

Abstract
Cancer is a complex disease and cancer development takes 10–50 years involving epigenetics. Evidence suggests that ~80% of human cancers are linked to environmental factors impinging upon genetics/epigenetics. Since advanced metastasized cancers are resistant to radiation/chemotherapeutic drugs, cancer prevention by relatively non-toxic chemopreventive “epigenetic modifiers” involving epigenetics/epigenomics is logical. Isothiocyanates are relatively non-toxic at low nutritional and even higher pharmacological doses, with good oral bioavailability, potent anti-oxidative stress/anti-inflammatory activities, possess epigenetic modifying properties, great anti-cancer efficacy in many in vitro cell culture and in vivo animal models. This review summarizes the latest advances on the role of epigenetics/epigenomics by isothiocyanates in prevention of skin, colon, lung, breast, and prostate cancers. The exact molecular mechanism how isothiocyanates modify the epigenetic/epigenomic machinery is unclear. We postulate “redox” processes would play important roles. Additionally, isothiocyanates sulforaphane and phenethyl-isothiocyanate, possess multifaceted molecular mechanisms would be considered as “general” cancer preventive agents not unlike chemotherapeutic agents like platinum-based or taxane-based drugs. Analogous to chemotherapeutic agents, the isothiocyanates would need to be used in combination with other non-toxic chemopreventive phytochemicals or drugs such as NSAIDs, 5-α-reductase/aromatase inhibitors targeting different signaling pathways would be logical for the prevention of progression of tumors to late advanced metastatic states.
Keywords: Isothiocyanates (ITCs), Cancer Chemoprevention, Epigenetics/epigenomics, skin, colon, lung, breast, prostate cancer
Introduction
Cancer is a complex chronic disease and cancer development is a multistep process (1, 2). In a simplified manner, it would involve initiation, promotion, to progression/metastasis (3, 4). Recent evidence suggests that many chronic illnesses including cancer are driven by epigenetics/epigenomics caused by environmental factors impinging upon the underlying genetic information (4–7). Since advanced metastasized cancer are resistant to radiation and chemotherapeutic drugs, prevention of early stages of cancer by relatively non-toxic dietary phytochemicals would be logical.
Role of epigenetics/epigenomics during the “long process” of cancer development
During the long process of cancer development particularly during the “promotion” stage, epigenetics has been postulated to play an important role in driving cellular transformation such as stem cells in forming benign microscopic tumor (8). Feinberg et al. has shown since the 1980s that most if not all tumors could be associated with widespread losses and some gains of DNA methylation throughout the genome (9, 10). This is reviewed recently in (6, 11), and elaborated on the role of epigenetics during cancer development However it is still unclear how epigenetics would be integrated with the different hallmarks of cancer as discussed by Weinberg et al. (12).
Epigenetics or in a more global context epigenomics is a very complex process involving modulation of epigenetic modifiers including the ‘readers’, ‘writers’ and ‘erasers’ with involvement of multiple events including methylation, acetylation, ubiquitination. (13). Accumulating evidence also suggests that multiple genetic and epigenetic alterations would occur concurrently during tumor development (6, 14). In this context, the epigenetic marks (DNA methylation, histone modifications, chromatin remodeling, and non-coding RNA) could be use as potential markers of different stages of cancer development including initiation, progression, and metastasis (15, 16).
From our recent UVB-induced skin carcinogenesis study, global CpG methylation changes more dramatically in early stages of 2 weeks as compared to 15 weeks and 25 weeks post UVB irradiation. The CpG methylome changes decreased as time progresses taken into account of the effect of aging (17). Imaging assisted evaluation of microscopic tumors may have suggested that during the early promotion stage of carcinogenesis, drastic epigenomic aberrations occurs particularly with the progenitor stem cells driving phenotypic gene expression, may occur before mutational events (6). These epigenomic alterations would involve stem cell dysregulation blocking differentiation, dysregulation of multiple pathways including DNA repair and cell cycle, apoptosis/autophagy, cellular defense, and inflammatory pathways. However, the underlying mechanism of UVB-induced global epigenomic changes in the skin is unclear. Acutely, UVB triggers oxidative stress, inflammation and DNA damage (18, 19), would be postulated to be some of the major drivers of epigenetic alterations.
Analyzing primary cutaneous melanoma samples for their global DNA methylation, found more than 98% loss of methylation and about 2% gain of methylation (20). In vitro study using melanoma cells showed that histone hypoacetylation is associated with downregulation of certain pro-apoptotic proteins, including Bak, Bim, and Bax, which belong to the BCL-2 family (21). Epigenetic reprogramming appears to play an increasing role in drug resistant in melanoma (22). In skin transformation model using epidermal JB6 cells, when JB6 cells were challenged with tumor promoter TPA (12-O-Tetradecanoylphorbol-13-acetate), significant alterations of DNA methylome coupled with transcriptome (23). In a recent UVB-induced mouse carcinogenesis epigenomic study, about 60% of the DNA-methylated-regions (DMRs) showed inversed relationship between DNA methylation and RNA expression, i.e., hypermethylation coupled with suppression of transcription or hypomethylation coupled with promotion of transcription, while the other 40% of these DMRs did not show such relationship (23).
In breast cancer, many studies have shown strong association of aberrant DNA hypomethylation with cancer development (24). Histone modifications such as loss of H4K16 acetylation has been implicated as an early event in breast cancer development and is associated with altered level of NAD-dependent histone deacetylases (HDACs); SIRT1 (25, 26).
In prostate cancer, up-regulation of EZH2 (Histone Methyltransferase, Enhancer of Zeste Homolog 2) appears common in both localized and metastatic prostate cancer, and associated with poor prognosis (27, 28). In early prostate carcinogenesis, during the transition from benign prostate epithelium to inflammatory lesions, DNA hypermethylation is observed in the promoter regions of key tumor suppresser genes such as GSTP1, RASSF1A, and APC (29, 30). In TRAMP mice and TRAMP-C1 cell line, hypermethylation of Nrf2’s CpGs occurred (31), and global alteration of epigenomic DNA methylation was found with profiling using MeDIP-seq in TRAMP prostate tumor (32). In a PTEN deletion mouse prostate carcinogenesis model, PTEN deletion drives aberrations of DNA methylome and transcriptome in different stages of prostate carcinogenesis (33). Prostate cancer exhibited global alterations of histone modifications, including histone acetylation (H3K9, H3K18, and H4K12) and methylation (H3K4me2) (34). Gerhauser et al. (35) investigated the molecular evolution of 292 early-onset prostate cancer patients using Illuminia 450K methylation array identified some unique DNA methylation patterns of 500 most discriminatory CpG sites between the different cell types (basal, stromal, normal luminal and tumor luminal) and found that the “purity-adjusted epigenetic prostate cancer index” (PEPCI) associated with increased Gleason score (36). By integrating the DNA methylation and RNA expression data from tumors diagnosed with early-onset, they were able to identify four robust subgroups which could stratify the patients into high- and low-risk groups(35).
Epigenetics plays an increasing role in the pathophysiology of colorectal cancer (CRC) (37). In an AOM-DSS induced colitis-accelerated colon cancer mouse model, differential CpG methylome and transcriptome occurs as compared to control mice (38). CpG island methylator phenotype (CIMP), has been identified as one of the molecular features of CRC (39, 40). Based on CIMP profiles, primary CRC may be clustered into three distinct but relatively homogeneous subclasses: CIMP1, characterized by intense methylation of multiple genes including MSI and BRAF mutations; CIMP2, increased methylation with age‐related genes, and mutations in KRAS; and CIMP negative, characterized by rare methylation with p53 mutation (39). CIMP1 and CIMP2 phenotypes are more often expressed in the proximal colon; CIMP1 has a good prognosis, whereas CIMP2 has a poor prognosis (41). In general, CIMP appears to be associated with significantly worse prognosis in CRC patients (42).
Overview of epigenetic mechanisms and their role in cancer prevention
Epigenetic mechanism appears to provide a robust means for organisms to respond to environmental cues through changes in gene expression (43), working in concert with other more rapid responders including channels/receptors mediated signaling. These post-translational modifications act in a coordinated fashion leading to chromatin conformational alterations that, in turn, regulate the genetic information accessed by transcription factors (44). The schematic representation of epigenetic pathways in stepwise carcinogenesis process and cancer prevention by isothiocyanates (ITCs) impinging upon and integrating with the different cellular processes are summarized in Figure 1. Epigenetic machinery would affect all the stages during carcinogenesis. Among the three epigenetic mechanisms, DNA CpG methylation is probably the most studied as reviewed by Feinberg (6). Specifically, DNA methylation entails the conjugation of methyl groups in cytosine (C) residues usually occur in CpG dinucleotide sequences, dispersed across the genome, leading to the formation of 5-methylcytosine (5mC) (45). DNA methylation is catalyzed by a class of enzymes known as DNA methyltransferases (DNMTs) the most predominate which include DNMT1, DNMT3A and DNMT3B. Unlike methylation, histone acetylation causes gene activation whereas histone methylation can result in either activation or repression of genes depending on the site of modifications (46). Histone deacetylases (HDACs), along with histone acetyltransferases (HAT), provide a significant mechanism of gene regulation involving removal and addition, of acetyl groups from an ε-N-acetyl lysine amino acid on histone proteins. The third epigenetic control would be regulated through non-coding-RNA-based mechanisms including micro-RNA. In particular, micro-RNA transcription can be regulated by both histone modification and DNA methylation, and micro-RNA themselves can, in turn, regulate key enzymes that enforce epigenetic remodeling (47).
Figure 1.
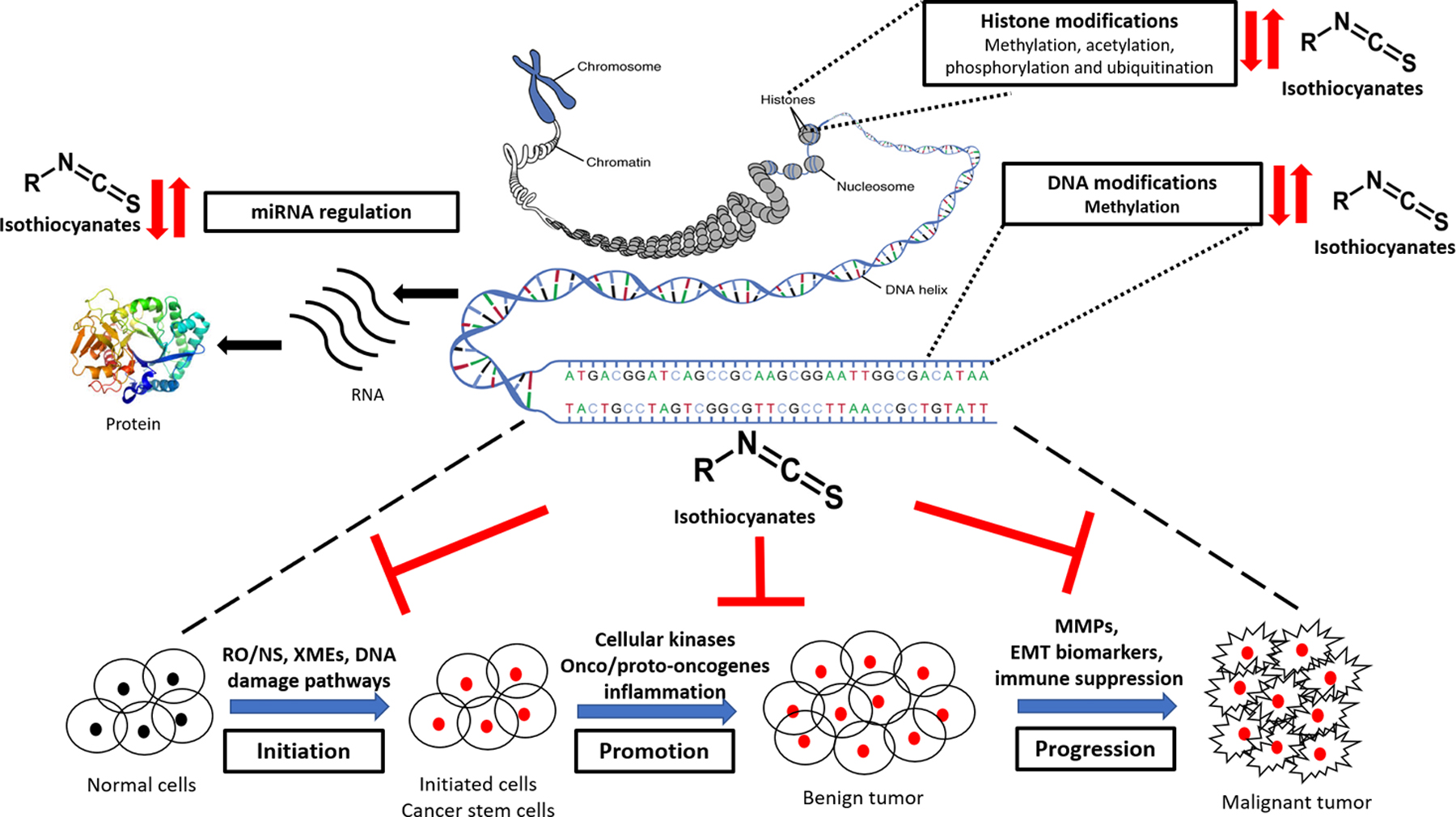
Schematic representation of epigenetic pathways in stepwise carcinogenesis process and chemopreventive effects of isothiocyanates (ITCs) impinging upon and integrating with the cellular processes during cancer development. Major epigenetic mechanism involved (A) Histone modification, (B) DNA modifications, and (C) miRNA regulation. The role of ITCs in modifying/reversing of these epigenetic mechanisms and signaling pathways including reactive oxygen/nitrogen species (RO/NS), xenobiotic metabolizing enzymes (XMEs), DNA damage/repair, cellular kinases, onco/proto-oncogenes, inflammatory, matrix metalloproteinases (MMPs), epithelial-mesenchymal transition (EMT) biomarkers, and immune suppression, among others, in the process of carcinogenesis viz. initiation, promotion and progression. (↑ activation, ↓ inhibition, ⊥ blocking)
Several chemotherapeutic drugs targeting epigenetic signaling have been approved by the FDA (14), and a growing list of dietary phytochemicals found naturally in food and plants have been studied targeting epigenetic mechanism during the carcinogenesis process for cancer prevention including modifications of histones and DNA methylations (48, 49). For instance, curcumin, a powerful anti-inflammatory/antioxidant agent, regulating multiple signaling pathways, is also found to modulate epigenetics/epigenomics pathways such as histones modifications, and DNA methylation (50, 51). Many natural products have been reported to inhibit the expression of HDACs, including curcumin (52, 53), ursolic acid (54), and SFN (55, 56). Triterpenoids such as cucurbitacin, ginsenoside Rh2, ginsenoside compound K and ursolic acid, have been shown to induce global hypomethylation, enhances hypermethylation/hypomethylation of the promoters of oncogenes and tumor suppressor genes, and modifying miRNA by targeting DNMT1 (48). SFN could regulates DNA demethylation by down-regulating the expression of DNMT1 and DNMT3B, thus inducing the demethylation of the cyclin D2 gene promoter and its expression in cancer cells (57). Question remains as to how all these chemopreventive phytochemicals modulate the epigenetic machinery. Since many of these compounds are redox active, therefore, we would like to postulate that redox signaling triggered by these compounds could play a role in modulating the epigenetic machinery, but further study would be needed.
Role of redox signaling in the modulation of epigenetic machinery
Cellular oxidative stress occurs when the cellular antioxidant/reductive capacity is overwhelmed by the oxidative challenges (58, 59), and could play a vital role in epigenetic reprogramming (60). Many epigenetic marks, including histone methylation, histone acetylation, ADP-ribosylation and DNA methylation, may be directly associated with central metabolism through redox intermediates (60). One mechanism by which reactive oxygen species (ROS) can affect DNA methylation is through its action on DNMTs activity or expression (61). In an oxidizing redox chromatin microenvironment and in the absence of S-adenosyl methionine (SAM), DNMT3a and DNMT3b can catalyze the direct conversion of 5mC or 5-hydroxymethylcytosine (5hmC) to an unmodified C, although the exact mechanisms are still unclear (62). Ye et al. have proposed an iron–redox–methylation hypothesis (63) in which excess amount of irons perturbs DNA methylation by at least two possible mechanisms: first, oxidative stress depletes GSH and drives SAM/SAH cycle to produce more SAH, which inhibits DNMT activity; and second, iron directly inhibits DNMT activity. The methylation process can be reversed by DNA demethylases such as ten-eleven translocation enzymes (TETs) (64). ROS are also thought to regulate DNA methylation by targeting the expression and/or activity of the TETs. Knockdown of TET1 significantly increase hydrogen peroxide-induced apoptosis of cerebellar granule cells, and cerebellar granule cells from Tet1 knockout mice are more sensitive to oxidative stress (65). In addition, mutagenic lesion of ROS-induced DNA damage of O6-methylguanine can inhibit binding of DNMT leading to hypomethylation with inhibition of methylation of adjacent cytosine nucleotides (66). Incorporation of 8-OHdG and the oxidation by-product of 5-mC in the Methyl-CpG binding protein (MBP) recognition sequence resulted in significant inhibition of the binding affinity of MBP (67).
In addition to epigenetic regulation of DNA methylation, redox also participates in histone acetylation and histone methylation. The histone deacetylation is mediated HDACs. A study has shown that the redox-regulating protein Thioredoxin 1 (Trx1) regulates the nucleocytoplasmic shuttling of class II HDACs through a redox-dependent mechanism (68). By forming a multiprotein complex with DnaJb5, a heat shock protein 40, and TBP-2, a Trx1-binding protein, Trx1 reduces HDAC4, a class II HDAC, at Cys-667 and Cys-669, which are easily oxidized to form a disulfide bond in response to hypertrophic stimuli(69). Hu, et al. showed that the catalytic activity of HDAC5 suppresses mitochondrial ROS generation and subsequent induction of NRF2-dependent antioxidant gene expression in cardiomyocytes (70). Collectively, there is strong evidence that ROS are capable to modulate chromatin accessibility by affecting histone acetylation state in particular via multiple modifications of HDAC expression and activity (71, 72). Histone acetylation is reversible and is regulated by a group of histone acetyltransferases (HATs) (73). Histone methylation is maintained by two classes of enzymes: histone methyltransferases (HMTs) and histone demethylases (HDMs) (74). HMTs and HDMs can also be subjected to redox regulation such that post-translational modifications of histone proteins by oxidants and environmental stresses can trigger gene transcription involved in chronic inflammatory events (75). Since many dietary chemopreventive phytochemicals possess redox (76, 77) and epigenetic/epigenomic properties (78), question remains the direct linkage of phytochemical’s redox signaling and epigenetics/epigenomics.
Isothiocyanates and cancer prevention
Isothiocyanates (ITCs) are a family of compounds derived almost exclusively from plants, although marine sponges and fungi also have been reported to produce a few ITCs (79, 80). ITCs are synthesized and stored as glucosinolates (β-thioglucoside N-hydroxysulfates) as secondary metabolites in plants including the cruciferous. Glucosinolates are relatively stable in plant cell. However, when the plant tissue containing glucosinolates is damaged, as in the case of preparation (cutting, chopping, mixing) or chewing food, a β-thioglucosidase called myrosinase is released and hydrolyzed the glucosinolates to various metabolites, including the ITCs (81). When cruciferous are cooked before consumption, myrosinase is inactivated and glucosinolates transit to the colon where they are hydrolyzed by the intestinal microbiota to ITCs (81). By far the most studied ITC is sulforaphane (SFN), derived from broccoli (80), although other ITCs, including phenylethyl ITC (PEITC), benzyl ITC (BITC) and allyl ITC (AITC), also possess chemopreventive properties. Multiple molecular mechanisms are involved in the chemopreventive effect of ITCs. Below, we will focus on epigenetic mechanisms in in vitro and in vivo studies in skin, breast, lung, colon and prostate cancer, as well as some clinical chemoprevention studies with ITCs.
Skin cancer
The ITCs show chemopreventive effects against UV- and chemically-induced skin carcinogenesis (82) and the involvement of the epigenetics in epidermal carcinogenesis has been studied in melanoma (83). The activity of SFN on epigenetic regulation of skin cancer has been reported on PcG proteins’ (e.g. Bmi1 and EZH2) function by means of blocking tumor progression through their inhibition thus decreasing H3K27me3 level. (84). In squamous cell carcinoma (SCC), SFN treatment suppressed cancer progression and metastasis in vivo through reduction in arginine methylation at H3 (85). As discussed earlier, UVB would drive perturbation of CpG methylome at initiation, promotion and progression stages of skin carcinogenesis, but importantly, SFN would block carcinogenesis more effectively at early stages than late stages and would attenuate some of the perturbations of UVB-induced CpG methylome (86). In cell culture model, SFN was also found to demethylate CpGs of Nuclear factor erythroid 2-related factor 2 (NRF2) promoter, resulting in re-activation of NRF2-dependent expression of detoxification enzymes heme oxygenase 1 (HO-1), NAD(P)H quinone oxidoreductase 1 (NQO-1), and UDP-glucronosyltransferase 1A1 (UGT1A1) potentially leading to inhibition of TPA-induced transformation in epidermal JB6 cells (87). Such induced NRF2 expression was attributed to promoter hypo-methylation through down-regulation of DNMTs and inhibition of overall HDAC enzyme activity (87). Professor Paul Talalay who first isolated and identified SFN (88), and he also advanced the concept of redox/electrophile-mediated induction of phase 2 detoxifying genes (89). Oxidants and electrophiles including SFN could modify cysteine’s sulfhydryl groups of Kelch-like ECH-associated protein-1 (KEAP1), prevent proteasomal degradation, allow newly synthesized NRF2 to enter the nucleus, and enhance NRF2-target gene transcription (reviewed in (90–92)). Interestingly, however, electrophile such as SFN and oxidant such as H2O2 may also regulate NRF2 via redox sensitive cysteine on the Nuclear Export Signal (NES) (93) as well as post-transcriptional/translation control of NRF2’s Internal Ribosomal Entry Sites (IRESs) (94). Question remains, how these different redox signaling of NRF2 would operate under different redox environments such as different redox agents, acute versus chronic redox agent’s stimulating conditions and pathological conditions.
Lung cancer
Several studies show an inverse relationship between consumption of dietary ITCs, and lung cancer risk (95). In vitro cell culture study shows the involvement of SFN and miR-616–5p in EMT and NSCLC metastasis (96). miR-616–5p directly targets GSK3β and decreased its expression, whereas SFN decreased miR-616–5p by histone modification, and followed by inactivation of the GSK3β/β-catenin signaling pathway and inhibition of EMT in NSCLC cells (96). SFN inhibits HDAC activity and increases acetylated histones H3 and H4 in A549 and H1299 lung cancer cells (97). In lung cancer stem cells (CSCs) miR-19a and miR-19b expression are up-regulated and SFN suppresses miR-19 and Wnt/β-catenin pathway resulting in inhibition of lung CSCs (98). In human lung cancer A549 cells, SFN epigenetically demethylates the CpG sites of the miR-9–3 promoter and reactivates miR-9–3 expression (99). Similarly, SFN was found to up-regulate miR-214 and mediate downregulation of c-MYC in non-small cell lung cancer, potentially results in inhibition of cancer stem-like cell properties and cisplatin resistance (100).
Colon cancer
In colon cancer model, SFN suppressed tumor development in Apc(min) mice, increased acetylated histones in the polyps, and inhibited HDAC in APC(min) mice (101). SFN treatment in human colon adenocarcinoma Caco-2 cells showed increased activation of NRF2 by demethylation of the NRF2 promoter region and reducing expression of DNMT1. (102). In polyposis in rat colon (Pirc) model, single oral administration of SFN and structurally related long-chain ITCs decreased HDAC3 expression and increased pH2AX levels in adenomatous colon polyps (103). HCT116 and SW480 colon cancer cells study showed that SFN or its analogs altered HAT/HDAC activities and histone acetylation status, lowered the expression of HDAC3, P300/CBP associated factor (PCAF) and lysine acetyltransferase 2A (KAT2A/GCN5), and attenuated homologous recombination (HR)/non-homologous end joining (NHEJ) repair activities (103). A 0.12% PEITC-enriched mouse-diet reduced mucosal and submucosal inflammation possibly via modulating regulating NFκB proteins and NFκB mRNA expression was inversely correlated with tri-methylation of lysine 27 on histone 3 near its promoter region in a time-dependent manner (104). Interestingly, colon cancer cells treated with low-dose PEITC for >1 month exhibited stable alterations in expression profile of epigenetic writers/erasers and chromatin-binding of HDACs and Polycomb-group (PcG) proteins (105). Sustained PEITC exposure not only blocked HDAC protein binding to euchromatin but was also associated with hypomethylation of PcG target genes that are typically hypermethylated in cancer (105). In human colon cancer cells ITCs inhibit HDAC enzyme activity and increase HDAC protein turnover and ITCs enhanced the acetylation and subsequent degradation of critical repair proteins, such as CtIP (106).
Breast cancer
Studies show that SFN modulates breast cancer risk at multiple stages of carcinogenesis through different biologic mechanisms including epigenetic modulations (107). SFN inhibited in dose- and time-dependent of hTERT in both MCF-7 and MDA-MB-231 human breast cancer cells. DNMT1 and DNMT3a, were decreased in SFN-treated breast cancer cells suggesting that SFN may repress hTERT by impacting epigenetic pathways (108). In combination studies, SFN and EGCG activated ERα MDA-MB-231 cells which was associated with significant reduction of DNMTs expression and activity, as well as of HDAC activity (109). Synergistic effect on MDA-MB-231 xenograft tumor growth, with combination of SFN and LSD1 inhibitor (110). In transplacental breast cancer chemoprevention study, SFN containing broccoli sprout diet upregulates tumor suppressor genes p53 and p16INK4a and downregulates tumor-promoting genes TERT and c-Myc potentially as a result of inhibition of HDAC1 expression (111). SFN in combination with genistein downregulates HDAC2, HDAC3 and KLF4 protein expression and reduced tumor volumes/sizes (112).
Prostate cancer
In prostate cancer, SFN reactivated and induced its downstream antioxidant stress pathway in TRAMP-C1 cells through epigenetic modification of NRF2 promoter, and could play a role in chemoprevention (113). The expression of long noncoding RNAs (lncRNAs) in prostate cancer cells was altered and SFN restored the altered expression of lncRNAs (114). Increased SFN consumption in mice with prostate cancer cell xenografts showed reduced HDAC enzyme activity in prostates, and peripheral blood mononuclear cells (115). SFN inhibited the expression and activity of human telomerase reverse transcriptase (hTERT). SFN treatment selectively decreased HDAC activity, and Class I and II HDAC protein’s expression increased acetylated histone H3 at the promoter for P21, induced p21 expression and increased tubulin acetylation in prostate cancer cells (116).
Cancer chemoprevention by isothiocyanates (ITCs) via non-epigenetic signaling pathways
Numerous studies have shown that many non-epigenetic mechanisms by which ITCs exert their biological anti-carcinogenic activities have been being well investigated including induction of carcinogen detoxication (117), enhanced DNA damage repair (118, 119), anti-inflammatory pathway through NF-κB regulation (120), elimination of cancer stem cells (121) and other cells (122), inhibition of cell cycle progression (123), induction of caspase/apoptosis (124), induction of autophagy (125), and MAPK signaling pathway (126), among others. As discussed above, the most studied and most relevant signaling pathway in the context of cancer prevention elicited by ITCs would be the NRF2 mediated signaling pathway. As reviewed by many scientists, and discussed above, ITCs react with sulfhydryl groups of C residues of Keap1, releasing NRF2 from Keap1 binding and then NRF2 translocating to the nucleus, partnering with sMafs and binding to the ARE found in many antioxidant/detoxifying enzymes including glutathione S-transferases (GSTs), thioredoxin, NQO-1, HO-1, among others (127–131).
Many studies have also shown that the ITCs are potent anti-inflammatory agents. SFN significantly reduced the nuclear translocation of the pro-inflammatory transcription factor nuclear factor (NF)-κB in pancreatic acinar cells, downregulating the expression of NF-κB target genes that code for pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1β), IL-6 and inflammatory enzymes cyclooxygenase-2 (COX-2), prostaglandin E (PGE) synthase, and inducible nitric oxide synthase (iNOS) (120, 132). ITCs can also modulate the activity of phase I metabolizing enzymes (133–135), such as downregulating CYP3A2 mRNA expression, as well as the activity of benzyloxyquinoline debenzylase, a marker of CYP3As and upregulating CYP1A1 and CYP1A2 mRNA expression and the activity of ethoxyresorufin-O-deethylase (EROD), a marker of CYP1A1/2 activities (136). Some procarcinogens require activation by phase I enzymes in order to become active carcinogens capable of binding DNA and forming cancer-causing DNA adducts (137). Inhibition of specific CYP enzymes involved in carcinogen activation has been found to prevent the development of cancer in animal models (138). In additional, ITCs have been shown to modulate the expression of cell cycle regulators, cyclins and cyclin-dependent kinases (CDK), and trigger apoptosis in cancer cell lines (139). For example, in a mouse model of CRC, PEITC administration reduced both the number and size of polyps and these alternations were associated with activation of the CDK inhibitor, p21, inhibition of various cyclins (A, D1, and E), and induction of apoptosis (140). ITCs also have been shown to suppress the formation of capillary-like structures from human umbilical endothelial cells and likely inhibit the expression of hypoxia inducible factors (HIFs) that control angiogenesis in endothelial cells and malignant cell lines inhibition of angiogenesis (141). As discussed above for BITC (142), SFN has been shown to induce autophagy in both in vitro cell culture models and in vivo models associated with decreased cell proliferation, alterations in protein levels of autophagy regulators Atg5 and phospho-mTOR (125, 143). Some other anticancer pathways such as inhibition of cell migration and invasion (144–146), have also been reported. Question remains how these non-epigenetic signaling pathways would be linked and integrated with epigenetic mechanisms elicited by ITCs in the context of cancer prevention necessitates further study.
Pharmacokinetics and Pharmacodynamics of dietary ITCs
Pharmacokinetics (PK) and pharmacodynamics (PD), a quantitative science, integrate and evaluate the dose/concentration-response relationships in the body following the administration of drugs (147, 148). Despite many reports studying the PK/PD of ITCs, there are not many showing the “direct quantitative relationship” between the dose/concentration of ITCs and the biological/pharmacological response(s) in in vivo animal models or in humans. In absorption, after oral administration of SFN in the rats the peak concentration (Cmax) were reported to be attained in 1 h, indicating rapid absorption (149). However, increasing the dose of SFN from 0.5–5 mg/kg, the oral absorption rate constant (kab) decreased and Cmax did not increase proportionally to the dose, suggesting non-linear absorption kinetics (149). In human, SFN was absorbed rapidly and achieved Cmax in 1 h after the consumption of broccoli sprout products (150, 151). Comparison of the area under the blood/plasma concentration time curve (AUC) AUC0–24 values between the intravenous (IV) and oral treatment groups, at low dose (2.8 μmol/kg) shows an absolute bioavailability of 82%, which decreased to 20% at the higher dose (28 μmol/kg) (149).
In drug distribution, a rapid decrease was observed in the plasma concentration of SFN after both oral and IV dosing indicating rapid and extensive cellular uptake into various tissues (149). This is consistent with studies showing ITCs achieve very high intracellular concentrations as a result of their interaction with glutathione (GSH) (152–154). Such extensive intracellular localization explains the very large apparent volume of distribution (Vd) of 102 L/kg after IV injection in rat. Additionally, the Vd of SFN decreased dramatically from 102 to 42 L/kg with increasing doses from 0.5 to 5.0 mg/kg (149), showing non-linear Vd. In another study of SFN PK, the volume of distribution at steady state (Vss) was around 3.7 L or 14.8 L/Kg after an IV dose of 25 mg/kg of SFN (155) and this result is consistent with Hanlon et al.’s findings (149).
Freeze-dried aqueous extract of broccoli sprouts inhibited bladder cancer development induced by N-butyl-N-(4-hydroxybutyl) nitrosamine significantly and dose-dependently, with over 70% of the ITCs present in the extract were excreted in the urine as ITC equivalents (ITCs + dithiocarbamates) within 12 h after a single oral dose, indicating substantial urinary excretion (156). Urinary concentrations of ITC equivalents were 2 to 3 orders of magnitude higher than those in plasma (156). In a cross-over human study of glucoraphanin-rich (GRR) versus SFN-rich (SFR), urinary excretion of SFN and its metabolites (in approximately 12-hour collections after dosing), was substantially greater with the SFN-rich (mean = 70%) than with GRR rich (mean = 5%) beverages (157). These studies again suggest that substantial renal excretion of ITC or its metabolites occur.
In terms of PD, several studies have reported the increased levels of Nrf2-targeted enzymes GSTs and NQO1in plasma (158) and saliva (159) in humans volunteers after consuming cruciferous vegetables. Similarly administration of SFN-rich preparations to healthy subjects led to increased mRNA or protein levels of NQO1 and GSTs in skin punch biopsies, blood cells, and buccal scrapings (160–162). As a proof of concept study, after IV administration of 25 mg/kg of SFN, moderate increase in mRNA (2–5 folds) of HO-1, NRF2, and NQO1, while significant increase (> 5 folds) for GSTT1, GPx1, and Maf in rat lymphocytes (155). These PK/PD effects of SFN could be modeled with indirect response (IDR) model directly linking plasma SFN concentrations with NRF2-mediated gene expression response in lymphocytes (155). Future studies involving integrating acute PK/PD and long-term cancer preventive effects of ITCs in human would be needed.
Ongoing clinical studies/trials of isothiocyanates and chemoprevention
Pre-clinical carcinogenesis studies of dietary ITCs have demonstrated extraordinary chemopreventive efficacy. Several clinical trials have illustrated that ITCs, administered orally as glucosinolate precursors or directly in its bioactive compound such as SFN, PEITC have preventive effects against cancers (163) (164). Specifically, SFN and PEITC have been studied in some clinical studies and briefly summarized in Table 1 (165–171). More ongoing ITCs clinical trials are listed in clinicaltrials.gov. In one particular clinical study involving male patient with recurrent prostate cancer, none of the patients experienced PSA doubling, a marker for disease severity, after serving SFN daily for 20 weeks (172). HDAC expression and activity inhibition has been used as a biomarker in some trials (173, 174) and one clinical study with healthy subjects showed that HDAC activity was significantly inhibited in PBMCs as early as 3 hours after consumption of 68 g of broccoli sprouts (115). Study using SFN in breast cancer patients showed an average decrease of 80.39 pmol/min/mg protein in blood HDAC activity from pre- to post-intervention (169). A clinical study of PEITC as an inhibitor of metabolic activation of a tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) for cancer prevention, the NNK metabolic activation ratio was significant reduced by 7.7% (171). ITCs have shown promising efficacy in various clinical cancer chemoprevention trials, however, future clinical trials involving epigenetic endpoints are warranted.
Table 1:
Summary of completed and ongoing clinical trials using SFN and PEITC
| Agent | Cancer type | Experiment detail | Outcome/Ongoing targets |
|---|---|---|---|
| SFN/Broccoli Sprout Extract | Lung | 291 healthy subjects; broccoli sprout beverages; GR (600 μmol) and SFN (40 μmol); 84 days | Increases urinary excretion of benzene (61%) and acrolein (23%) (165) |
| 50 healthy subjects; SFN-rich broccoli sprout beverage; 7 days | 20–50% ↑ in excretion levels of glutathione conjugates of acrolein, crotonaldehyde and benzene after SFN treatment (166) | ||
| 30 healthy subjects, young smokers; broccoli diet; 250 g/ day; 10 days | DNA repair activity ↑ in PBMC (167) | ||
| 72 former smokers with lung cancer; SFN; 120 μmol/time, 2 times/day; 12 months | Targets: lung cancer chemoprevention with SFN in former smokers | ||
| Prostate | 90 men with prostate cancer recurrence; 60 mg of SFN daily; 6 months | log PSA slope ↓ significantly (168) | |
| 98 men with prostate biopsy; 200 µmol of SFN, 2 capsules daily; 4 weeks | Targets: HDAC inhibition effects by SFN in human prostate cancer tissue/cells | ||
| 40 prostate cancer patients; 64 mg SFN daily, 2 times daily; 4 weeks | Epigenetic biomarkers measurement after SFN treatment | ||
| Breast | 54 breast biopsy subjects; 224 mg GR; Broccoli seed extract; | Ki67 ↓, HDAC3 ↓ in benign tissue, HDAC ↓ in PBMC (169) | |
| 34 DCIS breast cancer subjects; 100 µmols of SFN for 14 days | Change in Mean Proliferative Rate Measured by Ki67% | ||
| PEITC/Watercress | Lung | 11 healthy smokers; watercress; 56.8 g / meal for 3 meals/day; 3 days | ↑ Urinary NNAL plus NNAL-Gluc (33.5%) (170) |
| 82 healthy smokers; PEITC 10 mg 4 times/day for 5 weeks | ↓ NNK metabolic activation ratio (7.7%) (171) |
SFN, sulforaphane; PEITC, phenethyl isothiocyanate; GR, glucoraphanin; PBMC, Peripheral blood mononuclear cells; PSA, prostate-specific antigen; HDAC, Histone Deacetylase; NNAL-glu, 4-(methyInitrosamino)-l-(3-pyridyI)-l-butyl /3-D-glucopyranosiduronic acid; NNK, Nicotine-derived nitrosamine ketone; DCIS, Ductal carcinoma in situ; ↑, increase/activation; ↓, decrease/inhibition.
Conclusion and future prospective
ITCs found abundantly in the human diet and are derived from cruciferous vegetables, are highly promising cancer preventive agents. Multiple and multi-targeted mechanisms, including epigenetic reprograming, could be involved in the preventive and therapeutic effects of ITCs in cancer. Gene silencing through hypermethylation of CpG of tumor onco/proto-oncogenes, gene activation through hypomethylation of CpG of tumor suppressor genes, and other modifications including phosphorylation, acetylation, ubiquitination are potential cellular targets of epigenetic regulation by ITCs. Genome-wide DNA methylation analysis might help to better understand the targeted tissue specificity and global events in the regulation by ITCs. Extensive research on the extrapolation of preclinical to clinical dosages of ITCs for prevention studies needs to be further refined. Understanding of the PK/PD of parent/metabolites of the ITCs, its tissue levels and modulation of signaling mechanisms including epigenetic mechanisms is warranted in human studies.
Cancer is a complex chronic disease and cancer development is a multistep process (1, 2, 12, 175, 176). The ITCs as discussed above belongs to a very powerful class of biologically active food components, they are relatively non-toxic at low physiological and even higher pharmacological doses, with good oral bioavailability (149, 157), powerful anti-oxidative stress/anti-inflammatory (80) and possess epigenetic modifying properties as well as with great anti-cancer efficacy in many in vitro cell culture models and in vivo animal models. The exact molecular mechanism of how the ITCs as well as many other dietary chemopreventive phytochemicals modifying the epigenetic machinery is currently unclear. However it is highly likely through redox biological process playing an important role (60) analogous to the kinases/caspases activation as we have discussed back in the 1990s (177–179). Additionally, it would be tempting to speculate that the ITCs particularly SFN and PEITC, although differ in some aspects of signaling pathways (at same micromolar concentrations, SFN is a more potent NRF2 activator than PEITC, and SFN activates ERK more strongly compared to JNK, whereas PEITC would be reversed (126, 180, 181), they would be considered as “general” cancer preventive agents not unlike the chemotherapeutic agents such as platinum-based or the taxane-based drugs. Analogous to chemotherapeutic agents, for maximal efficacy, the ITCs would need to be used in combination with other cancer preventive agents including other less toxic dietary phytochemicals, natural products or drugs such as NSAIDs, SERMs, aromatase inhibitors, and 5‑α‑reductase inhibitors (182) targeting different signaling pathways. Together with other considerations and initiatives such as immunoprevention (183, 184) (185), the long-term control and prevention of progression of tumors to advanced and metastatic states may soon to be realized.
Acknowledgement:
This work was supported in part by institutional funds and by R01 AT009152 from the National Center for Complementary and Integrative Health (NCCIH) and R01 CA200129, from the National Cancer Institute (NCI) to A.N.K. X.L. and Z.L. are supported by China Scholarship Council, and A.A.F.S. by Yarmouk University scholarship.
Abbreviations:
- ITCs
Isothiocyanates
- SFN
Sulforaphane
- GS
glucosinolates
- EMT
Epithelial-Mesenchymal Transition
- NSCLC
Non-Small Cell Lung Carcinoma
- hTERT
human Telomerase Reverse Transcriptase
- OSCC
Oral Squamous Cell Carcinoma
- NQO1
NAD(P)H: quinone oxidoreductase
- HO-1
hemeoxygenase 1
- NRF2
Nuclear Factor Erythroid-2-related Factor-2
- NF-κB
Nuclear Factor Kappa B
- DNMT-1
DNA methyltransferase 1
- EGFR
Epidermal Growth Factor Receptor
References:
Uncategorized References
- 1.Varmus H, Unni AM, Lockwood WW. How Cancer Genomics Drives Cancer Biology: Does Synthetic Lethality Explain Mutually Exclusive Oncogenic Mutations? Cold Spring Harb Symp Quant Biol 2016;81:247–55. Epub 2017/01/27. doi: 10.1101/sqb.2016.81.030866. [DOI] [PubMed] [Google Scholar]
- 2.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res 2017;7(5):1016–36. [PMC free article] [PubMed] [Google Scholar]
- 3.Chen C, Kong AN. Dietary chemopreventive compounds and ARE/EpRE signaling. Free Radic Biol Med 2004;36(12):1505–16. Epub 2004/06/09. doi: 10.1016/j.freeradbiomed.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong AN. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol Ther 2013;137(2):153–71. Epub 2012/10/09. doi: 10.1016/j.pharmthera.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. Ilar j 2012;53(3–4):289–305. Epub 2013/06/08. doi: 10.1093/ilar.53.3-4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feinberg AP. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N Engl J Med 2018;378(14):1323–34. Epub 2018/04/05. doi: 10.1056/NEJMra1402513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature 2019;571(7766):489–99. Epub 2019/07/26. doi: 10.1038/s41586-019-1411-0. [DOI] [PubMed] [Google Scholar]
- 8.Scott RE, Wille JJ Jr., Wier ML. Mechanisms for the Initiation and Promotion of Carcinogenesis: A Review and a New Concept. Mayo Clinic Proceedings 1984;59(2):107–17. doi: 10.1016/S0025-6196(12)60244-4. [DOI] [PubMed] [Google Scholar]
- 9.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228(4696):187–90. Epub 1985/04/12. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 10.Feinberg AP, Vogelstein B. Alterations in DNA methylation in human colon neoplasia. Semin Surg Oncol 1987;3(3):149–51. Epub 1987/01/01. doi: 10.1002/ssu.2980030304. [DOI] [PubMed] [Google Scholar]
- 11.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016;17(5):284–99. Epub 2016/03/15. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg RA. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157(1):267–71. Epub 2014/04/01. doi: 10.1016/j.cell.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Esteller M Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007;8(4):286–98. Epub 2007/03/07. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 14.Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17(10):630–41. Epub 2016/09/16. doi: 10.1038/nrg.2016.93. [DOI] [PubMed] [Google Scholar]
- 15.Sapienza C, Issa JP. Diet, Nutrition, and Cancer Epigenetics. Annu Rev Nutr 2016;36:665–81. Epub 2016/03/30. doi: 10.1146/annurev-nutr-121415-112634. [DOI] [PubMed] [Google Scholar]
- 16.Kelly AD, Issa JJ. The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr Opin Genet Dev 2017;42:68–77. Epub 2017/04/17. doi: 10.1016/j.gde.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Wu R, Sargsyan D, Yin R, Kuo HC, Yang I, Wang L, Cheng D, Wang C, Li S, Hudlikar R, Lu Y, Kong AN. UVB drives different stages of epigenome alterations during progression of skin cancer. Cancer Lett 2019;449:20–30. Epub 2019/02/17. doi: 10.1016/j.canlet.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berube R, Drigeard Desgarnier MC, Douki T, Lechasseur A, Rochette PJ. Persistence and Tolerance of DNA Damage Induced by Chronic UVB Irradiation of the Human Genome. J Invest Dermatol 2018;138(2):405–12. Epub 2017/09/28. doi: 10.1016/j.jid.2017.08.044. [DOI] [PubMed] [Google Scholar]
- 19.Mirzoeva S, Tong X, Bridgeman BB, Plebanek MP, Volpert OV. Apigenin Inhibits UVB-Induced Skin Carcinogenesis: The Role of Thrombospondin-1 as an Anti-Inflammatory Factor. Neoplasia 2018;20(9):930–42. Epub 2018/08/18. doi: 10.1016/j.neo.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo X, Xu Y, Zhao Z. In-depth genomic data analyses revealed complex transcriptional and epigenetic dysregulations of BRAFV600E in melanoma. Mol Cancer. 2015;14:60. Epub 2015/04/19. doi: 10.1186/s12943-015-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther 2004;3(4):425–35. [PubMed] [Google Scholar]
- 22.Strub T, Ballotti R, Bertolotto C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020;10(4):1777–97. Epub 2020/02/12. doi: 10.7150/thno.36218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C, Wu R, Sargsyan D, Zheng M, Li S, Yin R, Su S, Raskin I, Kong AN. CpG methyl-seq and RNA-seq epigenomic and transcriptomic studies on the preventive effects of Moringa isothiocyanate in mouse epidermal JB6 cells induced by the tumor promoter TPA. J Nutr Biochem 2019;68:69–78. Epub 2019/04/29. doi: 10.1016/j.jnutbio.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rauscher GH, Kresovich JK, Poulin M, Yan L, Macias V, Mahmoud AM, Al-Alem U, Kajdacsy-Balla A, Wiley EL, Tonetti D, Ehrlich M. Exploring DNA methylation changes in promoter, intragenic, and intergenic regions as early and late events in breast cancer formation. BMC Cancer. 2015;15:816. Epub 2015/10/30. doi: 10.1186/s12885-015-1777-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, Grainge MJ, Ball GR, Abdelghany MK, Martinez-Pomares L, Heery DM, Ellis IO. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 2009;69(9):3802–9. Epub 2009/04/16. doi: 10.1158/0008-5472.CAN-08-3907. [DOI] [PubMed] [Google Scholar]
- 26.Linares A, Dalenc F, Balaguer P, Boulle N, Cavailles V. Manipulating protein acetylation in breast cancer: a promising approach in combination with hormonal therapies? J Biomed Biotechnol 2011;2011:856985. Epub 2010/12/29. doi: 10.1155/2011/856985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–9. Epub 2002/10/11. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 28.Saramaki OR, Tammela TL, Martikainen PM, Vessella RL, Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer. 2006;45(7):639–45. Epub 2006/04/01. doi: 10.1002/gcc.20327. [DOI] [PubMed] [Google Scholar]
- 29.Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004;64(6):1975–86. Epub 2004/03/18. doi: 10.1158/0008-5472.can-03-3972. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama M, Bennett CJ, Hicks JL, Epstein JI, Platz EA, Nelson WG, De Marzo AM. Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol 2003;163(3):923–33. Epub 2003/08/26. doi: 10.1016/s0002-9440(10)63452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, Foster BA, Kan YW, Kong AN. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One. 2010;5(1):e8579. Epub 2010/01/12. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Huang Y, Sargsyan D, Khor TO, Guo Y, Shu L, Yang AY, Zhang C, Paredes-Gonzalez X, Verzi M, Hart RP, Kong AN. Epigenetic alterations in TRAMP mice: epigenome DNA methylation profiling using MeDIP-seq. Cell Biosci 2018;8:3. Epub 2018/01/19. doi: 10.1186/s13578-018-0201-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C, Feng Y, Zhang C, Cheng D, Wu R, Yang Y, Sargsyan D, Kumar D, Kong AN. PTEN deletion drives aberrations of DNA methylome and transcriptome in different stages of prostate cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2020;34(1):1304–18. Epub 2020/01/10. doi: 10.1096/fj.201901205RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435(7046):1262–6. Epub 2005/07/01. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 35.Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, Heckmann D, Sidiropoulos N, Waszak SM, Hübschmann D, Urbanucci A, Girma EG, Kuryshev V, Klimczak LJ, Saini N, Stütz AM, Weichenhan D, Böttcher LM, Toth R, Hendriksen JD, Koop C, Lutsik P, Matzk S, Warnatz HJ, Amstislavskiy V, Feuerstein C, Raeder B, Bogatyrova O, Schmitz EM, Hube-Magg C, Kluth M, Huland H, Graefen M, Lawerenz C, Henry GH, Yamaguchi TN, Malewska A, Meiners J, Schilling D, Reisinger E, Eils R, Schlesner M, Strand DW, Bristow RG, Boutros PC, von Kalle C, Gordenin D, Sültmann H, Brors B, Sauter G, Plass C, Yaspo ML, Korbel JO, Schlomm T, Weischenfeldt J. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell. 2018;34(6):996–1011.e8. Epub 2018/12/12. doi: 10.1016/j.ccell.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russo JW, Balk SP. Initiation and Evolution of Early Onset Prostate Cancer. Cancer Cell. 2018;34(6):874–6. Epub 2018/12/12. doi: 10.1016/j.ccell.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 37.Jung G, Hernández-Illán E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nature Reviews Gastroenterology & Hepatology. 2020;17(2):111–30. doi: 10.1038/s41575-019-0230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Y, Wu R, Gaspar JM, Sargsyan D, Su ZY, Zhang C, Gao L, Cheng D, Li W, Wang C, Yin R, Fang M, Verzi MP, Hart RP, Kong AN. DNA methylome and transcriptome alterations and cancer prevention by curcumin in colitis-accelerated colon cancer in mice. Carcinogenesis. 2018;39(5):669–80. Epub 2018/03/17. doi: 10.1093/carcin/bgy043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harada S, Morlote D. Molecular Pathology of Colorectal Cancer. Adv Anat Pathol 2019. Epub 2019/09/11. doi: 10.1097/PAP.0000000000000247. [DOI] [PubMed] [Google Scholar]
- 40.Zeng Q, Lei F, Chang Y, Gao Z, Wang Y, Gao Q, Niu P, Li Q. An oncogenic gene, SNRPA1, regulates PIK3R1, VEGFC, MKI67, CDK1 and other genes in colorectal cancer. Biomed Pharmacother 2019;117:109076. Epub 2019/06/17. doi: 10.1016/j.biopha.2019.109076. [DOI] [PubMed] [Google Scholar]
- 41.Migheli F, Migliore L. Epigenetics of colorectal cancer. Clin Genet 2012;81(4):312–8. Epub 2012/01/24. doi: 10.1111/j.1399-0004.2011.01829.x. [DOI] [PubMed] [Google Scholar]
- 42.Juo YY, Johnston FM, Zhang DY, Juo HH, Wang H, Pappou EP, Yu T, Easwaran H, Baylin S, van Engeland M, Ahuja N. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Annals of Oncology. 2014;25(12):2314–27. doi: 10.1093/annonc/mdu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Portela A, Esteller M. Epigenetic modifications and human disease. Nature biotechnology. 2010;28(10):1057. [DOI] [PubMed] [Google Scholar]
- 44.Golbabapour S, Abdulla MA, Hajrezaei M. A concise review on epigenetic regulation: insight into molecular mechanisms. Int J Mol Sci 2011;12(12):8661–94. Epub 2012/01/25. doi: 10.3390/ijms12128661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet 2010;70:27–56. Epub 2010/10/06. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 46.Yan C, Boyd DD. Histone H3 acetylation and H3 K4 methylation define distinct chromatin regions permissive for transgene expression. Mol Cell Biol 2006;26(17):6357–71. Epub 2006/08/18. doi: 10.1128/MCB.00311-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malumbres M miRNAs and cancer: an epigenetics view. Mol Aspects Med 2013;34(4):863–74. Epub 2012/07/10. doi: 10.1016/j.mam.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S, Kuo H-CD, Yin R, Wu R, Liu X, Wang L, Hudlikar R, Peter RM, Kong A-N. Epigenetics/Epigenomics of Triterpenoids in Cancer Prevention and in Health. Biochemical Pharmacology. 2020:113890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hudlikar RR, Sargsyan D, Wu R, Su S, Zheng M, Kong AN. Triterpenoid corosolic acid modulates global CpG methylation and transcriptome of tumor promotor TPA induced mouse epidermal JB6 P+ cells. Chem Biol Interact 2020;321:109025. Epub 2020/03/07. doi: 10.1016/j.cbi.2020.109025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu R, Wang L, Yin R, Hudlikar R, Li S, Kuo HCD, Peter R, Sargsyan D, Guo Y, Liu X. Epigenetics/epigenomics and prevention by curcumin of early stages of inflammatory-driven colon cancer. Molecular Carcinogenesis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shanmugam MK, Arfuso F, Sng JC, Bishayee A, Kumar AP, Sethi G. Epigenetic effects of curcumin in cancer prevention. Epigenetics of Cancer Prevention: Elsevier; 2019. p. 107–28. [Google Scholar]
- 52.Cheng D, Li W, Wang L, Lin T, Poiani G, Wassef A, Hudlikar R, Ondar P, Brunetti L, Kong A-N. Pharmacokinetics, pharmacodynamics, and PKPD modeling of curcumin in regulating antioxidant and epigenetic gene expression in healthy human volunteers. Molecular pharmaceutics. 2019;16(5):1881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soflaei SS, Momtazi-Borojeni AA, Majeed M, Derosa G, Maffioli P, Sahebkar A. Curcumin: a natural pan-HDAC inhibitor in cancer. Current pharmaceutical design. 2018;24(2):123–9. [DOI] [PubMed] [Google Scholar]
- 54.Kim H, Ramirez CN, Su Z-Y, Kong A-NT. Epigenetic modifications of triterpenoid ursolic acid in activating Nrf2 and blocking cellular transformation of mouse epidermal cells. The Journal of nutritional biochemistry. 2016;33:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okonkwo A, Mitra J, Johnson GS, Li L, Dashwood WM, Hegde ML, Yue C, Dashwood RH, Rajendran P. Heterocyclic analogs of sulforaphane trigger DNA damage and impede DNA repair in colon cancer cells: interplay of HATs and HDACs. Molecular nutrition & food research. 2018;62(18):1800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang L-L, Zhou S-J, Zhang X-M, Chen H-Q, Liu W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomedicine & Pharmacotherapy. 2016;78:74–80. [DOI] [PubMed] [Google Scholar]
- 57.Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clinical epigenetics. 2011;3(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sies H Oxidative Stress 1 ed. London: Academic Press; 1985. 1/28/1985. 507 p. [Google Scholar]
- 59.Sies H Oxidative stress: a concept in redox biology and medicine. Redox Biol 2015;4:180–3. Epub 2015/01/16. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal 2011;15(2):551–89. Epub 2010/10/06. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kietzmann T, Petry A, Shvetsova A, Gerhold JM, Görlach A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol 2017;174(12):1533–54. Epub 2017/03/24. doi: 10.1111/bph.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Wijst MG, Venkiteswaran M, Chen H, Xu G-L, Plösch T, Rots MG. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 2015;10(8):671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye Q, Trivedi M, Zhang Y, Böhlke M, Alsulimani H, Chang J, Maher T, Deth R, Kim J. Brain iron loading impairs DNA methylation and alters GABAergic function in mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2019;33(2):2460–71. Epub 2018/10/03. doi: 10.1096/fj.201801116RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xin Y-J, Yuan B, Yu B, Wang Y-Q, Wu J-J, Zhou W-H, Qiu Z. Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Scientific reports. 2015;5:7645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hepburn PA, Margison GP, Tisdale MJ. Enzymatic methylation of cytosine in DNA is prevented by adjacent O6-methylguanine residues. J Biol Chem 1991;266(13):7985–7. [PubMed] [Google Scholar]
- 67.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res 2004;32(14):4100–8. Epub 2004/08/11. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133(6):978–93. [DOI] [PubMed] [Google Scholar]
- 69.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci U S A 2008;105(28):9633–8. Epub 2008/07/09. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu T, Schreiter FC, Bagchi RA, Tatman PD, Hannink M, McKinsey TA. HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J Biol Chem 2019;294(21):8640–52. Epub 2019/04/10. doi: 10.1074/jbc.RA118.007006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manea SA, Antonescu ML, Fenyo IM, Raicu M, Simionescu M, Manea A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol 2018;16:332–43. Epub 2018/03/28. doi: 10.1016/j.redox.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Licona C, Spaety ME, Capuozzo A, Ali M, Santamaria R, Armant O, Delalande F, Van Dorsselaer A, Cianferani S, Spencer J, Pfeffer M, Mellitzer G, Gaiddon C. A ruthenium anticancer compound interacts with histones and impacts differently on epigenetic and death pathways compared to cisplatin. Oncotarget 2017;8(2):2568–84. Epub 2016/12/10. doi: 10.18632/oncotarget.13711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138(5):1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang P, Torres K, Liu X, Liu C-g, Pollock RE. An overview of chromatin-regulating proteins in cells. Current Protein and Peptide Science. 2016;17(5):401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxidants & redox signaling. 2013;18(15):1956–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Surh YJ, Kundu JK, Na HK, Lee JS. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr 2005;135(12 Suppl):2993s–3001s. Epub 2005/12/01. doi: 10.1093/jn/135.12.2993S. [DOI] [PubMed] [Google Scholar]
- 77.Nair S, Li W, Kong AN. Natural dietary anti-cancer chemopreventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells. Acta Pharmacol Sin 2007;28(4):459–72. Epub 2007/03/23. doi: 10.1111/j.1745-7254.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 78.Li S, Kuo HD, Yin R, Wu R, Liu X, Wang L, Hudlikar R, Peter RM, Kong AN. Epigenetics/epigenomics of triterpenoids in cancer prevention and in health. Biochem Pharmacol 2020;175:113890. Epub 2020/03/03. doi: 10.1016/j.bcp.2020.113890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shapiro TA, Fahey JW, Wade KL, Stephenson KK, Talalay P. Chemoprotective Glucosinolates and Isothiocyanates of Broccoli Sprouts. Cancer Epidemiology Biomarkers & Prevention. 2001;10(5):501. [PubMed] [Google Scholar]
- 80.Yagishita Y, Fahey JW, Dinkova-Kostova AT, Kensler TW. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules. 2019;24(19). Epub 2019/10/09. doi: 10.3390/molecules24193593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barba FJ, Nikmaram N, Roohinejad S, Khelfa A, Zhu Z, Koubaa M. Bioavailability of Glucosinolates and Their Breakdown Products: Impact of Processing. Frontiers in Nutrition. 2016;3(24). doi: 10.3389/fnut.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Talalay P, Fahey JW, Healy ZR, Wehage SL, Benedict AL, Min C, Dinkova-Kostova AT. Sulforaphane mobilizes cellular defenses that protect skin against damage by UV radiation. Proc Natl Acad Sci U S A 2007;104(44):17500–5. Epub 2007/10/25. doi: 10.1073/pnas.0708710104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Penta D, Somashekar BS, Meeran SM. Epigenetics of skin cancer: Interventions by selected bioactive phytochemicals. Photodermatol Photoimmunol Photomed 2018;34(1):42–9. Epub 2017/10/05. doi: 10.1111/phpp.12353. [DOI] [PubMed] [Google Scholar]
- 84.Nandakumar V, Vaid M, Tollefsbol TO, Katiyar SK. Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice. Carcinogenesis. 2011;32(4):597–604. Epub 2010/12/28. doi: 10.1093/carcin/bgq282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Saha K, Fisher ML, Adhikary G, Grun D, Eckert RL. Sulforaphane suppresses PRMT5/MEP50 function in epidermal squamous cell carcinoma leading to reduced tumor formation. Carcinogenesis. 2017;38(8):827–36. Epub 2017/09/01. doi: 10.1093/carcin/bgx044. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 86.Li S, Yang Y, Sargsyan D, Wu R, Yin R, Kuo HD, Yang I, Wang L, Cheng D, Ramirez CN, Hudlikar R, Lu Y, Kong AN. Epigenome, transcriptome and protection by sulforaphane at different stages of UVB-induced skin carcinogenesis. Cancer Prev Res (Phila) 2020. Epub 2020/03/13. doi: 10.1158/1940-6207.Capr-19-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, Conney AH, Lu YP, Kong AN. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev Res (Phila) 2014;7(3):319–29. Epub 2014/01/21. doi: 10.1158/1940-6207.CAPR-13-0313-T. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A 1992;89(6):2399–403. Epub 1992/03/15. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogens. Proceedings of the National Academy of Sciences. 1993;90(7):2965–9. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014;39(4):199–218. Epub 2014/03/22. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018;34(1):21–43. Epub 2018/05/08. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baird L, Yamamoto M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol Cell Biol 2020;40(13). Epub 2020/04/15. doi: 10.1128/mcb.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem 2006;281(37):27251–63. Epub 2006/06/23. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 94.Li W, Thakor N, Xu EY, Huang Y, Chen C, Yu R, Holcik M, Kong AN. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res 2010;38(3):778–88. Epub 2009/11/26. doi: 10.1093/nar/gkp1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feskanich D, Ziegler RG, Michaud DS, Giovannucci EL, Speizer FE, Willett WC, Colditz GA. Prospective study of fruit and vegetable consumption and risk of lung cancer among men and women. J Natl Cancer Inst 2000;92(22):1812–23. Epub 2000/11/18. doi: 10.1093/jnci/92.22.1812. [DOI] [PubMed] [Google Scholar]
- 96.Wang DX, Zou YJ, Zhuang XB, Chen SX, Lin Y, Li WL, Lin JJ, Lin ZQ. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616–5p-mediated GSK3beta/beta-catenin signaling pathways. Acta Pharmacol Sin 2017;38(2):241–51. Epub 2016/11/29. doi: 10.1038/aps.2016.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang LL, Zhou SJ, Zhang XM, Chen HQ, Liu W. Sulforaphane suppresses in vitro and in vivo lung tumorigenesis through downregulation of HDAC activity. Biomed Pharmacother 2016;78:74–80. Epub 2016/02/24. doi: 10.1016/j.biopha.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 98.Zhu J, Wang S, Chen Y, Li X, Jiang Y, Yang X, Li Y, Wang X, Meng Y, Zhu M, Ma X, Huang C, Wu R, Xie C, Geng S, Wu J, Zhong C, Han H. miR-19 targeting of GSK3beta mediates sulforaphane suppression of lung cancer stem cells. J Nutr Biochem 2017;44:80–91. Epub 2017/04/22. doi: 10.1016/j.jnutbio.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 99.Gao L, Cheng D, Yang J, Wu R, Li W, Kong AN. Sulforaphane epigenetically demethylates the CpG sites of the miR-9–3 promoter and reactivates miR-9–3 expression in human lung cancer A549 cells. J Nutr Biochem 2018;56:109–15. Epub 2018/03/12. doi: 10.1016/j.jnutbio.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li QQ, Xie YK, Wu Y, Li LL, Liu Y, Miao XB, Liu QZ, Yao KT, Xiao GH. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 2017;8(7):12067–80. Epub 2017/01/12. doi: 10.18632/oncotarget.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20(3):506–8. Epub 2006/01/13. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou JW, Wang M, Sun NX, Qing Y, Yin TF, Li C, Wu D. Sulforaphane-induced epigenetic regulation of Nrf2 expression by DNA methyltransferase in human Caco-2 cells. Oncol Lett 2019;18(3):2639–47. Epub 2019/08/28. doi: 10.3892/ol.2019.10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Okonkwo A, Mitra J, Johnson GS, Li L, Dashwood WM, Hegde ML, Yue C, Dashwood RH, Rajendran P. Heterocyclic Analogs of Sulforaphane Trigger DNA Damage and Impede DNA Repair in Colon Cancer Cells: Interplay of HATs and HDACs. Mol Nutr Food Res 2018;62(18):e1800228. Epub 2018/06/21. doi: 10.1002/mnfr.201800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y, Dey M. Dietary Phenethyl Isothiocyanate Protects Mice from Colitis Associated Colon Cancer. Int J Mol Sci 2017;18(9). Epub 2017/09/08. doi: 10.3390/ijms18091908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park JE, Sun Y, Lim SK, Tam JP, Dekker M, Chen H, Sze SK. Dietary phytochemical PEITC restricts tumor development via modulation of epigenetic writers and erasers. Sci Rep 2017;7:40569. Epub 2017/01/13. doi: 10.1038/srep40569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rajendran P, Kidane AI, Yu TW, Dashwood WM, Bisson WH, Lohr CV, Ho E, Williams DE, Dashwood RH. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013;8(6):612–23. Epub 2013/06/19. doi: 10.4161/epi.24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fuentes F, Paredes-Gonzalez X, Kong AN. Dietary Glucosinolates Sulforaphane, Phenethyl Isothiocyanate, Indole-3-Carbinol/3,3’-Diindolylmethane: Anti-Oxidative Stress/Inflammation, Nrf2, Epigenetics/Epigenomics and In Vivo Cancer Chemopreventive Efficacy. Curr Pharmacol Rep 2015;1(3):179–96. Epub 2015/10/13. doi: 10.1007/s40495-015-0017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5(7):e11457. Epub 2010/07/14. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Meeran SM, Patel SN, Li Y, Shukla S, Tollefsbol TO. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS One. 2012;7(5):e37748. Epub 2012/06/05. doi: 10.1371/journal.pone.0037748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cao C, Wu H, Vasilatos SN, Chandran U, Qin Y, Wan Y, Oesterreich S, Davidson NE, Huang Y. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int J Cancer. 2018;143(6):1388–401. Epub 2018/04/11. doi: 10.1002/ijc.31419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li Y, Buckhaults P, Li S, Tollefsbol T. Temporal Efficacy of a Sulforaphane-Based Broccoli Sprout Diet in Prevention of Breast Cancer through Modulation of Epigenetic Mechanisms. Cancer Prev Res (Phila) 2018;11(8):451–64. Epub 2018/05/17. doi: 10.1158/1940-6207.CAPR-17-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paul B, Li Y, Tollefsbol TO. The Effects of Combinatorial Genistein and Sulforaphane in Breast Tumor Inhibition: Role in Epigenetic Regulation. Int J Mol Sci 2018;19(6). Epub 2018/06/15. doi: 10.3390/ijms19061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang C, Su Z-Y, Khor TO, Shu L, Kong A-NT. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochemical Pharmacology. 2013;85(9):1398–404. doi: 10.1016/j.bcp.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beaver LM, Kuintzle R, Buchanan A, Wiley MW, Glasser ST, Wong CP, Johnson GS, Chang JH, Löhr CV, Williams DE. Long noncoding RNAs and sulforaphane: a target for chemoprevention and suppression of prostate cancer. The Journal of nutritional biochemistry. 2017;42:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood). 2007;232(2):227–34. [PMC free article] [PubMed] [Google Scholar]
- 116.Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res 2011;55(7):999–1009. Epub 2011/03/05. doi: 10.1002/mnfr.201000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci U S A 1994;91(8):3147–50. Epub 1994/04/12. doi: 10.1073/pnas.91.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Charron CS, Clevidence BA, Albaugh GA, Kramer MH, Vinyard BT, Milner JA, Novotny JA. Assessment of DNA damage and repair in adults consuming allyl isothiocyanate or Brassica vegetables. J Nutr Biochem 2013;24(5):894–902. Epub 2012/08/21. doi: 10.1016/j.jnutbio.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fogarty MC, Hughes CM, Burke G, Brown JC, Davison GW. Acute and chronic watercress supplementation attenuates exercise-induced peripheral mononuclear cell DNA damage and lipid peroxidation. Br J Nutr 2013;109(2):293–301. Epub 2012/04/06. doi: 10.1017/S0007114512000992. [DOI] [PubMed] [Google Scholar]
- 120.Dong Z, Shang H, Chen YQ, Pan LL, Bhatia M, Sun J. Sulforaphane Protects Pancreatic Acinar Cell Injury by Modulating Nrf2-Mediated Oxidative Stress and NLRP3 Inflammatory Pathway. Oxid Med Cell Longev 2016;2016:7864150. Epub 2016/11/17. doi: 10.1155/2016/7864150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, Yu Y, Clouthier SG, Schwartz SJ, Wicha MS, Sun D. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res 2010;16(9):2580–90. Epub 2010/04/15. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Y The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis. 2012;33(1):2–9. Epub 2011/11/15. doi: 10.1093/carcin/bgr255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hasegawa T, Nishino H, Iwashima A. Isothiocyanates inhibit cell cycle progression of HeLa cells at G2/M phase. Anticancer Drugs. 1993;4(2):273–9. Epub 1993/04/01. doi: 10.1097/00001813-199304000-00021. [DOI] [PubMed] [Google Scholar]
- 124.Yu R, Mandlekar S, Harvey KJ, Ucker DS, Kong AN. Chemopreventive isothiocyanates induce apoptosis and caspase-3-like protease activity. Cancer Res 1998;58(3):402–8. [PubMed] [Google Scholar]
- 125.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res 2006;66(11):5828–35. Epub 2006/06/03. doi: 10.1158/0008-5472.Can-06-0139. [DOI] [PubMed] [Google Scholar]
- 126.Yu R, Jiao JJ, Duh JL, Tan TH, Kong AN. Phenethyl isothiocyanate, a natural chemopreventive agent, activates c-Jun N-terminal kinase 1. Cancer Res 1996;56(13):2954–9. [PubMed] [Google Scholar]
- 127.Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annual Review of Pharmacology and Toxicology. 2007;47(1):89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 128.Motohashi H, Yamamoto M. Carcinogenesis and transcriptional regulation through Maf recognition elements. Cancer Sci 2007;98(2):135–9. Epub 2006/11/30. doi: 10.1111/j.1349-7006.2006.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang DD. The Nrf2-Keap1-ARE signaling pathway: The regulation and dual function of Nrf2 in cancer. Antioxid Redox Signal. 2010;13(11):1623–6. Epub 2010/05/22. doi: 10.1089/ars.2010.3301. [DOI] [PubMed] [Google Scholar]
- 130.Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 2015;88(Pt B):108–46. Epub 2015/07/01. doi: 10.1016/j.freeradbiomed.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jeong WS, Jun M, Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006;8(1–2):99–106. Epub 2006/02/21. doi: 10.1089/ars.2006.8.99. [DOI] [PubMed] [Google Scholar]
- 132.Qi T, Xu F, Yan X, Li S, Li H. Sulforaphane exerts anti-inflammatory effects against lipopolysaccharide-induced acute lung injury in mice through the Nrf2/ARE pathway. Int J Mol Med 2016;37(1):182–8. Epub 2015/11/05. doi: 10.3892/ijmm.2015.2396. [DOI] [PubMed] [Google Scholar]
- 133.Nijhoff WA, Grubben MJ, Nagengast FM, Jansen JB, Verhagen H, van Poppel G, Peters WH. Effects of consumption of Brussels sprouts on intestinal and lymphocytic glutathione S-transferases in humans. Carcinogenesis. 1995;16(9):2125–8. Epub 1995/09/01. doi: 10.1093/carcin/16.9.2125. [DOI] [PubMed] [Google Scholar]
- 134.Nijhoff WA, Mulder TP, Verhagen H, van Poppel G, Peters WH. Effects of consumption of brussels sprouts on plasma and urinary glutathione S-transferase class-alpha and -pi in humans. Carcinogenesis. 1995;16(4):955–7. Epub 1995/04/01. doi: 10.1093/carcin/16.4.955. [DOI] [PubMed] [Google Scholar]
- 135.Hecht SS, Carmella SG, Murphy SE. Effects of watercress consumption on urinary metabolites of nicotine in smokers. Cancer Epidemiol Biomarkers Prev 1999;8(10):907–13. [PubMed] [Google Scholar]
- 136.La Marca M, Beffy P, Della Croce C, Gervasi PG, Iori R, Puccinelli E, Longo V. Structural influence of isothiocyanates on expression of cytochrome P450, phase II enzymes, and activation of Nrf2 in primary rat hepatocytes. Food Chem Toxicol 2012;50(8):2822–30. Epub 2012/06/06. doi: 10.1016/j.fct.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 137.Guo Z, Smith TJ, Wang E, Sadrieh N, Ma Q, Thomas PE, Yang CS. Effects of phenethyl isothiocyanate, a carcinogenesis inhibitor, on xenobiotic-metabolizing enzymes and nitrosamine metabolism in rats. Carcinogenesis 1992;13(12):2205–10. Epub 1992/12/01. doi: 10.1093/carcin/13.12.2205. [DOI] [PubMed] [Google Scholar]
- 138.SS H Promising Cancer Chemopreventive Agents, Volume 1: Cancer Chemopreventive Agents Totowa. Chemoprevention by Isothiocyanates In: Kelloff GJ, Hawk ET, Sigman CC, eds. 2004;21–35.
- 139.Kumar G, Tuli HS, Mittal S, Shandilya JK, Tiwari A, Sandhu SS. Isothiocyanates: a class of bioactive metabolites with chemopreventive potential. Tumour Biol 2015;36(6):4005–16. Epub 2015/04/04. doi: 10.1007/s13277-015-3391-5. [DOI] [PubMed] [Google Scholar]
- 140.Khor TO, Cheung WK, Prawan A, Reddy BS, Kong AN. Chemoprevention of familial adenomatous polyposis in Apc(Min/+) mice by phenethyl isothiocyanate (PEITC). Mol Carcinog 2008;47(5):321–5. Epub 2007/10/13. doi: 10.1002/mc.20390. [DOI] [PubMed] [Google Scholar]
- 141.Cavell BE, Syed Alwi SS, Donlevy A, Packham G. Anti-angiogenic effects of dietary isothiocyanates: mechanisms of action and implications for human health. Biochem Pharmacol 2011;81(3):327–36. Epub 2010/10/20. doi: 10.1016/j.bcp.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 142.Zhang QC, Pan ZH, Liu BN, Meng ZW, Wu X, Zhou QH, Xu K. Benzyl isothiocyanate induces protective autophagy in human lung cancer cells through an endoplasmic reticulum stress-mediated mechanism. Acta Pharmacol Sin 2017;38(4):539–50. Epub 2017/01/24. doi: 10.1038/aps.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vyas AR, Hahm ER, Arlotti JA, Watkins S, Stolz DB, Desai D, Amin S, Singh SV. Chemoprevention of prostate cancer by d,l-sulforaphane is augmented by pharmacological inhibition of autophagy. Cancer Res 2013;73(19):5985–95. Epub 2013/08/08. doi: 10.1158/0008-5472.CAN-13-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim M, Cho HJ, Kwon GT, Kang YH, Kwon SH, Her S, Park T, Kim Y, Kee Y, Park JH. Benzyl isothiocyanate suppresses high-fat diet-stimulated mammary tumor progression via the alteration of tumor microenvironments in obesity-resistant BALB/c mice. Mol Carcinog 2015;54(1):72–82. Epub 2014/04/15. doi: 10.1002/mc.22159. [DOI] [PubMed] [Google Scholar]
- 145.Gupta P, Adkins C, Lockman P, Srivastava SK. Metastasis of Breast Tumor Cells to Brain Is Suppressed by Phenethyl Isothiocyanate in a Novel In Vivo Metastasis Model. PLoS One. 2013;8(6):e67278. Epub 2013/07/05. doi: 10.1371/journal.pone.0067278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang L, Tian Z, Yang Q, Li H, Guan H, Shi B, Hou P, Ji M. Sulforaphane inhibits thyroid cancer cell growth and invasiveness through the reactive oxygen species-dependent pathway. Oncotarget 2015;6(28):25917–31. Epub 2015/08/28. doi: 10.18632/oncotarget.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mager DE, Jusko WJ. Development of translational pharmacokinetic-pharmacodynamic models. Clin Pharmacol Ther 2008;83(6):909–12. Epub 2008/04/05. doi: 10.1038/clpt.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Meibohm B, Derendorf H. Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modelling. Int J Clin Pharmacol Ther 1997;35(10):401–13. [PubMed] [Google Scholar]
- 149.Hanlon N, Coldham N, Gielbert A, Kuhnert N, Sauer MJ, King LJ, Ioannides C. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br J Nutr 2008;99(3):559–64. Epub 2007/09/18. doi: 10.1017/s0007114507824093. [DOI] [PubMed] [Google Scholar]
- 150.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta 2002;316(1–2):43–53. Epub 2001/12/26. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]
- 151.Al Janobi AA, Mithen RF, Gasper AV, Shaw PN, Middleton RJ, Ortori CA, Barrett DA. Quantitative measurement of sulforaphane, iberin and their mercapturic acid pathway metabolites in human plasma and urine using liquid chromatography-tandem electrospray ionisation mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2006;844(2):223–34. Epub 2006/08/26. doi: 10.1016/j.jchromb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 152.Zhang Y, Talalay P. Mechanism of differential potencies of isothiocyanates as inducers of anticarcinogenic Phase 2 enzymes. Cancer Res 1998;58(20):4632–9. [PubMed] [Google Scholar]
- 153.Ye L, Zhang Y. Total intracellular accumulation levels of dietary isothiocyanates determine their activity in elevation of cellular glutathione and induction of Phase 2 detoxification enzymes. Carcinogenesis 2001;22(12):1987–92. Epub 2001/12/26. doi: 10.1093/carcin/22.12.1987. [DOI] [PubMed] [Google Scholar]
- 154.Zhang Y, Callaway EC. High cellular accumulation of sulphoraphane, a dietary anticarcinogen, is followed by rapid transporter-mediated export as a glutathione conjugate. Biochem J 2002;364(Pt 1):301–7. Epub 2002/05/04. doi: 10.1042/bj3640301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang H, Khor TO, Yang Q, Huang Y, Wu TY, Saw CL, Lin W, Androulakis IP, Kong AN. Pharmacokinetics and pharmacodynamics of phase II drug metabolizing/antioxidant enzymes gene response by anticancer agent sulforaphane in rat lymphocytes. Mol Pharm 2012;9(10):2819–27. Epub 2012/08/31. doi: 10.1021/mp300130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Munday R, Mhawech-Fauceglia P, Munday CM, Paonessa JD, Tang L, Munday JS, Lister C, Wilson P, Fahey JW, Davis W, Zhang Y. Inhibition of urinary bladder carcinogenesis by broccoli sprouts. Cancer Res 2008;68(5):1593–600. Epub 2008/03/04. doi: 10.1158/0008-5472.Can-07-5009. [DOI] [PubMed] [Google Scholar]
- 157.Egner PA, Chen JG, Wang JB, Wu Y, Sun Y, Lu JH, Zhu J, Zhang YH, Chen YS, Friesen MD, Jacobson LP, Muñoz A, Ng D, Qian GS, Zhu YR, Chen TY, Botting NP, Zhang Q, Fahey JW, Talalay P, Groopman JD, Kensler TW. Bioavailability of Sulforaphane from two broccoli sprout beverages: results of a short-term, cross-over clinical trial in Qidong, China. Cancer Prev Res (Phila) 2011;4(3):384–95. Epub 2011/03/05. doi: 10.1158/1940-6207.Capr-10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bogaards JJ, Verhagen H, Willems MI, van Poppel G, van Bladeren PJ. Consumption of Brussels sprouts results in elevated alpha-class glutathione S-transferase levels in human blood plasma. Carcinogenesis 1994;15(5):1073–5. Epub 1994/05/01. doi: 10.1093/carcin/15.5.1073. [DOI] [PubMed] [Google Scholar]
- 159.Sreerama L, Hedge MW, Sladek NE. Identification of a class 3 aldehyde dehydrogenase in human saliva and increased levels of this enzyme, glutathione S-transferases, and DT-diaphorase in the saliva of subjects who continually ingest large quantities of coffee or broccoli. Clin Cancer Res 1995;1(10):1153–63. [PubMed] [Google Scholar]
- 160.Bauman JE, Zang Y, Sen M, Li C, Wang L, Egner PA, Fahey JW, Normolle DP, Grandis JR, Kensler TW, Johnson DE. Prevention of Carcinogen-Induced Oral Cancer by Sulforaphane. Cancer Prev Res (Phila) 2016;9(7):547–57. Epub 2016/06/25. doi: 10.1158/1940-6207.CAPR-15-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brown RH, Reynolds C, Brooker A, Talalay P, Fahey JW. Sulforaphane improves the bronchoprotective response in asthmatics through Nrf2-mediated gene pathways. Respir Res 2015;16:106. Epub 2015/09/16. doi: 10.1186/s12931-015-0253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Riedl MA, Saxon A, Diaz-Sanchez D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol 2009;130(3):244–51. Epub 2008/11/26. doi: 10.1016/j.clim.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Huh K, Iwata H, Yamamoto I. [Hepatic allopurinol oxidizing enzyme in mice]. Nihon Yakurigaku Zasshi 1975;71(2):175–83. [PubMed] [Google Scholar]
- 164.Palliyaguru DL, Yuan JM, Kensler TW, Fahey JW. Isothiocyanates: Translating the Power of Plants to People. Mol Nutr Food Res 2018;62(18):e1700965. Epub 2018/02/23. doi: 10.1002/mnfr.201700965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Egner PA, Chen JG, Zarth AT, Ng DK, Wang JB, Kensler KH, Jacobson LP, Munoz A, Johnson JL, Groopman JD, Fahey JW, Talalay P, Zhu J, Chen TY, Qian GS, Carmella SG, Hecht SS, Kensler TW. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: results of a randomized clinical trial in China. Cancer Prev Res (Phila) 2014;7(8):813–23. Epub 2014/06/11. doi: 10.1158/1940-6207.CAPR-14-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kensler TW, Ng D, Carmella SG, Chen M, Jacobson LP, Munoz A, Egner PA, Chen JG, Qian GS, Chen TY, Fahey JW, Talalay P, Groopman JD, Yuan JM, Hecht SS. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis 2012;33(1):101–7. Epub 2011/11/03. doi: 10.1093/carcin/bgr229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Riso P, Martini D, Moller P, Loft S, Bonacina G, Moro M, Porrini M. DNA damage and repair activity after broccoli intake in young healthy smokers. Mutagenesis 2010;25(6):595–602. Epub 2010/08/18. doi: 10.1093/mutage/geq045. [DOI] [PubMed] [Google Scholar]
- 168.Cipolla BG, Mandron E, Lefort JM, Coadou Y, Della Negra E, Corbel L, Le Scodan R, Azzouzi AR, Mottet N. Effect of Sulforaphane in Men with Biochemical Recurrence after Radical Prostatectomy. Cancer Prev Res (Phila) 2015;8(8):712–9. Epub 2015/05/15. doi: 10.1158/1940-6207.CAPR-14-0459. [DOI] [PubMed] [Google Scholar]
- 169.Atwell LL, Zhang Z, Mori M, Farris P, Vetto JT, Naik AM, Oh KY, Thuillier P, Ho E, Shannon J. Sulforaphane Bioavailability and Chemopreventive Activity in Women Scheduled for Breast Biopsy. Cancer Prev Res (Phila) 2015;8(12):1184–91. Epub 2015/10/30. doi: 10.1158/1940-6207.CAPR-15-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hecht SS, Chung FL, Richie JP Jr., Akerkar SA, Borukhova A, Skowronski L, Carmella SG. Effects of watercress consumption on metabolism of a tobacco-specific lung carcinogen in smokers. Cancer Epidemiol Biomarkers Prev 1995;4(8):877–84. [PubMed] [Google Scholar]
- 171.Yuan JM, Stepanov I, Murphy SE, Wang R, Allen S, Jensen J, Strayer L, Adams-Haduch J, Upadhyaya P, Le C, Kurzer MS, Nelson HH, Yu MC, Hatsukami D, Hecht SS. Clinical Trial of 2-Phenethyl Isothiocyanate as an Inhibitor of Metabolic Activation of a Tobacco-Specific Lung Carcinogen in Cigarette Smokers. Cancer Prev Res (Phila) 2016;9(5):396–405. Epub 2016/03/10. doi: 10.1158/1940-6207.CAPR-15-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Joshi J Alumkal RS, Motomi Mori, Jacob Schwartzman, Julie Nicole Graff, Tomasz M BeerChristopher W. Ryan, Dennis R Koop, Ganesh Cherala, Myrna Munar, Jason Frederick Flamiatos, Lina Gao, Erin Tucker. Sulforaphane treatment in men with recurrent prostate cancer. Journal of Clinical Oncology 2013, May 20;31. doi: DOI: 10.1200/jco.2013.31.15_suppl.5017. [DOI] [Google Scholar]
- 173.Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 2003;9(10 Pt 1):3578–88. [PubMed] [Google Scholar]
- 174.Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV, Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK, Goldspiel B, Fojo AT, Balcerzak SP, Bates SE. Phase I trial of the histone deacetylase inhibitor, depsipeptide (FR901228, NSC 630176), in patients with refractory neoplasms. Clin Cancer Res 2002;8(3):718–28. [PubMed] [Google Scholar]
- 175.Cho KR, Vogelstein B. Genetic alterations in the adenoma--carcinoma sequence. Cancer. 1992;70(6 Suppl):1727–31. Epub 1992/09/15. doi: 10.1002/1097-0142(19920915)70:4+<1727::aid-cncr2820701613>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 176.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. Epub 2000/01/27. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 177.Kong AN, Yu R, Lei W, Mandlekar S, Tan TH, Ucker DS. Differential activation of MAPK and ICE/Ced-3 protease in chemical-induced apoptosis. The role of oxidative stress in the regulation of mitogen-activated protein kinases (MAPKs) leading to gene expression and survival or activation of caspases leading to apoptosis. Restor Neurol Neurosci 1998;12(2–3):63–70. [PubMed] [Google Scholar]
- 178.Kong AN, Mandlekar S, Yu R, Lei W, Fasanmande A. Pharmacodynamics and toxicodynamics of drug action: signaling in cell survival and cell death. Pharm Res 1999;16(6):790–8. Epub 1999/07/09. doi: 10.1023/a:1011953431486. [DOI] [PubMed] [Google Scholar]
- 179.Kong AN, Yu R, Chen C, Mandlekar S, Primiano T. Signal transduction events elicited by natural products: role of MAPK and caspase pathways in homeostatic response and induction of apoptosis. Arch Pharm Res 2000;23(1):1–16. Epub 2000/03/23. doi: 10.1007/bf02976458. [DOI] [PubMed] [Google Scholar]
- 180.Kong AN, Yu R, Hebbar V, Chen C, Owuor E, Hu R, Ee R, Mandlekar S. Signal transduction events elicited by cancer prevention compounds. Mutat Res 2001;480–481:231–41. Epub 2001/08/17. doi: 10.1016/s0027-5107(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 181.Owuor ED, Kong AN. Antioxidants and oxidants regulated signal transduction pathways. Biochem Pharmacol 2002;64(5–6):765–70. Epub 2002/09/06. doi: 10.1016/s0006-2952(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 182.Umar A, Dunn BK, Greenwald P. Future directions in cancer prevention. Nat Rev Cancer. 2012;12(12):835–48. Epub 2012/11/16. doi: 10.1038/nrc3397. [DOI] [PubMed] [Google Scholar]
- 183.Meyskens FL Jr., Mukhtar H, Rock CL, Cuzick J, Kensler TW, Yang CS, Ramsey SD, Lippman SM, Alberts DS. Cancer Prevention: Obstacles, Challenges and the Road Ahead. J Natl Cancer Inst 2016;108(2). Epub 2015/11/10. doi: 10.1093/jnci/djv309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kensler TW, Spira A, Garber JE, Szabo E, Lee JJ, Dong Z, Dannenberg AJ, Hait WN, Blackburn E, Davidson NE, Foti M, Lippman SM. Transforming Cancer Prevention through Precision Medicine and Immune-oncology. Cancer Prev Res (Phila) 2016;9(1):2–10. Epub 2016/01/09. doi: 10.1158/1940-6207.Capr-15-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lippman SM, Abate-Shen C, Colbert Maresso KL, Colditz GA, Dannenberg AJ, Davidson NE, Disis ML, DuBois RN, Szabo E, Giuliano AR, Hait WN, Lee JJ, Kensler TW, Kramer BS, Limburg P, Maitra A, Martinez ME, Rebbeck TR, Schmitz KH, Vilar E, Hawk ET. AACR White Paper: Shaping the Future of Cancer Prevention - A Roadmap for Advancing Science and Public Health. Cancer Prev Res (Phila) 2018;11(12):735–78. Epub 2018/12/12. doi: 10.1158/1940-6207.Capr-18-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]