

Abstract
Objective-
Ornithine decarboxylase (ODC)-dependent putrescine synthesis promotes the successive clearance of apoptotic cells by macrophages, contributing to inflammation resolution. However, it remains unknown whether ODC is required for other arms of the resolution program.
Approach and Results-
RNA sequencing of ODC-deficient macrophages exposed to apoptotic cells showed increases in mRNAs associated with heightened inflammation and decreases in mRNAs related to resolution and repair compared with wild-type macrophages. In zymosan peritonitis, myeloid-ODC deletion led to delayed clearance of neutrophils and a decrease in the pro-resolving cytokine, IL-10. Nanoparticle-mediated silencing of macrophage ODC in a model of atherosclerosis regression lowered IL-10 expression, decreased efferocytosis, enhanced necrotic core area, and reduced fibrous cap thickness. Mechanistically, ODC deletion lowered basal expression of MerTK, an apoptotic cell receptor, via a histone-methylation-dependent transcriptional mechanism. Owing to lower basal MerTK, subsequent exposure to apoptotic cells resulted in lower MerTK-Erk1/2-dependent IL-10 production. Putrescine treatment of ODC-deficient macrophages restored the expression of both MerTK and apoptotic cell-induced IL-10.
Conclusions-
These findings demonstrate that ODC-dependent putrescine synthesis in macrophages maintains a basal level of MerTK expression needed to optimally resolve inflammation upon subsequent apoptotic cell exposure.
Keywords: macrophages, inflammation resolution, putrescine, efferocytosis, atherosclerosis
Subject Terms: Atherosclerosis, Cell Biology, Cell Signaling, Inflammation, Pathophysiology
Graphical Abstract

INTRODUCTION
Inflammation resolution is an active process guided by the production of protein, lipid, and gaseous mediators that dampen inflammation, promote repair, and enhance the phagocytic clearance (efferocytosis) of apoptotic cells (ACs) by resident and recruited macrophages.1–3 Interactions between ACs and macrophages, along with the subsequent degradation of ACs in the phagolysosome, drive the production of anti-inflammatory cytokines and pro-resolving mediators and repress the production of pro-inflammatory cytokines.2, 3 When efferocytosis fails, uncleared ACs become secondarily necrotic and the pro-resolving and anti-inflammatory responses are lost, leading to inflammation and tissue damage. Accordingly, impaired efferocytosis underlies a growing list of chronic inflammatory diseases, such as atherosclerosis, autoimmune diseases, neurodegenerative diseases, chronic lung disease, and inflammatory bowel disease.2, 3
In atherosclerosis, defective efferocytosis promotes features of clinically dangerous plaques, notably plaque necrosis and thinning of a protective, collagen-rich fibrous cap that overlies advanced plaques.4–6 Restoring efferocytosis or treatment with pro-resolving mediators decreases lesion size, reduces necrotic core formation, and enhances fibrous cap thickening.7–11 A widely-studied, atheroprotective resolving mediator that is produced by efferocytosing macrophages is interleukin-10 (IL-10).12, 13 IL-10 knockout mice show enhanced atherosclerosis progression compared with wild-type mice,14 and treatment with adeno-associated virus (AAV)-IL-10 reduces atherosclerosis in Western diet (WD)-fed Ldlr−/− mice.15, 16 Targeted delivery of IL-10 using nanoparticles (NPs) similarly reduces atherosclerosis,17 as does increasing regulatory T cells, which stimulate macrophages to secrete and then be activated by IL-10.18
Macrophages with pro-resolving properties are known to produce polyamines, which are low-molecular-weight, linear polycations that can interact with negatively charged DNA, RNA, and proteins.19 Polyamines, comprised of putrescine, spermidine, and spermine, are controlled by the rate-limiting enzyme ornithine decarboxylase (ODC), which directs many cellular functions, including proliferation, gene transcription, mRNA stability, and protein translation.20 In addition, polyamines, particularly putrescine, restrain macrophage polarization towards a pro-inflammatory phenotype and promote the successive clearance of apoptotic cells, termed continual efferocytosis.19, 21 However, much remains to be learned about the nature and mechanisms of polyamine-mediated processes in macrophages.
Herein, we demonstrate that mRNAs involved in resolution and repair are expressed at a lower level in apoptotic cell-exposed macrophages lacking ODC compared with wild-type macrophages. In vivo, myeloid deletion of ODC reduces the clearance of apoptotic neutrophils during acute peritonitis and prevents the production of IL-10. In vitro, IL-10 production is compromised in ODC-deficient versus wild-type macrophages after exposure to ACs. Mechanistically, ODC deletion prevents H3K9-di/trimethylation of the Mertk gene, which decreases basal MerTK expression. This decrease in basal MerTK attenuates Erk1/2-dependent IL-10 production by macrophages upon exposure to ACs. Delivery of ODC siRNA to lesional macrophages using targeted NPs in a model of atherosclerosis regression decreases efferocytosis, lowers IL-10 and macrophage phospho-Erk1/2, enhances necrotic core area, and decreases fibrous cap thickness in aortic root lesions. These findings point to a critical role for ODC-dependent putrescine synthesis in maintaining a basal level of MerTK expression required for macrophages to appropriately stimulate a resolution response after subsequent exposure to apoptotic cells.
MATERIALS AND METHODS
The data that support the findings of this study are available from the corresponding authors upon reasonable request. An expanded materials and methods section can be found in the online supplemental file.
Primary cell cultures
For bone marrow-derived macrophages (BMDMs), bone marrow cells from 8–12 week old mice (either male or female) were cultured for 7–10 days in DMEM supplemented with 10% (vol/vol) heat-inactivated (HI) fetal bovine serum (FBS), 10 U/mL penicillin,100 mg/mL streptomycin, and 20% (vol/vol) L-929 fibroblast-conditioned media. For human macrophages, peripheral human blood leukocytes were isolated from buffy coats of de-identified healthy adult volunteers (New York Blood Center) and subsequently purified in a discontinuous gradient of Histopaque solution. After 4 h of adhesion on 24-well plates, cells were rinsed, and the medium was changed to RPMI-1640 (GIBCO) containing 10% HI-FBS, 10 U/mL penicillin and 100 mg/mL streptomycin, and 10 ng/mL of M-CSF (Peprotech). These cells were then used for experiments after 7–10 days when they were more than 75% confluent.
Experimental animals
Animal protocols were approved by Columbia University’s institutional animal care and use committee. All mice were cared for according to the NIH guidelines for the care and use of laboratory animals, and all were in good general health based on appearance and activity. The mice were socially housed in standard cages at 22°C under a 12–12 h light-dark cycle in a barrier facility with ad libitum access to water and food. Odc1fl/fl and Mertkfl/fl mice were generated as described,21, 22 and Lyz2-Cre mice were a gift from Irmgard Förste.23 Littermate control mice were randomly assigned to experimental groups by investigators. Investigators were blinded for the atherosclerosis studies but were not blinded for the zymosan-induced sterile peritonitis experiments.
To test the effect of putrescine in atherosclerosis, 8-week-old male Ldlr−/− mice were placed on a Western diet for a total of 16 weeks. Between 8–16 weeks of Western diet feeding, drinking water of the experimental group was supplemented with 3 mM putrescine, whereas control mice remained on regular drinking water. Control and putrescine-supplemented water was exchanged every two or three days. Upon sacrifice, mice were perfused with PBS through left ventricular cardiac puncture, and aortic roots were collected and processed for histological immunostaining.
Tissue Collection and lesion analysis
Our experimental atherosclerosis studies complied with the guidelines set forth by the American Heart Association.24 For atherosclerosis studies, 8 week-old male Ldlr−/− were placed on a Western diet (Envigo, 88137) for 16 weeks. To induce plaque regression, mice were then switched back to normal laboratory diet for an additional six weeks while simultaneously receiving a helper-dependent adenovirus containing the human LDLR gene (HDAd-LDLR; 1×1011 viral particles per mouse). Tissue sections from aortic roots were stained with hematoxylin and eosin for morphometric lesion analysis. Atherosclerotic lesion area, defined as the space from the internal elastic lamina to the lumen, was quantified by taking the average of 6 sections spaced 30 mm apart, beginning at the base of the aortic root. Boundary lines were drawn around these regions, and the area measurements were obtained by image analysis software. Necrotic cores were quantified as areas that were negative for both eosin and hematoxylin and greater than 3,000-μm2. Collagen staining was performed using picrosirius red (Polysciences, catalog 24901A) per the manufacturer’s instructions. Collagen cap thickness was quantified at the lesional midpoint and both shoulder regions and then averaged and quantified as the ratio of collagen cap thickness to lesion area. Fasting blood glucose levels were measured using ONETOUCH Ultra after the food was withdrawn for 18 h. Total plasma cholesterol was measured using a kit from WAKO Diagnostics. Complete blood cell counts, including leukocyte differential, were obtained using a FORCYTE Hematology Analyzer (Oxford Science).
Tissue immunohistochemistry and immunofluorescence microscopy
Paraffin-embedded specimens were sectioned, de-paraffinized with xylene, and rehydrated in decreasing concentrations of ethanol. Sections were incubated with TUNEL staining reagents at 37°C for 60 min and then washed three times with PBS. Sections were then blocked for 60 min, incubated overnight at 4°C with the following antibodies: anti-ODC (1:200), anti-IL-10 (1:200), anti-P-Erk1/2, or anti-Mac2 (1:10,000) antibodies, incubated with fluorescently-labeled secondary antibodies, and counterstained with DAPI. Images were captured using a Leica epifluorescence microscope (DMI6000B).
RNA-sequencing
For RNA-sequencing, macrophages were rinsed twice with cold 1X PBS and lysed in TRIzol reagent (ThermoFisher). RNA was isolated using RNeasy kits (QIAGEN). RNA with a RIN value of > 8 was subjected to poly-dT pulldown using magnetic beads before RNA-seq, using RNA Ultra kits. Libraries were sequenced on a NextSeq 500 (Illumina) and reads were aligned to the mm10 transcriptome using HISAT225 after adaptor trimming using cutadapt.26 Reads counts per gene for RefSeq genes were computed using featureCounts.27 Counts were normalized to reads per kilobase per million (RPKM) and processed for pairwise differential expression analysis of selected conditions using DESeq228 with a False Discovery Rate (FDR)-adjusted p-value cutoff of 0.05. The PANTHER database was used for Gene Ontology analysis.29
Zymosan A-induced peritonitis
10-week-old Odcfl/fl or Odcfl/fl Lysz2-Cre+/− mice were injected intraperitoneally with 1 mg of zymosan A (Sigma-Aldrich) per mouse, and peritoneal exudates were collected at the indicated time intervals. Cells were resuspended in FACS staining buffer (PBS containing 2% FBS and 1 mM EDTA) at a density of 1 × 106 cells/100 μL and incubated with Fc block (anti-mouse CD16/32; Biolegend) for 30 min on ice. Cells were then immunostained for PE anti-Ly6G and FITC anti-F4/80 for 1 h on ice. Cells were washed in FACS buffer twice and then resuspended for analysis on a BD FACS Canto II flow cytometer. Data analysis was carried out using FlowJo software.
Statistical analysis
Data were tested for normality using either the D’Agostino-Pearson or Shapiro-Wilk test, and statistical significance was determined using GraphPad Prism software. P values for normally distributed data were calculated using either the Student’s t-test for two groups or 1-way ANOVA with Fisher’s LSD post hoc analysis when three or more groups were tested. Data that were not normally distributed were calculated using the non-parametric Mann-Whitney U test or the uncorrected Dunn’s test. Data are shown as mean values ± SEM. Differences were considered statistically significant at p % 0.05. Based on our previous mouse atherosclerosis studies and power calculations, the numbers of mice chosen for each cohort was sufficient to enable the testing of our hypotheses based on an expected 20%–30% coefficient of variations and an 80% chance of detecting a 33% difference in key plaque parameters (lesion size and necrotic core area). Exclusion criteria before the start of any of the in vivo studies were death, an injury requiring euthanasia, or weight loss > 15%.
RESULTS
Deletion of Macrophage ODC Lowers Expression of Resolution-Related Genes and Myeloid ODC Deletion Delays Inflammation Resolution in Zymosan-Induced Peritonitis
ODC-dependent putrescine synthesis from arginine promotes inflammation resolution by restraining pro-inflammatory macrophage polarization and enhancing the successive clearance of dead cells.19, 21 However, whether ODC-dependent putrescine synthesis contributes to other arms of the resolution program has yet to be examined. Therefore, we began our studies using RNA-sequencing (RNA-seq) as an unbiased approach to identify ODC-dependent genes that could be involved in resolution. Bone marrow-derived macrophages isolated from Odc1fl/fl and Odc1fl/fl Lysz2-Cre+/− mice, referred to as ODC-wild-type (ODC-WT) mice and myeloid (ODC-KO), respectively, were incubated with ACs for one hour followed by AC removal. After 6 hours of further incubation, the macrophages were lysed and then subjected to RNA-seq. Differences in gene expression between AC-incubated ODC-WT and ODC-KO macrophages were identified (Fig. 1 A), and PANTHER pathway analysis showed that ODC-KO macrophages had higher expression of genes associated with inflammation and lower expression of genes associated with resolution (Fig. 1 B).
Figure 1. Deletion of Macrophage ODC Lowers Expression of Resolution-Related Genes and Delays Inflammation Resolution in Zymosan-Induced Peritonitis.

(A-B) Bone marrow-derived macrophages from Odc1fl/fl Lysz2Cre−/− mice (ODC-WT Mfs) and Odc1fl/fl Lysz2Cre+/− mice (ODC-KO Mfs) were incubated for one h with ACs at a 10:1 AC:macrophage ratio, after which the ACs were removed by rinsing. Following an additional 6 h of incubation, the macrophages were lysed and subjected to RNA-sequencing (n = 4 biological replicates). (A) Heat map of upregulated or downregulated genes, colored by row-normalized Z scores. (B) PANTHER GO categories upregulated in ODC-KO versus ODC-WT macrophages (top), and PANTHER GO categories downregulated in ODC-KO versus ODC-WT macrophages (bottom). (C) Odc1fl/fl Lysz2Cre−/− and Odc1fl/fl Lysz2Cre+/− mice were injected intraperitoneally with 1 mg of zymosan A per mouse. Peritoneal exudates were assayed for total leukocyte number and for the percentage of Ly6G+ F4/80− neutrophils, and neutrophil number was calculated as total leukocytes × percentage of neutrophils. Resolution intervals (Ri) were calculated as described in the text (n = 3–5 mice per group). Values for all graphs are means ± S.E.M., *p < 0.05.
To determine whether the loss of ODC led to enhanced inflammation or defective resolution in vivo, we used the zymosan A model of acute peritonitis, which encompasses inflammation and resolution phases.30 Separate groups of ODC-WT and ODC-KO mice were injected with 1 mg of zymosan A for 6, 12, 24, or 48 hours and compared to mice before injection (0 hours). Mice were then sacrificed, and peritoneal exudates were collected and assayed for the number of neutrophils. Whereas peak inflammation was similar between ODC-WT and myeloid ODC-KO mice at 12 hours (Tmax), the decline in neutrophil numbers was slower in ODC-KO mice: the time to a 50% reduction from Tmax (T50-Tmax), known as resolution index, or Ri, was ~14 hours in ODC-WT mice as compared with ~23 hours in myeloid ODC-KO mice (Fig. 1 C and Fig. SI A). These data are consistent with the conclusion that myeloid ODC contributes to inflammation resolution.
Macrophage ODC is required for atherosclerosis regression
There is a growing appreciation for enhancing resolution as a treatment strategy for atherosclerosis, as resolution is impaired in progressing atherosclerotic plaques10, 31 and reawakened in regressing plaques.19, 32 Therefore, we decided to investigate whether atherosclerosis regression requires myeloid ODC. Ldlr−/− mice were fed a Western diet (WD) for 16 weeks (baseline cohort), and then some mice were switched to normal laboratory diet for an additional six weeks while simultaneously receiving a helper-dependent adenovirus containing the human LDLR gene (HDAd-LDLR) to lower plasma LDL cholesterol further (regression cohorts) (Fig. 2 A).19, 33 To reduce ODC expression in plaque macrophages at the time of regression, we utilized a validated, macrophage-targeting NP platform capable of carrying siRNA.34 These nanoparticles (NPs) are composed of a poly(lactic-co-glycolic acid) (PLGA) core containing G0-G14-complexed siRNA, and an outer surface composed of a stabilin-2 peptide ligand (S2P, CRTLTVRKC) conjugated to 1,2-distearoyl-sn-glycero-3-phosphoethanolamineN-[maleimide (polyethylene glycol)] (DSPE-PEG-Mal) polymer. Beginning at the time when WD was switched back to normal laboratory diet and HDAd-LDLR was delivered, we treated mice twice a week for six weeks with either ScrRNA-loaded NPs or siOdc1-loaded NPs. Blood glucose, body weight, total plasma cholesterol, and blood leukocytes, neutrophils, monocytes, lymphocytes, eosinophils, basophils, Ly6Chigh, and Ly6Clow cells were similar between the ScrRNA-NP and siOdc1-NP groups at the end of the regression period (Fig. SII, A–K). Moreover, macrophage ODC immunostaining was much higher in the lesions of the ScrRNA cohort versus the siOdc1 NP cohort (Fig. SIII A), indicating successful ODC silencing.
Figure 2. Treatment of WD-fed Ldlr−/− Mice with siOdc1-Loaded NPs Prevents a Reduction in Lesion Size and Necrotic Core Area, Inhibits Fibrous Cap Thickening, and Worsens Lesional Efferocytosis During Atherosclerosis Regression.

(A) Experimental design of atherosclerosis regression with NP treatment strategy. Ldlr−/− mice were placed on WD for 16 weeks (Basal), then some mice were switched to the regression protocol (WD to normal chow with a single injection of 1×1011 viral particles containing HDAd-LDLR) and injected twice a week for 6 weeks with either ScrRNA NPs or siOdc1 NPs. (B-D) (B) Aortic root cross-sections were quantified for total lesion size and necrotic core area (n = 8–10 mice per group). Representative images are shown, with necrotic cores outlined in dashed lines. Bar, 200 μm. (C) Collagen cap thickness was measured at the lesional midpoint and both shoulder regions and then averaged and quantified as the ratio of collagen cap thickness to lesion area. Data are presented relative to the average value obtained for the baseline group (n = 8–10 mice per group). Representative images are shown. Bar, 200 μm. (D) The ratio of macrophage-associated ACs:free ACs was quantified (n = 8–10 mice per group). Representative images are shown, with some of the free TUNEL+ cells indicated by arrows and some of the macrophage-associated TUNEL+ cells indicated by arrowheads. Bar, 50 μm. Values for all graphs are means ± S.E.M.; *p < 0.05.
As expected, six weeks of lowering plasma cholesterol reduced overall lesion size and necrotic core area in the ScrRNA-NP cohort, and, most importantly, these improvements were prevented in the siOdc1-NP cohort (Fig. 2 B). Similarly, fibrous cap thickness, an indicator of plaque stability in humans, and lesional macrophage efferocytosis, a key resolution process, were both enhanced during atherosclerosis regression in the ScrRNA-NP group but not in the siOdc1-NP group (Fig. 2, C and D). These data indicate that loss of macrophage ODC interrupts the resolution program required for atherosclerosis regression.
ODC-dependent putrescine synthesis in macrophages promotes IL-10 production upon exposure to ACs
Our RNA-seq dataset identified Il10 as a downregulated gene in ODC-KO macrophages. IL-10 drives resolution by promoting alternative macrophage activation, dampening inflammatory responses, and enhancing efferocytosis.35, 36 We therefore assayed IL-10 by ELISA in peritoneal exudates from ODC-WT and myeloid ODC-KO mice 24 hours after zymosan A treatment and found a ~50% reduction in the myeloid ODC-KO cohort (Fig. 3 A). Because IL-10 is upregulated in macrophages upon exposure to ACs,12, 13 we next tested whether targeting ODC inhibits AC-induced IL-10 production. Macrophages isolated from myeloid ODC-KO mice, or wild-type macrophages transfected with either of two separate ODC siRNAs, abolished AC-induced IL-10 at both the mRNA level at 6 hours and the protein level at 24 hours (Fig. 3, B–D, Fig. SIV A). Silencing ODC in human macrophages also lowered AC-induced IL-10 expression (Fig. 3 E and Fig. SIV B). In contrast, IL-1β, a key pro-inflammatory cytokine known to be downregulated after exposure to ACs37, was unaffected by the deletion of ODC (Fig. SIV C). Importantly, the defect in AC-induced IL-10 expression observed in ODC-KO macrophages could be rescued by exogenous putrescine, the downstream product of ODC activity (Fig. 3 F).
Figure 3. IL-10 Production Upon Exposure to ACs Requires ODC-Dependent Putrescine Synthesis.
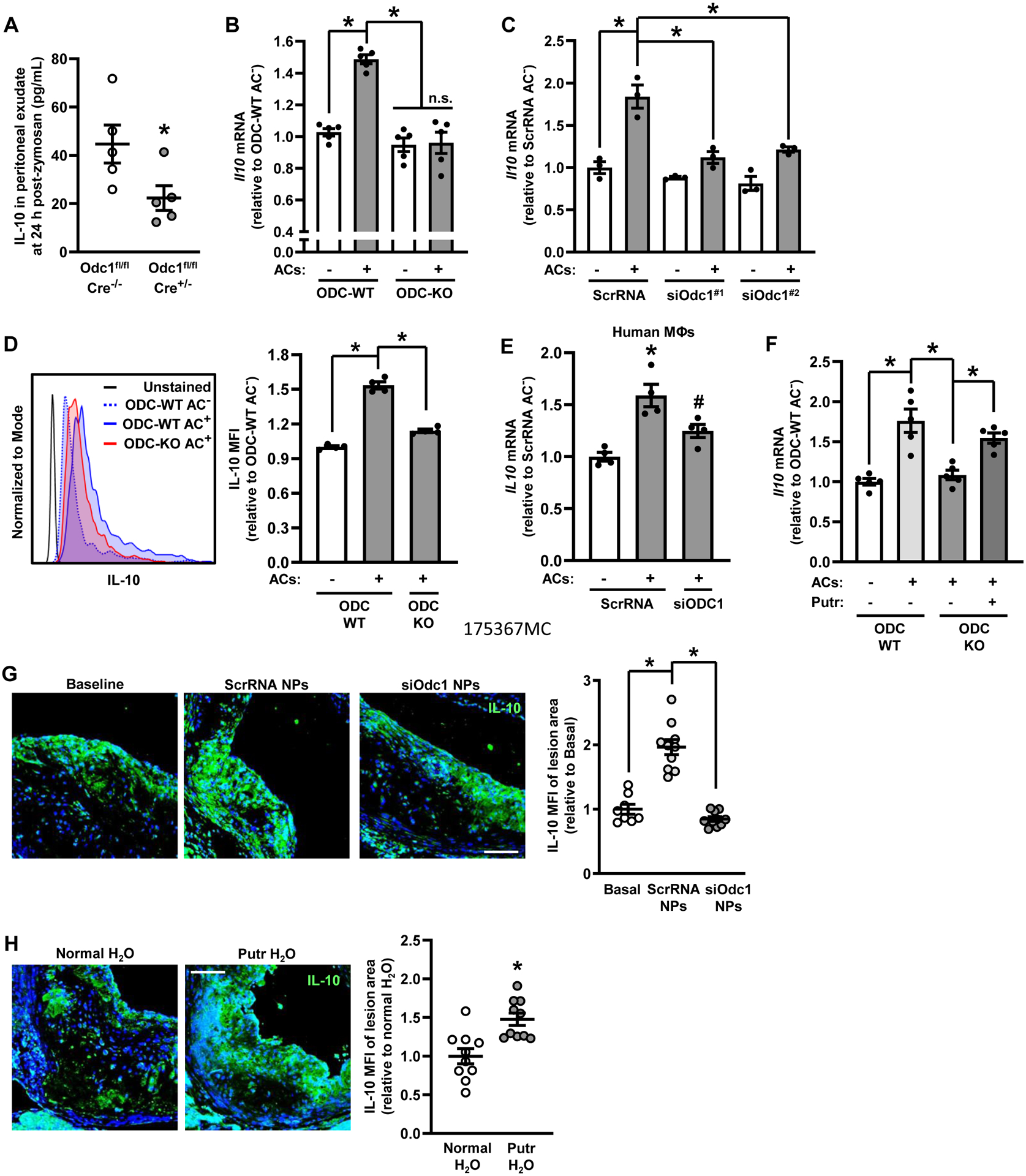
(A) Peritoneal exudates from 1C were assayed for IL-10 levels by ELISA (n = 5 mice per group). (B) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were incubated for one h with ACs at a 5:1 AC:macrophage ratio, after which the ACs were removed by rinsing. Following an additional 6 h of incubation, the macrophages were lysed and then assayed by qRT-PCR for Il10 (n = 5 biological replicates). (C) Bone marrow-derived macrophages were transfected with two separate Odc1-targeting siRNAs and assayed for IL10 mRNA as in B (n = 3 biological replicates). (D) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were incubated as in B except following the removal of ACs, macrophages were treated with GolgiStop (1:2000 dilution from commercial stock) and cultured for another 16 h. Cells were then detached and analyzed by flow cytometry for intracellular IL-10. A representative flow plot is shown (n = 4 biological replicates). (E) Human monocyte-derived macrophages were transfected with ScrRNA or siODC1 and assayed for IL10 mRNA as in B (n = 4 biological replicates). (F) Macrophages were treated as in B with the exception that exogenous putrescine was added to macrophages as indicated to achieve a concentration 100 μM for 2 days before the addition of ACs. Cells were lysed then analyzed for Il10 by qRT-PCR (n = 5 biological replicates). (G) Aortic root cross-sections from Figure 2A were immunostained for IL-10. Staining was quantified as MFI per lesion area. Data are presented relative to the average value obtained for the baseline specimens (n = 8–10 mice per group). Representative images are shown. Bar, 200 μm. (H) Ldlr−/− mice were fed a Western diet for 16 weeks. For some mice, drinking water was supplemented with 3 mM putrescine in the drinking water at 8 weeks of Western diet feeding. Aortic root cross-sections were immunostained for IL-10 as in G. Data are presented relative to the average value obtained for the normal H2O specimens (n = 10 mice per group). Representative images are shown. Bar, 200 μm. Values for all graphs are means ± S.E.M.; *p < 0.05; n.s., not significant.
Atherosclerotic lesional putrescine content, together with the macrophage enzyme that converts arginine to ornithine, arginase-1, is low in progressing lesions and elevated in regressing lesions.19 We found that macrophage IL-10 was also higher in regressing versus progressing plaques, which is consistent with a previous report,32 and, most importantly, show that this increase was prevented by siOdc1-NP treatment (Fig. 3 G). We previously reported that supplementation of the drinking water with putrescine during atherosclerosis progression reduced necrotic core area and lesion size, enhanced fibrous cap thickness, and raised efferocytosis,19 and we now show that putrescine supplementation enhances IL-10 production in progression lesions (Fig. 3 H). These combined in vitro and in vivo data show the importance of ODC and its enzymatic product putrescine in the production of IL-10 by macrophages exposed to ACs.
Putrescine drives AC-induced IL-10 expression through the activation of Erk1/2
Activation of mitogen-activated protein kinases (MAPKs), which include extracellular signal-regulated kinases 1 and 2 (Erk1/2), Jun N-terminal kinases (JNK), and p38, are required for IL-10 production by multiple stimuli.35 Moreover, ACs activate Erk1/2 in macrophages, and inhibiting Erk1/2 activation in vivo impairs inflammation resolution.38 Further, Erk1/2 activation in certain cell lines requires ODC.39–41 We therefore reasoned that ODC-mediated IL-10 production in macrophages exposed to ACs may involve a putrescine-Erk1/2 pathway. We first showed that ODC-KO macrophages had impaired Erk1/2 activation, i.e., lower phospho-Erk1/2, compared with WT macrophages 45 minutes after exposure to ACs (Fig. 4 A). Also, Mac2+ cells (macrophages) in regressing plaques showed enhanced phospho-Erk1/2 levels compared to baseline, which was blunted in mice that received siOdc1 NPs (Fig. 4 B). Treating macrophages with the small-molecule MEK inhibitor, U0126, or silencing Erk1 and 2 (Fig. SV), prevented AC-induced Il10 mRNA and IL-10 protein expression (Fig. 4, C–F). Furthermore, the loss in AC-induced Erk1/2 activation observed in ODC-KO macrophages could be reversed by exogenous putrescine (Fig. 4 G), and plaque macrophages from mice drinking putrescine-supplemented water showed elevated phospho-Erk1/2 levels in progressing atherosclerotic lesions (Fig. 4 H). Mechanistically, Erk1/2 activation can drive IL-10 expression through the transcription factor AP-1 (activator protein 1),36, 42, 43 which is comprised of c-Jun and c-Fos. In this context, we found that treating macrophages with T-5224, a small-molecule inhibitor that selectively blocks the DNA binding activity of c-Jun/c-Fos,44 or silencing c-Jun, lowered AC-induced Il10 expression (Fig. SVI, A–C).
Figure 4. Loss of ODC Blocks AC-Induced Erk1/2 Activation in the Setting of Efferocytosis, and Erk1/2 is Required for AC-Induced IL-10 Expression in Macrophages.

(A) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were incubated for 45 mins with ACs at a 5:1 AC:macrophage ratio, after which the ACs were removed by rinsing then lysed. Immunoblots were performed for P-Erk1/2, Erk1/2, and β-actin (n = 3 biological replicates). (B) Aortic root cross-sections from Figure 2A were immunostained for P-Erk1/2. Staining was quantified as MFI of P-Erk1/2 within Mac2+ regions (n = 8–10 mice per group). Bar, 200 μm. (C) Bone marrow-derived macrophages were incubated with ACs as in Figure 3C with the exception that some macrophages were treated with 10 μM U0126 for 1 h prior to addition of ACs. Cells were lysed then analyzed for Il10 by qRT-PCR (n = 3 biological replicates). (D) Bone marrow-derived macrophages were incubated with ACs as in Figure 3D with the exception that some macrophages were treated with U0126 for 1 h prior to addition of ACs. Cells were then detached and analyzed by flow cytometry for IL-10. (n = 4 biological replicates). (E) Bone marrow-derived macrophages were transfected with siErk1 and siErk2-targeting siRNAs and assayed for IL10 mRNA as in B (n = 3 biological replicates). (F) Bone marrow-derived macrophages were transfected with siErk1 and siErk2-targeting siRNAs and assayed for IL10 by flow cytometry (n = 3 biological replicates). (G) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were incubated with ACs as in A with the exception that some macrophages were treated with 100 μM exogenous putrescine for two days prior to the addition of ACs. Immunoblots were performed for P-Erk1/2, Erk1/2, and β-actin (n = 3 biological replicates). (H) Aortic root cross-sections from Figure 3H were immunostained for P-Erk1/2. Staining was quantified as MFI of P-Erk1/2 within Mac2+ regions (n = 10 mice per group). Bar, 200 μm. Values for all graphs are means ± S.E.M.; *p < 0.05.
Arginine from phagolysosomal degradation of ACs during efferocytosis can increase ornithine and, via ODC, putrescine in macrophages.19 We therefore considered the possibility that AC degradation plays a role in AC-induced Il10 expression. Silencing Rubicon, a key LC3-associated phagocytosis protein that is required for the fusion of lysosomes to phagosomes and AC degradation during efferocytosis,45, 46 partially blocked AC-induced IL-10 expression (Fig. SVI, D and E). However, inhibiting the release of AC-derived arginine from phagolysosomes by silencing the lysosomal arginine transporter Pqlc2 did not prevent AC-induced IL-10 expression (Fig. SVI, F and G). These data suggested to us a 2-hit model for AC-induced Il10: one hit is the ornithine-putrescine-ERK-AP1 pathway described here, which uses basal levels of cellular ornithine, not AC-arginine-ornithine, to produce ODC-derived putrescine; and the second hit would be a separate pathway that requires a non-arginine metabolite from degraded ACs. In considering this putative second hit, we turned to a recent study suggesting that phagolysosomal AC degradation liberates fatty acids to stimulate IL-10 production by activating the transcription factor Pbx1.13 This Pbx1 pathway also involves an AC-CD36 receptor signaling pathway.47 We therefore asked whether silencing both the ornithine-AP1 pathway and the Pbx1 pathway in AC-exposed macrophages would lead to additive inhibition of Il10. Consistent with the two-hit model, blocking the ornithine-AP1 pathway with T-5224 alone and silencing Pbx1 alone each caused a partial block of AC-induced Il10, whereas treating the macrophages with both T-5224 and siPbx1 decreased Il10 to the no-AC level (Fig. SVI, H and I). These data support the hypothesis that ACs induce Il10 by two independent and additive mechanisms: one pathway that requires AC degradation and Pbx1; and another pathway, newly described here, that uses basal levels of cellular ornithine and ODC to stimulate a putrescine-AP1 pathway of Il10 induction.
ODC-dependent putrescine synthesis controls basal levels of MerTK expression
We next turned our attention to the question of how putrescine enhances AC-induced activation of the ERK-AP1-Il10 pathway. AC-induced Erk1/2 activation and IL-10 production are known to be regulated by AC-binding receptors, specifically CD36 and MerTK.38, 47 We therefore considered the possibility that putrescine increased an AC receptor that would then activate Erk1/2 upon subsequent AC binding to the receptor. We surveyed our RNA-seq dataset for efferocytosis receptors that are downregulated in ODC-KO macrophages. We found that Mertk was lower in ODC-KO versus ODC-WT macrophages, which we confirmed by qRT-PCR, immunoblot, and cell-surface flow cytometry (Fig. 5, A–C). Importantly, the decrease in MerTK in ODC-KO macrophages could be rescued by putrescine treatment (Fig. 5 D).
Figure 5. ODC-Dependent Putrescine Synthesis Controls Basal Levels of MerTK Expression.

(A) Bone marrow-derived macrophages from ODC-WT and M-ODC-KO mice were lysed and then assayed by qRT-PCR for Mertk (n = 4 biological replicates). (B) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were lysed and immunoblots were performed for MerTK and β-actin (n = 4 biological replicates). (C) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were detached and analyzed for surface MerTK by flow cytometry. A representative flow plot is shown (n = 4 biological replicates). (D) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were treated as in B with the exception that 100 μM putrescine was added two days before lysis (n = 3 biological replicates). (E) Bone marrow-derived macrophages from Mertkfl/fl (MerTK-WT) and Mertkfl/fl Lysz2-Cre+/− (MerTK-KO) mice were incubated for with ACs as in Fig 4A with the exception that macrophages were detached, fixed, permeabilized, immunostained for P-Erk1/2, and analyzed by flow cytometry. A representative flow plot is shown (n = 4 biological replicates). (F) Bone marrow-derived macrophages from MerTK-WT and MerTK-KO mice were incubated with ACs and GolgiStop as in Fig 3D and analyzed for IL-10 by flow cytometry (n = 4 biological replicates). (G) Bone marrow-derived macrophages from MerTK-WT and MerTK-KO mice were incubated with ACs and GolgiStop as in Fig 3D, with the exception that a group of MerTK-KO macrophages were incubated with 100 μM putrescine, and analyzed for IL-10 by flow cytometry (n = 4 biological replicates). (H) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were either mock-electroporated or electroporated with a plasmid encoding wild-type MerTK. Two days after electroporation macrophages were treated with ACs and GolgiStop as in Fig 3D and analyzed for IL-10 by flow cytometry (n = 4 biological replicates). (I) Aortic root cross-sections from Figure 2A were immunostained for MerTK and Mac2. Staining was quantified as MFI of MerTK within Mac2+ regions (n = 8–10 mice per group). Bar, 200 μm. (J) Aortic root cross-sections from Figure 3H were immunostained for MerTK. Staining was quantified as MFI of MerTK within Mac2+ regions (n = 10 mice per group). Bar, 200 μm. Values for all graphs are means ± S.E.M.; *p < 0.05.
We next conducted an efferocytosis experiment under conditions that favor single-cell, not continual, efferocytosis, i.e., relatively low AC:macrophage ratio (5:1) and short incubation time (45 min). We predicted lower efferocytosis in ODC-KO macrophages owing to lower basal MerTK. However, ODC-KO macrophages showed similar levels of efferocytosis compared to ODC-WT macrophages (Fig. SVII A). To examine if there was compensatory upregulation of other efferocytosis-related molecules in ODC-KO macrophages that could explain this finding, we examined the expression of other AC receptors and several AC-macrophage bridging molecules. While most AC receptors and bridging molecules were not affected by ODC deletion, the efferocytosis receptor Axl was increased (Fig. SVII B). Moreover, the silencing of Axl lowered efferocytosis in WT macrophages and further lowered efferocytosis in ODC-KO macrophages (Fig. SVII C). Thus, the increase in Axl in ODC-KO macrophages provides a plausible explanation for why efferocytosis is not decreased in ODC-KO macrophages despite a decrease in MerTK.
As we and others have shown previously,38, 48 MerTK deletion in macrophages blocks AC-induced Erk1/2 activation (Fig. 5 E), and we found that MerTK depletion also lowered AC-induced IL-10 expression (Fig. 5 F). However, unlike the situation with ODC-KO macrophages, exogenous putrescine was unable to enhance IL-10 expression in MerTK-KO macrophages (Fig. 5 G), indicating that putrescine-mediated MerTK expression is necessary for IL-10 induction by putrescine (putrescine → MerTK → AC binding → IL-10). Consistent with this hypothesis, genetically elevating MerTK in ODC-KO macrophages rescued IL-10 production in AC-exposed macrophages (Fig. 5 H and Fig. SVII D). Also, Mac2+ cells (macrophages) in regressing plaques showed enhanced MerTK expression compared with Mac2+ cells in baseline plaques, and this increase was prevented in mice that received siOdc1 NPs (Fig. 5 I). Furthermore, putrescine supplementation enhanced MerTK expression in the macrophages of progressing lesions (Fig. 5 J). These combined in vitro and in vivo data show the importance of ODC-dependent putrescine in regulating MerTK expression.
Putrescine-dependent H3K9 di/trimethylation governs steady-state levels of MerTK expression
We next sought to elucidate how putrescine maintains basal Mertk mRNA expression in macrophages. Putrescine is known to alter chromatin structure by elevating histone 3- lysine 9 di/trimethylation (H3K9me2/3) and lowering H3K9 acetylation (H3K9ac)21. In support of this action of putrescine being operational in macrophages, we found that ODC-KO macrophages have reduced H3K9 di/trimethylation and slightly enhanced H3K9 acetylation, both of which could be reversed by putrescine treatment (Fig. 6 A). To determine if H3K9 modifications could alter Mertk expression, we treated macrophages with either 6-pentadecylsalicylic acid (6-PDSA), a histone acetyltransferase inhibitor,49 to block H3K9 acetylation or BIX 01294, an inhibitor of the histone-lysine N-methyltransferase G9a,50 to block H3K9 methylation. Whereas 6-PDSA had no impact on MerTK expression (Fig. 6 B), BIX 01294 reduced MerTK expression (Fig. 6 C), suggesting the importance of H3K9 methylation. This conclusion was further supported by directly silencing G9a, which reduced MerTK expression in a manner that could not be rescued by putrescine, (Fig. 6 D), as predicted by a putrescine → G9a/H3K9 methylation → Mertk pathway.
Figure 6. Putrescine-Dependent Di/Trimethylation Governs Steady-State Levels of MerTK Expression.

(A) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice were treated with 100 μM putrescine for two days then lysed and immunoblotted for H3K9me2/3, H3K9ac, and β-actin (n = 4 biological replicates). (B) Bone marrow-derived macrophages were treated with 10 μM 6-PDSA for 24 h then lysed and immunoblotted for MerTK and β-actin (n = 3 biological replicates). (C) Bone marrow-derived macrophages were treated with 5 μM BIX 01294 for 24hr then lysed and immunoblotted for MerTK and β-actin (n = 3 biological replicates). (D) Bone marrow-derived macrophages from ODC-WT and ODC-KO mice transfected with ScrRNA or siG9a, with some macrophages being treated with 100 μM putrescine for two days. Cells were then lysed and immunoblotted for MerTK, H3K9me2/3, and β-actin (n = 4 biological replicates). Representative immunoblots from two biological replicates are shown on the left and densitometric analysis is shown on the right. (E) Nuclear extracts from ODC-WT and ODC-KO macrophages were subjected to Mertk ChIP analysis using an anti-H3K9me3 antibody or IgG control. A region in the first intron containing a putative H3K9me3 motif was amplified by qRT-PCR and normalized to the values obtained from input DNA (n = 3–4 biological replicates). (F) Bone marrow-derived macrophages were transfected with either ScrRNA or siG9a and incubated with ACs as in Fig 4A. Cells were then lysed and immunoblotted for P-Erk1/2, Erk1/2, and β–actin (n = 3 biological replicates). (G) Bone marrow-derived macrophages were transfected with either ScrRNA or siG9a and treated with ACs and GolgiStop as in Fig 3D. Cells were detached, fixed, permeabilized, immunostained for IL-10, and analyzed by flow cytometry (n = 4 biological replicates). (H) Proposed model linking ODC-dependent putrescine to MerTK expression, IL-10, and inflammation resolution. Values for all graphs are means ± S.E.M. For panel D, *p < 0.05 for group 2, 3, 4, and 6 vs. group 1; ¶p < 0.05 for group 5 vs. group 4; and #p < 0.05 for group 6 vs. group 5. For panel E, *p < 0.05 for group 2 vs. group 1; and #p < 0.05 for group 2 vs. group 3.
H3K9me2/3 is frequently assigned as a repressor mark associated with closed chromatin, and thus its role in maintaining basal levels of Mertk could involve repression of a negative regulator of Mertk. However, H3K9me2/3 modification has also been described in transcriptional activation and mRNA elongation by RNA polymerase II.51–53 To seek evidence compatible with the latter possibility, we conducted a chromatin immunoprecipitation (ChIP) experiment in ODC-WT and ODC-KO macrophages using an anti-H3K9me3 antibody and PCR primers designed to amplify a region in intron 1 of Mertk, where a putative H3K9me3 motif sequence is located. We found that H3K9me3-chromosome immunoprecipitates contained this intronic region in an ODC-dependent manner (Fig. 6 E), consistent with the possibility that putrescine-dependent H3K9 di/trimethylation of the Mertk gene itself is responsible for maintaining basal levels of Mertk expression. Finally, in support of the role of this Mertk induction pathway in MerTK-Erk1/2-mediated induction of Il-10, we found that silencing G9a lowered AC-induced Erk1/2 activation and IL-10 expression (Fig. 6, F and G). Collectively, these results support a process through which putrescine-mediated H3K9 di/trimethylation maintains a basal level of MerTK expression, which is required to mount a resolution response upon subsequent AC-induced MerTK activation (Fig. 6H).
DISCUSSION
Along with the physical removal of dead cells, efferocytosis stimulates signaling pathways that drive inflammation resolution and reverse tissue dysfunction. A critical feature of the resolution program is the production of IL-10, which enhances efferocytosis, dampens the production of pro-inflammatory cytokines and chemokines, and promotes macrophage polarization towards a wound-resolving phenotype.36 A major source of IL-10 in the resolution phase arises from the interactions between ACs and cell-surface receptors on macrophages, and genetic deletion of many of the AC-binding receptors on macrophages leads to chronic, non-resolving inflammation.54, 55 We identified that basal levels of one of these AC-binding receptors, MerTK, is regulated by ODC-dependent putrescine synthesis in a manner that requires H3K9-di/trimethylation. Loss of ODC prevents MerTK-Erk1/2 dependent production of IL-10 upon exposure to ACs, which delays resolution in vivo and prevents atherosclerosis regression. These data provide a novel pathway for how MerTK expression is regulated to maintain an appropriate resolution response and also add growing support for the importance of therapeutically enhancing resolution as a treatment strategy to reverse chronic inflammatory diseases.
It is important to integrate the findings here and elsewhere as to the various ways ODC affects efferocytosis in vitro and in vivo. First, one must distinguish between single-cell efferocytosis, which occurs in vitro with relatively low AC:macrophage ratios and short incubation times, and continual efferocytosis, which occurs in vitro with high AC:macrophage ratios and long incubation times and in vivo in chronic settings like atherosclerosis.19, 56, 57 For single-cell efferocytosis, ODC-KO macrophages would be predicted to have lower efferocytosis owing to decreased MerTK expression. However, our data suggest that efferocytosis is maintained owing to an increase in Axl in ODC-KO macrophages. For continual efferocytosis, which would apply to atherosclerosis regression,19 efferocytosis was found to be lower when ODC was absent in macrophages, which can be explained by impairment of the previously described ODC-putrescine-Rac1 pathway19 and by lower IL-10.58 Moreover, the compensatory role of Axl observed in vitro would not be relevant, as our previous study showed that Axl does not play a role in macrophage efferocytosis in the setting of atherosclerosis.59
MerTK, a member of the Tyro-Axl-MerTK (TAM) family of receptor tyrosine kinases, mediates the binding and internalization of ACs. In addition to mediating efferocytosis, ligand-dependent activation of MerTK stimulates Erk1/2, which induces the production of 5-LOX-derived specialized pro-resolving lipid mediators (SPMs).38 SPMs, through the activation of their cognate G protein-coupled receptors, decrease leukocyte recruitment, enhance efferocytosis, and polarize macrophages towards a wound-resolving phenotype.60 In non-resolving inflammation, such as in advanced atherosclerosis, MerTK is cleaved and the ratio of SPMs to pro-inflammatory leukotrienes becomes imbalanced.7, 10 Our group has demonstrated that a cleavage-resistant form of MerTK prevents this imbalance in the SPM:leukotriene ratio and attenuates plaque necrosis.10 A critical component of the resolution program is the existence of beneficial, positive-feedback loops, and when executed successfully tissue function is restored. For example, pro-resolving macrophages release Gas6 and amplify IL-10 secretion via MerTK in a ligand-dependent manner.54 As another example, regulatory T cells, which accumulate in tissues during resolution, produce the anti-inflammatory cytokines IL-4 and IL-13, which causes macrophages to produce and respond to IL-10 in a paracrine and autocrine-dependent manner.18, 32
As stated above, IL-10 production has been attributed to AC-dependent activation of cell-surface receptors. However, a mechanism exists that also involves IL-10 production by the degradation of macromolecular components originating from the ingested AC itself, namely fatty acid oxidation. Fatty acids, presumably from hydrolysis of AC lipids by phagolysosomal degradation, fuel mitochondrial respiration and activate NAD+-dependent activation of the transcription factor Pbx1, which binds to the AC-response element of the Il10 promoter.13 However, binding of ACs to CD36 on macrophages is sufficient to stimulate Pbx1-dependent Il-10 production.47 Therefore, it may be possible that the metabolic activity of macrophages to drive fatty acid oxidation upon AC degradation may, in part, require ligand-dependent activation of AC-binding receptors.
Coordinated phases in inflammation and its resolution must be such that waning of the inflammatory response does not compromise host defense. A fascinating issue that arises from this work, therefore, is whether ODC activity might be suppressed in the initial stages of an anti-pathogen inflammatory response. If one considers that the loss of putrescine enhances LPS-stimulated inflammatory gene expression,21 lowering putrescine may provide a mechanism that helps the host neutralize pathogens soon after infection. In this context, ODC deletion enhances pathogen clearance in the setting of Helicobacter pylori and Citrobacter rodentium infection.21 It is interesting to consider that nitrite accumulates at high levels in the early stages of inflammation, whereas at later stages when inflammation is resolving, ornithine levels rise.61 This temporal regulation may occur because nitrite production by pro-inflammatory macrophages drives S-nitrosylation of a cysteine residue on ODC that significantly lowers its activity and prevents putrescine synthesis.62 Hypothetically, if ODC is not inactivated throughout the inflammatory phase, a pre-mature resolution response may compromise inflammation-induced pathogen neutralization. This scenario may help explain the finding that amastigotes of Trypanosoma cruzi, an intracellular parasite that causes Chagas disease, proliferate in polyamine-producing macrophages that have phagocytosed an AC.63
Polyamines in general, and putrescine in particular, control gene expression through a variety of mechanisms.64, 65 We have previously demonstrated that wound-resolving macrophages use putrescine to stabilize the mRNA of the RacGEF Mcf2 in a HuR-dependent manner.19 Here, we show that putrescine maintains basal levels of MerTK expression through H3K9 di/trimethylation. Along with mRNA stabilization and chromatin remodeling, polyamines can directly bind nucleic acids, and polyamine binding to DNA is known to regulate the rate of mRNA transcription by transitioning the conformation state of B-DNA into the Z-DNA state.66 In addition to its effects on DNA and RNA, polyamines can also be covalently linked to proteins by transglutaminase 2 (TG2), which catalyzes the formation of an isopeptide bond between glutaminyl residues and primary amines.67 These post-translational modifications, termed polyamination, lead to an increase in positive charges within the polyaminated regions, which are believed to alter protein structure. The consequence of protein polyamination can change protein-protein interactions, protein localization, and protein function.67 Future work investigating TG2-mediated protein polyamination during resolution is particularly interesting because TG2 inhibition results in defective efferocytosis,68 and hematopoietic deletion of TG2 in mice promotes atherosclerosis.69
In summary, we have demonstrated that a basal level of putrescine synthesis in macrophages controls the expression of MerTK through H3K9 di/trimethylation and that exposure to ACs drives a MerTK-Erk1/2 pathway that promotes IL-10 expression. Importantly, a failure to synthesize putrescine in macrophages impairs this pathway and impairs the resolution response. These findings add further support to the concept that enhancing resolution can be an effective therapeutic strategy to restore tissue function and thereby slow the progression of atherosclerosis and other chronic inflammatory diseases.
Supplementary Material
HIGHLIGHTS.
ODC-dependent putrescine synthesis drives IL-10 production, inflammation resolution, and atherosclerosis regression.
Putrescine promotes AC-induced IL-10 expression by maintaining basal levels of MerTK expression.
Putrescine-dependent MerTK expression is mediated by H3K9 di/trimethylation.
ACKNOWLEDGMENTS
We thank Dr. John Cleveland for the Odc1fl/fl mice, Dr. Carla Rothlin for the Mertkfl/fl mice, Dr. Matthew Molusky for assistance in analyzing and interpreting RNA-seq experiments, and Dr. Ray Birge for providing the Mertk plasmid. A.Y.J. and I.T. conceived, designed, and directed the research; N.K., W.T., and J.S. designed and engineered the targeted siRNA nanoparticles; A.Y.J., B.D.G., X.W., and P.A. performed experiments; G.K. carried out the histological analyses of atherosclerosis specimens; A.Y.J. and I.T. wrote the manuscript, and all authors contributed to its editing.
SOURCES OF FUNDING
This work was supported by NIH grants K99 HL145131 (A.Y.), HL007343-28 (B.D.G.), HL127464 (J.S., and I.T.), HL132412 (I.T.), and HL145228 (I.T.); a Liver Scholar Award (X.W.); and the Brigham and Women’s Hospital Khoury Innovation award no.122829 (W.T.). Flow cytometry was conducted in the Columbia Center for Translational Immunology Core Facility, funded by NIH grants DK063608 and OD020056.
Nonstandard Abbreviations and Acronyms:
- AC
apoptotic cell
- AP-1
activator protein 1
- BMDM
bone marrow-derived macrophage
- ERK
extracellular signal-regulated kinase
- IL
interleukin
- KO
knockout
- LDLR
LDL receptor
- MerTK
MER tyrosine-protein kinase
- NP
nanoparticle
- ODC
ornithine decarboxylase
- PBX1
PBX Homeobox 1
- 6-PDSA
6-pentadecylsalicylic acid
- WD
Western diet
- WT
wild-type
Footnotes
DISCLOSURES
The authors declare no competing financial interests.
REFERENCES
- 1.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol 2008;8:349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doran AC, Yurdagul A Jr., Tabas I Efferocytosis in health and disease. Nat Rev Immunol. 2020;20:254–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morioka S, Maueroder C, Ravichandran KS. Living on the edge: Efferocytosis at the interface of homeostasis and pathology. Immunity. 2019;50:1149–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yurdagul A Jr., Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage apoptosis and efferocytosis in the pathogenesis of atherosclerosis. Circ. J 2016;80:2259–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kojima Y, Weissman IL, Leeper NJ. The role of efferocytosis in atherosclerosis. Circulation. 2017;135:476–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, Spite M, Fredman G, Tabas I. Mertk receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Invest 2017;127:564–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler. Thromb. Vasc. Biol 2008;28:1421–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kojima Y, Volkmer JP, McKenna K, et al. Cd47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun 2016;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Milton J, Perretti M, Farokhzad OC, Tabas I. Targeted nanoparticles containing the pro-resolving peptide ac2–26 protect against advanced atheosclerosis in hypercholesterolemic mice. Sci. Transl. Med 2015;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351 [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, Ben-Sahra I, Gius DR, Yvan-Charvet L, Chandel NS, Schumacker PT, Thorp EB. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell metabolism. 2019;29:443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caligiuri G, Rudling M, Ollivier V, Jacob MP, Michel JB, Hansson GK, Nicoletti A. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein e knockout mice. Mol. Med 2003;9:10–17 [PMC free article] [PubMed] [Google Scholar]
- 15.Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Von Der Thüsen JH, Kuiper J, Fekkes ML, De Vos P, Van Berkel TJ, Biessen EA. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in ldlr−/− mice. Faseb j. 2001;15:2730–2732 [DOI] [PubMed] [Google Scholar]
- 17.Kamaly N, Fredman G, Fojas JJ, et al. Targeted interleukin-10 nanotherapeutics developed with a microfluidic chip enhance resolution of inflammation in advanced atherosclerosis. ACS Nano. 2016;10:5280–5292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, Islam MN, Rymond CC, Du J, Hook J, Kuriakose G, Bhattacharya J, Tabas I. Regulatory t cells promote macrophage efferocytosis during inflammation resolution. Immunity. 2018;49:666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yurdagul A Jr., Subramanian M, Wang X, et al. Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell metabolism. 2020;31:518–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casero RA Jr., Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat Rev Cancer. 2018;18:681–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardbower DM, Asim M, Luis PB, Singh K, Barry DP, Yang C, Steeves MA, Cleveland JL, Schneider C, Piazuelo MB, Gobert AP, Wilson KT. Ornithine decarboxylase regulates m1 macrophage activation and mucosal inflammation via histone modifications. Proc Natl Acad Sci U S A. 2017;114:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fourgeaud L, Través PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zagórska A, Rothlin CV, Nimmerjahn A, Lemke G. Tam receptors regulate multiple features of microglial physiology. Nature. 2016;532:240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res. 1999;8:265–277 [DOI] [PubMed] [Google Scholar]
- 24.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP III, Rosenfeld ME, Virmani R. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the american heart association. Arterioscler. Thromb. Vasc. Biol 2017;37:131–157 [DOI] [PubMed] [Google Scholar]
- 25.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat Biotechnol. 2019;37:907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17:10–12 [Google Scholar]
- 27.Liao Y, Smyth GK, Shi W. Featurecounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930 [DOI] [PubMed] [Google Scholar]
- 28.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A, Rabkin S, Vandergriff JA, Doremieux O. Panther: A browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bystrom J, Evans I, Newson J, Stables M, Toor I, Van RN, Crawford M, Colville-Nash P, Farrow S, Gilroy DW. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by camp. Blood. 2008;112:4117–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merched AJ, Ko K, Gotlinger KH, Serhan CN, Chan L. Atherosclerosis: Evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008;22:3595–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma M, Schlegel MP, Afonso MS, et al. Regulatory t cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. 2020;127:335–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willecke F, Yuan C, Oka K, Chan L, Hu Y, Barnhart S, Bornfeldt KE, Goldberg IJ, Fisher EA. Effects of high fat feeding and diabetes on regression of atherosclerosis induced by low-density lipoprotein receptor gene therapy in ldl receptor-deficient mice. PLoS. ONE 2015;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao W, Yurdagul A Jr., Kong N, Li W, Wang X, Doran AC, Feng C, Wang J, Islam MA, Farokhzad OC, Tabas I, Shi J. Sirna nanoparticles targeting camkiiγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci Transl Med. 2020;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving tgf-b, pge2, and paf. J. Clin. Invest 1998;101:890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saraiva M, O’Garra A. The regulation of il-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181 [DOI] [PubMed] [Google Scholar]
- 37.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving tgf-beta, pge2, and paf. J Clin Invest 1998;101:890–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. Mertk signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing camkii activity. Sci Signal. 2018;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu B, Jiang X, Cai L, Zhao X, Dai Z, Wu G, Li X. Putrescine mitigates intestinal atrophy through suppressing inflammatory response in weanling piglets. J Anim Sci Biotechnol. 2019;10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stefanelli C, Tantini B, Fattori M, Stanic I, Pignatti C, Clo C, Guarnieri C, Caldarera CM, Mackintosh CA, Pegg AE, Flamigni F. Caspase activation in etoposide-treated fibroblasts is correlated to erk phosphorylation and both events are blocked by polyamine depletion. FEBS Lett. 2002;527:223–228 [DOI] [PubMed] [Google Scholar]
- 41.Manni A, Wechter R, Gilmour S, Verderame MF, Mauger D, Demers LM. Ornithine decarboxylase over-expression stimulates mitogen-activated protein kinase and anchorage-independent growth of human breast epithelial cells. Int J Cancer. 1997;70:175–182 [DOI] [PubMed] [Google Scholar]
- 42.Kremer KN, Kumar A, Hedin KE. Haplotype-independent costimulation of il-10 secretion by sdf-1/cxcl12 proceeds via ap-1 binding to the human il-10 promoter. J Immunol. 2007;178:1581–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang ZY, Sato H, Kusam S, Sehra S, Toney LM, Dent AL. Regulation of il-10 gene expression in th2 cells by jun proteins. J Immunol. 2005;174:2098–2105 [DOI] [PubMed] [Google Scholar]
- 44.Tsuchida K, Chaki H, Takakura T, Kotsubo H, Tanaka T, Aikawa Y, Shiozawa S, Hirono S. Discovery of nonpeptidic small-molecule ap-1 inhibitors: Lead hopping based on a three-dimensional pharmacophore model. J Med Chem. 2006;49:80–91 [DOI] [PubMed] [Google Scholar]
- 45.Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, Green DR. Molecular characterization of lc3-associated phagocytosis reveals distinct roles for rubicon, nox2 and autophagy proteins. Nat. Cell Biol 2015;17:893–906 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (lc3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. U. S. A 2011;108:17396–17401 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, Han J, Silverstein R, Selleri L, Ma X. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins pbx1 and prep-1. Immunity. 2007;27:952–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishi C, Yanagihashi Y, Segawa K, Nagata S. Mertk tyrosine kinase receptor together with tim4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J Biol Chem. 2019;294:7221–7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mai A, Rotili D, Tarantino D, Ornaghi P, Tosi F, Vicidomini C, Sbardella G, Nebbioso A, Miceli M, Altucci L, Filetici P. Small-molecule inhibitors of histone acetyltransferase activity: Identification and biological properties. J Med Chem. 2006;49:6897–6907 [DOI] [PubMed] [Google Scholar]
- 50.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of h3k9me2 by a small-molecule inhibitor for the g9a histone methyltransferase. Mol Cell. 2007;25:473–481 [DOI] [PubMed] [Google Scholar]
- 51.Wiencke JK, Zheng S, Morrison Z, Yeh RF. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene. 2008;27:2412–2421 [DOI] [PubMed] [Google Scholar]
- 52.Monaghan L, Massett ME, Bunschoten RP, Hoose A, Pirvan PA, Liskamp RMJ, Jørgensen HG, Huang X. The emerging role of h3k9me3 as a potential therapeutic target in acute myeloid leukemia. Front Oncol. 2019;9:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone h3 lysine 9 methylation and hp1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. 2005;19:381–391 [DOI] [PubMed] [Google Scholar]
- 54.Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires m2c polarization and mertk induction. J Immunol. 2012;189:3508–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adomati T, Cham LB, Hamdan TA, et al. Dead cells induce innate anergy via mertk after acute viral infection. Cell Rep. 2020;30:3671–3681 [DOI] [PubMed] [Google Scholar]
- 56.Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, Collins S, Lysiak JJ, Hoehn KL, Ravichandran KS. Continued clearance of apoptotic cells critically depends on the phagocyte ucp2 protein. Nature. 2011;477:220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y, Subramanian M, Yurdagul A Jr., Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, Tabas I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. 2017;171:331–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD. Enhanced apoptotic cell clearance capacity and b cell survival factor production by il-10-activated macrophages: Implications for burkitt’s lymphoma. J. Immunol 2005;174:3015–3023 [DOI] [PubMed] [Google Scholar]
- 59.Subramanian M, Proto JD, Matsushima GK, Tabas I. Deficiency of axl in bone marrow-derived cells does not affect advanced atherosclerotic lesion progression. Sci Rep. 2016;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007;21:325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albina JE, Mills CD, Henry WL Jr., Caldwell MD. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol. 1990;144:3877–3880 [PubMed] [Google Scholar]
- 62.Bauer PM, Buga GM, Fukuto JM, Pegg AE, Ignarro LJ. Nitric oxide inhibits ornithine decarboxylase via s-nitrosylation of cysteine 360 in the active site of the enzyme. J Biol Chem. 2001;276:34458–34464 [DOI] [PubMed] [Google Scholar]
- 63.Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, DosReis GA, Lopes MF. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 2000;403:199–203 [DOI] [PubMed] [Google Scholar]
- 64.Miller-Fleming L, Olin-Sandoval V, Campbell K, Ralser M. Remaining mysteries of molecular biology: The role of polyamines in the cell. J Mol Biol. 2015;427:3389–3406 [DOI] [PubMed] [Google Scholar]
- 65.Gerner EW, Meyskens FL Jr. Polyamines and cancer: Old molecules, new understanding. Nat Rev Cancer. 2004;4:781–792 [DOI] [PubMed] [Google Scholar]
- 66.Thomas TJ, Gunnia UB, Thomas T. Polyamine-induced b-DNA to z-DNA conformational transition of a plasmid DNA with (dg-dc)n insert. J Biol Chem. 1991;266:6137–6141 [PubMed] [Google Scholar]
- 67.Yu CH, Chou CC, Lee YJ, Khoo KH, Chang GD. Uncovering protein polyamination by the spermine-specific antiserum and mass spectrometric analysis. Amino Acids. 2015;47:469–481 [DOI] [PubMed] [Google Scholar]
- 68.Eligini S, Fiorelli S, Tremoli E, Colli S. Inhibition of transglutaminase 2 reduces efferocytosis in human macrophages: Role of cd14 and sr-ai receptors. Nutr Metab Cardiovasc Dis. 2016;26:922–930 [DOI] [PubMed] [Google Scholar]
- 69.Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, Curtiss LK, Terkeltaub R. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler. Thromb. Vasc. Biol 2006;26:563–569 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.