOSUM62 similarity score between two peptides was then defined as the sum of the BLOSUM62 log-odds ratios for each amino acid substitution between the two peptides.
2.6 Measuring prediction performance
To quantify the performance of our cross-reactive antigen prediction methods, we first defined positive (binding) and negative (non-binding) peptide sets for each TCR based on the experimental deep-sequencing results from Birnbaum et al. We defined positive peptides as those peptides which were recovered after four rounds of TCR selection in the yeast display experiment and had sequence read counts in the 95th percentile. We defined negative peptides as peptides found in the preselection library that were not found after four rounds of selection. The deep sequencing results present only a sample of the total unique peptides in each selection round. It is thus important to note that, although the round-four peptides are a subset of the full experimental pre-selection library with >108 peptides, they are not a strict subset of the >105 unique peptides recovered from sequencing the pre-selection library. As our positive and negative sets were highly unbalanced with a small number of positive peptides (∼102) and a much larger set of negative peptides (∼105), we assessed prediction performance by calculating the area under the precision-recall curve (AUC), to assess directly the ability of our methods in identifying true positives among the peptides that we predict. Precision-recall curves and AUC values were generated using the R package ROCR (Sing et al., 2005).
3 Results
3.1 High-throughput modeling reproduces experimentally observed enrichment of binder peptides
Previously described experimental yeast display and deep sequencing generated libraries that were enriched for peptides specifically recognized by four TCRs (Birnbaum et al., 2014). Beginning with the crystallographic structures of the 2B4, 226, 5cc7 and Ob.1A12 TCRs in complex with their cognate pMHC complexes, we relaxed these template structures using the Rosetta suite of programs (Leaver-Fay et al., 2011; Nivon et al., 2013) (see Section 2). We then computationally modeled and scored the peptides with sequences in the preselection libraries and four sequential selection rounds—347 210 peptides for the 2B4 TCR, 811 481 peptides for the 226 TCR, 809 156 peptides for the 5cc7 TCR and 514 906 peptides for the Ob.1A12 TCR. In total, we modeled and scored 2 482 753 peptides (Birnbaum et al., 2014).
To increase the computational throughput in modeling the structures of these approximately 2.5 million peptides, we performed a restricted structural modeling procedure using Rosetta’s fixed backbone design application, fixbb, which optimizes side-chain conformations on a fixed backbone using the Rosetta energy function (Leaver-Fay et al., 2008) (see Section 2). We retained TCR and MHC side chains in the conformations adopted in the crystallographic structures with their cognate pMHCs (Fig. 1A and B). The mean runtime for a single peptide design by the fixbb application was ∼ 2.6 s.
Once each TCR-pMHC model was generated, we scored the full complex (GCOMPLEX) and isolated components (GTCR/MHC, GPEPTIDE) using Rosetta’s score application (see Section 2). These scores were combined to produce a binding score, ΔGBIND, which accounted for the peptide’s interaction energy with both the MHC and the TCR. The mean runtime to calculate a ΔGBIND score from a TCR-pMHC model was ∼ 4.7 s.
To quantitatively assess our peptide modeling and scoring approach, we examined sets of peptides for which experimental binding free energies were available for the 2B4, 226 and 5cc7 TCRs (Supplementary Table S1) (Birnbaum et al., 2014). Correlations between ΔGBIND and experimentally measured binding free energies were greater than 0.66 for all TCRs, and 0.69 for the entire set together (Fig. 1C).
We next examined the distributions of ΔGBIND across the four experimental selection rounds of the yeast display library where each successive round was further enriched in cross-reactive peptides via TCR selection. Indeed, ΔGBIND scoring of modeled complexes revealed an increasing enrichment of favorable energy scores for peptides in each subsequent selection round, in congruence with the subsequent enrichment of cross-reactive peptides for each round (Fig. 2). Thus, relying on a relatively simple structural modeling method to enable computational throughput permits the recovery of experimentally determined peptides bound by a TCR.
Fig. 2.

Distributions of ΔGBIND for peptides recovered from different selection rounds. We generated structural models of TCR-pMHC complexes using peptide sequences from all experimental selection libraries. For each unique peptide recovered in each selection round, we modeled its structure bound to MHC and TCR and computed ΔGBIND for the TCR-pMHC complex. The probability densities for ΔGBIND are plotted for each round of selection for the four TCRs analyzed in this study, 2B4, 226, 5cc7 and Ob.1A12. The probability density is defined such that the histogram has a total area of one. (n = total number of unique peptides in the given round; < 200: percent of peptides in the round with ΔGBIND less than 200)
3.2 Computational prediction of cross-reactive peptides by structural modeling
To examine the extent to which our modeling and scoring approach selected the peptides recognized by the TCRs with the strongest affinities, we compared the 50 peptides with the most favorable ΔGBIND to the 50 peptides with the most reads recovered by deep sequencing after the fourth round of selection for the 2B4, 226, 5cc7 and Ob.1A12 TCRs (Birnbaum et al., 2014). To assess predictive performance of our method we defined binding (positive) and non-binding (negative) peptide sets based on the deep sequencing selection results (see Section 2). Many of the recovered peptides from the fourth round of selection were represented by a single read count, and may not be a true binding peptide. To be conservative in defining positive and negative sets, we assigned only peptides from the fourth round of selection with read counts in the top 95th percentile as binding peptides. Non-binding peptides were then defined as peptides in the preselection library which were not identified in the fourth round of selection. We asked the computational method to identify top-scoring peptides from the pool of our small set of binding peptides (∼102) and large set of non-binding peptides (∼105). For a successful computational method, we would expect peptides with the most favorable ΔGBIND to be members of the positive set or to share amino acid preferences with these binding peptides. Among the 50 peptides that had the most favorable ΔGBIND according to our modeling and scoring method, 28, 27, 25 and 28 of these peptides were in the positive set of peptides for the 2B4, 226, 5cc7 and Ob.1A12 TCRs, respectively. Therefore, our scoring method was capable of identifying true binders within a large pool consisting primarily of non-binding peptides.
The amino acid preference generated using the 50 top experimentally selected peptides (50 peptides with the most abundant reads counts in round four) illustrated binding motifs distinct for each TCR (Birnbaum et al., 2014) (Fig. 3A). To further examine how the best scoring peptides compared to those identified experimentally after TCR selection, we compared heatmaps of amino acid preferences for the 50 top experimentally selected peptides and the top 50 computationally selected peptides (50 peptides with the most favorable ΔGBIND from the pool of positive and negative peptide sets) for each TCR (Fig. 3A and B). Many sequence features were shared between the top-scoring peptides and the peptides with the most abundant read counts. To quantify similarity between heatmaps, we flattened the heatmap matrices into vectors and calculated the Pearson correlation between them. Excluding those anchor positions restricted in the libraries for MHC binding, correlations between the heatmaps representing experimentally selected and computationally selected peptides were 0.91, 0.86, 0.84 and 0.53 for TCRs 2B4, 226, 5cc7 and Ob.1A12, respectively.
Given that our positive and negative sets are highly unbalanced (fewer binding than non-binding peptides), we utilized the area under the precision-recall curve (AUC) to quantitatively compare the performance of three structure-based scoring approaches: ΔGBIND, GCOMPLEX and ΔGBIND.2 (see Section 2) (Supplementary Fig. S1). While ΔGBIND represents interactions between the peptide and TCR and also the peptide and MHC, GCOMPLEX additionally accounts for deformations of the peptide itself. In contrast, ΔGBIND.2 removes interaction energies between peptide and MHC and only incorporates interaction energies between TCR and pMHC. ΔGBIND achieved highest AUC values for three out of four TCRs, while GCOMPLEX was slightly better in the case of the 5cc7 TCR (GCOMPLEX AUC = 0.23, ΔGBIND AUC = 0.21). The AUC values were especially different for Ob.1A12 TCR (GCOMPLEX AUC = 0.01, ΔGBIND AUC = 0.13), where binding peptides are one residue longer and contain fewer restricted anchor residues in the combinatorial libraries. Thus, we conclude that the inclusion of intra-peptide energies in GCOMPLEX is more detrimental than beneficial. ΔGBIND.2 showed the lowest overall performance, indicating that the inclusion of interaction energies between peptide and MHC as in ΔGBIND was critical to the success of cross-reactivity predictions. A possible reason is that some of the peptides in the preselection library may not bind stably to the MHC. Because a stable peptide-MHC interaction is a prerequisite for TCR binding, the incorporation of interaction energies between peptide and MHC improved prediction performance.
We note that in the case of the Ob.1A12 TCR, ΔGBIND was successful in assigning favorable scores to peptides carrying the ‘HF’ motif as was found experimentally (Birnbaum et al., 2014). It is evident from the experimental heatmap that Ob.1A12 is tolerant of amino acid substitutions outside the anchor residues and the central HF motif. This feature of Ob.1A12 is also captured by our modeling and scoring method, with the exceptions of a strong preference for Ala at position -4, Arg at -2, Lys at -1, and a few other less frequent substitutions in the experimental heatmap that ΔGBIND could not reproduce.
The 2B4, 226 and 5cc7 TCRs all recognize the MCC peptide (ADLIAYLKQATKG), which is presented in the TCR-pMHC crystal structures of 2B4 and 226, but the crystal structure for 5cc7 has a different peptide (5c1, ANGVAFFLTPFKA). Both the experimentally selected peptides and the top-scoring peptides by our modeling method revealed peptide motifs similar to these cognate peptides (their residues are in black boxes in Fig. 3A and B).
Because our modeling method started with the ternary complex structure containing cognate peptides, our method may simply favor peptides with similar sequences. Nevertheless, our scoring method does reproduce many amino acid substitutions seen in the top experimentally selected peptides (marked with amino acid frequency in Fig. 3A and B). We defined a substitution to be shared between the experimental and computed peptide sets if the frequency of the mutant amino acid at its peptide position was 2 fold higher than what would be expected by chance in both heatmaps (based on the NNK codon library used to design the yeast-display libraries). The following substitutions are shared between the two heatmaps for 2B4: L-1H, L-1Q, Y3F, Q6A and T8S. For 226, the shared substitutions are L-1W, A2G, Y3F, Q6A and Q6S. For 5cc7, the shared substitutions are F3Y, P7A and F8Y. For Ob.1A12, the shared substitutions are N-3H, P-2Q, N6A, I7Q, V8I, T9G, T9C and P10R. Ob.1A12 has an atypical docking mode with the TCR shifted toward the N-terminus of the peptide. It is surprising that in the wild-type crystal structure no peptide residues past residue 5 interact with the TCR, yet we see strong shared amino acid substitutions between the top 50 experimentally selected peptides and our top 50 computationally selected peptides for residues 6–10 at and near the C-terminus (Fig. 3A and B). We assume since we do not allow for changes in TCR binding conformation, these shared substitutions are likely favored due to interactions between peptide and MHC. In summary, although our modeling method may be biased toward the cognate peptide, our modeling and scoring method is still capable of identifying target peptides with beneficial or permissible mutations.
3.3 Comparison with an approach based on sequence similarity
Conscious that sequence motifs of top experimentally selected peptides closely matched that of the cognate peptide in the crystal structure, we asked whether scoring peptides solely on sequence similarity to cognate peptide would also perform well in identifying binding peptides. To provide a quantitative assessment of how our structural-based approach compared with an approach based on sequence similarity to the cognate peptide, we computed a BLOSUM62 similarity score (Henikoff and Henikoff, 1992) between the cognate peptide sequence and the sequence of each peptide in the pool of positive and negative peptides. We find that BLOSUM62 similarity outperforms ΔGBIND in classification of binding peptides for the three mouse TCRs, but ΔGBIND is higher performing in the case of the human Ob.1A12 TCR (Fig. 3C). This result is further illustrated by examining the amino acid frequencies in Figure 3A and B. For the mouse TCRs, the preferred amino acid for the majority of residues in the top experimentally selected peptides is that of the cognate peptide. However, for the Ob.1A12 TCR, only a three residue motif preference in the center of the peptide (HFF) is shared with the cognate peptide. Hence, while BLOSUM62 similarity performed well in prediction of peptide binding for TCRs with limited cross-reactivity, ΔGBIND showed stronger performance in prediction of peptides for a more degenerate TCR, capable of tolerating a range of amino acids near the N- and C-terminus of the peptide.
Given the different performance between the BLOSUM62 similarity score and ΔGBIND, we hypothesized that the two methods might complement one another. To test this, we examined the precision-recall curve of a score that combines BLOSUM62 similarity and ΔGBIND by averaging the rank for each prediction between the two methods. Indeed, the average rank method led to AUC of 0.57 and 0.41 for the 2B4 and 226 TCRs, respectively, showing large improvements in binding prediction performance over the better method for these TCRs (AUC = 0.31 and 0.33 for BLOSUM62; Fig. 3C). For the 5cc7 TCR, the average rank method performed as well as the BLOSUM62 and better than ΔGBIND, while for the Ob.1A12 TCR, the average rank method performed slightly worse than ΔGBIND but better than BLOSUM62 (Fig. 3C). The overall improvement in prediction performance using the average rank and the higher performance of ΔGBIND compared with BLOSUM62 in the case of the Ob.1A12 TCR highlight the value of incorporating structural information in next-generation peptide prediction algorithms.
4 Discussion
Numerous methods exist for the prediction of peptide binding to either class I or class II MHC molecules and have achieved high accuracy dependent upon the training and testing data utilized (Zhao and Sher, 2018). However, far fewer tools are available for prediction of TCR binding to pMHC and the accuracy of existing tools show room for improvement (Lanzarotti et al., 2018; Ogishi and Yotsuyanagi, 2019; Pierce and Weng, 2013; Schneidman-Duhovny et al., 2018; Tung et al., 2011). Utilizing structural information from four TCR-pMHC complexes, we present a high-throughput modeling and scoring approach capable of successfully selecting cross-reactive peptides from large pools of primarily non-binding peptides.
Several other groups incorporated structural information of the TCR-pMHC interface to aid in binding prediction. In a recent study, optimized FoldX and Rosetta energy terms were used to predict peptide binding given the sequences of MHC, TCR and a query peptide (Lanzarotti et al., 2018). We noted that the availability of a high-sequence-identity TCR structure template and successful prediction of peptide binding to MHC were vital to the success of their TCR-pMHC binding prediction. Similarly, the method ITCell utilizes atomic statistical potentials to predict a TCR’s peptide epitope from all possible peptides in the full-length parent protein when given sequences of class II MHC, the TCR variable region and the parent protein antigen as input (Schneidman-Duhovny et al., 2018). In the majority of test cases, ITCell ranked the correct peptide epitope among the top 20 peptides among all peptides that could result from the parent antigen.
Benchmarking sets for the aforementioned methods were generated by using overlapping peptides from the parent protein sequence of the cognate peptide as negatives (excluding the cognate), based on the assumption that parent protein sequence would harbor only a single peptide epitope for a given TCR, which resulted in ∼102–103 query peptides per TCR-pMHC test case (Lanzarotti et al., 2018; Schneidman-Duhovny et al., 2018). A more exact set of non-binding peptides would require experimental evidence for failed binding. Here, we present deep-sequencing results from yeast display as a robust and larger benchmarking tool for TCR epitope prediction. In particular, each preselection library provided >105 peptides, which were not selected by the TCR of interest and are likely negative non-binding peptides. Although 105 peptides is still a small subset of the theoretical diversity for the 13-mer (∼8.1 × 1016) and 14-mer (∼1.6 × 1018) peptides, they represent a larger challenge than previous benchmarks for predicting TCR epitopes.
Like our study, the success of both of the aforementioned methods relied on accurate template-based modeling of the TCR-pMHC complex (Lanzarotti et al., 2018; Schneidman-Duhovny et al., 2018). In our work, modeling of the TCR-pMHC was simplified as crystal structures of TCR-pMHC complexes existed for all four TCRs investigated and only structural changes resulting from the different peptide sequences needed to be accounted for. We note the success of our method requires template crystal structures and do not expect success with modeled structures unless they are structurally accurate. Future work examining TCR-pMHC modeling from the sequence in the context of our prediction method could broaden the applicability of our method.
Previous studies showed the TCR’s complementarity determining region (CDR) loops can be flexible and change their conformations upon ligand binding (Gagnon et al., 2006; Pierce and Weng, 2013; Reiser et al., 2002, 2003; Scott et al., 2011). Furthermore, it has been shown CDR flexibility can contribute to cross-reactivity (Hawse et al., 2014; Reiser et al., 2003). It may be surprising how well our modeling and scoring method performed without making any structural adjustments to the TCR molecules. It is unlikely our modeling method could predict antigens that require large backbone movements, or altered binding orientation, of the TCR for recognition. However, while our modeling method is conservative in terms of modeling any structural changes of the TCR's CDR loops, it appears to perform well in providing poor scores for unfavorable peptides.
Large conformational changes of the peptide can also occur upon TCR binding. For example, the DMF5 TCR that recognizes the MART-1 melanoma antigen presented by the class I MHC protein HLA-A2 was shown to cross-react with the DRG class of peptides that are chemically distinct from MART-1 (Gee et al., 2018). DMF5 TCR binding to an HLA-A2-presented DRG-class peptide led to a ‘register shift’ in the peptide, causing a C-terminal peptide extension from the MHC binding groove (Riley et al., 2018). Identification of cross-reactive peptides with such large structural adjustments relative to cognate peptide would be missed by our fixed-backbone peptide-modeling approach, as would instances in which MHC deformations are required (Borbulevych et al., 2009, 2011). However, peptides of class II pMHC complexes (i.e. those studied here) typically do not bulge from the groove and class II pMHC complexes are thus less prone to backbone rearrangements (Ayres et al., 2017; Tynan et al., 2005). Hence, the success seen here with class II complexes may not fully translate when predicting cross-reactivity in class I systems, although we should anticipate success with conformationally simpler modes of cross-reactivity that involve more commonly observed molecular mimicry mechanisms (Borbulevych et al., 2011; Macdonald et al., 2009).
Even when accounting for simple molecular mimicry mechanisms in cross-reactivity, peptide side-chain modeling must also be precise as a single erroneous side-chain conformation could lead to false positive or false negative predictions. While we do not have an estimate for the accuracy of side-chain modeling for our modeled peptides here, our previous work showed Rosetta’s side-chain optimization methods performed well, albeit on a limited set of TCR-pMHC point mutations (Borrman et al., 2017). Examining χ1 angle distributions for peptide residues in our models of the top binding peptides revealed limited χ1 angle variance at residues with strong amino acid preferences. Future mutational and structural assays could examine whether rotamer conservation for preferred amino acids at the TCR-pMHC interface is critical to binding. As advancements in technology allow for faster and more accurate modeling of larger conformational changes, future studies may focus on allowing for flexibility in CDR loops, MHC and peptide backbone to potentially identify cross-reactive peptides with distinct structural and chemical signatures.
To score the modeled TCR-pMHC structures, we accounted for the interactions made by the peptide with the TCR and the MHC using Rosetta’s scoring application with default weights for energy terms. Future work could potentially improve upon our results by optimizing energy term weights using machine learning approaches and taking advantage of structural and chemical trends in known TCR-pMHC complexes. For example, there is evidence that immunogenic peptides are enriched in hydrophobic amino acids at peptide centers (Calis et al., 2013), and structural modeling combined with neural-network optimized scoring has been used to predict neoantigen immunogenicity (Riley et al., 2019). Here, we employed time-saving modeling and scoring methods to efficiently interrogate large pools of peptides for binding. Future studies optimizing score functions and weights for predicting TCR cross-reactivity might take into account the consequences of the weak affinities TCRs have for their ligands, which can stem from the ‘imperfect’ interfaces that TCRs form, contributing to the difficulty in discriminating between potential ligands using default functions.
One potential application of our method is in cancer immunotherapy. Accurate identification and targeting of neoantigens (peptides derived from mutated tumor proteins) could lead to successful development of immuno-therapeutics. Recent work highlighted the importance of incorporating MHC binding strength, self-similarity to reference antigen and peptide-centric features to accurately predict neoantigen immunogenicity (Bjerregaard et al., 2017; Smith et al., 2019). Building on this and other work, structural modeling and scoring of peptide neoantigens in the context of the full TCR-pMHC complex rather than the MHC alone may provide additional insights beneficial to immunogenic prediction (Riley et al., 2019).
Many efforts have been made to enhance TCR affinity for tumor and viral antigens (Chervin et al., 2008; Holler et al., 2000; Li et al., 2005). However, enhanced affinity may lead to increased cross-reactivity (Hellman et al., 2019; Linette et al., 2013; Riley and Baker, 2018). To check for unwanted cross-reactivity of engineered TCRs, one may perform alanine scanning of the antigen to identify motifs essential for binding and then searching for possible self-antigens in a protein sequence database (Obenaus et al., 2015). The alanine scanning can be expedited using DNA barcode-labeled MHC multimers (Bentzen et al., 2016, 2018). A more direct approach is to interrogate all human peptides for cross-reactivity, like the recent T-Scan method which utilized the lentiviral delivery of an antigen library spanning the entire human proteome into antigen-presenting cells. Selected peptides confirmed the cognate MAGE-A3 epitope along with several novel cross-reactive endogenous self-peptides (Kula et al., 2019). Our modeling and scoring method could represent an in silico approach with a similarly broad coverage. We could scan all peptides of the entire human proteome computationally for possible binding to an engineered TCR granted a template TCR-pMHC crystal structure is available. The thus identified cross-reactive antigens could be further tested experimentally using binding assays or assays measuring immunogenic response.
Supplementary Material
Acknowledgements
The authors thank members of Weng and Baker labs for helpful discussions.
Funding
This project was partly funded by National Institutes of Health [R01GM103773, R01GM116960, R35GM118166 and R01GM126299].
Conflict of Interest: none declared.
References
- Adams J.J. et al. (2016) Structural interplay between germline interactions and adaptive recognition determines the bandwidth of TCR-peptide-MHC cross-reactivity. Nat. Immunol., 17, 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M., Nielsen M. (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class i system. Bioinformatics, 32, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres C.M. et al. (2017) Peptide and peptide-dependent motions in MHC proteins: immunological implications and biophysical underpinnings. Front. Immunol., 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen A.K. et al. (2018) T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide–MHC complexes. Nat. Biotechnol., 36, 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen A.K. et al. (2016) Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol., 34, 1037–1045. [DOI] [PubMed] [Google Scholar]
- Bhasin M., Raghava G.P. (2004) Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci., 13, 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum M.E. et al. (2014) Deconstructing the peptide-MHC specificity of T cell recognition. Cell, 157, 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerregaard A.M. et al. (2017) An analysis of natural T cell responses to predicted tumor neoepitopes. Front. Immunol., 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbulevych O.Y. et al. (2011) Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J. Immunol. (Baltimore, MD: 1950), 186, 2950–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbulevych O.Y. et al. (2009) T cell receptor cross-reactivity directed by antigen-dependent tuning of peptide-MHC molecular flexibility. Immunity, 31, 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrman T. et al. (2017) ATLAS: a database linking binding affinities with structures for wild-type and mutant TCR-pMHC complexes. Proteins Struct. Funct. Bioinf., 85, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calis J. J. a. et al. (2013) Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol., 9, e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervin A.S. et al. (2008) Engineering higher affinity T cell receptors using a T cell display system. J. Immunol. Methods, 339, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon S.J. et al. (2006) T cell receptor recognition via cooperative conformational plasticity. J. Mol. Biol., 363, 228–243. [DOI] [PubMed] [Google Scholar]
- Gee M.H. et al. (2018) Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell, 172, 549–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravano D.M., Hoyer K.K. (2013) Promotion and prevention of autoimmune disease by CD8+ T cells. J. Autoimmun., 45, 68–79. [DOI] [PubMed] [Google Scholar]
- Hahn M. et al. (2005) Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat. Immunol., 6, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawse W.F. et al. (2014) TCR scanning of peptide/MHC through complementary matching of receptor and ligand molecular flexibility. J. Immunol., 192, 2885–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman L.M. et al. (2019) Improving T cell receptor on-target specificity via structure-guided design. Mol. Therapy, 27, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Henikoff J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler P.D. et al. (2000) In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. USA, 97, 5387–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V. et al. (2017) NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol., 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kula T. et al. (2019) T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell, 178, 1016–1028.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzarotti E. et al. (2018) Identification of the cognate peptide-MHC target of T cell receptors using molecular modeling and force field scoring. Mol. Immunol., 94, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzarotti E. et al. (2019) T-cell receptor cognate target prediction based on paired α and β chain sequence and structural CDR loop similarities. Front. Immunol., 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A. et al. (2008) On-the-fly rotamer pair energy evaluation in protein design. In: Lecture Notes in Computer Science (Including Subseries Măndoiu,I. et al. (eds.) Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), 4983 LNBI Bioinformatics Research and Applications. ISBRA 2008. Lecture Notes in Computer Science, vol 4983. Springer, Berlin, Heidelberg, pp. 343–354.
- Leaver-Fay A. et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol., 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2005) Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol., 23, 349–354. [DOI] [PubMed] [Google Scholar]
- Linette G.P. et al. (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood, 122, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald W. a. et al. (2009) T cell allorecognition via molecular mimicry. Immunity, 31, 897–908. [DOI] [PubMed] [Google Scholar]
- Mason D. (1998) A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today, 19, 395–404. [DOI] [PubMed] [Google Scholar]
- Maynard J. et al. (2005) Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity, 22, 81–92. [DOI] [PubMed] [Google Scholar]
- Morgan R. a. et al. (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother., 36, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell E.W. et al. (2011) Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k). J. Immunol., 186, 5823–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M. et al. (2005) The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics, 57, 33–41. [DOI] [PubMed] [Google Scholar]
- Nivon L.G. et al. (2013) A pareto-optimal refinement method for protein design scaffolds. PLoS One, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell T.J. et al. (2018) MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst., 7, 129–132.e4. [DOI] [PubMed] [Google Scholar]
- Obenaus M. et al. (2015) Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol., 33, 402–407. [DOI] [PubMed] [Google Scholar]
- Ogishi M., Yotsuyanagi H. (2019) Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front. Immunol., 10, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce B.G., Weng Z. (2013) A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci., 22, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J.-B. et al. (2003) CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat. Immunol., 4, 241–247. [DOI] [PubMed] [Google Scholar]
- Reiser J.B. et al. (2002) A T cell receptor CDR3β loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity, 16, 345–354. [DOI] [PubMed] [Google Scholar]
- Riley T.P., Baker B.M. (2018) The intersection of affinity and specificity in the development and optimization of T cell receptor based therapeutics. Semin. Cell Dev. Biol., 84, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T.P. et al. (2018) T cell receptor cross-reactivity expanded by dramatic peptide–MHC adaptability. Nat. Chem. Biol., 14, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T.P. et al. (2019) Structure based prediction of neoantigen immunogenicity. Front. Immunol., 10, 2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D. et al. (2018) Predicting CD4 T-cell epitopes based on antigen cleavage, MHCII presentation, and TCR recognition. PLoS One, 13, e0206654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D.R. et al. (2011) Disparate degrees of hypervariable loop flexibility control T-cell receptor cross-reactivity, specificity, and binding mechanism. J. Mol. Biol., 414, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewell A.K. (2012) Why must T cells be cross-reactive? Nat. Publish. Group, 12, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T. et al. (2005) ROCR: visualizing classifier performance in R. Bioinformatics, 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Singh N.K. et al. (2017) Emerging concepts in TCR specificity: rationalizing and (Maybe) predicting outcomes. J. Immunol., 199, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.C. et al. (2019) Machine-learning prediction of tumor antigen immunogenicity in the selection of therapeutic epitopes. Cancer Immunol. Res., 7, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C.-W. et al. (2011) POPISK: t -cell reactivity prediction using support vector machines and string kernels. BMC Bioinformatics, 12, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tynan F.E. et al. (2005) T cell receptor recognition of a “super-bulged” major histocompatibility complex class I-bound peptide. Nat. Immunol., 6, 1114–1122. [DOI] [PubMed] [Google Scholar]
- Wooldridge L. et al. (2012) A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem., 287, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig K.W. et al. (1994) Clonal expansion and persistence of human T cells specific for an lmmunodominant myelin basic protein peptide. J. Immunol., 152, 5581–5592. [PubMed] [Google Scholar]
- Zhao W., Sher X. (2018) Systematically benchmarking peptide-MHC binding predictors: from synthetic to naturally processed epitopes. PLoS Comput. Biol., 14, e1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.