

Abstract
Here we report that N-phosphonium pyridinium intermediates are unusually reactive for pyridine SNAr reactions. Specifically, forming phosphonium salts from halopyridines typically requires elevated temperatures and Lewis acid additives. The alternative activation mode described in this paper permits C–P bond formation to occur at ambient temperatures in many cases, and functions across a broad range of substrates.
Keywords: pyridines, SNAr reaction, phosphonium salts, pyridinium salts
Graphical Abstract
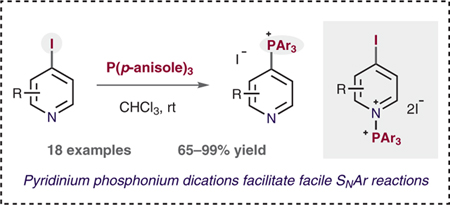
Pyridine-containing molecules are widely used in the chemical sciences as they have applications in pharmaceuticals and agrochemicals in addition to serving as ligands for coordination complexes and components of organic materials.1–3 Because of this prevalence, methods that derivatize simple pyridines into a diverse set of valuable products are highly sought after. Most pyridine-functionalization strategies involve transforming functional handles such as (pseudo)halides or boronic acids.4 We have focused on developing phosphonium salts as alternative functional groups for pyridine derivatization and we have shown that they are viable for the formation of a range of carbon–carbon and carbon–heteroatom bonds (Scheme 1A).5 In particular, phosphorus-ligand-coupling pathways are available through these salts, together with distinct ways of forming structures such as bipyridines.6 Here we report a new method for forming pyridinylphosphonium salts from iodopyridines that proceeds via an unusual dicationic N-phosphonium–pyridinium intermediate (Scheme 1B).
Scheme 1.
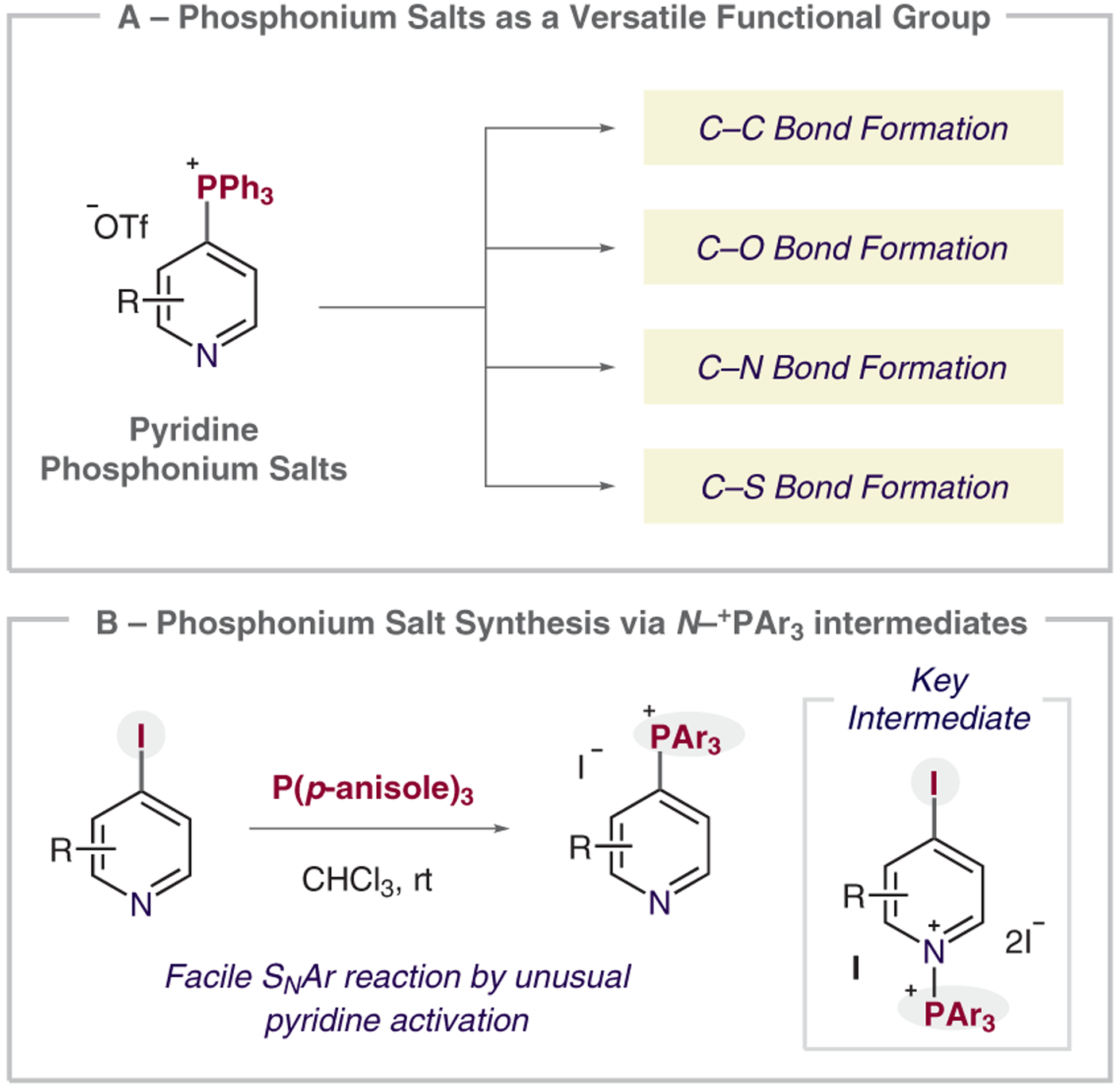
(A) Synthetic utility of heterocyclic phosphonium salts. (B) N + PAr3 activation strategy for facile phosphonium salt synthesis.
There are two common ways to form pyridinylphosphonium salts: first, an oxidative reaction of pyridine C–H pre-cursors, where phosphines attack N-triflylpyridinium salts (Scheme 2A);5a,7 and second, SNAr reactions between 2- and 4-halopyridines and phosphine nucleophiles (Scheme 2B). Although the latter approach requires a preinstalled halide, it sometimes has advantages over the C–H route. For example, the process tolerates such functional groups as alcohols, amines, or carboxylic acids. Also, there is an abundance of commercially available halopyridines. However, the SNAr process typically requires temperatures of more than 100 °C and Brønsted or Lewis acid additives.6,8
Scheme 2.
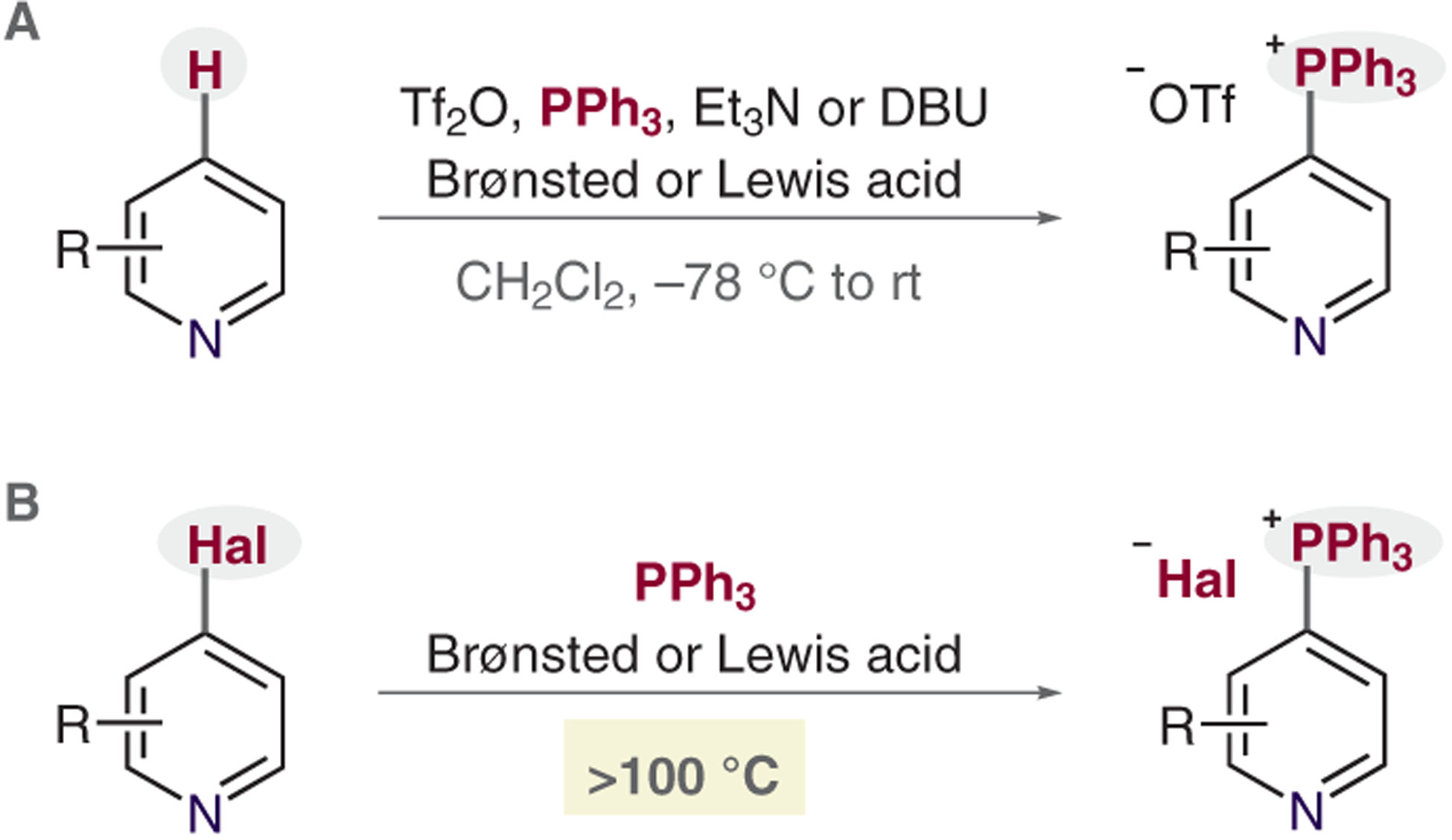
Common routes to pyridine phosphonium salts
We serendipitously found that two equivalents of PPh3 react with 4-iodopyridine to form salt 1a (Table 1, entry 1). Notably, the reaction occurred at room temperature and without exogenous acid. We decided to investigate a range of different phosphines to improve the yield of 1a. Surprisingly, PEt3 was similarly efficient to PPh3, but PPh2Et and PPhEt2 resulted in much higher conversion to 1a (Table 1, entries 2–4). The more electron-rich triaryl phosphine P(p-anisole)3 was similarly effective. Further optimization showed that the most effective protocol used one equivalent of phosphine with CHCl3 as a solvent (Table 1, entries 5–7; see the Supporting Information for further details).
Table 1.
Optimization of Pyridyl Phosphonium Salt Formation
 | ||||
|---|---|---|---|---|
| Entry | Phosphine (equiv) | Solvent | Residual 4-iodopyridinea (%) | Yield3 (%) |
| 1 | PPh3 (2.0) | CH2Cl2 | 71 | 5 |
| 2 | PEt3 (2.0) | CH2Cl2 | 64 | 8 |
| 3 | EtPPh2 (2.0) | CH2Cl2 | 10 | 79 |
| 4 | PhPEt2 (2.0) | CH2Cl2 | 4 | 86 |
| 5 | P(p-anisole)3 (2.0) | CH2Cl2 | 3 | 89 |
| 6 | P(p-anisole)3 (2.0) | CHCl3 | 1 | 85 |
| 7 | P(p-anisole)3 (1.0) | CHCl3 | 3 | 97 |
| 8b | P(p-anisole)3 (1.0) | CHCl3 | 34 | 66 |
Determined by 1H NMR with CHPh3 as internal standard.
Reaction performed with sublimed 4-iodopyridine.
We were intrigued by the mechanism of this substitution process, because the protocol occurs at ambient temperature and iodopyridines are typically the least reactive nucleofuges in SNAr reactions.9 There was a noticeable decrease in reaction efficiency when we used sublimed 4-iodopyridine (Table 1, entry 8). We hypothesized that a small amount of contaminant in the commercial material, or a minor amount of decomposition products, might function as a promoter for this reaction. 4-Chloro- and 4-bromopyridine did not react under the same conditions (Scheme 3A), but adding 10 mol% of iodine resulted in similar yields of phosphonium salt 1a. On the basis of these observations, we proposed the reaction mechanism shown in Scheme 3B. The phosphine first reacts with iodine resulting in an iodophosphonium salt.10 Next, a ligand-exchange reaction occurs to form the key N-phosphonium pyridinium dicationic salt I, which facilitates a subsequent SNAr reaction with the phosphine nucleophile. The I–+PAr3 cation is regenerated through this reaction in a manner consistent with the requirement for a catalytic amount of iodine. Support for intermediate I comes from two reports from the groups of Dutton and Weigland (Scheme 3C), who isolated and characterized DMAP versions of these unusual bis salts.11,12 To the best of our knowledge, there are no previous examples of the use of these dicationic species for synthetic transformations.
Scheme 3.
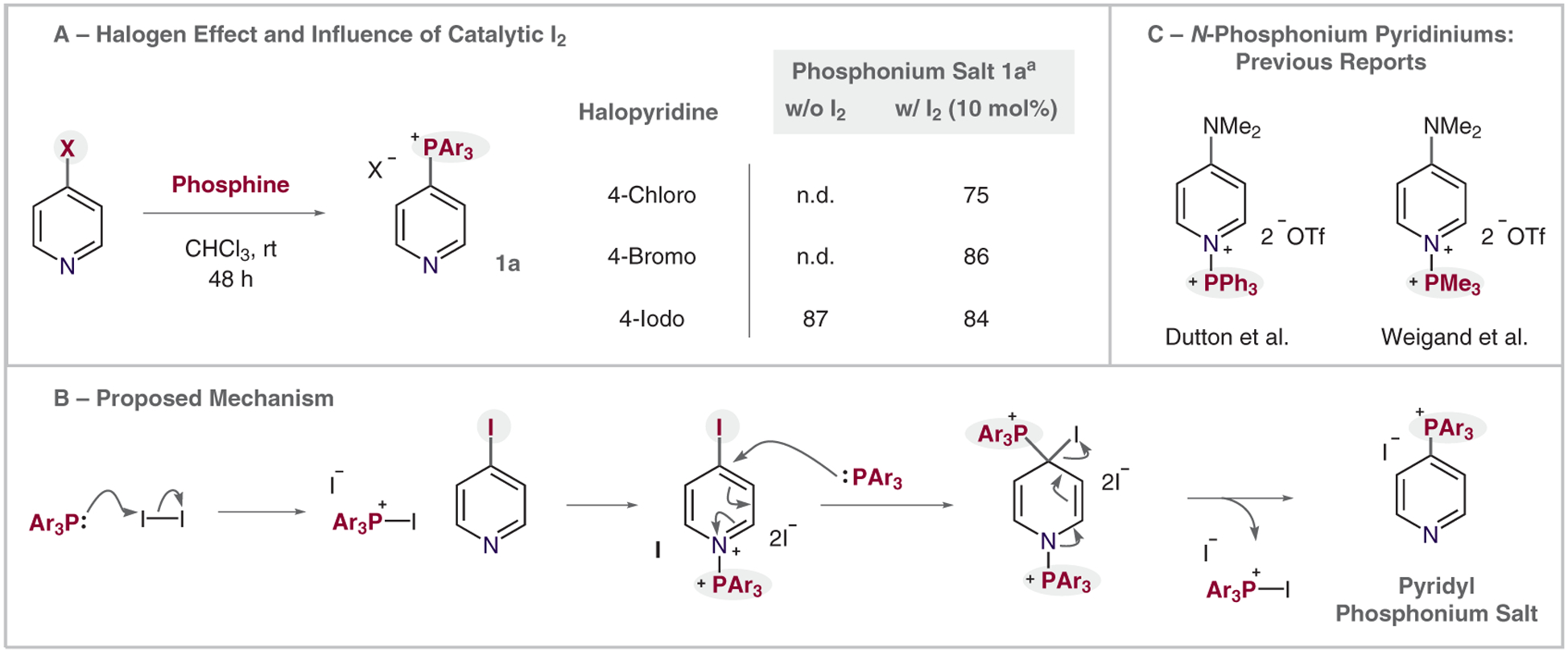
Experimental mechanistic insights and proposed mechanism. (A) Halogen effect and influence of catalytic I2. Yields were determined by 1H NMR with CHPh3 as internal standard. (B) Proposed reaction mechanism. (C) Previous reports of N-phosphonium pyridiniums.
Next, we explored the substrate scope for this C–P bond-forming reaction (Scheme 4). Note that the iodopyridine starting materials were either purchased commercially or were prepared in house, and it was not necessary to add exogenous I2. Also, we precipitated the phosphonium salt products from the reaction mixtures in high purity. Electron-withdrawing substituents at the 3-position worked well in the reaction, as did electron-donating groups when we raised the temperature to 50 °C (1b–e). It is notable that hydroxy and amino groups do not interfere with the reaction. We also obtained the 3,5-disubstituted salt 1f in excellent yield. The reaction tolerates 2-substituted pyridines, with pyridones, azaindoles and 2-arylpyridines performing well under the reaction conditions (1g–j). Importantly, SNAr-active halogens at other positions on the pyridine ring do not erode regioselectivity (1k and 1l). Salt 1m formed without evidence of cyclization onto the pendent alkene, thereby disfavoring a mechanism involving a radical at the 4-position of the pyridine ring.13 The reaction was extended to quinolines and 2-iodopyridines (1n–p), and we also obtained phosphonium salt derivatives of the pharmaceuticals etoricoxib and loratadine (1q and 1r, respectively). The most pertinent limitations of this reaction are the failures to react of 2,6-dihalopyridines or of pyridines with strongly electron-withdrawing groups, such as trifluoromethyl groups, at the 2-position. We found that SNAr-active iodoarenes did not function in this reaction.
Scheme 4.
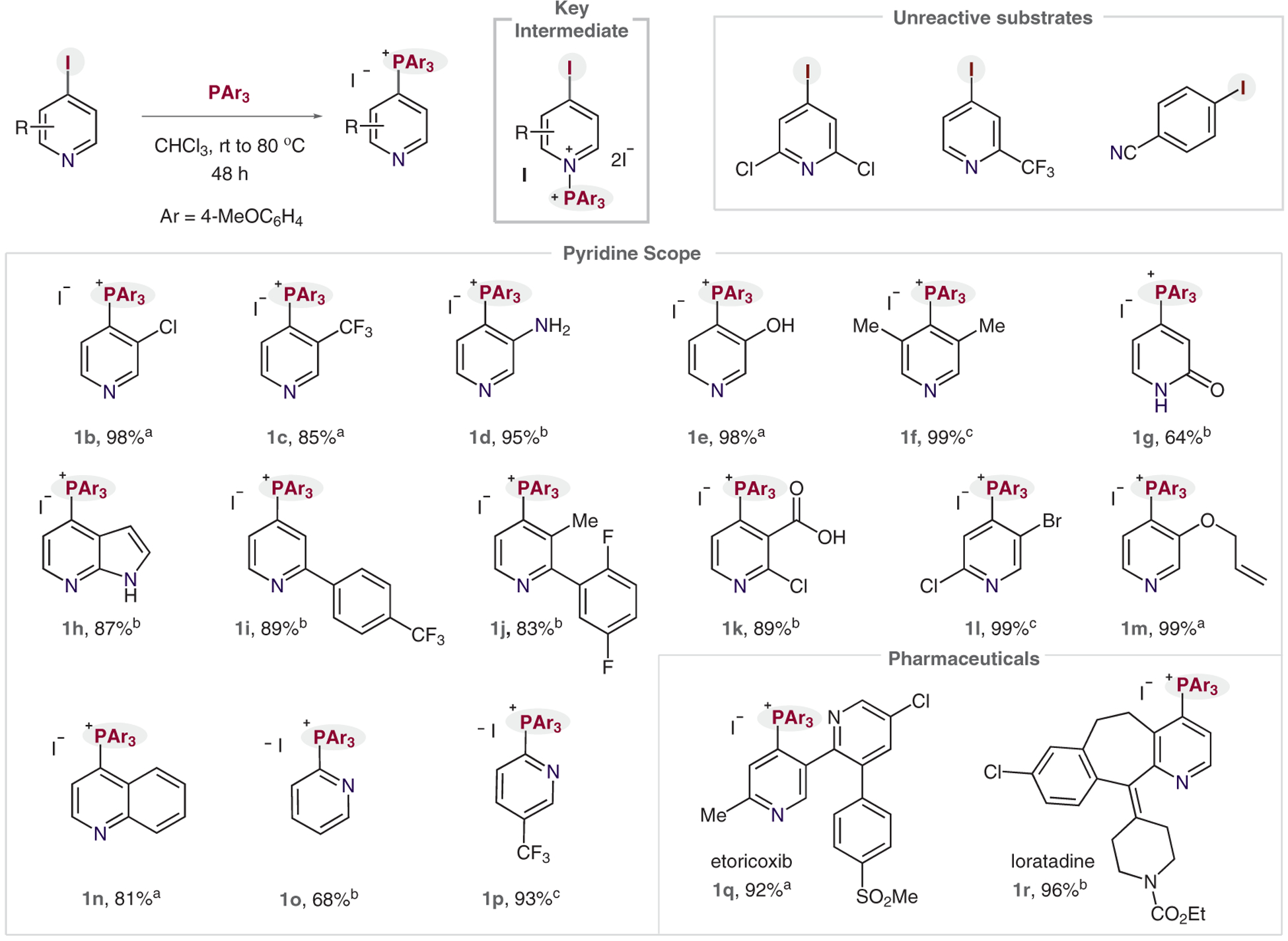
Reaction scope. Reagents and conditions: Iodopyridine (1.0 equiv), P(p-anisole)3 (1.0 equiv), CHCl3 (0.1 M), 0–80 °C, ≤48 h. Isolated yields shown. a rt. b 50 °C. c 80 °C.
Next, we attempted to detect the proposed N-phosphonium–pyridinium dication by using NMR spectroscopy (Figure 1). It was not possible to observe this intermediate from 4-iodopyridine, as the SNAr reaction occurs too quickly. However, use of pyridine as a substrate did provide some insight. Treating pyridine with 50 mol% I2 produced an up-field shift of the ortho and para protons in the 1H NMR spectra.14 The corresponding experiment with pyridine and 50 mol% of I–+PAr3 showed deshielding indicating the presence of a different species, which we assume is the N-phosphonium dication. We are currently performing further studies to investigate the mechanism of this reaction.
Figure 1.
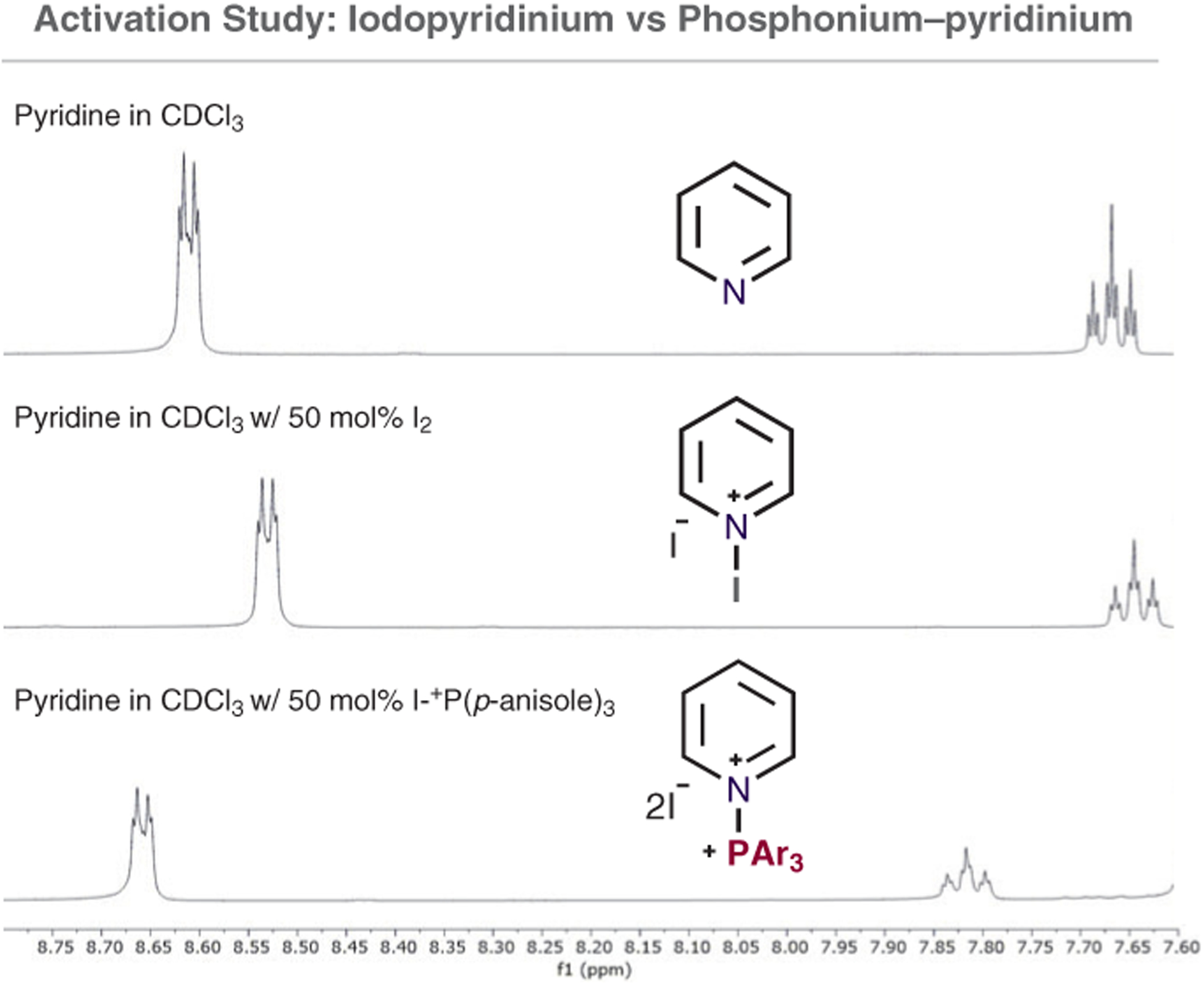
I2 and I–+P(p-anisole)3 spiking study for N-activation of pyridine
In summary, we have developed an unusual SNAr reaction of iodopyridines with phosphine nucleophiles. We postulate an unusual dicationic N-phosphonium pyridinium salt as a key intermediate that promotes rapid C–I-to-C–P substitution. We obtained a range of pyridyl phosphonium salts by using this method, including some examples that would be difficult to obtain by conventional SNAr methods.15 We are currently investigating other ways of exploiting this pyridine N-activating group in our laboratory, and the results will be reported in due course.
Supplementary Material
Funding Information
This work was supported by The National Institutes of Health (NIGMS) under Award Number R01 GM124094.National Institutes of Health (R01 GM124094)
Footnotes
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/a-1315-1279.
References and Notes
- (1).(a) Baumann M; Baxendale IR Beilstein J. Org. Chem 2013, 9, 2265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]; (c) Eicher T; Hauptmann S The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications, 2nd ed; Wiley-VCH: Weinheim, 2003. [Google Scholar]
- (2).(a) Zafar MN; Atif AH; Nazar MF; Sumrra SH; Gul-E-Saba, ; Paracha R Russ. J. Coord. Chem 2016, 42, 1. [Google Scholar]; (b) Wurz RP Chem. Rev 2007, 107, 5570. [DOI] [PubMed] [Google Scholar]; (c) Leclerc N; Sanaur S; Galmiche L; Mathevet F; Attias A-J; Fave J-L; Roussel J; Hapiot P; Lemaître N; Geffroy B Chem. Mater 2005, 17, 502. [Google Scholar]
- (3).Suh MP; Cheon YE; Lee EY Coord. Chem. Rev 2008, 252, 1007. [Google Scholar]
- (4).Campeau L-C; Fagnou K Chem. Soc. Rev 2007, 36, 1058. [DOI] [PubMed] [Google Scholar]
- (5).(a) Hilton MC; Dolewski RD; McNally AJ Am. Chem. Soc 2016, 138, 13806. [DOI] [PubMed] [Google Scholar]; (b) Anderson RG; Jett BM; McNally A Angew. Chem. Int. Ed 2018, 57, 12514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Patel C; Mohnike M; Hilton MC; McNally A Org. Lett 2018, 20, 2607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Koniarczyk JL; Hesk D; Overgard A; Davies IW; McNally AJ Am. Chem. Soc 2018, 140, 1990. [DOI] [PubMed] [Google Scholar]; (e) Zhang X; McNally A ACS Catal. 2019, 9, 4862. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang X; McNally A Angew. Chem. Int. Ed 2017, 56, 9833. [DOI] [PubMed] [Google Scholar]; (g) Che Y-Y; Yue Y; Lin L-Z; Pei B; Deng X; Feng C Angew. Chem. Int. Ed 2020, 59, 16414. [DOI] [PubMed] [Google Scholar]
- (6).(a) Hilton MC; Zhang X; Boyle BT; Alegre-Requena JV; Paton RS; McNally A Science 2018, 362, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boyle BT; Hilton MC; McNally A J. Am. Chem. Soc 2019, 141, 15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dolewski RD; Fricke PJ; McNally AJ Am. Chem. Soc 2018, 140, 8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mečiarová M; Toma Š; Loupy A; Horváth B Phosphorus, Sulfur Silicon Relat. Elem 2007, 183, 21. [Google Scholar]
- (9).Modern Nucleophilic Aromatic Substitution; Terrier F, Ed.; Wiley-VCH: Weinheim, 2013. [Google Scholar]
- (10).Cotton FA; Kibala PA J. Am. Chem. Soc 1987, 109, 3308. [Google Scholar]
- (11).Albayer M; Dutton JL J. Coord. Chem 2019, 72, 1307. [Google Scholar]
- (12).Weigand JJ; Burford N; Decken A; Schulz A Eur. J. Inorg. Chem 2007, 4868. [DOI] [PubMed] [Google Scholar]
- (13).Aycock RA; Wang H; Jui NT Chem. Sci 2017, 8, 3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Georgiou DC; Butler P; Browne EC; Wilson DJD; Dutton JL Aust. J. Chem 2013, 66, 1179. [Google Scholar]
- (15). Phosphonium Salts 1a–r: General Procedure An oven-dried 8 mL vial (<1.0 mmol) or 16 mL vial (1.0–4.0 mmol) equipped with a stirrer bar was charged with the appropriate iodopyridine (1.0 equiv), (p-anisole)3P (1.0 equiv), and CHCl3 (0.5 M). The mixture was then stirred at rt, 50 °C, or 80 °C for the appropriate time. The mixture was then diluted with CHCl3, and the product was precipitated with Et2O (100 mL per 1.0 mmol) at rt. (3-Chloropyridin-4-yl)[tris(4-methoxyphenyl)]phosphonium Iodide (1b) Prepared according to general procedure from 3-chloro-4-iodopyridine (72 mg, 0.30 mmol) and (p-anisole)3P (106 mg, 0.30 mmol) in CHCl3 (0.6 mL) at rt for 36 h to give a light-brown solid; yield: 174 mg (98%, 0.3 mmol); mp 91–93 °C. 1H NMR (400 MHz, CDCl3): δ = 9.12–8.59 (m, 2 H), 7.58 (dd, J = 12.7, 8.9, 6 H), 7.48–7.10 (m, 7 H), 3.95 (s, 9 H). 13C NMR (100 MHz, CDCl3) δ = 165.40 (d, J = 3.0), 151.98 (d, J = 5.0), 150.11 (d, J = 9.8), 136.35 (d, J = 12.4), 134.75 (d, J = 2.2), 130.32 (d, J = 8.4), 129.29 (d, J = 88.3), 117.02 (d, J = 14.5), 105.78 (d, J = 100.0), 56.51. 31P NMR (162 MHz, CDCl3) δ = 21.08. LRMS (ESI + APCI): m/z [M – I]+ calcd for C26H24ClNO3P: 464.1; found: 464.2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.