

Abstract
Background.—
Cerebrospinal fluid sodium concentration ([Na+]csf) increases during migraine, but the cause of the increase is not known.
Objective.—
Analyze biochemical pathways that influence [Na+]csf to identify mechanisms that are consistent with migraine.
Method.—
We reviewed sodium physiology and biochemistry publications for links to migraine and pain.
Results.—
Increased capillary endothelial cell (CEC) Na+, K+, -ATPase transporter (NKAT) activity is probably the primary cause of increased [Na+]csf. Physiological fluctuations of all NKAT regulators in blood, many known to be involved in migraine, are monitored by receptors on the luminal wall of brain CECs; signals are then transduced to their abluminal NKATs that alter brain extracellular sodium ([Na+]e) and potassium ([K+]e).
Conclusions.—
We propose a theoretical mechanism for aura and migraine when NKAT activity shifts outside normal limits: (1) CEC NKAT activity below a lower limit increases [K+]e, facilitates cortical spreading depression, and causes aura; (2) CEC NKAT activity above an upper limit elevates [Na+]e, increases neuronal excitability, and causes migraine; (3) migraine-without-aura may arise from CEC NKAT over-activity without requiring a prior decrease in activity and its consequent spreading depression; (4) migraine triggers disturb, and treatments improve, CEC NKAT homeostasis; (5) CEC NKAT-induced regulation of neural and vasomotor excitability coordinates vascular and neuronal activities, and includes occasional pathology from CEC NKAT-induced apoptosis or cerebral infarction.
Keywords: cerebrospinal fluid, Na+, K+ -ATPase transporter (NKAT), capillary endothelial cell (CEC)
A major problem in migraine pathophysiology is to understand the basis of symptoms. Migraine affects peripheral and central trigeminovascular pathways and central sensitization appears responsible for allodynia;1–10 cortical spreading depression (CSD) is associated with migraine aura;11–13 CSD can activate trigeminovascular pathways;13 and a large variety of medications have benefit in treating acute (and, less effectively, chronic) migraine.14–19 These findings delineate some of the anatomy, physiology, biochemistry, and pharmacology of migraine, but it is not clear what happens in neurons that causes CSD/aura or migraine.
The reason for a decreased CSD threshold among migraineurs is not known, though electrolyte changes during CSD include acute changes in [K+]e and [Na+]e.20,21 Some insight comes from mutations in 3 different genes identified in the rare familial hemiplegic migraine (FHM):22–26 CACNA1A (gain of function of a slow Ca2+ channel gene), ATP1A2 (loss of function of the α-2 isoform of the Na+, K+ -ATPase transporter [NKAT] gene), and SCN1A (gain of function of a voltage-gated Na+ channel gene). Elevation of extracellular glutamate and/or potassium has been suggested as a common mechanism for how 2 of these distinct genetic loci predispose a person to CSD.27 Alternatively, since these 3 genes have not been found mutated in the common forms of migraine to date,28 and since their dysfunction will influence potassium/sodium homeostasis, we propose that rather than the FHM mutations themselves it is their impact on potassium/sodium homeostasis that may reveal the common link in causing their migraine phenotypes.
We are investigating whether episodically increased neuronal excitability in migraine arises from a disturbance of brain sodium homeostasis, since we had found that [Na+]csf was altered in migraine, whereas Ca2+, K+, and Mg2+ were not.29 [Na+]csf rose significantly during the peak of migraine compared with the non-headache state of migraineurs and controls. This increase is found only in the cerebrospinal fluid (CSF) and not the plasma samples of migraineurs, which argues for a brain source and against a systemic origin. Radioactive Na+ distribution studies reveal that though the CSF Na+ composition is modified at different points along the neuraxis,30,31 it is reasonable to assume the [Na+]csf reflects [Na+]e, since equilibration of [Na+]e with lumbar CSF occurs rapidly,30,32,33 especially in ambulant people. We expect [Na+]e may be increased even more in specific brain regions in migraine, since the volume of CSF and its dispersion to the lumbar site of collection may have diluted the values we measured.
Elevated [Na+]e is important because it can lead to significant physiological effects by increasing neuronal excitability: Hodgkin and Katz34 demonstrated that the action potential rose at a rate roughly proportional to the rise of [Na+]e. When a neuron is at rest, the Na+ influx through voltage-gated Na+ channels is low, as these channels are usually closed or inactivated. However, the channel gate is displaced when [Na+]e increases.35 Higher [Na+]e speeded recovery from the inactivation state, enabling an earlier action potential and leading to hyperexcitability.35 Higher [Na+]csf caused a sympathetic hyperactivity response (increasing blood pressure and heart rate) through increasing ouabain-like substances and activating the brain renin-angiotensin-aldosterone system.36,37
We suggest that brain potassium/sodium homeostasis is disturbed in migraine because: (1) [K+]e is increased and [Na+]e is reduced during CSD/aura; (2) all 3 mutations in FHM affect potassium/sodium regulation; (3) [Na+]csf is increased in migraine; and (4) higher [Na+]e increases neuronal excitability. We derive a theory of migraine pathophysiology that may explain migraine symptoms resulting from a compromise in brain potassium/sodium homeostasis.
INCREASED CEC NKAT ACTIVITY IS THE LIKELY MECHANISM FOR INCREASED [Na+]csf
Routes of sodium transport across the cellular membrane include passage through voltage- or ligand-gated sodium channels, as well as by means of sodium transporters. We review herein the biochemistry of these 3 portals for sodium flux, and deduce that increased CEC NKAT activity is the likely cause of the increase in [Na+]csf. Review of NKAT regulators reveals that most have been implicated in migraine.
Voltage-Gated Sodium Channels.—
While there are 9 different types of voltage-gated sodium channels, only 5 exist in the central nervous system: NaV1.1, NaV1.2, NaV1.3, NaV1.6, and NaV1.7.38 NaV 1.1 function is relevant in migraine since SCN1A mutations have been reported in FHM, as described above. NaV1.3 has been implicated in the long-term effects of spinal cord injury, which leads to altered regulation of the NaV1.3 channel, resulting in hyperexcitability and central neuropathic pain.39 Similarly, alteration of sodium channels may be connected to the pain associated with migraine headaches.
Ligand-Gated Sodium Permeability.—
Ionotropic receptors are a group of transmembrane ion channels that are regulated by neurotransmitters.The ion channels are selective to one or more ions, including Na+, K+, Ca2+, or Cl−, so they are also responsible for sodium influx. This category includes glutamate (AMPA, kainate, and N-methyl-D-aspartic acid [NMDA]) and 5-HT3 receptors that allow sodium influx on receptor binding.
Sodium Transporters.—
These include (direction of ion flux in relation to the cell):
The NKAT (3Na+ out, 2K+ in).
The Na+/Ca2+ exchangers40 (sodium and calcium exchangers [NCXs] and NCKXs) (3–4 Na+, 1Ca2+, +/− 1K+, either in or out).
The Na+/glutamate symporter (3Na+ and 1 glutamate, in).
The Na+/H+ antiporter (1Na+, 1H+, either in or out).
Clearly, all 4 transporter groups could be involved in altering the [Na+]e, either by gain or loss of functions.
We can interpret an increase in [Na+]e during headache in 2 ways:
The first interpretation is that impairment of voltage- or ligand-gated sodium channels in migraineurs, or reduction in their density, would decrease sodium influx. Less sodium entering the cell would result in increased [Na+]e during migraine relative to the non-headache state and to controls. Since action potentials do not substantially change external ion concentrations, decreased function of voltage- or ligand-gated sodium channels will not cause the observed 4 mM increase in [Na+]csf, let alone higher values that may occur at local brain regions. Glutamate is known to increase in CSF in migraine41 and a glutamate-gated sodium influx would decrease [Na+]e, the opposite effect to that observed during migraine. Perhaps more importantly, while reduced ion flux through the Na+ channels might elevate [Na+]e, the increased neuronal excitability in migraine requires more, rather than less, ion flux through the channels. Thus, we exclude sodium channel block from causing increased [Na+]csf and [Na+]e during migraine.
The second interpretation is that a mechanism exists in migraineurs that causes excess sodium to be pumped into the extracellular space. This could result from sustained over-activation of sodium transporters, which are the only proteins capable of pumping sodium against its concentration and electrochemical gradients. The NKAT is the main exporter of Na+ and is reported to consume almost half of the brain’s energy.42,43 The other transporters consume much less energy and are therefore more likely involved in smaller modulations of Na+. For example, we suggest that potassium-dependent sodium and calcium exchanger (NCKX) has a role in CSD: the greater rise of [K+]e in ouabain-induced CSD in Ca2+ free solution44 is most likely from absence of the reverse mode NCKX that would export Na+ from the cell and import K+ and Ca2+ to the cell, if Ca2+ had not been removed.
Increased NKAT activity not only increases [Na+]e, but also decreases [K+]e. Thus, if NKAT over-activation contributes to the increased [Na+]e observed during migraine headache, one might expect an associated decrease in [K+]e. Our CSF data did not reveal a significant decrease in [K+]csf, though the overall [Na+]csf : [K+]csf ratio increased.29 We suggest that, during increased NKAT activity, the decrease in [K+]e/csf has been minimized by the strong glial regulation system for [K+].45
The magnitude of the NKAT role in the brain42,43 suggests that its increased activity is the most likely cause of the increased [Na+]csf and [Na+]e in migraine, though further studies of [Na+]e and [Na+]csf are required. If NKAT is involved in migraine pathophysiology, then the structural, functional, regional, and cellular heterogeneity of its 3 α-chain subunits, 3 β-chain subunits, and 7 γ-chain subunits is important.46 These are summarized in Table 1.
Table 1.—
The Cellular Locations, Effects of Gene Knockout, Known Human Mutations, and Functions of the 3 α, 3 β, and 5 γ Isoforms of Nkat That Are Found in the Brain
| Isoform | Brain/cell location multiple isoforms in same cell | Effect from gene knockout | Human mutations | Function | Refs |
|---|---|---|---|---|---|
| α1 | Neurons and glia | Homozygous die at blastocyst. Heterozygous normal | Catalytic, ion transport, phos-phorylation, and ATP binding. They differ in Na+, K+, and ATP binding, ouabain and Ca2+ inhibition. Eg, ouabain and ATP affinities are lowest for α1, highest for α2 and 3 | 106–111 | |
| α2 | Glial cells mainly, occasional neurons | Homozygous die at birth: ↓ breathing. Heterozygous have ↑ anxiety and ↓ long-term memory | FHM-2 | 106–109,111 | |
| α3 | Neurons | Homozygous die at birth. Heterozygous ↓ non-spatial learning and long-term memory | Rapid onset dystonia parkinsonism | 106–111 | |
| β1 | Mainly neurons | Not known | Membrane insertion, protein:protein interactions, tight junctions, cell polarity, and signaling | 112,113 | |
| β2 | Mainly glia | Deficient neural cell migration, die 15 d | 112–114 | ||
| β3 | Oligodendroglial cells and optic nerve | Not known | 115 | ||
| FXYD1 | Combines with α1–3 in cerebellum. Most abundant in Purkinje cells, and choroid plexus and ependymal cells. In neurons and glia. Small amount in Allan Atlas rat brain*. | ↑ NKAT activity in heart | ↓ Affinity of NKAT for Na+ and K+ | 46, 116–118 | |
| FXYD2 | Moderate amount in Allan Atlas rat brain*. | ↑ affinity of NKAT for Na+. | Renal hypo-magnesemia | ↓ Affinity of NKAT for Na+ | 46,119–121 |
| FXYD3 | None in Allan Atlas rat brain*. | Not done | ↓ Affinity of NKAT for Na+/K+ | 46 | |
| FXYD4 | None in Allan Atlas rat brain*. | Not known in brain ↓ Na+ resorption | 46 | ||
| FXYD5 | Small amount in Allan Atlas rat brain*. | Not done | ↑ NKAT Imax | 46 | |
| FXYD6 | Extensive, abundant expression in Allan Atlas rat brain*. Cerebellum and hippocampus. | Not done | ↓ Affinity of NKAT for Na+/K+ | 46,122 | |
| FXYD7 | Almost exclusive to brain. Extensive PTMs. Large amount in Allan Atlas rat brain*. | Not done | ↓ Affinity of NKAT for K+ | 46,87,123,124 |
FHM = familial hemiplegic migraine; NKAT = Na+, K+ -ATPase transporter.
Brain CEC NKAT.—
NKAT activity provides the mechanism for the higher concentration of Na+ and lower concentration of K+ in CSF and brain extracellular fluid compared with blood. This is achieved because a large excess of blood is delivered to the brain (blood flow is 1000-fold greater than interstitial fluid flow), the tight junctions between brain capillary endothelial cells separate CSF and extracellular fluid from blood, and NKAT is restricted to the abluminal surface of the CECs,47 as illustrated in Figure 1. Brain CECs have 500-fold more NKAT than peripheral CECs,48 thus brain CECs will respond with greater sensitivity to changes in NKAT regulators than elsewhere. The CEC NKAT-regulated interstitial fluid cations are the main source of [Na+]e in brain tissue. Minimal [Na+]e arises from the neurons and glial cells, since the [Na+]i is normally only 10–15 mM.
Fig 1.—

Illustration of brain capillary endothelium, luminal Na+, K+ -ATPase transporter (NKAT) regulator receptors, tight junctions, and the differences in flow and [Na+]e and [K+]e between blood and cerebrospinal fluid (CSF). Capillary endothelial cells (CECs) are exposed to circulating NKAT regulators on the blood luminal side, and if they detect a change, they signal to their abluminal NKATs to alter interstitial fluid [Na+]e and [K+]e that modulate neuronal and vasomotor excitability. Not shown in the figure (for simplicity) are the glial cell end-feet that wrap around the CECs and form part of the blood-brain-barrier, and the choroid plexus with its epithelial, abluminal NKAT that produces the majority of the [Na+]csf.
The NKAT on the ventricular/apical surface of the epithelial cells of the choroid plexus49 is supplied with its NKAT-rich capillary network, and is considered the primary source of [Na+]csf, though a significant contribution comes from CECs by way of the interstitial fluid.50–53 Since choroid and CSF are midline, perhaps the more lateralized tissue CEC NKATs, especially those in the cerebral cortex, are better placed to increase the [Na+]e, consistent with the common laterality in migraine.
Regulators of NKAT (Table 2).—
Table 2.—
Summary of Many of the Regulators of NKAT, Partitioned in Three Classes by Their Site of Modulation: Genetic, Direct, or Signal Transduction
| NKAT modulator | Site of modulation by class | Refs |
|---|---|---|
| Genetic class | ||
| FHM-1 | Gain of function in slow calcium channel. ↑ Ca2+ may modulate by dephosphorylating NKAT. | 72,125 |
| FHM-2 | Loss of function in α2 of NKAT. ↑ [Na+]i will alter NKAT expression. | 22 |
| FHM-3 | Gain of function in Na channel. ↑ [Na+]i will alter NKAT expression. | 25 |
| Dexamethasone | Increases NKAT via ↑RNA/protein. | 126 |
| Aldosterone | Increases NKAT via ↑RNA/protein. | 126 |
| TNFα | Down regulates NKAT via NF-κB. | 127 |
| Interleukin-1 | Increases NKAT localization and activity. | 128 |
| Thyroxine | Hyper- and hypothyroidism ↓ cerebellar NKAT. T3 ↑ muscle NKAT. | 129,130 |
| Direct class | ||
| Na/K | [Na+]i and [K+]e ↑NKAT by binding to a-chain. Inward Na currents can activate or inactivate NKAT. Resting membrane potential may modulate ion-binding. | 101,131–137 |
| Mg | Activates NKAT. Binds α-NKAT. Deprivation activates calcineurin-mediated Ca signaling. Dominant isolated renal Mg loss from γNKAT loss. | 101,138–141 |
| ATP and MgATP | Activates, binds α-NKAT. | 101 |
| V, Fe, Pb, Cu | Inhibits, binds NKAT. | 101 |
| Zn | Inhibits, binds NKAT. Involved in oxygen/glucose deprivation induced CSD. | 101,142 |
| Exogenous NKAT inhibitors | Specific inhibitors. Digoxin family. | 143–146 |
| Endogenous NKAT inhibitors | Specific endogenous inhibitors. Includes ouabain. | 147–153 |
| Phospholipids | Usually activate NKAT, but ↓ PE ↑ NKAT activity. | 103,154–156 |
| Cholesterol | ↓ NKAT. Lipid membrane fluidity and binds receptor. | 157,158 |
| Kinases | Generally inhibit NKAT, eg, PKA/PKC/PKG phosphorylation. | 159,160 |
| Phosphatases | Generally activate NKAT. | 159 |
| Signal transduction class | ||
| Ca | α1/2 NKAT interacts with NCX to regulate Ca2+. Release of membrane-bound intracellular Ca2+ ↑ NKAT by activating calcineurin-mediated dephosphorylation. Physiological Ca2+ levels do not change α-1, but inhibit α-2 and 3 NKAT. | 100,107,161 |
| Dopamine | D1-mediated (D1&5 receptors) via G protein, PKA, and adenyl cyclase, ↑NKAT. D2-mediated (D2,3,4 receptors) inhibits G proteins, via PKC, decreasing adenyl cyclase and ↓ α2 and 3 NKAT. | 162–164 |
| Norepinephrine | Activates NKAT via α1 adrenoreceptor, ↑Ca, activating calcineurin and dephosphorylating NKAT. Beta adrenergic receptors ↓ NKAT via cAMP-mediated inhibition of phosphatase PP1. | 157,165–168 |
| Serotonin | Agonists ↑the # of NKATs, antagonists inactivate NKAT. Activate 5HT1A in cortex, 5HT6 in cerebellum. Inactivate NKAT in choroid via 5HT2c, DAG and PKC-induced phosphorylation. Activates glial α2 NKAT. | 160,169,170 |
| Prostanoids, leukotrienes, and thromboxanes | Through GPCRs, G proteins, camp, and phosphorylation. Also prostaglandin responsive elements in NKAT transcription. | 171–173 |
| Nitric oxide | NO donors trigger migraine via cGMP, and blockade of NOS treats migraine with aura. | 174 |
| Estrogen | Activate 5HT2AR and SERT, may activate NKAT. | 175,176 |
| Glutamate | Activate NKAT via increased [Na+]i by ↑Na influx, and by G protein and PKG. | 101,177–179 |
| Cannabinoids | Activate NKAT via CB1 receptors via G proteins. Anandamide NKAT, possibly by inhibiting dopamine and 5HT uptake. | 180,181 |
| Hypoxia | Acutely degrades NKAT. 24 hours after hypoxia, ↑ α1 and 2 NKAT activity, via PKMζ. GM1 protects after ischemia. | 142,159,182,183 |
| CO | Induces persistent NKAT activation in Purkinje neurons, via G proteins and PKG. | 179 |
| Lipid peroxidation | MMA inhibits succinate dehydrogenase, increases TBARS, depresses NKAT in some locations, increases in others. 4HNE ↓ Dopamine uptake and NKAT activity. | 179,184–187 |
| Insulin | Increases α1 NKAT in cultured astrocytes via IGF-1R. | 126,188,189 |
| Endothelin | ↑NKAT in ciliary cells/brain capillary endothelium. | 190–192 |
| CGRP | Increases NKAT in depolarized (high [K+]e) muscle. | 193,194 |
| Capsaicin | Capsaicin releases CGRP at sensory nerve endings. Capsazepine inactivates NKAT, inhibits ATP hydrolysis. | 194,195 |
| Caffeine | Inhibits NKAT via GPCRs Adenosine A1a, A2a, A2b. | 196 |
CGRP = calcitonin G related peptide; CSD = cortical spreading depression; FHM = familial hemiplegic migraine; IGF-1 = insulin growth factor 1; NKAT = Na+, K+ -ATPase transporter.
The large number of molecular modulators of NKAT (a reflection of its importance in the brain) exert their effects by 3 distinct yet integrated mechanisms: genetically (polymorphism in NKAT or altered genetic regulation); directly (cations, ATP, membrane lipids, direct inhibitors or activators); or indirectly by signal transduction, usually from G-protein coupled receptors (GPCRs). Endothelial cell composition and functions are known to differ throughout the body and within tissues and are far from fully characterized, but receptors for many major NKAT regulators known to be involved in migraine have been demonstrated on brain CECs, including those for estrogen,54 serotonin,55 lysophospholipids,56 and GPCRs. Table 2 provides a framework of reference (a summary, but not exhaustive) for the extensive range of NKAT regulation pathways. We propose that the CEC is in a unique position to sense variation in any of these circulating NKAT regulators from blood, and signal changes to its abluminal NKAT to alter the brain interstitial fluid [Na+] and [K+]. This alters extracellular cations on the brain side of the CEC tight junctions and affects the excitability of neurons. The neuronal responses are manifest as aura or migraine.
THE CEC NKAT HOMEOSTASIS THEORY, AURA, AND MIGRAINE
We propose a potassium/sodium homeostasis theory for migraine pathophysiology based on the need to unify the known change in [K+]e and [Na+]e reported during CSD20,21 (that we assume also occurs during migraine aura), the observed increase in [Na+]csf during the peak of acute migraine pain29 (that we assume is accompanied by increased [Na+]e), that all 3 FHM mutations effect [K+]e and [Na+]e, and that increased [Na+]e increases neuronal excitability.
We hypothesize that NKAT regulators in the blood arrive at their receptors on the luminal side of CECs and transduce signals to NKATs on the abluminal CEC wall (Fig. 1). These altered CEC NKATs modify brain interstitial cations, causing symptoms:
Well State (Fig. 2).—
Fig 2.—
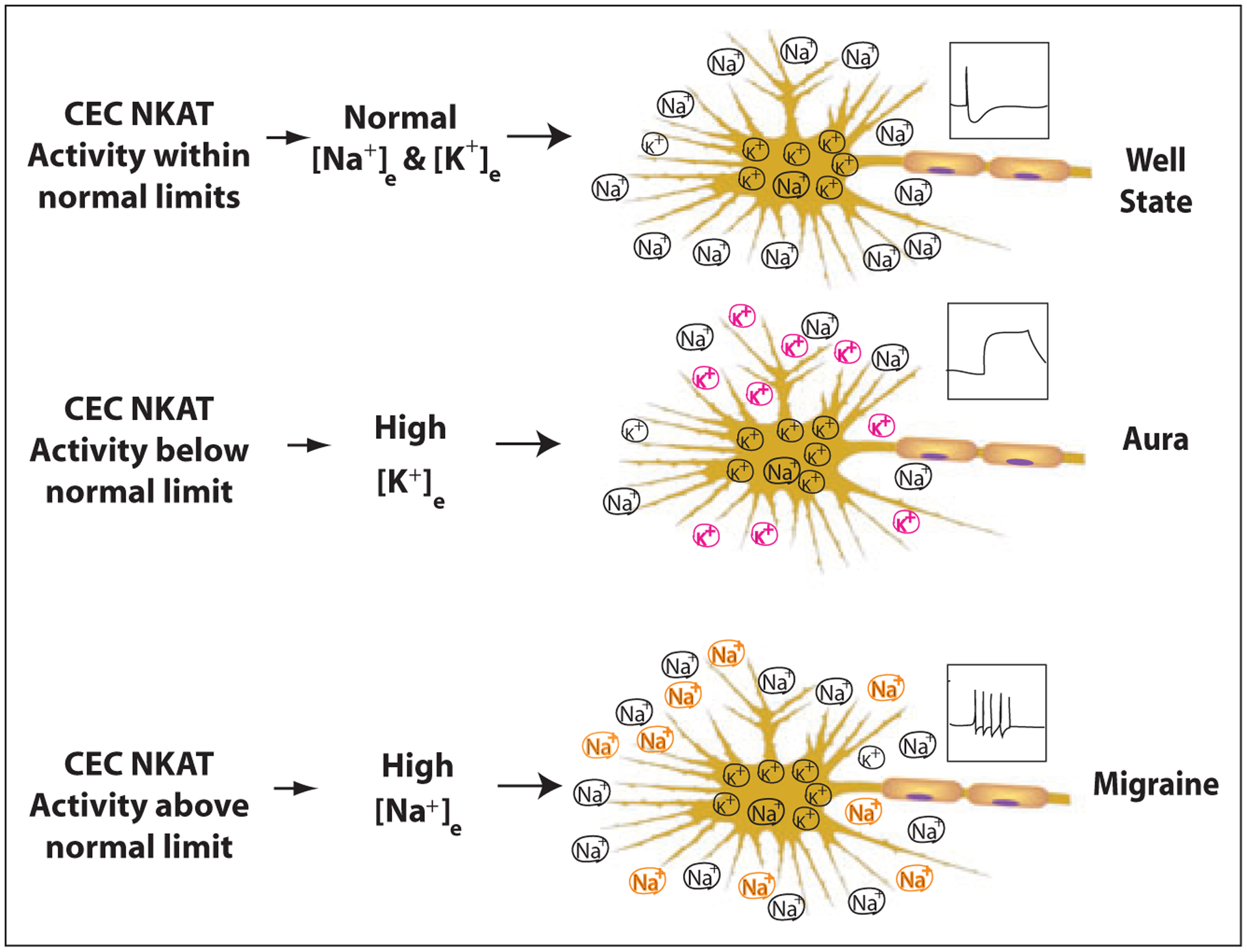
This schema represents the principal differences in [Na+]e and [K+]e predicted to occur at neurons for migraineurs in the Well State, during Aura, or during Migraine, based on the capillary endothelial cell Na+, K+ -ATPase transporter (CEC NKAT) theory. The boxed regions above each axon illustrate the effects from the Na+ changes on neuronal excitability: Normal action potentials in the Well State; cortical spreading depression (CSD) in Aura; increased action potential firing frequency in Migraine.
NKATs change the interstitial [Na+] and [K+] surrounding neurons and vary their excitability within normal homeostatic limits.
In migraine, the excitability shifts outside normal limits.
Aura (Fig. 2).—
When CEC NKAT activity falls below its normal lower limit, interstitial [K+]e rises, depolarizing the neuron, facilitating CSD that manifests clinically as aura. Reduced neuronal and glial NKAT activity along with potassium leak from cells may also contribute to the increased [K+]e at the start of CSD, since they have high [K+]i. Interestingly, decreased brain energy favors CSD57 and, because NKAT consumes a large amount of energy, this adds additional support to a role for NKAT in CSD. During CSD, extreme ion shifts occur at the neuron: [K+]e increases from 2.3 to 35 mM and [Na+]e decreases to 75 mM.20,21 Increases of [K+]e could induce persistent Na+ current (INap).58 Computer simulation demonstrates that INap and/or NMDA current could cause CSD-like depolarization.59 The elevated [K+]e induced depolarization could reverse NCX and NCKX, resulting in Ca2+ overloading, thus leading to apoptosis or excitotoxicity60 in neurons. CEC-induced vasoconstriction may also cause ischemia by 2 mechanisms: CEC NKATs change vasomotor tone by regulating the [K+] in interstitial fluid where it can alter excitability of local vascular smooth muscles.61–63 CECs may thus exert effects humorally by releasing mediators that include calcitonin G related peptide (CGRP), serotonin, and endothelin, which are altered in the blood during acute migraine.
We propose that repetitive and/or prolonged inhibition of CEC NKAT activity with prolonged increase of [K+]e may cause apoptosis of neurons and micro-infarction from vasoconstriction. We suggest that these neuronal and vasomotor effects are the basis for the increased frequency of hyperintense MRI signals and stroke observed in migraineurs with aura. This could be tested in an animal model by repeated and prolonged CEC NKAT inhibition with MRI and pathological evaluation.
In response to CSD and to avoid apoptosis and cerebral infarction, glial K+ re-uptake and increased NKAT activity rapidly restore the normal low [K+]e. Sustaining NKAT inhibition in cultured astrocytes with ouabain increases NKAT activity and leads to NKAT over-expression,64 and elevated [Na+]i has been shown to increase NKAT activity.65 We propose that if compensation is perfect, the aura ends without migraine. This is uncommon (see Aura Followed by Migraine, below).
The CSD moves at 3–6 mm per minute. Recovery of evoked potentials takes 15–30 minutes and ion redistribution takes at least 30 minutes.66 Leão initially described vascular changes in addition to the neuronal depression of CSD67 and the propagation of the CSD effect on cortical surface arterioles was recently reported to be separate from that in the brain parenchyma.68 The mechanism for propagation of either vascular or neuronal changes is not well understood. Based on the theory that decreased CEC NKAT induces CSD by increasing [K+]e in the interstitial fluid, we hypothesize that the “spreading” may be governed by the flow rates of K+ in the interstitial fluid.
Aura Followed by Migraine (Fig. 2).—
We propose that a more typical CEC NKAT response to the inhibition that caused aura is over-activation. This causes NKAT activity to exceed its normal upper limit, analogous to the cellular responses to NKAT inhibition,64,65 elevating [Na+]e and decreasing [K+]e. The high [Na+]e causes increased neuronal excitability, manifest clinically as migraine (Fig. 2).
Migraine Without Aura (Fig. 2).—
Independent of CSD, we propose that NKAT up-regulators cause CEC NKAT activity to rise above the normal upper limit, elevating [Na+]e and decreasing [K+]e. This causes varying degrees of increased neuronal excitability, manifest clinically as migraine (Fig. 2). It is possible that CSD may occur without obvious symptoms in patients who have migraine-without-aura, in the form of a silent aura.69 Genetic analysis may help to separate aura and headache.70
Deviation from NKAT Homeostasis.—
Homeostasis is disturbed enough to cause aura or migraine when any regulator (Table 2) or NKAT itself alters CEC NKAT activity outside normal levels. Mechanisms that may alter these regulators include mutations/polymorphisms, variations from the environment (such as diet, medications, exercise, or stress), or alterations in their transport, synthesis, signaling, metabolism, or clearance. The multiple redundancy of this system is understandable to regulate such an important enzyme.
Many molecular circuits known to be involved in the pathophysiology of migraine are regulators of NKAT (Table 2), including serotonin, CGRP, dopamine, estrogens, glutamate, cannabinoids, nitric oxide, noradrenaline, or caffeine. The CECs are known to have serotonin, estrogen, and phospholipid receptors, and GPCRs.54–56 This theory adds to the understanding of migraine pathophysiology by offering a mechanism for excessive fluctuations of neuronal excitability resulting from deviations in CEC NKAT activity caused by changes in its many regulatory inputs.
THE CEC NKAT HOMEOSTASIS THEORY AND GENERAL MIGRAINE FEATURES
Increased neuronal excitation in trigeminal neurons from increased [Na+]e could cause pain without an external stimulus and release of Substance P and CGRP.71 CEC NKAT disturbance is not limited to the trigeminal pathway, and when [Na+]e is elevated in any neural pathway, hyperexcitability would be manifest with pathway-specific symptoms. Each hyperexcitable pathway would have its own manifestation, consistent with the many symptoms during migraine. For instance, hyperexcitability of the vestibular pathway would cause nausea and/or ataxia, hyperexcitability of the auditory or visual pathways would cause phonophobia or photophobia, and hyperexcitability of the locus coeruleus would increase vigilance and alarm. Hyperexcitability of neurons that have inhibitory neurotransmission will further add to the variety of functional consequences. The range of possible symptoms are only limited by the neuronal circuits that can be affected by changes in the interstitial [Na+] and [K+]e.
We propose that deviation of CEC NKAT activity in migraine from normal limits can be short-lived or extended. As the [K+]e increases beyond tolerable levels during aura, glial re-uptake of [K+]e and increased NKAT activity is required to reduce [K+]e and avoid apoptosis. If there are sufficient intracellular stores of NKAT available for translocation to the cell membrane, this process can be rapid, within seconds. However, if not locally available, the transcription, translation, and translocation of new NKAT protein can take 30–60 minutes.73,74 Increased expression of CEC NKAT will restore the normal balance of [Na+]e and [K+]e and terminate the CSD. At this time, the migraineur returns to neuronal homeostasis and feels normal. If the required NKAT levels are perfectly attained, then there is no migraine. More often, there is widespread over-expression of the CEC NKAT and [Na+]e increases above the upper normal limits. Diffusely increased [Na+]e causes neuronal excitation that produces symptoms depending on the specific molecular isoforms and the location of the involved CECs, neurons, and vascular smooth muscles. These neuronal and vasomotor effects in migraine rapidly invoke many other cellular reactions, including activation of lipid mediators of inflammation and pain, disruption of oxidation control, etc. It may take hours to return these NKAT-derived activities to normal, a duration conforming to typical migraine.
We propose that when migraineurs are stressed and NKAT activity is close to the upper limit of normal, a single aggravating trigger such as sensory input (eg, sound, light, smell) can unmask the precarious CEC NKAT activity, with increased [Na+]e rapidly causing neuronal hyperexcitability and onset of migraine. In chronic migraine, we propose that chronically elevated CEC NKAT activity and the many reactive consequences of altered neuronal excitability fail to stabilize, and the high [Na+]e sustains neuronal hyperexcitability.
Aging.—
Migraine lessens with age, as do a number of NKAT parameters: NKAT activity and the neuronal resting membrane potential decrease with age in mice in a ouabain-dependent manner, considered to be due to lipid changes in the membrane.75,76 The mRNA levels of the NKAT α−3 isoform in neurons are reduced in normal aging77–79 (considered to increase the risk of Alzheimer’s disease with age80). NKAT activity is reduced with aging in synaptosome preparations from female rat brains.81 In a proteomic study of aging transgenic mice, NKAT levels dropped dramatically with aging.82 We propose that these multiple, independent studies of reduced NKAT activity with age are consistent with changes in migraine: (1) Inhibition of an already lowered NKAT activity will make it easier to drop below the lower activity limit, increasing [K+]e and causing spreading depression, consistent with observations that auras by themselves are more common in the elderly; (2) Less NKAT activity will make older people less capable of increasing [Na+]e to levels that cause migraine. This reduced NKAT activity may not be beneficial when faster reactions or cognition are desired from physiological increases in neuronal excitability, but it has the advantage of less migraine.
Diurnal Timing.—
Several studies have suggested that migraine has a circadian rhythm, with the time of onset either in the early morning or afternoon.83–86 Experimentally, R192Q migraine mice lack the physiological retardation in circadian adaption to phase-advance shifts.87 The NKAT theory is consistent with these observations, since its biochemistry has similar rhythms; 2 studies report a diurnal rhythm for NKAT expression that match the migraine onset times, consistent with predisposing migraineurs to attacks starting at these times: NKAT activity in the ventral suprachiasmatic nucleus neurons is diurnally regulated, with activity increasing during the day and decreasing at night.88 Inhibition by the NKAT inhibitor strophanthidin decreases at night.88 While other analytes also exhibit periodic rhythms, these clinical and NKAT diurnal correlates are consistent with, and may be responsible for, migraine chronovariations.
In humans, one can evaluate whether [Na+]csf has chronobiological variation similar to that of the time of onset of migraine and the diurnal NKAT variation. We found [Na+]csf does have such a rhythm in CSF sampled every 10 minutes for 24 hours from non-headache suffering controls.89 To extend this research, it is necessary to obtain less invasive samples than CSF; however, blood taken at the same time as CSF did not reveal changes in [Na+]. In searching for an alternative sample source with sodium regulation similar to that in the brain, salivary glands may be informative because they have significant origin from neuro-ectoderm.90 We are currently evaluating whether migraineurs have chronobiological variation in saliva [Na+] that is similar to the changes in [Na+]csf.
Known chronobiological variations in migraine time of onset, [Na+]csf, and NKAT suggest that chrono-pharmaceutical approaches may help. Specifically, timing of drug administration may be more effective if optimized to coincide with the circadian variations in [Na+]csf and NKAT, with particular attention to the higher risk times in early morning and mid-afternoon.
Triggers.—
Dehydration and over-hydration are both known migraine triggers. Such extreme ionic challenges will directly influence CEC NKAT activity and may lead to migraine. Salt intake is largely controlled by kidney NKAT, and blood plasma [Na+] varies much more than in brain or CSF, yet equilibration between plasma and brain/CSF is rapid (minutes).30,32,33 Thus, it is not surprising that salt intake can trigger headaches.91 Notably the elevated [Na+]csf we found was not reflected in blood plasma,29 perhaps emphasizing the substantial regulation (at CEC NKATs) that differentiates these body compartments.
Sleep disturbance, a known migraine trigger, is associated with NKAT dysfunction: Sleep deprivation induces overexpression of NKAT in rat brain synaptosomal preparations, and in the locus coeruleus, laterodorsal tegmentum, pedunculo pontine tegmentum, and medial preoptic area.92 Rapid eye movement sleep deprivation led to an increased ouabain binding Kd for NKAT in cultured fetal rat telencephalon neurons, which might contribute to the increased excitability in these rats.93 Migraine triggers of stress (eg, by noradrenergic receptors) and anoxia directly affect NKAT function by signal transduction pathways (Table 2).
Diet can trigger migraine in some migraineurs. We propose that any dietary component may influence the NKAT regulators (Table 2) and we single out lipids for specific comment. A reduction in polyunsaturated fatty acids (PUFAs) or increase in saturated fatty acids in membranes inhibit NKAT activity; we suggest that counteracting this inhibition with PUFA supplements may be the basis for the improved migraine symptoms reported from one open and one blinded trial,94,95 though another double-blinded trial failed to show that PUFA supplements prevented migraine.96
Hormonal changes, especially peri-menstrually, are known to trigger more frequent and severe migraine. We propose that 2 mechanisms from the NKAT theory may be responsible: Estrogens activate NKAT by signal transduction (Table 2); and, similar to cholesterol, estrogens influence membrane fluidity-based changes in NKAT activity.
Alterations of many if not all the pathways in Table 2 have been reported in migraine, adding further support for this theory. The fact that NKAT has different expression levels and isoforms in different brain regions would allow for almost endless specific variation in symptoms. This would be consistent with the endless unique aspects of each migraine between and within sufferers that are superimposed on the most characteristic symptoms (pulsatile, unilateral, headache, with nausea, photophobia, and phonophobia).
Treatments.—
We propose that existing or future medication will be successful when treatment re-establishes sodium homeostasis either directly or via signal transduction pathways of CEC NKATs. Non-pharmaceutical treatments, such as stress reduction, sleep hygiene, and avoiding dehydration or over-hydration may benefit by reducing NKAT perturbation. Since the gateway cell is the CEC, blood borne modifiers can affect CEC NKATs via their luminal receptors, thus removing the necessity for passage across the blood-brain-barrier. This may explain the central effects from well-established migraine medications that are known to have poor barrier penetrance, including triptans.
Cohen suggested that prophylactic drugs used to treat migraine do so through their affects on sodium regulation.97 Drug mechanisms in vivo are very complex but, based on the NKAT homeostasis theory, we can propose that blocking sodium channels may reduce the neuronal excitability that would otherwise result from excessively high [Na+]e. Prophylaxis takes many weeks before benefit, perhaps consistent with the time required to adjust all NKAT regulatory inputs for optimal balance and to stabilize (not flatten!) the interstitial fluid circulation of fluctuating [Na+]e and [K+]e.
The NKAT homeostasis theory affords useful directions to both evaluate and conceive of new treatment approaches. First, new medications can be evaluated on cell cultures, brain slices, CECs, and small blood vessels, or in vivo for their effect on CEC NKATs and sodium/potassium homeostasis. Second, we may help to personalize medical therapy by defining the most disruptive NKAT regulators (Table 2) in an individual. This could be evaluated by screening endothelial or lymph cells from an individual to identify regulators (Table 2) that most perturb their NKATs (NKAT changes in lymphocytes have been found in migraineurs98 and endothelial cell precursors have been identified in peripheral blood99). Third, in status migrainosis or severe chronic migraine, perhaps invasive efforts to correct the high [Na+]e and low [K+]e by blood plasma or CSF exchange may be worth considering. Finally, on a precautionary note, we predict the possibility of cellular apoptosis or stroke during aura if potassium homeostasis is not corrected quickly; aggressive efforts are therefore justified to lessen aura severity.
CAVEATS
There is a paucity of direct data at this stage on migraine sodium/potassium homeostasis, and much of this theory is deduced from basic science knowledge of NKAT biochemistry and functions. We propose that the principal caveats are due to the limited knowledge of brain molecular pathophysiology. There has only been our single study to report elevated CSF [Na+] and nothing is known about regional differences in brain or CSF [Na+]. Details of all molecules involved in migraine, including their isoform and cell distribution, amount and duration of change, and circadian variations are, as yet, poorly defined. As an example, the Allen Mouse Brain Atlas reveals the ubiquitous presence of transcripts of all NKAT isoforms in the brain, but does not reveal specific differential cellular details. We have tabulated many modulators of NKAT, but have not discussed many consequences that result from altered NKAT activity (eg,46,100–105). Defining the mechanisms of neuronal excitability with regard to sodium/potassium homeostasis requires further research, especially in neurons, glial cells, smooth muscle cells, and CECs. Defining vasomotor effects from altered [K+]e and NKAT-induced humoral vasoactive compounds requires further research. The effects of NKAT disturbance in neuron-to-neuron connections, excitatory vs inhibitory neurotransmission, and the interactions between altered NKAT activity in different brain regions and in surrounding non-neuronal cells are beyond the scope of this analysis.
CONCLUSIONS
To explain the increased [Na+]csf reported in migraine, we propose a theory based on deviations from CEC NKAT homeostasis. Migraineurs have genetic or environmental variations of the myriad regulators of NKATs that predispose them to disturbances in their homeostasis, specifically in CECs. Fluctuations of NKAT activity outside normal limits lead to neuronal and vasomotor effects. Activity below the normal limit increases [K+]e, depolarizes neurons, causing aura.Activity above the normal limit elevates [Na+]e, increases neuronal excitability, causing migraine. The same ionic disturbance from CEC NKATs also affects vasomotor tone by direct or humoral effects on smooth muscles.
This theory is consistent with the known events of migraine. We propose that deviation from normal CEC NKAT is a common pathway in the genesis of the distinct components of aura and migraine. There is much CEC NKAT regulation to define, but testing described herein, both in migraineurs and model systems, will help elucidate NKAT involvement in migraine, and help evaluate whether CEC NKAT modulation could more successfully manage migraine.
Acknowledgments:
We thank Jay Callaway and Katie Clark Vecchio for helpful discussions.The National Institutes of Health, the Norris, Lucas, and Glide Foundations, and HMRI funded this work.
Abbreviations:
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CEC
capillary endothelial cell
- CGRP
calcitonin G related peptide
- CSD
cortical spreading depression
- CSF
cerebrospinal fluid
- FHM
familial hemiplegic migraine
- GPCR
G-protein coupled receptor
- IGF1
insulin growth factor 1
- INap
persistent Na+ current
- [Na+]csf or [K+]csf
sodium or potassium ion concentration in CSF
- [Na+]e or [K+]e
brain tissue extracellular sodium or potassium ion concentration
- [Na+]i or [K+]i
intracellular sodium or potassium ion concentration
- NCX
sodium and calcium exchanger
- NCKX
potassium-dependent sodium and calcium exchanger
- NKAT
Na+, K+ -ATPase transporter
- NMDA
N-methyl-D-aspartic acid
- PKA
C, G protein kinase A, C, G
- PUFA
polyunsaturated fatty acid
- SD
spreading depression
Footnotes
Conflict of Interest: None
REFERENCES
- 1.May A, Goadsby PJ. The trigeminovascular system in humans: Pathophysiologic implications for primary headache syndromes of the neural influences on the cerebral circulation. J Cereb Blood Flow Metab. 1999;19:115–127. [DOI] [PubMed] [Google Scholar]
- 2.Lance JW, Adams RW, Lambert GA. Bulbo-cortical pathways and their possible relevance to migraine and epilepsy. Funct Neurol. 1986;1:357–361. [PubMed] [Google Scholar]
- 3.Adams RW, Lambert GA, Lance JW. Stimulation of brainstem nuclei in the cat: Effect on neuronal activity in the primary visual cortex of relevance to cerebral blood flow and migraine. Cephalalgia. 1989;9:107–118. [DOI] [PubMed] [Google Scholar]
- 4.Bartsch T, Levy MJ, Knight YE, Goadsby PJ. Inhibition of nociceptive dural input in the trigeminal nucleus caudalis by somatostatin receptor blockade in the posterior hypothalamus.Pain.2005;117:30–39. [DOI] [PubMed] [Google Scholar]
- 5.Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–982. [DOI] [PubMed] [Google Scholar]
- 6.Drummond PD, Lance JW. Extracranial vascular changes and the source of pain in migraine headache. Ann Neurol. 1983;13:32–37. [DOI] [PubMed] [Google Scholar]
- 7.Knight YE, Bartsch T, Kaube H, Goadsby PJ. P/Q-type calcium-channel blockade in the periaqueductal gray facilitates trigeminal nociception: A functional genetic link for migraine? J Neurosci. 2002;22:RC213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malick A, Strassman RM, Burstein R. Trigemino-hypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J Neurophysiol. 2000; 84:2078–2112. [DOI] [PubMed] [Google Scholar]
- 9.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. [DOI] [PubMed] [Google Scholar]
- 10.Moskowitz MA. Defining a pathway to discovery from bench to bedside: The trigeminovascular system and sensitization. Headache. 2008;48:688–690. [DOI] [PubMed] [Google Scholar]
- 11.Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA. 2001;98:4687–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauritzen M Cortical spreading depression in migraine. Cephalalgia. 2001;21:757–760. [DOI] [PubMed] [Google Scholar]
- 13.Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. [DOI] [PubMed] [Google Scholar]
- 14.Silberstein SD. Topiramate in migraine prevention: Evidence-based medicine from clinical trials. Neurol Sci. 2004;25(Suppl. 3):S244–S245. [DOI] [PubMed] [Google Scholar]
- 15.Leith JL, Wilson AW, Donaldson LF, Lumb BM. Cyclooxygenase-1-derived prostaglandins in the periaqueductal gray differentially control C-versus A-fiber-evoked spinal nociception. J Neurosci. 2007;27:11296–11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipton RB, Baggish JS, Stewart WF, Codispoti JR, Fu M. Efficacy and safety of acetaminophen in the treatment of migraine: Results of a randomized, double-blind, placebo-controlled, population-based study. Arch Intern Med. 2000;160:3486–3492. [DOI] [PubMed] [Google Scholar]
- 17.May A, Goadsby PJ. Pharmacological opportunities and pitfalls in the therapy of migraine. Curr Opin Neurol. 2001;14:341–345. [DOI] [PubMed] [Google Scholar]
- 18.Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–1110. [DOI] [PubMed] [Google Scholar]
- 19.Humphrey PP. The discovery and development of the triptans, a major therapeutic breakthrough. Headache. 2008;48:685–687. [DOI] [PubMed] [Google Scholar]
- 20.Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. [DOI] [PubMed] [Google Scholar]
- 21.Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience. 1978;3:1045–1059. [DOI] [PubMed] [Google Scholar]
- 22.De Fusco M, Marconi R, Silvestri L, et al. Haplo-insufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192–196. [DOI] [PubMed] [Google Scholar]
- 23.Vanmolkot KR, Kors EE, Hottenga JJ, et al. Novel mutations in the Na+, K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54:360–366. [DOI] [PubMed] [Google Scholar]
- 24.Terwindt GM, Ophoff RA, Haan J, Sandkuijl LA, Frants RR, Ferrari MD. Migraine, ataxia and epilepsy: A challenging spectrum of genetically determined calcium channelopathies. Dutch Migraine Genetics Research Group. Eur J Hum Genet. 1998;6:297–307. [DOI] [PubMed] [Google Scholar]
- 25.Dichgans M, Freilinger T, Eckstein G, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. [DOI] [PubMed] [Google Scholar]
- 26.Ducros A, Denier C, Joutel A, et al. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17–24. [DOI] [PubMed] [Google Scholar]
- 27.Moskowitz MA, Bolay H, Dalkara T. Deciphering migraine mechanisms: Clues from familial hemiplegic migraine genotypes. Ann Neurol. 2004;55:276–280. [DOI] [PubMed] [Google Scholar]
- 28.de Vries B, Freilinger T, Vanmolkot KR, et al. Systematic analysis of three FHM genes in 39 sporadic patients with hemiplegic migraine. Neurology. 2007;69:2170–2176. [DOI] [PubMed] [Google Scholar]
- 29.Harrington MG, Fonteh AN, Cowan RP, et al. Cerebrospinal fluid sodium increases in migraine. Headache. 2006;46:1128–1135. [DOI] [PubMed] [Google Scholar]
- 30.Sweet WH, Brownell GL, Scholl JA, Bowsher DR, Benda P, Stickley EE. The formation, flow and absorption of cerebrospinal fluid; newer concepts based on studies with isotopes. Res Publ Assoc Res Nerv Ment Dis. 1955;34:101–159. [PubMed] [Google Scholar]
- 31.Somjen GG. Neuroglia and spinal fluids. J Exp Biol. 1981;95:129–133. [DOI] [PubMed] [Google Scholar]
- 32.Bito LZ, Davson H. Local variations in cerebrospinal fluid composition and its relationship to the composition of the extracellular fluid of the cortex. Exp Neurol. 1966;14:264–280. [DOI] [PubMed] [Google Scholar]
- 33.Olsen NS, Rudolph GG. Transfer of sodium and bromide ions between blood, cerebrospinal fluid and brain tissue. Am J Physiol. 1955;183:427–432. [DOI] [PubMed] [Google Scholar]
- 34.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol. 1949;108:37–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuo CC, Liao SY. Facilitation of recovery from inactivation by external Na+ and location of the activation gate in neuronal Na+ channels. J Neurosci. 2000;20:5639–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Huysse JW, Hou X. Pressor response to CSF sodium in mice: Mediation by a ouabain-like substance and renin-angiotensin system in the brain. Brain Res. 2004;1021:219–231. [DOI] [PubMed] [Google Scholar]
- 37.Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol. 2006;21:295–304. [DOI] [PubMed] [Google Scholar]
- 38.Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: New windows on complex neurological diseases. J Neurosci. 2008;28:11768–11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwamoto T, Watanabe Y, Kita S, Blaustein MP. Na+/Ca2+ exchange inhibitors: A new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets. 2007;7:188–198. [DOI] [PubMed] [Google Scholar]
- 41.Martinez F, Castillo J, Rodriguez JR, Leira R, Noya M. Neuroexcitatory amino acid levels in plasma and cerebrospinal fluid during migraine attacks. Cephalalgia. 1993;13:89–93. [DOI] [PubMed] [Google Scholar]
- 42.Whittam R The dependence of the respiration of brain cortex on active cation transport. Biochem J. 1962;82:205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Astrup J, Sorensen PM, Sorensen HR. Oxygen and glucose consumption related to Na+-K+ transport in canine brain. Stroke. 1981;12:726–730. [DOI] [PubMed] [Google Scholar]
- 44.Menna G, Tong CK, Chesler M. Extracellular pH changes and accompanying cation shifts during ouabain-induced spreading depression. J Neurophysiol. 2000;83:1338–1345. [DOI] [PubMed] [Google Scholar]
- 45.Hertz L An intense potassium uptake into astrocytes, its further enhancement by high concentrations of potassium, and its possible involvement in potassium homeostasis at the cellular level. Brain Res. 1978;145:202–208. [DOI] [PubMed] [Google Scholar]
- 46.Geering K Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens. 2008;17:526–532. [DOI] [PubMed] [Google Scholar]
- 47.Betz AL, Firth JA, Goldstein GW. Polarity of the blood-brain barrier: Distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res. 1980;192:17–28. [DOI] [PubMed] [Google Scholar]
- 48.Eisenberg HM, Suddith RL. Cerebral vessels have the capacity to transport sodium and potassium. Science. 1979;206:1083–1085. [DOI] [PubMed] [Google Scholar]
- 49.Praetorius J, Nielsen S. Distribution of sodium transporters and aquaporin-1 in the human choroid plexus. Am J Physiol Cell Physiol. 2006;291:C59–C67. [DOI] [PubMed] [Google Scholar]
- 50.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochem Int. 2004;45:545–552. [DOI] [PubMed] [Google Scholar]
- 51.Cserr HF. Physiology of the choroid plexus. Physiol Rev. 1971;51:273–311. [DOI] [PubMed] [Google Scholar]
- 52.Go KG. The normal and pathological physiology of brain water. Adv Tech Stand Neurosurg. 1997;23:47–142. [DOI] [PubMed] [Google Scholar]
- 53.Kimelberg HK. Water homeostasis in the brain: Basic concepts. Neuroscience. 2004;129:851–860. [DOI] [PubMed] [Google Scholar]
- 54.Razmara A, Sunday L, Stirone C, et al. Mitochondrial effects of estrogen are mediated by estrogen receptor alpha in brain endothelial cells. J Pharmacol Exp Ther. 2008;325:782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brust P, Friedrich A, Krizbai IA, et al. Functional expression of the serotonin transporter in immortalized rat brain microvessel endothelial cells. J Neurochem. 2000;74:1241–1248. [DOI] [PubMed] [Google Scholar]
- 56.Takuwa Y, Takuwa N, Sugimoto N. The Edg family G protein-coupled receptors for lysophospholipids: Their signaling properties and biological activities. J Biochem. 2002;131:767–771. [DOI] [PubMed] [Google Scholar]
- 57.Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386–398. [DOI] [PubMed] [Google Scholar]
- 58.Somjen GG, Muller M. Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res. 2000;885:102–110. [DOI] [PubMed] [Google Scholar]
- 59.Kager H, Wadman WJ, Somjen GG. Simulated seizures and spreading depression in a neuron model incorporating interstitial space and ion concentrations. J Neurophysiol. 2000;84:495–512. [DOI] [PubMed] [Google Scholar]
- 60.Kiedrowski L High activity of K+-dependent plasmalemmal Na+/Ca2+ exchangers in hippocampal CA1 neurons. Neuroreport. 2004;15:2113–2116. [DOI] [PubMed] [Google Scholar]
- 61.Azin AL. Role of extracellular pO2 and pCO2 in membrane mechanisms regulating cerebral artery smooth muscle. Fiziol Zh SSSR Im I M Sechenova. 1981;67:1652–1660. [PubMed] [Google Scholar]
- 62.Ko EA, Han J, Jung ID, Park WS. Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res. 2008;44:65–81. [DOI] [PubMed] [Google Scholar]
- 63.Olesen SP, Munch E, Moldt P, Drejer J. Selective activation of Ca(2+)-dependent K+ channels by novel benzimidazolone. Eur J Pharmacol. 1994; 251:53–59 [DOI] [PubMed] [Google Scholar]
- 64.Hosoi R, Matsuda T, Asano S, et al. Isoform-specific up-regulation by ouabain of Na+,K+-ATPase in cultured rat astrocytes. J Neurochem. 1997;69:2189–2196. [DOI] [PubMed] [Google Scholar]
- 65.Senatorov VV, Stys PK, Hu B. Regulation of Na+,K+-ATPase by persistent sodium accumulation in adult rat thalamic neurones. J Physiol. 2000;525(Pt 2):343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lauritzen M Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. [DOI] [PubMed] [Google Scholar]
- 67.Leão A Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:391–396. [DOI] [PubMed] [Google Scholar]
- 68.Brennan KC, Beltran-Parrazal L, Lopez-Valdes HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–4151. [DOI] [PubMed] [Google Scholar]
- 69.Iadecola C From CSD to headache: A long and winding road. Nat Med. 2002;8:110–112. [DOI] [PubMed] [Google Scholar]
- 70.Barrett CF, van den Maagdenberg AM, Frants RR, Ferrari MD. Familial hemiplegic migraine. Adv Genet. 2008;63:57–83. [DOI] [PubMed] [Google Scholar]
- 71.Gulbenkian S, Uddman R, Edvinsson L. Neuronal messengers in the human cerebral circulation. Peptides. 2001;22:995–1007. [DOI] [PubMed] [Google Scholar]
- 72.van den Maagdenberg AM, Pietrobon D, Pizzorusso T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. [DOI] [PubMed] [Google Scholar]
- 73.Taormino JP, Fambrough DM. Pre-translational regulation of the (Na+ + K+)-ATPase in response to demand for ion transport in cultured chicken skeletal muscle. J Biol Chem. 1990;265:4116–4123. [PubMed] [Google Scholar]
- 74.Smith LC, Harrington MG, Britten RJ, Davidson EH. The sea urchin profilin gene is specifically expressed in mesenchyme cells during gastrulation. Dev Biol. 1994;164:463–474. [DOI] [PubMed] [Google Scholar]
- 75.Tanaka Y, Ando S. Synaptic aging as revealed by changes in membrane potential and decreased activity of Na+,K(+)-ATPase. Brain Res. 1990; 506:46–52. [DOI] [PubMed] [Google Scholar]
- 76.Tanaka Y, Ando S. Age-related changes in [3H] ouabain binding to synaptic plasma membranes isolated from mouse brains. J Biochem. 1992; 112:117–121. [DOI] [PubMed] [Google Scholar]
- 77.Chauhan NB, Siegel GJ. In situ analysis of Na, K-ATPase alpha1- and alpha3-isoform mRNAs in aging rat hippocampus. J Neurochem. 1996;66:1742–1751. [DOI] [PubMed] [Google Scholar]
- 78.Chauhan N, Siegel G. Na,K-ATPase: Increases in alpha1-messenger RNA and decreases in alpha3-messenger RNA levels in aging rat cerebral cortex. Neuroscience. 1997;78:7–11. [DOI] [PubMed] [Google Scholar]
- 79.Chauhan N, Siegel G. Differential expression of Na,K-ATPase alpha-isoform mRNAs in aging rat cerebellum. J Neurosci Res. 1997;47:287–299. [PubMed] [Google Scholar]
- 80.Chauhan NB, Lee JM, Siegel GJ. Na,K-ATPase mRNA levels and plaque load in Alzheimer’s disease. J Mol Neurosci. 1997;9:151–166. [DOI] [PubMed] [Google Scholar]
- 81.Fraser CL, Arieff AI. Na-K-ATPase activity decreases with aging in female rat brain synaptosomes. Am J Physiol Renal Physiol. 2001;281:F674–F678. [DOI] [PubMed] [Google Scholar]
- 82.Fu YJ, Xiong S, Lovell MA, Lynn BC. Quantitative proteomic analysis of mitochondria in aging PS-1 transgenic mice. Cell Mol Neurobiol. 2009;29:649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alstadhaug KB. Periodicity of migraine. Headache. 2006;46:532–533. [DOI] [PubMed] [Google Scholar]
- 84.Alstadhaug KB, Bekkelund S, Salvesen R. Circannual periodicity of migraine? Eur J Neurol. 2007;14:983–988. [DOI] [PubMed] [Google Scholar]
- 85.Solomon GD. Circadian rhythms and migraine. Cleve Clin J Med. 1992;59:326–329. [DOI] [PubMed] [Google Scholar]
- 86.Fox AW, Davis RL. Migraine chronobiology. Headache. 1998;38:436–441. [DOI] [PubMed] [Google Scholar]
- 87.van Oosterhout F, Michel S, Deboer T, et al. Enhanced circadian phase resetting in R192Q Cav2.1 calcium channel migraine mice. Ann Neurol. 2008;64:315–324. [DOI] [PubMed] [Google Scholar]
- 88.Wang HY, Huang RC. Diurnal modulation of the Na+/K+-ATPase and spontaneous firing in the rat retinorecipient clock neurons. J Neurophysiol. 2004;92:2295–2301. [DOI] [PubMed] [Google Scholar]
- 89.Harrington MG, Oborina E, Pogoda JM, et al. Circadian sodium rhythms in human cerebrospinal fluid. American Academy of Neurology Annual Meeting. 2008. [Google Scholar]
- 90.Denny PC, Ball WD, Redman RS. Salivary glands: A paradigm for diversity of gland development. Crit Rev Oral Biol Med. 1997;8:51–75. [DOI] [PubMed] [Google Scholar]
- 91.Kurlansky M Salt: A World History. New York: Penguin Books; 2002. [Google Scholar]
- 92.Majumdar S, Faisal M, Madan V, Mallick BN. Increased turnover of Na-K ATPase molecules in rat brain after rapid eye movement sleep deprivation. J Neurosci Res. 2003;73:870–875. [DOI] [PubMed] [Google Scholar]
- 93.Bignotto M, de Andrade UJ, de Carvalho JG, Benedito MA. Rapid eye movement sleep deprivation induces changes in the high-affinity binding of [3H]-ouabain to the rat cortical membranes. Neurosci Lett. 2006;396:143–147. [DOI] [PubMed] [Google Scholar]
- 94.Wagner W, Nootbaar-Wagner U. Prophylactic treatment of migraine with gamma-linolenic and alpha-linolenic acids. Cephalalgia. 1997;17:127–130;discussion 102. [DOI] [PubMed] [Google Scholar]
- 95.Harel Z, Gascon G, Riggs S, Vaz R, Brown W, Exil G. Supplementation with omega-3 polyunsaturated fatty acids in the management of recurrent migraines in adolescents. J Adolesc Health. 2002; 31:154–161. [DOI] [PubMed] [Google Scholar]
- 96.Pradalier A, Bakouche P, Baudesson G, et al. Failure of omega-3 polyunsaturated fatty acids in prevention of migraine: A double-blind study versus placebo. Cephalalgia. 2001;21:818–822. [DOI] [PubMed] [Google Scholar]
- 97.Cohen GL. Migraine prophylactic drugs work via ion channels. Med Hypotheses. 2005;65:114–122. [DOI] [PubMed] [Google Scholar]
- 98.Scarrone S, Podesta M, Cupello A, et al. Abnormalities of Na/K ATPase in migraine with aura. Cephalalgia. 2007;27:128–132. [DOI] [PubMed] [Google Scholar]
- 99.Wu H, Riha GM, Yang H, Li M, Yao Q, Chen C. Differentiation and proliferation of endothelial progenitor cells from canine peripheral blood mononuclear cells. J Surg Res. 2005;126:193–198. [DOI] [PubMed] [Google Scholar]
- 100.Horisberger JD, Lemas V, Kraehenbuhl JP, Rossier BC. Structure-function relationship of Na,K-ATPase. Annu Rev Physiol. 1991;53:565–584. [DOI] [PubMed] [Google Scholar]
- 101.Trachtenberg MC, Packey DJ, Sweeney T. In vivo functioning of the Na+, K+-activated ATPase. Curr Top Cell Regul. 1981;19:159–217. [DOI] [PubMed] [Google Scholar]
- 102.Blaustein MP, Zhang J, Chen L, Hamilton BP. How does salt retention raise blood pressure? Am J Physiol Regul Integr Comp Physiol. 2006;290:R514–R523. [DOI] [PubMed] [Google Scholar]
- 103.Lebel CP, Schatz RA. Altered synaptosomal phospholipid metabolism after toluene: Possible relationship with membrane fluidity, Na+,K(+)-adenosine triphosphatase and phospholipid methylation. J Pharmacol Exp Ther. 1990;253:1189–1197. [PubMed] [Google Scholar]
- 104.Liu XL, Miyakawa A, Aperia A, Krieger P. Na,K-ATPase generates calcium oscillations in hippocampal astrocytes. Neuroreport. 2007;18:597–600. [DOI] [PubMed] [Google Scholar]
- 105.Xie Z Molecular mechanisms of Na/K-ATPase-mediated signal transduction. Ann N Y Acad Sci. 2003;986:497–503. [DOI] [PubMed] [Google Scholar]
- 106.Berrebi-Bertrand I, Maixent JM, Christe G, Lelievre LG. Two active Na+/K+-ATPases of high affinity for ouabain in adult rat brain membranes. Biochim Biophys Acta. 1990;1021:148–156. [DOI] [PubMed] [Google Scholar]
- 107.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–F650. [DOI] [PubMed] [Google Scholar]
- 108.Jewell EA, Lingrel JB. Comparison of the substrate dependence properties of the rat Na,K-ATPase alpha 1, alpha 2, and alpha 3 isoforms expressed in HeLa cells. J Biol Chem. 1991;266:16925–16930. [PubMed] [Google Scholar]
- 109.Lingrel JB, Williams MT, Vorhees CV, Moseley AE. Na,K-ATPase and the role of alpha isoforms in behavior. J Bioenerg Biomembr. 2007;39:385–389. [DOI] [PubMed] [Google Scholar]
- 110.McGrail KM, Phillips JM, Sweadner KJ. Immuno-fluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K-ATPase. J Neurosci. 1991;11:381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Segall L, Daly SE, Blostein R. Mechanistic basis for kinetic differences between the rat alpha 1, alpha 2, and alpha 3 isoforms of the Na,K-ATPase. J Biol Chem. 2001;276:31535–31541. [DOI] [PubMed] [Google Scholar]
- 112.Peng L, Martin-Vasallo P, Sweadner KJ. Isoforms of Na,K-ATPase alpha and beta subunits in the rat cerebellum and in granule cell cultures. J Neurosci. 1997;17:3488–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watts AG, Sanchez-Watts G, Emanuel JR, Levenson R. Cell-specific expression of mRNAs encoding Na+,K(+)-ATPase alpha- and beta-subunit isoforms within the rat central nervous system. Proc Natl Acad Sci USA. 1991;88:7425–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Magyar JP, Bartsch U, Wang ZQ, et al. Degeneration of neural cells in the central nervous system of mice deficient in the gene for the adhesion molecule on glia, the beta 2 subunit of murine Na,K-ATPase. J Cell Biol. 1994;127:835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Martin-Vasallo P, Wetzel RK, Garcia-Segura LM, Molina-Holgado E, Arystarkhova E, Sweadner KJ. Oligodendrocytes in brain and optic nerve express the beta3 subunit isoform of Na,K-ATPase. Glia. 2000;31:206–218. [DOI] [PubMed] [Google Scholar]
- 116.Feschenko MS, Donnet C, Wetzel RK, Asinovski NK, Jones LR, Sweadner KJ. Phospholemman, a single-span membrane protein, is an accessory protein of Na,K-ATPase in cerebellum and choroid plexus. J Neurosci. 2003;23:2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arystarkhova E, Sweadner KJ. Splice variants of the gamma subunit (FXYD2) and their significance in regulation of the Na, K-ATPase in kidney. J Bioenerg Biomembr. 2005;37:381–386. [DOI] [PubMed] [Google Scholar]
- 118.Bell JR, Kennington E, Fuller W, et al. Characterization of the phospholemman knockout mouse heart: Depressed left ventricular function with increased Na-K-ATPase activity. Am J Physiol Heart Circ Physiol. 2008;294:H613–H621. [DOI] [PubMed] [Google Scholar]
- 119.Cairo ER, Friedrich T, Swarts HG, et al. Impaired routing of wild type FXYD2 after oligomerisation with FXYD2-G41R might explain the dominant nature of renal hypomagnesemia. Biochim Biophys Acta. 2008;1778:398–404. [DOI] [PubMed] [Google Scholar]
- 120.Meij IC, Koenderink JB, van Bokhoven H, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na(+),K(+)-ATPase gamma-subunit. Nat Genet. 2000;26:265–266. [DOI] [PubMed] [Google Scholar]
- 121.Jones DH, Li TY, Arystarkhova E, et al. Na,K-ATPase from mice lacking the gamma subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem. 2005; 280:19003–19011. [DOI] [PubMed] [Google Scholar]
- 122.Yamaguchi F, Yamaguchi K, Tai Y, Sugimoto K, Tokuda M. Molecular cloning and characterization of a novel phospholemman-like protein from rat hippocampus. Brain Res Mol Brain Res. 2001; 86:189–192. [DOI] [PubMed] [Google Scholar]
- 123.Crambert G, Beguin P, Uldry M, et al. FXYD7, the first brain- and isoform-specific regulator of Na,K-ATPase: Biosynthesis and function of its posttranslational modifications. Ann N Y Acad Sci. 2003; 986:444–448. [DOI] [PubMed] [Google Scholar]
- 124.Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000;68:41–56. [DOI] [PubMed] [Google Scholar]
- 125.Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. [DOI] [PubMed] [Google Scholar]
- 126.Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–C566. [DOI] [PubMed] [Google Scholar]
- 127.Kassardjian A, Kreydiyyeh SI. JNK modulates the effect of caspases and NF-kappaB in the TNF-alpha-induced down-regulation of Na+/K+ATPase in HepG2 cells. J Cell Physiol. 2008;216:615–620. [DOI] [PubMed] [Google Scholar]
- 128.Namekata K, Harada C, Kohyama K, Matsumoto Y, Harada T. Interleukin-1 stimulates glutamate uptake in glial cells by accelerating membrane trafficking of Na+/K+-ATPase via actin depolymerization. Mol Cell Biol. 2008;28:3273–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Banerjee B, Chaudhury S. Thyroidal regulation of different isoforms of NaKATPase in the primary cultures of neurons derived from fetal rat brain. Life Sci. 2002;71:1643–1654. [DOI] [PubMed] [Google Scholar]
- 130.Phakdeekitcharoen B, Phudhichareonrat S, Pookarnjanamorakot C, et al. Thyroid hormone increases mRNA and protein expression of Na+-K+-ATPase alpha2 and beta1 subunits in human skeletal muscles. J Clin Endocrinol Metab. 2007;92:353–358. [DOI] [PubMed] [Google Scholar]
- 131.Thompson SM, Prince DA. Activation of electrogenic sodium pump in hippocampal CA1 neurons following glutamate-induced depolarization. J Neurophysiol. 1986;56:507–522. [DOI] [PubMed] [Google Scholar]
- 132.Dunham ET, Glynn IM. Adenosinetriphosphatase activity and the active movements of alkali metal ions. J Physiol. 1961;156:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lingrel JB, Kuntzweiler T. Na+,K(+)-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- 134.Donnet C, Sweadner KJ. The mechanism of Na-K interaction on Na,K-ATPase. Ann N Y Acad Sci. 2003;986:249–251. [DOI] [PubMed] [Google Scholar]
- 135.Morth JP, Pedersen BP, Toustrup-Jensen MS, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. [DOI] [PubMed] [Google Scholar]
- 136.Fleidervish IA, Friedman A, Gutnick MJ. Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. J Physiol. 1996;493:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-D-aspartate: Role of electrogenic sodium pump. Science. 1992;258:665–667. [DOI] [PubMed] [Google Scholar]
- 138.Flashner MS, Robinson JD. Effects of Mg(2+) on activation of the (Na(+) + K(+)-dependent ATPase by Na(+1). Arch Biochem Biophys. 1979;192:584–591. [DOI] [PubMed] [Google Scholar]
- 139.Strugatsky D, Gottschalk KE, Goldshleger R, Karlish SJ. D443 of the N domain of Na+,K+-ATPase interacts with the ATP-Mg2+ complex, possibly via a second Mg2+ ion. Biochemistry (Mosc). 2005;44:15961–15969. [DOI] [PubMed] [Google Scholar]
- 140.Jacobsen MD, Pedersen PA, Jorgensen PL. Importance of Na,K-ATPase residue alpha 1-Arg544 in the segment Arg544-Asp567 for high-affinity binding of ATP, ADP, or MgATP. Biochemistry (Mosc). 2002;41:1451–1456. [DOI] [PubMed] [Google Scholar]
- 141.Meij IC, Koenderink JB, De Jong JC, et al. Dominant isolated renal magnesium loss is caused by misrouting of the Na+,K+-ATPase gamma-subunit. Ann N A Acad Sci. 2003;986:437–443. [DOI] [PubMed] [Google Scholar]
- 142.Dietz RM, Weiss JH, Shuttleworth CW. Zn2+ influx is critical for some forms of spreading depression in brain slices. J Neurosci. 2008;28:8014–8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hiatt A, McDonough AA, Edelman IS. Assembly of the (Na+ + K+)-adenosine triphosphatase. Post-translational membrane integration of the alpha subunit. J Biol Chem. 1984;259:2629–2635. [PubMed] [Google Scholar]
- 144.Palasis M, Kuntzweiler TA, Arguello JM, Lingrel JB. Ouabain interactions with the H5-H6 hairpin of the Na,K-ATPase reveal a possible inhibition mechanism via the cation binding domain. J Biol Chem. 1996;271:14176–14182. [DOI] [PubMed] [Google Scholar]
- 145.Kyte J Molecular considerations relevant to the mechanism of active transport. Nature. 1981; 292:201–204. [DOI] [PubMed] [Google Scholar]
- 146.Yoda A, Yoda S. Interaction between ouabain and the phosphorylated intermediate of Na,K-ATPase. Mol Pharmacol. 1982;22:700–705. [PubMed] [Google Scholar]
- 147.Greef K, Schadewaldt H. Introduction and Remarks on the History of Cardiac Glycosides. New York: Springer-Verlag; 1981. [Google Scholar]
- 148.Sole MJ, Benedict CR, Versteeg DH, de Kloet ER. Digitoxin therapy partially restores cardiac catecholamine and brain serotonin metabolism in congestive heart failure. J Mol Cell Cardiol. 1985;17:1055–1063. [DOI] [PubMed] [Google Scholar]
- 149.Balestrino M, Young J, Aitken P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res. 1999;838:37–44. [DOI] [PubMed] [Google Scholar]
- 150.Haglund MM, Schwartzkroin PA. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63:225–239. [DOI] [PubMed] [Google Scholar]
- 151.Durakovic Z, Plavsic F, Smalcelj A, Grgic V. [Blue color vision as a sign of digitalis poisoning]. Lijec Vjesn. 1992;114:132–134. [PubMed] [Google Scholar]
- 152.Beller GA, Smith TW, Abelmann WH, Haber E, Hood WB Jr. Digitalis intoxication. A prospective clinical study with serum level correlations. N Engl J Med. 1971;284:989–997. [DOI] [PubMed] [Google Scholar]
- 153.Johansson BW. Migraine: Effect of digoxin. J R Soc Med. 1982;75:215–216. [PMC free article] [PubMed] [Google Scholar]
- 154.Tanaka R, Strickland KP. Role of phospholipid in the activation of Na+, Ka+-activated adenosine triphosphatase of beef brain. Arch Biochem Biophys. 1965;111:583–592. [DOI] [PubMed] [Google Scholar]
- 155.Goldman SS, Albers RW. Sodium-potassium-activated adenosine triphosphatase. IX. The role of phospholipids. J Biol Chem. 1973;248:867–874. [PubMed] [Google Scholar]
- 156.Plataras C, Tsakiris S, Angelogianni P. Effect of CDP-choline on brain acetylcholinesterase and Na(+), K(+)-ATPase in adult rats. Clin Biochem. 2000;33:351–357. [DOI] [PubMed] [Google Scholar]
- 157.Hanson MA, Cherezov V, Griffith MT, et al. A specific cholesterol binding site is established by the 2.8: A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Maier GA, Robinson JD. Effects of local anesthetics and cholesterol on the (Na+ +K+)-dependent ATPase. Biochem Pharmacol. 1977;26:791–793. [DOI] [PubMed] [Google Scholar]
- 159.Tian D, Dmitrieva RI, Doris PA, et al. Protein kinase M zeta regulation of Na/K ATPase: A persistent neuroprotective mechanism of ischemic preconditioning in hippocampal slice cultures. Brain Res. 2008;1213:127–139. [DOI] [PubMed] [Google Scholar]
- 160.Fisone G, Snyder GL, Aperia A, Greengard P. Na+,K(+)-ATPase phosphorylation in the choroid plexus: Synergistic regulation by serotonin/protein kinase C and isoproterenol/cAMP-PK/PP-1 pathways. Mol Med. 1998;4:258–265. [PMC free article] [PubMed] [Google Scholar]
- 161.Das G, Gopalakrishnan A, Faisal M, Mallick BN. Stimulatory role of calcium in rapid eye movement sleep deprivation-induced noradrenaline-mediated increase in Na-K-ATPase activity in rat brain. Neuroscience. 2008;155:76–89. [DOI] [PubMed] [Google Scholar]
- 162.Fienberg AA, Hiroi N, Mermelstein PG, et al. DARPP-32: Regulator of the efficacy of dopaminergic neurotransmission. Science. 1998;281:838–842. [DOI] [PubMed] [Google Scholar]
- 163.Hazelwood LA, Free RB, Cabrera DM, Skinbjerg M, Sibley DR. Reciprocal modulation of function between the D1 and D2 dopamine receptors and the Na+,K+-ATPase. J Biol Chem. 2008;283:36441–36453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nishi A, Fisone G, Snyder GL, et al. Regulation of Na+, K+-ATPase isoforms in rat neostriatum by dopamine and protein kinase C. J Neurochem. 1999;73:1492–1501. [DOI] [PubMed] [Google Scholar]
- 165.Das G, Mallick BN. Noradrenaline acting on alpha1-adrenoceptor mediates REM sleep deprivation-induced increased membrane potential in rat brain synaptosomes. Neurochem Int. 2008;52:734–740. [DOI] [PubMed] [Google Scholar]
- 166.Mallick BN, Adya HV. Norepinephrine induced alpha-adrenoceptor mediated increase in rat brain Na-K ATPase activity is dependent on calcium ion. Neurochem Int. 1999;34:499–507. [DOI] [PubMed] [Google Scholar]
- 167.Mallick BN, Adya HV, Faisal M. Norepinephrine-stimulated increase in Na+, K+-ATPase activity in the rat brain is mediated through alpha1A-adrenoceptor possibly by dephosphorylation of the enzyme. J Neurochem. 2000;74:1574–1578. [DOI] [PubMed] [Google Scholar]
- 168.Aperia A, Ibarra F, Svensson LB, Klee C, Greengard P. Calcineurin mediates alpha-adrenergic stimulation of Na+,K(+)-ATPase activity in renal tubule cells. Proc Natl Acad Sci USA. 1992; 89:7394–7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Antonelli MC, Costa Lieste M, Mercado R, Hernandez RJ. Serotonin modulation of low-affinity ouabain binding in rat brain determined by quantitative autoradiography. Neurochem Res. 1998; 23:939–944. [DOI] [PubMed] [Google Scholar]
- 170.Pena-Rangel MT, Mercado R, Hernandez-Rodriguez J. Regulation of glial Na+/K+-ATPase by serotonin: Identification of participating receptors. Neurochem Res. 1999;24:643–649. [DOI] [PubMed] [Google Scholar]
- 171.Matlhagela K, Taub M. Regulation of the Na-K-ATPase beta(1)-subunit promoter by multiple prostaglandin-responsive elements. Am J Physiol Renal Physiol. 2006;291:F635–F646. [DOI] [PubMed] [Google Scholar]
- 172.Pascual J, Sterin-Borda L, Wald M, Borda ES. TXB2: Cardiostimulant effect that involves beta-adrenoceptor and Na+ + K+-ATPase activity. Prostaglandins Leukot Essent Fatty Acids. 1988;33: 53–59. [DOI] [PubMed] [Google Scholar]
- 173.Foley TD. 5-HPETE is a potent inhibitor of neuronal Na+, K(+)-ATPase activity. Biochem Biophys Res Commun. 1997;235:374–376. [DOI] [PubMed] [Google Scholar]
- 174.Olesen J The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol Ther. 2008;120:157–171. [DOI] [PubMed] [Google Scholar]
- 175.McQueen JK, Wilson H, Sumner BE, Fink G. Serotonin transporter (SERT) mRNA and binding site densities in male rat brain affected by sex steroids. Brain Res Mol Brain Res. 1999;63:241–247. [DOI] [PubMed] [Google Scholar]
- 176.Fink G, Sumner B, Rosie R, Wilson H, McQueen J. Androgen actions on central serotonin neurotransmission: Relevance for mood, mental state and memory. Behav Brain Res. 1999;105:53–68. [DOI] [PubMed] [Google Scholar]
- 177.Chang HH, Michaelis EK. Effects of L-glutamic acid on synaptosomal and synaptic membrane Na+ fluxes and (Na+-K+)-ATPase. J Biol Chem. 1980;255:2411–2417. [PubMed] [Google Scholar]
- 178.Chang HH, Michaelis EK. L-glutamate stimulation of Na+ efflux from brain synaptic membrane vesicles. J Biol Chem. 1981;256:10084–10087. [PubMed] [Google Scholar]
- 179.Nathanson JA, Scavone C, Scanlon C, McKee M. The cellular Na+ pump as a site of action for carbon monoxide and glutamate: A mechanism for long-term modulation of cellular activity. Neuron. 1995;14:781–794. [DOI] [PubMed] [Google Scholar]
- 180.Araya KA, David Pessoa Mahana C, Gonzalez LG. Role of cannabinoid CB1 receptors and Gi/o protein activation in the modulation of synaptosomal Na+,K+-ATPase activity by WIN55,212–2 and delta(9)-THC. Eur J Pharmacol. 2007;572:32–39. [DOI] [PubMed] [Google Scholar]
- 181.Steffens M, Feuerstein TJ. Receptor-independent depression of DA and 5-HT uptake by cannabinoids in rat neocortex—Involvement of Na(+)/K(+)-ATPase. Neurochem Int. 2004;44:529–538. [DOI] [PubMed] [Google Scholar]
- 182.Mahadik SP, Hawver DB, Hungund BL, Li YS, Karpiak SE. GM1 ganglioside treatment after global ischemia protects changes in membrane fatty acids and properties of Na+, K+-ATPase and Mg2+-ATPase. J Neurosci Res. 1989;24:402–412. [DOI] [PubMed] [Google Scholar]
- 183.Zhou G, Dada LA, Chandel NS, et al. Hypoxia-mediated Na-K-ATPase degradation requires von Hippel Lindau protein. FASEB J. 2008;22:1335–1342. [DOI] [PubMed] [Google Scholar]
- 184.Malfatti CR, Royes LF, Francescato L, et al. Intra-striatal methylmalonic acid administration induces convulsions and TBARS production, and alters Na+,K+-ATPase activity in the rat striatum and cerebral cortex. Epilepsia. 2003;44:761–767. [DOI] [PubMed] [Google Scholar]
- 185.Morel P, Tallineau C, Pontcharraud R, Piriou A, Huguet F. Effects of 4-hydroxynonenal, a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in rat striatal synaptosomes. Neurochem Int. 1998;33:531–540. [DOI] [PubMed] [Google Scholar]
- 186.Lu C, Chan SL, Fu W, Mattson MP. The lipid peroxidation product 4-hydroxynonenal facilitates opening of voltage-dependent Ca2+ channels in neurons by increasing protein tyrosine phosphorylation. J Biol Chem. 2002;277:24368–24375. [DOI] [PubMed] [Google Scholar]
- 187.Lu C, Chan SL, Haughey N, Lee WT, Mattson MP. Selective and biphasic effect of the membrane lipid peroxidation product 4-hydroxy-2,3-nonenal on N-methyl-D-aspartate channels. J Neurochem. 2001;78:577–589. [DOI] [PubMed] [Google Scholar]
- 188.Matsuda T, Murata Y, Kawamura N, et al. Selective induction of alpha 1 isoform of (Na+ + K+)-ATPase by insulin/insulin-like growth factor-I in cultured rat astrocytes. Arch Biochem Biophys. 1993;307:175–182. [DOI] [PubMed] [Google Scholar]
- 189.Sweeney G, Klip A. Regulation of the Na+/K+-ATPase by insulin: Why and how? Mol Cell Biochem. 1998;182:121–133. [PubMed] [Google Scholar]
- 190.Kawai N, Yamamoto T, Yamamoto H, McCarron RM, Spatz M. Endothelin 1 stimulates Na+,K(+)-ATPase and Na(+)-K(+)-Cl-cotransport through ETA receptors and protein kinase C-dependent pathway in cerebral capillary endothelium. J Neurochem. 1995;65:1588–1596. [DOI] [PubMed] [Google Scholar]
- 191.Krishnamoorthy RR, Prasanna G, Dauphin R, et al. Regulation of Na,K-ATPase expression by endothelin-1 in transformed human ciliary non-pigmented epithelial (HNPE) cells. J Ocul Pharmacol Ther. 2003;19:465–481. [DOI] [PubMed] [Google Scholar]
- 192.Spatz M, Kawai N, Merkel N, Bembry J, McCarron RM. Functional properties of cultured endothelial cells derived from large microvessels of human brain. Am J Physiol. 1997;272:C231–C239. [DOI] [PubMed] [Google Scholar]
- 193.Amann R, Donnerer J, Lembeck F. Ruthenium red selectively inhibits capsaicin-induced release of calcitonin gene-related peptide from the isolated perfused guinea pig lung. Neurosci Lett. 1989;101: 311–315. [DOI] [PubMed] [Google Scholar]
- 194.Andersen SL, Clausen T. Calcitonin gene-related peptide stimulates active Na(+)-K+ transport in rat soleus muscle. Am J Physiol. 1993;264:C419–C429. [DOI] [PubMed] [Google Scholar]
- 195.Mahmmoud YA. Capsazepine, a synthetic vanilloid that converts the Na,K-ATPase to Na-ATPase. Proc Natl Acad Sci USA. 2008; 105:1757–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tria E, Luly P, Tomasi V, Trevisani A, Barnabei O. Modulation by cyclic AMP in vitro of liver plasma membrane (Na+–K+)-ATPase and protein kinases. Biochim Biophys Acta. 1974;343:297–306. [DOI] [PubMed] [Google Scholar]