

Abstract
Gamma secretase inhibitors (GSIs), initially developed as Alzheimer's therapies, have been repurposed as anticancer agents given their inhibition of Notch receptor cleavage. The success of GSIs in preclinical models has been ascribed to induction of cancer stem‐like cell differentiation and apoptosis, while also impairing epithelial‐to‐mesenchymal transition and sensitizing cells to traditional chemoradiotherapies. The promise of these agents has yet to be realized in the clinic, however, as GSIs have failed to demonstrate clinical benefit in most solid tumors with the notable exceptions of CNS malignancies and desmoid tumors. Disappointing clinical performance to date reflects important questions that remain to be answered. For example, what is the net impact of these agents on antitumor immune responses, and will they require concurrent targeting of tumor‐intrinsic compensatory pathways? Addressing these limitations in our current understanding of GSI mechanisms will undoubtedly facilitate their rational incorporation into combinatorial strategies and provide a valuable tool with which to combat Notch‐dependent cancers. In the present review, we provide a current understanding of GSI mechanisms, discuss clinical performance to date, and suggest areas for future investigation that might maximize the utility of these agents.
Implications for Practice
The performance of gamma secretase inhibitors (GSIs) in clinical trials generally has not reflected their encouraging performance in preclinical studies. This review provides a current perspective on the clinical performance of GSIs across various solid tumor types alongside putative mechanisms of antitumor activity. Through exploration of outstanding gaps in knowledge as well as reasons for success in certain cancer types, the authors identify areas for future investigation that will likely enable incorporation of GSIs into rational combinatorial strategies for superior tumor control and patient outcomes.
Keywords: Gamma secretase inhibitors, Gamma secretase, Notch, Cancer, Solid tumors, Clinical trial
Short abstract
This review focuses on the current understanding of gamma secretase inhibitors (GSI), as well as clinical performance to date and areas for future investigation that might maximize the utility of these agents.
Development of Gamma Secretase Inhibitors
Efforts to curtail amyloid beta production in treatment of Alzheimer's disease led to development of gamma secretase inhibitors (GSIs) [1], which advanced to phase III trials [2]. Adverse events and lack of efficacy limited the utility of these agents [3, 4], but a shared proteolytic processing pathway between amyloid beta and the Notch family has been implicated in off‐target effects [5]. As a result, these agents have been repurposed for their ability to broadly inhibit the Notch pathway. In the current review, we discuss gamma secretase (GS) inhibition as a potential point of intervention to broadly target dysregulated Notch signaling in cancer.
Gamma Secretase Inhibitors and the Notch Pathway
Notch is an evolutionarily conserved pathway that transmutes extracellular information, through cell–cell interactions, into regulatory events guiding cell fate decisions. One of four Notch surface protein receptors (Notch 1–4 paralogs) binds to one of three delta‐like ligands (Dll) or one of two jagged ligands on itself (cis‐activation) or a neighboring cell (transactivation). Receptor‐ligand binding triggers sequential cleavage events by a disintegrin and metalloprotease (ADAM) and GS that culminate in release of the Notch intracellular domain (NICD) and translocation to the nucleus. Interaction with the RBP‐J (or CSL) transcription factor and recruitment of a transcriptional coactivator, mastermind‐like family (MAML), then enact changes in gene expression (Fig. 1) [6]. Many layers of complexity help explain this pathway's pleiotropic roles in embryonic development and cellular homeostasis, including variable ligand glycosylation [7] and dynamic ligand binding patterns that contribute to activation of distinct target genes [8]. Moreover, Notch signaling can block cell differentiation to preserve progenitor cells and maintain these pools through symmetric or asymmetric division [9]. In this manner, coordinated Notch signaling is integral to muscle, vasculature, cardiac, hematopoietic, nervous system, and pancreatic development [10].
Figure 1.
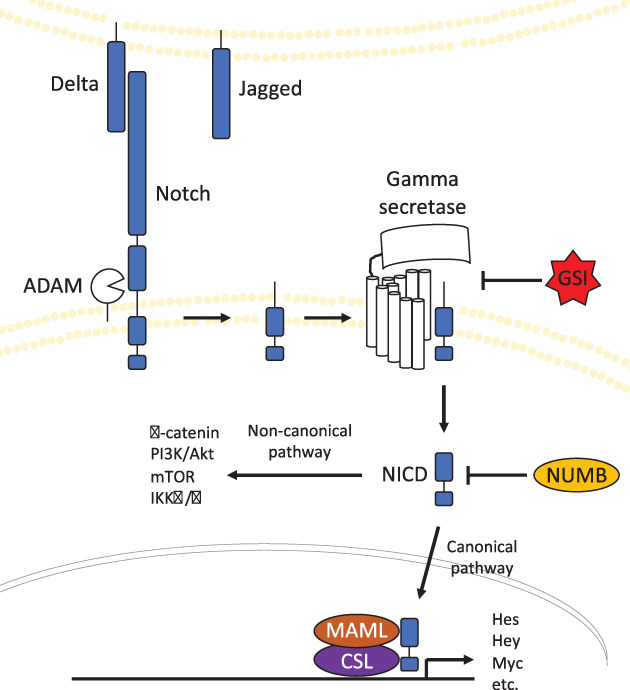
Schematic representation of Notch signaling pathway. A Notch receptor binds to cell‐bound or soluble Delta/Jagged ligands. Bound Notch is first cleaved by ADAM to release the extracellular portion. Gamma secretase then catalyzes a second cleavage event, liberating NICD into the intracellular space. NICD can interact with other pathways independent of transcriptional activity through noncanonical signaling or translocate to the nucleus, associate with CSL and MAML, and promote expression of target genes through canonical signaling.Abbreviations: GSI, gamma secretase inhibitor; MAML, mastermind‐like family; NICD, Notch intracellular domain.
Adaption of this pathway in malignant transformation was first recognized in T‐cell acute lymphoblastic leukemia (T‐ALL), wherein a t(7;9) chromosomal translocation positions a truncated Notch1 protein, similar to the NICD1, under control of the T‐cell receptor β locus [11]. In fact, activating mutations in Notch‐1 are present in the majority of T‐ALL cases [12]. Contribution of Notch signaling to tumorigenesis, however, is somewhat more nuanced, as coexpression of additional oncogenic alterations are typically required for transformation [13]. For example, truncated Notch1 can transform HC11 mouse mammary epithelial cells in vitro [13], whereas in transgenic mice Notch1 cooperates with c‐neu/erbB2 and c‐myc to induce transformation in vivo [13, 14]. Likewise, transformation of baby rat kidney cells in vivo required the presence of an additional oncogene, E1A [15]. NICD1 signaling was not sufficient to induce cancer initiation but could cooperate with AKT, Myc, and Ras/Raf/MAPK to drive formation of prostate adenocarcinoma in mice and promote more aggressive phenotypes in human prostate cancer cells [16]. Accordingly, although mutations in Notch regulatory sequences or domains (e.g., PEST [17]) may be sufficient to drive tumorigenesis in some instances, the majority of solid tumors with oncogenic Notch signaling might instead harbor aberrations in regulatory proteins (e.g., NUMB [18], TBC1D15 [19]), signaling partners, or relative dosage [20]. Thus, the role of Notch in tumorigenesis may be context dependent, acting most convincingly in cells already poised for transformation.
Contributions of Notch signaling to cancer progression extend beyond its roles in tumorigenesis. Cancer stem(‐like) cell proliferation and renewal are integral to disease progression and are promoted by Notch ligand expression in niche cells (in human glioblastoma [21], breast cancer [22], and colorectal cancer [23]) and broadly by dysregulated intracellular pathway signaling [24]. Activation of Notch promotes epithelial‐to‐mesenchymal transition (EMT), which then facilitates disease invasion and metastatic spread [25]. Further complicating its role in tumor progression, juxtacrine and paracrine Notch signaling help shape the tumor microenvironment (TME) [26]. Notch also participates as a coregulator of other tumorigenic pathways. For example, noncanonical Notch signaling, independent of its transcription factor function, titrates intracellular levels of active Wnt/β‐catenin in a context dependent manner [27]. Through these processes, the Notch pathway can instigate cancer resistance to traditional therapeutic strategies, rendering it a critical oncologic target.
Gamma Secretase: Structure and Function
Gamma secretase is a large heterotetrameric transmembrane protein complex composed of presenilin (PS), nicastrin (Nct), anterior pharynx defective‐1 (Aph‐1), and presenilin enhancer‐2 in an equimolar ratio. Two PS isoforms (PS1 or PS2) and three different Aph‐1 isoforms (Aph‐1aS, Aph‐1aL, Aph‐1ab) can be incorporated into the quaternary protein structure, each with tissue‐specific expression patterns [28]. Collectively, these proteins form a 19‐pass transmembrane disk structure organized around a central cleft with the large ectodomain of Nct located directly above. Following an initial cleavage event, termed "ectodomain shedding," substrates are recognized and positioned within the intermembrane cleft of GS by Nct, adjacent to the catalytic site of PS [29]. In the case of Notch proteins, this enables cleavage of the intracellular domain, translocation to the nucleus, and initiation of transcriptional changes. The activity of GS extends well beyond Notch, however, to encompass more than 90 substrates [30], including half of the human receptor tyrosine kinases [31].
Complexity is introduced to this process through several mechanisms. First, the specific combination of PS and Aph‐1 isoforms likely dictate distinct substrate and cleavage site preferences, influenced by tissue of origin and intracellular isoform equilibria. Activity of assembled complexes is then further regulated by GS modulators, nonessential proteins that interact with the GS quaternary structure [32].
Gamma Secretase Inhibitors and Cancer
GS cleaves Notch receptors 1–4; however, pharmacologic inhibition with GSIs does not block signaling of each Notch receptor equally. In fact, clinical GSIs are pharmacologically distinct, each with a discrete profile of Notch inhibition. Moreover, low concentrations of GSIs can actually potentiate cleavage of select Notch receptors [33]. It is logical to extrapolate these findings to infer that unique properties of individual GSIs likely influence utility for specific clinical indications. Thus far, observational bias has precluded a greater mechanistic understanding of GSIs in that many investigations have been limited to readouts of Notch and/or amyloid precursor protein (APP) cleavage [34]. Common GSIs having reached various stages of development with corresponding half‐maximal inhibitory concentration (IC50) values for different Notch receptors and APP are provided in Table 1 [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45].
Table 1.
Common GSIs at various stages of (pre‐)clinical development. IC50 values of each compound for different Notch receptors as well as those for APP (inhibition of amyloid beta production) are provided
| GSI | Names | IC50 (nM) | Clinical status | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| Notch1 | Notch2 | Notch3 | Notch4 | APP | ||||
| BMS‐906024 | AL101 | 0.29–1.6 | 0.14–0.7 | 0.32–3.4 | 0.71–2.9 | II | Gavai, 2015 [35]; Ran, 2017 [33]; Lessard, 2019 [36] | |
| BMS‐708163 | Avagacestat | 20.6–58 | 0.30–0.79 | II | Mitani, 2012 [38]; Gillman, 2010 [37] | |||
| BMS‐986115 | I | |||||||
| DAPT | GSI‐IX | 4.9 | 85.22 | 552.4–623.5 | 51.5 | 29 | preclinical | Ran, 2017 [33]; Lessard, 2019 [36]; Martone, 2009 [39] |
| MK‐0752 | 55–87.44 | 46.62 | 146.3–370 | 191 | 5 | II | Ran, 2017 [33]; Deangelo, 2006 [41]; Lessard, 2019 [36]; Cook, 2010 [40] | |
| PF‐03084014 | Nirogacestat | 0.6–13.3 | 0.002–0.01 | 1.21–15.07 | 0.81–10.77 | 1.2–6.2 | III | Wei, 2010 [43]; Ran 2017 [33]; Lessard, 2019 [36]; Lanz, 2010 [42] |
| RO4929097 | 0.46–4 | 2.24 | 19.8–28.31 | 3.4 | 14 | II | Ran, 2017 [33]; Luistro, 2009 [47]; Lessard, 2019 [36]; Gu, 2017 [44] | |
| GSI‐953 | Begacestat | 8 | I | Martone, 2009 [39] | ||||
| LY‐450139 | Semagacestat | 7.02–14.1 | 38.7 | 390.9–523.3 | 45.98 | 10.9–38 | III | Ran, 2017 [33]; Mitani, 2012 [38]; Lessard, 2019 [36]; Martone, 2009 [39] |
| LY‐411,575 | 0.129 | 0.119 | preclinical | Lewis, 2003 [45] | ||||
| LY‐900009 | 0.27 | I | Pant, 2012 [109] | |||||
Abbreviations: APP, amyloid precursor protein; GSI, gamma secretase inhibitor; IC50, half‐maximal inhibitory concentration.
Mechanisms of Gamma Secretase Inhibition in Cancer
Targeting the Notch pathway with GSIs has led to impaired cancer cell growth and tumor progression across a number of models, reflecting the common upregulation of this pathway. For example, GSI‐I and LY‐411,575 demonstrated ability to directly induce apoptosis in Kaposi sarcoma cells in vitro and in vivo, an effect that could be rescued by transfection and enforced expression of NICD [46]. Increased apoptosis following GSIs has also been suggested to be mediated by proteosomal inhibition. The combination of a GSI (GSI‐XII) plus the proteasome inhibitor bortezomib induced apoptosis in multiple myeloma cell lines and primary patient samples in vitro that could not be rescued by NICD overexpression [47]. However, restoring proteasome function with edaravone effectively abrogated apoptosis [47], a finding similarly observed in human breast cancer cell lines [48]. Interestingly, treatment of glioblastoma tumor‐initiating cells with GSI‐I–induced apoptosis. This was not the case with selective Notch inhibitors, suggesting a predominant contribution of proteasome inhibition relative to Notch inhibition in this model [49].
As mentioned, Notch is an important target of GS activity. It is a cell fate sensor and acts as a receptor to a variety of canonical and noncanonical ligands including the Delta, Jagged, Lag2 class of cell surface proteins [50]. Upon ligand binding, Notch isoforms are dependent upon cleavage by ADAM metalloproteases and GS. This allows the NICD to translocate to the nucleus, where it forms a transcriptional activation complex with CSL and coactivators of the MAML family [51]. One important downstream effector whose expression is activated by Notch is Hes1, a transcriptional repressor that prevents irreversible cell cycle exit [52]. Upregulated Notch signaling, therefore, might contribute to maintenance of cancer cell proliferative potential. Supporting this hypothesis, pancreatic adenocarcinoma (PDAC) cell lines downregulated Hes1 following treatment with the GSI MRK‐003 in vitro. Similarly, pretreatment of PDAC xenografts reduced tumor growth in nude mice by decreasing the number of tumor‐initiating cells [53]. GSI treatment also decreased Hes1 expression and tumor sphere formation in human ovarian cancer (SKOV3 and HO8910) [54] and a panel of melanoma cells [55] following treatment in vitro. Treatment of primary melanoma xenografts with the GSI RO4929097 led to decreased expression of stem cell markers and decreased tumor formation in serial xenograft transplants despite no additional treatment cycles [55]. Brief treatment of ERBB2 murine mammary tumor cells with MRK‐003 was sufficient to drive histologic changes and sustained impairment of tumor formation in syngeneic mice [56]. A durable impairment of proliferation following GS inhibitor exposure has also been observed in T‐cell acute lymphocytic leukemia [57], non‐small cell lung cancer [58], and prostate cancer [59]. These findings suggest that blocking Notch signaling with GSIs induces irreversible differentiation and cell cycle exit of tumor‐initiating cells.
Overcoming Treatment Resistance with Gamma Secretase Inhibitors
Although GS inhibitors can impair tumor growth through multiple mechanisms, combination with traditional therapeutic modalities might mitigate resistance mediated through Notch upregulation. Sequential treatment of A2780 and OVCAR3 human ovarian cancer cells with cisplatin followed by the GSI MK‐0725 significantly reduced growth in vitro (compared with concurrent administration or the reverse order) and in vivo [60]. Similarly, sequential administration of radiation followed by GSI in human NSCLC cells was concluded to more effectively impair Notch‐1 or Notch‐3 upregulation (pending cell line used), delaying tumor growth [61, 62]. In contrast, pretreatment with the GSI DAPT resensitized platinum resistant A2780/CP70 and OV2008/C13 cell lines to subsequent cisplatin treatment [63]. These discordant findings may reflect different Notch signaling equilibria at baseline, as resistant cell lines likely upregulated Notch signaling during acquisition of platinum resistance [64, 65].
More generally, concurrent treatment with GSIs and traditional chemotherapeutics has demonstrated superior tumor control in a variety of models. In prostate xenografts, PF‐0308014 plus docetaxel cotreatment significantly reduced tumor growth [59]. Likewise, coculture of human head and neck squamous cell carcinoma with DAPT or AG490 (impairs STAT3 activation) increased sensitivity to cisplatin in vitro [66]. Efficacy of these combinations was postulated to be a function PI3K/Akt pathway downregulation, potentially through disrupted Notch crosstalk, in preclinical models of human retinoblastoma [67], lung [68], gastric [69], colon [70], triple‐negative breast [71], and ERBB2‐driven murine breast [72] cancers. A somewhat disparate result was found in a study of multiple myeloma and non‐Hodgkin's lymphoma cell lines and patient samples, which nearly homogeneously expressed Notch‐1 and Notch‐2. MRK‐003 downregulated Notch targets and induced apoptosis in vitro but modestly increased pAkt; MRK‐003 and Akt inhibitor cotreatment then drove impressive apoptosis [73]. An observation that illustrates potential for compensatory pathways to subvert GSI monotherapy.
Enhanced efficacy of GSIs in combinatorial strategies is likely due, in part, to impairment of EMT, which commonly involves acquisition of stem cell features and upregulation of multidrug resistance transporters [74]. Specifically, upregulation of SNAI1/2, ZEB1/2, and TWIST1/2 regulates this process [75]. In the human hepatocellular carcinoma cell line MHCC97H, combination of sorafenib plus PF‐03084014 significantly reduced expression of SNAI1/2, Nanog, Oct4, and the multidrug resistance transporter ABCG2 [76]. Sensitization to chemotherapy was also demonstrated using MKN45 gastric cancer cells. Specifically, CD44+ cells, which strongly upregulated Notch1 signaling relative to CD44−, became sensitive to 5‐fluorouracil (5‐FU) by combination with DAPT. This correlated with loss of Snail, vimentin, and N‐cadherin protein expression alongside decreased migration and invasion in vitro [77]. In the case of ovarian cancer, a disease characterized by early and diffuse intraperitoneal metastases, EMT is driven by positive feedback between TGFβ/Smad and Notch signaling, a process in which each of the Notch1–4 receptors have been implicated [78, 79, 80]. GSI blockade of Notch signaling impaired TGFβ‐induced EMT in multiple cell lines, downregulating SNAI1/2, TWIST, and ZEB1 as well as migration and invasiveness [79, 80, 81]. Thus, GS inhibition of EMT might stymie acquisition of drug resistance and limit metastatic capacity of established tumors.
Clinical Trials with Gamma Secretase Inhibitors
Ability to drive tumor cell differentiation, reduce cancer stem cell burden, and sensitize tumors to traditional chemoradiotherapy strategies through multiple mechanisms in preclinical studies has spurred clinical investigation of GSIs in multiple cancer types (Table 2).
Table 2.
Clinical trials using gamma secretase inhibitors (GSIs) organized by cancer type of participating patients. Specific GSI used, and potential combinatorial agents, are provided alongside trial phase, number of patients with indicated cancer type, and trial outcomes
| Cancer and study cohort | GSI | Combination | Phase | Patients | n | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Ovarian | |||||||
| EOC: platinum resistant | RO4929097 | II | Recurrent or metastatic | 45 | PFS: 1.3 mo | Diaz‐Padilla et al. 2015 [112] | |
| Advanced solid tumors | RO4929097 | I | Refractory to standard therapy | 9 | 3 clinical benefit | Tolcher et al. 2012 [93] | |
| Advanced solid tumors | RO4929097 | Gemcitibine | I | Refractory to standard therapy | 2 | safe combination | Richter et al. 2014 [91] |
| Advanced solid tumors | RO4929097 | Temsirolimus | Ib | Refractory to standard therapy | 1 | safe combination | Diaz‐Padilla et al. 2013 [110] |
| Advanced solid tumors | RO4929097 | Cediranib | I | Refractory to standard therapy | safe combination | Sahebjam et al. 2013 [111] | |
| Advanced solid tumors | LY900009 | I | Refractory to standard therapy | 11 | MTD 30 mg 3×/wk | Pant et al. 2016 [109] | |
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 3 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [84] | |
| Colorectal | |||||||
| Advanced solid tumors | MK‐0752 | Dalotuzumab | I | Refractory to standard therapy | 9 | Safe combination | Brana et al. 2014 [122] |
| Advanced solid tumors | RO4929097 | Cediranib | I | Refractory to standard therapy | 6 | Safe combination | Sahebjam et al. 2013 [111] |
| Advanced solid tumors | PF‐03084014 | I | Refractory to standard therapy | 11 | Safe, response in desmoid tumors, overall PFS 1.6 mo | Messersmith et al. 2015 [85] | |
| Stage IV, metastatic | RO4929097 | II | Refractory to standard therapy | 37 | Safe, PFS 1.8 mo, dose/schedule have minimal activity | Strosberg et al. 2012 [121] | |
| Advanced solid tumors | RO4929097 | Capecitabine | I | Refractory to standard therapy | 18 | Safe, promising activity in colon cancer | LoConte et al. 2015 [123] |
| Advanced solid tumors | RO4929097 | I | Refractory to standard therapy | 12 | 3 clinical benefit | Tolcher et al. 2012 [93] | |
| Advanced solid tumors | LY900009 | I | Refractory to standard therapy | 5 | MTD 30 mg 3×/wk | Pant et al. 2016 [109] | |
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 16 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [88] | |
| Advanced solid tumors | BMS‐986115 | I | Refractory to standard therapy | 5 | Intermittent schedule likely preferable, better tolerated | Aung et al. 2018 [120] | |
| Desmoid | |||||||
| Advanced solid tumors | BMS‐986115 | I | Refractory to standard therapy | 1 | Intermittent schedule likely preferable/better tolerated | Aung et al. 2018 [120] | |
| Advanced solid tumors | PF‐03084014 | I | Refractory to standard therapy | 9 | Safe, response in desmoid tumors, overall PFS 1.6 mo | Messersmith et al. 2015 [85] | |
| Unresectable | PF‐03084014 | II | Progressive disease after more than one line | 17 | Promising strategy for desmoid tumors | Chen et al. 2016 [60] | |
| Breast | |||||||
| Advanced breast tumors | MK‐0752 | Docetaxel | I | Refractory to first‐line chemotherapy | 30 | Preliminary evidence of clinical efficacy | Schott et al. 2013 [107] |
| Advanced TNBC | PF‐03084014 | Docetaxel | Ib | Refractory to standard therapy | 29 | Median PFS 4.1 mo, 6 mo PFS 17.1% | Locatelli et al. 2017 [106] |
| Metastatic ER+ breast cancer | RO4929097 | Exemestane | Ib | Refractory to standard therapy | 15 | Safe combination, preliminary evidence of stable disease | Means‐Powell et al. 2012 [105] |
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 24 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [84] | |
| Advanced solid tumors | PF‐03084014 | I | Refractory to standard therapy | 7 | Safe combination, overall PFS 1.6 mo | Messersmith et al. 2015 [85] | |
| Advanced solid tumors | RO4929097 | Gemcitibine | I | Refractory to standard therapy | 5 | Safe combination | Richter et al. 2014 [91] |
| Advanced solid tumors | RO4929097 | I | Refractory to standard therapy | 10 | Safe combination | Tolcher et al. 2012 [93] | |
| Glioma | |||||||
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 42 | Modest level of clinical activity in gliomas | Krop et al. 2012 [84] | |
| CNS malignancies | MK‐0752 | I | Refractory pediatric tumors | 4 | Well tolerated, trial not completed | Hoffman et al. 2015 [99] | |
| CNS malignancies | MK‐0752 | I | Refractory pediatric tumors | 9 | Well tolerated | Fouladi et al. 2011 [98] | |
| Malignant gliomas | RO4929097 | Bevacizumab | I | Refractory to standard therapy | 13 | Well tolerated, PFS 3.7 mo | Pan et al. 2016 [100] |
| Glioblastoma, grade III AA | RO4929097 | Temozolomide, RT | I | Newly diagnosed | 21 | Well tolerated, glioblastoma median PFS 13 mo | Xu et al. 2016 [101] |
| Melanoma | |||||||
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 3 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [84] | |
| Advanced solid tumors | RO4929097 | I | Refractory to standard therapy | 24 | Safe combination | Tolcher et al. 2012 [93] | |
| Metastatic melanoma | RO4929097 | II | Chemotherapy naïve | 36 | Safe combination, PFS: 1.5 mo | Lee et al. 2015 [69] | |
| Pancreatic | |||||||
| Metastatic PDAC | RO4929097 | II | Refractory to standard therapy | 18 | Well‐tolerated, PFS: 1.5 mo | De Jesus‐Acosta et al. 2014 [89] | |
| Advanced solid tumors | RO4929097 | Gemcitibine | I | Refractory to standard therapy | 3 | Safe combination | Richter et al. 2014 [91] |
| Advanced solid tumors | PF‐03084014 | I | Refractory to standard therapy | 3 | Safe combination, overall PFS 1.6mo | Messersmith et al. 2015 [85] | |
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 2 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [84] | |
| Stage III/IV PDAC | MK‐0752 | Gemcitibine | I | Prior therapy acceptable | 44 | Well tolerated, PFS: 5.6 mo | Cook et al. 2018 [92] |
| NSCLC | |||||||
| Advanced solid tumors | MK‐0752 | I | Refractory to standard therapy | 3 | Inhibited Notch, minimal clinical activity as monotherapy | Krop et al. 2012 [84] | |
| Advanced solid tumors | PF‐03084014 | I | Refractory to standard therapy | 5 | Safe combination, overall PFS 1.6 mo | Messersmith et al. 2015 [85] | |
| NSCLC | RO4929097 | Erlotinib | I/II | Refractory to standard therapy | 16 | Safe combination, does not merit further eval | Gold et al. 2013 [86] |
Abbreviations: AA, anaplastic astrocytoma; CNS, central nervous system; EOC, epithelial ovarian cancer; ER, estrogen receptor; MTD, maximum tolerated dose; NSCLC, non‐small cell lung cancer; PDAC, pancreatic adenocarcinoma; PFS, progression‐free survival; TNBC, triple‐negative breast cancer.
Lung Cancer
A meta‐analysis encompassing over 3,600 patients from 19 studies revealed a significant correlation between overexpression of Notch1 and Notch3 receptors, as well as the Notch pathway components DLL3 and HES1, and poor outcome in patients with NSCLC [82]. Further suggesting a role for targeting Notch in NSCLC, GSIs can sensitize NSCLC cells to traditional chemotherapeutics and impair development of resistance [64, 83]. A small cohort of patients with lung cancer have been included in phase I trials using MK‐0752 or PF‐03084014 monotherapy to treat advanced‐stage solid tumors refractory to traditional measures; however, no evidence of clinical activity in patients with lung cancer was reported [84, 85]. In an effort to combat inevitable resistance to erlotinib, a phase I/II trial treated 16 patients with NSCLC with RO4929097 plus erlotinib. One patient had a partial response, four experienced stable disease at 6 weeks, and median progression‐free survival (PFS) was 42 days (64 days in patients with prior progression on erlotinib) [86]. Enrollment of heavily pretreated patients likely selected for aggressive disease and contributed to a lack of notable efficacy in these trials.
Pancreatic Cancer
Interestingly, constituent proteins of the GS tertiary complex, specifically APH1A, are upregulated in malignant relative to normal pancreatic tissue and higher expression may correlate with worse prognosis [87]. Correspondingly, Notch receptors 1–4 and the downstream target Hes1 are significantly increased in human malignant pancreatic tissue [88]. Significantly increased expression in PDAC and known contribution to tumor progression served as rationale for clinical investigation. Eighteen patients with metastatic refractory PDAC were treated with RO4929097 monotherapy in a phase II trial. Among 12 evaluable patients, three experienced stable disease as best response, and the median PFS was 1.5 months. A trend toward decreased HeyL expression was observed on pre‐ and post‐treatment biopsies [89]. Patients with pancreatic cancer included in phase I trials of advanced solid tumors treating with PF‐03084014 [85] or MK‐0752 [84] similarly did not experience clinical efficacy. Preclinical studies have suggested that antimetastatic activity of GSIs might be improved by combination with gemcitabine, providing rationale for further investigation of this combination in PDAC [90]. One of three patients with pancreatic cancer experienced prolonged stable disease in a phase I trial of RO4929097 plus gemcitabine [91]. However, in a phase I trial of exclusively stage III or IV pancreatic ductal adenocarcinoma, the clinical activity seen with MK‐0752 and gemcitabine combination was similar to what would be expected for gemcitabine alone. This led the authors conclude that the combination used does not warrant further evaluation [92].
Melanoma
Twenty‐four patients with melanoma were enrolled in a phase I trial of RO4929097 for those with advanced‐stage refractory solid tumors. Melanoma was among the most frequently benefited cancers, with one near complete response (by fluorodeoxyglucose–positron emission tomography) and one minor response (by RECIST). A total of four of 24 patients with melanoma were reported to have seen clinical benefit [93]. Following encouraging results, a phase II trial was initiated treating chemotherapy naïve patients with metastatic melanoma with RO4929097 monotherapy. One partial response and eight instances of stable disease were observed in 32 evaluable patients, with a median PFS of 1.5 months. Relative to patients with melanoma treated in the phase I trial, those in the phase II trial received a lower dose of RO4929097, which the authors postulate might contribute to poorer responses [94].
Gliomas
Notch signaling is frequently activated in human gliomas and maintains self‐renewal capacity of glioma stem cells [95]. It follows that expression of Notch‐1 predicts poor patient survival in proneural and classic glioblastomas [96]. Importantly, enrichment of Notch signaling components correlates with response to GSIs in glioma tumor‐initiating cells [97]. These findings served as the basis for clinical development of GSIs in patients with gliomas and other central nervous system (CNS) tumors. In a phase I trial of pediatric patients with CNS tumors refractory to solid tumors, MK‐0752 was found to be well tolerated; however, among the nine study patients with gliomas, no objective responses and one prolonged stable disease were observed. The study did identify consistent Hes1 and Hes5 staining across all tumors, suggesting Notch signaling is broadly activated in pediatric brain tumors [98]. A similar phase I trial treating pediatric patients with refractory CNS tumors found that, although MK‐0752 decreased NICD1 expression from baseline, the GSI had limited clinical activity. Ultimately, the sponsor withdrew support prior to trial completion [99]. Somewhat more encouraging results were observed when adult patients with gliomas were treated with MK‐0752 monotherapy. Here, among 42 patients with gliomas, a complete response of greater than a year was seen in anaplastic astrocytoma, as was stable disease greater than 1 year in glioblastoma multiform. A total of 10 patients with gliomas (24% of study patients with gliomas) experienced stable disease [84].
Rational combination of GSIs with existing treatment strategies has led to improved clinical outcomes. For instance, RO4929097 was used in combination with bevacizumab in an effort to preempt outgrowth of aggressive disease seen in bevacizumab refractory cases. Thirteen patients with malignant gliomas were treated with both agents in a phase I trial and, of 12 evaluable patients, 2 had radiographic responses, a complete response and a partial response. Median overall survival was 10.9 months, with a median PFS of 3.7 months [100]. Alternatively, combination of RO4929097 plus temozolomide and radiotherapy demonstrated a trend toward decreased intratumoral Ki67 staining and significantly decreased NICD1‐positive cells, the degree of which correlated with overall survival. GSI alone led to decreased glioma perfusion on dynamic contrast enhanced magnetic resonance imaging and significantly decreased CD133+ cells in tumor explants [101]. Reduced Notch signaling in these trials indicates GSIs can effectively cross the blood brain barrier and reach therapeutic concentrations. As such, further clinical investigations of GSIs in rational combinatorial strategies are warranted in malignant CNS tumors.
Breast Cancer
Oncogenic Notch signaling is also implicated in breast cancer, which represents approximately 30% of all new cancer diagnoses in female patients within the U.S. [102]. Here, increased expression of Notch‐1 and Jagged1 have been found to negatively correlate with overall survival [103]. Association of Notch signaling with disease progression led to the investigation of GSIs in clinical trials. A phase I trial of MK‐0752 in patients with advanced solid tumors, including 24 patients with breast cancer, demonstrated minimal clinical activity of this GSI in breast cancer [84]. Absence of significant clinical efficacy was also reported in phase I trials treating advanced solid tumors with PF‐03084014 [85] and RO4929097 [82]. Although GSIs are ineffective as a monotherapy, preclinical ability of GSIs to reduce cancer stem cell proliferative capacity and curtail chemoresistance has led to investigation of clinical combinatorial strategies [104]. A phase Ib trial of RO4929097 plus exemestane in 15 patients with estrogen receptor–positive metastatic breast cancer showed limited efficacy with one partial response, six stable disease, and seven progressive disease [105]. Combination of RO4929097 plus gemcitabine has also been investigated in a phase I trial of patients with advanced solid tumors, including five with breast cancer, although efficacy in this cohort was not reported [91]. A phase Ib clinical trial combining PF‐03084014 and docetaxel observed moderate clinical efficacy in patients with advanced triple‐negative breast cancer, with four partial responses (of 25 evaluable patients) and a median PFS of 4.1 months [106]. Encouragingly, a similar phase I clinical trial of MK‐0752 plus docetaxel combination therapy in patients with breast cancer resulted in 11 partial responses, 9 patients with stable disease, and 3 with progressive disease out of 24 evaluable. Combination therapy also reduced breast cancer stem cells and mammosphere‐forming efficacy in a subcohort of evaluable patients [107]. Although overall response rates have thus far been rather low, appropriate patient selection and mechanistic guided combinatorial strategies may guide future efficacy of GSIs in breast cancer.
Ovarian Cancer
Cytoplasmic NICD1 is highly expressed in human ovarian cancer and was found to be a poor prognostic factor for overall survival [108]. Following positive results in preclinical studies demonstrating prolonged response to cyclical GS inhibition, similar schedules have been adopted in multiple phase I clinical trials including patients with advanced‐stage ovarian cancer resistant to standard therapies. These include therapeutic intervention using RO4929097 [93], LY900009 [109], and MK‐0752 [84] alone or with RO4929097 in combination with gemcitabine [91], temsirolimus (mTOR inhibitor) [110], or cediranib (vascular endothelial growth factor inhibitor) [111]. These studies concluded these agents alone and in combination had an acceptable safety profile. Although three of nine patients with advanced ovarian cancer saw clinical benefit from RO4929097 monotherapy [93], the majority in these trials had brief stable or progressive disease. In a phase II study of 45 patients with recurrent or metastatic platinum resistant ovarian cancer, no objective responses were observed, and median progression‐free survival was 1.3 months on RO4929097, leading the authors to conclude this GSI monotherapy is not effective in ovarian cancer [112]. Interestingly, inhibition of Notch signaling with enoticumab, a fully human immunoglobulin G1 specific for Dll4, produced one partial response, six with stable disease, and two with reductions in cancer antigen‐125 that met response criteria among patients with ovarian cancer [113]. This suggests more specific targeting of Notch signaling, alone or in combination, might be a more favorable approach in this disease.
Colorectal Cancer
NICD1 and NICD3 signaling, denoted by nuclear localization on immunohistochemistry, correlates with recurrence and worse outcomes in patients with stage II and III colon cancer [114], whereas Notch 2 signaling might confer some degree of protection [115]. Downstream of Notch receptors, increased expression of Notch targets Hes1, Hey1, and Sox9 is associated with chemoresistance to 5‐FU, upregulated Wnt pathway signaling, metastases at the time of diagnosis, and worse overall survival [116, 117, 118]. In support of clinical investigation, the patient‐derived xenografts sensitive to PF‐03084014 were those with increased Notch and Wnt pathway signaling [119]. In a phase I trial of advanced solid tumors including 11 colorectal cancers, PF‐03084014 monotherapy was found to consistently reduce Hes4 expression but gave a median PFS of only 1.6 months [85]. Minimal antitumor activity was also observed in phase I trials of RO4929097 [93], LY900009 [109], MK‐0752 [84], and BMS‐986115 [120]. RO4929097 monotherapy in patients with metastatic, refractory colorectal cancer in a phase II study found no objective responses, 6 with stable disease, and 21 with progressive disease (a median PFS of 1.8 months), leading investigators to conclude minimal activity of this intervention [121]. Failure as a monotherapy has spurred combinatorial regimens. The combination of RO4929097 plus cediranib in a phase I trial of patients with advanced solid tumors, including 6 colorectal cancers of 20 total participants, found one partial response and prolonged disease stabilization in 11 patients [111]. An investigation of blocking Insulin‐like growth factor 1 receptor with dalotuzumab plus MK‐0752 was unsuccessful in the colorectal cancer cohort, as all evaluable patients on this combination had disease progression at the first radiologic evaluation [122]. Combination of RO4929097 with capecitabine proved somewhat more encouraging in a phase I evaluation of 30 advanced‐stage patients, 18 with colorectal cancer, as patients with colorectal cancer accounted for two of three observed partial responses. The investigators concluded this combination might be promising in fluoropyrimidine‐resistant metastatic colorectal cancer [123].
Desmoid Tumors
Although pathogenesis of desmoid tumors is frequently cited to involve perturbations in the Wnt pathway [124], upregulated Notch signaling is increasingly recognized as a contributor to disease progression [125]. In a phase I trial using PF‐03084014 for advanced‐stage solid tumors refractory to standard therapy, including nine patients with desmoid tumors (seven were evaluable), five patients had partial responses (by RECIST criteria) and two had prolonged disease stabilization [85]. On follow‐up of these same patients, all those that achieved a partial response continued to maintain duration of response for 47.9 to 73.6 months, and only one of seven had progressed. The mean clinical benefit of PF‐03084014 was 64 months compared with 13 for all prior interventions [126]. Encouraging results were also observed for a single patient with a desmoid tumor when treated with BMS‐986115 in a phase I trial [120]. Building upon these results, a phase II trial treating 17 patients with unresectable desmoid tumors having progressed on multiple lines of therapy PF‐03084014 was initiated. Of the 16 patients evaluable, 5 (29%) achieved a partial response (by RECIST criteria) and 11 patients experienced stable disease, with no cases of disease progression [127]. Success of the GSI PF‐03084014 (Nirogacestat) has spurred the phase III trial in adult patients with desmoid tumors (NCT03785964), recent breakthrough designation by the U.S. Food and Drug Administration, and orphan drug designation by the European Commission. Unravelling of mechanistic changes imposed by GSIs in these tumors may help translate their promise to other malignancies.
Current Limitations
A substantial limitation to our current understanding of these agents is the paucity of immune competent models used in their study. Although Notch inhibition may impede tumor progression, GSIs might conversely impair the antitumor immune response. CD8 T cells, the primary effectors of antitumor immunity, require Notch signaling for expression of canonical effector molecules, including interferon‐γ and granzyme B [128]. Lower levels of Notch‐1/2 in murine tumor‐infiltrating CD8 T cells then correlated with reduced cytotoxicity, whereas enforced Notch‐1 signaling increased cytotoxicity and led to superior tumor control [129]. Furthermore, activation of human T cells [94] and proliferation of murine CD8 T cells can be inhibited by GSIs in a dose‐dependent fashion [130]. Regulatory T cells (Tregs), a significant suppressive population within the TME, might also represent a substantial challenge. At baseline, Notch signaling destabilizes the Treg program; therefore, GSIs might promote Treg‐mediated suppressive functions and compromise antitumor immunity [131].
Elevated serum levels of interleukin (IL)‐6 and IL‐8, both signaling through STAT3, correlated with less‐favorable patient responses to RO4929097 [132]. Overexpressing these cytokines rendered a sensitive xenograft resistant to GSIs, possibly through abrogated Hes1 downregulation, an effect partially reversed by blocking IL‐8 [132]. Cytokine‐mediated resistance may be a factor of increased cancer stem cell activation (or formation) and proliferation. In a metastatic model of hormone therapy resistant human breast cancer, upregulation of IL‐6 increased pSTAT3 and Notch‐3 to increase CD133+ CD44− cancer stem cell renewal. Blockade of IL‐6 signaling effectively reversed the stem‐like features induced through this signaling cascade [133]. Stromal cells in the TME can also promote this resistance mechanism. Cancer‐associated fibroblasts increase cancer stem(‐like) cells through induction of IL‐6/pSTAT3/Notch signaling in human hepatocellular carcinoma. Nonselective Notch inhibition with RO4929097 or selective Notch‐1 inhibition with small interfering RNA impaired cancer‐associated fibroblast–induced stemness, as did inhibition of STAT3 phosphorylation [133]. Myeloid derived suppressor cells similarly induced stemness in human breast cancer xenografts, through IL‐6 and Notch‐2/3–driven STAT3 activation. Pathway inhibition with GSI‐I or pSTAT3 inhibition each partially reversed stemness, whereas the combination completely impaired stem cell formation [135]. Although apparently variable, which Notch receptor predominates likely depends on many factors including tissue of origin and Notch equilibria at baseline. Interestingly, in Notch3‐expressing breast cancer cells, MK‐0752 or RO4929097 inhibited Notch‐3, and thereby Hey2, significantly inducing IL‐6 and leading to increased numbers of breast cancer stem cells, an effect reversed by coadministration of GS inhibitor and anti–IL‐6R [136]. These findings seem to reiterate convergence of IL‐6, Notch, and STAT3 signaling; impairment of IL‐6 or Notch alone might be able to be rescued by upregulation of the other, whereas blocking both dramatically reduces stemness and tumor proliferation. Rational combination of GS inhibitors with other therapeutic modalities may preempt upregulation of such pathways responsible for eventual tumor resistance. More work is needed to explore the feasibility of incorporating these compounds into clinical treatment strategies as adjuvants.
Future Perspectives
Given the diversity of pathways in which GS participates, nonselective inhibition of this enzyme complex invites off‐target toxicity, including disruption of the gastrointestinal epithelium and abnormalities in lymphoid tissues in rodent models [137, 138] and humans [139]. It is also important to consider that certain GSIs might actually promote oncogenic transformation in the skin [140]. It might be that implementation of GSIs requires targeted administration. Specific tumor targeting with liposomes might improve overall utility, and brief cyclical dosing might mitigate impediments to antitumor immunity. Additionally, the structure of GS might lend itself to tailorable inhibition, as the Nct substrate binding site is located approximately 60Å from the catalytic site [29], suggesting potential inhibition at one or both sites. Targeting specific PS isoforms incorporated might then afford a semiselective inhibition of target pathways [141]. Optimization of GSIs should also include investigation of dosage de‐escalation, specific treatment schedules, and rational combinatorial strategies [34].
It is important to develop a profile by which to stratify tumors more or less likely to respond to GS inhibition, as treatment of resistant cell lines might actually increase the number of tumor‐initiating cells [53]. Although Notch‐1 expression alone does not correlate with response to GS inhibition, expression of Notch target genes (including Hes1, Hes4, Hes5, Hey2, HeyL, DTX1, and c‐Myc) correlated with response in T‐ALL cells [57]. Given this association, neuroendocrine tumors (NETs) may represent a type of cancers in which GSIs are well‐suited, as NETs derived from select tissues express Notch components frequently (pancreatic) or uniformly (rectal) [142]. Evaluation of NETs in clinical trials treating with GSIs thus far, however, has been minimal. Ten patients have been included in trials of advanced solid tumors treating with RO4929097 alone or in combination with some encouraging clinical responses [93, 110, 123].
Conclusion
There are many gaps in our knowledge that need to be answered to advance development of GSIs [143]. These agents have repeatedly demonstrated promising preclinical control of tumor progression; however, with the exception of desmoid tumors, this potential has not yet been harnessed clinically. Refinement of tumor Notch expression profiles and further mechanistic understanding of GSIs will necessarily assist in appropriate patient selection. Additionally, rational design of combinatoria'l strategies will maximize the potential of these agents by sensitizing tumors to traditional chemotherapeutics, while also compromising tumor ability to engage treatment resistance programs.
Author Contributions
Conception/design: Tyler R. McCaw, Evelyn Inga, Evelyn Inga, Renata Jaskula‐Sztul, Vikas Dudeja, James A. Bibb, Bin Ren, J. Bart Rose
Provision of study material or patients:
Collection and/or assembly of data: Tyler R. McCaw, Evelyn Inga, J. Bart Rose
Data analysis and interpretation:
Manuscript writing: Tyler R. McCaw, Evelyn Inga, Evelyn Inga, Renata Jaskula‐Sztul, Vikas Dudeja, James A. Bibb, Bin Ren, J. Bart Rose
Final approval of manuscript: Tyler R. McCaw, Evelyn Inga, Herbert Chen, Renata Jaskula‐Sztul, Vikas Dudeja, James A. Bibb, Bin Ren, J. Bart Rose
Disclosures
The authors indicated no financial relationships.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Wang R, Tang P, Wang P et al. Regulation of tyrosinase trafficking and processing by presenilins: Partial loss of function by familial Alzheimer's disease mutation. Proc Natl Acad Sci USA 2006;103:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coric V, van Dyck CH, Salloway S et al. Safety and tolerability of the γ‐secretase inhibitor avagacestat in a phase 2 study of mild to moderate alzheimer disease. Arch Neurol 2012;69:1430–1440. [DOI] [PubMed] [Google Scholar]
- 3. Doody RS, Raman R, Farlow M et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med 2013;369:341–350. [DOI] [PubMed] [Google Scholar]
- 4. Golde TE, Koo EH, Felsenstein KM et al. Γ‐secretase inhibitors and modulators. Biochim Biophy Acta 2013;1828:2898–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Woo HN, Park JS, Gwon AR et al. Alzheimer's disease and notch signaling. Biochem Biophys Res Commun 2009;390:1093–1097. [DOI] [PubMed] [Google Scholar]
- 6. Kovall RA, Blacklow SC. Mechanistic insights into notch receptor signaling from structural and biochemical studies. Curr Top Dev Biol 2010;92:31–71. [DOI] [PubMed] [Google Scholar]
- 7. Haines N, Irvine KD. Glycosylation regulates notch signalling. Nat Rev Mol Cell Biol 2003;4:786–797. [DOI] [PubMed] [Google Scholar]
- 8. Nandagopal N, Santat LA, LeBon L et al. Dynamic ligand discrimination in the Notch signaling pathway. Cell 2018;172:869–880.e819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimojo H, Ohtsuka T, Kageyama R. Dynamic expression of Notch signaling genes in neural stem/progenitor cells. Front Neurosci 2011;5:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev 2017;97:1235–1294. [DOI] [PubMed] [Google Scholar]
- 11. Ellisen LW, Bird J, West DC et al. Tan‐1, the human homolog of the drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991;66:649–661. [DOI] [PubMed] [Google Scholar]
- 12. Weng AP, Ferrando AA, Lee W et al. Activating mutations of Notch1 in human T cell acute lymphoblastic leukemia. Science 2004;306:269–271. [DOI] [PubMed] [Google Scholar]
- 13. Diévart A, Beaulieu N, Jolicoeur P. Involvement of Notch1 in the development of mouse mammary tumors. Oncogene 1999;18:5973–5981. [DOI] [PubMed] [Google Scholar]
- 14. Girard L, Hanna Z, Beaulieu N et al. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c‐myc and notch1 for oncogenesis. Genes Dev 1996;10:1930–1944. [DOI] [PubMed] [Google Scholar]
- 15. Capobianco AJ, Zagouras P, Blaumueller CM et al. Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol Cell Biol 1997;17:6265–6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stoyanova T, Riedinger M, Lin S et al. Activation of notch1 synergizes with multiple pathways in promoting castration‐resistant prostate cancer. Proc Natl Acad Sci USA 2016;113:E6457–E6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thompson BJ, Buonamici S, Sulis ML et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 2007;204:1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pece S, Serresi M, Santolini E et al. Loss of negative regulation by numb over notch is relevant to human breast carcinogenesis. J Cell Biol 2004;167:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Choi HY, Siddique HR, Zheng M et al. P53 destabilizing protein skews asymmetric division and enhances NOTCH activation to direct self‐renewal of TICS. Nature Commun 2020;11:3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mazzone M, Selfors LM, Albeck J et al. Dose‐dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci USA 2010;107:5012–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu TS, Costello MA, Talsma CE et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self‐renewal of cancer stem‐like cells. Cancer Res 2011;71:6061–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang W, Grivennikov SI. Top notch cancer stem cells by paracrine NF‐κB signaling in breast cancer. Breast Cancer Res 2013;15:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu J, Ye X, Fan F et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged‐1. Cancer Cell 2013;23:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Li Y, Banerjee S et al. Emerging role of Notch in stem cells and cancer. Cancer Lett 2009;279:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett 2013;341:41–45. [DOI] [PubMed] [Google Scholar]
- 26. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell 2018;34:536–548. [DOI] [PubMed] [Google Scholar]
- 27. Andersen P, Uosaki H, Shenje LT et al. Non‐canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol 2012;22:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beel AJ, Sanders CR. Substrate specificity of gamma‐secretase and other intramembrane proteases. Cell Mol Life Sci 2008;65:1311–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu P, Bai XC, Ma D et al. Three‐dimensional structure of human gamma‐secretase. Nature 2014;512:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haapasalo A, Kovacs DM. The many substrates of presenilin/γ‐secretase. J Alzheimers Dis 2011;25:3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merilahti JA, Elenius K. Gamma‐secretase‐dependent signaling of receptor tyrosine kinases. Oncogene 2019;38:151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gertsik N, Chiu D, Li YM. Complex regulation of gamma‐secretase: From obligatory to modulatory subunits. Front Aging Neurosci 2014;6:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ran Y, Hossain F, Pannuti A et al. Γ‐Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med 2017;9:950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. De Strooper B, Annaert W. Novel research horizons for presenilins and γ‐secretases in cell biology and disease. Ann Rev Cell Dev Bio 2010;26:235–260. [DOI] [PubMed] [Google Scholar]
- 35. Gavai AV, Quesnelle C, Norris D, et al. Discovery of clinical candidate bms‐906024: A potent pan‐notch inhibitor for the treatment of leukemia and solid tumors. ACS Medicinal Chem Lett 2015;6:523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lessard CB, Rodriguez E, Ladd TB, et al. Individual and combined presenilin 1 and 2 knockouts reveal that both have highly overlapping functions in hek293t cells. Int J Biol Chem 2019;294:11276‐11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gillman KW, Starrett JE Jr, Parker MF, et al. Discovery and evaluation of bms‐708163, a potent, selective and orally bioavailable γ‐secretase inhibitor. ACS Medicinal Chem Lett 2010;1:120‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mitani Y, Yarimizu J, Saita K, et al. Differential effects between γ‐secretase inhibitors and modulators on cognitive function in amyloid precursor protein‐transgenic and nontransgenic mice. J Neurosci 2012;32:2037‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martone RL, Zhou H, Atchison K, et al. Begacestat (gsi‐953): A novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein γ‐secretase for the treatment of alzheimer's disease. J Pharmacol 2009;331:598‐608. [DOI] [PubMed] [Google Scholar]
- 40. Cook JJ, Wildsmith KR, Gilberto DB, et al. Acute γ‐secretase inhibition of nonhuman primate cns shifts amyloid precursor protein (app) metabolism from amyloid‐β production to alternative app fragments without amyloid‐β rebound. J Neurosci 2010;30:6743‐6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deangelo D, Stone R, Silverman L, et al. A phase i clinical trial of the notch inhibitor mk‐0752 in patients with t‐cell acute lymphoblastic leukemia/lymphoma (t‐all) and other leukemias. J Clinl Oncol 2006;24:6585‐6585. [Google Scholar]
- 42. Lanz TA, Wood KM, Richter KE, et al. Pharmacodynamics and pharmacokinetics of the γ‐secretase inhibitor pf‐3084014. J Pharmacol 2010;334:269‐277. [DOI] [PubMed] [Google Scholar]
- 43. Wei P, Walls M, Qiu M, et al. Evaluation of selective γ‐secretase inhibitor pf‐03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol cancer Ther 2010;9:1618‐1628. [DOI] [PubMed] [Google Scholar]
- 44. Gu K, Li Q, Lin H, et al. Gamma secretase inhibitors: A patent review (2013‐2015). Expert Opinion Ther Pat 2017;27:851‐866. [DOI] [PubMed] [Google Scholar]
- 45. Lewis HD, Pérez Revuelta BI, Nadin A, et al. Catalytic site‐directed γ‐secretase complex inhibitors do not discriminate pharmacologically between notch s3 and β‐app cleavages. Biochemistry 2003;42:7580‐7586. [DOI] [PubMed] [Google Scholar]
- 46. Curry CL, Reed LL, Golde TE et al. Gamma secretase inhibitor blocks notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene 2005;24:6333–6344. [DOI] [PubMed] [Google Scholar]
- 47. Chen F, Pisklakova A, Li M et al. Gamma‐secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr) 2011;34:545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Han J, Ma I, Hendzel MJ et al. The cytotoxicity of gamma‐secretase inhibitor i to breast cancer cells is mediated by proteasome inhibition, not by gamma‐secretase inhibition. Breast Cancer Res 2009;11:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monticone M, Biollo E, Fabiano A et al. Z‐leucinyl‐leucinyl‐norleucinal induces apoptosis of human glioblastoma tumor‐initiating cells by proteasome inhibition and mitotic arrest response. Mol Cancer Res 2009;7:1822–1834. [DOI] [PubMed] [Google Scholar]
- 50. D'Souza B, Miyamoto A, Weinmaster G. The many facets of notch ligands. Oncogene 2008;27:5148–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang H, Zang C, Liu XS et al. The role of Notch receptors in transcriptional regulation. J Cell Physiol 2015;230:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor hes1. Science 2008;321:1095‐–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mizuma M, Rasheed ZA, Yabuuchi S et al. The gamma secretase inhibitor MRK‐003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther 2012;11:1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang LY, Zhang XL, Du P et al. Gamma‐secretase inhibitor, DAPT inhibits self‐renewal and stemness maintenance of ovarian cancer stem‐like cells in vitro. Chin J Cancer Res 2011;23:140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huynh C, Poliseno L, Segura MF et al. The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One 2011;6:e25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kondratyev M, Kreso A, Hallett RM et al. Gamma‐secretase inhibitors target tumor‐initiating cells in a mouse model of ERBB2 breast cancer. Oncogene 2012;31:93–103. [DOI] [PubMed] [Google Scholar]
- 57. Rao SS, O'Neil J, Liberator CD et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T‐cell acute lymphoblastic leukemia cells. Cancer Res 2009;69:3060–3068. [DOI] [PubMed] [Google Scholar]
- 58. Luistro L, He W, Smith M et al. Preclinical profile of a potent gamma‐secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 2009;69:7672–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cui D, Dai J, Keller JM et al. Notch pathway inhibition using PF‐03084014, a γ‐secretase inhibitor (GSI), enhances the antitumor effect of docetaxel in prostate cancer. Clin Cancer Res 2015;21:4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen X, Gong L, Ou R et al. Sequential combination therapy of ovarian cancer with cisplatin and γ‐secretase inhibitor MK‐0752. Gynecol Oncol 2016;140:537–544. [DOI] [PubMed] [Google Scholar]
- 61. Ikezawa Y, Sakakibara‐Konishi J, Mizugaki H et al. Inhibition of Notch and HIF enhances the antitumor effect of radiation in notch expressing lung cancer. Int J Clin Oncol 2017;22:59–69. [DOI] [PubMed] [Google Scholar]
- 62. Mizugaki H, Sakakibara‐Konishi J, Ikezawa Y et al. Γ‐secretase inhibitor enhances antitumour effect of radiation in Notch‐expressing lung cancer. Br J Cancer 2012;106:1953–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang M, Ma X, Wang J et al. Pretreatment with the gamma‐secretase inhibitor DAPT sensitizes drug‐resistant ovarian cancer cells to cisplatin by downregulation of notch signaling. Int J Oncol 2014;44:1401–1409. [DOI] [PubMed] [Google Scholar]
- 64. Liu YP, Yang CJ, Huang MS et al. Cisplatin selects for multidrug‐resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res 2013;73:406–416. [DOI] [PubMed] [Google Scholar]
- 65. Zhang S, Balch C, Chan MW et al. Identification and characterization of ovarian cancer‐initiating cells from primary human tumors. Cancer Rese 2008;68:4311–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gu F, Ma Y, Zhang Z et al. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep 2010;23:671–676. [DOI] [PubMed] [Google Scholar]
- 67. Xiao W, Chen X, He M. Inhibition of the Jagged/Notch pathway inhibits retinoblastoma cell proliferation via suppressing the PI3K/akt, Src, p38MAPK and Wnt/β‐catenin signaling pathways. Mol Med Rep 2014;10:453–458. [DOI] [PubMed] [Google Scholar]
- 68. Xie M, He J, He C et al. Γ secretase inhibitor BMS‐708163 reverses resistance to EGFR inhibitor via the PI3K/Akt pathway in lung cancer. J Cell Biochem 2015;116:1019–1027. [DOI] [PubMed] [Google Scholar]
- 69. Lee HW, Kim SJ, Choi IJ et al. Targeting notch signaling by γ‐secretase inhibitor I enhances the cytotoxic effect of 5‐FU in gastric cancer. Clin Exp Metastasis 2015;32:593–603. [DOI] [PubMed] [Google Scholar]
- 70. Meng RD, Shelton CC, Li YM et al. Γ‐secretase inhibitors abrogate oxaliplatin‐induced activation of the Notch‐1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res 2009;69:573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li ZL, Chen C, Yang Y et al. Gamma‐secretase inhibitor enhances sensitivity to doxorubicin in MDA‐MB‐231 cells. Int J Clin Pathol 2015;8:4378–5387. [PMC free article] [PubMed] [Google Scholar]
- 72. Efferson CL, Winkelmann CT, Ware C et al. Downregulation of Notch pathway by a γ‐secretase inhibitor attenuates AKT/mammalian target of rapamycin signaling and glucose uptake in an ERBB2 transgenic breast cancer model. Cancer Res 2010;70:2476–2484. [DOI] [PubMed] [Google Scholar]
- 73. Ramakrishnan V, Ansell S, Haug J et al. MRK003, a γ‐secretase inhibitor exhibits promising in vitro pre‐clinical activity in multiple myeloma and non‐Hodgkin's lymphoma. Leukemia 2012;26:340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saxena M, Stephens MA, Pathak H et al. Transcription factors that mediate epithelial–mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis 2011;2:e179–e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT‐inducing transcription factors. Nature Cell Bio 2014;16:488–494. [DOI] [PubMed] [Google Scholar]
- 76. Yang X, Xia W, Chen L et al. Synergistic antitumor effect of a γ‐secretase inhibitor PF‐03084014 and sorafenib in hepatocellular carcinoma. Oncotarget 2018;9:34996–35007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li LC, Wang DL, Wu YZ et al. Gastric tumor‐initiating CD44+ cells and epithelial‐mesenchymal transition are inhibited by γ‐secretase inhibitor DAPT. Oncology Lett 2015;10:3293–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gupta N, Xu Z, El‐Sehemy A et al. Notch3 induces epithelial–mesenchymal transition and attenuates carboplatin‐induced apoptosis in ovarian cancer cells. Gynecol Oncol 2013;130:200–206. [DOI] [PubMed] [Google Scholar]
- 79. Matsuno Y, Coelho AL, Jarai G et al. Notch signaling mediates TGF‐β1‐induced epithelial–mesenchymal transition through the induction of snai1. Int J Biochem Cell Biol 2012;44:776–789. [DOI] [PubMed] [Google Scholar]
- 80. Zhou J, Jain S, Azad AK et al. Notch and TGFΒ form a positive regulatory loop and regulate emt in epithelial ovarian cancer cells. Cell Signal 2016;28:838–849. [DOI] [PubMed] [Google Scholar]
- 81. Pazos M, Abramovich D, Bechis A et al. Gamma secretase inhibitor impairs epithelial‐to‐mesenchymal transition induced by TGF‐β in ovarian tumor cell lines. Mol Cell Endocrinol 2017;440:125–137. [DOI] [PubMed] [Google Scholar]
- 82. Yuan X, Wu H, Xu H et al. Meta‐analysis reveals the correlation of Notch signaling with non‐small cell lung cancer progression and prognosis. Sci Rep 2015;5:10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morgan KM, Fischer BS, Lee FY et al. Gamma secretase inhibition by BMS‐906024 enhances efficacy of paclitaxel in lung adenocarcinoma. Mol Cancer Ther 2017;16:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Krop I, Demuth T, Guthrie T et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK‐0752 in adult patients with advanced solid tumors. J Clin Oncol 2012;30:2307–2313. [DOI] [PubMed] [Google Scholar]
- 85. Messersmith WA, Shapiro GI, Cleary JM et al. A phase I, dose‐finding study in patients with advanced solid malignancies of the oral γ‐secretase inhibitor PF‐03084014. Clin Cancer Res 2015;21:60–67. [DOI] [PubMed] [Google Scholar]
- 86. Gold KA, Byers LA, Fan YH et al. A phase I/II trial combining erlotinib with gamma secretase inhibitor RO4929097 in advanced non‐small cell lung cancer (NSCLC). J Clin Oncol 2013;31:8104a. [Google Scholar]
- 87. Jeon YH, Ha M, Kim SW et al. Evaluation of the prognostic significances of γ‐secretase genes in pancreatic cancer. Oncol Lett 2019;17:4614–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Miyamoto Y, Maitra A, Ghosh B et al. Notch mediates TGF alpha‐induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003;3:565–576. [DOI] [PubMed] [Google Scholar]
- 89. De Jesus‐Acosta A, Laheru D, Maitra A et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs 2014;32:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yabuuchi S, Pai SG, Campbell NR et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett 2013;335:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Richter S, Bedard PL, Chen EX et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL‐078/CTEP 8575). Invest New Drugs 2014;32:243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cook N, Basu B, Smith DM et al. A phase I trial of the γ‐secretase inhibitor MK‐0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer 2018;118:793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tolcher AW, Messersmith WA, Mikulski SM et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol 2012;30:2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee SM, Moon J, Redman BG et al. Phase 2 study of RO4929097, a gamma‐secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer 2015;121:432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hu YY, Zheng MH, Cheng G et al. Notch signaling contributes to the maintenance of both normal neural stem cells and patient‐derived glioma stem cells. BMC Cancer 2011;11:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hai L, Zhang C, Li T et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF‐κB(p65) pathway. Cell Death Dis 2018;9:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Saito N, Fu J, Zheng S et al. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor‐initiating cells. Stem Cells 2014;32:301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fouladi M, Stewart CF, Olson J et al. Phase I trial of MK‐0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J Clin Oncol 2011;29:3529–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hoffman LM, Fouladi M, Olson J et al. Phase I trial of weekly MK‐0752 in children with refractory central nervous system malignancies: A pediatric brain tumor consortium study. Childs Nerv Syst 2015;31:1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pan E, Supko JG, Kaley TJ et al. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J Neurooncol 2016;130:571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Xu R, Shimizu F, Hovinga K et al. Molecular and clinical effects of Notch inhibition in glioma patients: A phase 0/I trial. Clin Cancer Res 2016;22:4786–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Committee on Practice Bulletins—Gynecology . Practice bulletin number 179: Breast cancer risk assessment and screening in average‐risk women. Obstet Gynecol 2017;130:e1–e16. [DOI] [PubMed] [Google Scholar]
- 103. Reedijk M, Odorcic S, Chang L et al. High‐level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 2005;65:8530–8537. [DOI] [PubMed] [Google Scholar]
- 104. Zhang CC, Yan Z, Zong Q et al. Synergistic effect of the γ‐secretase inhibitor PF‐03084014 and docetaxel in breast cancer models. Stem Cells Translational Medicine 2013;2:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Means‐Powell J, Minton S, Mayer I et al. A phase Ib dose escalation trial of RO4929097 (a γ‐secretase inhibitor) in combination with exemestane in patients with ER + metastatic breast cancer. Cancer Res 2012;72:P2‐14‐04a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Locatelli MA, Aftimos P, Dees EC et al. Phase I study of the gamma secretase inhibitor PF‐03084014 in combination with docetaxel in patients with advanced triple‐negative breast cancer. Oncotarget 2017;8:2320–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schott AF, Landis MD, Dontu G et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res 2013;19:1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Alniaimi AN, Demorest‐Hayes K, Alexander VM et al. Increased Notch1 expression is associated with poor overall survival in patients with ovarian cancer. Int J Gynecol Cancer 2015;25:208‐213. [DOI] [PubMed] [Google Scholar]
- 109. Pant S, Jones SF, Kurkjian CD et al. A first‐in‐human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur J Cancer 2016;56:1–9. [DOI] [PubMed] [Google Scholar]
- 110. Diaz‐Padilla I, Hirte H, Oza AM et al. A phase Ib combination study of RO4929097, a gamma‐secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs 2013;31:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sahebjam S, Bedard P, Castonguay V et al. A phase I study of the combination of RO4929097 and cediranib in patients with advanced solid tumours (PJC‐004/NCI 8503). Br J Cancer 2013;109:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Diaz‐Padilla I, Wilson MK, Clarke BA et al. A phase II study of single‐agent RO4929097, a gamma‐secretase inhibitor of Notch signaling, in patients with recurrent platinum‐resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. Gynecol Oncol 2015;137:216–222. [DOI] [PubMed] [Google Scholar]
- 113. Chiorean EG, LoRusso P, Strother RM et al. A phase I first‐in‐human study of enoticumab (REGN421), a fully human Delta‐like Ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res 2015;21:2695–2703. [DOI] [PubMed] [Google Scholar]
- 114. Ozawa T, Kazama S, Akiyoshi T et al. Nuclear Notch3 expression is associated with tumor recurrence in patients with stage II and III colorectal cancer. Ann Surg Oncol 2014;21:2650–2658. [DOI] [PubMed] [Google Scholar]
- 115. Chu D, Zhang Z, Zhou Y et al. Notch1 and Notch2 have opposite prognostic effects on patients with colorectal cancer. Ann Oncol 2011;22:2440–2447. [DOI] [PubMed] [Google Scholar]
- 116. Candy P, Phillips M, Redfern AD et al. Notch‐induced transcription factors are predictive of survival and 5‐fluorouracil response in colorectal cancer patients. Br J Cancer 2013;109:1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sun L, Ke J, He Z et al. HES1 promotes colorectal cancer cell resistance to 5‐FU by inducing of EMT and ABC transporter proteins. J Cancer 2017;8:2802–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yuan R, Ke J, Sun L et al. HES1 promotes metastasis and predicts poor survival in patients with colorectal cancer. Clin Exp metastasis 2015;32:169–179. [DOI] [PubMed] [Google Scholar]
- 119. Arcaroli J, Quackenbush K, Purkey A et al. Tumours with elevated levels of the Notch and WNT pathways exhibit efficacy to PF‐03084014, a γ‐secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer 2013;109:667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Aung KL, El‐Khoueiry AB, Gelmon K et al. A multi‐arm phase I dose escalating study of an oral notch inhibitor BMS‐986115 in patients with advanced solid tumours. Invest New Drugs 2018;36:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Strosberg JR, Yeatman T, Weber J et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 2012;48:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Brana I, Berger R, Golan T et al. A parallel‐arm phase I trial of the humanised anti‐IGF‐1R antibody dalotuzumab in combination with the AKT inhibitor MK‐2206, the mTOR inhibitor ridaforolimus, or the Notch inhibitor MK‐0752, in patients with advanced solid tumours. Br J Cancer 2014;111:1932–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. LoConte NK, Razak AR, Ivy P et al. A multicenter phase 1 study of γ‐secretase inhibitor RO4929097 in combination with capecitabine in refractory solid tumors. Invest New Drugs 2015;33:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Skubitz KM. Biology and treatment of aggressive fibromatosis or desmoid tumor. Mayo Clin Proc 2017;92:947–964. [DOI] [PubMed] [Google Scholar]
- 125. Shang H, Braggio D, Lee YJ et al. Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 2015;121:4088–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Villalobos VM, Hall F, Jimeno A et al. Long‐term follow‐up of desmoid fibromatosis treated with PF‐03084014, an oral gamma secretase inhibitor. Ann Surg Oncol 2018;25:768–775. [DOI] [PubMed] [Google Scholar]
- 127. Kummar S, Coyne GO, Do KT et al. Clinical activity of the γ‐secretase inhibitor PF‐03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol 2017;35:1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tsukumo SI, Yasutomo K. Regulation of CD8+ T cells and antitumor immunity by notch signaling. Front Immunol 2018;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sierra RA, Thevenot P, Raber PL et al. Rescue of Notch‐1 signaling in antigen‐specific CD8+ t cells overcomes tumor‐induced T‐cell suppression and enhances immunotherapy in cancer. Cancer Immunol Res 2014;2:800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Palaga T, Miele L, Golde TE et al. TCR‐mediated notch signaling regulates proliferation and IFN‐γ production in peripheral T cells. J Immunol 2003;171:3019–3024. [DOI] [PubMed] [Google Scholar]
- 131. Charbonnier LM, Wang S, Georgiev P et al. Control of peripheral tolerance by regulatory T cell–intrinsic notch signaling. Nat Immunol 2015;16:1162–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. He W, Luistro L, Carvajal D et al. High tumor levels of IL6 and IL8 abrogate preclinical efficacy of the gamma‐secretase inhibitor, RO4929097. Mol Oncol 2011;5:292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sansone P, Ceccarelli C, Berishaj M et al. Self‐renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun 2016;7:10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Xiong S, Wang R, Chen Q et al. Cancer‐associated fibroblasts promote stem cell‐like properties of hepatocellular carcinoma cells through IL‐6/STAT3/Notch signaling. Am J Cancer Res 2018;8:302–316. [PMC free article] [PubMed] [Google Scholar]
- 135. Peng D, Tanikawa T, Li W et al. Myeloid‐derived suppressor cells endow stem‐like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross‐talk signaling. Cancer Res 2016;76:3156–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Wang D, Xu J, Liu B et al. IL6 blockade potentiates the anti‐tumor effects of gamma‐secretase inhibitors in NOTCH3‐expressing breast cancer. Cell Death Differ 2018;25:330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Barten DM, Meredith JE, Zaczek R et al. Γ‐secretase inhibitors for Alzheimer's disease: Balancing efficacy and toxicity. Drugs R D 2006;7:87–97. [DOI] [PubMed] [Google Scholar]
- 138. Searfoss GH, Jordan WH, Calligaro DO et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma‐secretase inhibitor. J Biol Chem 2003;278:46107–46116. [DOI] [PubMed] [Google Scholar]
- 139. Schor NF. What the halted phase III gamma‐secretase inhibitor trial may (or may not) be telling us. Ann Neurol 2011;69:237–239. [DOI] [PubMed] [Google Scholar]
- 140. Li T, Wen H, Brayton C et al. Epidermal growth factor receptor and Notch pathways participate in the tumor suppressor function of γ‐secretase. J Biol Chem 2007;282:3226432273. [DOI] [PubMed] [Google Scholar]
- 141. Zhao B, Yu M, Neitzel M et al. Identification of gamma‐secretase inhibitor potency determinants on presenilin. J Biol Chem 2008;283:2927–2938. [DOI] [PubMed] [Google Scholar]
- 142. Wang H, Chen Y, Fernandez‐Del Castillo C et al. Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: A critical look at notch signaling pathway. Mod Pathol 2013;26:139–147. [DOI] [PubMed] [Google Scholar]
- 143. Janghorban M, Xin L, Rosen JM et al. Notch signaling as a regulator of the tumor immune response: To target or not to target? Front Immunl 2018;9:1649. [DOI] [PMC free article] [PubMed] [Google Scholar]