

Summary
Single-cell RNA sequencing technology promotes the profiling of single-cell transcriptomes at an unprecedented throughput and resolution. However, in scRNA-seq studies, only a low amount of sequenced mRNA in each cell leads to missing detection for a portion of mRNA molecules, i.e. the dropout problem which hinders various downstream analyses. Therefore, it is necessary to develop robust and effective imputation methods for the increasing scRNA-seq data. In this study, we have developed an imputation method (GraphSCI) to impute the dropout events in scRNA-seq data based on the graph convolution networks. Extensive experiments demonstrated that GraphSCI outperforms other state-of-the-art methods for imputation on both simulated and real scRNA-seq data. Meanwhile, GraphSCI is able to accurately infer gene-to-gene relationships and the inferred gene-to-gene relationships could also provide powerful assistance for imputation dynamically during the training process, which is a key promotion of GraphSCI compared with other imputation algorithms.
Subject areas: Genomics, Bioinformatics, Data Acquisition in Bioinformatics, Artificial Intelligence
Graphical abstract
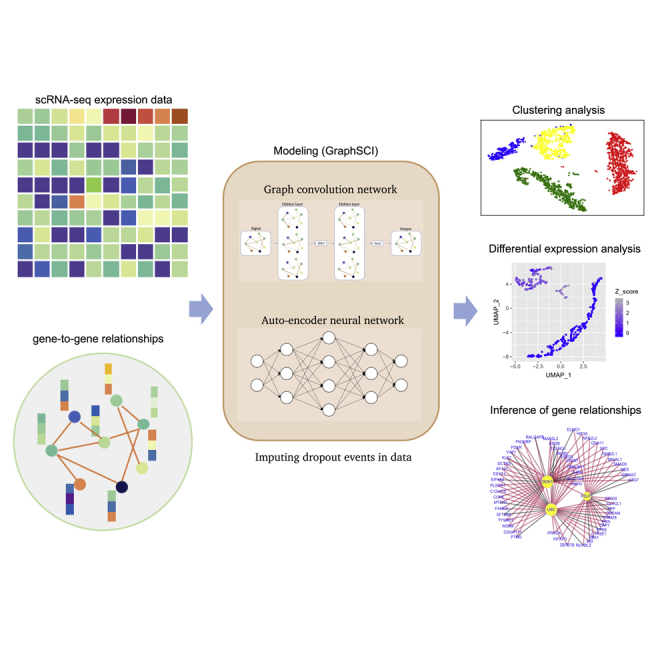
Highlights
-
•
Graph convolution network is used to impute the dropout events in scRNA-seq data
-
•
GraphSCI recovers transcriptome dynamics in scRNA-seq data sets effectively
-
•
GraphSCI improves various downstream analyses on scRNA-seq data significantly
-
•
GraphSCI is able to accurately infer gene-to-gene relationships during imputation
Genomics ; Bioinformatics ; Data Acquisition in Bioinformatics ; Artificial Intelligence
Introduction
Compared to bulk cell RNA sequencing (Wang et al., 2009) (RNA-seq), single-cell RNA sequencing technology (Kolodziejczyk et al., 2015) (scRNA-seq) has greatly promoted the profiling of transcriptomes at single-cell level and helped researchers to improve understanding of complex biological questions. It allows people to study cell-to-cell variability at a much higher throughput and resolution, such as studies of cell heterogeneity, differentiation and developmental trajectories (Saliba et al., 2014).
Despite its improvements, various technical deviations occurred due to the upgrade of sequencing techniques from bulk samples to single cells. Typically, the low RNA capture rate and sequencing efficiency lead to a large proportion of expressed genes with false zero counts in some cells, defined as ‘dropout’ event (Svensson et al., 2017; Kharchenko et al., 2014). For example, protocols based on droplet microfluidics (Zilionis et al., 2017) and Fluidigm C1 platform usually have a high dropout rate in the scRNA-seq data due to their technical limitations. And new droplet-based protocols, such as inDrop (Klein et al., 2015) and 10X Genomics (Zheng et al., 2017), have improved the detection rates but still have relatively low sensitivity, leading to the dropout events. On the other hand, although many of the zero counts represent the true absence of gene expression in specific cells, a considerable fraction is due to the dropout phenomenon where a truly expressed gene is undetected in some cells, resulting in zero or low read counts. Therefore, it is important to note the distinction between the truly expressed zeros and the false zeros in statistical analysis. Not all zeros can be considered as the missing values to be imputed. Imputation methods should impute the non-zero space but preserve the true-zero expression.
As a result, methods such as MAGIC (Van Dijk et al., 2018), SAVER (Huang et al., 2018), scImpute (Li and Li, 2018), scVI (Lopez et al., 2018), DCA (Eraslan et al., 2019), and DeepImpute (Arisdakessian et al., 2019) have been developed to correct the false zero read counts in order to recover true expression levels in scRNA-seq data. These approaches estimate “corrected” gene expressions by borrowing information across similar genes or cells. For example, MAGIC imputes gene expression data for each gene across similar cells based on Markov transition matrix, while SAVER takes advantage of gene-to-gene relationships by using Bayesian approach to infer the denoised expression. Both MAGIC and SAVER would recover the expression level of each gene in each cell including those unaffected by dropout events. ScImpute, on the other hand, determines the dropout entries based on a mixture model and imputes only the likely dropout entries across similar cells. However, MAGIC and SAVER fail to learn the non-linear relationships and the counting structures in the scRNA-seq data. Thus, with the development of deep learning, neural network-based imputation methods have been proposed such as SAVER-X (Wang et al., 2019), DCA and DeepImpute. By combining a deep autoencoder with a Bayesian model, SAVER-X extracts transferable gene−gene relationships to impute scRNA-seq data sets. DCA proposes an imputation method based on an Autoencoder, a kind of deep neural networks used to reconstruct data in an unsupervised manner. DeepImpute, in another way, constructs multiple sub-neural networks to impute genes in a divide-and-conquer approach, which utilizes dropout layers and loss functions to learn patterns in the data. In the imputation of each cell, DCA minimizes the zero-inflated negative binomial (ZINB) (Risso et al., 2018) model-based loss function to learn gene-specific distribution in scRNA-seq data.
However, these existing imputation methods for scRNA-seq aim at learning the similarity of cells or genes but not considering gene-to-gene relationships and cell-to-cell correlations simultaneously, resulting in the fact that they cannot retain biological variation across cells or genes. And decades of molecular biology research have taught us much about the principles of gene interaction and their influence on gene expression (Bhardwaj and Lu, 2005; Fraser et al., 2004). For example, the gene is truly not expressed due to gene regulation, but imputed by similar cells, which makes it difficult to study cell-to-cell variation and downstream analysis. This means that our imputation method not only needs to take advantage of the information between similar cells but also gene-to-gene relationships. More importantly, as imputation proceeds, the imputed gene expression matrix could infer more accurate gene-to-gene relationships while the inferred gene-to-gene relationship helps improve the accuracy of imputation. Therefore, our imputation method needs to be able to dynamically integrate the imputation of gene expressions and inference of the gene-to-gene relationships during the training process.
Accordingly, in this paper, we developed a Single-Cell Imputation method that combines Graph convolution network (GCN) and Autoencoder neural networks, called GraphSCI, to impute the dropout events in scRNA-seq by systematically integrating the gene expression with gene-to-gene relationships. We will use gene-to-gene relationships as prior knowledge to recover gene expression in a single cell because gene-to-gene interactions are likely to affect gene expression sensitively. And the combination of GCN and autoencoder neural networks makes it possible for us to dynamically utilize the increasingly accurate gene-to-gene relationships to impute gene expressions. By stacking the GCN and autoencoder network, GraphSCI is capable of exploring the gene-to-gene relationships in an explicit way, so as to impute the dropout events effectively. Furthermore, the deep generative model with gene-specific distribution such as ZINB and NB distribution could learn the true data distribution of scRNA-seq data and then impute the dropout events and avoid overfitting.
The gene-to-gene relationships can be regarded as a gene graph, in which the gene is the node and the edge is the relationship. As a consequence, the imputation task of gene expression can be converted into the node recovering problem on graphs. GCN (Kipf and Welling, 2017) is a very powerful neural network architecture for machine learning on graphs. It was designed to learn hidden layer representations that encode both local graph structure and features of nodes and edges. A number of recent studies describe applications of GCN such as node recovering problem (Meng et al., 2019; Gong et al., 2014; Chakrabarti et al., 2014; Yang et al., 2017). Inspired by the co-embedding attributed network (Meng et al., 2019), we combine GCN and autoencoder neural network to systematically learn the low-dimensional embedded representations of genes and cells. GCN exploits the spatial feature of gene-to-gene relationships effectively while Autoencoder neural network learns the non-linear relationships of cells and counting structures of scRNA-seq data, and thus the deep learning framework reconstructs gene expressions by integrating gene expressions and gene-to-gene relationships dynamically in the backward propagation of neural networks.
Our proposed method was shown to outperform competing methods over both simulated and real data sets by diverse downstream analyses. To assess the performance of the imputation methods, we evaluate their improvement on several downstream analyses. Firstly, we perform cell clustering and use clustering metrics to demonstrate their effectiveness to impute the dropout events. And then we also perform the differential expression analysis to evaluate their improvement of the identification of differentially expressed genes (DEGs). The evaluation performance illustrates the rationality and effectiveness of our proposed method. Furthermore, our method takes advantage of the gene-to-gene relationships in the framework that infers new reliable relationships simultaneously. Altogether, we demonstrate that our proposed method is highly scalable and parallelizable via graphical processing units (GPUs).
Results
Overview of the GraphSCI algorithm
GraphSCI is a deep neural network model that combines the GCN and autoencoder neural network to impute gene expression levels in scRNA-seq data. The overview of our method is shown in Figure 1 and the detailed model architecture is shown in Figure 2.
Figure 1.

The overview of GraphSCI algorithm
The input of GraphSCI framework is a gene expression matrix from scRNA-seq, and we construct the gene graph from the raw expression data through PCC. And GraphSCI combines the graph convolution network and autoencoder neural network to impute the dropout events in data. Finally, Extensive downstream analysis experiments demonstrated the effective and robustness of GraphSCI.
Figure 2.

The architecture of GraphSCI model
The input of GraphSCI is the gene expression matrix and the gene-to-gene relationships. The Inference model is to learn the low-dimensional representations of genes and cells based on a combination of graph convolution network and Autoencoder neural network. The Generative model utilizes the posterior distributions to reconstruct gene expression and gene-to-gene relationships respectively.
Usually, the expressions of genes are correlated by their related genes or interacting genes because the co-expressed genes are controlled by the same transcriptional regulatory program, functionally related, or members of the same pathway or protein complex (Weirauch, 2011). Therefore, given the log-normalized expression data , we first construct gene co-expression networks, called Gene Graph, from the raw expression data through the Pearson correlation coefficient (PCC). When the PCC between two genes is greater than 0.3 or less than −0.3, we assume that the two genes are co-expressed and there is an initial edge between them in the gene network. Obviously, our initial gene co-expression networks have a high rate of false positives because of dropout events in scRNA-seq data. We therefore combine the GCN and autoencoder neural network (AE) to dynamically integrate the imputation of gene expression and the inference of the gene co-expressed network where GCN encodes the gene co-expressed network with expression levels to the latent vector and then reconstructs the edges in gene co-expression network. AE encodes the gene expression matrix with gene co-expression network and finally sample from ZINB or NB distributions to reconstruct gene expression matrix.
This model enables us to utilize gene-to-gene relationships to impute the dropout events and further refine the gene co-expression network. The gene-to-gene network is an undirected graph, where each node corresponds to a gene and each edge between two genes indicates there is a significant co-expression relationship between them (Stuart et al., 2003). And the excellent characteristics of GCN allow us to regard the gene expression levels in different samples as node (gene) features in gene co-expression network and utilize them in the learning of gene network.
GraphSCI identifies cell types in simulated data
In order to assess our method, we followed the same way as the previous study (Eraslan et al., 2019) to construct two simulated data sets by Splatter (Zappia et al., 2017) package: (1) 2000 cells belonging to two types clustered by expression data of 3000 genes (namely SIM-T2) and (2) 3000 cells belonging to six types of cells clustered by expression data of 5000 genes (namely SIM-T6). On the SIM-T2 data with a simpler case, GraphSCI achieved imputed expression with a mean absolute error of 0.226, which is 21.2% lower than 0.274 by DCA. We further projected the imputed gene expression by t-SNE and clustered the cells by K-Means algorithm. As shown in Figure 3A, GraphSCI achieved 0.977, 0.994, 0.609 for ARI, CA, and SC values with standard deviation of 0.0038, 0.0043, 0.0024, respectively. These results are better than 0.920, 0.922 and 0.581 achieved by DeepImpute. Results with DCA, SAVER-X, scImpute are detailed in Table S1. By comparison, the clustering over original expression data without any imputation achieved 0.716, 0.508, 0.342 for ARI, CA and SC. Figure 3B shows 2000 cells by using the first two principle components obtained from t-SNE (Maaten and Hinton, 2008). Obviously, GraphSCI clearly separates two types of cells, while both DeepImpute and DCA have a small number of cells mixed together. The original data can't separate the cells at all.
Figure 3.

GraphSCI identifies cell types in simulated data with two cell groups (SIM-T2)
(A) The comparison of clustering performances of scRNA-seq, scImpute, SAVER, DCA, DeepImpute, and GraphSCI, measured by ARI, CA, and SC.
(B) The two principle components by t-SNE from simulated scRNA-seq data, imputed matrix by scImpute, SAVER, DCA, DeepImpute, GraphSCI. Each cell is colored by cell groups.
When tested on the SIM-T6 data set with six cell groups, similar results were obtained. As shown in Figure S1A, our method achieved 0.818, 0.859, and 0.34 for ARI, CA, and SC values, respectively. These are 5.1, 3.2, and 16.4% higher than those by DeepImpute, and 6.6%, 2.0%, and 19.7% higher than DCA. Table S1 details results by the raw data, SAVER-X and scImpute. The visualization consistently indicated that our method separates the six types of cells better than other methods (Figure S1B). Figure S2 shows the image of gene expression matrix (X) before and after imputation () in our simulated experiments. This comparison again demonstrates that GraphSCI could recover the original cell types effectively both in the Sim-T2 and the Sim-T6 data sets.
GraphSCI recovers transcriptome dynamics in real single-cell data
Another key criterion to evaluate the imputation methods is their ability to recover transcriptome dynamics in real single-cell data set. Therefore, we applied our method to three real scRNA-seq data sets and made comparisons with other methods. The first data set was obtained from mouse ES cells(Klein et al., 2015), which were measured to analyze the heterogeneity of mouse embryonic stem cells in different stages after leukemia inhibitory factor (LIF) withdrawal. We selected four different LIF withdrawal intervals (0, 2, 4, and 7 days) and put all cells together as the input of imputation. The imputed data were clustered by t-SNE. As shown by Figure 4A, GraphSCI separated the four stages of mouse ES cells clearly except that a few blue samples were mixed with the yellow. In comparison, the clustering obtained from the scImpute and DCA methods seriously mixed the blue samples with the yellow ones. As indicated by Figure 4B, ARI, CA, and SC of GraphSCI were significantly higher than DeepImpute and DCA. Results of ARI, CA, and SC with all methods are detailed in Table S2.
Figure 4.

The performances on Mouse embryonic stem cells data set
(A) shows the t-SNE visualization reproduced from scRNA-seq, scImpute, SAVER, DCA, DeepImpute, and GraphSCI from top to bottom, from left to right.
(B) The comparison of clustering performances of scRNA-seq, scImpute, SAVER, DCA, DeepImpute, and GraphSCI, measured by ARI, CA, and SC.
GraphSCI was further applied to two large data sets generated by the 10X scRNA-seq platform (Zheng et al., 2017), one of which is involved by the transcriptome of peripheral blood mononuclear cells (PBMCs) from a healthy donor. The data set contains 5247 PBMCs of 11 cell types. Because the same type of cells has similar expression profiles, we randomly selected 80% of PBMCs to train the model and used the remained for the independent test set. The imputed data on the independent set was conducted with dimension reduction results by t-SNE for visualizations. Figure 5A shows that the imputations by GraphSCI could separate brown and dark samples well in the low-dimension representation. The orange samples had a diving line with other samples. In comparison, the results obtained by DeepImpute and DCA didn't show obvious differences among the black, red, and green samples. The results of ARI, CA, and SC on the independent test set also showed that GraphSCI outperformed other methods (Figure 5B). In details, our method achieved 0.472, 0.552, and 0.177 for ARI, CA, and SC values, respectively. These are 1.7%, 0.7%, 4.7% and 14.0, 9.7, 34.1% greater than those by DeepImpute and DCA respectively. More detailed results are shown in Table S2.
Figure 5.

The performances on 5k peripheral blood mononuclear cells (PBMC) data set
(A) shows the t-SNE visualization reproduced from scRNA-seq, scImpute, SAVER, DCA, DeepImpute, and GraphSCI from top to bottom, from left to right.
(B) The comparison of clustering performances of scRNA-seq, scImpute, SAVER, DCA, DeepImpute, and GraphSCI, measured by ARI, CA, and SC.
In the E18 Mouse data set, ∼12,000 brain cells of 16 cell types were profiled from the 10X scRNA-seq platform. We applied GraphSCI to the large-scale scRNA-seq data set to demonstrate its robustness and scalability. As shown in Figure S3A, GraphSCI is able to separate cells of 16 cell types effectively in the low-dimension representation, while DeepImpute and DCA have mixed many subcellular types together. GraphSCI again achieved ARI, CA, and SC of 0.316, 0.422, and 0.030, respectively, consistently the greatest among all methods (Figure S3B and Table S2).
GraphSCI recovers gene expression levels in bulk RNA-seq data set
The efficacies of GraphSCI in recovering gene expression levels were further evaluated by a real RNA sequencing data set. The RNA sequencing data was obtained from C. elegans development experiments by Francesconi et al. (Francesconi and Lehner, 2014), which was used to simulate single-cell RNA-seq data with dropout rates ranging from 50% to 70%. The three data sets were generated by adding the single-cell specific noises through gene-wise subtracting values drawn from the exponential distribution. Since bulk RNA-seq data contains less noise than scRNA-seq, PCC was used to evaluate the effectiveness of imputation on real RNA-seq data set. As shown by Figure 6, GraphSCI outperformed DCA in recovering the gene expression levels in real RNA-seq data set. In details, the median of PCCs reached by GraphSCI in three data sets are 0.858, 0.807, and 0.788 respectively, consistently greater than 0.821, 0.765, and 0.742 achieved by DCA, and 0.787, 0.718, and 0.604 achieved by SAVER.
Figure 6.

GraphSCI recovers gene expression levels in bulk RNA-seq data
Box diagram (A–C) depict the Pearson correlation coefficient between simulated data or imputed data and original data. And the box represents the interquartile range, the horizontal line in the box is the median, and the whiskers represent 1.5 times the interquartile range.
GraphSCI improves differential expression analysis on scRNA-seq data
An effective imputation method should lead to an improvement in differential expression analysis because scRNA-seq data provide insight into gene expression in a single cell. To evaluate whether the identification of DEGs are more accurate after imputation, we utilized a scRNA-seq data set with corresponding bulk RNA-seq data to compared differential expression analysis results using DESeq2. This data set, generated by Chu et al. from H1 human embryonic stem cells (H1) differentiated into definitive endoderm cells (DEC), has six samples of bulk RNA-seq and 350 samples of scRNA-seq (212 for H1 ESC and 138 for DEC). We applied GraphSCI and DeepImpute to impute the gene expression on scRNA-seq data and performed DE analysis on the raw data and the imputed data respectively. The percentages of zero gene expression are 49.1% in raw scRNA-seq data that results in the lowest DEGs identification results. In contrast, GraphSCI and DeepImpute have improved the identification of DEGs and share more DEGs with bulk samples. In Figure 7A, we defined more quantitative evaluation metrics such as the area under the receiver operating characteristic curve (AUC), the accuracy (ACC), and F-scores for DEGs detection. In detail, our method achieved 0.913, 0.782, and 0.608 for AUC, ACC, and F-score values, respectively. Moreover, Figure 7B and Figure 7C show that the expression profiles of DEC and ESC marker genes (SOX2 and LEFTY1) after GraphSCI imputation could better reflect the gene expression signatures on recovering the expression patterns of signature genes. The performance of other signature genes including NANOG, DNMT3B, GATA6, and CXCR4 et al. has been shown in Figure S4.
Figure 7.

GraphSCI improves g differential expression analysis
(A) The bar plots show the performances of DEG detections from raw and imputed scRNA-seq data sets based on the gold standard defined by the bulk RNA-seq data set.
(B) The expression for selected signature genes (LEFTY1, SOX2) of H1 and DEC cells, respectively.
(C) The UMAP plots of the single cells overlaid by the expression of SOX2 and LEFTY1, which is the marker gene of H1 and DEC cells, respectively.
GraphSCI infers gene-to-gene relationships from scRNA-seq data
GraphSCI can not only impute gene expression data of scRNA-seq effectively, but also infer gene-to-gene relationships from the data. Due to the dropout events in raw scRNA-seq data (Zheng et al., 2017; Iacono et al., 2019), it is challenging to obtain accurate gene interactions directly from correlation coefficients between gene expression (Aibar et al., 2017). Here, we applied our method to raw scRNA-seq data sets and reconstructed gene relations during imputation. By compared with the known interactions from the STRINGdb (Szklarczyk et al., 2017), the gene interactions constructed by GraphSCI had a precision of 0.713 with the threshold of 0.5. Specifically, the true positive (TP) was 232647 and the false positive (FP) was 93,812. Figure 8 shows the imputed gene-to-gene relationships obtained by Cytoscape (Smoot et al., 2010). The true positive (TP) is an outcome where the model correctly predicts the gene-gene relationships and the false positive is an outcome where the model incorrectly predicts the gene-gene relationships. They showed the accuracy of our model to infer the gene-gene relationships from the raw scRNA-seq data. Similar results were also observed in previous experiments on the mouse ES cells data set. The constructed gene relations had a precision of 0.682 with 199291 true positives and 92,924 false positives. As a comparison, we utilized the PCC to infer gene-gene relationships from the raw scRNA-seq data, which obtains the precision of 0.598 and 0.492 respectively. It again verifies the effectiveness of our method, empirically showing that it facilitates the inference of the gene-to-gene relationships during the training process.
Figure 8.

The gene-to-gene relationships after reconstruction
We selected the three genes with the highest degree and their common interactive genes. Compared to STRINGdb, the edges colored by red represent the correct gene-to-gene relationships we inferred, and the black edges represent the false inferred relationships.
Discussion
In this study, we presented an imputation method, GraphSCI, based on GCNs, which are particularly suitable for single-cell RNA-seq data. Our method focused on imputing gene expression levels by integrating the gene expression with gene-to-gene relationships. By using gene-to-gene relationships as prior knowledge, this method avoided introducing excess biases during imputation and removed technical variations resulted from scRNA-seq.
To our best knowledge, this is the first study to integrate gene-to-gene relationships into deep learning framework for imputations on scRNA-seq. It is also the first attempt to employ GCN for learning the representation of gene-to-gene relationships in imputation study. Most importantly, extensive experiments were conducted on different kind of scRNA-seq data sets to demonstrate the superiority of our method.
GraphSCI was evaluated on both simulated and real data, which was identified with the best performances on diverse downstream analyses in comparison with other methods. In simulated data sets, GraphSCI was found to outperform other methods on the data with both small and large numbers of cells and cell types. The better performance of GraphSCI was further observed when it was applied in real data sets like bulk RNA-seq data and scRNA-seq data. In addition, another advantage of GraphSCI is its ability to infer the new gene-to-gene relationships, which is an absence of existing methods.
Applications of GCNs and exploiting gene-to-gene relationships for imputation, however, may also bring uncontrollable errors. For instance, the reliability of gene-to-gene relationships may influence the results of imputation. To solve this problem, we tried a variety of methods to build the gene-to-gene relationships, such as setting different thresholds to build edges or selecting original co-expressed samples to calculate PCC. We found that better performance could be achieved with the adjacency matrix obtained by selecting original co-expressed samples and PCC of >0.3 to determine edges. Figures S5 and S6 show the influence of the input gene-to-gene relationships on the overall results.
Another challenge for real data is that the evaluation of imputation may be difficult due to lack of ground truth. Therefore, we performed many clustering metrics, such as ARI, CA, and SC, to describe the effectiveness and robustness of competing methods, while we also utilized visualization to make the results clearer and more convincing. As shown in Figure S7, we could find that t-SNE showed better display results and GraphSCI consistently yields better performance with different clustering approaches.
The current GraphSCI was tested on data sets including simulated data and real data. The imputation power could be further improved with the increasing number of cells in the training set. Additionally, the deep learning networks by GraphSCI enable parallelization using GPUs to speed up training on large scRNA-seq data sets (Figure S8).
Resource availability
Lead contact
Yuedong Yang (yangyd25@mail.sysu.edu.cn) is the lead contact for this work.
Materials availability
This study does not generate any new materials.
Data and code availability
The scRNA-seq data sets used in this manuscript are publicly available and their details are summarized in Table S3. The C. elegans time course experimental data was provided by the supplementary material of Francesconi. et al. The mouse embryonic stem cells data was downloaded from GSE65525. The 5k PBMC from a healthy donor and 10K brain cells from an E18 Mouse were provided by the 10X scRNA-seq platform and the website of the data are https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.1.0/5k_pbmc_protein_v3 and https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/neuron_10k_v3. The human embryos cells scRNA-seq data was downloaded from GSE44183. The Human ESC scRNA-seq data set for differential expression analysis was downloaded from GSE75748. The code generated during this study is available at https://github.com/biomed-AI/GraphSCI. We tuned model hyper-parameters based on the experimental results on simulated data sets and used them across all data sets (Figures S9 and S10).
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
This study has been supported by the National Key R&D Program of China (2020YFB0204803), National Natural Science Foundation of China (61772566), Guangdong Key Field R&D Plan (2019B020228001 and 2018B010109006), Introducing Innovative and Entrepreneurial Teams (2016ZT06D211), Guangzhou S&T Research Plan (202007030010).
Author contributions
J.Rao, X.Zhou, and Y.Yang contributed concept and implementation. J.Rao and Y.Yang co-designed experiments. J.Rao was responsible for programming. All of the authors contributed to the interpretation of results. J.Rao and Y.Yang wrote the manuscript. All of the authors reviewed and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102393.
Supplemental information
References
- Aibar S., González-Blas C.B., Moerman T., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisdakessian C., Poirion O., Yunits B., Zhu X., Garmire L.X. DeepImpute: an accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol. 2019;20:1–14. doi: 10.1186/s13059-019-1837-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N., Lu H. Correlation between gene expression profiles and protein–protein interactions within and across genomes. Bioinformatics. 2005;21:2730–2738. doi: 10.1093/bioinformatics/bti398. [DOI] [PubMed] [Google Scholar]
- Chakrabarti D., Funiak S., Chang J., Macskassy S.A. Joint inference of multiple label types in large networks. ICML’14: Proceedings of the 31st International Conference on International Conference on Machine Learning. 2014;32:874–882. [Google Scholar]
- Eraslan G., Simon L.M., Mircea M., Mueller N.S., Theis F.J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019;10:390. doi: 10.1038/s41467-018-07931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi M., Lehner B. The effects of genetic variation on gene expression dynamics during development. Nature. 2014;505:208. doi: 10.1038/nature12772. [DOI] [PubMed] [Google Scholar]
- Fraser H.B., Hirsh A.E., Wall D.P., Eisen M.B. Coevolution of gene expression among interacting proteins. Proc. Natl. Acad. Sci. 2004;101:9033–9038. doi: 10.1073/pnas.0402591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong N.Z., Talwalkar A., Mackey L., Huang L., Shin E.C.R., Stefanov E., Shi E., Song D. Joint link prediction and attribute inference using a social-attribute network. ACM Trans. Intell. Syst. Technol. (Tist) 2014;5:1–20. [Google Scholar]
- Huang M., Wang J., Torre E., Dueck H., Shaffer S., Bonasio R., Murray J.I., Raj A., Li M., Zhang N.R. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods. 2018;15:539. doi: 10.1038/s41592-018-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono G., Massoni-Badosa R., Heyn H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 2019;20:110. doi: 10.1186/s13059-019-1713-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf T.N., Welling M. Semi-supervised classification with graph convolutional networks. 5th International Conference on Learning Representations (ICLR-17) 2017 [Google Scholar]
- Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk A.A., Kim J.K., Svensson V., Marioni J.C., Teichmann S.A. The technology and biology of single-cell RNA sequencing. Mol. Cel. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Li W.V., Li J.J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 2018;9:997. doi: 10.1038/s41467-018-03405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018;15:1053–1058. doi: 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaten L.v. d., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Meng, Z., Liang, S., Bao, H. & Zhang, X. Co-embedding attributed networks. Proceedings of the Twelfth ACM International Conference on Web Search and Data Mining, 2019. 393-401.
- Risso D., Perraudeau F., Gribkova S., Dudoit S., Vert J.-P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 2018;9:284. doi: 10.1038/s41467-017-02554-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba A.-E., Westermann A.J., Gorski S.A., Vogel J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. doi: 10.1093/nar/gku555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.-L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2010;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart J.M., Segal E., Koller D., Kim S.K.J.s. A gene-coexpression network for global discovery of conserved genetic modules. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Svensson V., Natarajan K.N., Ly L.-H., Miragaia R.J., Labalette C., Macaulay I.C., Cvejic A., Teichmann S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods. 2017;14:381. doi: 10.1038/nmeth.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A.J., Burdziak C., Moon K.R., Chaffer C.L., Pattabiraman D. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174:716–729.e27. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Agarwal D., Huang M., Hu G., Zhou Z., Ye C., Zhang N.R. Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods. 2019;16:875–878. doi: 10.1038/s41592-019-0537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch M.T. Vol. 1. 2011. pp. 215–250. (Gene Coexpression Networks for the Analysis of DNA Microarray Data). [Google Scholar]
- Yang, C., Zhong, L., Li, L.-J. & Jie, L. Bi-directional joint inference for user links and attributes on large social graphs. Proceedings of the 26th International Conference on World Wide Web Companion, 2017. 564-573.
- Zappia L., Phipson B., Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 2017;18:174. doi: 10.1186/s13059-017-1305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G.X., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R., Nainys J., Veres A., Savova V., Zemmour D., Klein A.M., Mazutis L. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc. 2017;12:44. doi: 10.1038/nprot.2016.154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data sets used in this manuscript are publicly available and their details are summarized in Table S3. The C. elegans time course experimental data was provided by the supplementary material of Francesconi. et al. The mouse embryonic stem cells data was downloaded from GSE65525. The 5k PBMC from a healthy donor and 10K brain cells from an E18 Mouse were provided by the 10X scRNA-seq platform and the website of the data are https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.1.0/5k_pbmc_protein_v3 and https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/neuron_10k_v3. The human embryos cells scRNA-seq data was downloaded from GSE44183. The Human ESC scRNA-seq data set for differential expression analysis was downloaded from GSE75748. The code generated during this study is available at https://github.com/biomed-AI/GraphSCI. We tuned model hyper-parameters based on the experimental results on simulated data sets and used them across all data sets (Figures S9 and S10).