

Abstract
Ovarian clear cell carcinoma (OCCC) is a rare, chemo-resistant subtype of ovarian cancer. To identify novel therapeutic targets and combination therapies for OCCC, we subjected a set of patient-derived ovarian cancer cell lines to arrayed high throughput siRNA and drug screening. The results indicated OCCC cells are vulnerable to knockdown of epigenetic gene targets such as bromodomain and extra-terminal domain (BET) proteins BRD2 and BRD3. Subsequent RNA interference assays, as well as BET inhibitor treatments validated these BET proteins as potential therapeutic targets. Because development of resistance to single targeted agents is common, we next performed sensitizer drug screens to identify potential combination therapies with the BET inhibitor CPI0610. Several PI3K or AKT inhibitors were among the top drug combinations identified and subsequent work showed CPI0610 synergized with alpelisib or MK2206 by inducing p53-independent apoptosis. We further verified synergy between CPI0610 and PI3K-AKT pathway inhibitors alpelisib, MK2206, or ipatasertib in tumor organoids obtained directly from OCCC patients. These findings indicate further preclinical evaluation of BET inhibitors, alone or in combination with PI3K-AKT inhibitors for OCCC is warranted.
Keywords: BRD2, BRD3, PI3K, ovarian cancer, organoids
Introduction
The mortality rate of patients with ovarian cancer is the highest among major gynecologic malignancies (1). With the exception of PARP inhibitors, ovarian cancer patients are uniformly treated with platinum-based chemotherapies. PARP inhibitors are active against high grade serous or endometrioid ovarian carcinoma, with the highest response rates seen in cancers with a BRCA1 or BRCA2 mutation and impairment in the homologous recombination pathway of DNA repair (2–4). However, effective targeted therapies for other ovarian cancer subtypes are lacking.
Ovarian clear cell carcinoma (OCCC) is a relatively rare subtype, representing about 5-25% of all ovarian cancer cases, depending on the geographic region (5). OCCC more often presents as early stage disease compared to the most frequent subtype, high-grade serous carcinoma (HGSC). In advanced stages, however, OCCC prognosis is significantly worse because of its poor response to platinum agents (6). A retrospective analysis showed 70% of OCCC patients had progressive disease while on primary platinum-based chemotherapy (7). Reported median overall survival in stage III/IV OCCC and HGSC were 12-21.3 months and 22-40.8 months, respectively (5, 7). These statistics indicate that the standard HGSC therapy is ineffective in OCCC and there is an unmet need for more effective therapeutic strategies.
Recent comprehensive genomic and molecular analyses have revealed that ovarian cancer is not only pathologically, but also genetically heterogenous. OCCC is often associated with endometriosis and the endometrium is the likely precursor tissue, as opposed to HGSC which frequently originates in distal fallopian tube epithelium (8, 9). OCCC is characterized by mutations in the SWI/SNF family member ARID1A, seen in ~66% of cases, and the oncogenic lipid kinase PIK3CA, seen in ~50% of cases, with less frequent mutations found in PPP2R1A and KRAS (10, 11). In contrast, HGSC is characterized by near universal mutation in the tumor suppressor TP53, frequent germline and somatic mutations in BRCA1 or BRCA2, or other genes involved in homologous recombination repair (12). Typically, OCCC has been grouped together with HGSC in clinical trials, but the distinct pathology and genetic profile suggest OCCC-specific therapeutic approaches may be more effective, in keeping with the goals of precision medicine.
Following the report of the first BET inhibitor, JQ1, bromodomain and extra-terminal domain (BET) proteins have been under active investigation as therapeutic targets for various diseases including cancer (13, 14). The BET family consists of BRD2, BRD3, BRD4, and BRDT and among these, BRD4 is the most well investigated as a therapeutic target (15). Initially, JQ1 was described as a BRD4 inhibitor, but JQ1 as well as most other currently available BET inhibitors inhibit multiple BET protein members due to the well-conserved bromodomain sequence (14).
In this study, we identified the BET proteins BRD2 and BRD3 as candidate therapeutic targets for OCCC by applying a custom high throughput siRNA screening platform to a unique set of patient-derived ovarian cancer cell lines that genomically and phenotypically resemble the primary tumors from which they were derived (16). Subsequent drug screening revealed synergy between BET protein inhibition and PI3K-AKT signaling inhibition, suggesting a potential drug combination therapeutic strategy for OCCC.
Materials and Methods
Cell culture and reagents
The derivation of OCI-C1p, OCI-C4p, OCI-C5x, and OCI-P5x cells was previously described (16). OCI-cells were cultured in OCMI-L, a specific growth medium developed for these cells (US Biological, Cat # 506390). The OCI cells are available from the Ince laboratory. The passage number of OCI cells at the time of each experiment was from 40 to 60. TOV21G cells were obtained from ATCC and cultured in 1:1 of Medium 199 (Gibco®) and MCDB105 (Cell Application Inc.) with 15% fetal bovine serum (FBS). RMG1 cells were obtained from JCRB cell bank and maintained in RPMI with 20% FBS. Dicer-substrate small interfering RNAs (dsiRNAs) targeting BRD2, BRD3, BRD4 as well as negative control dsiRNA were purchased from Integrated DNA Technologies. AllStars Hs Cell Death Control siRNA was purchased from QIAGEN. JQ1 (13), CP203 (13, 17), CPI0610 (18), Alpelisib (BYL719) (19) and MK2206 (20) were purchased from Selleckchem.
siRNA Screening
The siRNA screening method was previously described (21). Briefly, the same number of OCI-C5x or OCI-P5x cells were plated on 384 well plates 24 hours before siRNA transfection. Pooled siRNAs targeting each single gene were added to the designated wells with optimized concentrations of DharmaFECT1 (Dharmacon) for each cell line. Cell viability was measured using CellTiter-Glo (Promega) after 72 hours of transfection for OCI-C5x and 96 hours for OCI P5x. Each gene was tested in triplicate.
dsiRNA transfection
The same number of cells were plated on 96-well assay plates (Costar) 24 hours prior to dsiRNA transfection. Cells were transfected with 10nM pooled dsiRNAs targeting BRD2, BRD3 or BRD4. Optimized concentrations of DharmaFECT1 or RNAiMAX (Thermo Fisher Scientific) were used for transfection according to each cell line.
High-throughput combination drug screening
OCI-C5x or RMG1 cells were plated on 384 well plates 24 hours prior to drug administration. Cells were treated with a custom library of 135 FDA-approved chemotherapeutic agents or targeted inhibitors in the presence of IC30 of BET inhibitor CPI203 or DMSO as described (22). Each drug was tested in six different concentrations to obtain dose-response curves.
Drug combination assays
The same number of cells were plated on 96-well plates 24 hours prior to drug treatment. Cells were treated with drugs or DMSO at the indicated concentrations. Cell viability was assessed by CellTiter-Glo 2.0 after 6 days of drug treatment. Drug synergy was calculated using the Bliss method and graphed using Combenefit software (23,24). The Bliss Independence (BI) model predicts the inhibition rate if two inhibitors work independently and is determined by the following equation Ya+Yb−YaYb. where Ya and Yb represent the inhibition rate of drug A at dose a and the inhibition rate of drug B at dose b, respectively.
mRNA-Seq and Gene Set Enrichment Analysis
RNA was isolated from OCI-C5x cells using Direct-zol RNA kit (ZYMO Research®) after 24 hours of pooled dsiRNA transfection targeting BRD2, BRD3, BRD4 or negative control dsiRNA. Samples were prepared in triplicate. The RNA-Seq was outsourced to Novogene. Complementary DNA was sequenced after the sample quality check and library construction. The result was analyzed referring to hg19 and fragments per kilobase of exon per million mapped reads (FPKM) was determined for every gene. FPKM was statistically compared between each BET protein knockdown and control. Gene set enrichment was analyzed by GSEA software v4.0.3 (25). Hallmark gene sets collection was applied in this analysis.
ARID1A DNA sequence
cDNA was synthesized using Superscript III First-Strand Synthesis System (Invitrogen) after RNA extraction from OCI C1p, OCI C4p and OCI C5x cells by Direct-zol RNA Kits. Primer pairs were designed to split ARID1A cDNA into 9 fragments and each fragment was amplified by PCR amplification. Amplified cDNA was isolated by electrophoresis and DNA extracted with QIAquick Gel Extraction Kit (QIAGEN). Each DNA fragment was then cloned into TOPO-TA cloning vector (Invitrogen). TOP 10 competent cells (Invitrogen) were transformed with each vector and cultured at 37°C overnight. Vectors were harvested and purified using QIAprep Spin Miniprep kit (QIAGEN). Inserted cDNA sequence was determined by Sanger sequencing using BigDye Terminator v3.1 (Applied Biosystems) and M3 universal primer or specific sequencing primer. The information of primer pairs for cDNA amplification is described in Supplementary Fig. S1. The mutations detected by vector sequencing were verified in genomic DNA extracted from the cells by Sanger sequencing.
Western blotting
Cells were lysed in Cell Lysis Buffer (Cell Signaling Technology) supplemented with protease inhibitor (cOmplete, Mini Protease Inhibitor Cocktail by Roche) and phosphatase inhibitor (PhosSTOP by Sigma Aldrich). Protein was sonicated and collected by centrifugation. After measuring protein concentration by BCA assay, protein was boiled at 70°C in LDS sample buffer (Thermo fisher) with reducing agent (Thermo Fisher). The same amount of protein was loaded on each well of SDS-PAGE gels and subjected to electrophoresis. Proteins were transferred to PVDF membrane. After blocking with 5% non-fat milk, the membrane was incubated with the primary antibodies at optimized concentrations overnight at 4°C. The membrane was then incubated with secondary fluorescent antibodies (LI-COR) for 2 hours. Bands were detected using Li-Cor Odyssey NIR Scanner (LI-COR) or Chemidoc MP (Bio-Rad). Densitometry was performed with Fiji software (26). Antibodies used in this study are listed in the Supplementary Table S6.
Cell viability assay
Cells were plated on 96-well assay plates (Costar) and treated with dsiRNA or inhibitors. Cell viability was assessed using CellTiter-Glo® 2.0 (Promega) based on the protocol provided by the manufacturer.
Collection of fresh tissue from patients
All studies were conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS). Patient-derived fresh tissue and ascites samples were collected with informed patient consent according to Institutional Review Board (IRB) approved protocols. Samples were received from the Department of Obstetrics & Gynecology, University of Washington following confirmation of histopathological subtype by a pathologist.
Immunohistochemistry
Tissues were fixed in neutral buffered formalin and then processed to paraffin. Four-micron sections were deparaffinized and rehydrated and then stained using a three step ABC technique. Following antigen retrieval with citrate buffer, sections were blocked for peroxidase with 3% H2O2, and stained with primary antibody for AKT (1:300; Cell Signaling #4691) or p-AKT(Ser 473; 1:100; Cell Signaling #4060) overnight at 4°C. The next day, sections were stained with a biotinylated goat anti-rabbit secondary (Vector Labs) for one hour and then an avidin-biotin complex (Vectastain® Elite® ABC Peroxidase, Vector Labs). Sections were developed using DAB Quanto (Thermo Scientific) and counterstained with hematoxylin (Fisher Scientific), dehydrated and coverslipped with Histomount (National Diagnostics).
Patient-derived organoid establishment
OCCC Patient 1 tumor sample (1T) was processed within 2 hours after surgery and divided into three samples for organoid preparation, formalin fixation, or snap freezing and storage at −80°C. For organoid derivation, the tissue was minced and washed with base media (Advanced DMEM/F12 media containing 10mM HEPES, 1x Glutamax, and 1x Primocin (Invivogen)), transferred to a centrifuge tube and enzymatically digested using the Tumor Dissociation Kit (Miltenyi Biotec; Cat #130-095-929) according to the manufacturer’s instructions. The digested tissue suspension was strained over a 70 μm filter, centrifuged at 1000 rpm, and the pellet washed with base media. The pellet was incubated with 2 ml red blood cell lysis buffer (Roche) for 2 min at room temperature followed by an additional wash with base media and centrifugation at 1000 rpm. Organoids were cultured in Corning Matrigel Growth Factor Reduced Basement Membrane Matrix (Cat. No. 354230) in base media with 1x B27, 10mM Nicotinamide, 1.25mM N-Acetylcysteine, 10uM Y-27632, 50ng/mL rhEGF, 10 ng/mL rhFGF-10, 500nM A-83-01, 50mM beta-estradiol, 37.5ng/mL heregulin, 500ng/mL hydrocortisone, and 50% L-WRN conditioned media (ATCC Cat. No. CRL3276). OCCC Patient #1A and OCCC Patient #2A organoids derived from ascites samples were centrifuged at 1000 rpm, incubated with 2 ml red blood cell lysis buffer for 2 min at room temperature, washed with base media, and centrifuged at 1000 rpm. Media was changed every 3-4 days, with growth monitored until passaging was required. Organoids were passaged upon reaching 100-200 μm in diameter by digestion with Accutase (Stem Cell Technologies) and/or TrypLE (Gibco) at 37°C followed by trituration with a pipette. Drug assays and screens were performed on organoids within 2 weeks of sample collection.
High-throughput patient-derived organoid drug screens
One thousand cells per well were seeded into 384-well assay plates containing 50 μL media supplemented with 5% Matrigel (Corning). A panel of 42 oncology-focused drugs was acoustically administered (Labcyte Echo) as single agents using contactless, nanovolume liquid transfers to create a 3-log, 6-dose drug curve with drug concentrations ranging from 33 pmol/L to 200 mmol/L, depending on individual drug properties. Following a 6-day incubation with drugs, relative viability was determined by whole-well ATP quantification using Cell-Titer-Glo 2.0 (Promega) and normalized to vehicle-only controls (maximal DMSO concentration used was 0.2%). Top scoring drugs using Z score and relative AUC were identified as described (22).
Statistical analysis
False discovery rate (FDR) calculation and cluster analysis were performed by R 3.6.2. Other statistical analyses were performed using Graphpad Prism 7 (Graphpad Software). Unless otherwise mentioned, P values less than 0.05 were considered as significant.
Results
Arrayed siRNA screens utilizing patient-derived ovarian cancer cells identify BRD2 and BRD3 as candidate therapeutic targets for OCCC.
A major limitation in ovarian cancer research is the shortage of accurate model systems. Most commercially available ovarian cancer cell lines have undergone long term passage and do not accurately reflect the biology or genetics of the original tumor (27), and for some commonly used ovarian cancer cell lines, their original subtype is not clear. To address this, a set of patient-derived ovarian cancer cell line (OCI) models were established and when cultured in specific media, these retain the genetic and phenotypic characteristics of the original tumor(16). In this study we used three OCCC-derived cell lines, OCI-C1p, OCI-C4p, and OCI-C5x, one HGSC-derived cell line, OCI-P5x, as well as RMG1 and TOV21G cells. Key genomic features of these cells are summarized in Table 1. Some of these genomic alterations were previously reported (16, 28), and others were identified here, either by BROCA sequencing (29) to identify homologous recombination repair-related gene alterations, or by Sanger sequencing targeting ARID1A (Supplementary Fig. S1). Three out of five OCCC cell lines tested carried mutations in ARID1A and two carried mutations in PIK3CA. The single HGSC line, OCI-P5x, carried a mutation in TP53.
Table. 1.
Gene mutations detected or reported in OCIs, RMG1 and TOV21G cells.
| Cells | Subtype | ARID1A | PIK3CA | PTEN | p53 | BRCA1 | BRCA2 |
|---|---|---|---|---|---|---|---|
| OCI C1p | OCCC | p.N106Kfs*5 p.Y1523* |
p.Q456L p.P539R p.H1047R# |
WT | WT | WT | WT |
| OCI C5x | OCCC | p.P1898Hfs*25 | WT | WT | WT | p.Q451* | WT |
| TOV21G | OCCC | p.Y551Lfs*72 p.Q758Rfs*75 |
p.H1047Y | p.G143Afs*4 p.K267Rfs*9 |
WT | WT | WT |
| OCI C4p | OCCC | WT | WT | WT | p.R342* | WT | WT |
| RMG1 | OCCC | WT | WT | WT | W | WT | WT |
| OCI P5x | HGSC | WT | WT | WT | p.Y236N | WT | WT |
To identify potential therapeutic targets for OCCC, we applied an arrayed high throughput siRNA screening strategy to OCI-C5x cells (Figure 1a, b). OCI-P5x cells, derived from a HGSC, were subjected to the same screen to identify potential subtype-specific targets. The latter study identified SRC, BET, and BCL2 inhibitors as synergistic drug combinations with the PARP inhibitor rucaparib (21). We employed a custom arrayed siRNA library targeting 2187 genes, including the kinome (712 genes), epigenome (1192 genes), DNA damage and repair (318 genes), commonly aberrated cancer-associated genes in the TCGA HGSC cohort (mutated, copy number variation, or mRNA over/under-expressed) (123 genes), and previously identified MYC -synthetic lethal genes (48 genes) (30) (Supplementary Table S1 and NCI’s Cancer Target Discovery and Development (CTD2) Data Portal, https://ocg.cancer.gov/programs/ctd2/data-portal).
Figure 1. Arrayed siRNA screens identify bromodomain proteins as targetable vulnerabilities in OCCC.
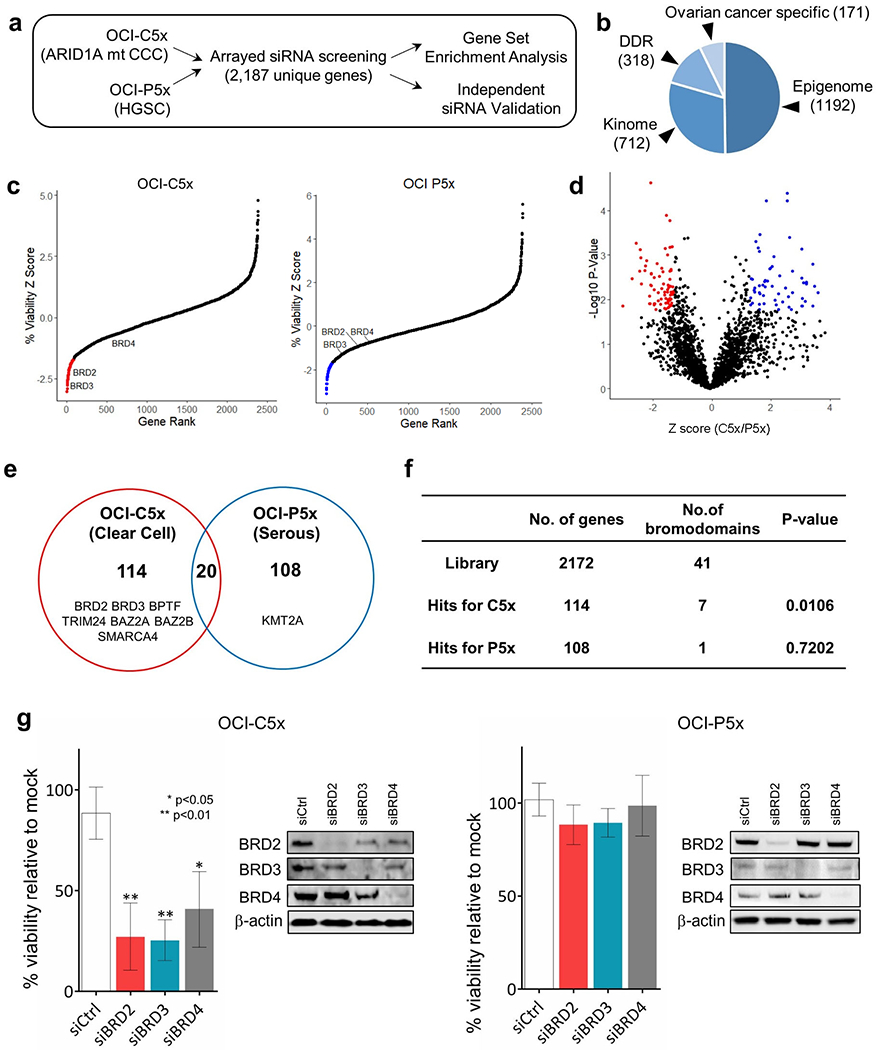
(a) Scheme of high throughput arrayed siRNA screens. (b) Makeup of custom siRNA library. (c) Z-score based target gene selection. Mean percentage cell viability relative to mock transfection was determined and transformed to Z-score. Genes were sorted by Z-score and plotted. Genes with Z-score less than −1.65 were selected as hits (red or blue) for each OCI cell type. (d) Volcano plot of cell viability comparison between OCI-C5x and OCI-P5x cells. Percentage cell viability was standardized in reference to the mean of each cell and fold change (OCI-C5x/OCI-P5x) was calculated and transformed to Z score. P-values for each gene were also determined by Student t-test. False discovery rate was calculated and FDR>1.29 or <−1.29 was used to select hits specific to OCI-C5x (red) and OCI-P5x (blue), respectively. (e) Venn diagram to summarize the hits from the siRNA screen. Genes encoding bromodomain proteins are listed. (f) Summary table for the number of bromodomain genes in the siRNA library and as hits. P-values were determined by Fisher’s exact test between the library and the hits for OCI C5x or OCI P5x, respectively. (g) Validation assay focusing on BET proteins BRD2, BRD3, and BRD4. Cells were transfected with 10nM of pooled dsiRNAs targeting each BET gene. Cell viability was assessed 72 hours after transfection. Values were the means ± SD of three independent assays. Knockdown efficiency and specificity were confirmed by western blotting.
Based on the selection criteria described in materials and methods, 134 and 128 candidate gene targets (hits) were identified for OCI-C5x and OCI-P5x cells, respectively (Figure 1c,d,e). The hits for each cell line are listed in Supplementary Table S2. Only 20 candidate gene targets were shared between the two cell lines. Hits specific to each cell line were subjected to Gene Set Enrichment Analysis (GSEA) referring to the Gene Ontology (GO) gene set collection (25, 31). This analysis revealed hits for OCI-C5x cells were enriched for genes related to chromatin or transcription regulation, indicating OCCC is potentially more vulnerable to epigenetic interventions compared to HGSC (Supplementary Fig. S2).
Among the hits specific to OCI-C5x cells were seven bromodomain-containing proteins out of 41 such proteins in the siRNA library. Fisher’s exact test revealed bromodomain proteins are significantly enriched in the hits for OCI-C5x (Figure 1f). The bromodomain motif recognizes lysine acetylation on a variety of proteins, including histones and transcription factors (32). Each bromodomain shows distinct affinities to lysine acetylations across histone tails, suggesting unique and complicated roles of bromodomains as readers of these post translational modifications. We focused on BET proteins BRD2 and BRD3 because both were among the top targets found in OCI-C5x cells and small molecule inhibitors to these proteins are available.
To confirm the results from the primary screen, pooled dsiRNAs targeting BRD2, BRD3, BRD4 as well as negative control dsiRNAs were introduced into OCI-C5x and OCI-P5x cells. dsiRNAs to all three genes significantly reduced survival of OCI-C5x but not OCI-P5x cells, consistent with the primary screen results (Figure 1g). Western blot analysis confirmed knockdown of BRD2, BRD3, and BRD4 protein with dsiRNAs (Figure 1g). The individual BRD2 or BRD3 siRNAs used for the pooled siRNAs were also tested in OCI-C5x cells. Individual siRNAs to both BRD2 and BRD3 reduced viability of OCI-C5x cells and again, western blot analysis confirmed knockdown of BRD2 and BRD3 protein (Supplementary Fig. S3).
BET inhibition as a novel therapeutic option for OCCC
The therapeutic potential of BET inhibition was investigated in additional OCCC cells, OCI-C1p, OCI-C4p, TOV21G, and RMG1. Because BET proteins and ARID1A are both involved in epigenetic regulation, and because OCI-C5x cells carry an ARID1A frameshift mutation, we asked if ARID1A mutation status correlated with sensitivity to BET inhibitors. OCI-C1p and TOV21G cells also carry mutations in ARID1A, while OCI-C4p, OCI-P5x, and RMG1 cells had no detectable ARID1A mutations (Table 1). Western blotting demonstrated loss of ARID1A protein in ARID1A mutated cells, suggesting these mutations destabilize the protein resulting in loss of ARID1A function (Figure 2a). ARID1A mutation status did not noticeably affect the sensitivity to BET protein knockdown, as BRD2 or BRD3 knockdown reduced viability by more than 50% in all OCCC cells regardless of ARID1A status (Figure 2b).
Figure 2. Validation of BET proteins as candidate targets for OCCC.
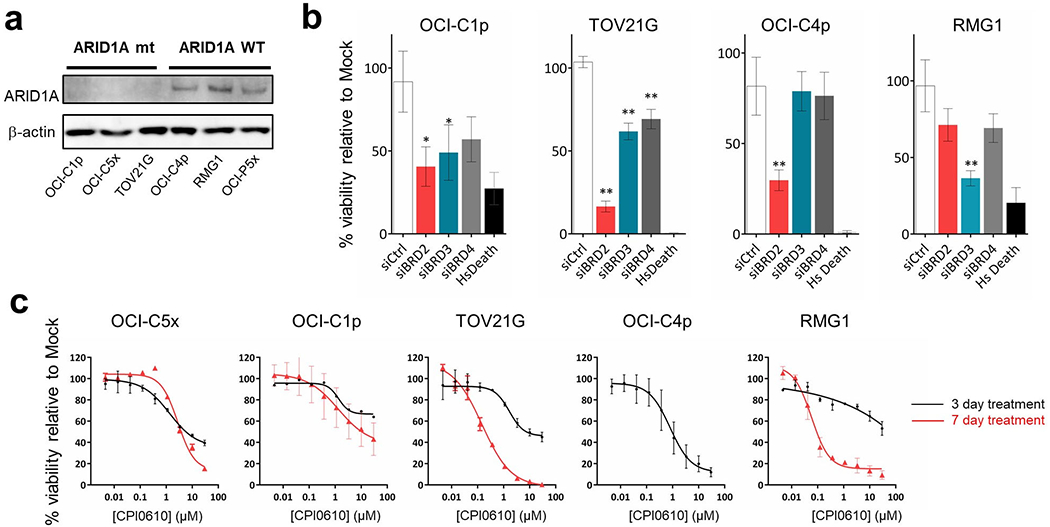
(a) Western blotting of ARID1A expression. OCCC cells harboring ARID1A mutations (OCI-C1p, OCI-C5x, TOV21G) or wild type ARID1A (OCI-C4p, RMG1, OCI P5x) were examined. The result was validated in three independent assays. (b) Knockdown of BET RNA in additional cell lines. 10nM of pooled dsiRNAs targeting BRD2, BRD3, BRD4 and negative control dsiRNAs were transfected into OCI-C1p, OCI-C4p, TOV21G and RMG1 cells. Cell viability was assessed 72 hours after the transfection. 10nM of AllStars Hs Cell Death Control siRNA (Hs Death) was transfected in parallel as a positive control for successful transfection. Values were the means ± SD of three independent assays. * represents P value <0.05 and ** represents P value < 0.01. (c) Dose-response curves of the BET inhibitor CPI0610 in OCCC cells. Cells were treated with the indicated concentrations of CPI0610 for 72 hours (black) or 7 days (red). Only a dose-response curve after 72 hours was obtained in OCI-C4p as the cells were unstable in long-term culture. Values were the means ± SD of three independent assays.
The OCCC cells were also treated with the BET inhibitor CPI0610, which is being tested in clinical trials (14). Except for RMG1, OCCC cells showed sensitivity to CPI0610 with an IC50 of up to 1.65 μM at 72 hours (Figure 2c, Supplementary Table S3). While RMG1 cells did not respond to short-term CPI0610 treatment, stronger sensitivity was observed after 7 days of treatment (Figure 2c). A similar trend was observed with the other two BET inhibitors, JQ1 and CPI203 (Supplementary Fig. S4, Supplementary Table S3). As seen with the siRNA knockdown, there was no clear correlation between the sensitivity to each BET inhibitor and ARID1A mutation status. Altogether, these results reveal a vulnerability of OCCC cells to BET inhibition, especially involving BRD2 and BRD3.
BET inhibitors synergize with PIK3-AKT pathway inhibitors in OCCC.
Although several BET inhibitors are now being tested in clinical trials for patients with solid tumors, the reported response rate is low (33, 34), a finding which motivated us to evaluate potential synergistic combination therapies. To identify drugs that might be effective in combination with BET inhibitors, we performed BET inhibitor sensitizer drug screens using two OCCC cells, OCI-C5x and RMG1 treated with 135 chemotherapeutics and targeted agents in the absence or presence of the BET inhibitor CPI203. The results are summarized in Figure 3a and Supplementary Table S4. We noted that several PI3K or AKT inhibitors were among the top drug combinations identified. Indeed, 10 out of 11 PI3K-AKT pathway inhibitors tested showed potentially synergistic interactions with CPI203 in OCI C5x cells and 5 out of 11 did so in RMG1 cells (Supplementary Fig. S5).
Figure 3. Drug combination assays with BET inhibitors and PI3K-AKT pathway inhibitors.
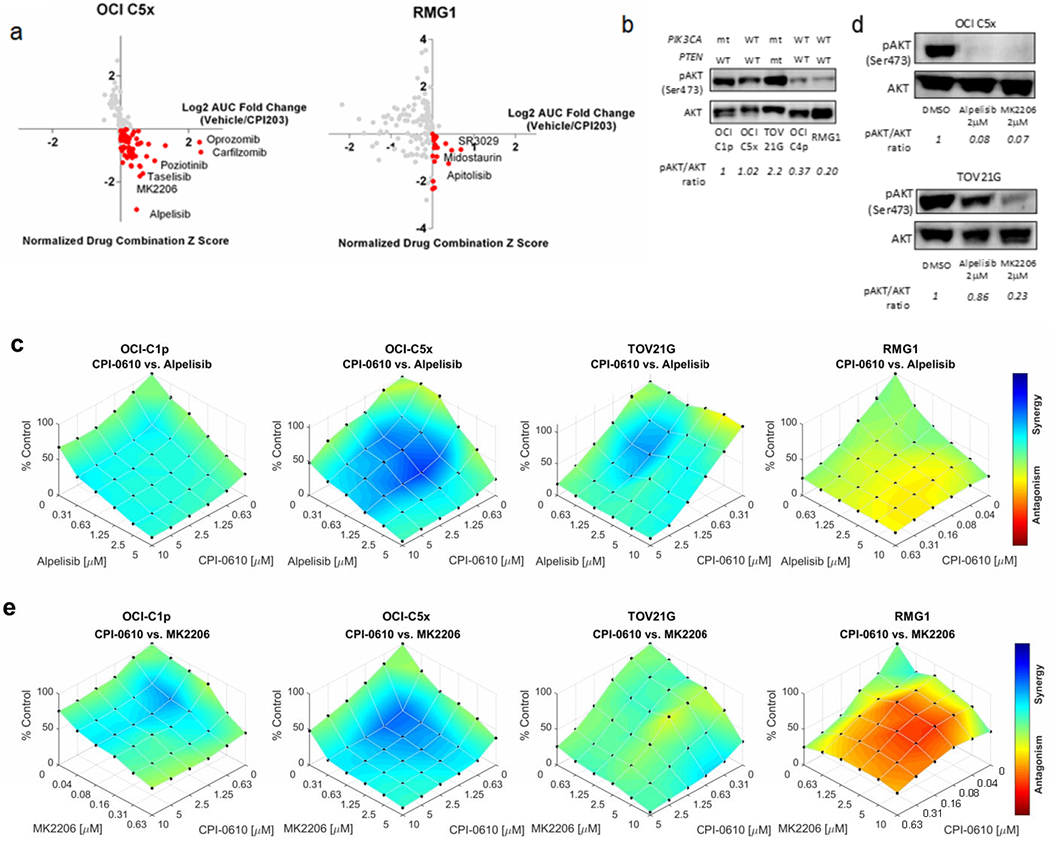
(a) The result of drug combination screens with CPI203 in OCI-C5x and RMG1 cells. X axis: AUC fold change (Vehicle/CPI203). Y axis: normalized combination Z scores. Inhibitors potentially synergistic with CPI203 are highlighted in red. (b) Western blotting for AKT phosphorylation (phospho-AKT) at serine 473 across multiple OCCC cells. Each blot was quantified by densitometry and relative pAKT/AKT ratio in reference to OCI C1p was determined as shown at the bottom. The reproducibility was verified in three independent assays. (c) Assessment of the synergistic interaction between CPI0610 and alpelisib using the Bliss Independence (BI) model. Cells were treated with CPI0610 and alpelisib at the indicated concentrations for 6 days. The maximum concentration was determined according to the sensitivity of each cell to the inhibitors up to 10μM. (d) Western blotting for phospho-AKT after 24 hours of the treatment with alpelisib or MK2206. Relative pAKT/AKT ratio in reference to DMSO treatment was determined by densitometry. Top; OCI-C5x, bottom; TOV21G. The experiment was repeated three times and the same trend was observed. (e) Assessment of the synergistic interaction between CPI0610 and MK2206 using the Bliss Independence (BI) model. Cells were treated with CPI0610 and MK2206 at the indicated concentrations for 6 days. The maximum concentration of MK2206 was determined as described above.
Based on previous reports showing high frequency PIK3CA mutations in OCCC and our preliminary genomic characterization, the PI3K-AKT signaling pathway is likely to be activated in many cases of OCCC (11, 35). We evaluated AKT phosphorylation at serine 473, a marker for AKT activation, across the OCCC cell lines. Western blotting clarified AKT is highly phosphorylated in PIK3CA-mutated OCI-C1p and TOV21G cells as expected, and interestingly, in OCI-C5x cells that do not have a PIK3CA mutation (Figure 3b). The result implies AKT is hyperactivated, not only by deleterious mutation of PIK3CA, but via other mutations or signaling aberrations. Based on these findings, we evaluated the combination of BET inhibitors and PI3K or AKT inhibitors in detail.
To evaluate potential synergistic interactions, three OCCC cells showing high AKT phosphorylation at serine 473 (OCI-C1p, OCI-C5x, TOV21G) were treated with the BET inhibitor CPI0610 and the FDA approved PI3K inhibitor alpelisib. RMG1 cells that do not show strong AKT phosphorylation were also tested as a comparison. We employed the Bliss Independence (BI) model to assess synergy (see materials and methods) (23). Briefly, if an observed percentage inhibition is more than the predicted inhibition determined by BI model, it indicates synergy, and the opposite suggests antagonism. As summarized in Figure 3c, clear synergistic interaction was observed in OCI-C1p, OCI-C5x and TOV21G cells but not in RMG1 cells, implying the combination of BET and PI3K inhibitors might be effective in OCCC presenting AKT hyperactivation.
Interestingly, we found that TOV21G cells were resistant to the PI3K inhibitor alpelisib. Since PTEN opposes PI3K in the AKT activation pathway, the loss of PTEN function can lead to aberrant AKT phosphorylation independently of PIK3CA. As TOV21G harbors both PIK3CA and PTEN mutations, we hypothesized that direct AKT inhibition may be more effective in TOV21G cells. Indeed, both alpelisib and the AKT inhibitor MK2206 reduced AKT phosphorylation in OCI C5x cells but TOV21G responded only to MK2206 (Figure 3d). Drug combination assessment by the BI model demonstrated TOV21G cells were sensitive to MK2206, and the combination of CPI0610 and MK2206 was synergistic. The other two AKT activated OCCC cells C1p and C5x also showed synergistic sensitivity to CPI0610 and MK2206 (Figure 3e).
BET inhibitor CPI0610 and PI3K-AKT inhibitors cooperate to trigger p53-independent apoptosis.
To understand how BET inhibition interacts with PI3K-AKT pathway inhibition, OCI-C5x and TOV21G cells were treated with alpelisib or MK2206 in the presence or absence of CPI0610 for 48 hours. As expected, AKT phosphorylation was reduced by alpelisib or MK2206 in a dose dependent manner (Figure 4a). However, AKT phosphorylation was reduced at lower concentrations of alpelisib or MK2206 when the cells were also treated with CPI0610, suggesting the observed synergism between BET inhibition and PI3K-AKT pathway inhibition may involve direct or indirect regulation of PI3K-AKT signaling by BET proteins.
Figure 4. BET inhibition sensitizes OCCC cells to PI3K-AKT inhibitors by promoting p53-independent apoptosis.
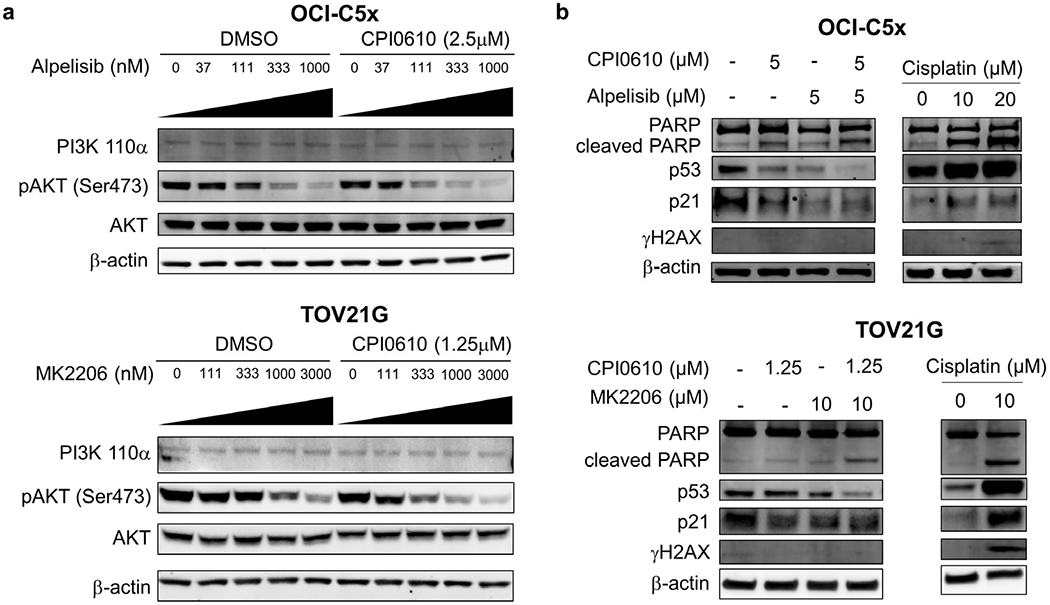
(a) OCI-C5x and TOV21G cells were treated with alpelisib or MK2206 at multiple concentrations in combination with CPI0610 or DMSO for 48 hours. Expression of PI3K p110α and phospho-AKT were examined by western blotting. Each experiment was repeated twice independently to validate the reproducibility. (b) Cells were treated with the indicated concentrations of CPI0610 and alpelisib (OCI-C5x) or MK2206 (TOV21G). PARP cleavage, p53 and p21 expression and phosphorylation of histone H2AX at serine 139 was determined by western blotting 48 hours after drug administration. Each experiment was repeated twice independently to validate the reproducibility.
Because PI3K inhibitors and AKT inhibitors are well known to lead to apoptosis in many cancer cells (36), we evaluated PARP cleavage, a marker for apoptosis, in OCI-C5x and TOV21G cells following alpelisib or MK2206 treatment in the presence or absence of CPI0610 (Figure 4b). The DNA damaging agent cisplatin was employed as a positive control for apoptosis. Cisplatin induced p53 and p21 expression in OCI-C5x and TOV21G cells, coinciding with enhanced histone H2AX phosphorylation at serine 139 and PARP cleavage, effects that are consistent with DNA damage activation of p53 and resultant apoptosis. In contrast, alpelisib or MK2206 did not induce p53 or p21 or increase H2AX phosphorylation. Cleaved PARP was increased slightly by single drug treatments and was further enhanced by drug combinations in both OCI-C5x and TOV21G cells. These findings suggest the apoptosis triggered by the combination of BET inhibition and PI3K-AKT signal inhibition is p53 independent and mechanistically distinct from that induced by cisplatin.
RNA-seq indicates BRD2, BRD3, and BRD4 are all involved in PI3K-AKT signaling.
BRD4 is known to regulate several important cancer promoting signals such as those downstream of MYC, Nf-κB and senescence-associated secretory phenotypes (15, 37). Based on the results above, we considered that BRD2 and BRD3 might have both unique and common roles in gene regulation related to OCCC cell survival. To understand the impact of each BET protein on gene expression in OCCC, we performed RNA-Seq and subsequent GSEA analysis following BET protein knockdown. Pooled dsiRNAs targeting BRD2, BRD3, BRD4 or negative control dsiRNAs were transfected into OCI-C5x cells and RNA was harvested at 24 hours. Compared to the negative control dsiRNA, 4038, 6442, and 3319 genes were up- or down-regulated by BRD2, BRD3, and BRD4 knockdown, respectively (Figure 5a and Supplementary Table S5). Whereas approximately 47% of the genes were shared by at least two of the BET protein knockdowns, more than half of the genes were specifically regulated by one of the BET proteins. Cluster analysis, shown as a heat map in Figure 5b, also indicates each BET protein has its own unique role in gene expression. GSEA analysis clarified the pathways that are regulated by each BET protein (Figure 5c). Focusing on the PI3K-AKT pathway, we found BRD2 knockdown decreases expression of genes involved in the PI3K-AKT-mTOR signaling pathway. BRD3 and BRD4 knockdown suppressed expression of genes related to MTORC1, which is immediately downstream of PI3K-AKT signaling. Furthermore, knockdown of all BET proteins resulted in a significant increase in expression of genes involved in apoptosis (Figure 5c). The GSEA results indicate disruption of all BET proteins could modify PI3K-AKT signaling and apoptotic sensitivity, in concordance with the findings in Figure 4.
Figure 5. mRNA-Seq indicates acute depletion of each BET protein alters the PI3K-AKT signaling pathway.
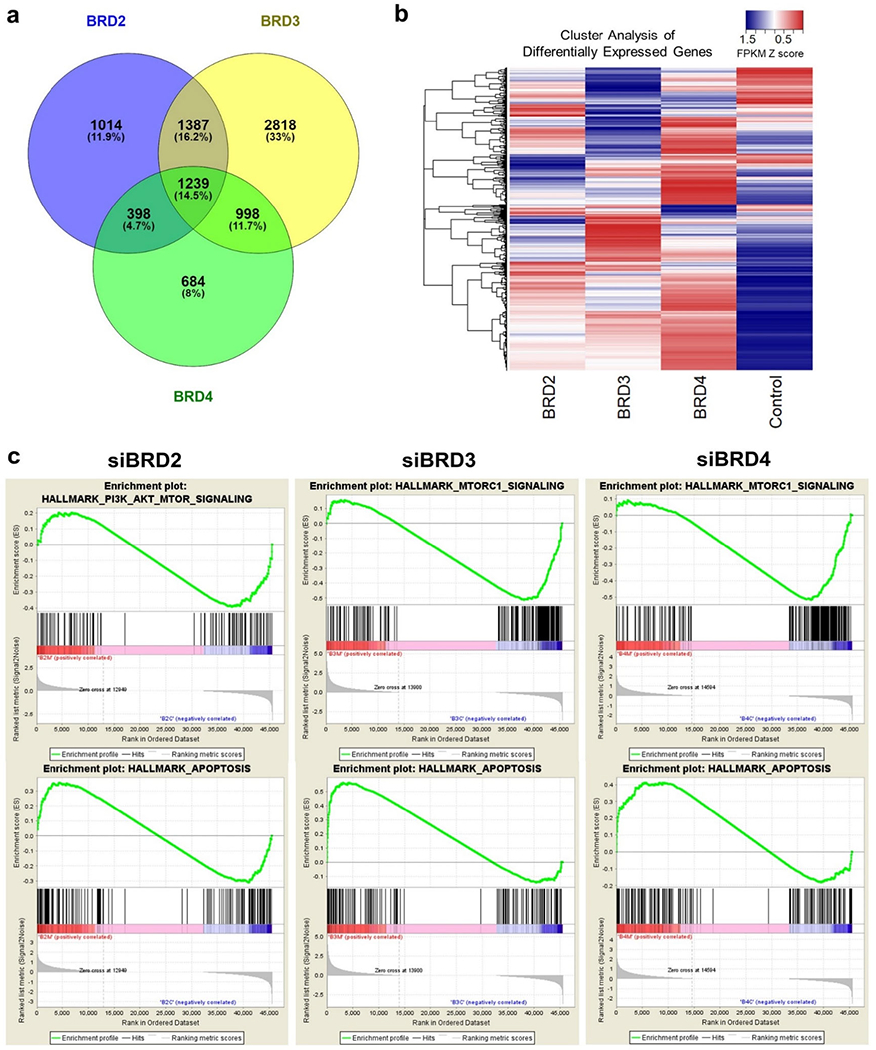
(a) Venn diagram to summarize the mRNA-Seq results. The number of genes up- or down-regulated by each BET protein knockdown is shown. (b) Overview of the cluster analysis focusing on differentially expressed genes. FPKM was transformed to Z score and cluster analysis was performed. The result was visualized as a heatmap. (c) Results of Gene Set Enrichment Analysis (GSEA). FPKM values in triplicates between each BET protein knockdown and control were compared. Top: The enrichment of PI3K-AKT-MTORC or MTORC1 signaling gene set. Bottom: Gene set enrichment related to apoptosis.
Tumor organoids cultured directly from patients with OCCC show synergistic sensitivity to BET and PI3K-AKT-mTOR inhibitors.
We isolated tumor organoids from ascites samples cultured from two OCCC patients (patient 1 and patient 2). These ascites-derived organoids (1A and 2A respectively) were subjected to a CLIA certified, high-throughput drug screening assay with a library of 42 clinically relevant oncology drugs that are FDA approved or in late-stage development (22, 38). Among the top scoring drugs for patient 1 was the BET inhibitor CPI0610, the PI3K inhibitor taselisib, and mTOR inhibitors everolimus and temsirolimus (Figure 6a). For patient 2, CPI0610, taselisib, everolimus, temsirolimus, and the AKT inhibitor ipatasertib scored as top drugs (Figure 6b). We also derived organoids from surgical resection of the primary tumor of patient 1 (Figure 6c). These tumor-derived organoids (1T) were tested with increasing concentrations of the BET inhibitor CPI-0610 in combination with alpelisib, MK2206, or ipatasertib. All three PI3K/AKT inhibitors demonstrated synergy with CPI-0610 (Figure 6d).
Figure 6. Sensitivity to combined BET and PI3K/AKT inhibitors in tumor organoids derived from OCCC patients.
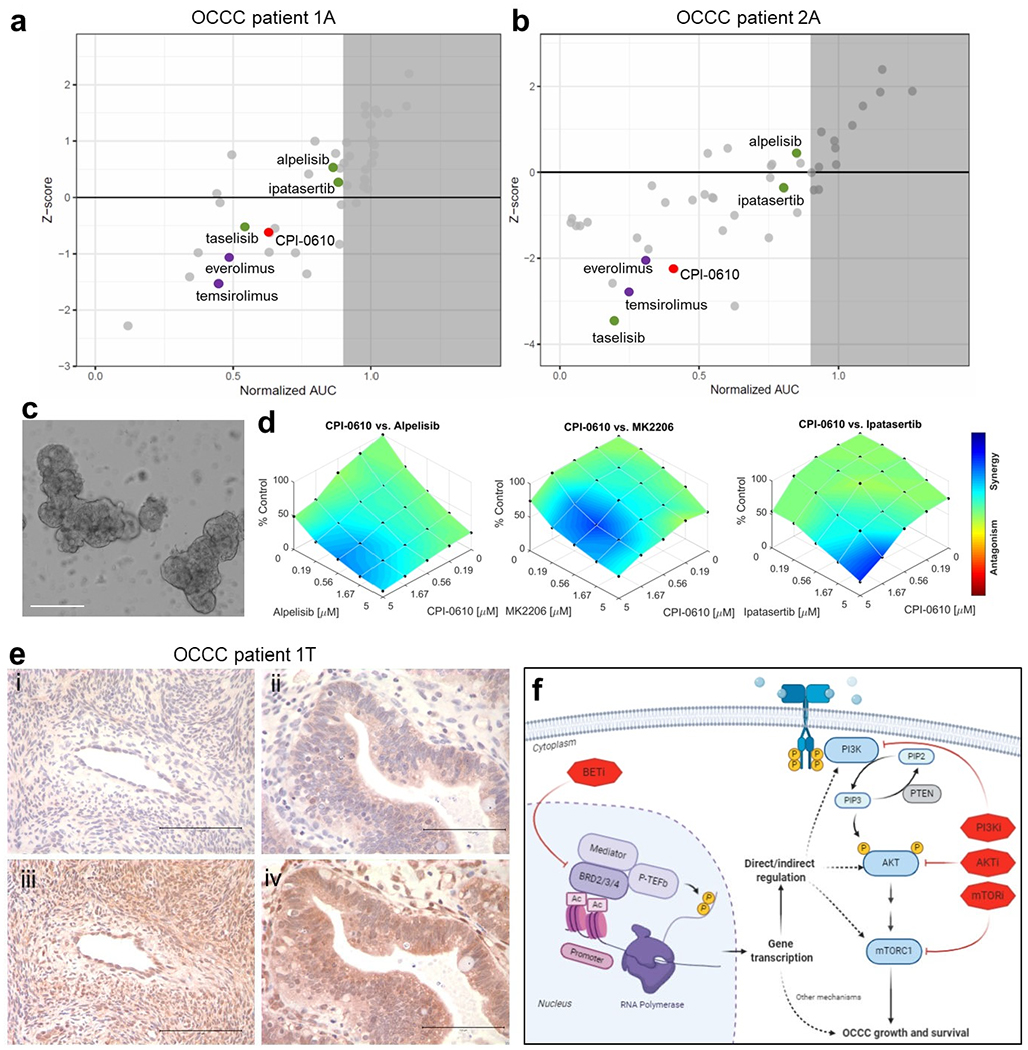
(a, b) The result of high-throughput drug screens in organoids derived from ascites of OCCC patient 1 (1A) and patient 2 (2A). (c) Representative brightfield image of organoids cultured from surgical resection of primary tumor of OCCC patient 1 (1T). Scale bar: 200 μm. (d) Dose-response plots of CPI0610 in combination with alpelisib, MK2206, or ipatasertib demonstrating synergistic effects in organoids derived from tumor sample of OCCC patient 1 (1T). (e) Immunohistochemical staining of tissue sections from OCCC patient 1 (1T): (i) normal ovary tissue stained for p-AKT (Ser473); (ii) tumor tissue stained for p-AKT (Ser473); (iii) normal ovary tissue stained for total AKT; (iv) tumor tissue stained for total AKT. Images are representative of staining across tissue sections and were captured using a 40x lens and 10x objective. Scale bar: 100 μm. (f) Schematic of mechanisms of PI3K and BET inhibitor synergy. Knockdown or inhibition of BET proteins directly or indirectly alters PI3K signaling which synergizes with PIK3CA or AKT inhibition leading to p53-independent apoptosis.
This functional data suggested tumors from both OCCC patients carried alterations in the PI3K/AKT pathway. Indeed, DNA sequencing identified known activating mutations in PIK3CA in patient 1 (p.Q546R) and patient 2 (p.E545K). Patient 2 also had PIK3CA copy gain and an ARID1A mutation (p.N116Tfs*116) with loss of heterozygosity (Table 2). Immunohistochemical staining demonstrated stronger cytoplasmic staining for p-AKT (Ser473) and total AKT in primary tumor tissue from patient 1 compared to normal ovarian tissue from the same patient (Figure 6e).
Table 2.
Gene mutations detected in OCCC patient derived organoids.
| Patient | Subtype | NGS Panel | Microsatellite instability | Mutations and variants identified |
|---|---|---|---|---|
| 1 | OCCC | BROCA* | No | • PIK3CA (p.Q546R, NM_006218.4:c.1637A>G) with copy gain • No other pathogenic somatic mutations were detected in BRCA1, BRCA2, or other homologous recombination DNA repair genes tested by this panel |
| 2 | OCCC | Oncoplex^ | No | • PIK3CA (p.Q546K, NM_006218.2:c.1636C>A) with copy gain • ARID1A (p.N116Tfs*116, NM_139135.2:c.347del) with LOH and/or copy gain • NOTCH1 (p.A2037S, NM_017617.3:c.6109G>T) • KMT2C (NM_170606:c.5009-1G>A) • U2AF2 (NM_001012478.1:c.1282-8C>T) • EML4 (p.N707S, NM_019063.5:c.2120A>G) • PHOX2B (NM_003924:c.429+5G>A) • SHH (p.L3M, NM_000193.2:c.7C>A) • Gains of portions of chromosomes 1 (ARID1A), 4, 5, 8, 10, 11, 15 (IDH2), 16, 17, 22, and X. • Losses of portions of chromosomes 14 and 19 are present. |
Mutations detected with a BROCA-HR panel as described (49).
University of Washington Department of Laboratory Medicine OncoPlex Cancer Gene Panel: https://testguide.labmed.uw.edu/public/view/OPX
Collectively, these results using clinical samples cultured as organoids in 3D verify our findings in the 2D culture models. BET and PI3K-AKT-mTOR pathway inhibition, alone or in combination, could be effective therapeutic strategies for OCCC patients with PI3K-AKT-mTOR pathway activation, irrespective of ARID1A mutation status. BRD2, BRD3, and BRD4 can directly or indirectly regulate PI3K-AKT signaling, and the therapeutic efficacy of inhibitors to these targets could be maximized when used in combination to achieve synergism (Figure 6f).
Discussion
In this study, we demonstrate the vulnerability of OCCC cells to the inhibition of BRD2 and BRD3 protein function by utilizing functional genomic and pharmacologic screens in patient-derived ovarian cancer cells and organoids. Although BET protein function is incompletely understood, BET proteins are being actively investigated as therapeutic targets for a variety of diseases. Silencing of BRD4, or inhibition of its activity by JQ1, downregulates expression of many genes that regulate cell growth and differentiation including the MYC oncogene, and impairs growth of a range of both liquid and solid tumor types (39,40). Among BET proteins, only BRD4 has a C-terminal domain (CTD) which serves to recruit the positive transcription elongation factor b (P-TEFb) complex to particular genomic locations to promote phosphorylation and activation of RNA Pol II (41). However, the significance of the other BET family members in ovarian cancer and, in particular, OCCC, is less clear. BRD2 and BRD3 also bind to acetylated lysine residues in histones and regulate gene transcription, but unlike BRD4 they do not possess a CTD. Berns et al. recently reported that ARID1A mutant OCCC cells are sensitive to BRD2 knockdown or JQ1, suggesting ARID1A mutation is synthetic lethal with BRD2 inhibition (42). We demonstrate that BRD2 knockdown was effective not only in ARID1A mutated but also in at least one ARID1A wild type OCCC cell line. Further, we show knockdown of BRD3 also resulted in significant growth inhibition in several OCCC cell lines, regardless of ARID1A status. Altogether, our results identify BRD2 and BRD3 as candidate therapeutic targets in OCCC.
We also provide evidence for distinct functional roles of BRD2, BRD3, and BRD4. Knockdown of any one of the three BET genes leads to altered expression of over 3,000 genes with approximately one half unique to each BET gene and one half that were shared among two or three BET genes. This implies different mechanisms of action underlie the sensitivity to different BET inhibitors even in the same cancer type. From a clinical perspective, selective inhibitors to each BET protein could potentially maximize clinical efficacy while minimizing toxicity. While several selective BRD4 inhibitors have been reported (43, 44), BRD2 or BRD3 specific BET inhibitors may be beneficial to OCCC patients based on the results of this study.
The sensitizer drug screens identified the combination of BET inhibitors and PI3K-AKT pathway inhibitors as a potential therapeutic option for the treatment of OCCC presenting with high AKT phosphorylation. Several previous reports have suggested similarly effective drug combinations in different cancer types. Stratikopoulos et al. demonstrated BET inhibition overcomes resistance to PI3K inhibitors in a broad range of cell lines (45) and Derenzini et al. reported increased vulnerability to PI3K inhibitors by BET inhibition in lymphoma (46), suggesting synergy between BET and AKT inhibitors may be a more general phenomenon.
Up to 50% of OCCCs harbor PIK3CA mutations which encodes the PI3K p110α catalytic subunit. Additional cases harbor PTEN mutations, PIK3R1 mutations, and AKT amplification, all of which can activate PI3K-AKT signaling (11). Genes regulating receptor tyrosine kinase/Ras signaling that lie upstream of PI3K/AKT are also altered in 29% of OCCC (47). Thus, a significant percentage of patients with OCCC could benefit from a combination of BET inhibitors and PI3K/AKT inhibitors. AKT phosphorylation could be a feasible marker for the use of this combination based on the experimental results presented here. Genomic information may also be required to select an appropriate PI3K/AKT inhibitor. Although PI3K inhibitors, including alpelisib, have an advantage of FDA approval, the insensitivity of TOV21G cells to alpelisib suggests that AKT should be directly inhibited in cases with mutation in PTEN or other downstream mediators of AKT signaling. Another possible target for PTEN mutated OCCC is the PI3K p110β subunit. Emerging evidence indicates cancers harboring PTEN mutations are dependent on signaling through the p110β subunit (48) and it is possible that a combination of BET inhibitors and PI3K p110β subunit selective inhibitors may be effective.
In addition, our research highlights the mTOR complex in the PI3K-AKT signaling cascade, as a possible alternative to PI3K inhibitors for use in combination therapy. FDA-approved mTOR complex inhibitors temsirolimus and everolimus showed mild but potentially synergistic interaction with CPI203 in the drug combination screens. Unfortunately, neither temsirolimus nor everolimus have significantly improved patient outcome in ovarian cancer clinical trials (49,50). Dual inhibition of mTOR and BET function may be an additional avenue to explore.
From the point of view of the mechanism of action, the combination of alpelisib or MK2206 and CPI0610 led to apoptosis without significant p53 induction or DNA damage, suggesting the combination could be effective in patients with p53 mutant, platinum resistant or refractory OCCC.
In conclusion, this study clarified the potential benefit of BET inhibition independently or in combination with PI3K or AKT inhibitors and provide rationale to launch clinical trials to examine the anti-tumor activity of BET inhibitors in combination with PI3K/AKT inhibitors in OCCC patients.
Supplementary Material
Acknowledgements
This project was supported by funding from Cure First, the NCI (U01 CA217883, R01 CA214428, and U54 CA132381), Seattle Tumor Translational Research, and the Rivkin Center for Ovarian Cancer.
Footnotes
Conflict of interest: CJK and CG are founders and equity holders in SEngine Precision Medicine. All other authors declare no conflicts of interest.
References
- 1.Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer. 2020;126(10):2225–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018;379(26):2495–505. [DOI] [PubMed] [Google Scholar]
- 3.Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol 2019;20(5):636–48. [DOI] [PubMed] [Google Scholar]
- 5.Anglesio MS, Carey MS, Kobel M, Mackay H, Huntsman DG, Vancouver Ovarian Clear Cell Symposium S. Clear cell carcinoma of the ovary: a report from the first Ovarian Clear Cell Symposium, June 24th, 2010. Gynecol Oncol. 2011;121(2):407–15. [DOI] [PubMed] [Google Scholar]
- 6.Oliver KE, Brady WE, Birrer M, Gershenson DM, Fleming G, Copeland LJ, et al. An evaluation of progression free survival and overall survival of ovarian cancer patients with clear cell carcinoma versus serous carcinoma treated with platinum therapy: An NRG Oncology/Gynecologic Oncology Group experience. Gynecol Oncol. 2017;147(2):243–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goff BA, Sainz de la Cuesta R, Muntz HG, Fleischhacker D, Ek M, Rice LW, et al. Clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy in stage III disease. Gynecol Oncol. 1996;60(3):412–7. [DOI] [PubMed] [Google Scholar]
- 8.Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case-control studies. Lancet Oncol. 2012;13(4):385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol. 2008;26(32):5284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibuya Y, Tokunaga H, Saito S, Shimokawa K, Katsuoka F, Bin L, et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer. 2018;57(2):51–60. [DOI] [PubMed] [Google Scholar]
- 12.The Cancer Genome Atlas Research. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018;8(1):24–36. [DOI] [PubMed] [Google Scholar]
- 15.White ME, Fenger JM, Carson WE 3rd. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell Immunol. 2019;337:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun. 2015;6:7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devaiah BN, Lewis BA, Cherman N, Hewitt MC, Albrecht BK, Robey PG, et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci U S A. 2012;109(18):6927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Côté A, Leblanc Y, et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J Med Chem. 2016;59(4):1330–9. [DOI] [PubMed] [Google Scholar]
- 19.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–8. [DOI] [PubMed] [Google Scholar]
- 20.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol cancer Ther. 2010;9(7):1956–67. [DOI] [PubMed] [Google Scholar]
- 21.Lui GYL, Shaw R, Schaub FX, Stork IN, Gurley KE, Bridgwater C, et al. BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine. 2020;60:102988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pauli C, Hopkins BD, Prandi D, Shaw R, Fedrizzi T, Sboner A, et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017;7(5):462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao W, Sachsenmeier K, Zhang L, Sult E, Hollingsworth RE, Yang H. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen. 2014;19(5):817–21. [DOI] [PubMed] [Google Scholar]
- 24.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(44):18032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109(24):9545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J Clin Oncol 2018;36(30):3007–14. [DOI] [PubMed] [Google Scholar]
- 34.Postel-Vinay S, Herbschleb K, Massard C, Woodcock V, Soria JC, Walter AO, et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur J Cancer. 2019;109:103–10. [DOI] [PubMed] [Google Scholar]
- 35.Sasano T, Mabuchi S, Kuroda H, Kawano M, Matsumoto Y, Takahashi R, et al. Preclinical Efficacy for AKT Targeting in Clear Cell Carcinoma of the Ovary. Mol Cancer Res. 2015;13(4):795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016;6(6):612–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narasimhan V, Wright JA, Churchill M, Wang T, Rosati R, Lannagan TRM, et al. Medium-throughput Drug Screening of Patient-derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clin Cancer Res. 2020;26(14):3662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting BET bromodomain proteins in solid tumors. Oncotarget. 2016;7(33):53997–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Z, He N, Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol. 2008;28(3):967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berns K, Caumanns JJ, Hijmans EM, Gennissen AMC, Severson TM, Evers B, et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene. 2018;37(33):4611–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niu Q, Liu Z, Alamer E, Fan X, Chen H, Endsley J, et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J Clin Invest. 2019;129(8):3361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Tian B, Chen H, Wang P, Brasier AR, Zhou J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur J Med Chem. 2018;151:450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou MM, et al. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell. 2015;27(6):837–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derenzini E, Mondello P, Erazo T, Portelinha A, Liu Y, Scallion M, et al. BET Inhibition-Induced GSK3beta Feedback Enhances Lymphoma Vulnerability to PI3K Inhibitors. Cell Rep. 2018;24(8):2155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itamochi H, Oishi T, Oumi N, Takeuchi S, Yoshihara K, Mikami M, et al. Whole-genome sequencing revealed novel prognostic biomarkers and promising targets for therapy of ovarian clear cell carcinoma. Br J Cancer. 2017;117(5):717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wee S, Wiederschain D, Maira S-M, Loo A, Miller C, deBeaumont R, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105(35):13057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tew WP, Sill MW, Walker JL, Secord AA, Bonebrake AJ, Schilder JM, et al. Randomized phase II trial of bevacizumab plus everolimus versus bevacizumab alone for recurrent or persistent ovarian, fallopian tube or peritoneal carcinoma: An NRG oncology/gynecologic oncology group study. Gynecol Oncol. 2018;151(2):257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emons G, Kurzeder C, Schmalfeldt B, Neuser P, de Gregorio N, Pfisterer J, et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol Oncol. 2016;140(3):450–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.