

Abstract
Both obesity and gestational diabetes mellitus (GDM) lead to poor maternal and fetal outcomes, including pregnancy complications, fetal growth issues, stillbirth, and developmental programming of adult-onset disease in the offspring. Increased placental oxidative/nitrative stress and reduced placental (trophoblast) mitochondrial respiration occur in association with the altered maternal metabolic milieu of obesity and GDM. The effect is particularly evident when the fetus is male, suggesting a sexually dimorphic influence on the placenta. In addition, obesity and GDM are associated with inflexibility in trophoblast, limiting the ability to switch between usage of glucose, fatty acids, and glutamine as substrates for oxidative phosphorylation, again in a sexually dimorphic manner. Here we review mechanisms underlying placental mitochondrial dysfunction: its relationship to maternal and fetal outcomes and the influence of fetal sex. Prevention of placental oxidative stress and mitochondrial dysfunction may improve pregnancy outcomes. We outline pathways to ameliorate deficient mitochondrial respiration, particularly the benefits and pitfalls of mitochondria-targeted antioxidants.
Keywords: placenta, obesity, gestational diabetes, mitochondria, oxidative stress, antioxidants
1. Effect of obesity and GDM in pregnancy
The World Health Organization recognizes obesity as a global epidemic. In 2014, approximately 13% of the world’s population was classified as obese (BMI ≥ 30); this figure is nearly 20% in developed countries with higher rates of obesity in women than men [1]. In the United States, the number of women of reproductive age who are obese is also on the rise: 39.7% of American women were obese in 2018 compared to 33.2% in 2004 and only 16.5% in 1980 [2–4].
Obesity is a significant health concern, linked to an increased risk of hypertension, diabetes, and atherosclerosis and leads to adverse outcomes in pregnancy, for both mother and fetus [5]. Obese women are 2-3 times more likely to develop preeclampsia, three times more at risk for gestational hypertension, 3-4 times more likely to have gestational diabetes mellitus (GDM) [6,7], are at increased risk for stillbirth [8], for excessive weight gain during pregnancy and have higher cesarean section rates [9]. Postpartum, obese women were found to be less likely to be able to lose weight or breastfeed successfully [10], and more likely to develop heart disease and hypertension later in life [11]. Babies born to obese women are twice as likely to be born with macrosomia and to suffer from congenital malformation, and 2.3 times more at risk for childhood obesity [11,12].
Gestational diabetes, a common metabolic disorder in pregnancy, occurs in approximately 7% of pregnancies in the United States and is associated with dysregulation of insulin and glucose. In obese women and women with GDM, insulin resistance increases, causing maternal hyperglycemia and neonatal hypoglycemia, which can instigate seizures and brain damage if glucose levels are low enough [13,14]. GDM increases the risk of pregnancy complications and postnatal consequences for both mother and fetus, including diabetes, obesity, and cardiovascular disease [15–17]. Maternal obesity and GDM developmentally program the fetus for obesity in a propagating cycle of dysfunction [18] (Figure 1). Not all obese women will develop GDM in pregnancy; however, as obesity increases the risk of GDM and other health issues, studying both combined and separate risks as well as early intervention is becoming a critical area of obstetric research.
Figure 1. The consequence of programming in utero.
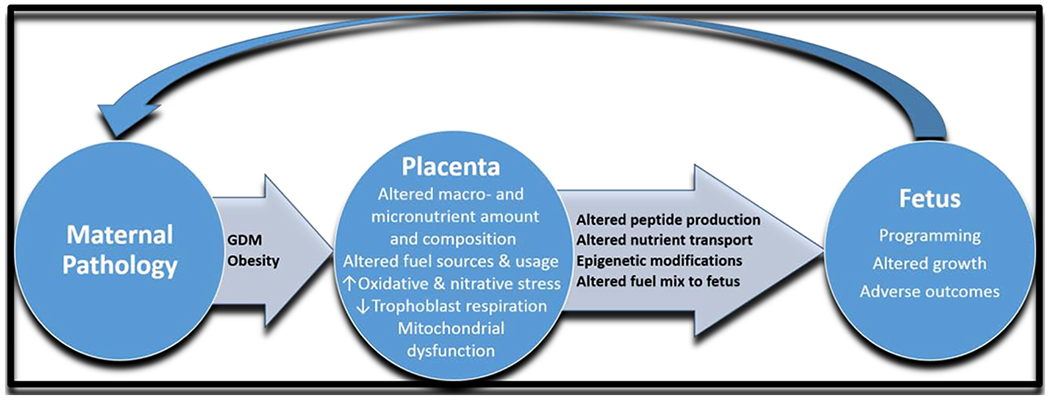
The maternal state affects placental and fetal development (programming). On reaching adulthood, the programmed individual may start this cycle anew, creating a pattern for future disease.
2. Placental structure and function: fuel consumption, storage, and energy production
The placenta is composed of three primary cell types: fetal vascular cells, including pericytes and endothelial cells; mesenchymal cells, including Hofbauer cells and fibroblasts; and trophoblasts [19]. Trophoblasts have several critical roles in the placenta: regulating signaling, apoptosis, steroid synthesis, amino acid transport [20], production of growth factors and hormones for fetoplacental growth, coordination of placental angiogenesis, and serving as a physical and biochemical barrier. The placenta has a high ATP demand to perform its many tasks, the most energetically-demanding among them being fetal nutrient transport and placental protein synthesis [21].
The primary trophoblast cells are cytotrophoblasts (CTBs) which continuously differentiate throughout pregnancy to two different cell types: multinucleated syncytiotrophoblasts (STBs), which cover the entire surface of the placental villous trees [22]; and extravillous trophoblasts (EVTs), which invade the maternal decidua and vasculature [23]. Trophoblast comprises 22-30% of the overall human placental villous volume at term [24–26]. Although there are conflicting reports on whether CTBs or STBs are the most metabolically active, STBs are widely considered to be the greater fuel consumers; however, it is agreed that trophoblasts as a whole constitute the vast majority of metabolic activity in the placenta [27–29].
The placenta itself consumes a significant portion of the nutrients in maternal blood delivered to it via the spiral arteries for rapid growth, angiogenesis, and syncytium formation [30–32]. It has an exceptionally high metabolic rate, comparable to that in brain, liver, kidney, and tumor cells [33,34] and having sixfold greater oxygen and glucose consumption per gram than the fetus. As fetal demand grows over gestation, the placenta still consumes proportionally more glucose and oxygen than it transports to the fetus in order to support its metabolism [33]. In a sheep model of pregnancy, only 55% of the oxygen and 28% of the glucose delivered to the uterus was transferred to the fetus [31]. A recent study in human pregnancy indicates that up to 70% of the glucose delivered to the uteroplacental interface may be effectively transferred to the fetus, with 30% remaining for placental consumption [35].
Although glucose and oxygen have long been considered the chief fuel sources for fetal and placental metabolism [36–38], recent studies indicate the placenta may use fatty acids and amino acids for metabolism more than previously thought. The capability to use and switch between multiple fuel sources is called fuel flexibility [39]. Only about 2-4% of fatty acids (FAs) in maternal circulation (mostly long-chain polyunsaturated fatty acids (LC-PUFA)) are transferred to the fetus at any given time in humans [40]. FAs can be converted for metabolism via β-oxidation, or stored by the placenta as lipid droplets [41]. Coenzyme A (CoA) couples to and metabolically activates FAs. The Acyl-carnitine shuttle system transfers acyl-CoA into the mitochondria using carnitine palmitoyltransferase 1 (CPT1) to join acyl-CoA to carnitine. Acyl-carnitine is transported across the inner mitochondrial membrane in exchange for carnitine by a translocase. Once in the matrix, CPT2 separates the carnitine from acyl-CoA, which can then be β-oxidized to form acetyl-CoA and thus enter the citric acid cycle [42].
Amino acids must undergo deamination before metabolism can proceed [21]. The amino acid with both the highest concentration in maternal and fetal plasma in mammals is glutamine. Glutamine is hydrolyzed to form glutamate, which is then converted by glutamate dehydrogenase to enter the citric acid cycle as α-ketoglutarate [43]. Serine and branched-chain amino acids valine, leucine, and isoleucine are found at higher levels in the placenta than in the maternal or fetal circulation in numerous animal models, indicating their use in placental-specific metabolism [44]. Deamination of these amino acids results in the production of a-keto acids or succinyl CoA, which can then be oxidized to produce ATP [45]. A summary of the major mechanisms for fuel usage by trophoblast mitochondria is illustrated in Figure 2.
Figure 2. Overview of major aerobic metabolic pathways.
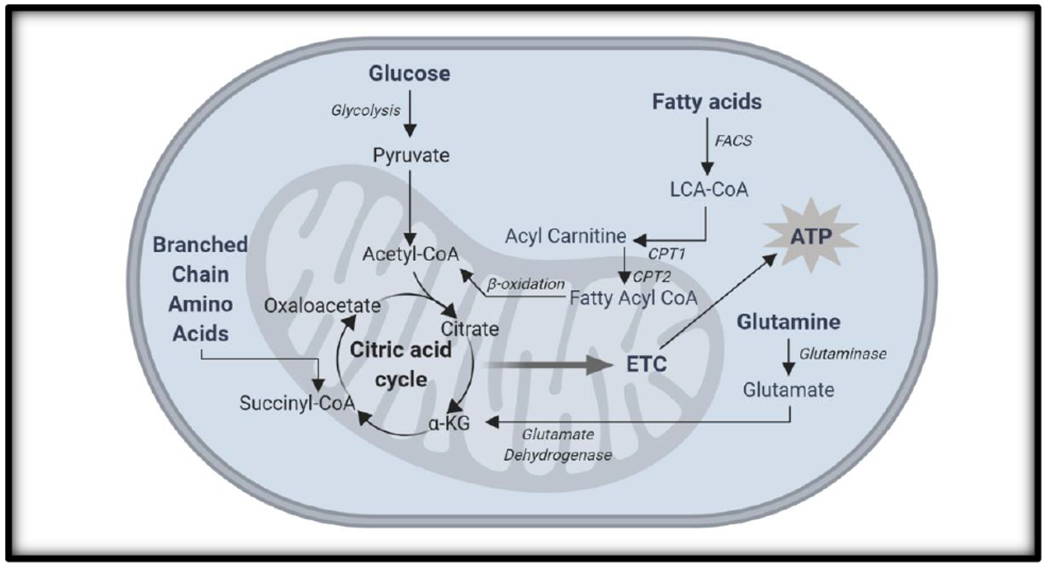
Glucose, fatty acids, and amino acids can all undergo oxidative metabolism in the mitochondria with one primary goal: ATP generation.
The release of human placental growth hormone (hPGH) and human placental lactogen (hPL) by the syncytiotrophoblast early in pregnancy drives an increase in maternal energy storage, particularly in the form of triglycerides and glycogen [46,47]. The mother can break down these stores in fasting states or near term when energy demand is highest, leaving more free glucose for fetoplacental development [48]. The placenta may also store nutrients, including vitamins and fatty acids. If vitamin levels are low, particularly B6, B12, and folate, the placenta will preferentially store them rather than transfer them to the fetus [49]. Storage of fatty acids as lipid droplets is common, primarily in STB, where excess placental fatty acids and insulin create more lipid droplets [50]. In summary, the placenta has several mechanisms to metabolize and store nutrients as needed to support fetal growth during gestation.
3. Mitochondrial function and trophoblast-specific roles
Mitochondria are complex, double-membraned organelles that generate adenosine triphosphate (ATP) using oxidative phosphorylation [51]. Oxidative phosphorylation begins in glycolysis with the conversion of glucose to pyruvate, which is then decarboxylated in the mitochondria to acetyl-coenzyme A (CoA), the first substrate for the citric acid cycle [52]. The resultant 10 NADH and 2 FADH2 generate 30-32 of the 36-38 ATP (dependent on the shuttling of the first two NADH) via proton donation to the mitochondrial electron transport chain (ETC). Electron shuttling through the ETC pumps protons from the mitochondrial matrix into the intermembrane space, creating a proton motive force that drives ATP Synthase (complex V) to join ADP and a free phosphate to form ATP [53]. ATP generation is dependent on the availability of substrates to oxidative phosphorylation, including oxygen, ADP, and inorganic phosphate [54,55].
3.1. Regulation of mitochondrial respiration
As oxidative phosphorylation is dependent upon mitochondrial electron transport chain functionality, any agent disrupting the activity of ETC complexes I-V similarly disrupts mitochondrial respiration. Mitochondrial respiration is highly regulated through several mechanisms, including nitric oxide (NO) inhibition, calcium (Ca2+) interactions, peroxisome proliferator-activated receptor γ coactivator 1 (PGC1) transcriptional activation, and 5′ adenosine monophosphate-activated protein kinase (AMPK) stimulation. NO primarily inhibits the activity of mitochondrial complex IV but may also inhibit complexes I and III, reducing the generation of ROS (reactive oxygen species). This inhibition at complex IV has been proposed to be a potential mechanism for redistributing oxygen intracellularly from regions of high concentration to regions of low concentration [56]. Calcium in the form of Ca2+ regulates the activity of several key metabolic enzymes, including FAD-glycerol phosphate dehydrogenase, pyruvate dehydrogenase phosphatase, NAD+-isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, and ATP synthase. Cyclooxygenase activity is also impacted indirectly as [Ca2+] affects cAMP signaling [57]. PGC1 increases transcription of genes in the oxidative phosphorylation pathway and mitochondrial biogenesis, notably in muscle and fat cells [58]. As AMPK increases, gluconeogenesis decreases; this promotes lipid and glucose metabolism as well as mitochondrial biogenesis [59]. In addition to endogenous regulators, multiple exogenous agents have been found or specifically developed to target mitochondrial complex elements, some of which are discussed further in Section 5. All these factors may affect the rates of reactive oxygen species formation, ATP synthesis, and substrate consumption.
Some specific mechanisms have been found to affect syncytiotrophoblast mitochondrial respiration, including less coupling control of oxidative phosphorylation compared to cytotrophoblast, reduced cardiolipin, and the presence of more superoxides compared to CTBs [20]. Selenium, which is discussed more at length in Section 5, upregulates mitobiogenesis and respiration in both human trophoblast cells and placental tissue [60]. Regulation of trophoblast mitochondrial respiration by microRNAs has been documented by our group: miR-210 upregulation suppresses trophoblast respiration via targeting of the iron-sulfur cluster (ISCU) in the setting of preeclampsia[61]. Respiration was significantly decreased in GDM placentas, which correlated with reduced expression of miR-143 whereas overexpressing miR-143 led to an increase in mitochondrial respiration [62].
3.2. Trophoblast oxidative and nitrative stress
During normal mitochondrial function, up to 2% of electrons “leak” from the ETC at complexes I and II, interact with oxygen, and form superoxide anions, which are a reactive oxygen species (ROS) [63]. ROS are present in normal pregnancy and necessary for cellular functions, including mitochondrial fusion/fission, autophagy, and cell signaling [64]. However, elevated levels of ROS, as observed in pathologic pregnancies, are associated with adverse outcomes: tissue and mitochondrial damage [65], decreased mitochondrial functional capacity [66], accelerated aging [67], and overall increased cellular oxidative stress [63]. Oxidative stress occurs when the production of ROS overwhelms antioxidant capacity[68]. This imbalance has been previously linked to placental tissue injury and spontaneous first-trimester abortion [69,70]. We have previously shown increased oxidative [63] and nitrative stress [71] in the placenta with maternal obesity coupled with a decrease in antioxidant defense enzymes [72].
Pregnancy itself is associated with persistent elevated oxidative and nitrative stress, but both obesity and gestational diabetes mellitus exacerbate these conditions [63,73,74]. Maternal obesity and GDM have both been found to increase placental ROS in both rodents and humans [74–76]. Maternal obesity led to more mitochondrial-specific ROS production in human term placentas [77]. Hyperglycemia stimulates aldose reductase to convert glucose to polyalcohol sorbitol, which then oxidizes further and increases the ratio of NADH to NAD+, inhibiting GAPDH, and increasing substrate availability to complex I [78]. In addition, hyperglycemia has been found to upregulate NADPH oxidase, which also generates ROS [79]. Specific products of reactive oxygen species action, including malondialdehyde and thiobarbituric acid-reactive substances, were found to be increased in GDM pregnancies, while antioxidants such as SOD and catalase were decreased [78,80]. Mitochondrial respiration is reduced in pregnancies complicated by obesity and GDM [62,73], as are antioxidant levels [78]. Obesity and GDM also both notably increase ROS, which may damage mitochondrial DNA and reduce electron transport chain activity, ATP production, and metabolic activity, particularly in ROS-sensitive syncytiotrophoblasts [84]. In GDM, elevated blood glucose levels increase ROS via membrane phospholipid or stress-activated signaling in rat models of diabetes [85,86]. Insulin resistance, a hallmark of GDM, has been found to reduce mitochondrial respiration, while the accompanying hyperglycemia reduced complex I activity [87,88]. Obesity decreases mitochondrial complex I-V expression in the placenta, but complex I activity increases, contributing to ROS levels already increased by the presence of fatty acids in maternal circulation [77,89]. Fuel flexibility is impaired in both obese and GDM pregnancies: syncytiotrophoblasts were unable to effectively switch from lipid to glucose oxidation [15].
Structural differences between cytotrophoblast and syncytiotrophoblast mitochondria may also affect mitochondrial respiration: CTB mitochondria are larger with lamellar cristae, while STB mitochondria are smaller with a dense matrix and vesicular cristae [90]. Unlike CTBs, STBs contain cholesterol side-chain cleavage enzyme (also known as P450scc, a member of the cytochrome P450 enzyme superfamily) in the inner mitochondrial membrane, allowing STBs to be more specialized for steroidogenesis. In addition to converting cholesterol to pregnenolone to begin steroidogenesis [90], cytochrome P450scc is also involved in superoxide formation, causing STBs to be more sensitive to ROS than CTBs [91]. This mainly occurs in early gestation when STBs do not express the antioxidant enzyme mitochondrial superoxide dismutase (SOD). However, after trophoblast plugs loosen around 12 weeks, the exposure to oxygen from the maternal circulation increases both oxidative stress and SOD expression [92]. Ultrastructural mitochondrial analysis of GDM syncytiotrophoblasts found mitochondrial swelling or destruction [93]. Pregnancies complicated by both obesity and GDM together showed this same mitochondrial disruption.
3.3. Consequences of trophoblast mitochondrial dysfunction
Dysfunctional mitochondria caused by maternal obesity and GDM have been linked to numerous immediate and long-term consequences in disease. As trophoblast mitochondria are involved in the provision of energy, their dysfunction affects energy-requiring trophoblast functions, including peptide synthesis and the transport of nutrients to the developing fetus and may ultimately lead to stillbirth [8,94]. Placental peptides regulate maternal metabolism and fetal growth and development; any irregularity in mitochondrial function may thus alter these effects together with negatively affecting placental transport. Changes in nutrient supply and composition to the placenta with obesity and GDM may alter their usage as fuel (fuel flexibility) for energy generation by mitochondria hence impacting what is available for transfer to the fetus. In addition to impairment of energy supply altered macro- and micronutrient composition reaching the placenta may affect one-carbon metabolism and cofactors that modulate epigenetic pathways related to fetal developmental programming [95]. Moreover, in a mouse model of obesity, a maternal diet high in fat and sucrose caused cardiac mitochondrial programming defects in the offspring, indicating a potential for multigenerational effects explicitly linked to mitochondrial dysfunction [96]. Fetal programming of many postnatal-onset diseases including early-life stress programming in the brain, renal disorders, neurodegenerative diseases, cardiovascular disease, Type 2 diabetes, and obesity have all been linked to placental mitochondrial dysfunction [97–102]. Future pregnancies to offspring impacted by fetal programming may have adverse outcomes, contributing to the feedback loop illustrated in Figure 1. Adverse maternal outcomes have also been associated with increased oxidative stress related to trophoblast mitochondrial function, including preterm delivery, stillbirth, and fetal growth restriction as well as higher risk for neurodegenerative diseases like Alzheimer’s and Parkinson’s later in life [103,104].
4. Sexual dimorphism in obese and GDM trophoblast function
Male fetuses are generally more at risk for pregnancy complications. Males are more susceptible to birth trauma [105], stillbirth [106], and developmental programming of adult-onset obesity, hypertension, and diabetes [32,107]. Abnormal fetal size (both fetal growth restriction and large-for-gestational-age diagnoses) occurs more frequently in male fetuses as well [17,108]. GDM is more likely to occur in a pregnancy with a male fetus, with marginally more significant risks for GDM in both the first and second pregnancy in one study, while another reported a 4% higher risk [109,110].
Maternal pathology magnifies these phenotypic fetal sex effects. Male fetuses from lean women had less placental oxidative and nitrative stress as well as more antioxidant SOD than males from obese women; females from obese women showed no difference compared to females from lean women [72]. In a rat study, females had less placental oxidative stress response to a high-fat diet with higher antioxidant defenses than male siblings [111]. Fetal sex affected neither the amount of mitochondrial DNA nor mitochondrial morphology, however [112]. Chronic hypoxia inhibited mitochondrial complexes I and IV in male guinea pig placentas, but there was no reduction in either complex in females [113]. In human studies, males from GDM women had fewer trophoblast mitochondria than females from GDM women, which correlated with reduced key antioxidant coactivator PGC-1α [114]. Trophoblast mitochondrial content was significantly reduced in placentas from obese mothers regardless of fetal sex [115]. Interestingly, both in an in vivo pregnant rat model and in human trophoblasts in vitro increased testosterone decreased trophoblast respiration and caused mitochondrial dysfunction which potentially could be a fetal sex-specific effect [116].
Recently, we found that fetal sex also affects fuel flexibility. In male STBs, flexibility to switch between using glucose, fatty acids, or glutamine for mitochondrial respiration significantly decreased from lean women to obese women to women with GDM. Male STBs from women with GDM were more dependent on fatty acids to maintain basal respiration than females, and less able to utilize glutamine [15].
Although the reasons for sexually dimorphic placental responses are not well understood, differences in trophoblast gene expression between males and females are well described. Female placentas had a higher expression of genes involved in immune regulation like JAK1, CXCL1, and IL2RB, which might benefit their response to infection and autoantigens [117]. Different mechanisms, including differential gene methylation [118] or escape from X-linked inactivation [119], may influence gene expression levels. Continued research into sexual dimorphism in the placenta and its driving causes is critical to understand the fetal impact on placental development.
5. Current and proposed treatments to improve mitochondrial function by reducing oxidative stress
Both obesity and GDM impact trophoblast mitochondrial function by increasing ROS and oxidative stress [120] and decreasing trophoblast respiration [115,121] and are associated with the adverse pregnancy outcomes seen in these conditions. Antioxidant treatments that focus on remediating trophoblast respiration to improve placental function and pregnancy outcomes are currently under investigation, including vitamins, general antioxidants, mitochondria-targeted antioxidants, and mitochondrial-derived peptides. A summary of the specific targets and effects of these treatments can be found in Figure 3 and Table 1.
Figure 3. Cellular sites of action of different agents for targeted reduction of oxidative stress.
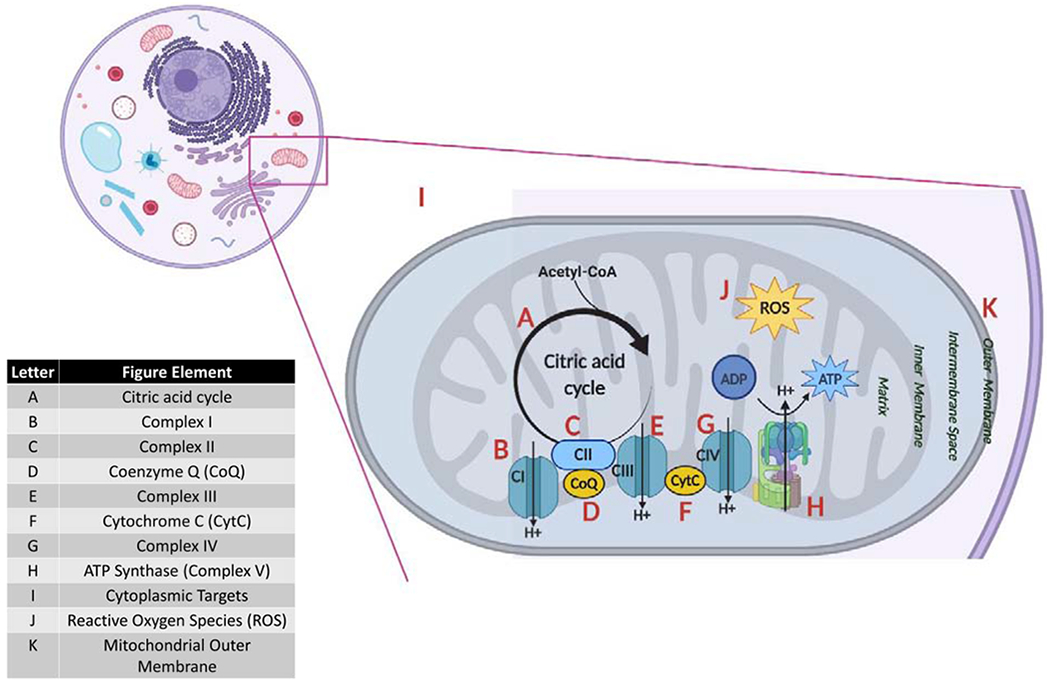
Each letter represents an antioxidant-targeted component of the mitochondria or the cytoplasm.
Table 1.
Cellular targets, and mechanistic effects of various antioxidant agents
| Agent | Location | Effect | Reference |
|---|---|---|---|
| Vitamin A | B, C | ↑ Complex I + II activity | [122] |
| Vitamin B complex | B1 – A, I B2 – B, C, D B3 – B, D B5, 6, 9, 12 – D |
B1 – cofactor in pyruvate, α-ketoglutarate and branched-chain ketoacid dehydrogenases B2: Complex I, II component; ↑CoQ B3: Complex I component; ↑CoQ B5, 6, 9, 12: ↑CoQ |
[126] |
| Vitamin C | D, I | ↓ ROS, ↑CoQ | [126] |
| Vitamin D | F | ↓ CytC | [134] |
| Vitamin E | J, K | ↓ ROS, fortifies membranes against ROS | [206] |
| Trolox | J | ↓ ROS | [145] |
| Vitamin K | D, F, G | Mediates electron transport to CoQ, CytC, Complex IV | [126,139] |
| Selenium | I | ↑ glutathione peroxidase, PGC-1α (mT biosynthesis), cell signaling cascades; necessary for antioxidant selenoproteins | [207,208] |
| MitoQ | D | ↑ CoQ | [164] |
| SkQ1 | J | ↓ ROS, ↓ mT membrane potential | [169] |
| Melatonin | I, J | ↑ glutathione reductase, SOD; ↓ ROS | [66] |
| Metformin | B | ↓ Complex 1 | [177] |
| SS31 | F, J | ↓ ROS, lipid peroxidase; prevents CytC from exiting to cytoplasm and signaling apoptosis | [173] |
| Tiron | J | ↓ ROS, permeabilizes mitochondrial membrane | [174] |
| Humanin | J | Restores mT glutathione; ↓ ROS | [186,209] |
| MOTS-C | I, J | Binds to NRF2 antioxidant gene promoter; ↓ ROS | [185] |
| SHLP2, SHLP3 | J | ↓ ROS | [185] |
| N-acetyl cysteine | I, J | ↑ glutathione, ↓ ROS | [210] |
| Sulforaphane | I, J | ↑ glutathione, ↓ ROS, ↓ mT membrane potential | [201] |
5.1. Vitamins to prevent ROS generation
Vitamin A (retinoic acid) increases the activity of both mitochondrial complexes I and II and is also critical to activating PKC-δ, which controls the flux of pyruvate into the citric acid cycle [122,123]. In a diabetic rat model, three times as much vitamin A was required for normal transcription of mitochondrial ATP6, which encodes for subunit 6 of ATP synthase, versus nondiabetic rats [124]. A deficiency of retinol (the less-active precursor to retinoic acid) led to embryo reabsorption and poor fetal outcomes in animal models that could be rescued by vitamin A supplementation; however, overdose also led to fetal demise, congenital defects, and trophoblast apoptosis in similar models [125].
The components of the vitamin B complex have different roles in oxidative phosphorylation as cofactors and their precursors. Vitamin B1 (thiamine) is a cofactor to ketoacid dehydrogenases acting on pyruvate, α-ketoglutarate, and branched-chain amino acids. B2 (riboflavin) is a component of mitochondrial complexes I and II, and a precursor to electron carrier flavin adenine dinucleotide (FAD) [126]. B3 (niacin) is critical to complex I function as a precursor to electron transporter nicotinamide adenine dinucleotide (NAD). B9 (folate) similarly increased NAD in vitro [127]. B2, B3, B5, B6, B9, and B12 are all involved in coenzyme Q (CoQ) synthesis [126,128]. The limited number of studies on the effect of B vitamins on trophoblasts have focused primarily on folate and B12 (cobalamin): deficiencies in both caused apoptosis and oxidative stress in vitro [125]. Imbalance in B vitamins can cause abnormalities in the one-carbon cycle, resulting in less production of thymidine, purines, ATP, NAD, and CoA, resulting in developmental programming for type 2 diabetes and obesity [129].
Vitamin D is best known for its involvement in aiding calcium absorption, but it also modifies placental mitochondrial function. Calcitriol (D3, the active form) is vital in pregnancy: it regulates decidualization, implantation, hormone secretion, and placental immune response via the vitamin D receptor VDR [130]. Trophoblasts readily convert inactive 25-hydroxyvitamin D to calcitriol, critical to STB use of vitamin D to transport calcium to the fetus [130]. However, excessive vitamin D induces nitric oxide production, which could be a root cause of GDM endothelial cell dysfunction [131].
Vitamin D concentrations directly influence mitochondrial function. The vitamin D response element VDRE can be found in the promoter region of some cytokines, including cytokine-regulator NF-κB, which also affects mitochondrial fusion, morphology, and ETC usage [132,133]. Diminishing cytochrome c subunits II and IV caused by vitamin D may inhibit mitochondrial respiration [134] but may help improve metabolism and cytokine production in obesity where vitamin D is depleted [135,136]. Placental apoptosis and oxidative stress declined and autophagy increased when treating pregnant rats with calcitriol [137].
Vitamin K (phylloquinone) is a fat-soluble vitamin involved in blood coagulation derived from plants and intestinal bacteria [126]. It has not been studied extensively in conjunction with pregnancy, as minimal vitamin K crosses the placenta to the fetus [138]. However, it does mediate electron transport from NADH to CoQ and cytochrome c, which may be of interest in regards to trophoblast treatment [139].
5.2. Vitamins as ROS scavengers
Vitamin C (ascorbic acid; AA) decreases ROS via its strong reducing potential [140,141]. The glucose transporter GLUT1 and sodium-dependent vitamin C transporter SVCT1 can transport vitamin C into the mitochondria as dehydroascorbic acid (DHA) and AA, respectively [140]. DHA is more effective against ROS but is present in lower amounts in the cell. Vitamin C also affects gene expression in several ways. For example, it is a competitive inhibitor for adenylyl cyclase, which would prevent the conversion of ATP to cAMP and, therefore, cAMP-regulated gene expression, and it decreased the apoptotic/anti-apoptotic ratio of BAX/BCL2 in first-trimester cultured EVTs [142,143].
Vitamin E (commonly α-tocopherol) is a ROS scavenger. As it is fat-soluble, vitamin E is incorporated into both the cellular membrane and the mitochondrial outer membrane to protect against ROS [144]. Trolox, a vitamin E analog, reduced ROS without altering gene expression or mitochondrial membrane potential [145], and it improved ATP synthase function and antioxidant levels when used as a pretreatment in rat cardiomyocytes prior to high glucose exposure [146].
When administered together, vitamin C can regenerate vitamin E’s antioxidant properties [147]. There is no current evidence that early supplementation with vitamins C and E improves trophoblast mitochondrial function in healthy pregnancy [148]. However, it may help reduce insulin resistance, increase SOD, and improve neonatal blood sugar levels in GDM in humans [149].
5.3. Selenium
Selenium is located at the catalytic site of several antioxidant and regulatory enzymes, including glutathione peroxidase and thioredoxin reductase [150]. SOD activity increased and ROS decreased in hypoxic conditions after 72 hours of selenium treatment on human EVT cell cultures [151]. Selenium deficiency resulted in reduced selenium-dependent thyroid hormone converting enzymes, causing fetal growth restriction and associated programming issues [152]. Protein levels of selenoprotein H, PGC-1α, and NRF1 were elevated after selenium treatment in trophoblasts in vitro, protecting against oxidative stress by stimulating mitochondrial biogenesis [153].
5.4. Melatonin
Melatonin is a potent antioxidant, acting as a ROS scavenger, in non-tumor cells but is cytotoxic in tumor cells [154,155]. In non-trophoblast studies, melatonin enhanced gene expression of antioxidant enzymes catalase, glutathione peroxidase, and SOD [156]. Thioredoxin, NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), and glutamate-cysteine ligase gene expression were all upregulated in placental tissue following melatonin treatment; sFLT secretion was reduced in primary trophoblasts treated with melatonin, but it did not change sFLT-related endothelial dysfunction [157].
In trophoblasts affected by hypoxia/reoxygenation, melatonin successfully repaired oxidative damage [158], and mitochondrial respiration was improved in syncytiotrophoblasts from obese women after melatonin treatment [66]. Interestingly, combining melatonin with vitamins A and C demonstrated an additive inhibition of lipid peroxidation and effective antioxidant treatment in isolated human placental mitochondria [159].
5.5. Synthetic antioxidants
One of the best-studied mitochondria-targeted synthetic antioxidants is MitoQ, coenzyme Q covalently bound to a lipophilic triphenylphosphonium cation (TPP+). As lipophilic cations do not require specific transport mechanisms, TPP+ and its ubiquinone cargo can accumulate very quickly in the mitochondria [160]. MitoQ is reduced in the mitochondria by the ETC to active ubiquinol, which prevents lipid peroxidation and mitochondrial damage [161]. In vivo, MitoQ has been successfully used in human clinical trials to treat cardiovascular disease and is well tolerated [161,162]. MitoQ also prevented trophoblast mitochondrial stress in early hypoxic rat pregnancies [160]. In vitro, however, MitoQ has been shown to cause mitochondrial swelling and depolarization [163,164]. Small amounts of MitoQ are sufficient to kill cells in vitro, including human trophoblast (unpublished observation), likely due to the rapid accumulation of TPP+ permitted by increased mitochondrial membrane permeability. It is presumed that in vivo, TPP+ can be metabolized or cleared before toxicity can occur [165].
Research on gene expression changes in pregnancy with MitoQ treatment has primarily focused on ameliorating the effects of hypoxia. Gene expression of growth factors Vegfa and Igf2 in the hypoxic placenta increased after placental-targeted nanoparticle-encapsulated MitoQ (nMitoQ) treatment in a rat model of pregnancy, but only when the fetus was female [166]. Using nMitoQ also decreased placental release of miRNA and subsequently decreased mRNA expression of genes related to fetal brain injury in hypoxic conditions [167].
Analogs of MitoQ have shown similar effects but with varying levels of efficacy. Idebenone is a non-mitochondria-targeted coenzyme Q10 structural analog not bound to TPP+ and exhibits weaker antioxidant ability, while SkQ1 (10-(6’-plastoquinonyl)decyltriphenylphosphonium, aka Visomitin) is a more powerful antioxidant combining TPP+ and plastoquinone. Idebenone prevented a reduction of cytochrome c oxidase and mRNA levels of PPARG1 and NRF1 in diabetic rats; it also reduced apoptosis in the embryos of these same animals [168]. SkQ1 exclusively targeted the mitochondria, reduced ROS and senescence in cell culture and rodent models, and slowed aging in a fungus and two animal models [169]. MitoQ and idebenone reduce oxidative and inflammatory markers in cardiovascular disease in a sexually dimorphic manner: female fetuses respond better to both than males in GDM in rats [170,171].
Other potential mitochondria-targeted treatments have not yet been applied to trophoblasts but may be of future interest based on their results in other tissues. SS31, part of the Szeto-Schiller group of peptides, prevents cardiolipin from converting cytochrome c into a peroxidase while preserving its ETC function [172]. Previously used in downregulating CD36 in brain injury, SS31 is now finding success as a ROS scavenger, improving mitochondrial function in traumatic brain injury, Parkinson’s Disease, cancer, and diabetes [172,173]. Tiron, an iron chelator found to cross the mitochondrial membrane, functions similarly to the SS class of peptides and can permeabilize the cell membrane, scavenge ROS, and prevent ADP depletion [174]. Gramicidin S-based JP4-039 and XJB-5-131 have targeting sequences for selective mitochondrial accumulation. Although quite recently created, both already show promise in reducing oxidative stress and preventing lipid peroxidation and apoptosis [175].
Metformin, a synthetic biguanide, can effectively reduce insulin resistance and cardiovascular risk factors and has antioxidant effects as it increases SOD and reduces ROS in diabetic patients [176,177]. It is reported to primarily affect mitochondrial ROS formation by inhibiting complex I [178]. However, it also upregulates gene expression of the ROS scavenger thioredoxin and inhibits the expression of pro-inflammatory genes IL-6 and TNFα [179,180]. Although metformin has been used for more than sixty years to treat both gestational and non-gestational diabetes [181], limited research is available on its effects on the placenta, and none yet published on trophoblast mitochondrial function. Recent studies suggest, however, that metformin can cross the placenta to the fetus [182]. More data is needed regarding safe dosage levels.
5.6. Mitochondrial-derived peptides
Peptides generated by the mitochondria themselves, including humanin, MOTS-c (mitochondrial open reading frame of 12s rRNA-c), and SHLP (small humanin-like proteins), help maintain mitochondrial function and cell viability in pathologic conditions [183]. Humanin is a 24-amino-acid cytoprotective peptide that initially found success in preventing β-amyloid plaques in Alzheimer’s [184]. Humanin acts as an antioxidant by restoring mitochondrial glutathione levels and increasing mitobiogenesis [185]. Humanin also stimulates insulin secretion, which may help increase glucose uptake; however, circulating humanin levels decrease with age, which may lead to eventual insulin resistance [186,187]. MOTS-c, a 16-amino-acid peptide that controls mitochondrial insulin regulation and homeostasis, increases AMPK and insulin sensitivity and promotes the expression of Nrf2 antioxidant genes [188,189]. SHLP1-6, most notably SHLP2 and 3, increase oxygen consumption rate and reduce apoptosis and ROS [190]. Mice on high-fat diets treated with these peptides demonstrated reduced weight gain, decreased ROS, and protection against ischemic injury [185].
5.7. Targeted superoxide dismutase conjugates
Treatments attempting to deliver antioxidant superoxide dismutase directly are challenging as SOD has a half-life of approximately six minutes in the circulation [191]. Compensating by increasing SOD dosage paradoxically exacerbated oxidative damage [192]. Due to its instability, rapid excretion, and low cellular uptake due to poor permeability across cell membranes, clinical applications of SOD alone as a therapeutic agent are limited. SOD conjugates have been developed with better stability and longer half-lives, as recombinant SOD conjugated to nanoparticles is protected from being degraded in serum [193].
Some of the earliest concepts involved SOD conjugation to pyran polymers, catalase, and gelatin [192,194,195]. However, SOD-conjugated liposomes were quickly found to be more effective in reducing ROS and inflammation. Long-circulating polyethylene glycol (PEG) liposomes loaded with SOD accumulated in inflammatory sites with greater efficiency and specificity than conventional stearylamine liposomes [196]. Polymers have regained some popularity. Both carboxymethylcellulose (CMC) and polymethyl vinyl ether-co-maleic anhydride (PMVE/MA) conjugates significantly increased SOD enzyme activity in diabetic rats and protected them from degenerative changes in brain, kidney, and liver tissue [197].
Several types of nanoparticles conjugated to SOD have been successful at reducing oxidative stress. SOD loaded on poly (D,L-lactide-co-glycolide) nanoparticles protected rat brain cells from oxidative stress related to ischemia-reperfusion injury, reducing infarcts by 65% over controls with a 75% survival rate (compared to 0% in controls) at 28 days [198]. Silica nanoparticles conjugated with Cu/Zn SOD were engineered with an HIV transactivator protein domain to enhance transmembrane transport [199]. However, the generation of these conjugates often results in toxic byproducts; a greener approach incubating Cu/Zn with glucose under heat-induced aggregation created a stable product that efficiently crossed cell membranes [191].
5.8. Global and indirect mitochondrial antioxidants
Global and indirect mitochondrial antioxidants are not explicitly targeted to remediate mitochondrial dysfunction but still reduce cellular oxidative stress. N-acetyl cysteine is a global antioxidant, as it has several roles: a precursor to glutathione, ROS scavenger, and direct interactor with NO2 and HOX [200]. Sulforaphane, an indirect mitochondrial antioxidant derived from cruciferous vegetables, similarly raises glutathione levels and decreases ROS, but it also diminishes mitochondrial membrane potential [201]. Sulforaphane prevented maleic acid-induced oxidative damage, improved function of mitochondrial complex I, and decreased ROS in non-pregnant rats [202]. Although sulforaphane protects mitochondrial function in both cancerous and non-cancerous cells, it paradoxically induces mitochondrial biogenesis, demonstrating that its mechanism is still not fully understood [203].
5.9. Behavioral modification
Finally, there is some evidence indicating that modifying certain lifestyle behaviors may reduce oxidative stress. Restricting caloric intake without inducing malnutrition reduces overall oxidative stress, which may have developed as a defense mechanism to survive in adverse conditions [204]. Moderate exercise may reduce metabolic rate by presenting regular oxidative stress challenge to the muscles and boosts insulin resistance and endogenous antioxidant levels, as seen in rat skeletal muscle [205]. Excessive consumption of antioxidant supplements is detrimental as low amounts of ROS are necessary to maintain mitochondrial function [204]. However, moderate antioxidant supplement consumption paired with behavioral modifications may be considered a part of an overall treatment plan to improve cellular response to oxidative stress.
6. Conclusion
The highly metabolic placenta relies on efficient mitochondrial function to produce the requisite energy for supporting a developing fetus through the transport of nutrients, gases, and wastes. Both maternal obesity and GDM generate placental oxidative stress, impair trophoblast mitochondrial function, impact fetal growth and development, developmentally program adult-onset disease, and are associated with a range of short and long term adverse fetal and maternal outcomes. Fetal sex influences trophoblast mitochondrial response to gestational pathology, with males generally responding less well to adverse pregnancy conditions, resulting in more severe health risks at birth and beyond. Both global and mitochondria-targeted antioxidants and mitochondrial-derived peptides may be a potential answer to correcting trophoblast mitochondrial dysfunction with obesity and GDM. Further investigation is warranted to identify the best agents, dosages, and timing of administration to improve both maternal and fetal gestational and long-term health outcomes.
Highlights.
Maternal obesity and gestational diabetes adversely affect pregnancy outcomes
Increased oxidative stress leads to decreased trophoblast mitochondrial respiration
Effects are sexually dimorphic with male placenta being more severely affected
Mitochondria-targeted treatments may improve trophoblast respiration
8. Acknowledgments
The authors would like to thank Dr. Luis Sobrevia of Pontificia Universidad Catolica de Chile for his invitation to submit this review. Figures 2 and 3 were created with assistance from the OHSU academic license of Biorender (http://www.biorender.com).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Conflicts of Interest
The authors have no conflicts of interest to disclose.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
9. References
- [1].Ng M, et al. , Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013., Lancet (London, England). 384 (2014) 766–81. 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Flegal K, Carroll M, Kuczmarski R, Johnson C, Overweight and obesity in the United States: prevalence and trends, 1960–1994, Int. J. Obes 22 (1998) 39–47. 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- [3].Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM, Prevalence of Overweight and Obesity Among US Children, Adolescents, and Adults, 1999-2002, JAMA. 291 (2004) 2847. 10.1001/jama.291.23.2847. [DOI] [PubMed] [Google Scholar]
- [4].Hales C, Carroll M, Fryar C, Ogden C, Prevalence of Obesity and Severe Obesity Among Adults: United States, 2017-2018, NCHS Data Brief. 360 (2020) 1–8. http://www.ncbi.nlm.nih.gov/pubmed/26633046. [PubMed] [Google Scholar]
- [5].Bray GA, Complications of Obesity, Ann. Intern. Med 103 (1985). 10.7326/0003-4819-103-6-1052. [DOI] [PubMed] [Google Scholar]
- [6].Baeten JM, Bukusi EA, Lambe M, Pregnancy complications and outcomes among overweight and obese nulliparous women., Am. J. Public Health. 91 (2001) 436–40. 10.2105/ajph.91.3.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ramachenderan J, Bradford J, McLean M, Maternal obesity and pregnancy complications: A review, Aust. New Zeal. J. Obstet. Gynaecol 48 (2008) 228–235. 10.1111/j.1479-828X.2008.00860.x. [DOI] [PubMed] [Google Scholar]
- [8].Yao R, Ananth CV, Park BY, Pereira L, Plante LA, Obesity and the risk of stillbirth: A population-based cohort study, Am. J. Obstet. Gynecol 210 (2014) 457.e1–457.e9. 10.1016/j.ajog.2014.01.044. [DOI] [PubMed] [Google Scholar]
- [9].Dennedy MC, Avalos G, O’Reilly MW, O’Sullivan EP, Dunne FP, The impact of maternal obesity on gestational outcomes., Ir. Med. J 105 (2012) 23–5. http://www.ncbi.nlm.nih.gov/pubmed/22838105. [PubMed] [Google Scholar]
- [10].Marchi J, Berg M, Dencker A, Olander EK, Begley C, Risks associated with obesity in pregnancy, for the mother and baby: a systematic review of reviews, Obes. Rev 16 (2015) 621–638. 10.1111/obr.12288. [DOI] [PubMed] [Google Scholar]
- [11].Leddy MA, Power ML, Schulkin J, The impact of maternal obesity on maternal and fetal health., Rev. Obstet. Gynecol 1 (2008) 170–8. http://www.ncbi.nlm.nih.gov/pubmed/19173021. [PMC free article] [PubMed] [Google Scholar]
- [12].Stubert J, Reister F, Hartmann S, Janni W, The Risks Associated With Obesity in Pregnancy., Dtsch. Arztebl. Int 115 (2018) 276–283. 10.3238/arztebl.2018.0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Catalano PM, Obesity, insulin resistance, and pregnancy outcome., Reproduction. 140 (2010) 365–71. 10.1530/REP-10-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Smirnakis KV, Martinez A, Blatman KH, Wolf M, Ecker JL, Thadhani R, Early pregnancy insulin resistance and subsequent gestational diabetes mellitus, Diabetes Care. 28 (2005) 1207–1208. 10.2337/diacare.28.5.1207. [DOI] [PubMed] [Google Scholar]
- [15].Wang Y, Bucher M, Myatt L, Use of Glucose, Glutamine, and Fatty Acids for Trophoblast Respiration in Lean Women, Women With Obesity, and Women With Gestational Diabetes, J. Clin. Endocrinol. Metab 104 (2019) 4178–4187. 10.1210/jc.2019-00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barker DJ, The fetal and infant origins of adult disease., BMJ. 301 (1990) 1111. 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barker DJP, Hales CN, Fall CHD, Osmond C, Phipps K, Clark PMS, Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth, Diabetologia. 36 (1993) 62–67. 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- [18].Catalano PM, Obesity and Pregnancy—The Propagation of a Viscous Cycle?, J. Clin. Endocrinol. Metab 88 (2003) 3505–3506. 10.1210/jc.2003-031046. [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Vascular Biology of the Placenta, Colloq. Ser. Integr. Syst. Physiol. From Mol. to Funct 2 (2010) 1–98. 10.4199/C00016ED1V01Y201008ISP009. [DOI] [Google Scholar]
- [20].Holland O, Dekker Nitert M, Gallo LA, Vejzovic M, Fisher JJ, Perkins AV, Review: Placental mitochondrial function and structure in gestational disorders., Placenta. 54 (2017) 2–9. 10.1016/j.placenta.2016.12.012. [DOI] [PubMed] [Google Scholar]
- [21].Vaughan O, Fowden A, Placental metabolism: substrate requirements and the response to stress, Reprod. Domest. Anim 51 (2016) 25–35. 10.1111/rda.12797. [DOI] [PubMed] [Google Scholar]
- [22].Ellery PM, Cindrova-Davies T, Jauniaux E, Ferguson-Smith AC, Burton GJ, Evidence for Transcriptional Activity in the Syncytiotrophoblast of the Human Placenta, Placenta. 30 (2009) 329–334. 10.1016/J.PLACENTA.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].He N, van Iperen L, de Jong D, Szuhai K, Helmerhorst FM, van der Westerlaken LAJ, de Sousa Lopes S.M. Chuva, Human Extravillous Trophoblasts Penetrate Decidual Veins and Lymphatics before Remodeling Spiral Arteries during Early Pregnancy., PLoS One. 12 (2017) e0169849. 10.1371/journal.pone.0169849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boyd PA, Brown RA, Coghill GR, Slidders W, Stewart WJ, Measurement of the mass of syncytiotrophoblast in a range of human placentae using an image analysing computer, Placenta. 4 (1983) 255–262. 10.1016/S0143-4004(83)80004-6. [DOI] [PubMed] [Google Scholar]
- [25].Mayhew T, Ohadike C, Baker P, Crocker I, Mitchell C, Ong S, Stereological Investigation of Placental Morphology in Pregnancies Complicated by Pre-eclampsia with and without Intrauterine Growth Restriction, Placenta. 24 (2003) 219–226. 10.1053/PLAC.2002.0900. [DOI] [PubMed] [Google Scholar]
- [26].Knobil E, Neill JD, Knobil and Neill’s Physiology of Reproduction, Elsevier, 2006. [Google Scholar]
- [27].Barnea ER, Placental Biochemistry, in: The First Twelve Weeks of Gestation, Springer Berlin Heidelberg, 1992: pp. 111–127. 10.1007/978-3-642-84385-3_7. [DOI] [Google Scholar]
- [28].Bax BE, Bloxam DL, Energy metabolism and glycolysis in human placental trophoblast cells during differentiation, Biochim. Biophys. Acta - Bioenerg 1319 (1997) 283–292. 10.1016/S0005-2728(96)00169-7. [DOI] [PubMed] [Google Scholar]
- [29].Kolahi KS, Valent AM, Thornburg KL, Cytotrophoblast, Not Syncytiotrophoblast, Dominates Glycolysis and Oxidative Phosphorylation in Human Term Placenta., Sci. Rep 7 (2017) 42941. 10.1038/srep42941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Carter AM, Placental oxygen consumption. Part I: In vivo studies - A review, Placenta. 21 (2000) S31–S37. 10.1053/plac.1999.0513. [DOI] [PubMed] [Google Scholar]
- [31].Meschia G, Battaglia FC, Hay WW, Sparks JW, Utilization of substrates by the ovine placenta in vivo., Fed. Proc 39 (1980) 245–9. http://www.ncbi.nlm.nih.gov/pubmed/7353681. [PubMed] [Google Scholar]
- [32].Myatt L, Placental adaptive responses and fetal programming., J. Physiol 572 (2006) 25–30. 10.1113/jphysiol.2006.104968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hay WW, Energy and substrate requirements of the placenta and fetus, Proc. Nutr. Soc 50 (1991) 321–336. 10.1079/PNS19910042. [DOI] [PubMed] [Google Scholar]
- [34].Bloxam DL, Human Placental Energy Metabolism: Its Relevance to in vitro Perfusion, in: Karger Publishers, 1985: pp. 59–69. 10.1159/000410670. [DOI] [PubMed] [Google Scholar]
- [35].Michelsen TM, Holme AM, Holm MB, Roland MC, Haugen G, Powell TL, Jansson T, Henriksen T, Uteroplacental Glucose Uptake and Fetal Glucose Consumption: A Quantitative Study in Human Pregnancies, J. Clin. Endocrinol. Metab 104 (2019) 873–882. 10.1210/jc.2018-01154. [DOI] [PubMed] [Google Scholar]
- [36].Ericsson A, Hamark B, Powell TL, Jansson T, Glucose transporter isoform 4 is expressed in the syncytiotrophoblast of first trimester human placenta, Hum. Reprod 20 (2005) 521–530. 10.1093/humrep/deh596. [DOI] [PubMed] [Google Scholar]
- [37].Türkay Korgun E, Demir R, Hammer A, Dohr G, Desoye G, Skofitsch G, Hahn T, Glucose Transporter Expression in Rat Embryo and Uterus During Decidualization, Implantation, and Early Postimplantation1, Biol. Reprod 65 (2001) 1364–1370. 10.1095/biolreprod65.5.1364. [DOI] [PubMed] [Google Scholar]
- [38].Hay WW Jr, Placental-fetal glucose exchange and fetal glucose metabolism., Trans. Am. Clin. Climatol. Assoc 117 (2006) 321–40. http://www.ncbi.nlm.nih.gov/pubmed/18528484. [PMC free article] [PubMed] [Google Scholar]
- [39].Aibibula M, Naseem KM, Sturmey RG, Glucose metabolism and metabolic flexibility in blood platelets, J. Thromb. Haemost 16 (2018) 2300–2314. 10.1111/jth.14274. [DOI] [PubMed] [Google Scholar]
- [40].Gil-Sánchez A, Koletzko B, Larqué E, Current understanding of placental fatty acid transport, Curr. Opin. Clin. Nutr. Metab. Care 15 (2012) 265–272. 10.1097/MCO.0b013e3283523b6e. [DOI] [PubMed] [Google Scholar]
- [41].Biron-Shental T, Schaiff WT, Ratajczak CK, Bildirici I, Nelson DM, Sadovsky Y, Hypoxia regulates the expression of fatty acid-binding proteins in primary term human trophoblasts, Am. J. Obstet. Gynecol 197 (2007) 516.e1–516.e6. 10.1016/J.AJOG.2007.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McGarry JD, Brown NF, The Mitochondrial Carnitine Palmitoyltransferase System - From Concept to Molecular Analysis, Eur. J. Biochem 244 (1997) 1–14. 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- [43].Wu X, Xie C, Zhang Y, Fan Z, Yin Y, Blachier F, Glutamate-glutamine cycle and exchange in the placenta-fetus unit during late pregnancy, Amino Acids. 47 (2015) 45–53. 10.1007/s00726-014-1861-5. [DOI] [PubMed] [Google Scholar]
- [44].Battaglia F, Regnault T, Placental Transport and Metabolism of Amino Acids, Placenta. 22 (2001) 145–161. 10.1053/PLAC.2000.0612. [DOI] [PubMed] [Google Scholar]
- [45].Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, Schmidt AF, Imamura F, Stewart ID, Perry JRB, Marney L, Koulman A, Karoly ED, Forouhi NG, Sjogren RJO, Naslund E, Zierath JR, Krook A, Savage DB, Griffin JL, Chaturvedi N, Hingorani AD, Khaw KT, Barroso I, McCarthy M.l., O’Rahilly S, Wareham NJ, Langenberg C, Genetic Predisposition to an Impaired Metabolism of the Branched-Chain Amino Acids and Risk of Type 2 Diabetes: A Mendelian Randomisation Analysis, PLOS Med. 13 (2016) e1002179. 10.1371/journal.pmed.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Newbern D, Freemark M, Placental hormones and the control of maternal metabolism and fetal growth., Curr. Opin. Endocrinol. Diabetes. Obes 18 (2011) 409–16. 10.1097/MED.0b013e32834c800d. [DOI] [PubMed] [Google Scholar]
- [47].Jameson JL, DeGroot LJ, M. D (De Kretser David M., Giudice L, Grossman A, Melmed S, Potts JT, Weir GC, Endocrinology : adult & pediatric, Elsevier Inc., 2016. https://www.sciencedirect.com/book/9780323189071/endocrinology-adult-and-pediatric. [Google Scholar]
- [48].Burton GJ, Fowden AL, The placenta: a multifaceted, transient organ, Philos. Trans. R. Soc. B Biol. Sci 370 (2015) 20140066. 10.1098/rstb.2014.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Baker H, Frank O, Deangelis B, Feingold S, Kaminetzky HA, Role of placenta in maternal-fetal vitamin transfer in humans, Am. J. Obstet. Gynecol 141 (1981) 792–796. 10.1016/0002-9378(81)90706-7. [DOI] [PubMed] [Google Scholar]
- [50].Lewis RM, Desoye G, Placental Lipid and Fatty Acid Transfer in Maternal Overnutrition, Ann. Nutr. Metab 70 (2017) 228–231. 10.1159/000463397. [DOI] [PubMed] [Google Scholar]
- [51].Olszewska A, Szewczyk A, Mitochondria as a pharmacological target: Magnum overview, IUBMB Life. 65 (2013) 273–281. 10.1002/iub.1147. [DOI] [PubMed] [Google Scholar]
- [52].McCommis KS, Finck BN, Mitochondrial pyruvate transport: a historical perspective and future research directions., Biochem. J 466 (2015) 443–54. 10.1042/BJ20141171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD, Mitochondrial proton and electron leaks., Essays Biochem. 47 (2010) 53–67. 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Barbiroli B, lotti S, Lodi R, In vivo assessment of human skeletal muscle mitochondria respiration in health and disease, Mol. Cell. Biochem 174 (1997) 11–15. 10.1023/A:1006839403270. [DOI] [PubMed] [Google Scholar]
- [55].Chance B, Williams GR, Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria, J. Biol. Chem 221 (1956)477–489. [PubMed] [Google Scholar]
- [56].Tengan CH, Moraes CT, NO control of mitochondrial function in normal and transformed cells, Biochim. Biophys. Acta - Bioenerg 1858 (2017) 573–581. 10.1016/j.bbabio.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tarasov AI, Griffiths EJ, Rutter GA, Regulation of ATP production by mitochondrial Ca2+, Cell Calcium. 52 (2012) 28–35. 10.1016/j.ceca.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM, Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1, Cell. 98 (1999) 115–124. 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- [59].Herzig S, Shaw RJ, AMPK: Guardian of metabolism and mitochondrial homeostasis, Nat. Rev. Mol. Cell Biol 19 (2018) 121–135. 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Khera A, Dong LF, Holland O, Vanderlelie J, Pasdar EA, Neuzil J, Perkins AV, Selenium supplementation induces mitochondrial biogenesis in trophoblasts, Placenta. 36 (2015) 863–869. 10.1016/j.placenta.2015.06.010. [DOI] [PubMed] [Google Scholar]
- [61].Muralimanoharan S, Guo C, Myatt L, Maloyan A, Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity., Int. J. Obes. (Lond). 39 (2015) 1274–81. 10.1038/ijo.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Muralimanoharan S, Maloyan A, Myatt L, Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: role of miR-143., Clin. Sci 130 (2016) 931–41. 10.1042/CS20160076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Myatt L, Cui X, Oxidative stress in the placenta, Histochem. Cell Biol. 122 (2004) 369–382. 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- [64].Mannaerts D, Faes E, Cos P, Briedé JJ, Gyselaers W, Cornette J, Gorbanev Y, Bogaerts A, Spaanderman M, Van Craenenbroeck E, Jacquemyn Y, Oxidative stress in healthy pregnancy and preeclampsia is linked to chronic inflammation, iron status and vascular function., PLoS One. 13 (2018) e0202919. 10.1371/journal.pone.0202919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Guo C, Sun L, Chen X, Zhang D, Oxidative stress, mitochondrial damage and neurodegenerative diseases., Neural Regen. Res. 8 (2013) 2003–14. 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ireland KE, Maloyan A, Myatt L, Melatonin Improves Mitochondrial Respiration in Syncytiotrophoblasts From Placentas of Obese Women, Reprod. Sci 25 (2018) 120–130. 10.1177/1933719117704908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Harman D, The Biologic Clock: The Mitochondria?, J. Am. Geriatr. Soc 20 (1972) 145–147. 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- [68].Burton GJ, Jauniaux E, Oxidative stress, Best Pract. Res. Clin. Obstet. Gynaecol 25 (2011) 287–299. 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jauniaux E, Watson AL, Hempstock J, Bao Y-P, Skepper JN, Burton GJ, Onset of Maternal Arterial Blood Flow and Placental Oxidative Stress: A Possible Factor in Human Early Pregnancy Failure, Am. J. Pathol 157 (2000) 2111–2122. 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jauniaux E, Greenwold N, Hempstock J, Burton GJ, Comparison of ultrasonographic and Doppler mapping of the intervillous circulation in normal and abnormal early pregnancies, Fertil. Steril 79 (2003) 100–106. 10.1016/S0015-0282(02)04568-5. [DOI] [PubMed] [Google Scholar]
- [71].Roberts VHJ, Smith J, McLea SA, Heizer AB, Richardson JL, Myatt L, Effect of increasing maternal body mass index on oxidative and nitrative stress in the human placenta., Placenta. 30 (2009) 169–75. 10.1016/j.placenta.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Evans L, Myatt L, Sexual dimorphism in the effect of maternal obesity on antioxidant defense mechanisms in the human placenta., Placenta. 51 (2017) 64–69. 10.1016/j.placenta.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Myatt L, Maloyan A, Obesity and Placental Function, Semin. Reprod. Med 34 (2016) 042–049. 10.1055/s-0035-1570027. [DOI] [PubMed] [Google Scholar]
- [74].Roberts VHJ, Smith J, McLea SA, Heizer AB, Richardson JL, Myatt L, Effect of Increasing Maternal Body Mass Index on Oxidative and Nitrative Stress in the Human Placenta, Placenta. 30 (2009) 169. 10.1016/J.PLACENTA.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Plows JF, Stanley JL, Baker PN, Reynolds CM, Vickers MH, The Pathophysiology of Gestational Diabetes Mellitus., Int. J. Mol. Sci 19 (2018). 10.3390/ijms19113342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Spada APM, Damasceno DC, Sinzato YK, Campos KE, Faria PA, Dallaqua B, Calderon IMP, Rudge MVC, Rodrigues T, Oxidative Stress in Maternal Blood and Placenta From Mild Diabetic Rats., Reprod. Sci 21 (2014) 973–977. 10.1177/1933719113519175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hastie R, Lappas M, The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity, Placenta. 35 (2014) 673–683. 10.1016/J.PLACENTA.2014.06.368. [DOI] [PubMed] [Google Scholar]
- [78].Lappas M, Hiden U, Desoye G, Froehlich J, De Mouzon SH, Jawerbaum A, The role of oxidative stress in the pathophysiology of gestational diabetes mellitus, Antioxidants Redox Signal. 15 (2011) 3061–3100. 10.1089/ars.2010.3765. [DOI] [PubMed] [Google Scholar]
- [79].Xia L, Wang H, Munk S, Kwan J, Goldberg HJ, Fantus IG, Whiteside CI, High glucose activates PKC-ζ and NADPH oxidase through autocrine TGF-β 1 signaling in mesangial cells, Am. J. Physiol. Physiol 295 (2008) F1705–F1714. 10.1152/ajprenal.00043.2008. [DOI] [PubMed] [Google Scholar]
- [80].Tiwari BK, Pandey KB, Abidi AB, S.l. Rizvi, Markers of Oxidative Stress during Diabetes Mellitus., J. Biomarkers. 2013 (2013) 378790. 10.1155/2013/378790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fernandez-Twinn DS, Gascoin G, Musial B, Carr S, Duque-Guimaraes D, Blackmore HL, Alfaradhi MZ, Loche E, Sferruzzi-Perri AN, Fowden AL, Ozanne SE, Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity, Sci. Rep 7 (2017) 1–11. 10.1038/srep44650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wallace JG, Bellissimo CJ, Yeo E, Fei Xia Y, Petrik JJ, Surette MG, Bowdish DME, Sloboda DM, Obesity during pregnancy results in maternal intestinal inflammation, placental hypoxia, and alters fetal glucose metabolism at mid-gestation, Sci. Rep 9 (2019) 1–16. 10.1038/s41598-019-54098-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Escobar J, Teramo K, Stefanovic V, Andersson S, Asensi MA, Arduini A, Cubells E, Sastre J, Vento M, Amniotic Fluid Oxidative and Nitrosative Stress Biomarkers Correlate with Fetal Chronic Hypoxia in Diabetic Pregnancies, Neonatology. 103 (2013) 193–198. 10.1159/000345194. [DOI] [PubMed] [Google Scholar]
- [84].Hung T-H, Skepper JN, Charnock-Jones DS, Burton GJ, Hypoxia-Reoxygenation, Circ. Res 90 (2002) 1274–1281. 10.1161/01.RES.0000024411.22110.AA. [DOI] [PubMed] [Google Scholar]
- [85].Reece EA, Obesity, diabetes, and links to congenital defects: A review of the evidence and recommendations for intervention, J. Matern. Neonatal Med 21 (2008) 173–180. 10.1080/14767050801929885. [DOI] [PubMed] [Google Scholar]
- [86].Reece EA, Ma XD, Wu YK, Dhanasekaran D, Aberrant patterns of cellular communication in diabetes-induced embryopathy I. Membrane signalling, J. Matern. Neonatal Med 11 (2002) 249–253. 10.1080/jmf.11.4.249.253. [DOI] [PubMed] [Google Scholar]
- [87].Phielix E, Schrauwen-Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, Kooi ME, Moonen-Kornips E, Sels JP, Hesselink MKC, Schrauwen P, Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients, Diabetes. 57 (2008) 2943–2949. 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Holland OJ, Hickey AJR, Alvsaker A, Moran S, Hedges C, Chamley LW, Perkins AV, Changes in mitochondrial respiration in the human placenta over gestation, Placenta. 57 (2017) 102–112. 10.1016/j.placenta.2017.06.011. [DOI] [PubMed] [Google Scholar]
- [89].Shirasuna K, Takano H, Seno A, Ohtsu A, Karasawa T, Takahashi M, Ohkuchi A, Suzuki H, Matsubara S, Iwata H, Kuwayama T, Palmitic acid induces interleukin-1β secretion via NLRP3 inflammasomes and inflammatory responses through ROS production in human placental cells, J. Reprod. Immunol 116 (2016) 104–112. 10.1016/j.jri.2016.06.001. [DOI] [PubMed] [Google Scholar]
- [90].Martínez F, Kiriakidou M, Strauss JF, Structural and Functional Changes in Mitochondria Associated with Trophoblast Differentiation: Methods to Isolate Enriched Preparations of Syncytiotrophoblast Mitochondria 1, Endocrinology. 138 (1997) 2172–2183. 10.1210/endo.138.5.5133. [DOI] [PubMed] [Google Scholar]
- [91].Bustamante J, Ramírez-Vélez R, Czerniczyniec A, Cicerchia D, Aguilar de Plata AC, Lores-Arnaiz S, Oxygen metabolism in human placenta mitochondria, J. Bioenerg. Biomembr 46 (2014) 459–469. 10.1007/s10863-014-9572-x. [DOI] [PubMed] [Google Scholar]
- [92].James JL, Stone PR, Chamley LW, The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy, Hum. Reprod. Update. 12 (2006) 137–144. 10.1093/humupd/dmi043. [DOI] [PubMed] [Google Scholar]
- [93].Meng Q, Shao L, Luo X, Mu Y, Xu W, Gao C, Gao L, Liu J, Cui Y, Ultrastructure of Placenta of Gravidas with Gestational Diabetes Mellitus, Obstet. Gynecol. Int 2015 (2015) 1–9. 10.1155/2015/283124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mele J, Muralimanoharan S, Maloyan A, Myatt L, Impaired mitochondrial function in human placenta with increased maternal adiposity., Am. J. Physiol. Endocrinol. Metab 307 (2014) E419–25. 10.1152/ajpendo.00025.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Rodriguez-Cano AM, Calzada-Mendoza CC, Estrada-Gutierrez G, Mendoza-Ortega JA, Perichart-Perera O, Nutrients, Mitochondrial Function, and Perinatal Health, Nutrients. 12 (2020). 10.3390/nul2072166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ferey JLA, Boudoures AL, Reid M, Drury A, Scheaffer S, Modi Z, Kovacs A, Pietka T, DeBosch BJ, Thompson MD, Diwan A, Moley KH, A maternal high-fat, high-sucrose diet induces transgenerational cardiac mitochondrial dysfunction independently of maternal mitochondrial inheritance, Am. J. Physiol. Circ. Physiol 316 (2019) H1202–H1210. 10.1152/ajpheart.00013.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Leduc L, Levy E, Bouity-Voubou M, Delvin E, Fetal programming of atherosclerosis: Possible role of the mitochondria, Eur. J. Obstet. Gynecol. Reprod. Biol 149 (2010) 127–130. 10.1016/j.ejogrb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- [98].Hoffmann A, Spengler D, The mitochondrion as potential interface in early-life stress brain programming, Front. Behav. Neurosci 12 (2018). 10.3389/fnbeh.2018.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Sukjamnong S, Chan YL, Zakarya R, Nguyen LT, Anwer AG, Zaky AA, Santiyanont R, Oliver BG, Goldys E, Pollock CA, Chen H, Saad S, MitoQ supplementation prevent long-term impact of maternal smoking on renal development, oxidative stress and mitochondrial density in male mice offspring, Sci. Rep 8 (2018). 10.1038/s41598-018-24949-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lei F, Wang W, Fu Y, Wang J, Zheng Y, Oxidative stress and mitochondrial dysfunction in parafacial respiratory group induced by maternal cigarette smoke exposure in rat offspring, Free Radic. Biol. Med 129 (2018) 169–176. 10.1016/j.freeradbiomed.2018.09.003. [DOI] [PubMed] [Google Scholar]
- [101].Bansal A, Rashid C, Simmons RA, Impact of fetal programming on mitochondrial function and susceptibility to obesity and type 2 diabetes, in: Mitochondria Obes. Type 2 Diabetes Compr. Rev. Mitochondrial Funct. Involv. Metab. Dis, Elsevier, 2019: pp. 325–345. 10.1016/B978-0-12-811752-1.00014-6. [DOI] [Google Scholar]
- [102].Rodríguez-Rodríguez P, Ramiro-Cortijo D, Reyes-Hernández CG, López de Pablo AL, Carmen Gonzalez M, Arribas SM, Implication of oxidative stress in fetal programming of cardiovascular disease, Front. Physiol 9 (2018) 602. 10.3389/fphys.2018.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Morén C, Hernández S, Guitart-Mampel M, Garrabou G, Mitochondrial toxicity in human pregnancy: An update on clinical and experimental approaches in the last 10 years, Int. J. Environ. Res. Public Health. 11 (2014) 9897–9918. 10.3390/ijerph110909897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Fallatah AM, Babatin HM, Nassibi KM, Banweer MK, Fayoumi MN, Oraif AM, Maternal and Neonatal Outcomes among Obese Pregnant Women in King Abdulaziz University Hospital: A Retrospective Single-Center Medical Record Review, Med. Arch. (Sarajevo, Bosnia Herzegovina). 73 (2019) 425–432. 10.5455/medarh.2019.73.425-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Ferrara A, Kim C, Gestational Diabetes Mellitus: Diagnosis, Maternal and Fetal Outcomes, and Management, in: Diabetes in Women, Humana Press, Totowa, NJ, 2009: pp. 239–253. 10.1007/978-1-60327-250-6_13. [DOI] [Google Scholar]
- [106].Gabbay-Benziv R, Baschat AA, Gestational diabetes as one of the “great obstetrical syndromes” - the maternal, placental, and fetal dialog, Best Pract. Res. Clin. Obstet. Gynaecol 29 (2015) 150–155. 10.1016/J.BPOBGYN.2014.04.025. [DOI] [PubMed] [Google Scholar]
- [107].Le Moullec N, Fianu A, Maillard O, Chazelle E, Naty N, Schneebeli C, Gérardin P, Huiart L, Charles M-A, Favier F. Sexual dimorphism in the association between gestational diabetes mellitus and overweight in offspring at 5-7 years: The OBEGEST cohort study., PLoS One. 13 (2018) e0195531. 10.1371/journal.pone.0195531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Ehrenberg HM, Mercer BM, Catalano PM, The influence of obesity and diabetes on the prevalence of macrosomia, Am. J. Obstet. Gynecol 191 (2004) 964–968. 10.1016/j.ajog.2004.05.052. [DOI] [PubMed] [Google Scholar]
- [109].Retnakaran R, Shah BR, Fetal Sex and the Natural History of Maternal Risk of Diabetes During and After Pregnancy, J. Clin. Endocrinol. Metab 100 (2015) 2574–2580. 10.1210/jc.2015-1763. [DOI] [PubMed] [Google Scholar]
- [110].Jaskolka D, Retnakaran R, Zinman B, Kramer CK, Sex of the baby and risk of gestational diabetes mellitus in the mother: a systematic review and meta-analysis, Diabetologia. 58 (2015) 2469–2475. 10.1007/s00125-015-3726-1. [DOI] [PubMed] [Google Scholar]
- [111].Priego T, Sánchez J, Picó C, Palou A, Sex-differential Expression of Metabolism-related Genes in Response to a High-fat Diet, Obesity. 16 (2008) 819–826. 10.1038/oby.2007.117. [DOI] [PubMed] [Google Scholar]
- [112].Mandò C, Anelli GM, Novielli C, Panina-Bordignon P, Massari M, Mazzocco MI, Cetin I, Impact of Obesity and Hyperglycemia on Placental Mitochondria, Oxid. Med. Cell. Longev 2018 (2018) 1–10. 10.1155/2018/2378189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Song H, Telugu BP, Thompson LP, Sexual dimorphism of mitochondrial function in the hypoxic guinea pig placenta., Biol. Reprod 100 (2019) 208–216. 10.1093/biolre/ioy167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jiang S, Teague AM, Tryggestad JB, Aston CE, Lyons T, Chernausek SD, Effects of maternal diabetes and fetal sex on human placenta mitochondrial biogenesis, Placenta. 57 (2017) 26–32. 10.1016/J.PLACENTA.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Mele J, Muralimanoharan S, Maloyan A, Myatt L, Impaired mitochondrial function in human placenta with increased maternal adiposity, Am. J. Physiol. - Endocrinol. Metab 307 (2014) E419–E425. 10.1152/ajpendo.00025.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Mishra JS, Blesson CS, Kumar S, Testosterone Decreases Placental Mitochondrial Content and Cellular Bioenergetics, Biology (Basel). 9 (2020) 176. 10.3390/biology9070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Sood R, Zehnder JL, Druzin ML, Brown PO, Gene expression patterns in human placenta, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 5478–5483. 10.1073/pnas.0508035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Gong S, Johnson MD, Dopierala J, Gaccioli F, Sovio U, Constância M, Smith GC, Charnock-Jones DS, Genome-wide oxidative bisulfite sequencing identifies sex-specific methylation differences in the human placenta., Epigenetics. 13 (2018) 228–239. 10.1080/15592294.2018.1429857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Gong S, Sovio U, Aye IL, Gaccioli F, Dopierala J, Johnson MD, Wood AM, Cook E, Jenkins BJ, Koulman A, Casero RA, Constancia M, Charnock-Jones DS, Smith GC, Smith GCS, Placental polyamine metabolism differs by fetal sex, fetal growth restriction, and preeclampsia., JCI Insight. 3 (2018). 10.1172/jci.insight.120723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Myatt L, Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta., Placenta. 31 Suppl (2010) S66–9. 10.1016/j.placenta.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Muralimanoharan S, Maloyan A, Myatt L, Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: role of miR-143., Clin. Sci. (Lond). 130 (2016) 931–41. 10.1042/CS20160076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Estornell E, Tormo JR, Marin P, Renau-Piqueras J, Timoneda J, Barber T, Effects of vitamin A deficiency on mitochondrial function in rat liver and heart, Br. J. Nutr 84 (2000) 927–934. 10.1017/s0007114500002567. [DOI] [PubMed] [Google Scholar]
- [123].Acin-Perez R, Hoyos B, Zhao F, Vinogradov V, Fischman DA, Harris RA, Leitges M, Wongsiriroj N, Blaner WS, Manfredi G, Hammerling U, Control of oxidative phosphorylation by vitamin A illuminates a fundamental role in mitochondrial energy homoeostasis, FASEB J. 24 (2010) 627–636. 10.1096/fj.09-142281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Berdanier CD, Everts HB, Hermoyian C, Mathews CE, Role of vitamin A in mitochondrial gene expression, in: Diabetes Res. Clin. Pract, Elsevier, 2001: pp. S11–S27. 10.1016/S0168-8227(01)00331-X. [DOI] [PubMed] [Google Scholar]
- [125].Baker BC, Hayes DJL, Jones RL, Effects of micronutrients on placental function: Evidence from clinical studies to animal models, Reproduction. 156 (2018) R69–E82. 10.1530/REP-18-0130. [DOI] [PubMed] [Google Scholar]
- [126].Kucharská J, Vitamins in mitochondrial function, in: Mitochondrial Med. Mitochondrial Metab. Dis. Diagnosis Ther., Springer; Netherlands, 2008: pp. 367–384. 10.1007/978-1-4020-6714-3_21. [DOI] [Google Scholar]
- [127].Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD, Quantitative flux analysis reveals folate-dependent NADPH production, Nature. 510 (2014) 298–302. 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Depeint F, Bruce WR, Shangari N, Mehta R, O’Brien PJ, Mitochondrial function and toxicity: Role of the B vitamin family on mitochondrial energy metabolism, Chem. Biol. Interact 163 (2006) 94–112. 10.1016/j.cbi.2006.04.014. [DOI] [PubMed] [Google Scholar]
- [129].Finer S, Saravanan P, Hitman G, Yajnik C, The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity, Diabet. Med 31 (2014) 263–272. 10.1111/dme.12390. [DOI] [PubMed] [Google Scholar]
- [130].Knabl J, Vattai A, Ye Y, Jueckstock J, Hutter S, Kainer F, Mahner S, Jeschke U, Role of placental VDR expression and function in common late pregnancy disorders, Int. J. Mol. Sci 18 (2017). 10.3390/ijms18112340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Knabl J, Hiittenbrenner R, Hutter S, Giinthner-Biller M, Riedel C, Hiden U, Kainer F, Desoye G, Jeschke U, Gestational diabetes mellitus upregulates vitamin D receptor in extravillous trophoblasts and fetoplacental endothelial cells, Reprod. Sci 22 (2015) 358–366. 10.1177/1933719114542020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Van Etten E, Mathieu C, Immunoregulation by 1,25-dihydroxyvitamin D3: Basic concepts, in: J. Steroid Biochem. Mol. Biol, Pergamon, 2005: pp. 93–101. 10.1016/j.jsbmb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- [133].Albensi BC, What Is Nuclear Factor Kappa B (NF-κΒ) Doing in and to the Mitochondrion?, Front. Cell Dev. Biol 7 (2019) 154. 10.3389/fcell.2019.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Consiglio M, Destefanis M, Morena D, Foglizzo V, Forneris M, Pescarmona G, Silvagno F, The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation, PLoS One. 9 (2014). 10.1371/journal.pone.0115816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Shin JS, Choi MY, Longtine MS, Nelson DM, Vitamin D effects on pregnancy and the placenta., Placenta. 31 (2010) 1027–34. 10.1016/j.placenta.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mata-Greenwood E, Huber HF, Li C, Nathanielsz PW, Role of pregnancy and obesity on vitamin D status, transport, and metabolism in baboons, Am. J. Physiol. Metab 316 (2019) E63–E72. 10.1152/ajpendo.00208.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Tian X, Ma S, Wang Y, Hou L, Shi Y, Yao M, Wang X, Zhang H, Jiang L, Effects of Placental Ischemia Are Attenuated by 1,25-Dihydroxyvitamin D Treatment and Associated with Reduced Apoptosis and Increased Autophagy, DNA Cell Biol. 35 (2016) 59–70. 10.1089/dna.2015.2885. [DOI] [PubMed] [Google Scholar]
- [138].Greer FR, Vitamin K the basics-What’s new?, Early Hum. Dev 86 (2010) 43–47. 10.1016/j.earlhumdev.2010.01.015. [DOI] [PubMed] [Google Scholar]
- [139].Marriage B, Clandinin MT, Glerum DM, Nutritional cofactor treatment in mitochondrial disorders, J. Am. Diet. Assoc 103 (2003) 1029–1038. 10.1016/S0002-8223(03)00476-0. [DOI] [PubMed] [Google Scholar]
- [140].KC S, Cárcamo JM, Golde DW, Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glutl) and confers mitochondrial protection against oxidative injury, FASEB J. 19 (2005) 1657–1667. 10.1096/fj.05-4107com. [DOI] [PubMed] [Google Scholar]
- [141].Guaiquil VH, Vera JC, Golde DW, Mechanism of Vitamin C Inhibition of Cell Death Induced by Oxidative Stress in Glutathione-depleted HL-60 Cells, J. Biol. Chem 276 (2001) 40955–40961. 10.1074/jbc.M106878200. [DOI] [PubMed] [Google Scholar]
- [142].Kawashima A, Sekizawa A, Koide K, Hasegawa J, Satoh K, Arakaki T, Takenaka S, Matsuoka R, Vitamin C induces the reduction of oxidative stress and paradoxically stimulates the apoptotic gene expression in extravillous trophoblasts derived from first trimester tissue, Reprod. Sci 22 (2015) 783–790. 10.1177/1933719114561561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Belin S, Kaya F, Burtey S, Fontes M, Ascorbic Acid and Gene Expression: Another Example of Regulation of Gene Expression by Small Molecules?, Curr. Genomics. 11 (2009) 52–57. 10.2174/138920210790217936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Wang X, Quinn PJ, The location and function of vitamin E in membranes (Review), Mol. Membr. Biol 17 (2000) 143–156. 10.1080/09687680010000311. [DOI] [PubMed] [Google Scholar]
- [145].Distelmaier F, Valsecchi F, Forkink M, Van Emst-De Vries S, Swarts HG, Rodenburg RJT, Verwiel ETP, Smeitink JAM, Willems PHGM, Koopman WJH, Trolox-sensitive reactive oxygen species regulate mitochondrial morphology, oxidative phosphorylation and cytosolic calcium handling in healthy cells, Antioxidants Redox Signal. 17 (2012) 1657–1669. 10.1089/ars.2011.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Davargaon RS, Sambe AD, Muthangi V V S, Toxic effect of high glucose on cardiomyocytes, H9c2 cells: Induction of oxidative stress and ameliorative effect of trolox, J. Biochem. Mol. Toxicol 33 (2019) e22272. 10.1002/jbt.22272. [DOI] [PubMed] [Google Scholar]
- [147].Guan Z, fang Li H, li Guo L, Yang X, Effects of vitamin C, vitamin E, and molecular hydrogen on the placental function in trophoblast cells, Arch. Gynecol. Obstet 292 (2015) 337–342. 10.1007/s00404-015-3647-8. [DOI] [PubMed] [Google Scholar]
- [148].Poston L, Igosheva N, Mistry HD, Seed PT, Shennan AH, Rana S, Karumanchi SA, Chappell LC, Role of oxidative stress and antioxidant supplementation in pregnancy disorders, Am. J. Clin. Nutr 94 (2011) 1980S–1985S. 10.3945/ajcn.110.001156. [DOI] [PubMed] [Google Scholar]
- [149].Maged AM, Torky H, Fouad MA, GadAllah SH, Waked NM, Gayed AS, Salem AK, Role of antioxidants in gestational diabetes mellitus and relation to fetal outcome: a randomized controlled trial, J. Matern. Neonatal Med 29 (2016) 4049–4054. 10.3109/14767058.2016.1154526. [DOI] [PubMed] [Google Scholar]
- [150].Zhong L, Holmgren A, Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations., J. Biol. Chem 275 (2000) 18121–8. 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- [151].Na JY, Seok J, Park S, Kim JS, Kim GJ, Effects of selenium on the survival and invasion of trophoblasts., Clin. Exp. Reprod. Med 45 (2018) 10–16. 10.5653/cerm.2018.45.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hofstee P, Bartho LA, McKeating DR, Radenkovic F, McEnroe G, Fisher JJ, Holland OJ, Vanderlelie JJ, Perkins AV, Cuffe JSM, Maternal selenium deficiency during pregnancy in mice increases thyroid hormone concentrations, alters placental function and reduces fetal growth, J. Physiol 597 (2019) 5597–5617. 10.1113/JP278473. [DOI] [PubMed] [Google Scholar]
- [153].Khera A, Dong L, Holland O, Vanderlelie J, Pasdar EA, Neuzil J, Perkins AV, Selenium supplementation induces mitochondrial biogenesis in trophoblasts, Placenta. 36 (2015) 863–869. 10.1016/J.PLACENTA.2015.06.010. [DOI] [PubMed] [Google Scholar]
- [154].Sagrillo-Fagundes L, Bienvenue-Pariseault J, Vaillancourt C, Melatonin: The smart molecule that differentially modulates autophagy in tumor and normal placental cells, PLoS One. 14 (2019). 10.1371/journal.pone.0202458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Zang LY, Cosma G, Gardner H, Vallyathan V, Scavenging of reactive oxygen species by melatonin, Biochim. Biophys. Acta - Gen. Subj 1425 (1998) 469–477. 10.1016/S0304-4165(98)00099-3. [DOI] [PubMed] [Google Scholar]
- [156].Fischer TW, Kleszczyhski K, Hardkop LH, Kruse N, Zillikens D, Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2’-deoxyguanosine) in ex vivo human skin, J. Pineal Res 54 (2013) 303–312. 10.1111/jpi.12018. [DOI] [PubMed] [Google Scholar]
- [157].Hannan NJ, Binder NK, Beard S, Nguyen TV, Kaitu’u-Lino TJ, Tong S, Melatonin enhances antioxidant molecules in the placenta, reduces secretion of soluble fms-like tyrosine kinase 1 (sFLT) from primary trophoblast but does not rescue endothelial dysfunction: An evaluation of its potential to treat preeclampsia, PLoS One. 13 (2018). 10.1371/journal.pone.0187082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Lanoix D, Lacasse A-A, Reiter RJ, Vaillancourt C, Melatonin: The watchdog of villous trophoblast homeostasis against hypoxia/reoxygenation-induced oxidative stress and apoptosis, Mol. Cell. Endocrinol 381 (2013) 35–45. 10.1016/J.MCE.2013.07.010. [DOI] [PubMed] [Google Scholar]
- [159].Milczarek R, Hallmann A, Sokotowska E, Kaletha K, Klimek J, Melatonin enhances antioxidant action of α-tocopherol and ascorbate against NADPH- and iron-dependent lipid peroxidation in human placental mitochondria, J. Pineal Res 49 (2010) no–no. 10.1111/j.1600-079X.2010.00779.x. [DOI] [PubMed] [Google Scholar]
- [160].Nuzzo AM, Camm EJ, Sferruzzi-Perri AN, Ashmore TJ, Yung H-W, Cindrova-Davies T, Spiroski A-M, Sutherland MR, Logan A, Austin-Williams S, Burton GJ, Rolfo A, Todros T, Murphy MP, Giussani DA, Placental Adaptation to Early-Onset Hypoxic Pregnancy and Mitochondria-Targeted Antioxidant Therapy in a Rodent Model., Am. J. Pathol 188 (2018) 2704–2716. 10.1016/j.ajpath.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RAJ, Cocheme HM, Murphy MP, Dominiczak AF, Mitochondria-targeted antioxidant MitoQlO improves endothelial function and attenuates cardiac hypertrophy., Hypertens. (Dallas, Tex. 1979). 54 (2009) 322–8. 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- [162].Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA, Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury, FASEB J. 19 (2005) 1088–1095. 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- [163].Gottwald EM, Duss M, Bugarski M, Haenni D, Schuh CD, Landau EM, Hall AM, The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue., Physiol. Rep 6 (2018) e13667. 10.14814/phy2.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RAJ, Murphy MP, Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties, J. Biol. Chem 276 (2001) 4588–4596. 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- [165].Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V, Mitochondrially targeted compounds and their impact on cellular bioenergetics., Redox Biol. 1 (2013) 86–93. 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ganguly E, Aljunaidy MM, Kirschenman R, Spaans F, Morton JS, Phillips TEJ, Case CP, Cooke C-LM, Davidge ST, Sex-Specific Effects of Nanoparticle-Encapsulated MitoQ (nMitoQ) Delivery to the Placenta in a Rat Model of Fetal Hypoxia, Front. Physiol 10 (2019) 562. 10.3389/fphys.2019.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Phillips TJ, Scott H, Menassa DA, Bignell AL, Sood A, Morton JS, Akagi T, Azuma K, Rogers MF, Gilmore CE, Inman GJ, Grant S, Chung Y, Aljunaidy MM, Cooke CL, Steinkraus BR, Pocklington A, Logan A, Collett GP, Kemp H, Holmans PA, Murphy MP, Fulga TA, Coney AM, Akashi M, Davidge ST, Case CP, Treating the placenta to prevent adverse effects of gestational hypoxia on fetal brain development, Sci. Rep 7 (2017). 10.1038/s41598-017-06300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Higa R, Roberti S, Mazzucco MB, White V, Jawerbaum A, Effect of the antioxidant idebenone on maternal diabetes-induced embryo alterations during early organogenesis, Reprod. Biomed. Online. 37 (2018) 397–408. 10.1016/j.rbmo.2018.05.006. [DOI] [PubMed] [Google Scholar]
- [169].Skulachev VP, Anisimov VN, Antonenko YN, Bakeeva LE, Chernyak BV, Erichev VP, Filenko OF, Kalinina NI, Kapelko VI, Kolosova NG, Kopnin BP, Korshunova GA, Lichinitser MR, Obukhova LA, Pasyukova EG, Pisarenko OI, Roginsky VA, Ruuge EK, Senin II, Severina II, Skulachev MV, Spivak IM, Tashlitsky VN, Tkachuk VA, Vyssokikh MY, Yaguzhinsky LS, Zorov DB, An attempt to prevent senescence: A mitochondrial approach, Biochim. Biophys. Acta - Bioenerg 1787 (2009) 437–461. 10.1016/j.bbabio.2008.12.008. [DOI] [PubMed] [Google Scholar]
- [170].Higa R, Roberti S, Mazzucco MB, White V, Jawerbaum A, Effect of the antioxidant idebenone on maternal diabetes-induced embryo alterations during early organogenesis., Reprod. Biomed. Online. 37 (2018) 397–408. 10.1016/j.rbmo.2018.05.006. [DOI] [PubMed] [Google Scholar]
- [171].Teran E, Hernández I, Tana L, Teran S, Galaviz-Hernandez C, Sosa-Macias M, Molina G, Calie A, Mitochondria and Coenzyme Q10 in the Pathogenesis of Preeclampsia., Front. Physiol 9 (2018) 1561. 10.3389/fphys.2018.01561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Szeto HH, First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics., Br. J. Pharmacol 171 (2014) 2029–50. 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT, A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36., J. Biol. Chem 282 (2007) 4634–42. 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- [174].Reelfs O, Abbate V, Hider RC, Pourzand C, A Powerful Mitochondria-Targeted Iron Chelator Affords High Photoprotection against Solar Ultraviolet A Radiation., J. Invest. Dermatol 136 (2016) 1692–1700. 10.1016/j.jid.2016.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Seminotti B, Leipnitz G, Karunanidhi A, Kochersperger C, Roginskaya VY, Basu S, Wang Y, Wipf P, Van Houten B, Mohsen A-W, Vockley J, Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers., Hum. Mol. Genet 28 (2019) 928–941. 10.1093/hmg/ddy403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Dehkordi AH, Abbaszadeh A, Mir S, Hasanvand A, Metformin and its antiinflammatory and anti-oxidative effects; new concepts, J. Ren. Inj. Prev 8 (2018) 54–61. 10.15171/jrip.2019.11. [DOI] [Google Scholar]
- [177].Rena G, Hardie DG, Pearson ER, The mechanisms of action of metformin., Diabetologia. 60 (2017) 1577–1585. 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F, Cellular and molecular mechanisms of metformin: an overview., Clin. Sci. (Lond). 122 (2012) 253–70. 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hou X, Song J, Li XN, Zhang L, Wang XL, Chen L, Shen YH, Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOX03 pathway, Biochem. Biophys. Res. Commun 396 (2010) 199–205. 10.1016/j.bbrc.2010.04.017. [DOI] [PubMed] [Google Scholar]
- [180].Alhaider AA, Korashy HM, Sayed-Ahmed MM, Mobark M, Kfoury H, Mansour MA, Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression, Chem. Biol. Interact 192 (2011) 233–242. 10.1016/j.cbi.2011.03.014. [DOI] [PubMed] [Google Scholar]
- [181].Romero R, Erez O, Hüttemann M, Maymon E, Panaitescu B, Conde-Agudelo A, Pacora P, Yoon BH, Grossman L.l., Metformin, the aspirin of the 21st century: its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity., Am. J. Obstet. Gynecol 217 (2017) 282–302. 10.1016/j.ajog.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Lindsay RS, Loeken MR, Metformin use in pregnancy: promises and uncertainties, Diabetologia. 60 (2017) 1612–1619. 10.1007/s00125-017-4351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Yang Y, Gao H, Zhou H, Liu Q, Qi Z, Zhang Y, Zhang J, The role of mitochondria-derived peptides in cardiovascular disease: Recent updates, Biomed. Pharmacother 117 (2019) 109075. 10.1016/j.biopha.2019.109075. [DOI] [PubMed] [Google Scholar]
- [184].Yen K, Lee C, Mehta H, Cohen P, The emerging role of the mitochondrial-derived peptide humanin in stress resistance., J. Mol. Endocrinol 50 (2013) R11–9. 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Kim S-J, Xiao J, Wan J, Cohen P, Yen K, Mitochondrially derived peptides as novel regulators of metabolism., J. Physiol 595 (2017) 6613–6621. 10.1113/JP274472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kuliawat R, Klein L, Gong Z, Nicoletta-Gentile M, Nemkal A, Cui L, Bastie C, Su K, Huffman D, Surana M, Barzilai N, Fleischer N, Muzumdar R, Potent humanin analog increases glucose-stimulated insulin secretion through enhanced metabolism in the cell, FASEB J. 27 (2013) 4890–4898. 10.1096/fj.13-231092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH, Poduval A, Hwang D, Barzilai N, Cohen P, Humanin: A novel central regulator of peripheral insulin action, PLoS One. 4 (2009). 10.1371/journal.pone.0006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim S-J, Mehta H, Hevener AL, de Cabo R, Cohen P, The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance., Cell Metab. 21 (2015) 443–54. 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Lee C, Nuclear transcriptional regulation by mitochondrial-encoded MOTS-c, Mol. Cell. Oncol 6 (2019) 1–2. 10.1080/23723556.2018.1549464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, Wan J, Muzumdar R, Barzilai N, Cohen P, Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers., Aging (Albany. NY). 8 (2016) 796–809. 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Cai L, Lin C, Yang N, Huang Z, Miao S, Chen X, Pan J, Rao P, Liu S, Preparation and characterization of nanoparticles made from co-incubation of SOD and glucose, Nanomaterials. 7 (2017). 10.3390/nano7120458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Mao GD, Thomas PD, Lopaschuk GD, Poznansky MJ, Superoxide dismutase (SOD)-catalase conjugates. Role of hydrogen peroxide and the Fenton reaction in SOD toxicity, J. Biol. Chem 268 (1993) 416–420. [PubMed] [Google Scholar]
- [193].Mauricio MD, Guerra-Ojeda S, Marchio P, Valles SL, Aldasoro M, Escribano-Lopez I, Herance JR, Rocha M, Vila JM, Victor VM, Nanoparticles in Medicine: A Focus on Vascular Oxidative Stress, Oxid. Med. Cell. Longev 2018 (2018) 6231482. 10.1155/2018/6231482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Kakimoto K, Kojima Y, Ishii K, Onoue K, Maeda H, The suppressive effect of gelatin-conjugated superoxide dismutase on disease development and severity of collagen-induced arthritis in mice, Clin. Exp. Immunol 94 (2008) 241–246. 10.1111/j.1365-2249.1993.tb03438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H, Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD, Science (80-). 244 (1989) 974–976. 10.1126/science.2543070. [DOI] [PubMed] [Google Scholar]
- [196].Cruz MEM, Gaspar MM, Martins MBF, Corvo ML, Liposomal superoxide dismutases and their use in the treatment of experimental arthritis, Methods Enzymol. 391 (2005) 395–413. 10.1016/S0076-6879(05)91022-7. [DOI] [PubMed] [Google Scholar]
- [197].Mansuroǧlu B, Derman S, Yaba A, Kizilbey K, Protective effect of chemically modified SOD on lipid peroxidation and antioxidant status in diabetic rats, Int. J. Biol. Macromol 72 (2015) 79–87. 10.1016/j.ijbiomac.2014.07.039. [DOI] [PubMed] [Google Scholar]
- [198].Reddy MK, Labhasetwar V, Nanoparticle-mediated delivery of superoxide dismutase to the brain: an effective strategy to reduce ischemia-reperfusion injury, FASEB J. 23 (2009) 1384–1395. 10.1096/fj.08-116947. [DOI] [PubMed] [Google Scholar]
- [199].Chen YP, Chen CT, Hung Y, Chou CM, Liu TP, Liang MR, Chen CT, Mou CY, A new strategy for intracellular delivery of enzyme using mesoporous silica nanoparticles: Superoxide dismutase, J. Am. Chem. Soc 135 (2013) 1516–1523. 10.1021/ja3105208. [DOI] [PubMed] [Google Scholar]
- [200].Aldini G, Altomare A, Baron G, Vistoli G, Carini M, Borsani L, Sergio F, N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why, Free Radic. Res 52 (2018) 751–762. 10.1080/10715762.2018.1468564. [DOI] [PubMed] [Google Scholar]
- [201].Greco T, Shafer J, Fiskum G, Sulforaphane inhibits mitochondrial permeability transition and oxidative stress, Free Radic. Biol. Med 51 (2011) 2164–2171. 10.1016/j.freeradbiomed.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Briones-Herrera A, Avila-Rojas SH, Aparicio-Trejo OE, Cristóbal M, León-Contreras JC, Hernández-Pando R, Pinzón E, Pedraza-Chaverri J, Sanchez-Lozada LG, Tapia E, Sulforaphane prevents maleic acid-induced nephropathy by modulating renal hemodynamics, mitochondrial bioenergetics and oxidative stress, Food Chem. Toxicol 115 (2018) 185–197. 10.1016/j.fct.2018.03.016. [DOI] [PubMed] [Google Scholar]
- [203].Negrette-Guzmán M, Huerta-Yepez S, Tapia E, Pedraza-Chaverri J, Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: A seemingly contradictory dual role and an integrative hypothesis, Free Radic. Biol. Med 65 (2013) 1078–1089. 10.1016/j.freeradbiomed.2013.08.182. [DOI] [PubMed] [Google Scholar]
- [204].Poljsak B, Strategies for reducing or preventing the generation of oxidative stress., Oxid. Med. Cell. Longev 2011 (2011) 194586. 10.1155/2011/194586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Radák Z, Nakamura A, Nakamoto H, Asano K, Ohno H, Goto S, A period of anaerobic exercise increases the accumulation of reactive carbonyl derivatives in the lungs of rats, Pfliigers Arch. Eur. J. Physiol 435 (1998) 439–441. 10.1007/s004240050537. [DOI] [PubMed] [Google Scholar]
- [206].Kagan VE, Serbinova EA, Stoyanovsky DA, Khwaja S, Packer L, Assay of ubiquinones and ubiquinols as antioxidants, Methods Enzymol. 234 (1994) 343–354. 10.1016/0076-6879(94)34104-4. [DOI] [PubMed] [Google Scholar]
- [207].Mehta SL, Kumari S, Mendelev N, Li PA, Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia., BMC Neurosci. 13 (2012) 79. 10.1186/1471-2202-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Tinggi U, Selenium: Its role as antioxidant in human health, in: Environ. Health Prev. Med, BioMed Central, 2008: pp. 102–108. 10.1007/s12199-007-0019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Matsunaga D, Sreekumar PG, Ishikawa K, Terasaki H, Barron E, Cohen P, Kannan R, Hinton DR, Humanin Protects RPE Cells from Endoplasmic Reticulum Stress-Induced Apoptosis by Upregulation of Mitochondrial Glutathione, PLoS One. 11 (2016) e0165150. 10.1371/journal.pone.0165150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Wright DJ, Renoir T, Smith ZM, Frazier AE, Francis PS, Thorburn DR, McGee SL, Hannan AJ, Gray LJ, N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease, Transl. Psychiatry. 5 (2015) e492. 10.1038/tp.2014.131. [DOI] [PMC free article] [PubMed] [Google Scholar]