

Abstract
Small extracellular vesicles (sEVs) derived from antigen-presenting cells such as macrophages can induce therapeutically relevant immune responses. Anti-inflammatory miRNAs are elevated in sEVs secreted by RAW 264.7 mouse macrophages after lipopolysaccharide (LPS) stimulation. We observed uptake of these sEVs by primary mouse cortical neurons, microglia and astrocytes followed by downregulation of proinflammatory miRNA target genes in recipient cells. Pre-treating primary microglia with these sEVs decreased pro-inflammatory gene expression. A single intrathecal injection of sEVs derived from LPS stimulated RAW 264.7 cells attenuated mechanical hyperalgesia in the complete Freund’s adjuvant (CFA) mouse model of inflammatory pain and formalin induced acute pain. Importantly, sEVs did not alter the normal pain threshold in control mice. RNA sequencing of dorsal horn of the spinal cord showed sEVs-induced modulation of immune regulatory pathways. Further, a single prophylactic intrathecal injection of sEVs two weeks prior, attenuated CFA-induced pain hypersensitivity and was ineffective in formalin model. This indicates that prophylactic sEVs administration can be beneficial in attenuating chronic pain without impacting responses to the protective physiological and acute inflammatory pain. Prophylactic administration of sEVs could form the basis for a safe and novel vaccine-like therapy for chronic pain or as an adjuvant, potentially reducing the dose of drugs needed for pain relief.
1. Introduction
Chronic pain is the most prevalent, disabling, and expensive public health condition in the United States (Gereau Iv et al., 2014). Though primarily driven by neuronal dysfunction, the development of chronic pain also involves activated microglia and astrocytes as well as peripheral immune cells. Gene expression changes in these cells and the dysregulation of pro- and anti-inflammatory mediators further exacerbates chronic pain (Baral et al., 2019; Ji et al., 2016). Since crosstalk amongst these cells is mediated through cytokines, chemokines, and other signaling molecules, understanding this system can help elucidate mechanisms of nociceptive signaling in chronic pain. Recent studies have shown that small extracellular vesicles (sEVs) contribute to signaling events underlying physiological and pathological processes (El Andaloussi et al., 2013). Vesicles secreted by cells are heterogenous and classified based on their biogenesis and size. Exosomes are nanosized sEVs originating from multivesicular bodies transporting miRNAs, mRNAs, lipids, and proteins through bodily fluids. However, since it is difficult to separate exosomes from microvesicles that are formed by an outward budding from the plasma membrane, the term sEVs is used to encompass all classes of secreted lipid membrane vesicles ranging from 30–150 nm in size (Théry et al., 2018).
The contents of sEVs change depending on cell types and stimuli thereby facilitating intercellular communication upon sEVs uptake by recipient cells. Immune cell-derived sEVs play an important role in innate and acquired immune responses and can confer immunoprotective or immunosuppressive effects on target cells. sEVs released by antigen-presenting cells (APC) such as dendritic cells, macrophages, and B cells possess immunomodulatory properties via their ability to present peptide/major histocompatibility complex (MHC) to immune cells including CD8+ and CD4+ T cells (Robbins and Morelli, 2014). This form of antigen presentation is thought to produce anti-inflammatory effects, as supported by studies showing that dendritic cell–derived sEVs suppress the onset of collagen-induced arthritis in mice and reduce the severity of established arthritis (Kim et al., 2005).
The pleotropic function of macrophages enables them to mediate both pro- and anti-nociceptive effects during nerve injury or inflammation (Raoof et al., 2018). We have previously shown that RAW 264.7 macrophages stimulated with lipopolysaccharide (LPS) secrete sEVs with elevated levels of miR-21–3p, miR-146a, and miR-146b miRNAs (McDonald et al., 2014). These miRNAs can prevent overactivation of the innate immune response and repress NF-κB1 and other mRNAs involved in Toll-like receptor (TLR) signaling resulting in the inhibition of proinflammatory cytokine transcription and translation, a necessary step in resolution of inflammation (Alam and O’Neill, 2011; Boldin and Baltimore, 2012; Taganov et al., 2006). miR-21 is also an NF-κB-induced suppressor of inflammation and functions as a molecular switch regulating the synthesis of the anti-inflammatory cytokines IL-10 and IL-4 (Alam and O’Neill, 2011; Boldin and Baltimore, 2012). In addition to miRNAs, LPS-stimulation altered the macrophage sEVs transcriptome. Pathway analysis of the altered RNAs in sEVs showed significant changes in in both adaptive and innate immune processes, including pathways related to NF-κB activation and TLR cascades (McDonald et al., 2014). A single injection of sEVs derived from RAW 264.7 mouse macrophage cells into hind paw attenuated thermal hyperalgesia but not mechanical hypersensitivity in complete Freund’s adjuvant (CFA) induced mouse model of inflammatory pain suggesting an immunoprotective role for sEVs derived from RAW 264.7 cells (McDonald et al., 2014). These data suggested that uptake of macrophage-derived sEVs can regulate molecular events in recipient cells, including the resolution of inflammation and pain.
To obtain insights into the molecular mechanisms underlying sEVs-mediated information transfer in neurons and glia, the role of sEVs-miRNAs in mediating chronic pain, and the application of sEVs as therapeutic agents, we tested the hypotheses that uptake of RAW 264.7 cells-derived sEVs can induce protective gene expression changes in vitro in primary neuronal and glial cells, and in vivo in mouse models of inflammatory pain.
2. Materials and Methods
2.1. RAW 264.7 mouse macrophage cell line culture
RAW 264.7 mouse macrophage cells (ATCC TIB-71) were cultured in complete DMEM (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS (Gibco), 100U/mL penicillin-streptomycin (Gibco) at 37°C with 5% CO2. At 70–80% confluence, cells were washed with PBS without ions (Gibco) and the medium was changed to DMEM medium supplemented with 10% exosome-deplete heat inactivated FBS, 100U/mL penicillin-streptomycin in the presence or absence of 1μg/ml LPS (Sigma-Aldrich) at 37°C with 5% CO2. Conditioned media was collected at 24 hours for sEVs isolation.
2.2. Isolation and purification of sEVs from RAW 264.7 cell conditioned media
sEVs were purified as previously described (McDonald et al., 2013; McDonald et al., 2014). Cells were grown in exosome-deplete media for 24 hours. Conditioned media was collected and centrifuged at 300 x g for 10 min at 4°C to pellet dead cells followed by centrifugation at 12,000 x g for 30 min at 4°C to remove debris and large vesicles. Supernatant was filtered through a 0.22 µm syringe filter and sEVs were pelleted by centrifugation at 110,000 x g for 70 min at 4°C and washed with PBS containing 1U/ml RNAsin Plus RNase inhibitor (Promega). sEVs were resuspended in PBS and subjected to a second spin at 110,000 x g for 70 min at 4°C. The resulting pellets were resuspended in PBS for subsequent experiments. Protein was determined by Bradford assay (Bio-Rad).
2.3. Nanoparticle tracking analysis (NTA)
The size distribution and particle number/concentration of purified sEVs from RAW 264.7 cells were measured by NTA using a NanoSight NS300 system with blue 488 nm laser (Malvern Instruments). NTA tracks the Brownian motion of individual particles, enabling particle size distribution using the Stokes-Einstein equation. sEVs were diluted in filtered PBS to obtain 20–60 vesicles per field of view for optimal tracking. The diluted sample was introduced into flow cell using a syringe pump with a constant flow rate and three videos of 30 s each were recorded. Shutter speed and gain were adjusted followed by manual focusing to enable the optimum visualization of a maximum number of vesicles. The samples were advanced between each recording to perform replicate measurements. Each video was analyzed using the NanoSight NTA 3.2 software to obtain the mean, mode, and median vesicle size together with an estimate of the concentration. All NTA measurements were performed with identical system settings for consistency.
2.4. Transmission electron microscopy (TEM)
Isolated sEVs were fixed by resuspending in 2% paraformaldehyde (PFA, Electron Microscopy Sciences) in 0.1 M phosphate buffer (PB). Carbon-coated Formvar grids (Electron Microscopy Sciences) were floated on a drop of 10 μl sEVs suspension on clean parafilm in a dry environment for 20 min. Grids were washed with drops of 100 μl PB thrice for 2 min. Samples were fixed on grids with a 50 μl of 1% glutaraldehyde (Sigma-Aldrich) for 5 min and washed with 8 drops of distilled water. Samples were contrasted with 100 μl of 1% uranyl acetate (UA, Electron Microscopy Sciences) and embedded in 50 μl of 0.2% UA in 0.16% methylcellulose (Sigma-Aldrich) on a parafilm-covered ice dish. Excessive fluid was blotted on Whatman NO.1 filter paper. Grids were observed under a FEI Tecnai 12 digital TEM (FEI Company) at 80 kV.
For immune-gold labeling TEM, after washing with 3 drops of PB, grids were transferred to 4 drops of 50 mM glycine (Sigma-Aldrich) for 3 min each. Non-specific sites were blocked by introducing 5% (w/v) bovine serum albumin (BSA, AMRESCO) in PB for 10 min. Grids were incubated with 10 μl of CD81 primary antibody (1:100, #sc-166029, Santa Cruz Biotechnology) at room temperature for 30 min, washed with 0.1% BSA/PB, incubated with 10 nm gold-conjugated secondary antibody (1:25, #25815, Electron Microscopy Sciences) at room temperature for 20 min, and washed with 8 drops of PB. Samples were contrasted, embedded, dried, and imaged as above.
2.5. Dose dependent uptake of sEVs by Neuro-2a cells
Neuro-2a cells (ATCC CCL-131) were plated at density of 3 x 105 in 6-well plate in total 3 mL complete DMEM media. The complete medium was replaced by DMEM exosome-depleted medium next day. For the dose response studies, 0, 0.5, 1, 2.5, and 5 µg sEVs (Exo(−) and Exo(+)) were added in each well for 24 h. Equal volume of PBS was used as control. After 24 h cells were washed thrice with PBS and cell pellets used for miRNA qPCR as described below.
2.6. Timed-pregnant mice
All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care & Use Committee of Drexel University College of Medicine. Mice were maintained in a 12-hour light/dark cycle and provided food and water ad libitum. For in vitro experiments using primary cells, timed-pregnant CD-1 mice were purchased from Charles River Laboratories. All dams were received approximately 14 days after impregnation. Our experiments utilized pups born the same day or postnatal day one (P0-P1) for primary neuronal cultures. P1-P3 mice were used to generate primary glial cultures.
2.7. Primary neuronal and glial culture
Primary cultures for neurons and glial cells were prepared from cortex of postnatal P0-P1 (for neurons) and P1-P3 (microglia or astrocytes) CD-1 mice. Mice were deeply anesthetized by inducing hypothermia on ice and decapitated. Cortices were dissected out aseptically under a dissecting microscope in cold Hank’s balance salt solution without Ca2+ and Mg2+ (HBSS−/−) (Invitrogen) containing 10mM HEPES buffer (Invitrogen). Tissues were minced and incubated for 20 min at 37°C in papain enzyme solution (7.5U/mL; Worthington Biochemical) in HBSS (3mL/brain). 1 mg/mL DNAse I (Sigma-Aldrich) was added to stop non-neuronal cells from dividing. Papain was neutralized by addition of an equal volume of complete plating medium consisting of either Neurobasal-A media (Gibco) supplemented with 2% fetal bovine serum (FBS) (Gibco), 2% horse serum, 2% B-27 supplement (Gibco), 0.5% gentamicin and 0.5mM GlutaMAX-I (Invitrogen) for neurons or Dulbecco’s modified eagle’s medium (DMEM) (Sigma-Aldrich) supplemented with 10% heat inactivated FBS, 100U/mL penicillin-streptomycin (Gibco) for glia. Cell suspensions were further dissociated mechanically by trituration through a glass 5mL pipette and subsequently through a flame polished Pasteur pipette. Dissociated cells were passed through a 40µm nylon mesh strainer and centrifuged at 800 RPM for neurons or 1200 RPM for glia at 4°C for 5 mins. Resultant cell pellets were re-suspended in complete plating medium on 0.05% poly-D-lysine (PDL, Sigma-Aldrich)/1ug/mL laminin (Sigma-Aldrich) coated coverslips or 12-well plates for neurons and uncoated flasks for glia. All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. For neuronal culture, after 4 hours of incubation, the medium was changed to complete maintenance media of Neurobasal-A containing 2% B27 supplement and 0.5mM GlutaMAX-I (Gibco). Harvested cells were stained against primary antibodies NeuN (Millipore, 1:500) neuronal nuclear marker or MAP2 (Millipore, 1:500) dendritic marker and 1ng/ml DAPI nuclear stain to determine purity. Glia were grown to confluency undisturbed for 8–10 days, then microglia were shaken off using a plate shaker at 200 rpm for 3 hours. Detached microglial cells are pelleted at 1200 RPM for 5 mins and seeded onto fresh 0.05% PDL coated plates. Purity of the culture was determined by staining for microglial marker CD11b-AF488 (Abcam, 1:250) and DAPI. Morphology was also used to determine cell purity and resting state of cell using phase/contrast microscopy. Astrocyte monolayer were enzymatically detached by incubation with 1X TryPLE express (Gibco). Detached cells were pelleted at 1200 RPM and sub-cultured. Media was changed every 2–3 days. After 8–10 days in culture, when astrocytes were confluent, they were enzymatically detached and reseeded into new 0.05% PDL coated plates for experimentation. Purity and maturity of astrocytes were assessed with antibodies against astrocyte marker GFAP-AF647 (BD pharma, 1:250) and DAPI to determine the proportion of GFAP-positive astrocytes. Neuronal and microglial experiments were conducted 3–4 days after plating in exosome-free media. To minimize residual microglial or oligodendrocyte precursor cell contamination of primary astrocyte cultures, experiments were performed soon after astrocyte purification at 1–2 days following second reseeding.
2.8. Fluorescent labeling of sEVs
For immunofluorescent studies, sEVs were labeled with general lipophilic fluorescent membrane dye, PKH26, using the PKH26 labeling kit according to manufactures instructions (Sigma-Aldrich). Briefly, purified aliquots of 20 μg sEVs were resuspended in 180 μl diluent C and mixed with 20 μl PKH26 dye diluted in diluent C for 5 min. Staining was stopped by washing labeled exosomes with 0.1% BSA in nuclease free water and pelleted by centrifugation at 110,000 x g for 70 min at 4°C. Labeled sEVs pellet was resuspended in 50 μl PBS and stored in −80°C until use.
2.9. sEVs uptake studies using primary CNS cells in vitro
To confirm the uptake of sEVs in vitro, primary CNS cells were incubated with sEVs for 24 h. The seeding densities for 6 well plate was 300,000 cells/well with 3 mL media and 3 µg sEVs/well and for 12 well plates, 100,000 cells/well in 1.5 ml media and 1.5 µg sEVs/well were used. For detection of labeled sEVs in recipient cells, the cells on coverslips were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.1% Triton X-100 for 10 min then blocked with appropriate blocking buffer for 1 hour at room temperature. Cells were incubated overnight at 4°C with primary antibodies against MAP2 (Millipore, 1:500) for neurons. Neurons were counterstained with goat-anti mouse IgG1 AF488 secondary antibody. For microglia and astrocytes, cells were incubated with CD11b-AF488 (Abcam, 1:250) for microglia or GFAP-AF647 (BD Pharma, 1:250) for astrocytes at room temperature for 2 hours. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Coverslips were mounted onto slides with ProLong Glass Antifade mountant (Invitrogen). Cells were visualized using 10x or 60x objective using a laser scanning confocal microscope (ZEISS LSM 700). Images were captured and processed by laser scanning microscope and FluoView Imaging software (Olympus FV1000).
2.10. sEVs uptake in vivo
To track internalization of RAW 264.7 macrophage-derived sEVs in vivo, C57BL/6 mice (Jackson Laboratories) were intrathecally injected with 10µL of 1µg PKH26-labeled sEVs in PBS. After 18 h, mice were sacrificed, transcardially perfused with 0.9% saline solution followed by 4% paraformaldehyde (PFA) 0.1 M phosphate buffer (PB). Lumbar spinal cords and DRGs were dissected, post-fixed for 24 h, and cryopreserved in 30% sucrose then embedded in OCT compound (Sakura). Embedded tissue was frozen and stored at −80°C. Spinal cords were sectioned at 30 µm using a microtome (Thermo Fisher Scientific) and DRG were sectioned using a cryostat (Leica). Sections were blocked with 5% NGS in 0.3% Triton/PB for 2 h at room temperature (RT). Sections were stained with MAP2A (1:200, Sigma-Aldrich, MAB378), GFAP (1:1000, Sigma-Aldrich, MAB360), Iba1 (1:2000, Wako Chemicals, 019–197410) overnight at 4°C, followed by Alexa Fluor conjugated secondary antibodies (Invitrogen) for 2 h at RT. Sections were washed and counter stained with DAPI and mounted onto glass slides. Sections were visualized with laser scanning confocal microscope (Olympus FV3000). Images were acquired using FluoView Imaging software and processed with ImagJ.
2.11. RNA isolation and qRT-PCR
Total RNA was isolated using the mirVana RNA isolation kit (Ambion) following manufactures instructions. RNA concentrations were determined using a NanoDrop ND1000 spectrophotometer (NanoDrop Technology Inc). The Maxima cDNA synthesis kit (Thermofisher Scientific) was used to generate cDNA and 2 µl cDNA was used for Taqman based quantitative real time mRNA analysis containing 10 µl Taqman Fast Universal PCR master mix (2×) no AmpErase UNG (Life Technologies), 1 µl Taqman primer probe (20×), in up to 20 µl nuclease-free water. Gapdh was used as the normalizer. Relative expression was calculated from raw cycle threshold (CT) values using the 2-ΔΔCT method (Schmittgen and Livak, 2008). The primer probes were the following: Irak1(assay ID: Mm01193538_m1), Traf6 (assay ID: Mm00493836_m1), Il-12a (assay ID: Mm00434169_m1), Pdcd4 (assay ID: Mm01266062_m1), Il-10 (assay ID: Mm01288386_m1), and Il-6 (assay ID: Mm01210733_m1), and Gapdh (assay ID: Mm99999915_g1) (Applied Biosystems).
For miRNAs, RNAs were reversely transcribed with the miRNA Reverse Transcription kit (Applied Biosystems). To analyze miRNAs following sEVs uptake by CNS cells, isolated RNAs were pre-amplified with the TaqMan miRNA assay (Applied Biosystems) and miRNAs High Capacity cDNA reverse transcription kit was used to generate cDNA for miRNAs (Applied Biosystems). The miRNAs assessed were: miR-146a (assay ID: mmu478399_mir), miR-146b (assay ID: mmu478513_mir), miR-21 (assay ID: 477975_mir) and U6 snRNA (assay ID: 001973). One microliter cDNA was used for Taqman based quantitative real time mRNA analysis containing 5 µl Taqman Universal PCR master mix (2×) no AmpErase UNG (Life Technologies), 0.5 µl TaqMan microRNA assay (Applied Biosystems), in up to 10 µl nuclease-free water. Relative expression was normalized to U6 snRNA.
2.12. Enzyme-linked immunosorbent assay (ELISA)
To determine circulating cytokine concentrations, recipient cell culture supernatants were analyzed for IL-6, IL-10, IL-12 and TNFα using ELISA kits according to manufacturer’s protocol (Bio-legend).
2.13. Western blot analysis for sEVs markers and CNS cells
CNS cells were washes in ice-cold PBS and lysed in radioimmunoprecipitation assay buffer (RIPA) containing Halt protease inhibitor cocktail (Thermo Scientific). The concentrations of total protein were determined using a DC Protein Assay kit (Bio-Rad) following manufacturer’s instructions. Following denaturation at 85°C for 3 min, protein lysates were loaded and separated on a 10% SDS-PAGE (Novex/Life Technologies). Following transfer, PVDF membranes were blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline and 0.1% Tween-20 (TBS-T) for 1 h at room temperature, then incubated with primary antibodies overnight at 4°C. Primary antibodies used were mouse TRAF6 (1:250, Santa Cruz), rabbit IRAK-1 (1:500, Cell Signaling Technology) or mouse GAPDH (1:1000, Santa Cruz). Blots were washed an incubated at room temperature with goat anti-mouse IgG-HRP anti-mouse IgG kappa BP-HRP (1:2500) or donkey anti-rabbit IgG-HRP conjugated secondary antibodies (1:5000, Abcam). Proteins were detected using western HRP substrate (Immobilon Forte, Thermo Scientific) and blots analyzed by enhanced chemiluminescence using the FluorChem M system (ProteinSimple).
For sEVs samples, the Exo(−) and Exo(+) samples and RAW 264.7 cells were processed similarly. The primary antibodies used were mouse anti-CD81 (1:1000, Santa Cruz), rabbit anti-Alix (1:1000, Abcam), rabbit anti-Calnexin (1:1000, Abcam) and rabbit anti-GAPDH (1:1000, Abcam). Blots were washed and incubated with goat anti-mouse IgG-HRP (1:10000) or donkey anti-rabbit IgG-HRP conjugated secondary antibodies (1:10000, Abcam).
2.14. Mice
Behavioral tests were performed using C57BL/6J mice (8–10 weeks old) obtained from Jackson Laboratories. Mice were housed in 12 h light/dark cycles. Behavioral assays were performed by researchers blinded to the treatment received.
2.15. Complete Freund’s Adjuvant (CFA) inflammatory pain model
Baseline measurements were obtained before CFA injection. Mice were injected subcutaneously with 20 µl of 50% emulsified Complete Freund’s Adjuvant (CFA; Difco Laboratories) in PBS into the plantar hind paw. Mechanical sensitivity was measured using a series of von Frey filaments (North Coast Medical). Static mechanical paw withdrawal thresholds of the ipsilateral paw were assessed by applying calibrated/graded von Frey monofilaments to the surface of the CFA-injected paw. Paw withdrawal threshold was determined and evaluated by increasing or decreasing the stimulus (fiber) using Dixons up-down method (Chaplan et al., 1994; Dixon, 1980). The pattern of responses was used to determine the animals paw withdrawal threshold. Thermal sensitivity was measured using the Hargreaves method. The baseline latencies were set to approximately 10 seconds with a maximum of 20 seconds as the cutoff to prevent potential injury. Animals were placed on a heated plexiglass platform (30°C). Thermal stimulation was applied to the paw using an infrared light source under the plexiglass platform. Paw withdrawal was measured by the von Frey and Hargreaves tests 1h, 3 h, 24 h 48 h, 7 days and 10 days.
2.16. Formalin model of acute inflammatory pain
Mice were injected with 15 µl of 5% formalin (Sigma Aldrich) subcutaneously into the plantar hind paw. Immediately following formalin injection, the spontaneous pain behaviors as time spent licking/flicking the injected paw was recorded in 5-minute intervals for 60 min.
2.17. Administration of sEVs
Ten µL sEVs solution or saline was administered under the dura mater at the lumbar enlargement using a Hamilton syringe and 30-gauge needle and a tail flick indicating successful puncture. For CFA model sEVs were injected immediately after or for prophylactic studies, 14 days before CFA administration. For the formalin model i.t. injection of sEVs was performed 3 hours or 14 days prior formalin injection.
2.18. RNA sequencing
Total RNA was isolated using the miRVana kit (Applied biosystems) from the dorsal horn of the lumbar spinal cord tissue of sEVs or PBS injected mice seven days following CFA or saline treatment. The RNA concentration and integrity were determined using the Agilent RNA 6000 nanochip on an Agilent Bioanalyzer 2100. RNA was reverse transcribed to double-strand cDNA. The resulting cDNA was end-repaired, 3’ adenylated, adaptor ligated and purified. Purified products were PCR amplified, denatured and cyclized. Samples sequenced as 50 bp single reads using BGISEQ-500 platform, generating on an average around 30 million reads per sample. Raw sequencing reads were run through SOAPnuke software to filter out the low-quality reads, reads with adaptors and unknown bases. Clean reads were assembled into Unigenes, followed with Unigene functional annotation and single sequence repeat (SSR) detection to calculate the Unigene expression levels and SNPs of each sample. Reads were mapped using unbiased HISAT (Hierarchical Indexing for Spliced Alignment of Transcripts). Low-expressed genes with transcriptper-million TPM ≤ 1 in all samples were filtered out. Differentially expressed genes (DEGs) between sample groups were identified from the normalized TPM values, using a two-tailed t-test p-value ≤ 0.01 and a fold change ≥ 2. Statistical enrichment of Gene Ontology (GO) terms and KEGG pathways was done using DAVID bioinformatics tool (Huang da et al., 2009)(p-value ≤ 0.05) and the enriched terms were summarized using REVIGO (Supek et al., 2011). Enrichment of miRNA targets was assessed by a hypergeometric test, against the set of computationally predicted target genes available from TargetScan (Lewis et al., 2005). Proteins interacting with Irak1, Traf6, and Pdcd4 were obtained from the APID database (Prieto and De Las Rivas, 2006).
2.19. Statistical analysis
For in vitro and in vivo experiments, statistical analyses were performed using Graph Pad Prism 7.0 (Graph Pad Software). For formalin testing the sessions were divided into two phases: 0–10 min and 10–60 min the average time spent licking/flicking the paw was recorded every 5 min for 60 min. All data are presented as averages ± SEM and were analyzed using Student’s t test (for experiments with two groups), one-way ANOVA followed by Tukey’s multiple comparisons test (for experiments with three or more groups) or repeated measures two-way ANOVA followed by Bonferroni’s post hoc test for behavioral data. Differences between averages were considered statistically significant when p<0.05.
2.20. Data availability
Gene-level expression values for all samples are provided as FPKM (fragments per kilobase of transcript per million mapped reads) and TPM (transcript per million) in the Supp Table 2. Raw RNA sequencing data will be made available in NCBI Sequence Retrieval System (SRS) upon publication.
3. Results
3.1. Isolation and characterization of RAW 264.7 cells-derived sEVs.
Isolation and characterization of sEVs derived from RAW 264.7 cells with and without LPS stimulation was performed as reported previously by ultracentrifugation. (McDonald et al., 2014). TEM images of sEVs showed the expected size range and integrity (Figure 1A). Immunogold labeling (Figure 1B) and western blot analysis (Figure 1C) of sEVs lysate from unstimulated (Exo−) or stimulated (Exo+) and RAW 264.7 cell lysate were used to probe for positive sEVs markers Alix and CD81. The absence of the negative marker Calnexin in sEVs and its presence in whole cell lysate was also confirmed by western blot. The absence of negative marker suggests purity of sEVs. NTA analysis of sEVs showed particles had a mean diameter of 113.6 ± 7.9 nm for Exo(−) and 143.3 ± 0.8 nm for Exo(+). The particle concentration were 8.6 x 108 ± 9.8 x 107 particles/ml for Exo(−) and 2.6 x 109 ± 3.2 x 107 particles/ml for Exo(+) (Figure 1D). On average we found 1 µg sEVs protein equals to 1 x 109 particles.
Figure 1. Characterization of sEVs derived from RAW 264.7 cells.
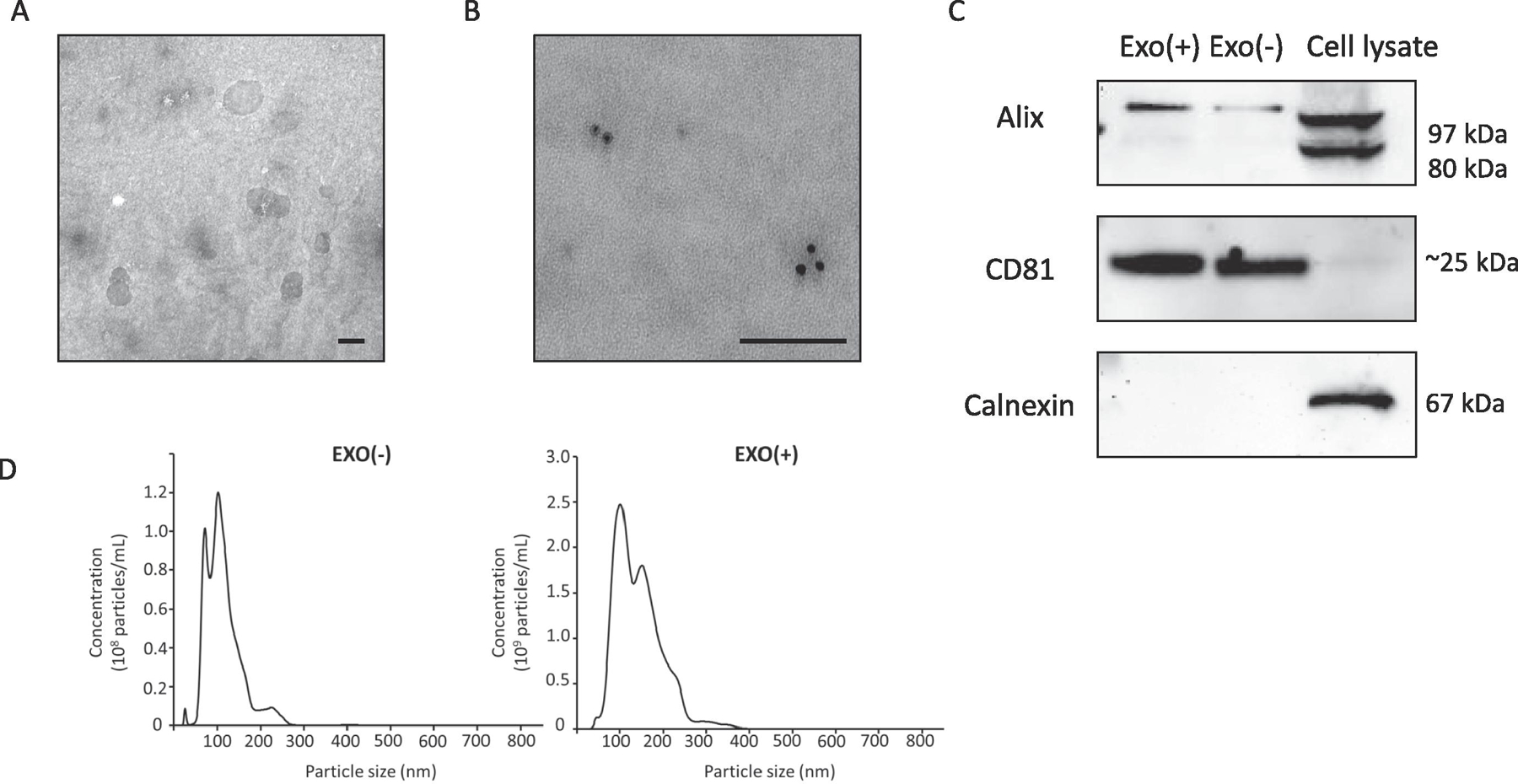
(A) Transmission electron microscopy (TEM) images of sEVs purified from culture media of RAW 264.7 cells showing size range and integrity (scale bar = 100 nm). (B) Immunogold labeling for CD81 (scale bar = 100 nm). (C) Western blot analysis of sEVs lysate from unstimulated (Exo−) or stimulated (Exo+) and RAW 264.7 cell lysate indicates that two commonly detected sEVs proteins, Alix and CD81 are present in sEVs. The negative marker Calnexin is absent in sEVs but present in whole cell lysate. The absence of negative marker suggests purity of our sEVs preparations. (D) Nanoparticle tracking analysis (NTA) of sEVs showed Exo(−) particles with a mean diameter of 113.6 ± 7.9 nm and a particle concentration of 8.6 x 108 ± 9.8 x 107 particles/ml and for Exo(+) the mean diameter was 143.3 ± 0.8 nm and a particle concentration 2.6 x 109 ± 3.2 x 107 particles/ml. On average we found 1 µg sEVs protein equals to 1 x 109 particles.
3.2. miRNA changes induced by dose response uptake of Exo(+) sEVs in recipient Neuro-2a cells.
To determine the relative expression of three miRNAs in recipient cells upon uptake of sEVs, a dose response study was performed by incubating Neuro-2a cells with PBS, 0.5, 1, 2.5 and 5 µg sEVs for 24 h. The qPCR analysis for the three miRNAs (miR-146a, miR-146b and miR-21) that were significantly upregulated in Exo(+) showed that the uptake of 1µg Exo(+) consistently resulted in maximum levels of all three miRNAs in the recipient cells (Figure 2). This suggests a potential for a saturation effect in recipient cells after the uptake of exogenous miRNAs. We used 1µg sEVs in all our studies.
Figure 2. miRNA changes induced by dose response uptake of Exo(+) sEVs in recipient Neuro-2a cells.
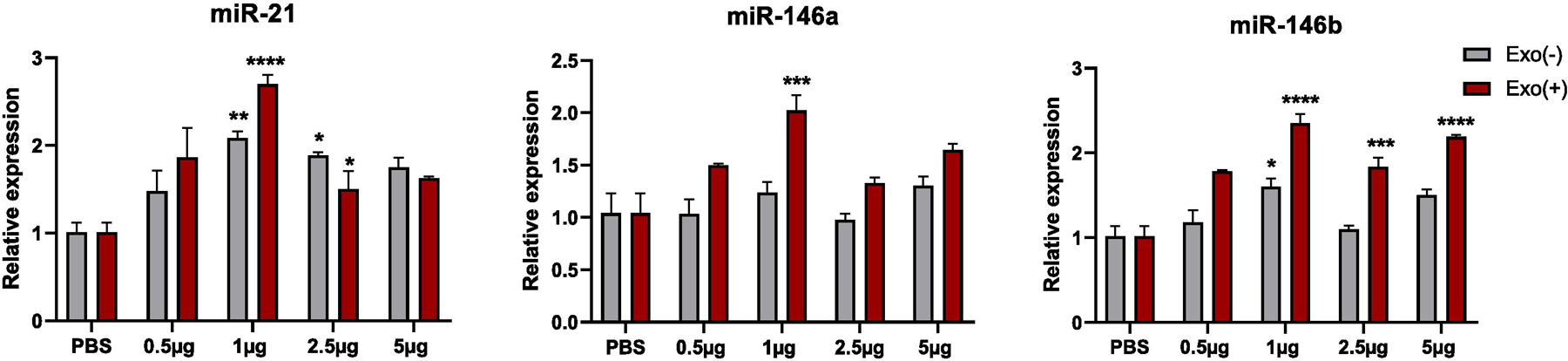
Neuro-2a cells were treated with Exo(−) or Exo(+) at a concentration of 0, 0.5, 1, 2.5 and 5 µg sEVs for 24 h. RNA from Neuro-2a cells were used for qPCR analysis to measure the relative levels of miR-21, miR-146a and miR-146b (n=3). All data shown as mean ± SEM and analyzed with two-way ANOVA followed by Sidak post test p < 0.05, **p <0.01, ***p <0.001, ****p <0.0001.
3.3. Primary CNS cells take up functional miRNAs from macrophage-derived sEVs.
We first determined whether mouse primary neurons and glia cultured in exosome-free media could take up RAW 264.7-derived sEVs that we isolated as previously described (McDonald et al., 2014). sEVs were labeled with PKH26, a lipophilic membrane dye, then incubated with primary cortical microglia, neurons, or astrocytes. Labeled sEVs were efficiently taken up by all three cell types (Figure 3A). We then investigated the cellular uptake of three miRNAs previously reported as upregulated in secreted sEVs after cellular LPS treatment (miR-146a, miR-146b, and miR-21–3p) because of their known anti-inflammatory roles (Boldin and Baltimore, 2012). Purified sEVs (1 µg/mL) from LPS stimulated or unstimulated RAW 264.7 cells (Exo(+) and Exo(−), respectively) were again incubated with primary microglia, neurons, or astrocytes for 24 h. Each cell type treated with Exo(+) sEVs had elevated levels of miR-146a, miR-146b, and miR-21, indicating the transfer of miRNAs from Exo(+) sEVs to target cells (Figure 3B).
Figure 3. Uptake of sEVs transferred miRNAs to primary microglia, neurons, and astrocytes.
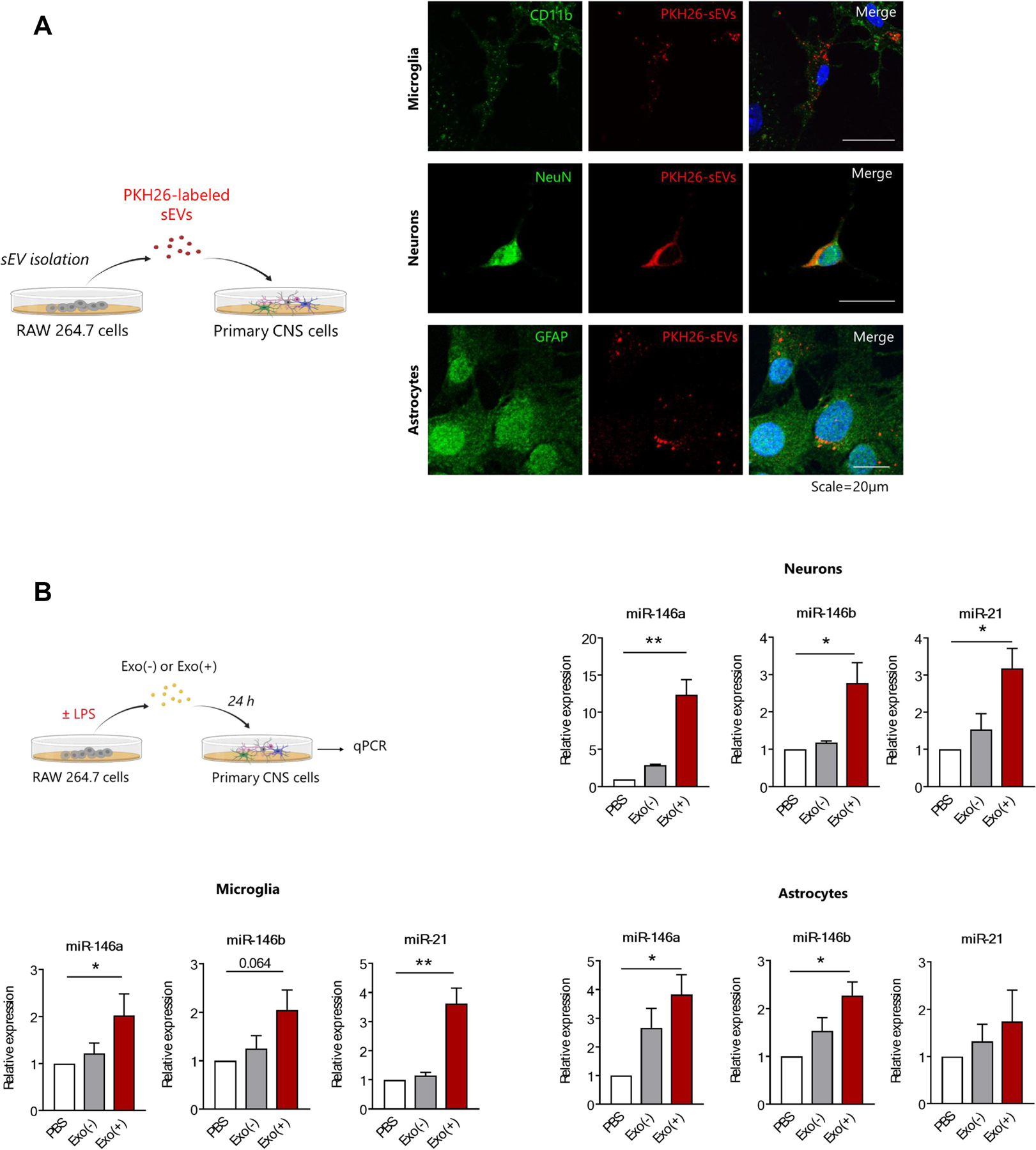
(A) Experimental design and representative images of immunostaining showing sEVs are internalized by primary microglia, neurons, and astrocytes. PKH26 labeled sEVs (red) are taken up by cells labeled for specific markers; primary microglia (anti-CD11b), neurons (anti-NeuN), and astrocytes (anti-GFAP). Scale bar = 20 µm. (B) Schematic of in vitro experiment and qPCR analysis of miRNAs in recipient cells incubated with sEVs. The sEVs isolated from RAW 264.7 cell conditioned media from naïve (Exo(−) or after 24 h stimulation with 1µg LPS (Exo(+) were incubated with primary cells for 24 hours. Exo(+) sEVs treatment increased relative levels of miR-146a, miR-146b and miR-21 in recipient cells. Expression of miR-146a, miR-146b and miR-21 were normalized to U6 snRNA (n=3). All data shown as mean ± SEM and analyzed with one-way ANOVA *p < 0.05, **p <0.01.
We then determined if the internalized miRNAs altered mRNA and protein expression of their known proinflammatory targets. miR-21 targets the proinflammatory cytokine interleukin-12a (IL12a) in microglia (Lu et al., 2011) and programmed cell death receptor 4 (Pdcd4) in neurons and astrocytes (Sheedy et al., 2010), while miR-146a and miR-146b, involved in NF-κB activation and release of proinflammatory cytokines, target IRAK-1 and TRAF6 in all three cell types (Boldin and Baltimore, 2012). All target mRNA levels were reduced by Exo(+) treatment regardless of cell type (Figure 4A). Additionally, Exo(+) treatment also reduced IRAK-1 and TRAF6 protein expression in all cell types (Figure 4B) and quantification shown in Supplementary Figure 1. Thus, sEV-derived miRNAs were functional, reduced target mRNA and protein levels in recipient cells, and the magnitude of these effects were dependent on cell type and endogenous expression levels of target mRNA.
Figure 4. sEV-derived miRNAs are functional and decrease their target mRNA and protein in in primary microglia, neurons and astrocytes.
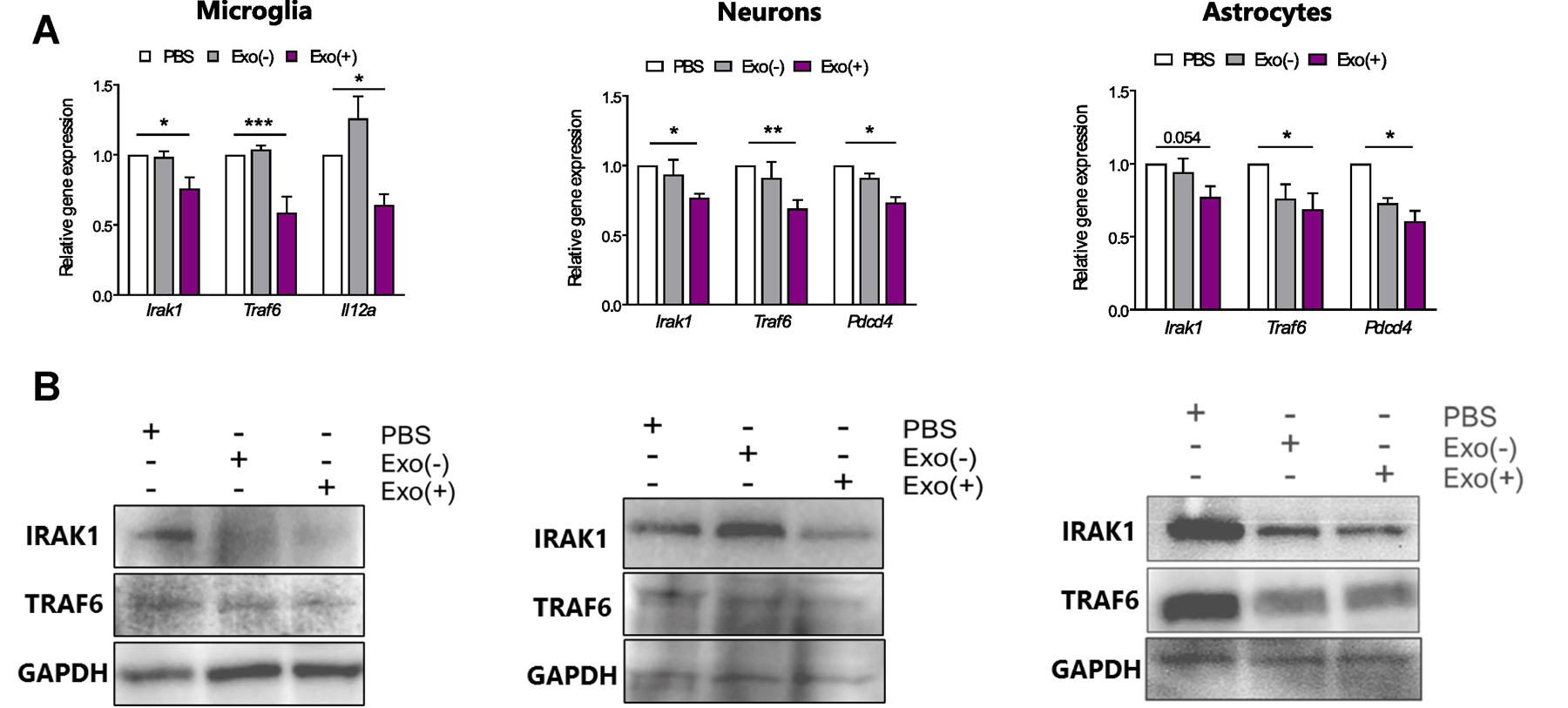
Exo(+) treatment reduced relative mRNA and protein levels of genes targeted by Exo(+) enriched miRNAs in all cell types. (A) qRT-PCR was used to measure relative mRNA levels of miR-146a, miR-146b, and miR-21 targets, Irak-1, Traf6, and Il12a or Pdcd4 respectively, in microglia, neurons and astrocytes following 24 h incubation with sEVs (n=3). Data shown are mean ± SEM. Statistical analysis was determined by one-way ANOVA *p < 0.05, **p < 0.01, ***p < 0.001. (B) Representative western blots of IRAK-1, TRAF6 and GAPDH in sEVs treated microglia, neurons and astrocytes.
We next investigated whether pre-treatment with sEVs could alter the inflammatory response of cells exposed to LPS. Microglia pre-treated with either Exo(+) and Exo(−) sEVs showed reduced proinflammatory Tnfα and Il-6 mRNA expression after LPS stimulation. However, only Exo(+) sEVs decreased secreted TNFα protein and increased anti-inflammatory cytokine IL-10 release from these cells (Figure 5), further supporting their enhanced anti-inflammatory activity. Exo(−) sEVs also contain these miRNAs, albeit less, and could contribute to reduction of Tnfα and Il-6 mRNA.
Figure 5. Pre-incubating microglia with RAW 264.7-derived sEVs reduced LPS-induced proinflammatory gene expression.
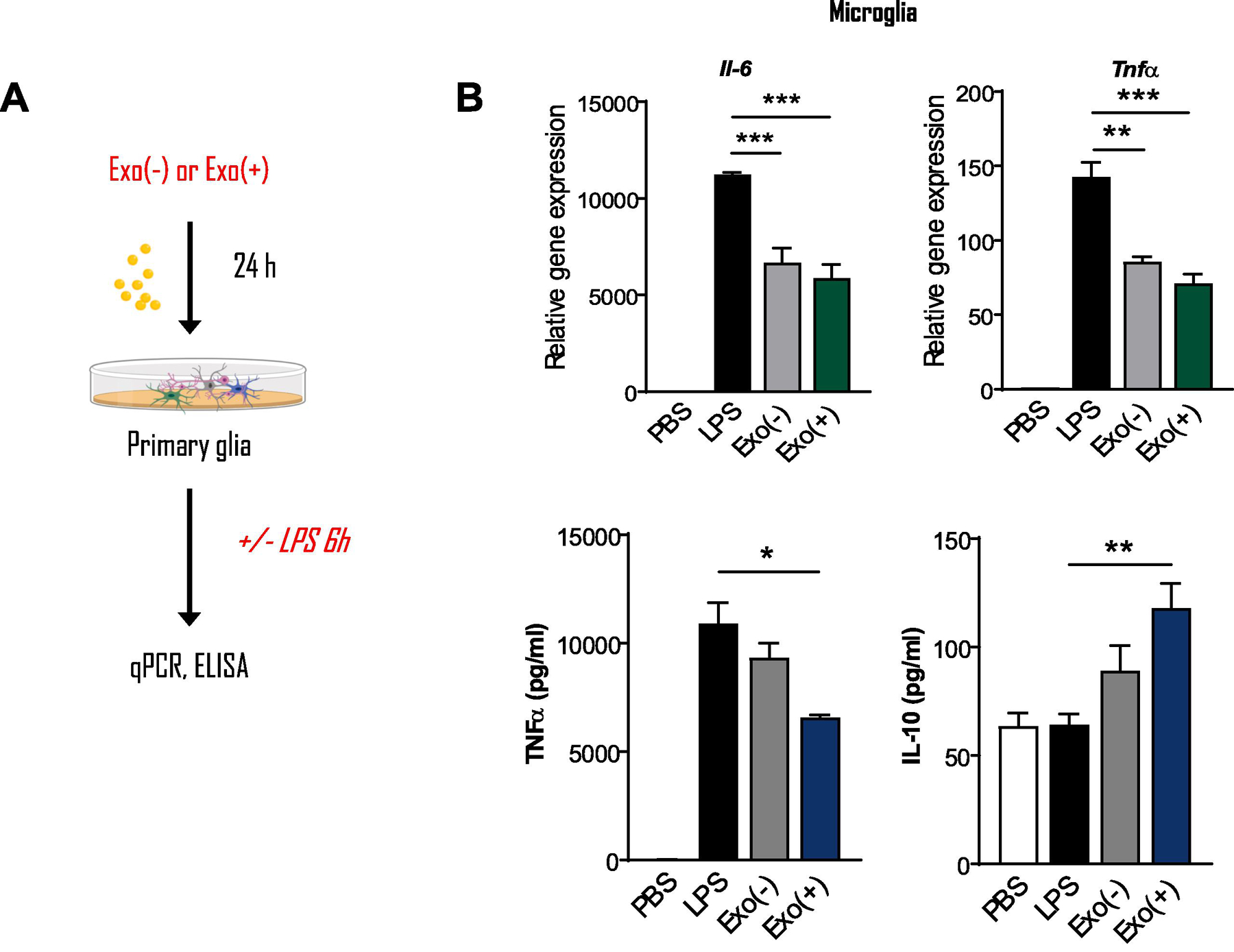
Exo(−) and Exo(+) treatment decreased mRNA levels of Tnf and Il6 in primary microglia stimulated with LPS as determined by qPCR. ELISA for secreted cytokines showed that Exo(+) treatment decreased TNF and increased anti-inflammatory IL-10 secretion from these cells, (n=3). All data shown as mean ± SEM and analyzed with one-way or two-way ANOVA followed by Dunnett’s post-test *p < 0.05, **p <0.01, ***p <0.001.
3.4. Exo(+) sEVs relieve mechanical allodynia and alter gene expression in CFA-injected mice
As our in vitro studies demonstrated that sEVs have multifaceted anti-inflammatory activity, we next investigated if these sEVs attenuate chronic inflammatory pain in vivo using the complete Freund’s adjuvant (CFA) mouse model of persistent inflammatory pain. We first examined sEV uptake in vivo by injecting 1μg PKH26-labeled sEVs intrathecally in the lumbar region of the spinal cord. This resulted in strong fluorescent labeling of spinal cord and dorsal root ganglion cells, indicating that these cells could take up sEVs in vivo (Figure 6A). Co-staining with markers for neurons, microglia, and astrocytes did not show cell specific uptake (Figure 6B). Larger cells take up more sEVs suggesting that this is not a mechanism specific phenomenon, but rather availability of larger surface area for sEV uptake by bigger recipient cells (Figure 6A, 6B, and Supplementary Figure 2).
Figure 6. Uptake of intrathecally injected sEVs in mice.
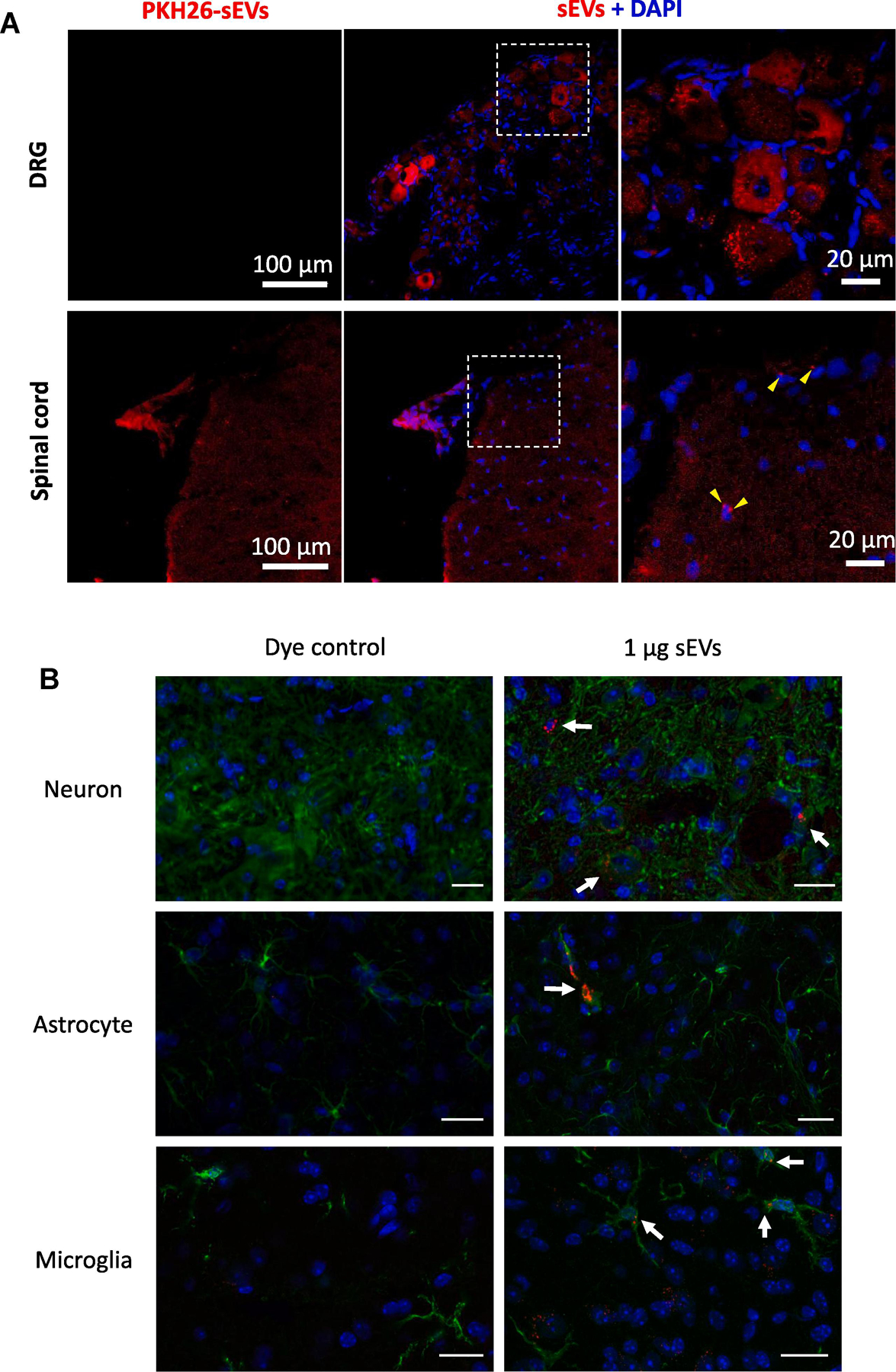
(A) Intrathecal (i.t.) injection of sEVs isolated from conditioned media of RAW 264.7 cells labeled with PKH26 showed uptake by dorsal root ganglion (DRG) (top) and spinal cord (SC) (bottom) (1 μg in 10 μl PBS). Red PKH26, Blue DAPI, Scale bar =100µm. (B) Uptake of sEVs in vivo by neurons, astrocytes and microglia in vivo. One µg PKH26 labeled sEVs were injected intrathecally and after 18 h of sEVs injection, mice were perfused with 4% PFA. Spinal cord was isolated and 30 µm section were stained for MAP2A as neuronal marker, GFAP as astrocytic marker, and Iba1 as microglial marker. Nuclei were stained with DAPI and PKH26 labeled sEVs are shown in red. A dye control was included for all cell types. sEV uptake was observed under a confocal laser scanning microscope. Scale bar = 20 µm. Arrows show the labeled sEVs uptake.
Next, mice were injected with CFA or PBS, and three hours later received an intrathecal injection either 1µg Exo(+) or Exo(−) sEVs or saline (Figure 7A). Following CFA and/or sEVs injection, mice were tested for mechanical allodynia and thermal hyperalgesia using von Frey filaments or the Hargreaves test, respectively. A single intrathecal injection of Exo(+), but not Exo(−), attenuated mechanical allodynia at 48 h post CFA injection and this effect lasted for 10 days (Figure 7B). Additionally, both types of sEVs had no effect on the basal pain threshold in PBS-treated control mice. Thermal hypersensitivity was not altered in animals intrathecally-injected with either type of sEVs (Figure 7C), in contrast to our previous studies showing that a single local, intraplantar injection of sEVs from RAW 264.7 cells can attenuate thermal but not mechanical hypersensitivity in the CFA model (McDonald et al., 2014) . Together, these suggests that route of administration as well as the anti-inflammatory signature of Exo(+) sEVs plays an important role in their efficacy. A previous study showed that exosomes derived from human umbilical cord mesenchymal stem cell reversed neuropathic pain induced by spinal nerve ligation and that the analgesic effects may involve the actions of exosomes on neuron and glial cells (Shiue et al., 2019). They also observed that the analgesic effects remained 7 days after administering 1.2 mg/ml intrathecal exosome was discontinued (Shiue et al., 2019).
Figure 7. Therapeutic administration of sEVs Exo(+) (i.t.) reverse mechanical allodynia induced by CFA in male mice.
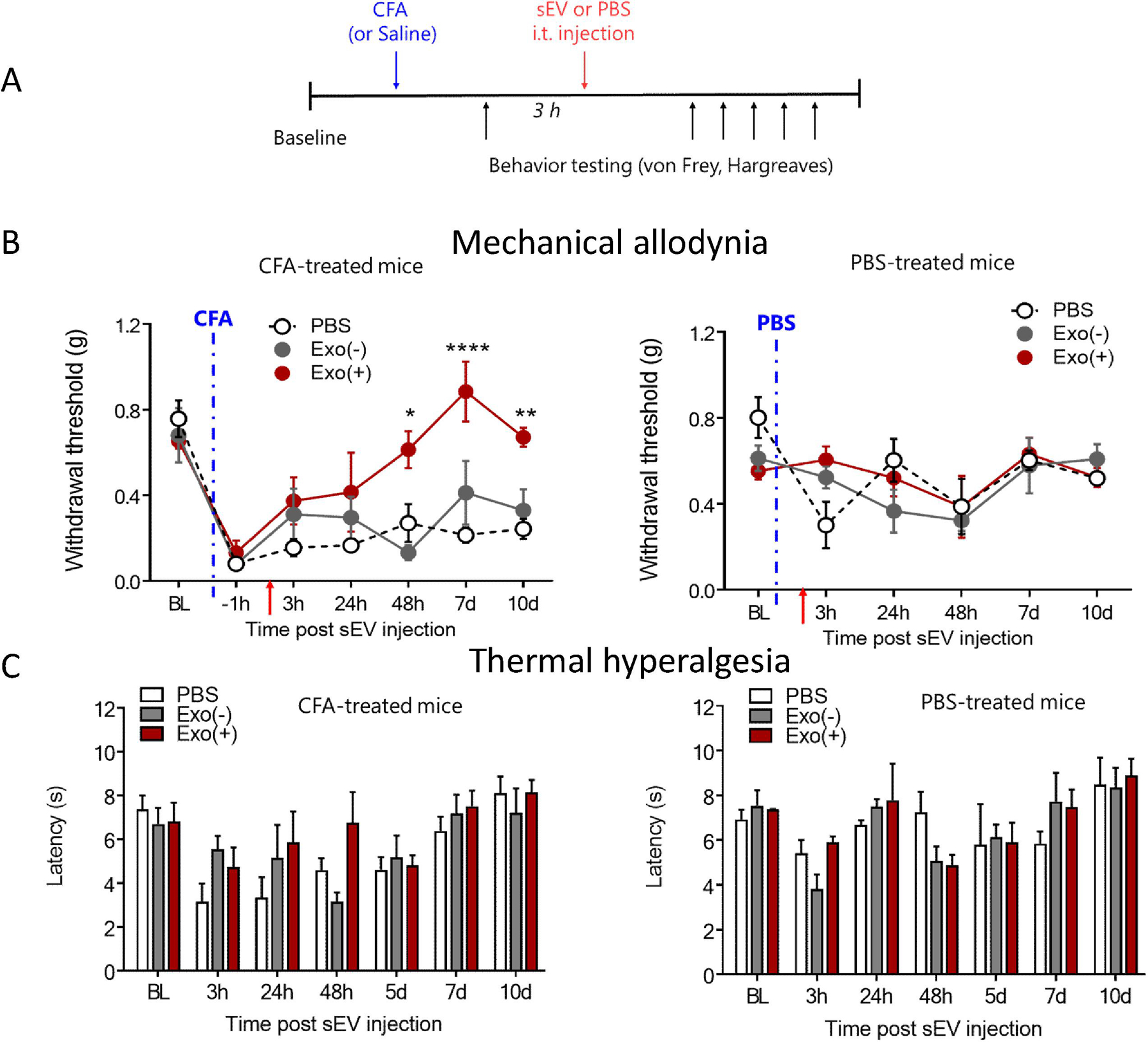
(A) Exo(+) treatment attenuated mechanical pain but did not alter thermal sensitivity in the CFA model. Schematic of in vivo experiment; 8-week-old male C57BL/6 mice injected with CFA followed by an i.t. injection of saline, or 1µg Exo(−) or Exo(+). Mechanical sensitivity was determined using von Frey filaments in CFA or saline treated mice. At 48 h, Exo(+) reduced mechanical allodynia induced by CFA which lasted for 8 days (n=3). sEVs injections did not alter responses in saline treated mice. No significant difference was seen for thermal sensitivity. Data shown are mean ± SEM. Statistical analysis was determined by repeated measures two-way ANOVA with Bonferroni post-test *p < 0.05, **p < 0.01, ****p < 0.0001.
Spinal cord dorsal horn represents the first relay of pain signaling from the periphery to the CNS and changes in spinal excitability and amplification of signals in this area contributes to inflammatory pain (Muley et al., 2016). Since the most significant changes in behavior were observed 7 days post sEV administration, we sequenced RNA from the dorsal horn of the lumbar spinal cord at this time. A total of 1355 genes were differentially expressed between the experimental groups (Figure 8A; p-value ≤ 0.01 and fold change ≥ 2). The transcription profile of Exo(−) samples was markedly different from PBS samples, with 1116 differentially expressed genes (Supplementary Table 1). In comparison the Exo(+) samples had only 301 genes differentially expressed compared with PBS samples. Interestingly, the same set of genes were up and down regulated in both Exo(−) and Exo(+) samples compared to PBS samples, but the magnitude of differential expression was more pronounced in the Exo(−) samples (Figure 8B). The complete list of differentially expressed genes in dorsal horn spinal cord tissue from sEVs and PBS treated groups, 7-days post-CFA treatment is shown in Supplementary Table 2. There was no significant enrichment of genes targeted by miR-146a, miR-146b, and miR-21. Furthermore, the genes Irak-1, Traf6, and Pdcd4, were not differentially expressed in Exo(−) or Exo(+) samples, although several genes whose protein products interact with Irak-1, Traf6, and Pdcd4 were among the differentially expressed genes. This may reflect the long-term effects of sEVs in the spinal cord compared to the short-term effects of sEVs in cell culture. Furthermore, Figure 8C highlights a list of several differentially expressed genes involved in the inflammatory response (Gene Ontology GO:0006954) as well as the genes interacting with Irak-1, Traf6, and Pdcd4 and whether they are up or down regulated in Exo(−) and Exo(+) treated samples. These results suggest that the biomolecular cargo within RAW 264.7-derived sEVs can regulate multiple genes in parallel and can collectively contribute to lowering inflammatory pain hypersensitivity.
Fig. 8. Gene expression changes in dorsal horn of spinal cord following therapeutic administration of sEVs.
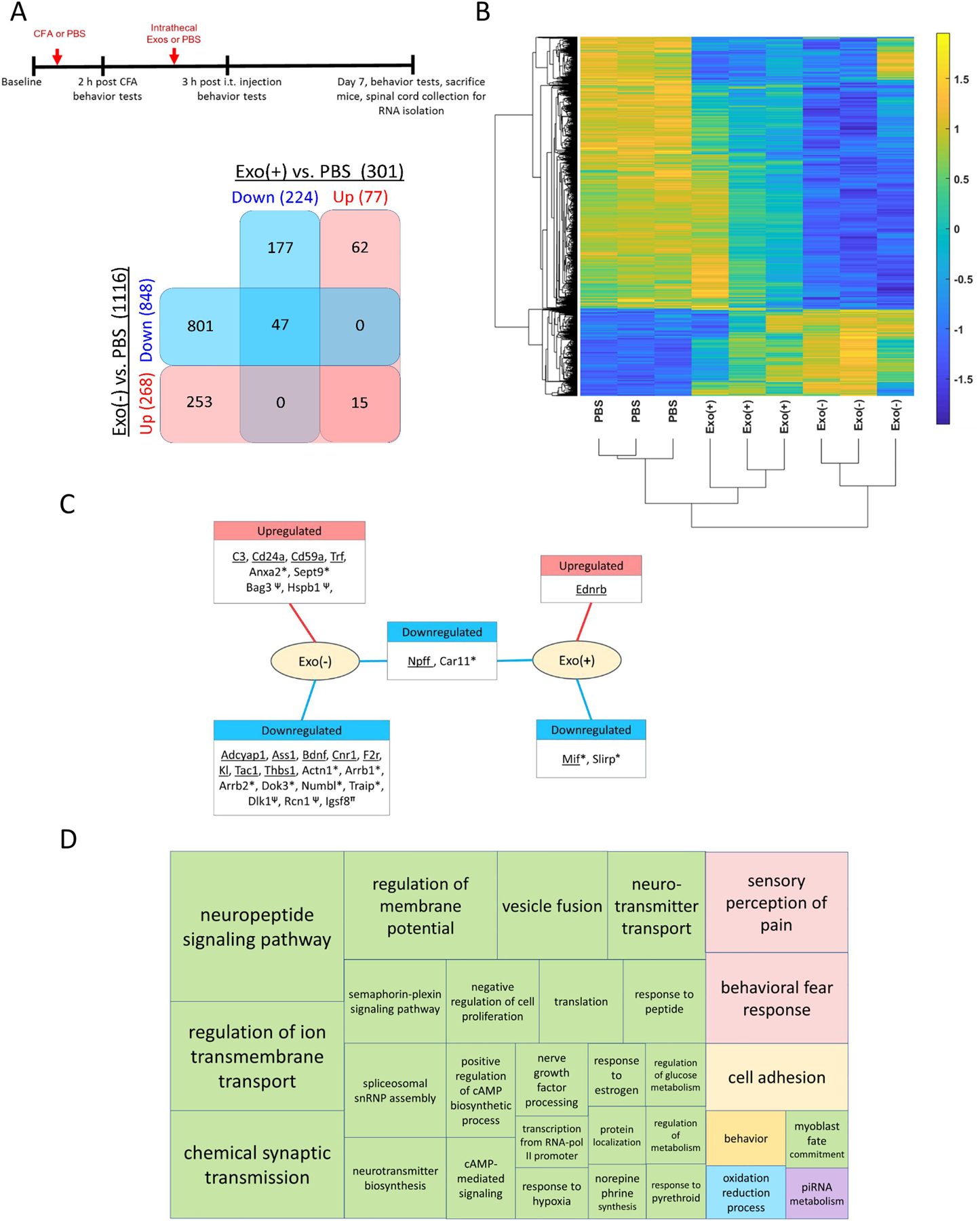
(A) Schematic representation of the experimental design and Venn diagram of differentially expressed genes (DEGs) in spinal cord at 7-days post-CFA treatment (n=3). (B) Gene clustering using single linkage of the Pearson correlation of log2-TPM values. (C) DEGs grouped by up- or down-regulation in Exo(−) and Exo(+) samples. GO:0006954 (underlined) and the DEGs whose protein products interact with Traf6*, Irak1 ψ, Pdcd4π are shown. (D) TreeMap of the Gene Ontology (GO) biological processes enriched with the DEGs.
Further, the differentially expressed genes in Exo(−) and Exo(+) samples were enriched in 157 and 21 biological processes, respectively (enrichment p-value ≤ 0.01) (Supplementary Table 3), and a summary of the enriched biological processes is depicted in Figure 8D. Both Exo(−) and Exo(+) sEVs regulated a subset of common biological processes in recipient cells, suggesting either an intrinsic regulatory property or a fundamental mechanism of action of these sEVs. The significantly higher number of genes and biological processes in Exo(−) vs. Exo(+) sEVs treated mice suggests that LPS treatment of donor RAW 264.7 cells causes significant changes in the composition of sEVs and consequently affect the type and magnitude of transcriptional changes observed in sEVs-treated spinal cord. The enriched Gene Ontology biological processes included neurotransmitter processes, sensory perception of pain, response to pain, neurodegenerative diseases such as Parkinson’s and Alzheimer’s, and opioid addiction.
3.5. sEVs attenuates formalin induced late phase pain response.
We also tested sEVs in a model of acute inflammatory pain induced by formalin injection. Two distinct phases of formalin pain behavior can be observed in this model. The first phase starts immediately after injection and lasts for 10 min. This phase appears to be mediated by C-fiber activation in response to direct chemical stimulation of nociceptors. The second phase, starting at 10 min and lasting for the next 45 min, represent peripheral nerve inflammation-induced changes in central sensitization to noxious stimuli by altering dorsal horn signaling. Pain response scoring includes licks, twitches, raising or shaking of the injected paw (Le Bars et al., 2001). Since the behavior testing in this model is completed in an hour, 1μg sEVs was injected intrathecally 48 h prior to formalin into the hind paw. Mice that received an intrathecal injection of either Exo(+) or Exo(−) 48 h prior to formalin injection showed a significant reduction in the late phase formalin-induced nocifensive response (Figure 9A; two-way ANOVA), further suggesting sEVs are protective. Importantly, Exo(+) induced more robust attenuation of spontaneous pain, suggesting a potential role for their enriched miRNAs.
Figure 9. (A) Intrathecal injection of sEVs attenuates formalin induced late phase pain response.
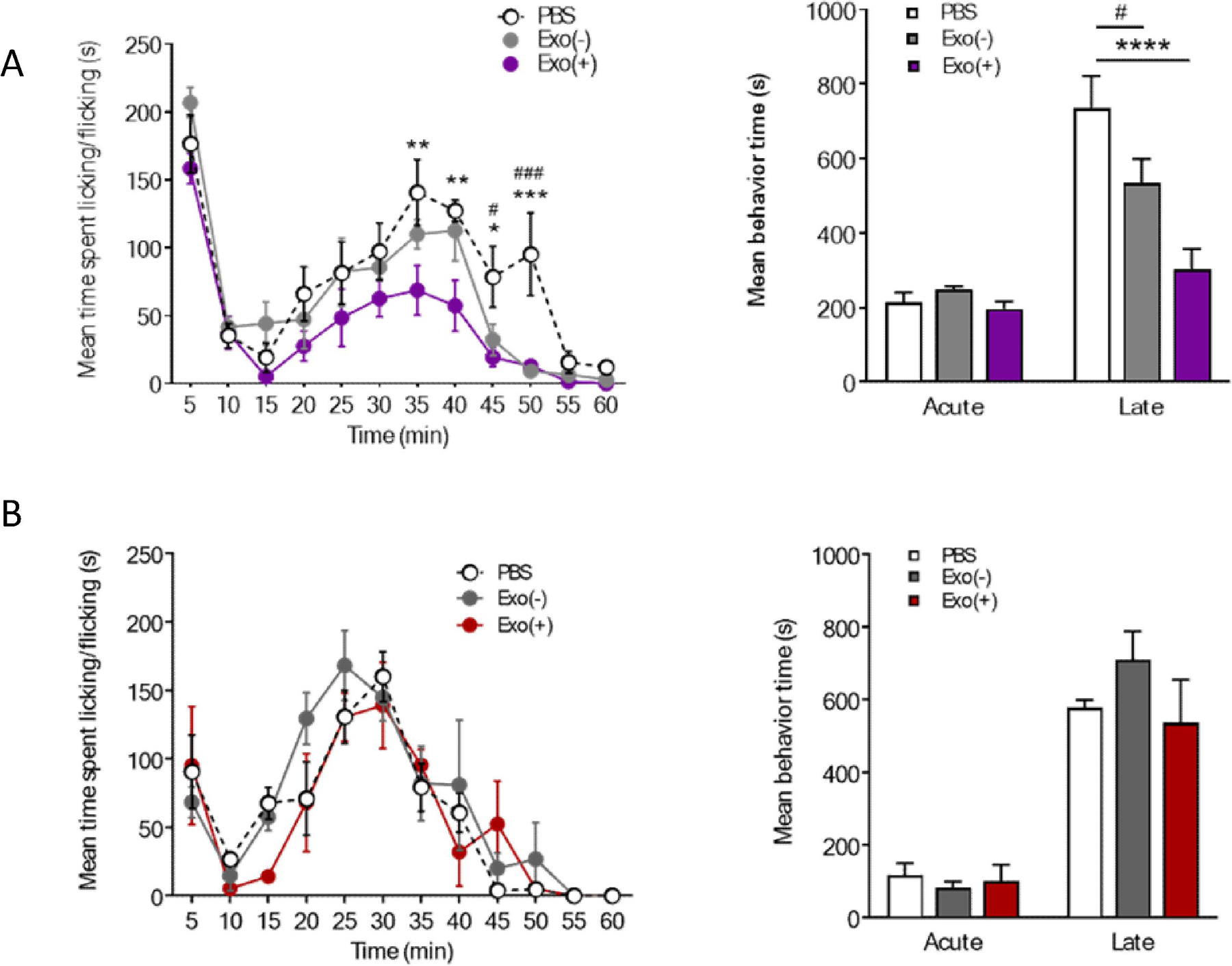
sEVs administered intrathecally 48 h prior attenuated late phase nocifensive behaviors in the formalin model of both sexes. Formalin (5%) was subcutaneously injected 48 h following i.t. injection of 1µg sEVs or vehicle. Exos(+) reduced late phase nocifensive behaviors in 8-week-old male and female C57BL/6 mice (n=6). Data shown are mean ± SEM. Statistical significance was determined by two-way ANOVA. *, #p < 0.05;**p < 0.01; ***, ###p < 0.001; ****p < 0.0001. (B) Prophylactic sEVs treatment did not alter pain responses in the formalin-induced pain model. Two weeks after intrathecal injection of 1 µg Exo(−), Exo(+) or saline, formalin (5%) was injected into hind paw of C57BL/6 female mice (n=3). Data shown as mean ± SEM and analyzed by two-way ANOVA.
3.6. Prophylactic administration of Exo(+) and Exo(−) sEVs attenuates chronic mechanical allodynia and thermal hyperalgesia in CFA-injected mice but not formalin induced acute inflammatory pain.
Based on these results, we hypothesized that prophylactic administration of sEVs may also confer a protective effect in the formalin and CFA models. Thus, we injected mice intrathecally with sEVs or PBS two weeks prior to CFA injection. Treatment with sEVs did not block the induction of mechanical hypersensitivity in the CFA model, but strikingly, both types of sEVs reversed thermal and mechanical hypersensitivity starting at 24 h after CFA injection lasting 10 days post-injection (Figure 10). Control mice injected with sEVs did not display altered pain thresholds throughout the duration of the experiment. Using the same experimental design in the formalin injection model, we did not observe any differences in spontaneous pain behaviors in sEV-injected mice (Figure 9B), suggesting that prophylactic use of sEVs impact inflammatory pain pathways that are activated by CFA over time and thus, could be effective against chronic pain. Attenuation of hypersensitivity by both Exo(−) and Exo(+) sEVs in CFA model mice suggest the role of macrophage-derived cargo in mediating these effects.
Figure 10. Prophylactic intrathecal administration of sEVs attenuates thermal and mechanical pain in CFA model in male mice.
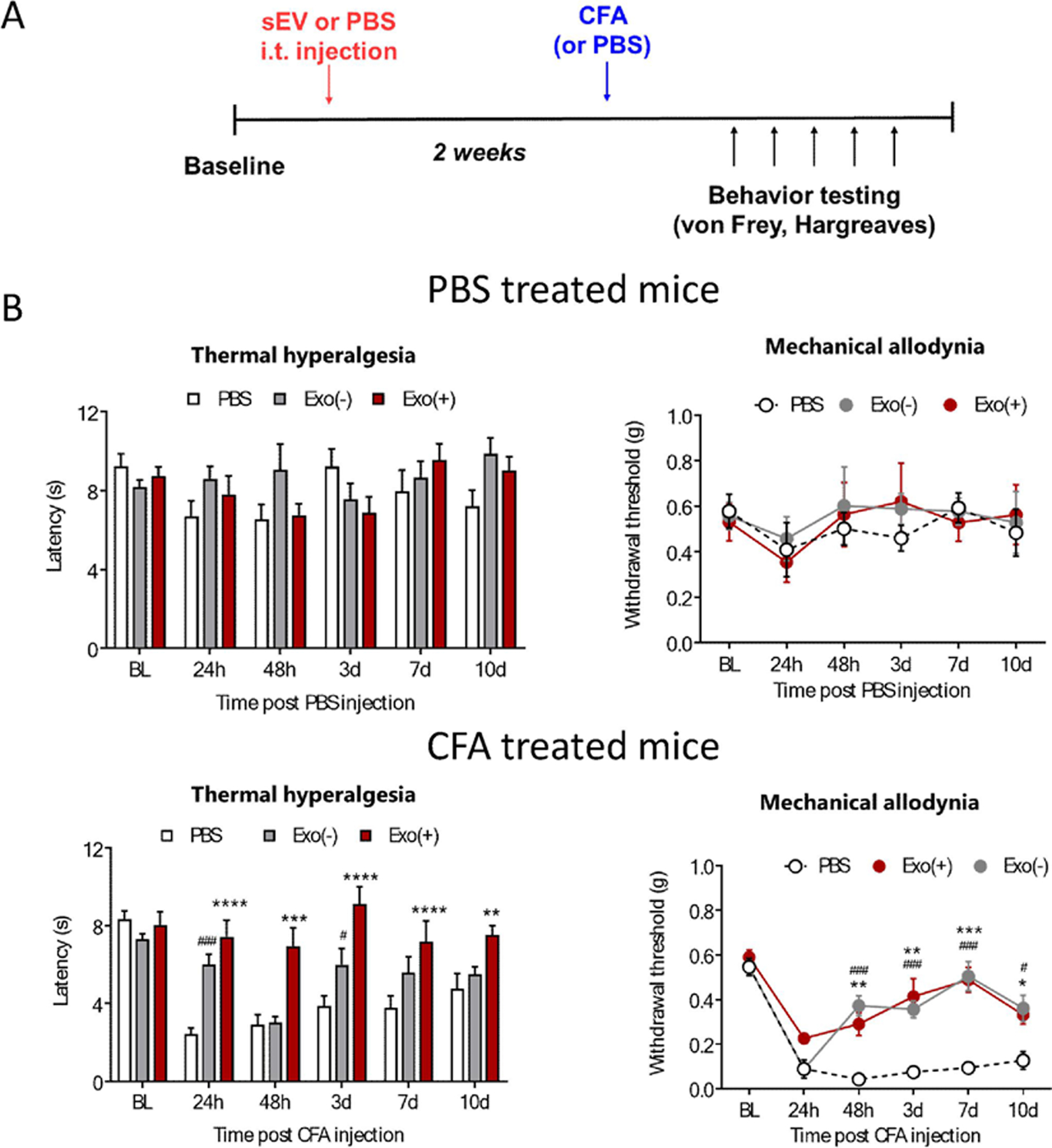
Schematic representation of the experimental design. Two weeks after intrathecal injection of 1 µg Exo(−), Exo(+) or saline, CFA was injected into hind paw to induce inflammation Prophylactic sEV treatment of male mice alleviates mechanical and thermal hyperalgesia in the CFA model and does not alter normal pain sensitivity in control animals, (n= 6). Data shown as mean ± SEM and analyzed by repeated measures two-way ANOVA with Bonferroni post-test, *,#p < 0.05; **p < 0.01; ***, ###p < 0.001; ****,####p < 0.0001.
4. Discussion
APC release sEVs with immunomodulatory functions (Lindenbergh and Stoorvogel, 2018). Here we show that transferred miRNAs are functional in recipient CNS cells by confirming changes in target gene expression linked to inflammation. miR-146 mediates immunosuppressive actions by targeting TLR downstream effectors IRAK-1 and TRAF6, leading to reduced secretion of proinflammatory mediators. miR-21 can affect pro-inflammatory responses through silencing of Il12a. Through its negative regulation of PDCD4, miR-21 indirectly upregulates the anti-inflammatory cytokine Il-10; miR-21 also suppresses LPS-induced TNFα and IL-12 expression along with NF-κB activation while enhancing levels of IL-10 (Das et al., 2014; Sheedy et al., 2010). A pronociceptive role has been attributed to miR-21 functioning as a ligand for TLR8, resulting in the generation of inflammatory mediators and neuronal hyperexcitability in rodent model of neuropathic pain (Zhang et al., 2018). These observations suggest additional roles for miR-21 that can differ from traditional mRNA regulation. miR-21 was shown to activate TLR7 signaling pathway leading to neurotoxicity in simian immunodeficiency virus induced neuropathogenesis (Yelamanchili et al., 2015). Expression of target genes could also govern the cell and model specific impact of individual miRNAs. Our in vitro studies show that recipient cells incubated with Exo(+), downregulated target genes involved in downstream TLR signaling, a major pathway involved in the inflammatory response, suggesting a potential mechanism for Exo(+) mediated effects.
Pre-treatment with Exo(+) and Exo(−) sEVs reduced LPS-induced proinflammatory cytokines and enhanced the expression of anti-inflammatory IL-10 in microglia. We postulate two reasons: 1) Exo(−) sEVs also contain miRNAs and negatively regulate gene expression in LPS stimulated recipient cells, and 2) sEVs released by macrophages carry a variety of functional anti-inflammatory biomolecules. While we chose to focus our in vitro studies on miR-146a, miR-146b and miR-21 for their known involvement in targeting proinflammatory mediators, the RAW 264.7 derived sEVs also contain additional miRNAs, mRNA, proteins, and lipids. Though difficult to attribute function to a single component in the compendium of biomolecules packaged, our data suggests miRNAs can contribute to the effectiveness of sEVs in reducing inflammation in microglia.
sEV-induced effects differed depending on the recipient cells. The immunosuppressive effect was more robust in microglia, suggesting differences in molecular reprogramming in recipient cells. Microglia are of myeloid origin and thus it is possible that macrophage derived sEVs are more efficacious in microglia compared to other CNS cells. From our in vitro studies, we conclude the macrophage derived sEVs may be acting to maintain the balance between pro- and anti-inflammatory cytokines in order to suppress the immune response in recipient cells.
Our in vivo studies show that an intrathecal injection of sEVs from LPS stimulated macrophages could only reverse the already established CFA-induced mechanical allodynia with no change in thermal hypersensitivity in mice. This suggests that route of administration may also play a role in the efficacy of the sEVs. The fact that Exo(+) but not Exo(−) sEVs from unstimulated macrophages are effective when administered after CFA, along with our in vitro data suggests that the effects produced by Exo(+), at least in part, may be mediated by their anti-inflammatory miRNA signature.
To investigate the efficacy in a second model of inflammatory pain, we intrathecally administered sEVs and injected formalin into their hind paw after 48 h. The late phase spontaneous pain response was reduced by both Exo(−) and Exo(+) suggesting biomolecules in RAW 264.7 cells-derived sEVs can be efficacious when administered 48 h prior to formalin injection. This prophylactic effect of sEVs led us to our next study where we intrathecally administered sEVs two weeks prior to CFA injection. When administered prophylactically, both Exo(−) and Exo(+) were protective, attenuated both thermal and mechanical hypersensitivity induced by CFA, and did not alter the normal pain threshold in control mice. Prophylactic sEVs also did not impact the development of mechanical or thermal hypersensitivity. However, intrathecal administration of sEVs two weeks prior to formalin did not alter the spontaneous pain response. This could be due to temporal differences in eliciting immune responses needed to attenuate pain induced by different agents. The observation that sEVs administration did not block the induction of hypersensitivity in CFA model, but reversed hypersensitivity starting at 24h after CFA injection suggest that sEVs modulate pain once established.
RNA sequencing of dorsal horn tissue from saline and CFA treated mice revealed several genes associated with anti-inflammatory pathways were upregulated while proinflammatory genes were downregulated, thereby contributing to attenuation of inflammation and pain. We postulate that sEVs from macrophages could be playing a homeostatic role and elicit a functional response only when there is an insult (CFA in vivo or LPS stimulation of recipient cells in vitro). Though the use of sEVs is more therapeutically relevant as treatment, prophylaxis would be appropriate when painful situations are inevitable such as in combat or post-surgical pain.
APC employ multiple mechanisms to generate immune responses to foreign antigens while avoiding responses to self-antigens (Blander and Medzhitov, 2006). This process can be regulated by TLRs (Kumar et al., 2019), as ligands for several TLR family members can enhance the ability to present antigens by upregulating MHC class II molecules in APC (Bakhru et al., 2014; Blander and Medzhitov, 2006; Mantegazza et al., 2012). MHC class II molecules are ligands for CD4+ T cells and crucial for initiating the adaptive immune response. sEVs from APC can also present peptide/MHC complexes to T cells (Robbins et al., 2016). The contribution of leukocytes in regulating pain varies at the different steps of the inflammatory process in the CFA model. A 6-day latency after CFA injection is required for antigen-specific priming, clonal expansion, and differentiation of CD4+ T lymphocytes and their recruitment in sufficient number at the site of inflammation (Basso et al., 2016). The ability of CD4+ T lymphocytes to produce opioids and thus confer analgesia is a property linked to the effector phase of adaptive T cell response (Boue et al., 2012) and may contribute to the stimulatory potency of sEVs from APC. Additional studies are needed to investigate sEVs induced alterations in MHC class II molecules.
TLR agonists including LPS have been investigated as potential vaccine adjuvants. Adjuvants have diverse mechanisms of action and are selected based on the route of administration and the type of immune response desired for a particular vaccine (Vogel, 2000). It is possible that in addition to the differences in composition of sEVs released by donor cells stimulated with LPS, the route of administration and thus the recipient cells impacted also contributed to the differences in pain behavioral outcome. Though miR-146a was first documented to regulate TLR signaling through targeting IRAK1 and TRAF6, regulation of additional target genes such as ICOS (inducible T cell costimulatory) by miR-146a (Pratama et al., 2015) can also serve as crucial molecular brakes to control B and T cells, two key arms in orchestrating protective humoral immunity (Cho et al., 2018). It is conceivable that sEVs administration resulted in protective autoimmunity as part of a reparative mechanism that can be induced; injury acts as a stress signal that evokes beneficial autoimmunity in the form of a corrective immune response directed against self (Arnold et al., 2007; Shechter et al., 2009; Yoles et al., 2001).
Hyperalgesic priming, a concept developed by Dr. Jon Levine and colleagues expounds that a prior acute inflammatory or neuropathic insult can produce hyperresponsiveness of nociceptors to a subsequent insult, thereby contributing to the transition from acute to chronic pain (Reichling and Levine, 2009). Thus, hyperalgesic priming arises from an initial injury and results in susceptibility to develop a prolonged pain state following a subsequent normally subthreshold noxious stimuli in primed animals. Can there be a phenomenon that is antithetical to hyperalgesic priming? Here we hypothesize that sEVs from APC will attenuate hyperalgesia only when the pain threshold is persistently low (i.e. experiencing more pain or hypersensitivity) and chronic (or at least not transient or acute). We propose that the concept of “analgesic priming” induced by exogenous administration of sEVs from APC and an adaptation which is not manifested as a behavioral outcome, unless under conditions of persistent pain.
In conclusion, by exerting multitude of different effects in parallel, sEVs can be a therapeutic or prophylactic agent without affecting the basal threshold of pain perception. As a therapeutic agent or adjuvant, these sEVs can provide acquired immunity in attenuating chronic pain by decreasing the intensity and or duration of suffering. Pain prophylactic can be highly beneficial especially for patients undergoing surgery or workers in sectors prone to higher occupational injuries. Since these and other situations often call for relieving pain with opioids, sEVs could represent the basis for a novel therapeutic approach that could significantly reduce the use of opioids. Prophylactic sEVs administration could form the basis for a safe and long-term therapy for chronic pain.
Supplementary Material
Supplementary table 1 Significant differentially expressed genes (DEGs) in dorsal horn spinal cord tissue from Exo(−) vs. PBS; and Exo(+) vs. PBS experimental groups, 7-days post-CFA treatment (n=3, p-value≤0.01, absolute fold change ≥2.0).
Supplementary table 2 The complete list of differentially expressed genes in dorsal horn spinal cord tissue from sEVs and PBS treated groups, 7-days post-CFA treatment (n=3, p-value≤0.01, absolute fold change ≥2.0).
Supplementary table 3 The differentially expressed genes in Exo(−) and Exo(+) samples were enriched in 157 and 21 biological processes, respectively (enrichment p-value ≤ 0.01).
Supplementary figure 1
sEV-derived miRNAs decrease their target protein IRAK1 and TRAF6 in primary microglia, neurons and astrocytes. Quantification of representative western blots of IRAK-1, TRAF6 normalized to GAPDH in sEVs treated microglia, neurons and astrocytes (n=2).
Supplementary figure 2
Uptake of sEVs in vivo by neurons, astrocytes and microglia showing sEVs and cellular markers. One µg PKH26 labeled sEVs were injected intrathecally and after 18 h of sEVs injection, mice were perfused with 4% PFA. Spinal cord was isolated and 30 µm section were stained for MAP2A as neuronal marker, GFAP as astrocytic marker, and Iba1 as microglial marker. Nuclei were stained with DAPI and PKH26 labeled sEVs are shown in red. A dye control was included for all cell types. sEV uptake was observed under a confocal laser scanning microscope. Scale bar = 20 µm. Arrows show the labeled sEVs uptake.
Highlights.
Macrophage-derived exosomes transfer anti-inflammatory miRNAs to primary CNS cells
Intrathecal exosomes reduce mechanical hypersensitivity in inflammatory pain model
Prophylactic exosomes two weeks prior attenuate pain hypersensitivity in mice
Exosomes did not impact the induction of hypersensitivity in inflammatory pain model
These small extracellular vesicles could be used as pain therapeutic or prophylactic
Acknowledgments:
We thank Dr. Bradley Nash for critical reading and editing of the manuscript.
All data is available in the main text or the supplementary materials.
Funding:
This study was funded by NIH NINDS R01NS102836 to Seena K. Ajit.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alam MM, O’Neill LA, 2011. MicroRNAs and the resolution phase of inflammation in macrophages. European journal of immunology 41, 2482–2485. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B, 2007. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. The Journal of experimental medicine 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhru P, Sirisaengtaksin N, Soudani E, Mukherjee S, Khan A, Jagannath C, 2014. BCG vaccine mediated reduction in the MHC-II expression of macrophages and dendritic cells is reversed by activation of Toll-like receptors 7 and 9. Cell Immunol 287, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baral P, Udit S, Chiu IM, 2019. Pain and immunity: implications for host defence. Nat Rev Immunol 19, 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso L, Boué J, Mahiddine K, Blanpied C, Robiou-du-Pont S, Vergnolle N, Deraison C, Dietrich G, 2016. Endogenous analgesia mediated by CD4(+) T lymphocytes is dependent on enkephalins in mice. Journal of neuroinflammation 13, 132–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R, 2006. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440, 808–812. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Baltimore D, 2012. MicroRNAs, new effectors and regulators of NF-kappaB. Immunological reviews 246, 205–220. [DOI] [PubMed] [Google Scholar]
- Boue J, Blanpied C, Djata-Cabral M, Pelletier L, Vergnolle N, Dietrich G, 2012. Immune conditions associated with CD4+ T effector-induced opioid release and analgesia. Pain 153, 485–493. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Cho S, Lee H-M, Yu IS, Choi YS, Huang H-Y, Hashemifar SS, Lin L-L, Chen M-C, Afanasiev ND, Khan AA, Lin S-W, Rudensky AY, Crotty S, Lu L-F, 2018. Differential cell-intrinsic regulations of germinal center B and T cells by miR-146a and miR-146b. Nature communications 9, 2757–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Ganesh K, Khanna S, Sen CK, Roy S, 2014. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. Journal of immunology (Baltimore, Md. : 1950) 192, 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ, 1980. Efficient analysis of experimental observations. Annual review of pharmacology and toxicology 20, 441–462. [DOI] [PubMed] [Google Scholar]
- El Andaloussi S, Mager I, Breakefield XO, Wood MJA, 2013. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 12, 347–357. [DOI] [PubMed] [Google Scholar]
- Gereau Iv RW, Sluka KA, Maixner W, Savage SR, Price TJ, Murinson BB, Sullivan MD, Fillingim RB, 2014. A Pain Research Agenda for the 21st Century. The Journal of Pain 15, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA, 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Ji R-R, Chamessian A, Zhang Y-Q, 2016. Pain regulation by non-neuronal cells and inflammation. Science (New York, N.Y.) 354, 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lechman ER, Bianco N, Menon R, Keravala A, Nash J, Mi Z, Watkins SC, Gambotto A, Robbins PD, 2005. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J Immunol 174, 6440–6448. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sunagar R, Gosselin E, 2019. Bacterial Protein Toll-Like-Receptor Agonists: A Novel Perspective on Vaccine Adjuvants. Frontiers in Immunology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bars D, Gozariu M, Cadden SW, 2001. Animal Models of Nociception. Pharmacological Reviews 53, 597. [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP, 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Lindenbergh MFS, Stoorvogel W, 2018. Antigen Presentation by Extracellular Vesicles from Professional Antigen-Presenting Cells. Annu Rev Immunol 36, 435–459. [DOI] [PubMed] [Google Scholar]
- Lu TX, Hartner J, Lim EJ, Fabry V, Mingler MK, Cole ET, Orkin SH, Aronow BJ, Rothenberg ME, 2011. MicroRNA-21 limits in vivo immune response-mediated activation of the IL-12/IFN-gamma pathway, Th1 polarization, and the severity of delayed-type hypersensitivity. J Immunol 187, 3362–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Guttentag SH, El-Benna J, Sasai M, Iwasaki A, Shen H, Laufer TM, Marks MS, 2012. Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity 36, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MK, Capasso KE, Ajit SK, 2013. Purification and microRNA profiling of exosomes derived from blood and culture media. Journal of visualized experiments : JoVE, e50294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MK, Tian Y, Qureshi RA, Gormley M, Ertel A, Gao R, Aradillas Lopez E, Alexander GM, Sacan A, Fortina P, Ajit SK, 2014. Functional significance of macrophage-derived exosomes in inflammation and pain. PAIN® 155, 1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muley MM, Krustev E, McDougall JJ, 2016. Preclinical Assessment of Inflammatory Pain. CNS neuroscience & therapeutics 22, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, Hutloff A, Jordan MA, Zhao JL, Casellas R, Athanasopoulos V, Vinuesa CG, 2015. MicroRNA-146a regulates ICOS-ICOSL signalling to limit accumulation of T follicular helper cells and germinal centres. Nature communications 6, 6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto C, De Las Rivas J, 2006. APID: Agile Protein Interaction DataAnalyzer. Nucleic acids research 34, W298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoof R, Willemen HLDM, Eijkelkamp N, 2018. Divergent roles of immune cells and their mediators in pain. Rheumatology (Oxford, England) 57, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Levine JD, 2009. Critical role of nociceptor plasticity in chronic pain. Trends in neurosciences 32, 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PD, Dorronsoro A, Booker CN, 2016. Regulation of chronic inflammatory and immune processes by extracellular vesicles. Journal of Clinical Investigation 126, 1173+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PD, Morelli AE, 2014. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ, 2008. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M, 2009. Infiltrating Blood-Derived Macrophages Are Vital Cells Playing an Anti-inflammatory Role in Recovery from Spinal Cord Injury in Mice. PLOS Medicine 6, e1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA, 2010. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nature immunology 11, 141–147. [DOI] [PubMed] [Google Scholar]
- Shiue SJ, Rau RH, Shiue HS, Hung YW, Li ZX, Yang KD, Cheng JK, 2019. Mesenchymal stem cell exosomes as a cell-free therapy for nerve injury-induced pain in rats. Pain 160, 210–223. [DOI] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T, 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D, 2006. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America 103, 12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach J-M, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, Bedina Zavec A, Benmoussa A, Berardi AC, Bergese P, Bielska E, Blenkiron C, Bobis-Wozowicz S, Boilard E, Boireau W, Bongiovanni A, Borràs FE, Bosch S, Boulanger CM, Breakefield X, Breglio AM, Brennan MÁ, Brigstock DR, Brisson A, Broekman MLD, Bromberg JF, Bryl-Górecka P, Buch S, Buck AH, Burger D, Busatto S, Buschmann D, Bussolati B, Buzás EI, Byrd JB, Camussi G, Carter DRF, Caruso S, Chamley LW, Chang Y-T, Chen C, Chen S, Cheng L, Chin AR, Clayton A, Clerici SP, Cocks A, Cocucci E, Coffey RJ, Cordeiro-da-Silva A, Couch Y, Coumans FAW, Coyle B, Crescitelli R, Criado MF, D’Souza-Schorey C, Das S, Datta Chaudhuri A, de Candia P, De Santana EF, De Wever O, del Portillo HA, Demaret T, Deville S, Devitt A, Dhondt B, Di Vizio D, Dieterich LC, Dolo V, Dominguez Rubio AP, Dominici M, Dourado MR, Driedonks TAP, Duarte FV, Duncan HM, Eichenberger RM, Ekström K, El Andaloussi S, Elie-Caille C, Erdbrügger U, Falcón-Pérez JM, Fatima F, Fish JE, Flores-Bellver M, Försönits A, Frelet-Barrand A, Fricke F, Fuhrmann G, Gabrielsson S, Gámez-Valero A, Gardiner C, Gärtner K, Gaudin R, Gho YS, Giebel B, Gilbert C, Gimona M, Giusti I, Goberdhan DCI, Görgens A, Gorski SM, Greening DW, Gross JC, Gualerzi A, Gupta GN, Gustafson D, Handberg A, Haraszti RA, Harrison P, Hegyesi H, Hendrix A, Hill AF, Hochberg FH, Hoffmann KF, Holder B, Holthofer H, Hosseinkhani B, Hu G, Huang Y, Huber V, Hunt S, Ibrahim AG-E, Ikezu T, Inal JM, Isin M, Ivanova A, Jackson HK, Jacobsen S, Jay SM, Jayachandran M, Jenster G, Jiang L, Johnson SM, Jones JC, Jong A, Jovanovic-Talisman T, Jung S, Kalluri R, Kano S. i., Kaur S, Kawamura Y, Keller ET, Khamari D, Khomyakova E, Khvorova A, Kierulf P, Kim KP, Kislinger T, Klingeborn M, Klinke DJ, Kornek M, Kosanović MM, Kovács ÁF, Krämer-Albers E-M, Krasemann S, Krause M, Kurochkin IV, Kusuma GD, Kuypers S, Laitinen S, Langevin SM, Languino LR, Lannigan J, Lässer C, Laurent LC, Lavieu G, Lázaro-Ibáñez E, Le Lay S, Lee M-S, Lee YXF, Lemos DS, Lenassi M, Leszczynska A, Li ITS, Liao K, Libregts SF, Ligeti E, Lim R, Lim SK, Linē A, Linnemannstöns K, Llorente A, Lombard CA, Lorenowicz MJ, Lörincz ÁM, Lötvall J, Lovett J, Lowry MC, Loyer X, Lu Q, Lukomska B, Lunavat TR, Maas SLN, Malhi H, Marcilla A, Mariani J, Mariscal J, Martens-Uzunova ES, Martin-Jaular L, Martinez MC, Martins VR, Mathieu M, Mathivanan S, Maugeri M, McGinnis K, McVey MJ, Meckes DG, Meehan KL, Mertens I, Minciacchi VR, Möller A, Møller Jørgensen M, Morales-Kastresana A, Morhayim J, Mullier F, Muraca M, Musante L, Mussack V, Muth DC, Myburgh KH, Najrana T, Nawaz M, Nazarenko I, Nejsum P, Neri C, Neri T, Nieuwland R, Nimrichter L, Nolan JP, Nolte-’t Hoen ENM, Noren Hooten N, O’Driscoll L, O’Grady T, O’Loghlen A, Ochiya T, Olivier M, Ortiz A, Ortiz LA, Osteikoetxea X, Østergaard O, Ostrowski M, Park J, Pegtel DM, Peinado H, Perut F, Pfaffl MW, Phinney DG, Pieters BCH, Pink RC, Pisetsky DS, Pogge von Strandmann E, Polakovicova I, Poon IKH, Powell BH, Prada I, Pulliam L, Quesenberry P, Radeghieri A, Raffai RL, Raimondo S, Rak J, Ramirez MI, Raposo G, Rayyan MS, Regev-Rudzki N, Ricklefs FL, Robbins PD, Roberts DD, Rodrigues SC, Rohde E, Rome S, Rouschop KMA, Rughetti A, Russell AE, Saá P, Sahoo S, Salas-Huenuleo E, Sánchez C, Saugstad JA, Saul J, Schiffelers RM, Schneider R, Schøyen TH, Scott A, Shahaj E, Sharma S, Shatnyeva O, Shekari F, Shelke GV, Shetty AK, Shiba K, Siljander PRM, Silva AM, Skowronek A, Snyder OL, Soares RP, Sódar BW, Soekmadji C, Sotillo J, Stahl PD, Stoorvogel W, Stott SL, Strasser EF, Swift S, Tahara H, Tewari M, Timms K, Tiwari S, Tixeira R, Tkach M, Toh WS, Tomasini R, Torrecilhas AC, Tosar JP, Toxavidis V, Urbanelli L, Vader P, van Balkom BWM, van der Grein SG, Van Deun J, van Herwijnen MJC, Van Keuren-Jensen K, van Niel G, van Royen ME, van Wijnen AJ, Vasconcelos MH, Vechetti IJ, Veit TD, Vella LJ, Velot É, Verweij FJ, Vestad B, Viñas JL, Visnovitz T, Vukman KV, Wahlgren J, Watson DC, Wauben MHM, Weaver A, Webber JP, Weber V, Wehman AM, Weiss DJ, Welsh JA, Wendt S, Wheelock AM, Wiener Z, Witte L, Wolfram J, Xagorari A, Xander P, Xu J, Yan X, Yáñez-Mó M, Yin H, Yuana Y, Zappulli V, Zarubova J, Žėkas V, Zhang J. y., Zhao Z, Zheng L, Zheutlin AR, Zickler AM, Zimmermann P, Zivkovic AM, Zocco D, Zuba-Surma EK, 2018. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of extracellular vesicles 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel FR, 2000. Improving Vaccine Performance with Adjuvants. Clinical Infectious Diseases 30, S266–S270. [DOI] [PubMed] [Google Scholar]
- Yelamanchili SV, Lamberty BG, Rennard DA, Morsey BM, Hochfelder CG, Meays BM, Levy E, Fox HS, 2015. MiR-21 in Extracellular Vesicles Leads to Neurotoxicity via TLR7 Signaling in SIV Neurological Disease. PLOS Pathogens 11, e1005032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, Kuchroo V, Cohen IR, Weiner H, Schwartz M, 2001. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci 21, 3740–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Guo JS, Li SS, Wu XB, Cao DL, Jiang BC, Jing PB, Bai XQ, Li CH, Wu ZH, Lu Y, Gao YJ, 2018. TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. The Journal of experimental medicine 215, 3019–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1 Significant differentially expressed genes (DEGs) in dorsal horn spinal cord tissue from Exo(−) vs. PBS; and Exo(+) vs. PBS experimental groups, 7-days post-CFA treatment (n=3, p-value≤0.01, absolute fold change ≥2.0).
Supplementary table 2 The complete list of differentially expressed genes in dorsal horn spinal cord tissue from sEVs and PBS treated groups, 7-days post-CFA treatment (n=3, p-value≤0.01, absolute fold change ≥2.0).
Supplementary table 3 The differentially expressed genes in Exo(−) and Exo(+) samples were enriched in 157 and 21 biological processes, respectively (enrichment p-value ≤ 0.01).
Supplementary figure 1
sEV-derived miRNAs decrease their target protein IRAK1 and TRAF6 in primary microglia, neurons and astrocytes. Quantification of representative western blots of IRAK-1, TRAF6 normalized to GAPDH in sEVs treated microglia, neurons and astrocytes (n=2).
Supplementary figure 2
Uptake of sEVs in vivo by neurons, astrocytes and microglia showing sEVs and cellular markers. One µg PKH26 labeled sEVs were injected intrathecally and after 18 h of sEVs injection, mice were perfused with 4% PFA. Spinal cord was isolated and 30 µm section were stained for MAP2A as neuronal marker, GFAP as astrocytic marker, and Iba1 as microglial marker. Nuclei were stained with DAPI and PKH26 labeled sEVs are shown in red. A dye control was included for all cell types. sEV uptake was observed under a confocal laser scanning microscope. Scale bar = 20 µm. Arrows show the labeled sEVs uptake.
Data Availability Statement
Gene-level expression values for all samples are provided as FPKM (fragments per kilobase of transcript per million mapped reads) and TPM (transcript per million) in the Supp Table 2. Raw RNA sequencing data will be made available in NCBI Sequence Retrieval System (SRS) upon publication.