

Abstract
A highly strained covalent organic cage compound was synthesized from hexahydroxy tribenzotriquinacene (TBTQ) and a meta‐terphenyl‐based diboronic acid with an additional benzoic acid substituent in 2’‐position. Usually, a 120° bite angle in the unsubstituted ditopic linker favors the formation of a [4+6] cage assembly. Here, the introduction of the benzoic acid group is shown to lead to a perfectly preorganized circular hydrogen‐bonding array in the cavity of a trigonal‐bipyramidal [2+3] cage, which energetically overcompensates the additional strain energy caused by the larger mismatch in bite angles for the smaller assembly. The strained cage compound was analyzed by mass spectrometry and 1H, 13C and DOSY NMR spectroscopy. DFT calculations revealed the energetic contribution of the hydrogen‐bonding template to the cage stability. Furthermore, molecular dynamics simulations on early intermediates indicate an additional kinetic effect, as hydrogen bonding also preorganizes and rigidifies small oligomers to facilitate the exclusive formation of smaller and more strained macrocycles and cages.
Keywords: boronate esters, cage compounds, density functional calculations, dynamic covalent chemistry, hydrogen bonding
Cages put under tension: Endohedral hydrogen bonding stabilizes a highly strained covalent organic cage compound. The introduction of carboxylic acid groups at the concave side of a curved diboronic acid linker switches cage geometry from tetrahedral [4+6] to trigonal‐bipyramidal [2+3] assembly. Thermodynamic stability and kinetic preorganization of early intermediates have been analyzed by DFT calculations.
Introduction
Subcomponent self‐assembly [1] is a powerful tool for the one‐pot‐synthesis of complex nanoarchitectures directly from simple precursors. Depending on the interactions that connect the individual building blocks, supramolecular, [2] metal–organic [3] or dynamic covalent [4] structures have been reported. For design purposes, geometrical concepts such as the directional bonding approach [5] allow control over geometry and topology of the obtained scaffolds, as these properties are directly encoded [6] in the molecular structure of the building blocks. In recent years, a large variety of covalent organic cages [7] with different size, shape [8] and topology [9] have been designed and synthesized. Potential applications for these porous molecular materials [10] range from molecular recognition, [11] sensing, [12] gas storage[ 8i , 13 ] and separation, [14] to self‐sorting, [15] encapsulation [16] and reactivity control [17] of large aromatic guests. In terms of complexity, rigid cages offer the tempting potential to hierarchically organize multiple functionalities with high spatial precision around the cage scaffold. Exohedral functionalization at the outer surface typically impacts the solubility [18] or solid‐state packing[ 8f , 19 ] of these modular porous units. Ultimately, the implementation of cross‐linkable functions in the cage periphery can lead to covalently linked ‘cage‐to‐framework’ materials. [20] On the other hand, endohedral functionalization of cages [21] is less explored. Besides chemical stabilization via post‐synthetic modification, [22] sixfold ether synthesis within an imine cage [23] and alkyne cages with inward‐pointing pyridines [24] have been reported. Besides chemical modulation of the pores, cavity‐directing functional sites might be utilized to template specific cage geometries or could interfere with each other during the assembly process. To date, template‐assisted synthesis [25] of covalent organic cage compounds has been rarely addressed. With proper choice of functional precursors however, switching between different cage geometries might be realized.
Results and Discussion
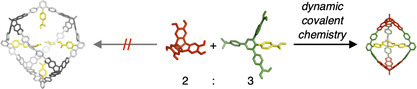
Here, we report on the serendipitous formation of a highly strained [2+3] molecular cage A2CPhCOOH 3 from hexahydroxy tribenzotriquinacene [26] (TBTQ) A [18] and terphenyl diboronic acid derivative CPhCOOH through the formation of six boronate esters under water‐removing conditions. DFT calculations suggest that the three endohedral PhCOOH groups in the trigonal‐bipyramidal cage are perfectly preorganized for intramolecular hydrogen bonding, which overcompensates the additional strain energy in relation to larger [4+6] assemblies (Figure 1).
Figure 1.
Supramolecular templation of strained covalent organic cages.
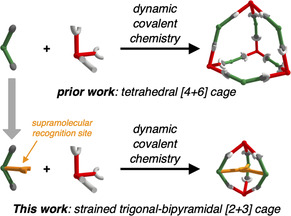
In prior work, we reported on a series of shape‐selective covalent organic cage compounds derived from orthogonal TBTQ A and diboronic acids with varying bite angles.[ 15b , 18 ] For 2′‐methyl‐[1,1’:3’,1’’‐terphenyl]‐4,4’’‐diboronic acid (CMe) possessing a 120° bite angle at the central phenylene unit, tetrahedral A4CMe 6 cages were obtained as the only detectable self‐assembly product. Since the six bent CMe linkers are octahedrally distributed around the cage pore, all Me groups in 2’‐position are located within the cage cavities, whereas any substituent in 5’‐position would point to the outside of the cages. Aiming for functionalized cavities, we envisioned the attachment of supramolecular binding sites, for example, COOH groups within the cage cavities. Based on a procedure introduced by Höger and co‐workers, [27] we applied a modular synthetic approach (Scheme 1) that allows for easy modifications of linkers C at both 2’‐ and 5’‐position by proper choice of aldehyde (1) and carboxylic acid (4) starting materials. Here, we synthesized a novel linker CPhCOOH possessing a PhCOOH substituent in 2’‐position as an endohedral recognition site and a tBuPh group in 5’‐position to further enhance the solubility of the final assemblies. Diiodide 5[ 26 , 27 ] was synthesized in two literature‐known steps and reacted to bispinacol ester 6 by twofold Miyaura borylation. Treatment of 6 with BBr3 followed by aqueous workup resulted in simultaneous cleavage of both pinacol and methyl ester protecting groups to give diboronic acid CPhCOOH in 76 % yield. As CMe and CPhCOOH have very similar bite angles between the two boronic acids, we initially expected a similar behavior in dynamic covalent reactions with catechol‐containing precursors such as A. However, subtle effects caused by slight differences in solubility or rotational freedom for the attached phenylene units cannot be ruled out entirely.
Scheme 1.
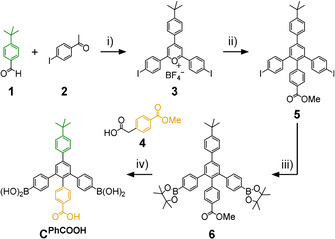
Modular synthesis of diboronic acid CPhCOOH: i) BF3⋅OEt2, 100 °C, 2 h, 38 %; [27] ii) 4 [29] /NaOH, MeOH, 30 min, then Ac2O, 160 °C, 2 h, 55 %; [28] iii) B2pin2/KOAc/Pd(dppf)Cl2, DMF, reflux, 2.5 h, 71 %; iv) BBr3, CH2Cl2, 0 °C→rt, 3.75 h, 76 %.
For initial investigations regarding cage formation, we applied our established protocol [15b] and monitored the reaction of A and CPhCOOH in THF in the presence of 4Å molecular sieves by 1H NMR spectroscopy (Figure S15). In contrast to A4CMe 6, we did not observe the quantitative formation of a closed boronate ester assembly. Instead, rather broad signals and significant amounts of free catechols were observed even after prolonged reaction times and further addition of molecular sieves. MALDI‐TOF mass spectrometry (MS) of the reaction mixture showed the two most prominent signals at m/z=1883.24 and 2382.70 (Figure S17), which correspond to smaller [A2CPhCOOH 2+Na]+ and [A2CPhCOOH 3+Na]+ assemblies containing only two TBTQ units in a [2+3] cage or [2+2] macrocycle, respectively. This somewhat unexpected finding contradicts the directional bonding approach, [5] which predicts a tetrahedral [4+6] assembly for the combination of a tritopic 90° and a ditopic 120° linker as it was realized for A4CMe 6. [15b] As we did not observe notable precipitation in both reactions with CMe and CPhCOOH, it is rather unlikely that the different outcome is caused by solubility issues. For CPhCOOH instead, a closer look at the structure of the molecular components and the MALDI‐TOF MS data for the reaction mixture suggests a strong preorganization of two or three CPhCOOH molecules via hydrogen bonding between the COOH groups, which favors the formation of the strained [2+2] macrocycles and [2+3] cage assemblies. In order to break these hydrogen bonds and to induce the formation of larger assemblies, e.g., [4+6] cages, we also attempted cage synthesis in the presence of up to two equivalents of AcOH, referred to CPhCOOH. However, 1H NMR (Figure S16) and MALDI‐TOF mass spectra looked identical as for the reaction without AcOH (Figure S15), thus indicating the strong driving force for supramolecular preorganization. However, no full conversion to the stoichiometric cage product was achieved, as usually observed for binary mixtures in 2:3 ratio.[ 15b , 18 ] Both [2+2] macrocycles and [2+3] cages were simultaneously obtained as the two main products, accompanied by significant amounts of open oligomers. Presumably, additional strain is induced when closing the macrocyclic [2+2] intermediate with a third linker CPhCOOH, thus shifting the equilibrium towards the macrocyclic fragment, which still possesses two unreacted catechol units. In order to facilitate full conversion and isolation of A2CPhCOOH 3, we screened for optimized reaction conditions. Finally, reaction of A and CPhCOOH in MeCN/THF 10:1 under reflux for 24 hours (Scheme 2) resulted in precipitation of a crude product. Resolvation of the isolated material in [D4]MeOH induced complete disassembly into the monomeric building blocks. Based on 1H NMR integration, the measured ratio of A/CPhCOOH=2:3 indicates the predominant formation of a closed‐shell [2+3] or [4+6] assembly rather than open [2+2] macrocycles or other oligomeric side products. MALDI‐TOF MS for the isolated product (Figure 2 c) revealed only one set of signals at m/z=2323.29 [A2CPhCOOH 3‐C4H9+Na]+, 2340.29 [A2CPhCOOH 3‐C4H9+K]+, 2357.32 [A2CPhCOOH 3]+, 2364.27 [A2CPhCOOH 3+Li]+ and 2381.31 [A2CPhCOOH 3+Na]+ that were all assigned to A2CPhCOOH 3 and various monocationic adducts. Remarkably, the very high tendency for adduct formation with alkali metal ions indicates that the endohedral array of COOH groups might serve as an efficient binding station for metal ions.
Scheme 2.
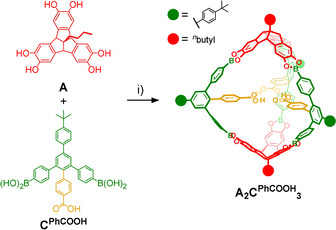
Synthesis of A2CPhCOOH 3: i) THF/MeCN 10:1, reflux, 24 h, 64 %.
Figure 2.
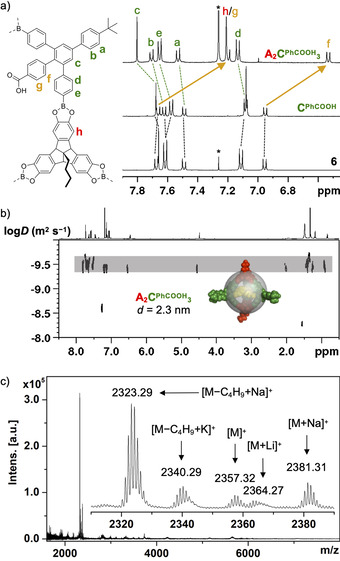
a) Aromatic region of 1H NMR spectrum (400 MHz, rt) for A2CPhCOOH 3 (CDCl3), CPhCOOH ([D8]THF) and 6 (CDCl3), b) DOSY NMR (600 MHz, CDCl3, rt; inset shows PM6‐minimized space‐filling model with the solvodynamic diameter indicated as a semi‐transparent gray sphere) and c) MALDI‐TOF MS for A2CPhCOOH 3.
In contrast to the reaction in THF under equilibrating conditions (Figure S17), the absence of signals for other assemblies confirms the isolation of pure A2CPhCOOH 3 cages from reaction in MeCN. Presumably, closed‐shell cages in MeCN are significantly less soluble than the more polar open intermediates, thus driving the dynamic formation of boronate esters towards completion. Finally, A2CPhCOOH 3 cages are kinetically trapped by precipitation. After filtration and subsequent washing with MeCN and n‐hexane, the raw product was further purified by suspending it in dry acetone for four hours at 60 °C to obtain cage A2CPhCOOH 3 in pure form. After work‐up, the isolated cage was soluble in CHCl3 and the 1H NMR spectrum (Figure 2 a) shows only one single peak at 4.55 ppm for the TBTQ bridgehead proton, thus further confirming the formation of one single cage species. Integration of protons corresponding to either TBTQ A or linker CPhCOOH confirmed the expected ratio of A/CPhCOOH=2:3, whereas the occurrence of one single set of peaks for all chemically distinguishable protons argues for a highly symmetrical assembly.
In comparison to precursors 6 (in CDCl3) and CPhCOOH (in [D8]THF), there is a striking upfield shift of roughly 0.5 ppm for the aromatic protons of the PhCOOH substituents that are located in the cage cavity. We attribute this effect to additional shielding by the π‐surface of the cage interior and, primarily, efficient hydrogen bonding between the closely arranged PhCOOH groups. For the acidic protons, a broad and barely detectable signal at 10.9 ppm was observed. To estimate the cage size and ultimately differentiate between a [2+3] or [4+6] assembly, the solvodynamic diameter was determined via DOSY NMR measurements (Figure 2 b). In CDCl3, a diffusion constant of 3.43×10−10 m2 s−1 was obtained for the monodisperse cage species. According to the Stokes‐Einstein equation, this value correlates to a solvodynamic diameter of 2.3 nm, which is in very good agreement with a PM6 [30] ‐minimized space filling model (depicted as a semi‐transparent sphere in Figure 2 b). Cage A2CPhCOOH 3 shows a considerably smaller diameter than the previously reported tetrahedral cage A4CMe 6 (d=3.0 nm), [15b] even though the protruding tBuPh substituents in 5’‐position at the bent linkers CPhCOOH are expected to result in even larger values for the tetrahedral [4+6] assemblies, thus ultimately proving the formation of the smaller [2+3] cage.
Remarkably, the simple exchange from Me to PhCOOH as endohedral substituent completely switched the cage size from a [4+6] to a [2+3] assembly and the geometrical arrangement of the linkers C from octahedral to trigonal planar, respectively. To evaluate if these selectivities are driven by different thermodynamic stabilities, we performed geometry optimizations employing DFT calculations with the ωB97XD [31] functional and the def2‐SVP [32] basis set for cages in tetrahedral [4+6] and trigonal‐bipyramidal [2+3] geometry for both CMe and CPhCOOH. In order to decrease the computational demand, the apical nBu substituents in A were replaced by Me groups and the exohedral tBuPh groups in 5’‐position for CPhCOOH were replaced by H atoms, as these groups are located on the outer cage surface and should not influence the inherent cage stability or preference for a specific topology. For CMe, a formal transformation of one A4CMe 6 into two A2CMe 3 cages revealed a difference in electronic energies of +60 kJ mol−1 in the gas phase, indicating a significantly higher thermodynamic stability for the larger cage (Figure 3 a). To adjust for any solvation effects, we simulated MeCN as solvent by applying a self‐consistent field method using the integral equation formalism model (IEFPCM) [33] as implemented in the Gaussian 16 [34] quantum chemical software package. Again, an energy difference of +62 kJ mol−1 was calculated, indicating that the equilibrium between the two cages is hardly affected by solvation but rather a consequence of the inherent stability of the individual cage geometries. In accordance with the directional bonding approach, this selectivity is matched in the synthetic experiment and is most presumably related to the inherent strain energy induced by the larger mismatch in the bite angles and a significant bending in the three struts for CMe (see Figure 3 a). For CPhCOOH however, DFT optimization in the gas phase revealed a completely reversed thermodynamic driving force, as the theoretical results indicate A2CPhCOOH 3 to be −207 kJ mol−1 more stable in electronic energy than the larger [4+6] assembly (Figure 3 a). For calculations with the MeCN continuum model, this energy difference is reduced to −150 kJ mol−1, indicating the pronounced stabilization of polar COOH groups in polar aprotic solvents such as MeCN. The geometry‐optimized structures for both A4C6 cages show a very good overlap for the cage backbone (Figure S21 a). As a response to the pore filling with the internal hydrogen bonding array of the PhCOOH groups, a slight deviation was observed for the A2C3 cages (Figure S21 b). Therefore, we conclude that the inherent strain induced by the rigid scaffold of a specific cage stoichiometry does not depend on the inner substituents. However, attractive supramolecular interactions between the endohedral PhCOOH groups in the cage pores overcompensate the higher strain energy for the A2CPhCOOH 3 cage.
Figure 3.
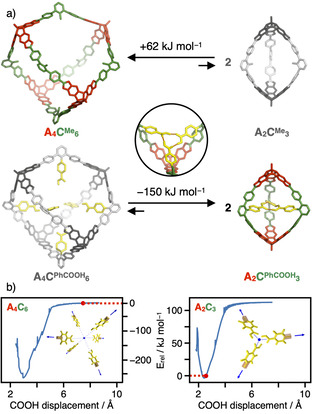
a) Equilibria between tetrahedral A4C6 and trigonal‐bipyramidal A2C3 cages with endohedral Me (top) and PhCOOH (bottom) functionalization (electronic energies and geometry‐optimized models derived from DFT calculations, H atoms are omitted for clarity); b) potential energy plots for the assembly of six or three PhCOOH moieties as realized in A4CPhCOOH 6 and A2CPhCOOH 3, respectively (the actual distance in the DFT‐optimized cages is indicated in red).
To further probe this stabilizing contribution, we performed relaxed scans for the endohedral substituents in both geometries (Figure 3 b). For this purpose, we constructed model systems containing six and three benzoic acid molecules in the respective octahedral (A4C6) or trigonal‐planar (A2C3) arrangement. To artificially adjust for varying cage sizes, we systematically scanned the distance of all COOH groups from the center of mass for both model systems. During these constrained optimizations, all benzoic acid molecules were kept fixed at the C−H unit in 4‐position (indicated as orange boxes in Figure 3 b) to simulate the rigid character of the cage pores, while the molecules itself were allowed to structurally relax. The obtained potential energy profiles presented in Figure 3 b show that the actual arrangement of the three PhCOOH groups in A2CPhCOOH 3 is very close to the energetically most favorable distance, while the separation in the larger A4CPhCOOH 6 is very far from the potential minimum. Essentially, there is no attractive interaction between the COOH groups at a distance of 7.49 Å from the focal point in A4CPhCOOH 6, whereas the A2C3 scaffold preorganizes the COOH groups almost perfectly for a circular threefold hydrogen‐bonding motif (see inset in Figure 3 a). To quantify this stabilizing interaction, we reoptimized the threefold array with DFT and the MeCN continuum model at both the distance realized in the cage and the most distant arrangement as reference. From the electronic energies, a binding energy of −104 kJ mol−1 was calculated. Assuming that the inherent strain in A2CPhCOOH 3 is the same as for the empty A2CMe 3, a stabilizing contribution of −106 kJ mol−1 per cage was estimated from the reaction equations in Figure 3 a. The very good agreement between these two values further corroborates the subtle balance between the geometrical strain distributed in the cage backbone and any stabilizing interactions within the cage cavities.
Besides the hydrogen‐bond mediated thermodynamic driving force for the formation of the more strained [2+3] cages, kinetic effects might also play a role, as hydrogen bonding could already fixate early intermediates during cage formation, thus directing any further reactions towards more strained smaller assemblies. As a key intermediate, we identified bisboronate ester ACPhCOOH 2, since hydrogen bonding between the PhCOOH moieties might severely restrict rotational motions in this molecule. To test this hypothesis, we performed molecular dynamics (MD) simulations for both ACMe 2 and ACPhCOOH 2 with the semi‐empirical PM6 [30] method along with the D3H4 [35] correction for an adequate description of hydrogen bonding and dispersion as implemented in the MOPAC2016 [36] program suite. As a measure for preorganization, we plotted the B‐B distance of the two remaining unfunctionalized boronic acid moieties. For both structures, ten MD trajectories at 298 K with randomly generated starting configurations were calculated for 1 ns (see Figure 4 for one selected example for each structure and Figure S22 for all trajectories for ACPhCOOH 2). For ACMe 2, the structure appeared to be rather flexible and a wriggling motion with a large variation of the B‐B distance for the boronic acids ranging from 14 to 35 Å was observed. For ACPhCOOH 2 however, after a short time period of 100 ps, most trajectories were trapped in a U‐shaped structure with a fixed B‐B distance of around 13 Å (Figure 4 a). Few simulations resulted in the population of an even more compact structure (d B−B=5.3 Å), with one of the PhCOOH units forming a hydrogen bond to the opposite boronate ester (Figure S24). However, as this structure is approximately 18 kJ mol−1 higher in energy than the U‐shaped arrangement, this metastable conformer is presumably only populated in neglectable amounts. For a more specific analysis, we used one representative starting configuration for ACPhCOOH 2 to calculate an ensemble of ten trajectories with randomly generated initial velocities based on a Maxwell–Boltzmann distribution at room temperature (Figure S23). Again, all simulations were quickly trapped in the two fixed geometries and the distance of the free boronic acids stayed constant for the remaining simulation. The average B−B distance for the prearranged U‐shaped conformer of intermediate ACPhCOOH 2 is very close to the value of 9.6 Å calculated for the DFT‐optimized structures of the two isomers of the macrocycle A2CPhCOOH 2, which is assumed to act as the next intermediate within the mechanism of cage formation.
Figure 4.
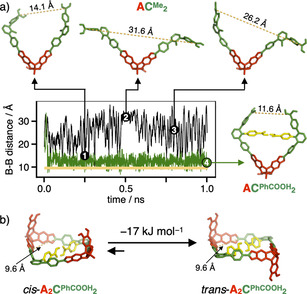
a) Molecular dynamics calculations of ACMe 2 (black) and ACPhCOOH 2 (green) for 1 ns at 298 K (1–3 display structures for ACMe 2 at 250, 500 and 800 ps, 4 displays structure for ACPhCOOH 2 at 1 ns; yellow dashed lines indicate the B‐B distance for the free boronic acids, the yellow line in the graph indicates the B‐B distance in macrocyclic A2CPhCOOH 2); b) equilibrium between cis‐ and trans‐A2CPhCOOH 2 (structures and electronic energies derived from DFT calculations, H atoms are omitted for clarity).
Therefore, we postulate an additional kinetic effect that favors the preorganization of the building blocks towards the facile synthesis of the strained [2+2] macrocycle. Hereby, the formation of larger macrocycles, for example, [3+3] or [4+4] is prevented, which are essential on‐pathway intermediates in the synthesis of the larger tetrahedral [4+6] cages. Interestingly, DFT calculations showed that the trans‐isomer is −17 kJ mol−1 more stable than the cis‐isomer (Figure 4 b). Since the [2+3] cage can only be formed via the less preferred cis‐isomer, the trans‐macrocycle serves as a resting state, thus preventing further ring closure to the even more strained [2+3] cage assemblies. These subtle energy differences might be the explanation for the pronounced occurrence of the [2+2] macrocycles while performing the reactions under equilibrium conditions in THF (see Figures S15 and S16). Dynamic covalent reaction of A/CPhCOOH in a 1:1 ratio in THF further confirmed the inherent stability of the macrocyclic intermediates. MALDI‐TOF MS of the reaction mixture predominantly showed signals at m/z=1843.37 and 3745.94, which can be assigned to [2+2] assemblies [A2CPhCOOH 2‐H2O+H]+ and [2⋅A2CPhCOOH 2+Na]+, respectively (Figure S18). In particular, no evidence for A2CPhCOOH 3 or other larger assemblies was found.
For the A/CMe reaction mixture, A4CMe 6 is the only observable product despite a significant mismatch between optimal (141°) and actual (120°) bite angle for the linker CMe. Therefore, strain energy is released in a social self‐sorting experiment for a A/CMe/D mixture (D=2,5‐di‐nbutyl‐1,4‐diboronic acid) by forming the mixed A4CMe 4 D2 cage as the predominant product.[ 15a , 15b ] To probe the effect of the endohedral PhCOOH groups on the formation of mixed cages, we performed a similar self‐sorting experiment for A/CPhCOOH/D by dissolving the building blocks in [D8]THF in a 4:4:2 ratio, followed by the addition of 4Å molecular sieves as water removing agent. Progress of the reaction was monitored by 1H NMR spectroscopy and MALDI‐TOF MS. In contrast to A/CMe/D, no formation of mixed cages was observed but rather narcissistic self‐sorting into binary assemblies (Figure 5). MALDI‐TOF MS of the reaction mixture confirms the concurrent formation of the cubic cage A8D12 [18] (m/z=5953.83 for [A8D12+Na]+), macrocycle A2CPhCOOH 2 (m/z=1882.95 for [A2CPhCOOH 2+Na]+) and closed‐shell cage A2CPhCOOH 3 (m/z=2382.35 for [A2CPhCOOH 3+Na]+) with no evidence for any mixed assemblies. Apparently, molecular recognition of the COOH groups via the pronounced stabilization of CPhCOOH‐containing assemblies by intramolecular hydrogen bonding enforces narcissistic self‐sorting of the linkers CPhCOOH and D into separated hydrogen‐bonded structures and cubic cages, respectively. Formation of A8D12 was also observed in the 1H NMR spectrum of the reaction mixture (Figure S19). Alongside, linker CPhCOOH was distributed between the closed A2CPhCOOH 3 cage and open oligomers such as A2CPhCOOH 2 in a similar manner as for the A/CPhCOOH mixture, thus indicating that the self‐sorted system is under thermodynamic equilibrium.
Figure 5.
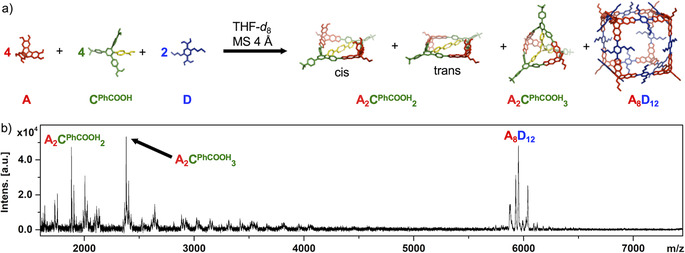
a) Narcissistic self‐sorting for A/CPhCOOH/D reaction mixture; b) MALDI‐TOF MS for reaction of A, CPhCOOH and D in 4:4:2 ratio in THF at room temperature and in the presence of 4Å molecular sieves.
Based on both experimental and computational results, we rationalize the observed formation of the strained A2CPhCOOH 3 cage as follows. For reactions in THF, we assume reversible boronate ester formation, at least for the initial steps. For CPhCOOH, smaller assemblies, e.g., [2+3] cage, [2+2] macrocycles or [1+2] open fragments, are significantly stabilized compared to larger [4+6] cages due to intramolecular hydrogen bonding. However, the subtle balance between hydrogen bonding and inherent strain energy results in a rather complex equilibrium mixture and no clear preference for one specific product. From reactions in MeCN, A2CPhCOOH 3 is isolated in pure form as the only kinetic product due to significant lower solubility of the closed cage compared to other fragments with unreacted polar catechol or boronic acid groups. Just recently, a detailed mechanistic study on [2+3] imine cages [37] revealed the influence of different solvents on the exchange dynamics between isostructural cages indicating that kinetic trapping of the final assemblies might play a much bigger role as initially assumed in dynamic covalent cage formation.
Conclusions
A highly strained [2+3] trigonal‐bipyramidal boronate ester cage has been synthesized by reacting hexahydroxy TBTQ A with 120° diboronic acid CPhCOOH in 2:3 molar ratio in MeCN. As we have shown in previous work, the bite angle of 120° for the ditopic linker C would usually trigger the assembly of tetrahedral [4+6] cages. For CPhCOOH however, pre‐organization in dimers or trimers via intermolecular hydrogen‐bonding between the COOH groups induces the formation of strained boronate ester bonds in smaller macrocycles and cages. MALDI‐TOF MS and 1H, 13C and DOSY NMR confirmed the formation of a highly symmetrical closed‐shell assembly with the hydrodynamic radius being in good agreement with a PM6‐minimized model for A2CPhCOOH 3. DFT calculations with a MeCN continuum model revealed that there is indeed a stabilizing contribution of −106 kJ mol−1 for the circular threefold hydrogen bonding motif in the cavity of A2CPhCOOH 3, which overcompensates the implemented strain energy of +62 kJ mol−1 when formally transforming one [4+6] into two [2+3] cages. This model system illustrates the limitations of the directional bonding approach for the prediction of cage geometry and topology through geometrical considerations for the molecular precursors. Supramolecular interactions between attached functional units and with solvent molecules can strongly influence the stability and formation pathways for complex dynamic covalent assemblies, thus giving access to structures that are normally not preferred. MD simulations on small oligomeric intermediates showed that there is also a kinetic contribution to the observed selectivity, since intramolecular hydrogen bonding in ACPhCOOH 2 already facilitates the formation of strained [2+2] macrocycles via fixation of preferential conformers. The pronounced tendency of A2CPhCOOH 2 and A2CPhCOOH 3 to form adducts with monocations in MALDI‐TOF MS experiments showcases the potential of these cages as ionic receptors. In future work, the implementation of switchable recognition sites within the cage pores could be utilized for a stimuli‐responsive assembly of cages with different shapes and geometries.
Experimental Section
Chemicals and solvents were purchased from commercial suppliers Alfa Aesar, Merck, Acros, Abcr, Fischer and Sigma–Aldrich and were used without further purification. Solvents were distilled prior to use. CH2Cl2 and DMF were dried with the solvent purification system “PureSolv MD 7” from Inert Technology. TLC sheets ALUGRAM Xtra SIL G/UV254 were purchased from Macherey–Nagel. Column chromatography was carried out with individually packed glass‐columns of different sizes (silica, grain‐size 40–63 μm, Macherey–Nagel). NMR spectra were recorded on a Bruker Avance III 400 or 600 spectrometer. Chemical shifts are indicated in ppm using the residual protonated solvent signal as internal standard (1H NMR: 7.26 ppm for CDCl3 and 1.72 ppm for [D8]THF; 13C NMR: 77.16 ppm for CDCl3 and 67.21 ppm for [D8]THF). Signal multiplicities are denoted as s (singlet), d (doublet), t (triplet), m (multiplet) and br (broad). Processing of the raw data was performed with the program Topspin 3.0. [36] MALDI‐TOF mass spectra were recorded on a ultrafleXtreme Bruker Daltonics (matrix: trans‐2‐(3‐(4‐tertButylphenyl)‐2‐methyl‐2‐propenylidene)‐malononitrile, DCTB). ESI mass spectra were recorded on a micrOTOF focus spectrometer from Bruker Daltonics GmbH. Infrared spectra were taken on a Jasco FT/IR‐430. Elemental analyses were performed on an Elementar CHNS 932 analyzer.
All geometry‐optimizations for the cages, macrocycles and intermediates were performed with density functional theory (DFT), using the ωB97XD [31] functional and the def2‐SVP [32] basis set. Additionally, a self‐consistent reaction field method using the integral equation formalism model (IEFPCM) was used to simulate the solvent MeCN [33] as implemented in the Gaussian 16 [34] quantum chemical software package. Relaxed scans and molecular dynamics simulations were performed in the framework of the semi‐empirical PM6 [30] method along with the D3H4 [35] correction for an adequate description of hydrogen bonds and dispersion by using the MOPAC2016 [36] program suite. In order to decrease the computational demand, the tBuPh substituents in C were replaced by H atoms and the nBu chains in TBTQ A were replaced by Me groups in all DFT and molecular dynamics calculations.
Compounds 3, [27] 4, [29] 5 [28] and A [18] have been synthesized according to previously published procedures.
Bispinacol ester (6): Under nitrogen atmosphere, B2pin2 (1.19 g, 4.68 mmol, 2.5 equiv), Pd(dppf)Cl2 (205 mg, 281 μmol, 0.15 equiv), KOAc (1.10 g, 11.2 mmol, 6.0 equiv) and diiodide 5 (1.40 g, 1.87 mmol, 1.0 equiv) were dissolved in dry DMF (120 mL) and stirred at 90 °C for two hours and 30 minutes. Afterwards, DMF was removed under reduced pressure. The remaining solid was suspended in water (170 mL), treated in an ultrasonic bath for 40 minutes, filtrated and washed with water (150 mL) to remove the remaining DMF. Then, the solid was dissolved in EtOAc and filtrated over celite to remove the catalyst. The solvent was again removed under reduced pressure before the remaining solid was dissolved in a minimum amount of CHCl3 and impurities were precipitated by addition of n‐hexane. The precipitate was filtered over a membrane filter and the solvent of the mother liquor was removed under reduced pressure. The remaining solid was dissolved in a minimum amount of EtOAc and the product was precipitated with n‐hexane, filtrated, and carefully washed by dropwise addition of cold EtOAc (3 mL) to obtain bispinacol ester 6 as a beige solid (1.00 g, 1.34 mmol, 71 %). m.p. 193.5 °C; 1H NMR (400 MHz, CDCl3, rt): δ = 7.67 (m, 4 H, MeOOCPh‐H/H 4’/6’), 7.62 (m, 6 H, tBuPh‐H/H 3/5/3’’/5’’), 7.49 (d, 3 J HH=8.6 Hz, 2 H, tBuPh‐H), 7.11 (d, 3 J HH=8.2 Hz, 4 H, H 2/6/2’’/6’’), 6.95 (d, 3 J HH=8.6 Hz, 2 H, MeOOCPh‐H), 3.87 (s, 3 H, COOCH 3), 1.37 (s, 9 H, C(CH 3)3), 1.34 (s, 24 H, BOC(CH 3)2) ppm; 13C NMR (101 MHz, CDCl3, rt): δ=167.23, 150.91, 144.54, 144.51, 142.30, 140.73, 137.43, 136.74, 134.35, 131.82, 129.40, 128.85, 128.56, 127.78, 126.98, 126.01, 83.91, 52.07, 34.73, 31.49, 25.02 ppm; FTIR (ATR): =660 (s), 829 (m), 1087 (m), 1143 (m), 1276 (m), 1359 (s), 1608 (m), 1719 (m), 2977 (w) cm−1; MS (MALDI‐TOF, DCTB in CHCl3, pos): m/z=748.41 [M]+, 771.40 [M+Na]+; elemental analysis calcd (%) for C48H54B2O6: C 77.02, H 7.27; found: C 76.52, H 7.43.
Diboronic acid CPhCOOH: Under nitrogen atmosphere, bispinacol ester 6 (800 mg, 1.07 mmol, 1.0 equiv) was dissolved in dry CH2Cl2 (88 mL) and cooled to 0 °C. BBr3 (910 μL, 2.41 g, 9.62 mmol, 9.0 equiv, d=2.65 g mL−1) was added dropwise and the solution was stirred for one hour at 0 °C, then two hours and 45 minutes at room temperature. Water (100 mL) was added to quench the reaction and the mixture was stirred overnight at room temperature. The mixture was extracted with EtOAc (3×50 mL) and the combined organic phases were washed with water (100 mL) and brine and dried over Na2SO4. The solvent was removed under reduced pressure and the remaining solid was suspended in CH2Cl2 (150 mL) and treated in an ultrasonic bath, before it was filtrated and washed with CH2Cl2 (40 mL) and n‐hexane (40 mL) to obtain CPhCOOH as a beige solid (465 mg, 815 μmol, 76 %). m.p. 222.1 °C (decomposition); 1H NMR (400 MHz, [D8]THF, rt): δ=11.19 (b, 1 H, COOH), 7.68 (s, 2 H, H 4’/6’), 7.66 (d, 3 J HH=8.6 Hz, 2 H, HOOCPh‐H), 7.62 (d, 3 J HH=8.6 Hz, 2 H, tBuPh‐H), 7.58 (d, 3 J HH=8.2 Hz, 4 H, H 3/5/3’’/5’’), 7.49 (d, 3 J HH=8.6 Hz, 2 H, tBuPh‐H), 7.08 (m, 8 H, H 2/6/2’’/6’’ /B(OH)2), 6.95 (d, 3 J HH=8.6 Hz, 2 H, HOOCPh‐H), 1.35 (s, 9 H, C(CH 3)3); 13C NMR (101 MHz, [D8]THF, rt): δ=167.48, 151.24, 145.56, 144.22, 143.50, 141.48, 138.50, 137.81, 134.28, 132.61, 129.77, 129.44, 129.25, 128.89, 127.59, 126.51, 35.18, 31.68 ppm; FTIR (ATR): =555 (m), 657 (m), 704 (m), 826 (m), 1017 (m), 1335 (s), 1607 (m), 1694 (m), 2960 (w), 3393 (br) cm−1; MS (ESI‐TOF, MeOH/MeCN, neg): m/z=569.22 [M−H]−; elemental analysis calcd (%) for C35H32B2O6⋅0.5 H3CCOOCH2CH3: C 72.34, H 5.91; found: C 72.16, H 5.93.
Cage A2CPhCOOH 3: TBTQ A (20.0 mg, 46.3 μmol, 1.0 equiv) and diboronic acid CPhCOOH (39.6 mg, 69.4 μmol, 1.5 equiv) were dissolved in THF (0.5 mL). MeCN (5 mL) was added and a violet precipitate formed. The suspension was stirred for 24 hours at 90 °C. After cooling down to room temperature, the solid was filtrated and washed with MeCN (5 mL) and n‐hexane (10 mL). Then, the raw product was stirred in dry acetone (9 mL) for 4 hours at 60 °C and the remaining solid was filtrated to obtain cage A2CPhCOOH 3 as a light pink solid (34.8 mg, 14.7 μmol, 64 %). m.p. >230 °C (decomposition); 1H NMR (400 MHz, CDCl3, rt): δ=10.92 (s br, 3 H, COOH), 7.80 (s, 6 H, H 4’/6’), 7.71 (d, 3 J HH=8.5 Hz, 6 H, tBuPh‐H), 7.65 (d, 3 J HH=8.3 Hz, 12 H, H 3/5/3’’/5’’), 7.53 (d, 3 J HH=8.5 Hz, 6 H, tBuPh‐H), 7.20 (m, 18 H, HOOCPh‐H/TBTQ‐Ar‐H), 7.13 (d, 3 J HH=8.3 Hz, 12 H, H 2/6/2’’/6’’), 6.53 (d, 3 J HH=8.5 Hz, 6 H, HOOCPh‐H), 4.56 (s, 6 H, TBTQ‐CH), 2.01 (m, 4 H, CH 2CH2CH2CH3), 1.38 (m, 35 H, C(CH 3)3/CH 2CH 2CH3), 0.90 (t, 3 J HH=7.1 Hz, 6 H, CH2CH 3) ppm; 13C NMR (151 MHz, CDCl3, rt): δ=169.30, 151.01, 148.42, 145.16, 145.07, 142.25, 140.26, 140.02, 139.73, 137.56, 134.04, 130.77, 129.92, 128.22, 127.09, 126.10, 125.71, 125.53, 124.20, 107.87, 67.21, 60.51, 40.06, 34.78, 31.52, 26.55, 23.55, 14.27 ppm; FTIR (ATR): =553 (s), 662 (s), 831 (m), 1017 (m), 1067 (m), 1328 (s), 1607 (m), 1689 (m), 2964 (w), 3384 (br) cm−1; MS (MALDI‐TOF, DCTB in CHCl3, pos): m/z=2323.29426 [M‐C4H9+Na]+, 2340.28800 [M‐C4H9+K]+, 2357.31661 [M]+, 2381.47 [M+Na]+; elemental analysis calcd (%) for C157H120B6O18⋅8 H2O: C 75.32, H 5.48; found: C 75.24, H 5.45.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support from the DFG (BE 4808/2‐1) is gratefully acknowledged. M.I.S.R. and M.B. wish to thank the Klaus Tschira Stiftung gGmbH (GSO/KT 21) for funding. Open access funding enabled and organized by Projekt DEAL.
N. Schäfer, M. Bühler, L. Heyer, M. I. S. Röhr, F. Beuerle, Chem. Eur. J. 2021, 27, 6077.
In memory of Carsten Schmuck
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.13353257.v1).
References
- 1. Campbell V. E., Nitschke J. R., Synlett 2008, 3077–3090. [Google Scholar]
- 2.
- 2a. Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15599–15603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15828–15832; [Google Scholar]
- 2b. Luo J., Wang J.-W., Zhang J.-H., Lai S., Zhong D.-C., CrystEngComm 2018, 20, 5884–5898. [Google Scholar]
- 3.
- 3a. Smulders M. M. J., Riddell I. A., Browne C., Nitschke J. R., Chem. Soc. Rev. 2013, 42, 1728–1754; [DOI] [PubMed] [Google Scholar]
- 3b. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 3c. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Jin Y., Yu C., Denman R. J., Zhang W., Chem. Soc. Rev. 2013, 42, 6634–6654; [DOI] [PubMed] [Google Scholar]
- 4b. Beuerle F., Gole B., Angew. Chem. Int. Ed. 2018, 57, 4850–4878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4942–4972. [Google Scholar]
- 5. Stang P. J., Olenyuk B., Acc. Chem. Res. 1997, 30, 502–518. [Google Scholar]
- 6. Lehn J. M., Chem. Eur. J. 2000, 6, 2097–2102. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947; [DOI] [PubMed] [Google Scholar]
- 7b. Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053; [Google Scholar]
- 7c. Mastalerz M., Acc. Chem. Res. 2018, 51, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Liu X., Liu Y., Li G., Warmuth R., Angew. Chem. Int. Ed. 2006, 45, 901–904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 915–918; [Google Scholar]
- 8b. Liu X., Warmuth R., J. Am. Chem. Soc. 2006, 128, 14120–14127; [DOI] [PubMed] [Google Scholar]
- 8c. Liu Y., Liu X., Warmuth R., Chem. Eur. J. 2007, 13, 8953–8959; [DOI] [PubMed] [Google Scholar]
- 8d. Xu D., Warmuth R., J. Am. Chem. Soc. 2008, 130, 7520–7521; [DOI] [PubMed] [Google Scholar]
- 8e. Mastalerz M., Chem. Commun. 2008, 4756–4758; [DOI] [PubMed] [Google Scholar]
- 8f. Tozawa T., Jones J. T. A., Swamy S. I., Jiang S., Adams D. J., Shakespeare S., Clowes R., Bradshaw D., Hasell T., Chong S. Y., Tang C., Thompson S., Parker J., Trewin A., Bacsa J., Slawin A. M. Z., Steiner A., Cooper A. I., Nat. Mater. 2009, 8, 973–978; [DOI] [PubMed] [Google Scholar]
- 8g. Sun J., Warmuth R., Chem. Commun. 2011, 47, 9351–9353; [DOI] [PubMed] [Google Scholar]
- 8h. Schneider M. W., Oppel I. M., Mastalerz M., Chem. Eur. J. 2012, 18, 4156–4160; [DOI] [PubMed] [Google Scholar]
- 8i. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 1516–1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1542–1546; [Google Scholar]
- 8j. Elbert S. M., Rominger F., Mastalerz M., Chem. Eur. J. 2014, 20, 16707–16720; [DOI] [PubMed] [Google Scholar]
- 8k. Slater A. G., Little M. A., Pulido A., Chong S. Y., Holden D., Chen L., Morgan C., Wu X., Cheng G., Clowes R., Briggs M. E., Hasell T., Jelfs K. E., Day G. M., Cooper A. I., Nat. Chem. 2017, 9, 17–25; [DOI] [PubMed] [Google Scholar]
- 8l. Lauer J. C., Zhang W. S., Rominger F., Schroder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 5126–5130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5226–5230; [Google Scholar]
- 9b. Zhang L., Jin Y., Tao G. H., Gong Y., Hu Y., He L., Zhang W., Angew. Chem. Int. Ed. 2020, 59, 20846–20851; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21032–21037; [Google Scholar]
- 9c. Sun Z., Li P., Xu S., Li Z.-Y., Nomura Y., Li Z., Liu X., Zhang S., J. Am. Chem. Soc. 2020, 142, 10833–10840. [DOI] [PubMed] [Google Scholar]
- 10. Slater A. G., Cooper A. I., Science 2015, 348, aaa8075. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Ferrand Y., Crump M. P., Davis A. P., Science 2007, 318, 619–622; [DOI] [PubMed] [Google Scholar]
- 11b. Mitra T., Jelfs K. E., Schmidtmann M., Ahmed A., Chong S. Y., Adams D. J., Cooper A. I., Nat. Chem. 2013, 5, 276–281. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Adv. Mater. 2012, 24, 6049–6052; [DOI] [PubMed] [Google Scholar]
- 12b. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Chem. Commun. 2013, 49, 8398–8400. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Mastalerz M., Chem. Eur. J. 2012, 18, 10082–10091; [DOI] [PubMed] [Google Scholar]
- 13b. Kunde T., Nieland E., Schröder H. V., Schalley C. A., Schmidt B. M., Chem. Commun. 2020, 56, 4761–4764. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Elbert S. M., Regenauer N. I., Schindler D., Zhang W.-S., Rominger F., Schröder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 11438–11443; [DOI] [PubMed] [Google Scholar]
- 14b. Liu M., Zhang L., Little M. A., Kapil V., Ceriotti M., Yang S., Ding L., Holden D. L., Balderas-Xicohténcatl R., He D., Clowes R., Chong S. Y., Schütz G., Chen L., Hirscher M., Cooper A. I., Science 2019, 366, 613–620. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Beuerle F., Klotzbach S., Dhara A., Synlett 2016, 27, 1133–1138; [Google Scholar]
- 15b. Klotzbach S., Beuerle F., Angew. Chem. Int. Ed. 2015, 54, 10356–10360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10497–10502; [Google Scholar]
- 15c. Acharyya K., Mukherjee S., Mukherjee P. S., J. Am. Chem. Soc. 2013, 135, 554–557; [DOI] [PubMed] [Google Scholar]
- 15d. Acharyya K., Mukherjee P. S., Chem. Eur. J. 2014, 20, 1646–1657; [DOI] [PubMed] [Google Scholar]
- 15e. Acharyya K., Mukherjee P. S., Chem. Commun. 2015, 51, 4241–4244; [DOI] [PubMed] [Google Scholar]
- 15f. Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2017, 56, 1244–1248; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1264–1268; [Google Scholar]
- 15g. Wagner P., Rominger F., Zhang W.-S., Gross J. H., Elbert S. M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2021, 10.1002/anie.202016592; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 10.1002/ange.202016592; [DOI] [Google Scholar]
- 15h. Greenaway R. L., Santolini V., Pulido A., Little M. A., Alston B. M., Briggs M. E., Day G. M., Cooper A. I., Jelfs K. E., Angew. Chem. Int. Ed. 2019, 58, 16275–16162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16421–16427; [Google Scholar]
- 15i. Abet V., Szczypiński F. T., Little M. A., Santolini V., Jones C. D., Evans R., Wilson C., Wu X., Thorne M. F., Bennison M. J., Cui P., Cooper A. I., Jelfs K. E., Slater A. G., Angew. Chem. Int. Ed. 2020, 59, 16755–16763; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 20447; [Google Scholar]
- 15j. Kołodziejski M., Stefankiewicz A. R., Lehn J.-M., Chem. Sci. 2019, 10, 1836–1843; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15k. Jiao T., Wu G., Chen L., Wang C.-Y., Li H., J. Org. Chem. 2018, 83, 12404–12410. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Zhang C., Wang Q., Long H., Zhang W., J. Am. Chem. Soc. 2011, 133, 20995–21001; [DOI] [PubMed] [Google Scholar]
- 16b. Yu C., Jin Y., Zhang W., Chem. Rec. 2015, 15, 97–106; [DOI] [PubMed] [Google Scholar]
- 16c. Ortiz M., Cho S., Niklas J., Kim S., Poluektov O. G., Zhang W., Rumbles G., Park J., J. Am. Chem. Soc. 2017, 139, 4286–4289; [DOI] [PubMed] [Google Scholar]
- 16d. Sun N., Wang C., Wang H., Yang L., Jin P., Zhang W., Jiang J., Angew. Chem. Int. Ed. 2019, 58, 18011–18016; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18179–18184. [Google Scholar]
- 17. Leonhardt V., Fimmel S., Krause A.-M., Beuerle F., Chem. Sci. 2020, 11, 8409–8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klotzbach S., Scherpf T., Beuerle F., Chem. Commun. 2014, 50, 12454–12457. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Bojdys M. J., Briggs M. E., Jones J. T. A., Adams D. J., Chong S. Y., Schmidtmann M., Cooper A. I., J. Am. Chem. Soc. 2011, 133, 16566–16571; [DOI] [PubMed] [Google Scholar]
- 19b. Schneider M. W., Oppel I. M., Ott H., Lechner L. G., Hauswald H.-J. S., Stoll R., Mastalerz M., Chem. Eur. J. 2012, 18, 836–847. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Jin Y., Voss B. A., McCaffrey R., Baggett C. T., Noble R. D., Zhang W., Chem. Sci. 2012, 3, 874–877; [Google Scholar]
- 20b. Kim Y., Koo J., Hwang I.-C., Mukhopadhyay R. D., Hong S., Yoo J., Dar A. A., Kim I., Moon D., Shin T. J., Ko Y. H., Kim K., J. Am. Chem. Soc. 2018, 140, 14547–14551; [DOI] [PubMed] [Google Scholar]
- 20c. Zhu Q., Wang X., Clowes R., Cui P., Chen L., Little M. A., Cooper A. I., J. Am. Chem. Soc. 2020, 142, 16842–16848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. McCaffrey R., Long H., Jin Y., Sanders A., Park W., Zhang W., J. Am. Chem. Soc. 2014, 136, 1782–1785; [DOI] [PubMed] [Google Scholar]
- 21b. Qiu L., McCaffrey R., Jin Y., Gong Y., Hu Y., Sun H., Park W., Zhang W., Chem. Sci. 2018, 9, 676–680; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Yang J., Chatelet B., Hérault D., Dutasta J.-P., Martinez A., Eur. J. Org. Chem. 2018, 5618–5628; [Google Scholar]
- 21d. Yang J., Chatelet B., Dufaud V., Hérault D., Michaud-Chevallier S., Robert V., Dutasta J.-P., Martinez A., Angew. Chem. Int. Ed. 2018, 57, 14212–14215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14408–14411. [Google Scholar]
- 22.
- 22a. Liu M., Little M. A., Jelfs K. E., Jones J. T. A., Schmidtmann M., Chong S. Y., Hasell T., Cooper A. I., J. Am. Chem. Soc. 2014, 136, 7583–7586; [DOI] [PubMed] [Google Scholar]
- 22b. Hu X.-Y., Zhang W.-S., Rominger F., Wacker I., Schröder R. R., Mastalerz M., Chem. Commun. 2017, 53, 8616–8619; [DOI] [PubMed] [Google Scholar]
- 22c. Bhat A. S., Elbert S. M., Zhang W.-S., Rominger F., Dieckmann M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 8819–8823; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8911–8915; [Google Scholar]
- 22d. Alexandre P.-E., Zhang W.-S., Rominger F., Elbert S. M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2020, 59, 19675–19679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 19843–19847. [Google Scholar]
- 23. Schneider M. W., Oppel I. M., Griffin A., Mastalerz M., Angew. Chem. Int. Ed. 2013, 52, 3611–3615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3699–3703. [Google Scholar]
- 24. Burgun A., Valente P., Evans J. D., Huang D. M., Sumby C. J., Doonan C. J., Chem. Commun. 2016, 52, 8850–8853. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Stefankiewicz A. R., Sambrook M. R., Sanders J. K. M., Chem. Sci. 2012, 3, 2326–2329; [Google Scholar]
- 25b. Sun L. Y., Sinha N., Yan T., Wang Y. S., Tan T. T. Y., Yu L., Han Y. F., Hahn F. E., Angew. Chem. Int. Ed. 2018, 57, 5161–5165; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5256–5261. [Google Scholar]
- 26.
- 26a. Kuck D., Chem. Rev. 2006, 106, 4885–4925; [DOI] [PubMed] [Google Scholar]
- 26b. Kuck D., Chem. Rec. 2015, 15, 1075–1109; [DOI] [PubMed] [Google Scholar]
- 26c. Dhara A., Beuerle F., Synthesis 2018, 50, 2867–2877. [Google Scholar]
- 27. Klyatskaya S., Dingenouts N., Rosenauer C., Müller B., Höger S., J. Am. Chem. Soc. 2006, 128, 3150–3151. [DOI] [PubMed] [Google Scholar]
- 28. Vliegenthart A. B., Welling F. A. L., Roemelt M., Klein Gebbink R. J. M., Otte M., Org. Lett. 2015, 17, 4172–4175. [DOI] [PubMed] [Google Scholar]
- 29. Breslow R., Marks P., Mahendran A., Yao Y. (Columbia University), US20180118709, 2018.
- 30. Stewart J. J. P., J. Mol. Model. 2007, 13, 1173–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chai J.-D., Head-Gordon M., Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- 32. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 33. Tomasi J., Mennucci B., Cammi R., Chem. Rev. 2005, 105, 2999–3093. [DOI] [PubMed] [Google Scholar]
- 34.Gaussian 16, revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford, CT (USA), 2016.
- 35.
- 35a. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 35b. Řezáč J., Hobza P., J. Chem. Theory Comput. 2012, 8, 141–151. [DOI] [PubMed] [Google Scholar]
- 36.J. J. P. Stewart, MOPAC2016, Stewart Computational Chemistry, Colorado Springs, CO (USA), http://OpenMOPAC.net, 2016.
- 37. Schick T. H. G., Rominger F., Mastalerz M., J. Org. Chem. 2020, 85, 13757–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.TopSpin 3.0, Bruker, www.bruker.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary