

ABSTRACT
Fungal secondary metabolites are small molecules that exhibit diverse biological activities exploited in medicine, industry and agriculture. Their biosynthesis is governed by co-expressed genes that often co-localize in gene clusters. Most of these secondary metabolite gene clusters are inactive under laboratory conditions, which is due to a tight transcriptional regulation. Modifications of chromatin, the complex of DNA and histone proteins influencing DNA accessibility, play an important role in this regulation. However, tinkering with well-characterised chemical and genetic modifications that affect chromatin alters the expression of only few biosynthetic gene clusters, and thus the regulation of the vast majority of biosynthetic pathways remains enigmatic. In the past, attempts to activate silent gene clusters in fungi mainly focused on histone acetylation and methylation, while in other eukaryotes many other post-translational modifications are involved in transcription regulation. Thus, how chromatin regulates the expression of gene clusters remains a largely unexplored research field. In this review, we argue that focusing on only few well-characterised chromatin modifications is significantly hampering our understanding of the chromatin-based regulation of biosynthetic gene clusters. Research on underexplored chromatin modifications and on the interplay between different modifications is timely to fully explore the largely untapped reservoir of fungal secondary metabolites.
Keywords: secondary metabolites, fungi, chromatin, transcriptional regulation
This review focuses on the chromatin-based regulation of fungal secondary metabolite biosynthesis and argues that the incomplete picture of chromatin modifications in fungi significantly hampers the activation and exploitation of fungal secondary metabolites in medicine, industry and agriculture.
INTRODUCTION
The fungal kingdom is the source of a wealth of small bioactive compounds called secondary metabolites (SMs). SMs exhibit a wide range of biological activities, some of which had and have significant impact on human societies. For instance, penicillin was accidentally discovered in 1928 by Alexander Fleming from the mould Penicillium chrysogenum through its anti-bacterial activity. This compound was subsequently developed into the first broad-spectrum antibiotic that provided the first effective medication against bacterial infections and started the antibiotic era in modern medicine (Aly, Debbab and Proksch 2011). Similarly, strobilurins were discovered in the forest mushroom fungus Strobilurus tenacellus in 1977 and are now widely used broad-spectrum fungicides to control pathogens mainly on cereals and soybeans (Balba 2007). Other fungal SMs with immunosuppressive (e.g. cyclosporin), anticholesterolemic (e.g. pravastatin), anticancer (e.g. taxol), or dye (e.g. monascorubrin) activity have been discovered, collectively highlighting the wide medical and industrial potential of fungal SMs. However, fungal SMs can also be mycotoxins that cause severe health issues on human and livestock, of which aflatoxins produced by Aspergillus species are among the most carcinogenic food contaminants (Verheecke, Liboz and Mathieu 2016). Fungal SMs can act as virulence factors when produced by plant pathogenic fungi, thereby contributing to agricultural losses (Collemare and Lebrun 2011). While SMs are generally non-essential for normal fungal growth and reproduction, they play key roles in morphological development, signalling, interactions with other organisms (Demain and Fang 2000) and protection against environmental stresses (Eisenman and Casadevall 2012; Griffiths et al. 2018).
Despite the importance of SMs to human societies, fungi have not been extensively exploited for SMs production (Keller 2018), and most efforts thus far focused on plants and bacteria, especially on members of the bacterial genus Streptomyces (Katz and Baltz 2016). However, the genomic era has revealed that fungal genomes encode many more biosynthetic pathways than previously thought (Collemare et al. 2008a; Keller 2018), reviving the interest in exploiting the fungal kingdom to find novel compounds. The discrepancy between the very low number of known compounds produced by a given species and the high number of SM biosynthetic pathways in the genome is likely explained by the tight transcriptional control of SM biosynthetic pathways (Brakhage 2013). Fungal SMs are only produced under very specific ecological conditions that remain difficult to determine in the laboratory. Approaches such as co-culture with other organisms and growth on diverse media (also known as OSMAC for One Strain Many Compounds) successfully induced the production of novel compounds (Baral, Akhgari and Metsä-Ketelä 2018), but their implementation is not straightforward. Thus, understanding the transcriptional regulation of SM production in fungi remains key in order to access the hidden fungal resource of SMs.
Here, after a brief introduction on SM biosynthetic pathways and their regulation in fungi, we will focus on a specific type of transcriptional regulation that has been the subject of extensive research in the last decade: chromatin modifications. Despite great progress in this research field, we argue that our knowledge is still limited, mainly because our efforts have focused on only few chromatin modifications. With the example of higher eukaryotes and the availability of novel experimental approaches, we propose new research directions that might lead to unlocking the full potential of fungal SMs.
GENE CLUSTER ORGANISATION OF BIOSYNTHETIC PATHWAYS
SMs are generally synthesised from just a few precursors originating from primary metabolism via biochemical pathways that can be classified based on the core enzyme involved in the biosynthesis of the first stable intermediate (Fig. 1A): polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), hybrid PKS-NRPS enzymes, dimethylallyl tryptophan synthases and terpene cyclases are associated with the production of polyketides, non-ribosomal peptides, PKS-NRPS hybrids, indole alkaloids, and terpenes, respectively (Keller 2018). PKSs and NRPSs core enzymes are large multidomain proteins, in which all domains operate conjointly to support the synthesis of the growing intermediate. The products synthesised by core enzymes are usually further modified by tailoring or decorating enzymes to produce the final active compound(s) (Fig. 1A) (Keller 2018). In fungi, genes encoding the core and tailoring enzymes of a given biosynthetic pathway are often physically linked in the genome and are co-regulated, defining a so-called gene cluster organization (Keller and Hohn 1997) (Fig. 1B). In addition to genes encoding these biosynthetic enzymes, gene clusters may also comprise genes that encode transporters involved in SM efflux or self-protection, and transcription factors that regulate the expression of genes involved in the biosynthetic pathway (Gardiner, Jarvis and Howlett 2005; Keller, Turner and Bennett 2005; Bergmann et al. 2010; Keller 2018) (Fig. 1B).
Figure 1.
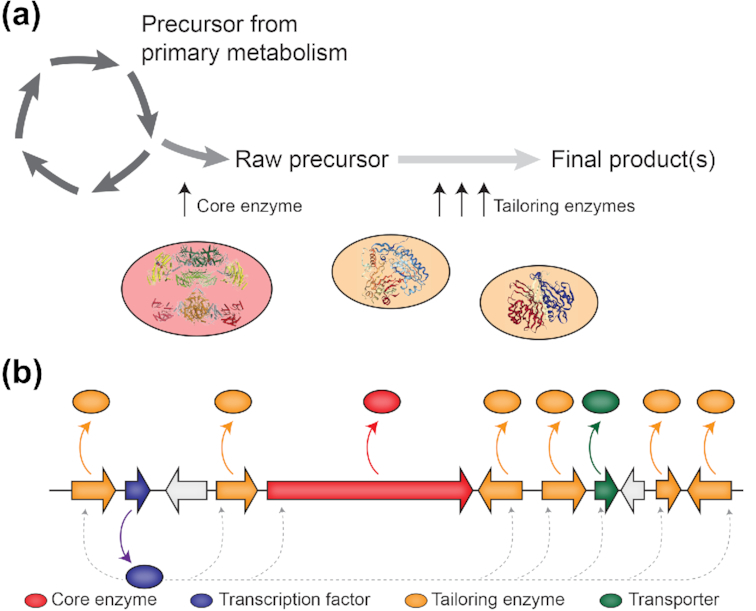
Secondary metabolites are synthesised by biosynthetic pathways that are encoded by gene clusters. (A) Secondary metabolites (SMs) are synthesised from few precursors by biochemical pathways centred around characteristic core enzymes (red). Core enzymes often contain multiple domains or modules that operate conjointly to support the synthesis of the precursor. The precursor is usually further modified by tailoring or decorating enzymes (orange) to produce the final active compound, which is subsequently exported by transporters (green). (B) In fungi, genes encoding core (red) and tailoring enzymes (orange) as well as transporters (green) involved in SM efflux or self-protection are often physically inked in the genome, defining a so-called gene cluster organization (Keller and Hohn 1997). Additionally, gene cluster may also comprise transcription factors (blue) that directly regulate the expression of genes involved in the biosynthetic pathway (dashed arrows) (Keller, Turner and Bennett 2005; Keller 2018).
The evolutionary forces that are acting on the formation and maintenance of SM genes in physically linked gene clusters remain unclear (Rokas, Wisecaver and Lind 2018). Clustering of SM genes may reduce the risk for dissociation during meiotic recombination or chromosomal rearrangements (Hurst, Pál and Lercher 2004; Seidl and Thomma 2014; Thomma et al. 2016), which may lead to incomplete gene clusters that could form toxic intermediates (McGary, Slot and Rokas 2013; Rokas, Wisecaver and Lind 2018). Examination of 100 fungal genomes revealed a strong bias for the physical pairing of genes encoding enzymes that share a toxic intermediate (McGary, Slot and Rokas 2013), and most of these enzyme pairs share promoter regions (McGary, Slot and Rokas 2013; Galazka and Freitag 2014; Thomma et al. 2016). However, intermediates are not necessarily more toxic than the final compound (Griffiths et al. 2016). Tight genetic linkage between SM genes could favour co-adaptation, optimizing protein–protein interactions and metabolic fluxes in a given biosynthetic pathway (Hurst, Pál and Lercher 2004; Santoni, Castiglione and Paci 2013; Slot 2017). Finally, the physical clustering of SM genes may provide the potential for tight co-regulation, thereby enabling highly coordinated interactions between enzymes involved in the same biosynthetic pathway (McGary, Slot and Rokas 2013; Galazka and Freitag 2014; Thomma et al. 2016).
LOCAL AND GLOBAL TRANSCRIPTIONAL REGULATION OF FUNGAL SECONDARY METABOLITE GENE CLUSTERS
Secondary metabolite biosynthesis is very tightly regulated in order to ensure that the potent active compounds are only produced when required. One of the most striking examples is the ACE1 gene cluster in the rice blast fungus Pyricularia oryzae that is exclusively expressed for only a few hours during penetration into the first cell of a rice leaf (Collemare et al. 2008b). Such a fine transcriptional co-regulation of genes within a given SM biosynthetic pathway is complex and involves several interconnected layers of regulation, including gene cluster-specific transcription factors (TFs) and global regulators (Brakhage 2013; Macheleidt et al. 2016; Keller 2018).
Local regulation: gene cluster-specific transcription factors
Many SM gene clusters comprise one or several genes that encode TFs and additional co-regulators, which locally govern co-expression of the genes belonging to the pathway (Fig. 1B). The best studied TF in a SM gene cluster is aflR, which regulates the expression of the aflatoxin and sterigmatocystin genes in Aspergillus species (Woloshuk et al. 1994; Yu et al. 1996). However, this regulation appears more complex as AflR functions together with a putative co-activator encoded within the gene cluster, AflJ (Chang 2003). AflR also regulates the genes of the aflatoxin orthologous gene cluster in Dothistroma septosporum, which produces dothistromin (Chettri et al. 2013). In this fungus, however, the orthologue of AflJ is not co-regulated and does not seem to play a role in the regulation of the dothistromin gene cluster (Chettri et al. 2013). Similarly, the regulation of the biosynthesis gene cluster responsible for the red pigment bikaverin in the rice pathogen Fusarium fujikuroi is likely dependent on additional co-regulators as deletion of the cluster-specific TF bik5 only led to the loss of expression of some but not all of the genes within the gene cluster (Wiemann et al. 2009). The involvement of co-activators has also been suggested for other SM gene clusters such as those controlling the production of the anthraquinones monodictyphenone and cladofulvin in Aspergillus nidulans and Cladosporium fulvum, respectively (Chiang et al. 2010; Griffiths et al. 2016). These putative co-activators often do not contain known conserved domains and their occurrence in fungal SM gene clusters could be more frequent than initially expected. In A. nidulans, the expression of afoA, which encodes the transcription factor that regulates the asperfuranone gene cluster, was found to be under the direct control of another transcription factor, ScpR (Bergmann et al. 2010). Notably, the ScpR gene is located at another gene cluster that comprises two NRPS genes (Bergmann et al. 2010), suggesting the existence of cross-talk between different gene clusters. However, such cross-talk has only been reported in a single case, and thus the prevalence of such complex regulation remains unknown.
Global regulation: response to environmental signals
SM biosynthesis is responsive to many environmental stimuli, including shifts in nitrogen or carbon sources, pH, temperature, or light as well as the presence of other organisms (Brakhage 2013; Macheleidt et al. 2016; Keller 2018). Fungal responses to these stimuli often rely on global transcriptional regulators that, in contrast to gene cluster-specific TFs, affect the expression of a large number of target genes throughout the genome (Bayram et al. 2008; Macheleidt et al. 2016). For instance, AreA not only regulates large sets of nitrogen catabolic genes in response to the quality and quantity of nitrogen, but also impacts the production of a broad range of SMs including gibberellins (Tudzynski et al. 1999; Michielse et al. 2014; Tudzynski 2014). The expression of six of the seven genes of the gibberellin biosynthesis gene cluster in F. fujikuroi is dependent on AreA, which is able to bind GATA motifs in their promoters (Mihlan et al. 2003; Michielse et al. 2014). Similarly, PacC is a key factor in the fungal pH regulation (Tilburn et al. 1995), and it activates, for instance, penicillin biosynthesis at alkaline pH in P. chrysogenum (Then Bergh and Brakhage 1998) as well as represses the expression of sterigmatocystin biosynthesis genes in A. nidulans (Keller and Hohn 1997). The carbon catabolite repression transcription factor CreA also regulates the production of aflatoxins (Fasoyin et al. 2018), consistent with the presence of CreA binding sites present at the aflR locus (Bhatnagar, Ehrlich and Cleveland 2003).
The velvet complex is highly conserved in ascomycetes and basidiomycetes where it controls sexual development and SM gene cluster expression in response to light (Bayram and Braus 2012). The heterotrimeric velvet complex consists of VeA and VelB, two of the four members of the velvet family (Bayram and Braus 2012), as well as the non-velvet protein LaeA (Bayram et al. 2008). It associates in the nucleus only under dark conditions where it coordinates expression of genes involved in development and SM production (Bayram and Braus 2012). In A. nidulans, laeA deletion impairs the expression of the sterigmatocystin, penicillin and lovastatin biosynthetic gene clusters (Bok and Keller 2004). LaeA collectively affects up to nearly 50% of SM gene clusters, and thus can be considered a master regulator of fungal SM gene cluster expression (Perrin et al. 2007; Macheleidt et al. 2016). LaeA exhibits location-dependent control of gene expression (Bok and Keller 2004; Bok et al. 2006; Perrin et al. 2007) as it specifically controls the expression of genes within but not outside of the ∼60 kb sterigmatocystin cluster in A. nidulans (Brown et al. 1996; Bok et al. 2006). Additionally, genes placed into the context of the gene cluster are LaeA-dependently regulated (Bok et al. 2006). LaeA encodes a putative methyltransferase-domain protein that was suggested to play a role in regulating DNA accessibility (Bok and Keller 2004; Bok et al. 2006; Perrin et al. 2007).
CHROMATIN-BASED TRANSCRIPTIONAL REGULATION IN EUKARYOTES
DNA in the nucleus is organized by a complex of DNA and (histone) proteins – the chromatin. The basic ‘unit’ of chromatin is the nucleosome, which is formed by DNA wrapped around a histone octamer of two copies of each of the four core histone proteins (H2A, H2B, H3 and H4) (Armeev et al. 2018). Chromatin is dynamic in response to different environmental signals and can transition between different states: the ‘open’, loosely packed and transcriptionally active euchromatin, and the ‘closed’, tightly packed and transcriptionally silent heterochromatin (Fig. 2). The combination of DNA methylation together with different post-translational modifications (PTMs) of histones – the histone code (Strahl and Allis 2000) – influences chromatin packing and thus DNA accessibility for the transcriptional machinery, which, in turn, impacts the regulation of gene expression.
Figure 2.

Chromatin impacts the transcriptional regulation of secondary metabolite clusters. Secondary metabolite (SM) gene cluster can be located in transcriptionally repressive heterochromatic genomic regions (orange region). In response to different environmental signals, chromatin can transit (dark arrows) from the heterochromatic to the transcriptionally active euchromatic state (green regions), thereby allowing SM biosynthetic activity. Genes associated with the SM cluster are coloured (see Fig. 1), while genes not part of the cluster are shown in grey.
DNA methylation
Methylation of DNA (mainly of cytosines; 5-mC) has been observed in most animals, plants and fungi (Feng et al. 2010; Zemach and Zilberman 2010; Zemach et al. 2010; Seidl 2017). In many plants and vertebrates, DNA methylation plays an important role in regulating gene expression (Zemach and Zilberman 2010). Similarly, DNA methylation of transposons and other types of highly repetitive DNA is considered an important genome defense mechanism, in which DNA methylation is associated with the formation of heterochromatin and thus suppresses transposon expression and transposition, thereby mitigating the impact of transposons on host genomes (Feng et al. 2010; Zemach et al. 2010; Seidl 2017; Seidl and Thomma 2017).
5-mC methylation has been extensively studied in animals and plants. In contrast, genome-wide DNA methylation patterns have only been characterised in few fungal species where DNA methylation is mainly associated with heterochromatic regions at transposons (Selker et al. 2003; Zemach et al. 2010; Montanini et al. 2014; Jeon et al. 2015). For instance, DNA methylation in the model fungus Neurospora crassa is restricted to transposable elements that have been targeted by a genome defence system (Repeat Induced Point mutations; RIP) (Selker et al. 2003; Lewis et al. 2009). Nevertheless, gene methylation has also been observed in few fungi (Zemach et al. 2010; Mishra, Baum and Carbon 2011; Jeon et al. 2015). In the human pathogen, Candida albicans, gene methylation modulates the transcriptional activity of genes associated with morphological plasticity and iron metabolism, which is important for signalling and pathogenicity (Mishra, Baum and Carbon 2011). Similarly, exons in the saprophytic fungus Uncinocarpus reesii can be methylated in certain genomic contexts, and this methylation is correlated with transcription (Zemach et al. 2010).
Genome-wide 5-mC methylation in N. crassa is mediated by Dim-2 (defective in methylation) (Kouzminova and Selker 2001), which is a DNA methyltransferase (DMT) belonging to the Dnmt1 family (Zemach and Zilberman 2010). Next to Dim-2, N. crassa encodes an additional DNA methyltransferase called RID, a member of the fungal DMT-like family that also contains Masc1 from Ascobolus immersus (Malagnac et al. 1997; Freitag et al. 2002). RID and Masc1 play important roles in genome defense processes targeting transposons; however, a DNA methyltransferase activity has not yet been directly demonstrated for either of these proteins (Malagnac et al. 1997; Freitag et al. 2002). Aspergillus flavus and A. nidulans genomes encode a single DMT, DmtA, which is closely related to sequences belonging to the DMT-like family (Liu et al. 2012; Yang et al. 2016). Genome-wide 5-mC levels in A. flavus are low, and thus the functional significance of DNA methylation in this fungus remains controversial (Liu et al. 2012; Yang et al. 2016). Therefore, in most fungi, the occurrence, abundance, and influence of DNA methylation on the regulation of gene expression have not been investigated in detail.
Histone post-translational modifications
Next to DNA methylation, multiple amino acid residues of histone proteins can be post-translationally modified (PTM)1 (Turner 2007). In higher eukaryotes, at least 15 different histone modifications have been reported (Ballard et al. 2002; Chen et al. 2007; Fujiki et al. 2011; Tan et al. 2011; Xie et al. 2012; Rothbart and Strahl 2014; Brehove et al. 2015; Hottiger 2015; Goudarzi et al. 2016; Clancy et al. 2017; Wei et al. 2017; Ohkuni et al. 2018; Shanmugam et al. 2018) (Table 1). Histone acetylation and methylation were discovered first (Allfrey, Faulkner and Mirsky 1964), which likely reflects their abundance in eukaryotic cells and their significant roles in regulating chromatin. Histone lysine acetylation has been particularly studied as it was associated with transcriptional activation (Castillo, López-Rodas and Franco 2017). In contrast, histone lysine methylation and ubiquitination are associated with both transcriptional activation and repression (Santos-Rosa et al. 2002; Strahl et al. 2002; Zhang et al. 2003; Morillon et al. 2005; Shilatifard 2006; Lewis et al. 2009; Connolly, Smith and Freitag 2013; Jamieson et al. 2013; Wiemann et al. 2013; Gacek-Matthews et al. 2016; Freitag 2017). All PTMs contribute to transcriptional regulation, but a few appear to play more specific roles in other cellular processes, including DNA repair, apoptosis, cell cycle and pathogenicity (Clarke et al. 1999; Shroff et al. 2004; Giannattasio et al. 2005; Ahn et al. 2005a; Soyer, Rouxel and Fudal 2015; Seidl, Cook and Thomma 2016). For example, lysine formylation and ubiquitination are involved in the DNA damage response (Jiang et al. 2007; Deem, Li and Tyler 2012; Hunt et al. 2013), and serine and threonine phosphorylation as well as biotinylation are related to chromatin compaction during the cell cycle (Hendzel et al. 1997; De Souza et al. 2000; Stanley, Griffin and Zempleni 2001; Neurohr et al. 2011; Singh, Wijeratne and Zempleni 2013).
Table 1.
Overview of the post-translational modifications (PTMs) of histone identified in eukaryotes.
| PTMs (abbreviation) | Writer/Eraser proteinsa | Function in transcription | Reported in fungi | Reference |
|---|---|---|---|---|
| Methylation (me) | HMTs/HDMs | Activation/repression | Yes | Strauss and Reyes-Dominguez 2011 |
| Acetylation (ac) | HATs/HDACs | Activation | Yes | Strauss and Reyes-Dominguez 2011 |
| Butyrylation (buty) | HATs/HDACs | Activation | Yes | Zhang et al. 2009 |
| Propionylation (prop) | HATs/HDACs | Activation | Yes | Zhang et al. 2009 |
| Crotonylation (cr) | HATs/HDACs | Activation | Yes | Andrews et al. 2016 |
| Succinylation (suc) | unknown/HDACs | Activation | Yes | Xie et al. 2012 |
| Malonylation (mal) | unknown/HDACs | Activation | Yes | Xie et al. 2012 |
| Phosphorylation (ph) | Kinase/phosphatase | Repression | Yes | Ahn et al. 2005a,b; Hsu et al. 2000; De Souza et al. 2000 |
| Ubiquitination (ub) | ubiquitin-conjugating enzymes/isopeptidase | Activation/repression | Yes | Trujillo et al. 2011; Robzyk, Recht and Osley 2000 |
| Sumoylation (su) | SUMO-conjugating enzymes/isopeptidase | Repression | Yes | Trujillo et al. 2011; Nathan et al. 2006 |
| Citrullination (cit) | deiminase | Repression | No | - |
| ADP-ribosylation (ar) | ART/hydrolase | Activation | No | - |
| Biotinylation (bio) | Biotinyl ligases/unknown | Repression | Yes | Hasim et al. 2013 |
| Proline isomerization (iso) | Isomerases | Repression | Yes | Monneau et al. 2013 |
| Formylation (fo) | - | Activation/repression | No | - |
HMT: histone methyltransferase; HDM: histone demethylase; HAT: histone acetyltransferase; HDAC: histone deacetylase; ART: ADP-ribosyltransferase
The removal or addition of specific histone PTM is catalysed by writer and eraser proteins (Table 1). For example, histone acetylation and methylation are catalysed by histone acetyl transferases (HATs) and histone methyl transferases (HMTs), respectively, and the removal of acetyl and methyl groups is catalysed by histone deacetylases (HDACs) and histone demethylases (HDMs), respectively (Lee and Workman 2007; Freitag 2017). The activity of these enzymes is characterised by both histone and amino acid specificity (Zhang et al. 2003; Wapenaar and Dekker 2016), allowing tight control of the histone code. In contrast, writers and erasers for other modifications are less well known. Interestingly, HATs could also catalyse crotonylation, butyrylation and propionylation (Chen et al. 2007; Liu et al. 2009; Sabari et al. 2015), while enzymes performing succinylation and malonylation have not yet been identified (Castillo, López-Rodas and Franco 2017). Functional characterisation of HDACs suggested that they can act as eraser proteins for propionylation (Liu et al. 2009), succinylation and malonylation (Du et al. 2011). However, the occurrence of these different acylation modifications seems to be correlated to the cellular concentration of acyl-CoA, and thus specific activities of writer/eraser proteins might not be required (Liu et al. 2009; Sabari et al. 2015).
In fungi, most studies into PTMs have so far merely focused on histone acetylation and methylation (Fig. 2). In N. crassa, H3K9me3 co-localizes with DNA methylation and forms constitutive heterochromatin at telomeres, centromeres and transposon-rich regions throughout the genome (Lewis et al. 2009; 2010). Notably, heterochromatin formation through DNA methylation and H3K9me3 is catalysed by DCDC (the Dim-5/-7/-9, Cul4/Ddb1 complex), containing the histone methyltransferase (KMT1) Dim-5 (a homolog of the Schizosaccharomyces pombe Clr4), which, in turn, directs cytosine methylation by recruitment of heterochromatin protein 1 (HP1) and the DNA methyltransferase Dim-2 (Tamaru and Selker 2001; Tamaru et al. 2003; Freitag et al. 2004; Lewis et al. 2010; Freitag 2017). In contrast to H3K9me3, H3K27me3 is mainly localised at transcriptionally silent, often sub-telomerically localised genes and forms facultative heterochromatin (Jamieson et al. 2013; Freitag 2017). Actively transcribed genes in euchromatin have been often associated with several different histone PTMs, mostly acetylation (H3K4ac, H3K9ac, H3K14ac, H4K12ac or H4K16ac) and methylation (H3K4me2/3 or H3K36me3) of lysine residues (Shilatifard 2006; Bhaumik, Smith and Shilatifard 2007; Lewis et al. 2009; Gacek-Matthews et al. 2015; Freitag 2017) (Fig. 2). Unsurprisingly, H3K9me3 in A. nidulans and F. fujikuroi as well as H3K27me3 in the cereal pathogen Fusarium graminearum generally do not overlap with H3K4me2/3 (for example A. nidulans Fig. 3) (Connolly, Smith and Freitag 2013; Wiemann et al. 2013; Gacek-Matthews et al. 2016). Methylation of H3K4 and H3K36 seems to activate transcription through the recruitment of HATs at specific methylated nucleosomes (Martin et al. 2006; Ginsburg et al. 2014; Martin et al. 2017) However, besides histone acetylation and methylation, other PTMs such as butyrylation, propionylation, phosphorylation, proline isomerisation, sumoylation, and ubiquitination have also been reported in fungi (Table 1) (De Souza et al. 2000; Hsu et al. 2000; Robzyk, Recht and Osley 2000; Ahn et al. 2005b; Nathan et al. 2006; Zhang et al. 2009; Strauss and Reyes-Dominguez 2011; Trujillo et al. 2011; Xie et al. 2012; Hasim et al. 2013; Monneau et al. 2013; Andrews et al. 2016), therefore suggesting that these additional PTMs can influence chromatin accessibility and thereby transcriptional regulation.
Figure 3.

Secondary metabolite clusters are associated with heterogeneous chromatin landscapes. Secondary metabolite (SM) gene cluster in Aspergillus nidulans are located along the eight chromosomes and are associated with euchromatic (H3K4me3 and H3K36me3; dark-green and light-green, respectively) as well as with heterochromatic (H3K9me3; orange) regions. Publicly available chromatin immunoprecipitation sequencing samples (Gacek-Matthews et al. 2016) were mapped to the A. nidulans genome assembly (Galagan et al. 2005) using BWA (Li and Durbin 2009) and normalised using deepTools (Ramírez et al. 2014) (counts per million, window length 300 nucleotides, smooth length 900 nucleotides). The location SM gene cluster are indicated in dark blue (Inglis et al. 2013), and characterised SM gene cluster are highlighted (Inglis et al. 2013; Macheleidt et al. 2016).
CHROMATIN-BASED TRANSCRIPTIONAL REGULATION OF FUNGAL SECONDARY METABOLITE GENES
The first evidence that chromatin contributes to SM gene clusters regulation was obtained a decade ago from the deletion of the HDAC hdaA in A. nidulans, leading to the activation of two SM gene clusters producing sterigmatocystin and penicillin (Shwab et al. 2007). The same study demonstrated that application of the HDAC inhibitor trichostatin A to Alternaria alternata and Penicillium expansum induced the production of several unknown compounds (Shwab et al. 2007), indicating that both genetic and chemical manipulation of chromatin modifications can lead to the activation of SM gene clusters. Two histone PTMs have been extensively studied in relation to SM gene expression, acetylation and methylation.
Histone acetylation and regulation of fungal secondary metabolite gene clusters
First characterised in A. nidulans, histone acetylation plays important roles in regulating SM gene cluster activity in many fungi (Fig. 2). Accumulation of histone acetylation in promoters of genes involved in the aflatoxin biosynthetic gene cluster correlates with protein accumulation and production of aflatoxin (Roze et al. 2007). Similarly, enrichment of H3K9ac at active SM gene clusters, for instance, at the gibberellin or the fusarin C gene clusters, has been observed in F. fujikuroi (Niehaus et al. 2013; Studt et al. 2013; Wiemann et al. 2013). In line with these observations, deletion of HATs can impair SM production (Nützmann et al. 2011; Kong et al. 2018). Deletion of 36 of the 40 HATs encoded in the A. nidulans genome and screening for loss of orsellinic acid production led to the identification of the HAT GcnE, which is part of the Saga/Ada complex (Nützmann et al. 2011). This complex is responsible for histone 3 acetylation (H3K9ac and H3K14ac) and plays a major role in activating SM biosynthetic gene clusters (e.g. of sterigmatocystin, terrequinone and penicillin) (Nützmann et al. 2011, 2013). Increase of SM production is correlated with global increase of H3K14ac and SM gene cluster-specific H3K9ac (Nützmann et al. 2011, 2013). Consequently, overexpression of HATs can lead to an increase of SM production. For instance, overexpression of the A. nidulans HAT esaA increases H4K12ac specifically at SM gene cluster loci and enhances the production of sterigmatocystin, penicillin, terrequinone and orsellinic acid (Soukup et al. 2012). As histone acetylation is generally associated with SM gene cluster activity, deletion of HDACs causes constitutive or hyperacetylation of SM gene clusters. Therefore, genetic manipulation of HDACs has been proven successful for activation of SM gene clusters in Aspergillus fumigatus (Lee et al. 2009), Aspergillus oryzae (Kawauchi, Nishiura and Iwashita 2013), F. fujikuroi (Niehaus et al. 2016), P. oryzae (Maeda et al. 2017), Fusarium asiaticum (Maeda et al. 2017), P. chrysogenum (Guzman-Chavez et al. 2018) and A. nidulans (Albright et al. 2015; Henke et al. 2016). In contrast, over-expression of HATs has been thus far much less commonly exploited.
The general model in which HDAC deletion causes increased SM gene cluster activity, however, does not always apply. HDAC deletion can also lead to repression of SM production as shown for gliotoxin in A. fumigatus (Lee et al. 2009), bikaverin, fusarubin, gibberellins and fusaric acid in F. fujikuroi (Studt et al. 2013), deoxynivalenol in F. graminearum (Li et al. 2011), aflatoxins in A. flavus (Lan et al. 2016) and naphtha-γ-pyrone and chrysogine in P. chrysogenum (Guzman-Chavez et al. 2018). The impact of histone acetylation on transcription is often more complex due to functional complementarity between different HDAC genes. In F. fujikuroi, deletion of hda1 repressed the production of bikaverin, fusarubin, gibberellin and fusaric acid but not of fusarins (Studt et al. 2013). Deletion of hda2 repressed the production of bikaverin, gibberellins, fusaric acid and fusarins, but not of fusarubin (Studt et al. 2013). A metabolomics study in A. nidulans comparing a wild-type and a strain with a down-regulated HDAC gene revealed a nearly equal number of up-regulated and down-regulated metabolites (Albright et al. 2015). Consistently, functional analysis of the H4K16-specific HDAC HosA in A. nidulans revealed converse regulatory effects depending on the specific SM gene cluster (Pidroni et al. 2018). Consequently, even though histone acetylation has been commonly associated with transcriptional activation, its functional consequence for transcription is rather complex and seems to be often SM-gene-cluster-specific.
Mechanisms of histone acetylation-mediated regulation fungal SM gene clusters have recently received additional attention due to co-cultivation experiments with bacteria. Following co-cultivation, Streptomyces rapamycinicus triggered modification of fungal histones in A. nidulans and thereby elicited the production of orsellinic acid and its derivatives (Nützmann et al. 2011). In particular, the presence of bacteria altered H3K9ac levels at 890 different loci, 593 of these showed significant higher acetylation including the gene basR that encodes a Myb-like transcription factor (Fischer et al. 2018). BasR activates the expression of the orsellinic gene cluster as well as other biosynthetic pathways (Fischer et al. 2018). Although BasR is not conserved in all fungi, it is conceivable that similar responses could be driven by different transcription factors in other fungal species, suggesting that chromatin-based activation of silent SM gene clusters may involve the co-activation of additional global regulators.
Histone methylation and regulation of fungal secondary metabolite gene clusters
Histone methylation, in particular the repressive H3K9me3 and H3K27me3 marks, is another PTM that has been extensively studied in relation to regulation of SM production in fungi (Fig. 2). During active growth of A. nidulans, silent SM gene clusters are marked by H3K9me3 and the HP-1 homolog HepA, while in stationary phase and during activation of SM production H3K9me3 and HepA levels decrease (Reyes-Dominguez et al. 2010). Consequently, interfering with H3K9me3 by deletions of hepA and the histone methyltransferase Dim-5 homolog clrD led to an increased expression of multiple SM gene clusters, including those involved in the biosynthesis of sterigmatocystin, penicillin and terrequinone A (Reyes-Dominguez et al. 2010). H3K27me3 in F. graminearum as well as in F. fujikuroi is located at species-specific, often sub-telomeric, regions that are enriched for genes involved in SM biosynthesis (Connolly, Smith and Freitag 2013; Studt et al. 2016). Notably, deletion or silencing of histone methyltransferases in N. crassa (set7), F. graminearum (kmt6), or F. fujikuroi (kmt6) led to the loss of H3K27me3 and to the activation of hundreds of genes, many of which are involved in SM biosynthesis (Connolly, Smith and Freitag 2013; Jamieson et al. 2013; Studt et al. 2016). Knock-down of kmt6 in F. fujikuroi led to the transcription of four otherwise silent SM gene clusters and to the accumulation of novel SMs, including beauvericin (Niehaus et al. 2016; Studt et al. 2016), and the kmt6 deletion mutant in F. graminearum resulted in the expression of 32 out of 45 SM gene clusters (Connolly, Smith and Freitag 2013). Similarly, H3K27me3 and H3K9me3 contribute to SM biosynthesis regulation in the fungal endophyte Epichloë festucae that forms symbiotic association with grass hosts (Chujo and Scott 2014). During plant colonisation and compared to in vitro culture, H3K27me3 and H3K9me3 levels were reduced at SM gene clusters responsible for the biosynthesis of ergot alkaloids (eas) and lolitrems (ltm), two SMs with bioprotective activity against herbivores (Chujo and Scott 2014). Deletion of the EfclrD (kmt1) or EfezhB (kmt6) methyltransferases led to a reduction of H3K9me3 and H3K27me3, respectively, and to the de-repression of eas and ltm gene expression in vitro (Chujo and Scott 2014).
In contrast to these repressive marks, the role of the activating mark H3K4me2/3 in the regulation of fungal SM gene clusters is much less clear. Di- and tri-methylation of lysine 4 is catalysed by the methyltransferase Set1 (KMT2), which is the catalytic subunit of the COMPASS complex (Miller et al. 2001; Krogan et al. 2002). Deletion of bre2 (cclA), which encodes another COMPASS component, resulted in strong reduction of H3K4me2/3 and changes in SM profiles, for instance the activation of the monodictyphenone and F9775A/F9775B SM gene clusters in A. nidulans (Bok et al. 2009) and the gliotoxin SM gene cluster in A. fumigatus (Palmer et al. 2013). Notably, loss of H3K4me at these SM gene clusters is associated with loss of H3K9me3, likely due to crosstalk between these histone modifications (Bok et al. 2009; Strauss and Reyes-Dominguez 2011). These observations differ from the intuitive expectations for H3K4me-marked genes, as here H3K4me is seemingly associated with SM gene repression. Similarly, H3K4me2 was only enriched at a few genes within the gibberellin gene cluster under environmental conditions that stimulate SM production in F. fujikuroi (Wiemann et al. 2013).
Altering H3K36 methylation, another activating histone mark, also showed a strong contrasting effect on SM production in F. fujikuroi (Janevska et al. 2018). Deletions of the euchromatin H3K36 methylase set2 and the subtelomeric H3K36 methylase ash1 resulted in decreased production of gibberellins, and increased production of fusarins and fusaric acid (Janevska et al. 2018). The absence of ash1 additionally resulted in decreased production of bikaverin, but increased production of fusarubin (Janevska et al. 2018). Some SM gene clusters, e.g. fusarin C, are highly induced in F. graminearum mutants lacking H3K27me3, yet no enrichment of H3K4me3 or H3K36me3 was observed (Connolly, Smith and Freitag 2013), while accumulation of H3K36me3 was observed for multiple active SM gene clusters in A. nidulans (Gacek-Matthews et al. 2015). De-methylation of H3K36me3 and H3K4me3, which in A. nidulans is catalysed by KdmA and KdmB, respectively, positively and negatively influence genome-wide gene expression patterns, in particular of SM genes (Gacek-Matthews et al. 2015; 2016). In kdmB mutant strains, 50% of SM genes were mis-regulated in the mutant under SM inducing conditions, the majority of which showed lower expression (Gacek-Matthews et al. 2016). These observations suggest that KdmB activity is required for the normal induction of the majority of SM gene clusters in A. nidulans (Gacek-Matthews et al. 2016). While deletion of kmt6 induced a considerable number of SM gene clusters in F. graminearum, four gene clusters were found to be down-regulated (Connolly, Smith and Freitag 2013). Collectively, studies on histone methylation and acetylation in fungi suggest that individual histone PTMs contribute to a more complex regulatory system that control SM gene clusters activity and that chromatin-based regulation cannot be restricted to few activating or repressing modifications.
Is DNA methylation involved in the regulation of fungal secondary metabolism?
Due to the low abundance of DNA methylation in fungal genomes, its role in the regulation of SM gene clusters received only little attention. However, several examples have clearly demonstrated its importance in the regulation of gene expression in fungi (Zemach et al. 2010; Lin et al. 2013; Jeon et al. 2015). Deletion of the A. flavus DNA methyltransferase dmtA resulted in significant reduction of aflatoxin production (Yang et al. 2016), suggesting that gene methylation is important for gene activation rather than repression in this fungal species. Experimental evidence for a role of DNA methylation in the regulation of SM production was also found in several other fungi such as A. niger (Fisch et al. 2009) and F. fujikuroi, but not P. oryzae where it only affects development (Jeon et al. 2015). Treatment with DNA methyltransferase inhibitors like 5-azacytidine has led to the identification of SM compounds not present in control treatments in 10 out of 12 fungi (Williams et al. 2008). In A. flavus, 5-azacytidine-treatment modified the expression of genes involved in fungal development as well as in SM regulation and biosynthesis. Reduced aflatoxin biosynthesis was correlated with lower expression of most genes within the SM cluster, and particularly on the complete inhibition of the expression of aflQ, aflI and aflLa (Lin et al. 2013; Yang et al. 2015), which is consistent with the phenotype of the dmtA deletion mutant (Yang et al. 2016). However, considering the overall low abundance of DNA methylation is mostly fungal, its role in regulating SM gene clusters remains unclear. Future characterisation of DNA methylation in additional fungal genomes might reveal specific genomic environments, for instance, the proximity to transposable elements, or association to additional histone modification that might link this chromatin modification to SM gene cluster regulation.
Chromatin-based tools for activation of silent gene clusters
Activation of silent gene clusters has been an active area of investigation leading to the development of various strategies (Brakhage and Schroeckh, 2011). The most commonly employed strategies are overexpression or targeted deletion of local or global transcription factors, induction of a pathway through promoter exchange or cultivation under diverse conditions (OSMAC approach), including co-culture with other microorganisms. Another approach is to transfer gene clusters to activate their expression in a heterologous host (Clevenger et al. 2017; see Skellam 2019 for a recent review about this strategy). However, the OSMAC approach is tedious because many different culture conditions need to be tested in order to activate the production of a very limited number of new compounds, while the other strategies are restricted to a given target gene cluster. The CRISPR/Cas9 genome editing tool now allows the modification (for instance deletion, single nucleotide substitutions and/or promotor swaps) of several genes or pathways at once. However, establishing this tool in different fungal systems is tedious and technically demanding, which will likely hamper its exploitation in many non-model systems and thus prevent access to their SM diversity.
Pioneering experiments on the regulation of SM gene clusters by chromatin modifications opened the way to use them as a tool for accessing the large SM potential of the fungal kingdom. HDACs have been successfully targeted to induce or increase the production of SMs (Pfannenstiel and Keller 2019). Genetic approaches are limited to genetically tractable fungi. Thus, the use of chemical inhibitors has been a preferred approach to induce silent SM gene clusters in non-model fungi. The most commonly used compounds are inhibitors of HDACs, including trichostatin A and suberoylanilide hydroxamic acid (SAHA), and inhibitors of DNA methyltransferases, including 5-azacytidine. Both types of inhibitors were found to affect the expression of fungal SM gene clusters in different fungi (Fisch et al. 2009; Zutz et al. 2013). Notably, the effect of HDAC inhibitors are often consistent with the observed phenotypes of HDAC deletion mutants as shown in A. nidulans (Shwab et al. 2007) and F. fujikuroi (Studt et al. 2013), validating these approaches to induce silent SM gene clusters. SAHA induced the production of new cladochromes and of calphostin B in Cladosporium cladosporioides (Williams et al. 2008) and resulted in the identification of new compounds like nygerone A in A. niger (Henrikson et al. 2009), cyclodepsipeptides in Beauveria felina (Chung et al. 2013) as well as in Microascus sp. (Vervoort, Drašković and Crews 2011), and prenylated luteorides A–C metabolites from Torrubiella luteorostrata (Asai, Yamamoto and Oshima 2011). The use of DNA methyltransferase inhibitors suggests that this chromatin modifications plays an important role in regulating certain SM gene clusters in several fungi (Fisch et al. 2009; Asai et al. 2012; Zutz et al. 2013). The combination of both HDAC and DNA methyltransferase inhibitors induced the production of tenuipyrone in Isaria tenuipes (Asai et al. 2012) and of many derivatives of tenellin in Beauveria bassiana (Yakasai et al. 2011). Chemical approaches are nowadays widely exploited to activate SM production in non-model species, especially with endophytes and marine isolates that are promising species for drug discovery (González-Menéndez et al. 2016; Qadri et al. 2017; Demers et al. 2018; Siless et al. 2018; Triastuti et al. 2019).
The strategy to alter chromatin modifications to activate silent SM gene clusters is successful; however, the effect of these modifications on gene expression is often pleiotropic. In addition, in some fungal species these genetic modifications do not lead to the activation of any silent gene cluster (Griffiths et al. 2015). Moreover, the effect of chemical inhibitors on SM gene cluster expression depends on the composition of the medium used to grow fungi (Zutz et al. 2013). The accumulation of studies that employed chemical and genetic alteration of chromatin states shows that this approach can be successfully used to induce SM gene clusters, but the outcome for a given fungus is unpredictable and there is no single approach that will activate SM production in a large number of fungal species. Instead, the observed pleiotropic effect on gene expression and the fact that the efforts mentioned above have not resulted in activating all biosynthetic pathways in the tested fungi suggest that the histone code is not conserved among fungi or that histone modifications are read differently between SM gene clusters. Modification of several chromatin modifications at once could provide better results, but such an approach requires a more detailed characterisation of all histone modifications and DNA methylation at SM gene cluster loci in diverse fungi (Pfannenstiel and Keller 2019).
KEY CHALLENGES TO ACTIVATE SILENT FUNGAL SECONDARY METABOLITE CLUSTER USING CHROMATIN MODIFIERS
Significant efforts have been invested in manipulating the chromatin-based regulation to harness the wealth of SM gene clusters in fungal genomes. Nevertheless, the vast majority of fungal SM gene clusters remain silent, highlighting that our understanding of their regulation is far from complete. To advance our understanding of SM gene cluster regulation, we here identified key challenges that await to be addressed in the future.
Chromatin modifications: more than just methylation and acetylation
In fungi, studies on chromatin modifications and their roles in regulating SM production have mostly been restricted to histone acetylation and methylation. Such focus is sensible considering that these PTMs modulate transcription. However, these PTMs do not affect all SM gene clusters in a given fungus (Gacek and Strauss 2012; Connolly, Smith and Freitag 2013; Jamieson et al. 2013; Wiemann et al. 2013; Gacek-Matthews et al. 2016), which might at least partially be explained by the involvement of specific global regulators such as BasR in Aspergillus species (Fischer et al. 2018). Additionally, the histone code in fungi is not restricted to only two histone PTMs: at least 15 different histone PTMs have been reported in eukaryotes, and most of them also occur in yeast (Table 1). These diverse PTMs have been experimentally reported in human to affect 75 different amino acid positions across the four core histones, representing an overall total of 216 different modifications (Fig. 4). In contrast, only 75 published histone modifications affecting 43 different amino acid positions have been reported in the yeast Saccharomyces cerevisiae, likely reflecting that histone PTMs have been more extensively studied in human rather than yeast. Interestingly, butyrylation is the most abundant modification in human and crotonylation is as abundant as acetylation (Fig. 4A). In yeast, methylation and acetylation are the most abundant histone PTMs, likely reflecting the research efforts in fungi so far. The distribution of methylation, acetylation and succinylation across the four histone proteins is similar in human and yeast, with the highest number of methylation and acetylation found for histone H3, while the less-well studied PTMs differ between human and yeast histones which is likely due to the lower number of studies reporting on these PTMs (Fig. 4B). In sharp contrast to yeast and human, only 39 PTMs affecting 21 amino acids are predicted in the secondary metabolite producer A. nidulans (Fig. 4a); only five types of PTMs are predicted to occur on three histones, of which acetylation and methylation represent 82% (Fig. 4). However, since most known PTMs also occur in fungi (Table 1), it can be anticipated that PTMs other than methylation and acetylation similarly play a role in regulating SM production. Deletion of the sumO gene in A. nidulans resulted in increased production of asperthecin and in decreased production of austinol/dehydroaustinol and sterigmatocystin (Szewczyk et al. 2008), indicating that also sumoylation is involved in regulation of SM gene clusters. Considering the role of butyrylation, crotonylation and succinylation in active transcription in higher eukaryotes (Table 1), a similar situation may also occur in fungi. Additionally, the histone code in fungi may even be more complex and involves as of yet unreported PTMs. For example, 14 novel mass shifts that do not correspond to any known modifications have been reported on lysine residues at histones H2B, H3 and H4 in S. cerevisiae (Zhang et al. 2009). Obtaining an unbiased and comprehensive overview of all potential PTMs in fungi is therefore imperative to resolve their function and potential involvement in regulating SM gene cluster activity.
Figure 4.

Histone post-translational modifications in human, yeast, and the secondary metabolite producing Aspergillus nidulans. The relative number of the different histone post-translational modifications (PTMs) were retrieved for the four core histone proteins from Homo sapiens (accession numbers: H2A P0C0S8; H2B P62807; H3 P84243; H4 P62805), Saccharomyces cerevisiae (accession numbers: H2A P04911; H2B P02293; H3 P61830; H4 P02309) and Aspergillus nidulans (accession numbers: H2A P08844; H2B P23754; H3 P23753; H4 P23750) from the reviewed (Swiss-Prot) Uniprot database (The UniProt Consortium 2017). The PTMs from A. nidulans are only predictions based on similarity. (A) Proportion of each experimentally validated or predicted histone PTM indicates prevalent modifications. (B) Distribution of each histone PTM across the four core histones indicates where the modifications have been identified or predicted.
Acetylation, butyrylation, propionylation and crotonylation are related acylation PTMs that seem to involve the same writer and eraser proteins (Table 1), and it has been suggested that their use reflects the concentration of the different precursors in the cell (Liu et al. 2009; Du et al. 2011; Castillo, López-Rodas and Franco 2017). Such an observation would argue that different related modifications actually have the same function, the choice of the modification depending on the available pool of metabolic precursors. However, they could also represent different signals, bringing another level of subtleties in the histone code if diverse combinations of related histone PTMs impact transcription differently. Similar additional subtleties also concern DNA methylation. 5-mC is the major reported modification; however, several variants resulting from the demethylation process also exist, including 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxycytosine (Kumar, Chinnusamy and Mohapatra 2018). In addition to 5-mC DNA methylation that in fungi primarily occurs at transposable elements, adenine can also be methylated (N6-mA) at transcriptionally active genes in early-branching fungi (Mondo et al. 2017; Seidl 2017). Even though its function is still largely unknown (Mondo et al. 2017; Seidl 2017), this DNA modification is nevertheless a candidate to probe its role in regulating SM gene clusters. Similar to 5-mC, several variants of adenine methylation are found with N6-hydroxymethyladenine and N6-formyladenine (Kumar, Chinnusamy and Mohapatra 2018). These intermediates in the demethylation pathway could be part of a more complex histone code than previously expected. Based on the accumulating evidence of the occurrence and abundance of these additional histone PTMs and DNA methylation, together with the observation that only few gene clusters seem to be solely regulated by histone acetylation and methylation, it is timely to shift focus and study underexplored chromatin modifications to unravel the complex regulation of SM gene clusters expression.
Crosstalk between chromatin modifications modulates gene expression
Chromatin-based gene expression is a complex process modulated by combinations and interactions (the crosstalk) between different modifications (Macheleidt et al. 2016; Atlasi and Stunnenberg 2017), yet this aspect of the histone code remains poorly understood. For instance, H3K9me3 in N. crassa is a mark for subsequent DNA methylation through binding of HP1 and recruitment of the Dim-2 DNA methyltransferase leading to the formation of heterochromatin (Tamaru and Selker 2001; Tamaru et al. 2003; Rose and Klose 2014). Loss of H3K4me caused by deletion of the COMPASS component bre2 is associated with decrease of H3K9me3 (Bok et al. 2009), which could suggest a thus far underappreciated crosstalk between H3K4 and H3K9 methylation in fungi (Freitag 2017). Deletion of set7 and loss of H3K27me3 in F. graminearum led to activated expression of many genes, yet a considerable number of genes that lost this modification remained silent, suggesting that other factors such as transcription factor activity or crosstalk with other chromatin modifications are required to regulate expression (Connolly, Smith and Freitag 2013). Similarly, in P. oryzae and F. asiaticum, deletion of the HDAC hda-1 resulted in an increased production of SMs, but the overall degree of H3 and H4 acetylation was similar to wild type (Maeda et al. 2017), suggesting that local changes or crosstalk between different modifications regulate SM gene cluster activity. The interplay between methylation and acetylation as shown for H3K4 and H3K36 to activate transcription (Martin et al. 2006; Ginsburg et al. 2014; Martin et al. 2017) is another example highlighting the need for functional analyses to evaluate how different chromatin modifications interact. New genome editing tools such as CRISPR/Cas9 will facilitate multiple gene or promoter replacements, allowing to target diverse chromatin modifications in a controlled manner at once. Such a knowledge is important since it will provide the basis for more advanced genetic and chemical manipulations that might activate a larger number of SM gene clusters in the future.
The localization of secondary metabolite gene clusters and its relation to chromatin-based regulation
Chromatin immunoprecipitation together with next-generation sequencing (ChIP-seq) facilitated the genome-wide exploration of major chromatin modifications, mainly histone acetylation and methylation (Connolly, Smith and Freitag 2013; Jamieson et al. 2013; Wiemann et al. 2013; Basenko et al. 2015; Schotanus et al. 2015; Gacek-Matthews et al. 2016). These studies revealed the common occurrence of heterochromatic regions (H3K9me3 and/or H3K27me3) at transposon-rich regions that are localized at sub-telomers, at centromeres, or along the chromosomal arms (for example A. nidulans, Fig. 3). Many SM gene clusters are localized in or at close proximity to sub-telomeric regions, and SM gene clusters are often flanked by or embedded in heterochromatic regions (Bok et al. 2006; Palmer and Keller 2010; Connolly, Smith and Freitag 2013; Wiemann et al. 2013; Gacek-Matthews et al. 2016). F. graminearum mutants lacking H3K27me3 (kmt6 deletion mutants) display de-regulation of many SM gene clusters that are localized within sub-telomeric regions (Connolly, Smith and Freitag 2013). Similarly, in A. nidulans many SM gene clusters are located in proximity to (often sub-telomeric) H3K9me3-rich regions (Gacek-Matthews et al. 2016). For example, the penicillin cluster is located at a transposons-rich region 30 kb from the telomere of chromosome VI (Shaaban et al. 2010) (Fig. 3). Reduction of H3K9me3 levels due to the deletion of the Dim-5 homolog crlD activates the expression of multiple SM gene clusters including penicillin (Reyes-Dominguez et al. 2010). Notably, the removal of a distal 3.7 kb PbIa transposable element resulted in decreased penicillin production, suggesting that transposable elements also play a positive role in regulating SM gene cluster expression (Shaaban et al. 2010). Therefore, the localisation of SM gene clusters at or in proximity of transposon-rich sub-telomeric regions seems to play an important role in regulating their expression. Nevertheless, a considerable number of SM gene clusters are located outside of sub-telomeric regions (Fig. 3) and many of these are regulated by chromatin modifications (Gacek and Strauss 2012). For example, SM gene clusters in F. fujikuroi located outside of sub-telomeric regions are associated with heterochromatic regions (Wiemann et al. 2013). The PKS19 gene cluster producing fujikurins is embedded within an H3K9me3-rich region on the long arm of chromosome VIII (Wiemann et al. 2013). Importantly, not all SM clusters are associated with heterochromatin. For instance, a considerable number of SM gene clusters in A. nidulans and in F. fujikuroi are not located in close proximity to heterochromatin (Fig. 3) (Wiemann et al. 2013; Gacek-Matthews et al. 2016). Therefore, the expression of these SM gene clusters might be regulated by the occurrence of activating or not yet studied chromatin modifications, as well as by the spatial architecture of chromatin (Wiemann et al. 2013).
The recent development of chromatin conformation capture (3C) methods allows to investigate the 3D architecture of genomes. Hi-C sequencing revealing contacts between genomic loci at the whole-genome scale in an untargeted fashion (Lieberman-Aiden et al. 2009; Rao et al. 2014). These contact maps confirmed that genomes are organised in compartments corresponding to the euchromatin and heterochromatin states (Lieberman-Aiden et al. 2009; Mizuguchi et al. 2014; Galazka et al. 2016; Rowley et al. 2017; Winter et al. 2018). Higher resolution contact maps revealed that genomes are partitioned in several sub-compartments that correspond to different combinations of chromatin modification marks, including for instance H3K36me3, H3K27me3, H3K9me3 and H4K20me3 in humans (Rao et al. 2014). Similar in N. crassa, the genome-wide contact map in wild-type strongly correlates with H3K9me3, and to a lower extent to H3K27me3 (Galazka et al. 2016). A Hi-C contact map generated for the S. pombe deletion mutant of the sole H3K4 methyltransferase Clr4, which is affected in heterochromatin formation, revealed significantly fewer sub-compartments and increased inter- and intra-chromosomal interactions (Mizuguchi et al. 2014). Such effects differ for distinct histone modifications. For example, in N. crassa, targeted deletion of dim-5 (loss of H3K9me3) or hp1 affects chromatin architecture only mildly, while deletion of set-7 (loss of H3K27me2/3) or the importin alpha dim-3 (involved in Dim-5 localization) decreased interactions between constitutive heterochromatin compartments (Galazka et al. 2016; Klocko et al. 2016). In E. festucae, the eas gene cluster forms part of an interaction sub-compartment, in which genes display induced expression in planta compared with in vitro conditions (Winter et al. 2018), suggesting that chromatin and 3D structure influence gene co-expression, especially during interaction with other organisms. Unfortunately, this study did not include histone modifications and a contact map during in planta colonization, which is the only known condition to induce eas expression. The increase or reduction of interactions between different genomic compartments when histone modifications are altered provides a mechanistic explanation for the activation of only certain SM gene clusters by chromatin modifications. Thus, the combination of Hi-C with ChIP-seq has the potential to characterise the interaction compartments in which SM gene clusters reside. Such knowledge may improve our predictions on which histone modification should be modified in order to activate silent SM gene clusters.
Chromatin dynamics during inter-species crosstalk
Interactions between species occur in diverse ecological niches. In particular, fungi compete with other microorganisms that live in the same niche and thus produce antimicrobial compounds, both peptides and SMs, to colonise their niche. For instance, the co-cultivation of Bacillus subtilis or Escherichia coli with the mushroom Coprinopsis cinerea leads to the activation of fungal defense responses, including the production of antimicrobials (Kombrink et al. 2019; Stöckli et al. 2019). Similarly, co-cultivation of A. nidulans and Streptomyces bacteria activate otherwise silent SM gene clusters, a process that is mediated by bacterial-triggered changes in the chromatin landscape (Fischer et al. 2018), demonstrating that inter-species crosstalk is an important trigger for chromatin dynamics. Thus, experiments that specifically address chromatin in co-cultures between fungi and other microorganisms are needed to determine whether the mechanism observed in A. nidulans to activate the production of antimicrobial compounds is conserved. Additionally, many fungal species engage in symbiotic interactions, ranging from mutualistic to parasitic, with different hosts (e.g. plants, insects or nematodes). Fungal symbionts often produce specific chemical compounds to establish and support symbioses. For instance, the ectomycorrhizal basidiomycete fungus Laccaria bicolor produces sesquiterpenes to promote lateral root formation in plant hosts (Ditengou et al. 2015). Similarly, many plant pathogens are known to activate the expression of a plethora of SM gene clusters at specific stages during host colonisation to promote virulence (Collemare, O'Connell and Lebrun 2019), and thus further insights into chromatin dynamics and its roles in transcriptional regulation during host interaction are crucial.
Unfortunately, studying histone and DNA modifications during host interaction suffers from technical challenges. In early stages of host colonisation, when SM gene clusters are typically expressed, fungal biomass is low compared with abundant host biomass. Unsurprisingly, chromatin dynamics during host interactions thus far focused on few specific SM gene clusters using targeted approaches such as ChIP-PCR assays (e.g. Chujo and Scott 2014), while genome-wide analyses using ChIP-seq only considered differences between in vitro conditions (e.g. Connolly et al. 2013; Wiemann et al. 2013). Similarly, genome-wide analyses of the 3D arrangement (Hi-C) of chromatin during host interaction is significantly hampered by low fungal biomass, explaining why, for instance, in planta contact maps were not studied in E. festucae (Winter et al. 2018), or any other fungal plant symbiont. Consequently, methods to enrich for fungal biomass in these complex samples are needed in the future to be able to study chromatin and its relationship with SM gene cluster regulation. For example, fungal histone proteins could be tagged (e.g. by biotin or V5 tags; Kolodziej et al. 2009) and enriched by immunoprecipitation, prior to traditional ChIP experiments with antibodies aimed at specific histone modifications. Furthermore, enrichment methods could exploit differences in physical and/or biochemical properties between fungal and host nuclei or cells prior to ChIP experiments. However, to our knowledge, no enrichment method has yet been successfully exploited to study (genome-wide) fungal chromatin during host colonisation. Unravelling how changes in chromatin, both in terms of modifications and spatial organisation, translate to changes in expression during host interaction therefore remains a significant challenge for the future. In contrast, studies of fungal interactions with other microorganisms are typically more tractable, and an increasing number of co-culture experiments is expected to be published in the coming years. These studies will provide key knowledge about the dynamics of the chromatin modification in response to diverse organisms and might reveal mechanisms awaiting to be exploited to produce fungal natural compounds with yet unknown activities.
CONCLUDING REMARKS
The discovery of chromatin as a central regulator of fungal SM production significantly impacted on our understanding of the complex transcriptional regulation of biosynthetic gene clusters. The rapid progress in identifying the genetic determinants underlying this chromatin-based regulation provided new tools to activate silent gene clusters. Thus far, most efforts have focused on two well-known chromatin modifications, histone methylation and histone acetylation. However, most SM gene clusters remain untouched by any genetic or chemical modification of chromatin, and thus we still lack a comprehensive understanding on how chromatin modifications regulate SM gene clusters. Based on examples from other eukaryotes and the availability of novel technologies that allow to comprehensively study the composition and organisation of chromatin, we are now entering a new decade during which several outstanding questions can be answered. What is the contribution of other chromatin modifications to the regulation of SM gene clusters? How do chromatin modifications interact with each other to provide a tight and subtle regulation of SM gene clusters? How are chromatin modifications orchestrated in the different genomic sub-compartments, and what is the chromatin dynamic during interactions with other organisms? Answering these questions is important to advance our basic knowledge on chromatin-based regulation, but it will also provide the needed tools to successfully activate the wide diversity of fungal SM gene clusters. Exploitation of the fungal kingdom to the discovery and development of novel bioactive compounds would then enter a completely new era.
Footnotes
The nomenclature of histone PTMs systematically describes: the modified histone protein (e.g. H3), followed by the single letter code and position in the protein of the modified amino acid (e.g. K9), and the description of the modification (e.g. ac for acetylation, me2 for di-methylation, etc.; see Table 1) (Turner 2005).
Contributor Information
Jérôme Collemare, Westerdijk Fungal Biodiversity Institute, Uppsalalaan 8, 3584 CT Utrecht, the Netherlands.
Michael F Seidl, Laboratory of Phytopathology, Wageningen University & Research, Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands.
FUNDING
This work was supported by the Research Council Earth and Life Sciences (ALW) of the Netherlands Organization for Scientific Research (NWO) [863.15.005 to M.F.S.].
Conflict of interest. None declared.
REFERENCES
- Ahn S-H, Cheung WL, Hsu J-Yet al.. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell. 2005a;120:25–36. [DOI] [PubMed] [Google Scholar]
- Ahn S-H, Henderson KA, Keeney Set al.. H2B (Ser10) phosphorylation is induced during apoptosis and meiosis in S. cerevisiae. Cell Cycle. 2005b;4:780–3. [DOI] [PubMed] [Google Scholar]
- Albright JC, Henke MT, Soukup AAet al.. Large-scale metabolomics reveals a complex response of Aspergillus nidulans to epigenetic perturbation. ACS Chem Biol. 2015;10:1535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly AH, Debbab A, Proksch P. Fifty years of drug discovery from fungi. Fungal Diversity. 2011;50:3. [Google Scholar]
- Andrews FH, Shinsky SA, Shanle EKet al.. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armeev GA, Gribkova AK, Pospelova Iet al.. Linking chromatin composition and structural dynamics at the nucleosome level. Curr Opin Struct Biol. 2018;56:46–55. [DOI] [PubMed] [Google Scholar]
- Asai T, Chung Y-M, Sakurai Het al.. Tenuipyrone, a novel skeletal polyketide from the entomopathogenic fungus, Isaria tenuipes, cultivated in the presence of epigenetic modifiers. Org Lett. 2012;14:513–5. [DOI] [PubMed] [Google Scholar]
- Asai T, Yamamoto T, Oshima Y. Histone deacetylase inhibitor induced the production of three novel prenylated tryptophan analogs in the entomopathogenic fungus, Torrubiella luteorostrata. Tetrahedron Lett. 2011;52:7042–5. [Google Scholar]
- Atlasi Y, Stunnenberg HG. The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. 2017;18:643–58. [DOI] [PubMed] [Google Scholar]
- Balba H. Review of strobilurin fungicide chemicals. J Environ Sci Health B. 2007;42:441–51. [DOI] [PubMed] [Google Scholar]
- Ballard TD, Wolff J, Griffin JBet al.. Biotinidase catalyzes debiotinylation of histones. Eur J Nutr. 2002;41:78–84. [DOI] [PubMed] [Google Scholar]
- Baral B, Akhgari A, Metsä-Ketelä M. Activation of microbial secondary metabolic pathways: Avenues and challenges. Synth Syst Biotechnol. 2018;3:163–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basenko EY, Sasaki T, Ji Let al.. Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. Proc Natl Acad Sci USA. 2015;112:E6339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram O, Braus GH. Coordination of secondary metabolism and development in fungi: The velvet family of regulatory proteins. FEMS Microbiol Rev. 2012;36:1–24. [DOI] [PubMed] [Google Scholar]
- Bayram O, Krappmann S, Ni Met al.. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 2008;320:1504–6. [DOI] [PubMed] [Google Scholar]
- Bergmann S, Funk AN, Scherlach Ket al.. Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster. Appl Environ Microbiol. 2010;76:8143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar D, Ehrlich KC, Cleveland TE. Molecular genetic analysis and regulation of aflatoxin biosynthesis. Appl Microbiol Biotechnol. 2003;61:83–93. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–16. [DOI] [PubMed] [Google Scholar]
- Bok JW, Chiang Y-M, Szewczyk Eet al.. Chromatin-level regulation of biosynthetic gene clusters. Nat Chem Biol. 2009;5:462–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok JW, LaeA Keller NP., a regulator of secondary metabolism in Aspergillus spp. Eukaryotic Cell. 2004;3:527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok JW, Noordermeer D, Kale SPet al.. Secondary metabolic gene cluster silencing in Aspergillus nidulans. Mol Microbiol. 2006;61:1636–45. [DOI] [PubMed] [Google Scholar]
- Brakhage AA. Regulation of fungal secondary metabolism. Nat Rev Microbiol. 2013;11:21–32. [DOI] [PubMed] [Google Scholar]
- Brakhage AA, Schroeckh V. Fungal secondary metabolites - strategies to activate silent gene clusters. Fungal Genet Biol. 2011;48:15–22. [DOI] [PubMed] [Google Scholar]
- Brehove M, Wang T, North Jet al.. Histone core phosphorylation regulates DNA accessibility. J Biol Chem. 2015;290:22612–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DW, Yu JH, Kelkar HSet al.. Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. Proc Natl Acad Sci USA. 1996;93:1418–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo J, López-Rodas G, Franco L. Histone post-translational modifications and nucleosome organisation in transcriptional regulation: Some open questions. Adv Exp Med Biol. 2017;966:65–92. [DOI] [PubMed] [Google Scholar]
- Chang P-K. The Aspergillus parasiticus protein AFLJ interacts with the aflatoxin pathway-specific regulator AFLR. Mol Genet Genomics. 2003;268:711–9. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sprung R, Tang Yet al.. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chettri P, Ehrlich KC, Cary JWet al.. Dothistromin genes at multiple separate loci are regulated by AflR. Fungal Genet Biol. 2013;51:12–20. [DOI] [PubMed] [Google Scholar]
- Chiang Y-M, Szewczyk E, Davidson ADet al.. Characterization of the Aspergillus nidulans monodictyphenone gene cluster. Appl Environ Microbiol. 2010;76:2067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chujo T, Scott B.. Histone H3K9 and H3K27 methylation regulates fungal alkaloid biosynthesis in a fungal endophyte-plant symbiosis. Mol Microbiol. 2014;92:413–34. [DOI] [PubMed] [Google Scholar]
- Chung Y-M, El-Shazly M, Chuang D-Wet al.. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, induces the production of anti-inflammatory cyclodepsipeptides from Beauveria felina. J Nat Prod. 2013;76:1260–6. [DOI] [PubMed] [Google Scholar]
- Clancy KW, Russell A-M, Subramanian Vet al.. Citrullination/methylation crosstalk on histone H3 regulates ER-target gene transcription. ACS Chem Biol. 2017;12:1691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AS, Lowell JE, Jacobson SJet al.. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol. 1999;19:2515–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger KD, Bok JW, Ye Ret al.. A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat Chem Biol. 2017;13:895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collemare J, Billard A, Böhnert HUet al.. Biosynthesis of secondary metabolites in the rice blast fungus Magnaporthe grisea: The role of hybrid PKS-NRPS in pathogenicity. Mycol Res. 2008a;112:207–15. [DOI] [PubMed] [Google Scholar]
- Collemare J, Lebrun M-H. Fungal secondary metabolites: Ancient toxins and novel effectors in plant–microbe interactions. Oxford, UK: John Wiley & Sons, Ltd, 2011:377–400. [Google Scholar]
- Collemare J, Pianfetti M, Houlle A-Eet al.. Magnaport h e grisea avirulence gene ACE1 belongs to an infection-specific gene cluster involved in secondary metabolism. New Phytol. 2008b;179:196–208. [DOI] [PubMed] [Google Scholar]
- Collemare J, O'Connell R, Lebrun MH. Nonproteinaceous effectors: the terra incognita of plant-fungal interactions. New Phytol. 2019;223:590–6.. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Connolly LR, Smith KM, Freitag M. The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet. 2013;9:e1003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CP, Osmani AH, Wu LPet al.. Mitotic histone H3 phosphorylation by the NIMA kinase in Aspergillus nidulans. Cell. 2000;102:293–302. [DOI] [PubMed] [Google Scholar]
- Deem AK, Li X, Tyler JK. Epigenetic regulation of genomic integrity. Chromosoma. 2012;121:131–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demain AL, Fang A. The natural functions of secondary metabolites. Adv Biochem Eng Biotechnol. 2000;69:1–39. [DOI] [PubMed] [Google Scholar]
- Demers DH, Knestrick MA, Fleeman Ret al.. Exploitation of mangrove endophytic fungi for infectious disease drug discovery. Mar Drugs. 2018;16:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditengou FA, Müller A, Rosenkranz Met al.. Volatile signalling by sesquiterpenes from ectomycorrhizal fungi reprogrammes root architecture. Nat Commun. 2015;23;6:6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su Xet al.. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman HC, Casadevall A. Synthesis and assembly of fungal melanin. Appl Microbiol Biotechnol. 2012;93:931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasoyin OE, Wang B, Qiu Met al.. Carbon catabolite repression gene creA regulates morphology, aflatoxin biosynthesis and virulence in Aspergillus flavus. Fungal Genet Biol. 2018;115:41–51. [DOI] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang Xet al.. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch KM, Gillaspy AF, Gipson Met al.. Chemical induction of silent biosynthetic pathway transcription in Aspergillus niger. J Ind Microbiol Biotechnol. 2009;36:1199–213. [DOI] [PubMed] [Google Scholar]
- Fischer J, Müller SY, Netzker Tet al.. Chromatin mapping identifies BasR, a key regulator of bacteria-triggered production of fungal secondary metabolites. Elife. 2018;7:1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag M, Hickey PC, Khlafallah TKet al.. HP1 is essential for DNA methylation in neurospora. Mol Cell. 2004;13:427–34. [DOI] [PubMed] [Google Scholar]
- Freitag M, Williams RL, Kothe GOet al.. A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci USA. 2002;99:8802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag M. Histone methylation by SET domain proteins in fungi. Annu Rev Microbiol. 2017;71:413–39. [DOI] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine Het al.. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek A, Strauss J. The chromatin code of fungal secondary metabolite gene clusters. Appl Microbiol Biotechnol. 2012;95:1389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek-Matthews A, Berger H, Sasaki Tet al.. KdmB, a jumonji histone H3 demethylase, regulates genome-wide H3K4 trimethylation and is required for normal induction of secondary metabolism in Aspergillus nidulans. PLoS Genet. 2016;12:e1006222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek-Matthews A, Noble LM, Gruber Cet al.. KdmA, a histone H3 demethylase with bipartite function, differentially regulates primary and secondary metabolism in Aspergillus nidulans. Mol Microbiol. 2015;96:839–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Cuomo Cet al.. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–15. [DOI] [PubMed] [Google Scholar]
- Galazka JM, Freitag M. Variability of chromosome structure in pathogenic fungi-of “ends and odds.” Curr Opin Microbiol. 2014;20C:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galazka JM, Klocko AD, Uesake Met al.. Neurospora chromosomes are organized by blocs of importin alpha-dependent heterochromatin that are largely independent of H3K9me3. Genome Res. 2016:gr.203182.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner DM, Jarvis RS, Howlett BJ. The ABC transporter gene in the sirodesmin biosynthetic gene cluster of Leptosphaeria maculans is not essential for sirodesmin production but facilitates self-protection. Fungal Genet Biol. 2005;42: 257–63. [DOI] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Plevani Pet al.. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem. 2005;280:9879–86. [DOI] [PubMed] [Google Scholar]
- Ginsburg DS, Anlembom TE, Wang Jet al.. NuA4 links methylation of histone H3 lysines 4 and 36 to acetylation of histones H4 and H3. J Biol Chem. 2014;289:32656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Menéndez V, Pérez-Bonilla M, Pérez-Victoria Iet al.. Multicomponent analysis of the differential induction of secondary metabolite profiles in fungal endophytes. Molecules. 2016;21:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudarzi A, Zhang D, Huang Het al.. Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol Cell. 2016;62:169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths S, Mesarich CH, Overdijk EJRet al.. Down-regulation of cladofulvin biosynthesis is required for biotrophic growth of Cladosporium fulvum on tomato. Mol Plant Pathol. 2018;19:369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths S, Mesarich CH, Saccomanno Bet al.. Elucidation of cladofulvin biosynthesis reveals a cytochrome P450 monooxygenase required for anthraquinone dimerization. Proc Natl Acad Sci USA. 2016;113:6851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths S, Saccomanno B, de Wit PJet al.. Regulation of secondary metabolite production in the fungal tomato pathogen Cladosporium fulvum. Fungal Genet Biol. 2015;84:52–61. [DOI] [PubMed] [Google Scholar]
- Guzman-Chavez F, Salo O, Samol Met al.. Deregulation of secondary metabolism in a histone deacetylase mutant of Penicillium chrysogenum. Microbiologyopen. 2018;8:e00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasim S, Tati S, Madayiputhiya Net al.. Histone biotinylation in Candida albicans. FEMS Yeast Res. 2013;13:529–39. [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MAet al.. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–60. [DOI] [PubMed] [Google Scholar]
- Henke MT, Soukup AA, Goering AWet al.. New aspercryptins, lipopeptide natural products, revealed by HDAC inhibition in Aspergillus nidulans. ACS Chem Biol. 2016;11:2117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrikson JC, Hoover AR, Joyner PMet al.. A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Org Biomol Chem. 2009;7:435–8. [DOI] [PubMed] [Google Scholar]
- Hottiger MO. Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu Rev Biochem. 2015;84:227–63. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Sun ZW, Li Xet al.. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102: 279–91. [DOI] [PubMed] [Google Scholar]
- Hunt CR, Ramnarain D, Horikoshi Net al.. Histone modifications and DNA double-strand break repair after exposure to ionizing radiations. Radiat Res. 2013;179:383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Pál C, Lercher MJ. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5:299–310. [DOI] [PubMed] [Google Scholar]
- Inglis DO, Binkley J, Skrzypek MSet al.. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013;13:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson K, Rountree MR, Lewis ZAet al.. Regional control of histone H3 lysine 27 methylation in Neurospora. Proc Natl Acad Sci USA. 2013;110:6027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janevska S, Baumann L, Sieber CMKet al.. Elucidation of the two H3K36me3 histone methyltransferases Set2 and Ash1 in Fusarium fujikuroi unravels their different chromosomal targets and a major impact of Ash1 on genome stability. Genetics. 2018;208:153–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J, Choi J, Lee G-Wet al.. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci Rep. 2015;5:8567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Zhou X, Taghizadeh Ket al.. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc Natl Acad Sci USA. 2007;104:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L, Baltz RH. Natural product discovery: Past, present, and future. J Ind Microbiol Biotechnol. 2016;43:155–76. [DOI] [PubMed] [Google Scholar]
- Kawauchi M, Nishiura M, Iwashita K. Fungus-specific sirtuin HstD coordinates secondary metabolism and development through control of LaeA. Eukaryotic Cell. 2013;12: 1087–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller N, Hohn T. Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol. 1997;21:17–29. [PubMed] [Google Scholar]
- Keller NP, Turner G, Bennett JW. Fungal secondary metabolism - From biochemistry to genomics. Nat Rev Microbiol. 2005;3:937–47. [DOI] [PubMed] [Google Scholar]
- Keller NP. Fungal secondary metabolism: Regulation, function and drug discovery. Nat Rev Microbiol. 2018;195:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocko AD, Ormsby T, Galazka JMet al.. Normal chromosome conformation depends on subtelomeric facultative heterochromatin in Neurospora crassa. Proc Natl Acad Sci USA. 2016;113:15048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej KE, Pourfarzad F, de Boer Eet al.. Optimal use of tandem biotin and V5 tags in ChIP assays. BMC Mol Biol. 2009;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombrink A, Tayyrov A, Essig Aet al.. Induction of antibacterial proteins and peptides in the coprophilous mushroom Coprinopsis cinerea in response to bacteria. ISME J. 2019;13:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, van Diepeningen AD, Van der Lee TAJet al.. The Fusarium graminearum histone acetyltransferases are important for morphogenesis, DON biosynthesis, and pathogenicity. Front Microbiol. 2018;9:654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzminova E, Selker EU. dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001;20:4309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami Set al.. COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem. 2002;277:10753–5. [DOI] [PubMed] [Google Scholar]
- Kumar S, Chinnusamy V, Mohapatra T. Epigenetics of modified DNA bases: 5-methylcytosine and beyond. Front Genet. 2018;9:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H, Sun R, Fan Ket al.. The Aspergillus flavus histone acetyltransferase AflGcnE regulates morphogenesis, aflatoxin biosynthesis, and pathogenicity. Front Microbiol. 2016;7:1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Oh J-H, Shwab EKet al.. HdaA, a class 2 histone deacetylase of Aspergillus fumigatus, affects germination and secondary metabolite production. Fungal Genet Biol. 2009;46:782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL. Histone acetyltransferase complexes: One size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–95. [DOI] [PubMed] [Google Scholar]
- Lewis ZA, Adhvaryu KK, Honda Set al.. DNA methylation and normal chromosome behavior in Neurospora depend on five components of a histone methyltransferase complex, DCDC. PLoS Genet. 2010;6:e1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis ZA, Honda S, Khlafallah TKet al.. Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res. 2009;19:427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang C, Liu Wet al.. The HDF1 histone deacetylase gene is important for conidiation, sexual reproduction, and pathogenesis in Fusarium graminearum. Mol Plant Microbe Interact. 2011;24:487–96. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams Let al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J-Q, Zhao X-X, Zhi Q-Qet al.. Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet Biol. 2013;56:78–86. [DOI] [PubMed] [Google Scholar]
- Liu B, Lin Y, Darwanto Aet al.. Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J Biol Chem. 2009;284:32288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S-Y, Lin J-Q, Wu H-Let al.. Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. Pellegrini M (ed.). PLoS One. 2012;7:e30349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheleidt J, Mattern DJ, Fischer Jet al.. Regulation and role of fungal secondary metabolites. Annu Rev Genet. 2016;50:371–92. [DOI] [PubMed] [Google Scholar]
- Maeda K, Izawa M, Nakajima Yet al.. Increased metabolite production by deletion of an HDA1-type histone deacetylase in the phytopathogenic fungi, Magnaporthe oryzae (Pyricularia oryzae) and Fusarium asiaticum. Lett Appl Microbiol. 2017;65:446–52. [DOI] [PubMed] [Google Scholar]
- Malagnac F, Wendel B, Goyon Cet al.. A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell. 1997;91:281–90. [DOI] [PubMed] [Google Scholar]
- Martin BJE, McBurney KL, Maltby VEet al.. Histone H3K4 and H3K36 methylation independently recruit the NuA3 histone acetyltransferase in Saccharomyces cerevisiae. Genetics. 2017;205:1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DGE, Grimes DE, Baetz Ket al.. Methylation of histone H3 mediates the association of the NuA3 histone acetyltransferase with chromatin. Mol Cell Biol. 2006;26:3018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary KL, Slot JC, Rokas A. Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc Natl Acad Sci USA. 2013;110:11481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielse CB, Pfannmüller A, Macios Met al.. The interplay between the GATA transcription factors AreA, the global nitrogen regulator and AreB in Fusarium fujikuroi. Mol Microbiol. 2014;91:472–93. [DOI] [PubMed] [Google Scholar]
- Mihlan M, Homann V, Liu T-WDet al.. AREA directly mediates nitrogen regulation of gibberellin biosynthesis in Gibberella fujikuroi, but its activity is not affected by NMR. Mol Microbiol. 2003;47:975–91. [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover Jet al.. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA. 2001;98:12902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra PK, Baum M, Carbon J. DNA methylation regulates phenotype-dependent transcriptional activity in Candida albicans. Proc Natl Acad Sci USA. 2011;108:11965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi T, Fudenberg G, Mehta Set al.. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature. 2014;516:432–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondo SJ, Dannebaum RO, Kuo RCet al.. Widespread adenine N6-methylation of active genes in fungi. Nat Genet. 2017;49: 964–8. [DOI] [PubMed] [Google Scholar]
- Monneau YR, Soufari H, Nelson CJet al.. Structure and activity of the peptidyl-prolyl isomerase domain from the histone chaperone Fpr4 toward histone H3 proline isomerization. J Biol Chem. 2013;288:25826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanini B, Chen P-Y, Morselli Met al.. Non-exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 2014;15:411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, Nair Aet al.. Dynamic lysine methylation on histone H3 defines the regulatory phase of gene transcription. Mol Cell. 2005;18:723–34. [DOI] [PubMed] [Google Scholar]
- Nathan D, Ingvarsdottir K, Sterner DEet al.. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006;20:966–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurohr G, Naegeli A, Titos Iet al.. A midzone-based ruler adjusts chromosome compaction to anaphase spindle length. Science. 2011;332:465–8. [DOI] [PubMed] [Google Scholar]
- Niehaus E-M, Kleigrewe K, Wiemann Pet al.. Genetic manipulation of the Fusarium fujikuroi fusarin gene cluster yields insight into the complex regulation and fusarin biosynthetic pathway. Chem Biol. 2013;20:1055–66. [DOI] [PubMed] [Google Scholar]
- Niehaus E-M, Studt L, Bargen von KWet al.. Sound of silence: The beauvericin cluster in Fusarium fujikuroi is controlled by cluster-specific and global regulators mediated by H3K27 modification. Environ Microbiol. 2016;18:4282–302. [DOI] [PubMed] [Google Scholar]
- Nützmann H-W, Fischer J, Scherlach Ket al.. Distinct amino acids of histone H3 control secondary metabolism in Aspergillus nidulans. Appl Environ Microbiol. 2013;79:6102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nützmann H-W, Reyes-Dominguez Y, Scherlach Ket al.. Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires Saga/Ada-mediated histone acetylation. Proc Natl Acad Sci USA. 2011;108:14282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuni K, Levy-Myers R, Warren Jet al.. N-terminal sumoylation of centromeric histone H3 variant Cse4 regulates its proteolysis to prevent mislocalization to non-centromeric chromatin. G3. 2018;8:1215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JM, Bok JW, Lee Set al.. Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus. PeerJ. 2013;1:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JM, Keller NP. Secondary metabolism in fungi: Does chromosomal location matter? Curr Opin Microbiol. 2010;13:431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RM, Fedorova ND, Bok JWet al.. Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog. 2007;3:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannenstiel BT, Keller NP. On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol Adv. 2019, pii: S0734-9750(19)30012-6. DOI: 10.1016/j.biotechadv.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidroni A, Faber B, Brosch Get al.. A Class 1 Histone deacetylase as major regulator of secondary metabolite production in Aspergillus nidulans. Front Microbiol. 2018;9:2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadri M, Nalli Y, Jain SKet al.. An insight into the secondary metabolism of Muscodor yucatanensis: Small-molecule epigenetic modifiers induce expression of secondary metabolism-related genes and production of new metabolites in the endophyte. Microb Ecol. 2017;73:954–65. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl Set al.. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NCet al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Dominguez Y, Bok JW, Berger Het al.. Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol Microbiol. 2010;76:1376–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robzyk K, Recht J, Osley MA. Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000;287:501–4. [DOI] [PubMed] [Google Scholar]
- Rokas A, Wisecaver JH, Lind AL. The birth, evolution and death of metabolic gene clusters in fungi. Nat Rev Microbiol. 2018;16:731–44. [DOI] [PubMed] [Google Scholar]
- Rose NR, Klose RJ. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim Biophys Acta. 2014;1839:1362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839:627–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu Xet al.. Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell. 2017;67:837–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze LV, Arthur AE, Hong S-Yet al.. The initiation and pattern of spread of histone H4 acetylation parallel the order of transcriptional activation of genes in the aflatoxin cluster. Mol Microbiol. 2007;66:713–26. [DOI] [PubMed] [Google Scholar]
- Sabari BR, Tang Z, Huang Het al.. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni D, Castiglione F, Paci P. Identifying correlations between chromosomal proximity of genes and distance of their products in protein-protein interaction networks of yeast. PLoS One. 2013;8:e57707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJet al.. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–11. [DOI] [PubMed] [Google Scholar]
- Schotanus K, Soyer JL, Connolly LRet al.. Histone modifications rather than the novel regional centromeres of Zymoseptoria tritici distinguish core and accessory chromosomes. Epigenetics Chromatin. 2015;8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Cook DE, Thomma BPHJ. Chromatin biology impacts adaptive evolution of filamentous plant pathogens. PLoS Pathog. 2016;12:e1005920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Thomma BPHJ. Sex or no sex: Evolutionary adaptation occurs regardless. Bioessays. 2014;36:335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl MF, Thomma BPHJ. Transposable elements direct the coevolution between plants and microbes. Trends Genet. 2017;33:842–51. [DOI] [PubMed] [Google Scholar]
- Seidl MF. Adenine N6-methylation in diverse fungi. Nat Genet. 2017;49:823–4. [DOI] [PubMed] [Google Scholar]
- Selker EU, Tountas NA, Cross SHet al.. The methylated component of the Neurospora crassa genome. Nature. 2003;422:893–7. [DOI] [PubMed] [Google Scholar]
- Shaaban M, Palmer JM, El-Naggar WAet al.. Involvement of transposon-like elements in penicillin gene cluster regulation. Fungal Genet Biol. 2010;47:423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam MK, Arfuso F, Arumugam Set al.. Role of novel histone modifications in cancer. Oncotarget. 2018;9:11414–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–69. [DOI] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch Det al.. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwab EK, Bok JW, Tribus Met al.. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryotic Cell. 2007;6:1656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siless GE, Gallardo GL, Rodriguez MAet al.. Metabolites from the dark septate endophyte Drechslera sp. evaluation by LC/MS and principal component analysis of culture extracts with histone deacetylase inhibitors. Chem Biodivers. 2018;15:e1800133. [DOI] [PubMed] [Google Scholar]
- Singh MP, Wijeratne SSK, Zempleni J. Biotinylation of lysine 16 in histone H4 contributes toward nucleosome condensation. Arch Biochem Biophys. 2013;529:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skellam E. Strategies for engineering natural product biosynthesis in fungi. Trends Biotechnol. 2019;37:416–27. [DOI] [PubMed] [Google Scholar]
- Slot JC. Fungal gene cluster diversity and evolution. Adv Genet. 2017;100:141–78. [DOI] [PubMed] [Google Scholar]
- Soukup AA, Chiang Y-M, Bok JWet al.. Overexpression of the Aspergillus nidulans histone 4 acetyltransferase EsaA increases activation of secondary metabolite production. Mol Microbiol. 2012;86:314–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soyer JL, Rouxel T, Fudal I. Chromatin-based control of effector gene expression in plant-associated fungi. Curr Opin Plant Biol. 2015;26:51–6. [DOI] [PubMed] [Google Scholar]
- Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells. Effects of cell proliferation. Eur J Biochem. 2001;268:5424–9. [DOI] [PubMed] [Google Scholar]
- Stöckli M, Morinaka BI, Lackner Get al.. Bacteria-induced production of the antibacterial sesquiterpene lagopodin B in Coprinopsis cinerea. Mol Microbiol. 2019. doi: 10.1111/mmi.14277. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Grant PA, Briggs SDet al.. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss J, Reyes-Dominguez Y. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet Biol. 2011;48:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt L, Rösler SM, Burkhardt Iet al.. Knock-down of the methyltransferase Kmt6 relieves H3K27me3 and results in induction of cryptic and otherwise silent secondary metabolite gene clusters in Fusarium fujikuroi. Environ Microbiol. 2016;18:4037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt L, Schmidt FJ, Jahn Let al.. Two histone deacetylases, FfHda1 and FfHda2, are important for Fusarium fujikuroi secondary metabolism and virulence. Appl Environ Microbiol. 2013;79:7719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk E, Chiang Y-M, Oakley CEet al.. Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl Environ Microbiol. 2008;74:7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414:277–83. [DOI] [PubMed] [Google Scholar]
- Tamaru H, Zhang X, McMillen Det al.. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat Genet. 2003;34:75–9. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee Set al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Then Bergh K, Brakhage AA. Regulation of the Aspergillus nidulans penicillin biosynthesis gene acvA (pcbAB) by amino acids: Implication for involvement of transcription factor PACC. Appl Environ Microbiol. 1998;64:843–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucl Acids Res. 2017;45:D158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomma BPHJ, Seidl MF, Shi-Kunne Xet al.. Mind the gap; Seven reasons to close fragmented genome assemblies. Fungal Genet Biol. 2016;90:24–30. [DOI] [PubMed] [Google Scholar]
- Tilburn J, Sarkar S, Widdick DAet al.. The Aspergillus PacC zinc finger transcription factor mediates regulation of both acid- and alkaline-expressed genes by ambient pH. EMBO J. 1995;14:779–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triastuti A, Vansteelandt M, Barakat Fet al.. How histone deacetylase inhibitors alter the secondary metabolites of Botryosphaeria mamane, an endophytic fungus isolated from Bixa orellana, L. Chem Biodivers. 2019;; 16::e1800485. DOI: 10.1002/cbdv.201800485 [DOI] [PubMed] [Google Scholar]
- Trujillo KM, Tyler RK, Ye Cet al.. A genetic and molecular toolbox for analyzing histone ubiquitylation and sumoylation in yeast. Methods. 2011;54:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudzynski B, Homann V, Feng Bet al.. Isolation, characterization and disruption of the areA nitrogen regulatory gene of Gibberella fujikuroi. Mol Gen Genet. 1999;261:106–14. [DOI] [PubMed] [Google Scholar]
- Tudzynski B. Nitrogen regulation of fungal secondary metabolism in fungi. Front Microbiol. 2014;5:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12:110–2. [DOI] [PubMed] [Google Scholar]
- Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9:2–6. [DOI] [PubMed] [Google Scholar]
- Verheecke C, Liboz T, Mathieu F. Microbial degradation of aflatoxin B1: Current status and future advances. Int J Food Microbiol. 2016;237:1–9. [DOI] [PubMed] [Google Scholar]
- Vervoort HC, Drašković M, Crews P. Histone deacetylase inhibitors as a tool to up-regulate new fungal biosynthetic products: Isolation of EGM-556, a cyclodepsipeptide, from Microascus sp. Org Lett. 2011;13:410–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapenaar H, Dekker FJ. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Liu X, Chen Jet al.. Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017;27:898–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann P, Sieber CMK, Bargen von KWet al.. Deciphering the cryptic genome: Genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog. 2013;9:e1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann P, Willmann A, Straeten Met al.. Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: Genes, their function and regulation. Mol Microbiol. 2009;72:931–46. [DOI] [PubMed] [Google Scholar]
- Williams RB, Henrikson JC, Hoover ARet al.. Epigenetic remodeling of the fungal secondary metabolome. Org Biomol Chem. 2008;6:1895–7. [DOI] [PubMed] [Google Scholar]
- Winter DJ, Ganley ARD, Young CAet al.. Repeat elements organise 3D genome structure and mediate transcription in the filamentous fungus Epichloë festucae. PLoS Genet. 2018;14:e1007467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloshuk CP, Foutz KR, Brewer JFet al.. Molecular characterization of aflR, a regulatory locus for aflatoxin biosynthesis. Appl Environ Microbiol. 1994;60:2408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Dai J, Dai Let al.. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. 2012;11: 100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakasai AA, Davison J, Wasil Zet al.. Nongenetic reprogramming of a fungal highly reducing polyketide synthase. J Am Chem Soc. 2011;133:10990–8. [DOI] [PubMed] [Google Scholar]
- Yang K, Liang L, Ran Fet al.. The DmtA methyltransferase contributes to Aspergillus flavus conidiation, sclerotial production, aflatoxin biosynthesis and virulence. Sci Rep. 2016;6:23259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Zhuang Z, Zhang Fet al.. Inhibition of aflatoxin metabolism and growth of Aspergillus flavus in liquid culture by a DNA methylation inhibitor. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2015;32:554–63. [DOI] [PubMed] [Google Scholar]
- Yu JH, Butchko RA, Fernandes Met al.. Conservation of structure and function of the aflatoxin regulatory gene aflR from Aspergillus nidulans and A. flavus. Curr t. 1996;29:549–55. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva Pet al.. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–9. [DOI] [PubMed] [Google Scholar]
- Zemach A, Zilberman D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol. 2010;20:R780–5. [DOI] [PubMed] [Google Scholar]
- Zhang K, Chen Y, Zhang Zet al.. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J Proteome Res. 2009;8:900–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang Z, Khan SIet al.. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 2003;12:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutz C, Gacek A, Sulyok Met al.. Small chemical chromatin effectors alter secondary metabolite production in Aspergillus clavatus. Toxins. 2013;5:1723–41. [DOI] [PMC free article] [PubMed] [Google Scholar]