

Abstract
Most genetic alterations that drive melanoma development and resistance to targeted therapy have been uncovered. In contrast, and despite their increasingly recognized contribution, little is known about the non‐genetic mechanisms that drive these processes. Here, we performed in vivo gain‐of‐function CRISPR screens and identified SMAD3, BIRC3, and SLC9A5 as key actors of BRAFi resistance. We show that their expression levels increase during acquisition of BRAFi resistance and remain high in persister cells and during relapse. The upregulation of the SMAD3 transcriptional activity (SMAD3‐signature) promotes a mesenchymal‐like phenotype and BRAFi resistance by acting as an upstream transcriptional regulator of potent BRAFi‐resistance genes such as EGFR and AXL. This SMAD3‐signature predicts resistance to both current melanoma therapies in different cohorts. Critically, chemical inhibition of SMAD3 may constitute amenable target for melanoma since it efficiently abrogates persister cells survival. Interestingly, decrease of SMAD3 activity can also be reached by inhibiting the Aryl hydrocarbon Receptor (AhR), another druggable transcription factor governing SMAD3 expression level. Our work highlights novel drug vulnerabilities that can be exploited to develop long‐lasting antimelanoma therapies.
Keywords: Aryl hydrocarbon Receptor, CRISPR‐SAM, melanoma, SMAD3, targeted therapy resistance
Subject Categories: Cancer, Skin
Using a CRISPR activation screening, we identified genes involved in BRAF inhibitor (BRAFi) resistance in cutaneous melanoma. Their upregulation promoted tumour growth of therapy‐naïve melanoma cells and BRAFi‐resistance. Inhibition of these genes (not mutated) may be useful for therapy.

The paper explained.
Problem
Precision medicine has greatly improved the survival of patients with cutaneous melanoma, but relapse limits the long‐lasting responses. Relapse is explained by the capability of persister cells to survive despite targeted therapies; however, the molecular mechanism conferring resistance is not fully understood
Results
We performed a genetic screen (CRISPR activation) to identify genes conferring resistance to BRAF inhibitors, promoting survival of persister cells and relapse. The current study finds that genes that enable tumor growth of therapy‐naïve melanoma cells are also enriched during acquisition of resistance to BRAFi. SMAD3 targeted genes (SMAD3‐signature) could be useful to find subpopulations of pre‐existing BRAFi‐resistant cells within therapy‐naïve lesions. Moreover, chemical inhibition of SMAD3 decreases persister cells population.
Impact
We established in a clinical context that a high expression level of several tumor‐promoting genes fuels cancer cell plasticity, therapy resistance, and relapse. By highlighting the role of tumor‐promoting genes, this work expands our understanding of the biology underlying tumor growth. Moreover, we identify novel drug vulnerabilities that can be exploited to develop long‐lasting anticancer therapies.
Introduction
Identifying molecular cancer drivers is critical for precision oncology. Last year, the cancer genome atlas (TCGA) identified 299 driver genes by focusing on point mutations and small indels across 33 cancer types (Bailey et al, 2018). It represents the most comprehensive effort thus far to identify cancer driver mutations. Complementary studies are required to elucidate the role of copy‐number variations, genomic fusions, and methylation events in the 33 TCGA projects.
Moreover, there is increasing evidence that non‐genetic reprogramming leading to cancer cell dedifferentiation, stemness, invasiveness also contribute to tumor growth and therapy resistance (Puisieux et al, 2014; Bai et al, 2019). Thus, deciphering the signaling pathways that drive such processes may also lead to innovative cancer therapies. Recent gene expression quantifications performed at single‐cell level by single‐cell RNA sequencing (scRNA‐Seq) demonstrated that cancer cells operate a dedifferentiation process, for instance in glioblastoma and melanoma (Patel et al, 2014; Tirosh et al, 2016; Rambow et al, 2018), promoting tumor growth, stemness, and therapy resistance. This “onco‐dedifferentiation” seems to be independent of de novo mutations and could offer new targets/strategies to cure cancer. However, these scRNA‐Seq studies are mainly descriptive; the tumor growth capability of each gene/RNA is not yet investigated at the genome‐scale. Such functional analyses are nowadays feasible using clustered regularly interspaced short palindromic repeats (CRISPR)‐Cas9 screens (Shalem et al, 2015). The majority of the CRISPR‐Cas9 screens is based on the invalidation of coding genes but modulation of gene expression is reachable with the CRISPR‐Cas9 synergistic activation mediator (SAM) approach (Konermann et al, 2015). It corresponds to an engineered protein complex for the transcriptional activation of endogenous genes. Importantly, SAM can further be combined with a human genome‐wide library to activate all known coding isoforms from the RefSeq database (23,430 isoforms) for gain‐of‐function screening without a priori. To date, CRISPR screens are mainly performed in vitro using cell lines or primary cultures (Meyers et al, 2017). A pan‐cancer CRISPR‐Cas9 knockout screen was performed in vitro (324 human cancer cell lines from 30 cancer types) to identify essential genes for cancer cell fitness (defined as genes required for cell growth or viability) and to prioritize candidates for cancer therapeutics (Behan et al, 2019). However, because the contribution of the tumor environment in tumor growth is increasingly recognized, it seems important to perform such screens in the relevant patho‐physiological context and, for instance, take advantage of animal models.
We selected cutaneous melanoma as a paradigm since novel therapeutic strategies are critically needed (Bai et al, 2019). Targeted therapies such as BRAF inhibitors (BRAFi) initially showed great promise in patients with BRAF(V600)‐mutated metastatic melanoma. Unfortunately, the vast majority of patients that initially respond to these drugs, almost inevitably develop resistance. Although combination therapies (BRAF and MEK inhibitors) enhance the response and delay relapse, the overall survival remains unsatisfactory highlighting the need of new therapeutic targets (Ascierto et al, 2016).
The mechanisms underlying resistance are numerous and probably not mutually exclusive (Sullivan & Flaherty, 2013; Welsh et al, 2016; Song et al, 2017). Resistance can be driven by a small pre‐existing subpopulation, harboring‐specific genetic alterations that confer them with resistance to the inhibitors (Wagle et al, 2011). Such alterations may also occur de novo, during treatment (Welsh et al, 2016). In addition, there is increased evidence that non‐genetic reprogramming may confer drug‐tolerant and/or resistant phenotypes to melanoma cells (Rambow et al, 2018; Corre et al, 2018; Tsoi et al, 2018; Hugo et al, 2015a; Talebi et al, 2018; Rapino et al, 2018; preprint: Marin‐Bejar et al, 2020). Earlier works demonstrated that phenotype switching from a proliferative to an invasive/mesenchymal‐like state is also likely to contribute to therapy resistance (Hoek & Goding, 2010; Konieczkowski et al, 2014; Müller et al, 2014; Verfaillie et al, 2015; Boshuizen et al, 2018). Paradoxically, MITF‐induced differentiation into a slow cycling, pigment‐producing state was also reported to confer tolerance to BRAFi (Müller et al, 2014; Smith et al, 2016). It therefore seems that various drug‐tolerant subpopulations can emerge under therapeutic pressure and that these cells can provide a pool from which resistance develops. Targeting these populations of persister cells is therefore crucial to achieve effective personalized therapies (Nassar & Blanpain, 2016).
Here, we performed unbiased screens to identify genes promoting tumor growth from persister cells and conferring resistance to BRAFi using CRISPR‐Cas9 SAM methodology. We demonstrate that, in addition to promote melanoma development, Mothers against decapentaplegic homolog 3 (SMAD3), Baculoviral IAP repeat‐containing protein 3 (BIRC3), and Sodium/hydrogen exchanger 5 (SLC9A5) also support relapse since they promote both BRAFi‐resistance and tumor growth capability of persister cells. Their expression levels correlated with BRAFi resistance and relapse. Consequently, their inhibition strongly reduced the number of persister cells. Moreover, we demonstrate that the transcription factor AhR governs SMAD3 expression levels and in turn SMAD3 drives the expression of a set of genes associated with BRAFi resistance and mesenchymal phenotype. These experiments identify integrated AhR‐SMAD3 signaling as a key driver of melanoma growth and relapse, pointing to a new therapeutic vulnerability in melanoma.
Results
Identification of tumor‐promoting genes by in vivo gain‐of‐function CRISPR screen
Since the tumor environment influences, at least in part, the tumor growth capability of cancer cells, we performed an in vivo genome‐wide CRISPR‐Cas9 SAM screen to identify in vivo tumor‐promoting genes, defined as genes whose expression support tumor growth (in contrast to driver genes bearing a driver mutation such as BRAF (V600E)). To select the most appropriate cellular model, we classified melanoma biopsies from The Cancer Genome Atlas (TCGA) cohort (n = 458) in function of differentiation states according to the most recent melanoma profiling data (Fig 1A; Tsoi et al, 2018). As anticipated, the vast majority of these tumors, which are almost all drug naive, exhibited a differentiated profile (89%; melanocytic and transitory). We selected the 501Mel cell line since (i) these cells display a melanocytic differentiation state as the majority of diagnosticated melanoma, (ii) they harbor the BRAF(V600E) mutation as ~50% of cutaneous melanoma, (iii) they are highly sensitive to BRAFi with an IC50 value of 0.45 µM to vemurafenib [PLX4032] (Halaban et al, 2010; Corre et al, 2018), and importantly (iv) they are unable to generate tumor in nude mice (Ohanna et al, 2011). This latter characteristic may allow to identify tumor‐promoting genes.
Figure 1. Identification of tumor‐promoting genes by in vivo gain‐of‐function CRISPR screen.
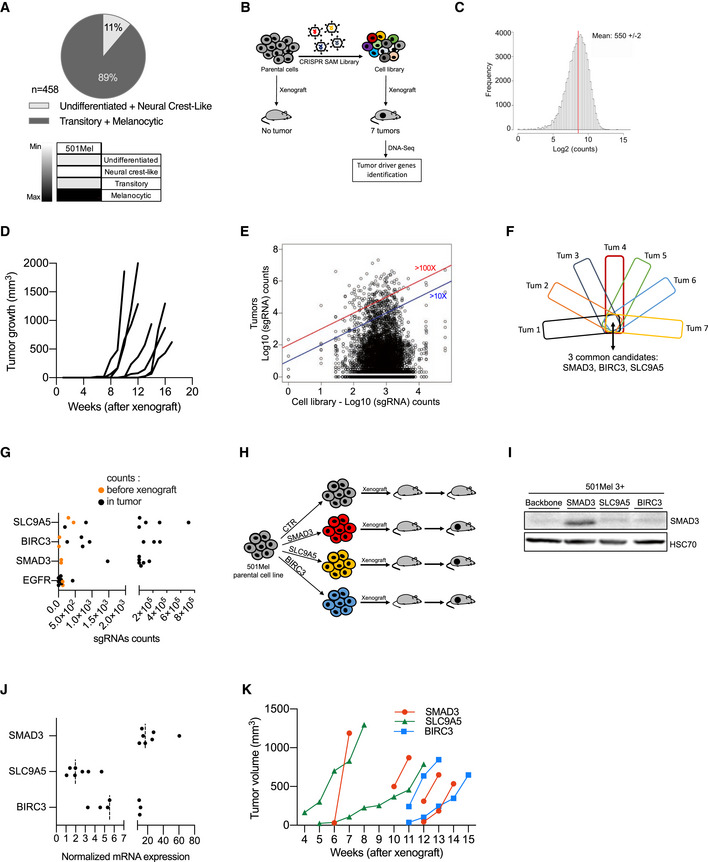
- Determination of differentiation states of skin cutaneous melanoma (SKCM) biopsies from the TCGA cohort (n = 458) according to (Tsoi et al, 2018). The vast majority of these tumors exhibited a differentiated profile (89%; melanocytic and transitory states). The others display a dedifferentiated profile (11%; neural crest‐like cells and undifferentiated states). Human melanoma 501Mel cell line is classified as differentiated melanoma cells (melanocytic cells), according to the gene expression profile (four categories were defined by (Tsoi et al, 2018); melanocytic, transitory, neural crest‐like, and undifferentiated cells). 501mel cell line was selected for the CRISPR screens.
- Workflow depicting the in vivo CRISPR‐SAM screen to identify tumor‐promoting genes. Parental cells and CRISPR‐SAM cell library were xenografted on nude mice (3 × 106 cells/mouse, n = 6 and n = 10, respectively) and tumor growth was monitored during 5 months. Seven tumors were collected and analyzed by DNA‐Seq to identify the sgRNAs (Tables EV1 and EV2).
- sgRNAs distribution in the cell library. sgRNA mean reaches 550 ± 2.
- Tumor growth curves for the 7 tumors arising from the CRISPR‐SAM‐engineered cells xenografted in nude mice as detailed in panel B. (No tumor for the parental cell line (501Mel cells)) (Table EV2).
- Distribution of sgRNAs in cell library and in the 7 tumors (log10(sgRNAs counts)). Blue and red lines indicated the enrichment ≥ 10 fold or ≥ 100 fold (tumors vs cell library (in vitro)) (Table EV1).
- From the seven tumors, the top hundred genes (enriched) have been selected and common genes are SMAD3, BIRC3, and SLC9A5 (Table EV3).
- sgRNA counts in tumors versus in CRISPR‐SAM cell library (respectively, black and orange points). Each black point corresponds to one tumor. Two replicates have been shown for CRISPR‐SAM cell library (orange points).
- Workflow depicting the validation step: 501Mel cells overexpressing SMAD3, BIRC3, or SLC9A5 (obtained by CRISPR‐SAM) were xenografted on nude mice and tumor volume was monitored using caliper. 3 × 106 cells/mouse. n = 7, 6, and 6 mice, respectively.
- SMAD3 expression levels in 501Mel cells overexpressing the cofactors for CRISPR‐SAM approach and the control sgRNA (501Mel 3 + backbone) or the SMAD3 sgRNA. SLC9A5 and BIRC3 sgRNAs are used as controls to show the specificity of the SMAD3 overexpression. HSC70 serve as loading control.
- SMAD3, BIRC3, and SLC9A5 mRNA expression levels in melanoma cell lines described in H and I. n = 7 independent biological experiments. Dashed lines for medians.
- Tumor growth curves from 501Mel cells overexpressing SMAD3, BIRC3, or SLC9A5.
Data information: Western blot results are representative of at least two independent experiments. Source data and unprocessed original blots are available in Appendix Fig S1 source data. See also Fig EV1.
Source data are available online for this figure.
To generate the CRISPR‐SAM cell library, we modified the 501Mel cells, to express constitutively defective‐Cas9 and the required cofactors for CRISPR‐SAM technology (Konermann et al, 2015). These engineered cells were infected with the single‐guide RNA (sgRNA) lentivirus library that contained at least three different guides per coding gene (Konermann et al, 2015; Fig 1B). The infection was performed at a multiplicity of infection (MOI) of 0.2 ensuring that only one guide is expressed per infected cell. Infected cells were positively selected using antibiotic selection during 7 days. By DNA sequencing, we observed a normal distribution of the sgRNAs in two cell library replicates (Fig 1C). Only 78 sgRNAs were not detected in our cell library, which validate our protocol and the cell library (>70,100 sgRNAs were detected; Table EV1). Thus, the controls were proper to identify in vivo tumor‐promoting genes.
The cell library (30 × 106 cells) was fractionated and subcutaneously xenografted in 10 nude mice (3 × 106 cells/mouse) and tumor growth was monitored using caliper over a 5‐month period (Fig 1D and Table EV2). As previously demonstrated (Ohanna et al, 2011), we confirmed that parental 501Mel are unable to form tumors in nude mice (n = 6). In contrast, seven tumors were obtained from the CRISPR‐engineered cells xenografted in 10 mice (Fig 1D). The nature of the sgRNAs, their abundance and occurrence across these 7 tumors were determined by DNA‐Seq (Fig 1E and F, and Table EV3).
By comparing the most represented genes (sgRNAs) in each tumor (Tum), we identified 3 common genes (Fig 1F). An enrichment of SMAD3, BIRC3, and SLC9A5 sgRNAs was found in tumors when compared to their starting abundance (cell library; orange points; Fig 1G), in contrast to EGFR sgRNAs. Thirty‐six other genes were recurrently retrieved in the tumors but not in all (Table EV3). Interestingly, YAP1 which has already been identified as melanoma growth‐promoting gene was also found (Table EV3). This supports the robustness of the screen (Lamar et al, 2012; Verfaillie et al, 2015; Hugo et al, 2015b). The majority of the tumor‐promoting genes (Table EV4) identified here are not considered as genes required for cell growth or viability (except the essential genes YAP1, SLC25A41, and TGIF1; Behan et al, 2019) and are not frequently altered in melanoma (Fig EV1A). Moreover, the transforming growth factor (TGF)‐β pathway seemed well‐represented among the tumor‐promoting genes (Table EV4). Since our results suggest that a high expression level of these tumor‐promoting genes is sufficient to promote melanoma tumor growth, we evaluated the biological consequences of the inhibition of one tumor‐promoting gene, BIRC3. The chemical inhibitor Birinapant reduced the SKMel28R and Me1402 cell density (Fig EV1B and C), suggesting that the BIRC3 protein is involved in cell proliferation of these melanoma cells as previously demonstrated (Krepler et al, 2013; Vetma et al, 2017).
Figure EV1. Tumor‐promoting genes are not frequently altered in cutaneous melanoma. Related to Fig 1.
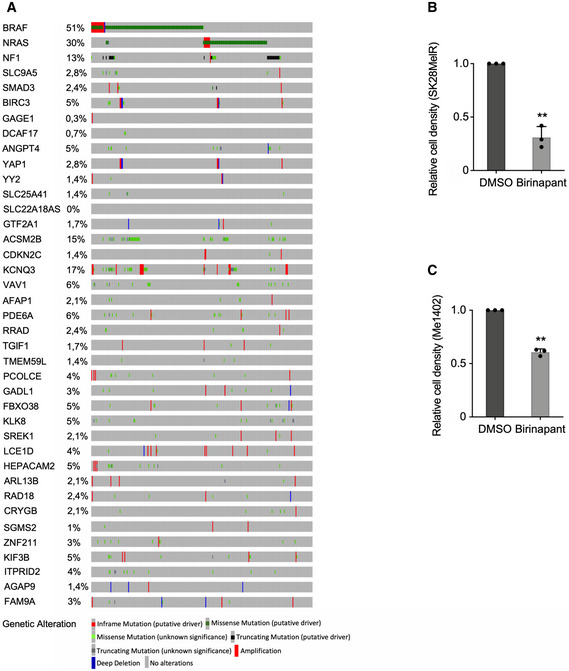
- Genomic alterations and 36 tumor‐promoting genes identified in Fig 1. BRAF, NRAS, and NF1 have been added to the list of tumor‐promoting genes according to melanoma classification (Akbani et al, 2015). Analyses were performed using the webtool available at www.cbioportal.org.
- Birinapant, an inhibitor of BIRC proteins (BIRC3 and BIRC2), decreased cell density (SKMel28R cells, Birinapant 100 nM, 96 h).
- Birinapant, an inhibitor of BIRC proteins (BIRC3 and BIRC2), decreased cell density (Me1402 cells, Birinapant 100 nM, 96 h).
Data information: Each histogram represents the mean ± s.d. from three independent experiments. Bilateral Student test (with non‐equivalent variances) **P < 0.01.
Source data are available online for this figure.
Next, we examined in vivo the ability of these tumor‐promoting genes to promote tumor growth by xenografting cell populations overexpressing the sgRNAs individually. We focused on the top 3 tumor‐promoting genes: SMAD3, BIRC3, and SCL9A5 (Fig 1H–K). We generated three new CRISPR‐engineered cell lines, and we evaluated the overexpression levels of these genes by Western blot experiments (SMAD3) and RT–qPCR (SMAD3, BIRC3, and SLC9A5) (Fig 1I and J). Finally, we confirmed that they independently foster tumor development (Fig 1K). Altogether, our results demonstrated that in vivo CRISPR‐SAM screen identifies new tumor‐promoting genes, which may constitute amenable target for melanoma.
Genome‐wide CRISPR activation screen identifies BRAFi‐resistance genes in cutaneous melanoma
BRAFi provokes tumor shrinkage in the vast majority of patients with BRAF(V600)‐mutated metastatic melanoma but resistance almost inevitably occurs (Bai et al, 2019). To examine the genes promoting BRAFi resistance and relapse, we performed an in cellulo screen using the same cell library in the presence of BRAFi (Fig 2). Briefly, the CRISPR‐SAM 501Mel cell library (40 × 106 cells) was treated for 14 days with BRAFi (2 µM), using either the BRAFi used in clinical practice (vemurafenib), the next‐generation inhibitor that is still under investigation in clinical trials (PLX8394), or the solvent (dimethyl sulfoxide (DMSO)) as control (Fig 2A). This procedure allows for the enrichment of sgRNAs (genes) conferring resistance. The nature of the sgRNA present in the resistant population and their abundance was determined by DNA‐Seq (Fig 2B). The best hit was the Epidermal growth factor receptor gene (EGFR), a well‐known BRAFi‐resistance gene (Sun et al, 2014; Shaffer et al, 2017). By examining the enrichment of sgRNAs targeting EGFR promoter (Fig 2B), we decided to retain genes with at least two sgRNAs among the enriched sgRNAs present in BRAFi‐exposed cells (with a false discovery rate, FDR < 0.05) (Tables EV5 and EV6, Appendix Fig S1) since sometimes one of the three sgRNAs designed per gene is not detected or not enriched as observed for EGFR and BIRC3 (Fig 2C). A recent publication confirmed that sgRNAs are not all functional in CRISPRa libraries and it could be interesting to increase the number of sgRNAs per target and to cover more TSS per gene (Sanson et al, 2018).
Figure 2. Genome‐wide CRISPR activation screen identifies BRAFi‐resistance genes in cutaneous melanoma.
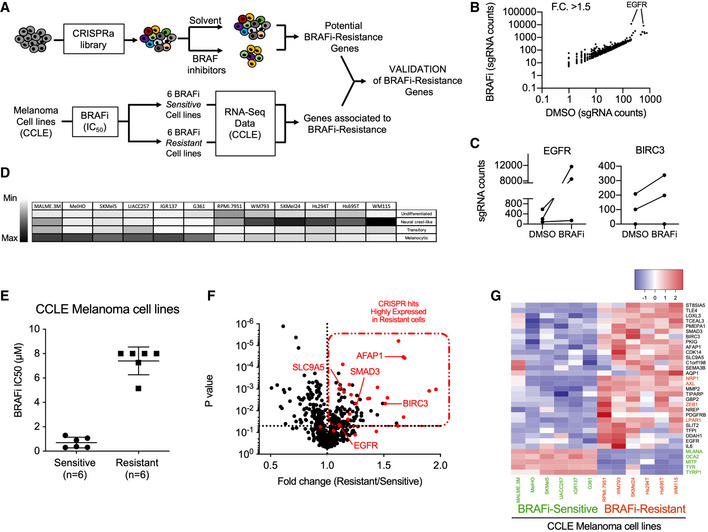
- CRISPR‐SAM workflow. Cell library was exposed to DMSO (solvent of BRAFi) or BRAFi 2 µM (vemurafenib (Vem) or PLX8394 (PB, Paradox Breaker)) during 14 days. 40 × 106 cells per arm. Experiments were done in duplicate.
- sgRNAs counts in BRAFi‐resistant cells versus in DMSO‐exposed cells for EGFR and BIRC3 (BRAFi for PBV, Appendix Fig S1)
- Determination of differentiation states of 12 human BRAF(V600) melanoma cell lines from CCLE according to (Tsoi et al, 2018).
- BRAF(V600) melanoma cell lines from CCLE were distributed in two groups (Sensitive and “intrinsically” Resistant, n = 6 cell lines per group) according to their BRAFi IC50 (µM, half maximal inhibitory concentrations) (Barretina et al, 2012). Error bars reflect mean ± s.d.
- Volcano plot showing the expression levels of BRAFi‐resistant genes (identified in 501Mel by CRISPR‐SAM screen) in 12 human melanoma cell lines. The fold change corresponds to the ratio of expression levels found in resistant and sensitive cell lines. In red: selected genes. EGFR is considered as positive control and SMAD3, BIRC3, and SLC9A5; selected as favorite genes for the next steps. SMAD3, BIRC3, SLC9A5, and AFAP1 are BRAFi‐resistance genes and potent tumor‐promoting genes (Fig 1). Raw data are available in Table EV5 (BRAFi for PBV, Appendix Fig S1)
- Heat map recapitulating the expression levels of the selected hits (red dots in Fig 2F) in BRAFi‐resistant and BRAFi‐sensitive cell lines. Markers of differentiation (MITF, MLANA, OCA2, TYRP1, TYR). In red: resistance genes already published (NRP1, AXL, ZEB1, LPAR1). Scale corresponds to Z scores.
Source data are available online for this figure.
Apart sgRNAs targeting the EGFR promoter, the sgRNAs enrichment in BRAFi‐exposed cells was unexpectedly weak (Tables EV5 and EV6). Thus, to select the best BRAFi‐resistance genes, we examined the gene expression levels of these potential BRAFi‐resistance genes identified by CRISPR screen in 12 melanoma cell lines (Fig 2D and E, Appendix Fig S1). We postulated that BRAFi‐resistance genes are highly expressed in BRAFi‐resistant cells (n = 6) as already demonstrated by other approaches for NRP1, AXL, and NGFR. To this end, we confronted CRISPR‐SAM candidates (identified in 501Mel cells) to gene expression data from six melanoma cell lines that were highly resistant to BRAFi according to the Cancer Cell Lines Encyclopedia (CCLE) versus six sensitive cell lines (Barretina et al, 2012; Fig 2E). It is important to note that the CCLE BRAFi‐resistant cell lines are therapy‐naïve and intrinsically resistant.
We next focused on candidate genes which were both enriched in CRISPR screen and highly expressed in the majority of BRAFi‐resistance cell lines (Fig 2F and G). To better evaluate the validity of our candidate genes identified using this workflow, we added well‐established and validated BRAFi‐resistant genes (NRP1, AXL, ZEB1, and LPAR1) (Müller et al, 2014; Konermann et al, 2015; Rizzolio et al, 2018) and five genes associated with melanoma cell differentiation (MITF, OCA2, MLANA, TYR, and TYRP1) (Levy et al, 2006). All BRAFi‐resistant cell lines presented a dedifferentiated profile as anticipated. Importantly, our candidate genes including SMAD3, SLC9A5, and BIRC3 (Fig 2G, black color) displayed a similar expression profile than observed for the well‐established and validated BRAFi‐resistant genes (Fig 2G, red color), strongly suggesting that these genes may also confer BRAFi resistance. EGFR and platelet‐derived growth factor receptor (PDGFR)‐β showed high expression only in a few BRAFi‐resistant cell lines, as previously observed in patients (Sun et al, 2014).
Together, our results confirmed the robustness of the functional in cellulo CRISPR‐SAM screen.
Validation of BRAFi SAM‐selected resistance genes
To evaluate the contribution of the CRISPR‐SAM‐selected genes in BRAFi resistance, we examined the transcriptome of the differentiated cell line M229 (transitory cell state) at different stages during acquisition of resistance (Fig 3A; Song et al, 2017). As described by Song et al (2017), cell lines were exposed to chronic exposure to BRAFi and analyzed at different days of treatment (P: parental cells, 2D: two days of treatment, DTP: drug‐tolerant persister cells, DTPP: drug‐tolerant proliferating persister cells, SDR: single‐drug‐resistant cell). We observed a sequential upregulation in the expression of BRAFi SAM‐selected genes: a group of genes (MMP2, SEMA3B, BIRC3, TIPARP, etc) being expressed earlier than a 2nd group (IL6, EGFR, AFAP1, etc). The majority of the candidates were overexpressed while the cells exhibited resistance to a single BRAFi agent (single‐drug resistance, SDR). Comparable results were obtained with the M238 melanoma cell line (Figs 3B and EV2A). Importantly, we found that combining the BRAFi with MEKi (DDR) led to comparable upregulation of the BRAFi SAM‐selected genes than observed in cells exposed to BRAFi alone (SDR) (Fig 3C). These results indicate that a common gene expression program can confer resistance to inhibitors of MAPK pathway as previously reported (Moriceau et al, 2015; Corre et al, 2018; Lee et al, 2020).
Figure 3. Validation of BRAFi SAM‐selected resistance genes.
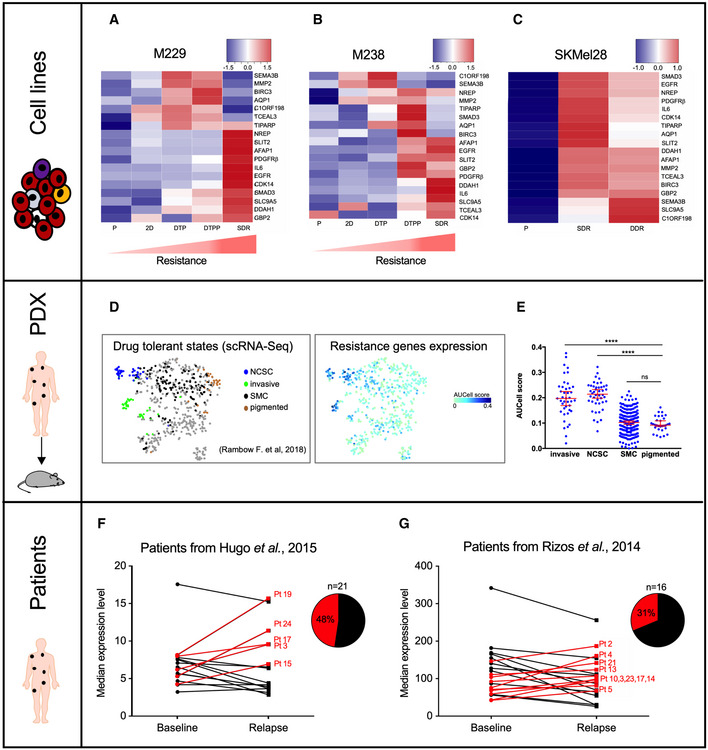
- Expression levels of our hits in M229 melanoma cells during the BRAFi‐resistance acquisition. P: parental cells, 2D: two days of treatment, DTP: drug‐tolerant persister cells, DTPP: drug‐tolerant proliferating persister cells, SDR: single‐drug‐resistant cells (BRAFi) (Song et al, 2017). Scale corresponds to Z scores.
- Expression levels of our hits in M238 melanoma cells (Song et al, 2017) during the BRAFi‐resistance acquisition. Scale corresponds to Z scores.
- Expression levels of our hits in parental, single‐drug‐resistant (SDR, BRAFi), or dual drug‐resistant (DDR, BRAFi + MEKi) SKMel28 melanoma cell lines (Song et al, 2017). Scale corresponds to Z scores.
- T‐distributed Stochastic Neighbor Embedding (t‐SNE) plot showing the 4 drug‐tolerant states (NCSC (neural crest stem cells), invasive, SMC (starved‐like melanoma cells) and pigmented cells) according to single‐cell RNA‐seq (scRNA‐Seq) performed in PDX MEL006 model exposed to BRAFi + MEKi (Rambow et al, 2018). Our BRAFi‐resistance genes are mainly expressed in NCSC and invasive cells.
- AUCell score for each drug‐tolerant states. ****P < 0.0001, Mann–Whitney test.
- Expression level of our genes (median) in cutaneous melanoma biopsies before the BRAFi treatment (baseline) and during the relapse. Cohort from (Hugo et al, 2015b). The expression level of our BRAFi‐resistance genes is increased in relapse samples (48%, n = 21).
- Expression level of our genes (median) in cutaneous melanoma biopsies before the BRAFi treatment and during the relapse. Cohort from (Rizos et al, 2014). The expression level of our BRAFi‐resistance genes is increased in relapse samples (31%, n = 16).
Data information: See also Fig EV2.
Source data are available online for this figure.
Figure EV2. BRAFi‐resistance genes are associated with invasive phenotype and resistance to BRAFi in vitro and in vivo. Related to Fig 3.
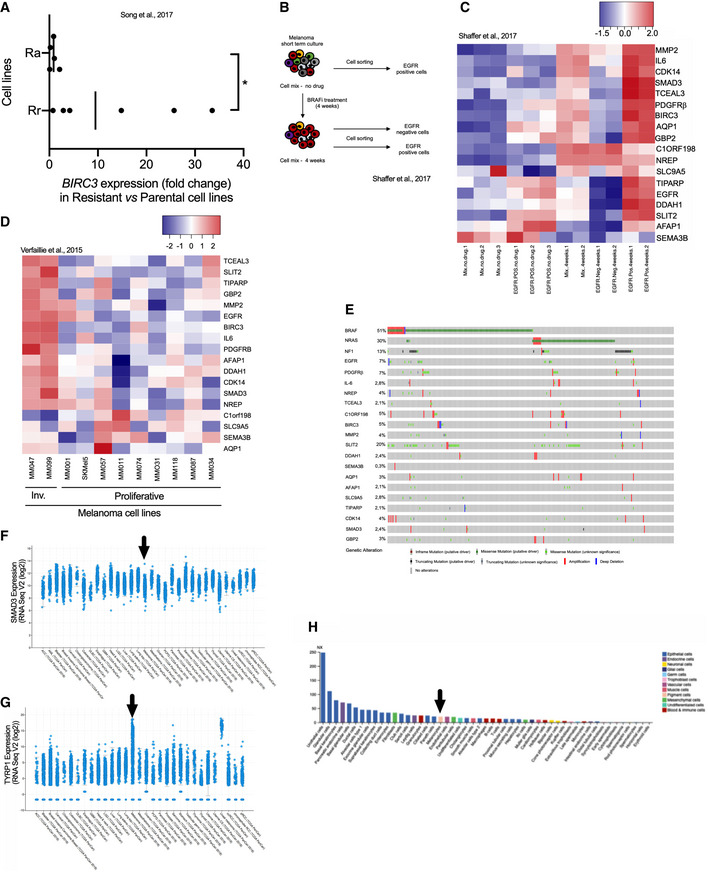
- BIRC3 expression levels (fold change) in melanoma Rr and Ra cell lines. The fold change has been calculated by comparing BIRC3 expression levels in resistant/parental cell lines (Song et al, 2017). Two groups have been made: Rr (n = 6) and Ra (n = 5) cell lines; Ra for resistance and MAPK reactivation and Rr for resistance and MAPK redundant. BRAFi resistance in Ra cell lines is due to additional genetic alterations such as BRAF splicing events for M395 SDR. BRAFi resistance in Rr cell lines is due to gene expression reprogramming. Two‐tailed Mann–Whitney test, P = 0.0303. Vertical bars correspond to the medians.
- Schematic representation of the workflow. As described in the original paper (Shaffer et al, 2017), fresh melanoma cells were obtained by patient tumor dissociation. Cells mix was treated with BRAFi. Cell sorting (EGFR positive and/or negative) was performed at two time points: before treatment (no drug) and after 4 weeks of treatment. Cells “mix” corresponds to the unsorted population. The EGFR‐positive cells (exposed to BRAFi) are able to produce colonies.
- Heat map depicting mRNA expression of BRAFi‐resistance genes, using an RNA‐Seq dataset obtained from BRAFi‐treated melanoma cells (Shaffer et al, 2017). Scale corresponds to Z scores.
- Heat map illustrating the expression levels of BRAFi‐resistance genes in invasive (Inv.) and proliferative melanoma cell lines (n = 2 and n = 9, respectively) (Verfaillie et al, 2015). Scale corresponds to Z scores.
- Genomic alterations in 18 BRAFi‐resistance genes identified in Fig 3 (from www.cbioportal.org). BRAF, NRAS, and NF1 have been added according to melanoma classification (Akbani et al, 2015).
- SMAD3 expression levels (RNA Seq V2 (log2) in the TCGA dataset. Picture was downloaded from www.cbioportal.org. Arrow indicates cutaneous melanoma.
- TYRP1 expression levels (RNA Seq V2 (log2) in the TCGA data set. Pictures were downloaded from www.cbioportal.org. Arrow indicates cutaneous melanoma. TYRP1 is highly expressed in drug‐naive tumors; cutaneous and uveal melanoma in contrast to SMAD3.
- SMAD3 expression and cell types (https://www.proteinatlas.org/ENSG00000166949‐SMAD3/celltype). The consensus normalized expression (NX) value for SMAD3 and organ/tissue represents the maximum NX value in the three data sources (HPA, GTEx, and FANTOM5). Black arrow highlights the melanocytes.
Source data are available online for this figure.
Having shown that combination of BRAFi and MEKi promotes sequential upregulation of BRAFi SAM‐selected genes, we monitored their expression levels in an in vivo preclinical patient‐derived xenograft (PDX) model (Rambow et al, 2018). Using scRNA‐Seq, our collaborators reported the presence of dedifferentiated drug‐tolerant cells exhibiting a neural crest stem cell (NCSC) and invasive profiles at minimal residual disease isolated from the MEL006 PDX model (Rambow et al, 2018). Here, we showed that the BRAFi SAM‐selected genes were highly and selectively expressed in both of these cell populations at mRNA level (Fig 3D and E). To reinforce these observations, we performed SMAD3 immunostainings in four BRAF‐mutant PDXs exposed to BRAF/MEK inhibitors until resistance (recently characterized in (preprint: Marin‐Bejar et al, 2020), Appendix Fig S2A and B). Immunostainings showed the emergence of SMAD3+ cells in Dabrafenib + Trametinib resistant lesions from the MEL003 and MEL006 PDXs in contrast to PDXs characterized by an intrinsic resistance mechanism (MEL007 and MEL037).
Moreover, we found that EGFR‐expressing cells sorted from melanoma tumors displayed comparatively high expression levels of the BRAFi SAM‐selected genes (Fig EV2B and C). Importantly, these EGFR‐positive cells are able to proliferate in the presence of BRAFi and generate BRAFi‐resistant colonies (Shaffer et al, 2017). In addition, high expression levels of the BRAFi SAM‐selected genes have been found in invasive cells when compared to proliferative melanoma cell lines (Verfaillie et al, 2015; Fig EV2D). Together, these data confirm the upregulated expression of BRAFi SAM‐selected genes in cells shown to contribute to relapse, suggesting their involvement in establishing drug‐tolerant and/or resistant phenotypes in vivo.
To evaluate the clinical relevance of the SAM‐selected BRAFi‐resistance genes, we compared their expression levels (median) in two independent BRAFi drug naive/drug‐resistant patient cohorts (Fig 3F and G, n = 21 (Hugo et al, 2015b) and n = 16 (Rizos et al, 2014) patients, respectively). The expression levels of the selected resistant candidate genes increased during relapse in drug‐resistant patients (48 and 31%). Notably, none of these genes have been implicated in recurrent gene‐amplification events that are sometimes identified in drug‐naive lesions (cBioPortal, TCGA; Gao et al, 2013) (Fig EV2, EV3, EV4, EV5). These data indicate that the increase in expression of the SAM‐selected genes in BRAFi‐resistant cells is likely associated with a (non‐genetic) dedifferentiation process of melanoma cells induced by the therapy. Together, these in vitro and in vivo gene expression analyses strongly support a BRAFi‐resistance function for the SAM‐selected genes.
Figure EV3. BIRC3 and EGFR are potent BRAFi‐resistance genes. Related to Fig 5.
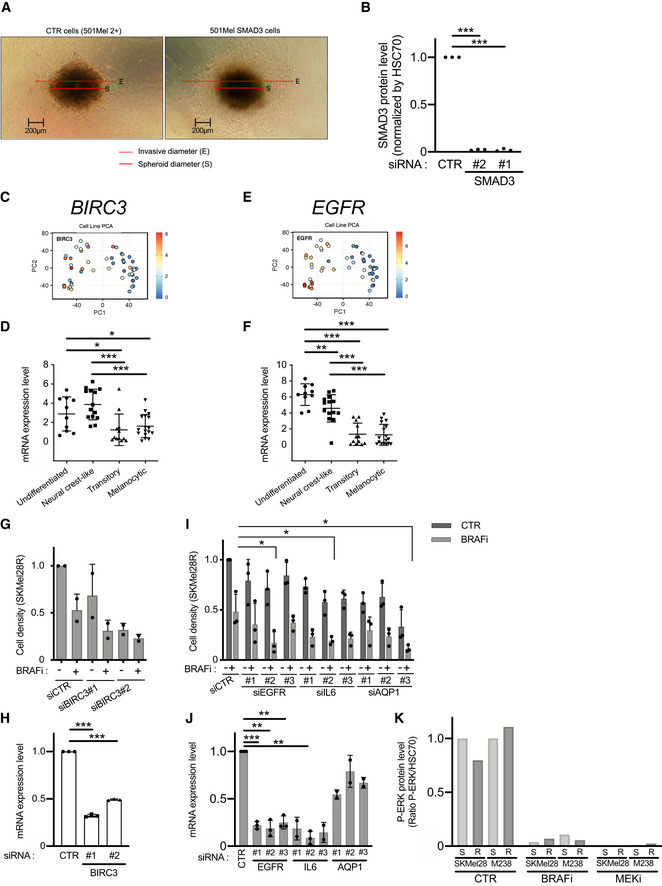
- Pictures illustrating the ratio (invasive diameter/spheroid diameter; E/S) used in Fig 5E (Invasion assay, melanoma spheroids). Two cell lines are illustrated: CTR cells correspond to 501Mel cells expressing dCas9 and HSF1‐p65‐MS2 (named here 501Mel 2+) and the SMAD3 cells (overexpressing SMAD3).
- SMAD3 Knock‐down efficiency of two siRNAs targeting SMAD3 in SKMel28R (Fig 5G). SMAD3 protein expression levels have been quantified in three independent experiments by Western blots. Expression levels have been compared to HSC70. Values obtained with the control siRNA (CTR) have been set at 1. Bilateral Student test (with non‐equivalent variances) ***P < 0.001.
- PCA analysis of BIRC3 expression in melanoma cell lines in function of their dedifferentiation states (generated by the webtool http://systems.crump.ucla.edu/dediff/index.php). Scale: red color corresponds to a high BIRC3 expression level (unit of the scale: Log2 FPKM).
- BIRC3 expression in these four subtypes of melanoma cells (undifferentiated = 10; neural crest‐like = 14; transitory = 12; melanocytic = 17) (Tsoi et al, 2018). Error bars represents the mean ± s.d. Multiple comparisons have been done using ordinary one‐way ANOVA, *P < 0.05, ***P < 0.001.
- PCA analysis of EGFR expression in melanoma cell lines in function of their dedifferentiation states (unit of the scale: Log2 FPKM).
- EGFR expression in these four subtypes of melanoma cells (undifferentiated = 10; neural crest‐like = 14; transitory = 12; melanocytic = 17). Error bars represents the mean ± s.d. Multiple comparisons have been done using ordinary one‐way ANOVA, **P < 0.01, ***P < 0.001.
- BIRC3 depletion (siRNA#1 & #2) reduced cell density and restored BRAFi (vemurafenib) effect on BRAFi‐resistant cells (SKMel28R). CTR for non‐targeting siRNA. Vem for the BRAFi vemurafenib and DMSO for dimethylsulfoxide (solvent of Vem). n = 2 biologically independent experiments. Each histogram represents the mean ± s.d.
- BIRC3 Knock‐down efficiency of two siRNAs targeting BIRC3 in SKMel28R (Fig EV3G). BIRC3 mRNA expression levels have been quantified in three independent experiments by RT–qPCR. Values obtained with the control siRNA (CTR) have been set at 1. Each histogram represents the mean ± s.d. n = 3 biologically independent experiments. Bilateral Student test (with non‐equivalent variances) ***P < 0.001.
- Knock‐down of EGFR, IL6, or AQP1 (siRNA#1, #2, and #3) restored BRAFi (vemurafenib) effect on BRAFi‐resistant cells (SKMel28R). n = 3 biologically independent experiments are presented. Each histogram represents the mean ± s.d.. Bilateral Student test (with non‐equivalent variances) *P < 0.05.
- Knock‐down efficiency of siRNAs targeting EGFR, IL6, or AQP1 in SKMel28R (Fig EV3I). mRNA expression levels have been quantified in two or three independent experiments by RT–qPCR. Each histogram represents the mean ± s.d. n = 3 biologically independent experiments for siCTR, siEGFR#1,2,4, IL6#3, and n = 2 for the other siRNAs. Values obtained with the control siRNA (CTR) have been set at 1. Bilateral Student test (with non‐equivalent variances) **P < 0.01, ***P < 0.001
- Efficiency of BRAFi (Vem) and MEKi (Cobi) to inhibit the MAPK signaling pathway (P‐ERK) (as used in Fig 5O). Values obtained with the solvent (CTR, here DMSO) have been set at 100%.
Source data are available online for this figure.
Figure EV4. SMAD3 signature is associated with mesenchymal phenotype in liver cancer cell lines. Related to Fig 7.
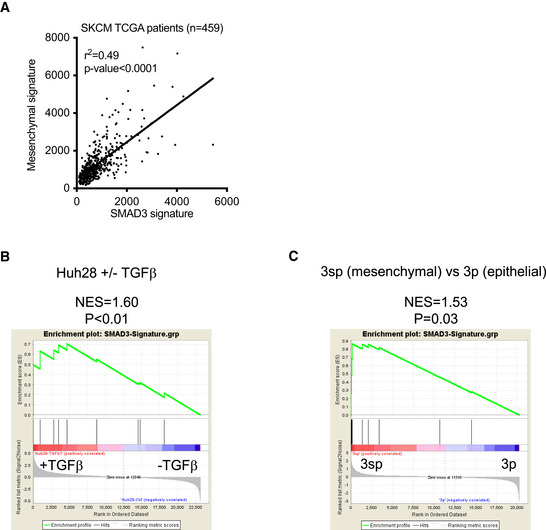
- Plot showing correlation between SMAD3‐signature and mesenchymal signature (Mak et al, 2016) in the TCGA dataset (SKCM). Pearson correlation test: P < 0.0001
- Gene Set Enrichment Analysis (GSEA) of the SMAD3 signature in TGF‐β treated versus non‐treated Huh‐28 cholangiocarcinoma cells (Merdrignac et al, 2018).
- Gene Set Enrichment Analysis of the SMAD3 signature in hepatoma cell lines : mesenchymal (3sp cells) versus epithelial (3p cells) (van Zijl et al, 2011).
Figure EV5. sgRNA targeting four isoforms of SMAD3 and BRAFi resistance.
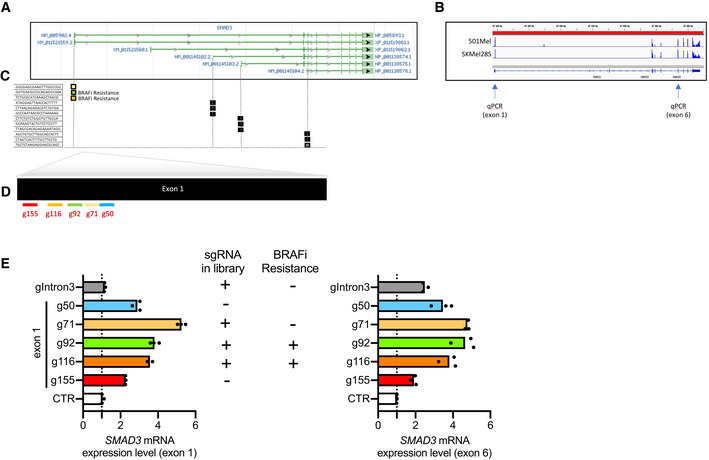
- SMAD3 mRNA isoforms according to NCBI. The longest isoform NM_005902.4 encodes SMAD3 protein (NP_005893.1). Three shorter isoforms of SMAD3 mRNA encode shorter SMAD3 proteins.
- Snapshot of SMAD3 mRNA expression in 501Mel and SKMel28S cells using IGV tool. Note that exon 1 is detectable in these melanoma cell lines. Arrows indicate the binding sites of primers for RT–qPCR (exon 1 or 6 for NM_005902.4).
- Ability of six sgRNAs to transactivate SMAD3 expression have been compared. Among the five sgRNAs targeting the first exon, three are found in the library (g71, g92, and g116). We designed g50 and g155 to evaluate the impact of sgRNA position on exon 1. sgRNA gIntron3 has been designed as a control (unable to promote the longest SMAD3 isoform but efficient to transactivate the smallest isoform). These two isoforms contain the exon 6 used for RT–qPCR.
- SMAD3 quantification by RT–qPCR (exon 1 or 6) in HEK293 cells transiently transfected with the three plasmids required for the CRISPR‐SAM as previously described (Konermann et al, 2015). CTR for empty plasmid (no sgRNA cloned into lenti sgRNA(MS2)_zeo backbone). RT–qPCR data are representative of two independent experiments. Source data are available in Fig EV5 source data. Each histogram represents the mean of three technical replicates from one representative experiment. No statistical analyses were thus performed. Dotted line highlights the value of 1 (corresponding to the effect obtained by the control sgRNA).
Source data are available online for this figure.
BRAFi‐resistance genes promote tumor growth
Long‐term effect of BRAFi is reduced by the ability of persister cells to resist to BRAFi but also to promote the tumor growth (relapse). Thus, we investigated the capability of the BRAFi‐persister cells (obtained from the in vitro CRISPR screen, Fig 2) to promote tumor growth into nude mice. The subset of BRAFi‐resistant/persister cells (Fig 4A) was engrafted (36 × 106 cells, 3 × 106/mouse), and tumor growth was monitored (Fig 4B).
Figure 4. BRAFi‐resistance genes promote tumor growth.
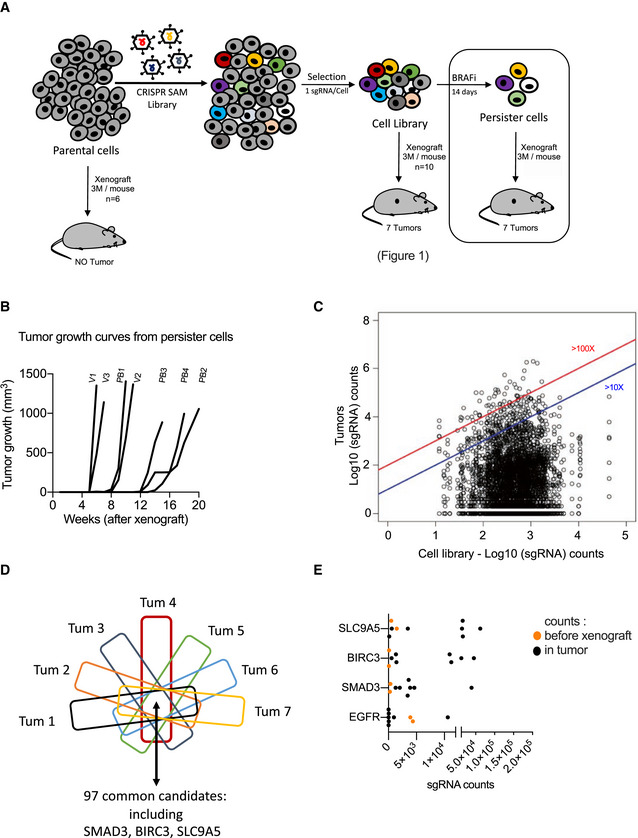
- Tumor growth curves from BRAFi‐persister cells (monitored during 5 months; Table EV2). V for Vem‐resistant cells and PB for PLX8394‐resistant cells.
- Distribution of sgRNAs in BRAFi‐resistant cells (before xenograft) and in the 7 tumors emerging from the BRAFi‐resistant cells (log10(sgRNAs counts)). Blue and red lines indicated the enrichment ≥ 10 fold or ≥ 100 fold (tumors versus BRAFi‐resistant cells (in vitro)). Raw data are available in Table EV7.
- From the seven tumors arising from BRAFi‐resistant cells, the common genes (enriched) have been extracted. Ninety‐seven genes including SMAD3, BIRC3, and SLC9A5 are detected in all these 7 tumors. Raw data are available in Table EV7.
- sgRNAs counts in tumors versus sgRNA detected in BRAFi‐resistant cell library (in vitro) (respectively, black and orange points) for selected candidates. Each black point corresponds to one tumor. EGFR was the most potent BRAFi‐resistant gene (Fig 2). Two replicates have been shown for CRISPR‐SAM cell library (orange points). SMAD3, BIRC3, and SLC9A5 were identified as hits in the three screens (Figs 1, 2, and 4).
Source data are available online for this figure.
These BRAFi‐persister cells formed tumors, in contrast to parental 501Mel cells (Ohanna et al, 2011). We determined the nature and abundance of sgRNAs present in each emerging tumor (Fig 4C and Table EV7), and we detected 97 genes (sgRNAs) detected in all tumors. Interestingly, we recovered the tumor‐promoting genes SMAD3, BIRC3, and SLC9A5 identified above (Fig 4D). We looked for the enrichment of these sgRNAs in each tumor developed from persister cells (Fig 4E). EGFR sgRNA was not frequently enriched in tumors (as previously described for human melanoma tumors (Prahallad et al, 2012; Sun et al, 2014; Shaffer et al, 2017)) in contrast to SMAD3, BIRC3, and SLC9A5 (Fig 4E and Appendix Fig S3). Together this suggests that these 3 genes are potential interesting targets for antimelanoma therapy.
Functional validation of genes involved in BRAFi resistance and relapse
We focused on the transcription factor SMAD3 as a model gene for monitoring BRAFi resistance and relapse due to its critical function downstream of the TGFβ pathway. Although this pathway is known to promote melanoma phenotype switching/dedifferentiation (Sun et al, 2014), to support melanoma growth (Berking et al, 2001) and metastasis, little is known about the role of SMAD3 in melanoma biology and as a modulator of resistance to targeted therapy.
We confirmed that SMAD3 mRNA is highly expressed in dedifferentiated cells (Fig 5A–C) and in BRAFi‐resistant cells (Fig 2G).
Figure 5. Functional validation of genes involved in BRAFi resistance and relapse.
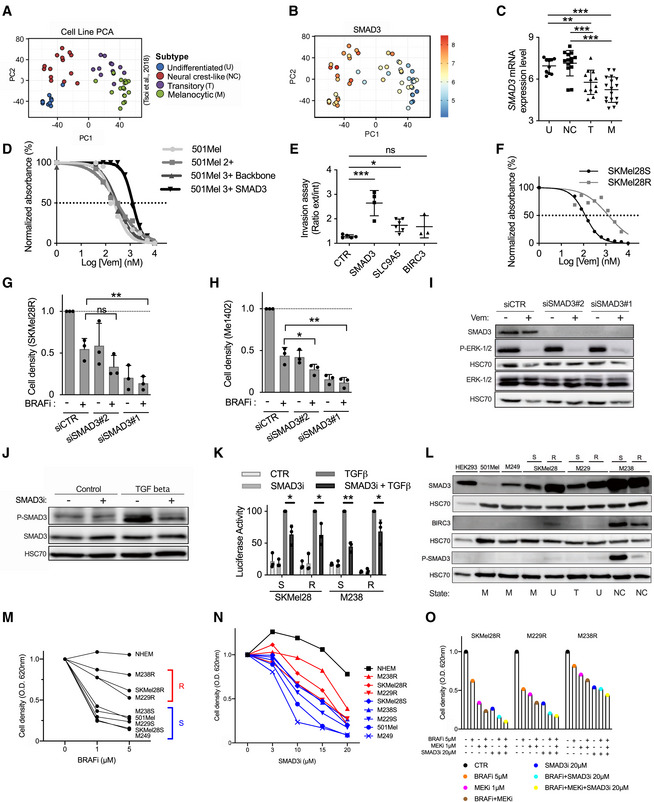
- PCA analysis of melanoma cell lines in function of their dedifferentiation states (generated by the webtool http://systems.crump.ucla.edu/dediff/index.php).
- SMAD3 expression increases with melanoma cell dedifferentiation. PCA analysis of SMAD3 expression in melanoma cell lines in function of their dedifferentiation states. Scale: red color corresponds to a high SMAD3 expression level (unit of the scale: Log2 FPKM).
- SMAD3 expression in these four subtypes of melanoma cells (U, undifferentiated; NC, neural crest‐like; T, transitory; M, melanocytic). Number in each group: U = 10, NC = 14, T = 12, M = 17. Error bars reflect mean ± s.d. Multiple comparisons have been done using ordinary one‐way ANOVA **P < 0.01, ***P < 0.001.
- SMAD3 gain‐of‐function increases BRAFi resistance. Determination of BRAFi half maximal inhibitory concentrations (IC50 values) for 501Mel cell lines (Log[Vem] (nM)). Parental 501Mel cells (n = 5), 501Mel cells expressing dCas9 and HSF1‐p65‐MS2 (named here 501Mel 2+, n = 5), the 501Mel 2 + cells expressing a control guide (backbone, n = 5) and the 501Mel 2 + cells overexpressing SMAD3 (n = 3 biologically independent experiments). A representative experiment has been chosen. Dotted line is used to determine the IC50 values.
- SMAD3 and SLC9A5 gain‐of‐function increases invasion capability of melanoma cells. Invasion assays for engineered cell lines (melanoma spheroids): CTR; 501Mel 2+, SMAD3; 501Mel 2 + cells overexpressing SMAD3. Two other cell lines overexpressing SLC9A5 or BIRC3 have been tested. Explanation for ratio calculation is detailed in Fig EV3A. Results obtained from two biologically independent experiments. (n = 5, 4, 6, and 3 spheroids, respectively). Error bars reflect mean ± s.d. Multiple comparisons have been done using ordinary one‐way ANOVA, *P < 0.05, ***P < 0.001.
- Characterization of SKMel28 sensitive and resistant cell lines (SKMel28S and SKMel28R). Determination of BRAFi half maximal inhibitory concentrations (IC50 values) (Log[Vem] (nM)). A representative experiment has been chosen among two experiments. Dotted line is used to determine the IC50 values.
- SMAD3 depletion (siRNA#1 & #2) decreased cell density and increased BRAFi effect (vemurafenib) on BRAFi‐resistant cells (SKMel28R). CTR for non‐targeting siRNA. DMSO for dimethylsulfoxide (solvent of vemurafenib; BRAFi). n = 3 biologically independent experiments. Each histogram represents the mean ± s.d.; Multiple comparisons have been done using ordinary one‐way ANOVA, **P < 0.01.
- SMAD3 depletion (siRNA#1 & #2) decreased cell density and increased BRAFi effect (vemurafenib, BRAFi) on BRAFi‐resistant cells (Me1402). CTR for non‐targeting siRNA. DMSO for dimethylsulfoxide (solvent of vemurafenib). n = 3 biologically independent experiments. Each histogram represents the mean ± s.d.; multiple comparisons have been done using ordinary one‐way ANOVA, *P < 0.05, ** P < 0.01.
- Validation of SMAD3 knock‐down by Western blot experiments in Me1402 cells exposed or not to vemurafenib (BRAFi (Vem) 5 µM, 2 days). Cells were exposed to BRAFi 24 h after siRNA transfection. Vemurafenib inhibitory effect on mutated BRAF was evaluated by analyzing the phospho‐ERK1/2 levels. ERK and HSC70 serves as loading control.
- Validation of SMAD3 inhibitor (SIS3, SMAD3i). Effect of SMAD3i (10 µM) on the level of phospho‐SMAD3 Ser423/425 in response to TGFβ (2 ng/ml, 1 h) (or solvent: HCl 4 mM + Bovine serum albumin 1 mg/ml) in SKMel28R cells (expressing a high endogenous level of SMAD3 mRNA). Serum starved cells (500,000 per well) were pretreated with SMAD3i 10 µM (or control solvent) during 2 h before TGFβ addition. SMAD3 and HSC70 serve as loading control for Western blot experiments.
- Inhibitory effect of SMAD3i (SIS3) on the SMAD‐responsive luciferase activity. Vector encodes the Firefly luciferase reporter gene under the control of a minimal (m)CMV promoter and tandem repeats of the SMAD Binding Element (SBE). Cells (10,000 per well) were pretreated with SMAD3i 10 µM or control solvent during 1.5 h, and next cells were exposed to TGFβ 10 ng/ml (or solvent: HCl 4 mM + Bovine serum albumin 1 mg/ml) for 6 h. n = 3 biologically independent experiments. Each histogram represents the mean ± s.d.; Bilateral Student test (with non‐equivalent variances); TGFβ vs TGFβ+SMAD3i, *P < 0.05, **P < 0.01.
- SMAD3, Phospho‐SMAD3 Ser423/425, and BIRC3 expression levels in melanoma cell lines. Four subtypes of melanoma cells (U, undifferentiated; NC, neural crest‐like; T, transitory; M, melanocytic) have been compared. 501Mel and M249 cells are melanocytic cells in contrast to dedifferentiated BRAFi‐resistant cells (R). Three couples of melanoma cell lines (Sensitive (S) and (R)) have been used to illustrate the SMAD3 and BIRC3 increases in BRAFi‐resistant cell lines. HEK293 cells are used as control (kidney). HSC70 serves as loading control for Western blot experiments. Each antibody has been evaluated on individual membrane (explaining the three HSC70, loading controls).
- The vemurafenib decreased melanoma cell density. Cell lines have been exposed to Vem (1 or 5 µM, 84 h) to define two groups of cell lines (sensitive (blue) and “resistant” (red) cell lines). Normal human melanocytes (NEHM) have been used to evaluate the effect of Vem on normal cells (mean of 3 independent donors). Representative values (mean of biological triplicates) of n = 3 biologically independent experiments.
- The SMAD3 inhibitor decreased melanoma cell density. Cell lines have been exposed to SMAD3i (0, 3, 10, 15, or 20 µM, 84 h). The cell lines defined as S and R to BRAFi in the item 5 M are indicated in blue and red. Normal human melanocytes (NEHM) have been used to evaluate the effect of SMAD3i on normal cells (n = 3 independent donors). The SMAD3i effect on NHEMs is weak and manageable for these normal cells (NHEM). Representative values (mean of biological triplicates) of n = 2 biologically independent experiments.
- The chemical inhibition of SMAD3 by SIS3 (SMAD3i) improved current therapy effect (BRAFi + MEKi; Vem 5 µM and Cobi 1 µM) on BRAFi‐resistant cells (SKMel28R, M229R & M238R). Cells have been treated as detailed for panel M. Representative values (mean of biological triplicates) of n = 2 biologically independent experiments.
Data information: Western blot results are representative of at least two independent experiments. Source data and unprocessed original blots are available in Appendix Fig S2 source data. See also Fig EV3.
Source data are available online for this figure.
To reinforce the role of SMAD3 in BRAFi resistance, we showed that gain‐of‐function of SMAD3 significantly increases the BRAFi resistance of melanoma cells when compared to different control cells (parental 501Mel cells and the CRISPR‐engineered cells: 501Mel cells expressing dCas9 and HSF1‐p65‐MS2 (named here 501Mel 2+) and the 501Mel 2+ cells expressing a control guide (named 3+ backbone; Fig 5D). In addition, we showed that gain‐of‐function of SMAD3 also promotes the three‐dimensional (3D) tumor spheroid invasion capability of melanoma cells. Similar results were obtained for SLC9A5 (Figs 5E and EV3A). These results strongly suggest that a high expression level of SMAD3 confers BRAFi resistance and invasion capability in melanoma cells.
Thus, we investigated if SMAD3 impairment re‐sensitizes cells to BRAF inhibitor, using small‐interfering RNA (siRNA) (Fig 5F–I). As the dedifferentiation status correlated with BRAFi resistance, we selected two BRAFi‐resistant cell lines, SKMel28 BRAFi‐resistant cells (SKMel28R, Fig 5F and G; Hugo et al, 2015b) and Me1402 melanoma cells (Fig 5H). In contrast to the SKMel28R, the resistance of which was created by chronic exposure to non‐lethal doses of BRAFi, Me1402 cells are intrinsically resistant.
Surprisingly, the single SMAD3 depletion decreased the cell density in a similar magnitude than BRAFi treatment in these BRAFi‐resistant cells (Figs 5G and H, and EV3B). The SMAD3 depletion did not modify the ERK pathway (Fig 5I) in contrast to the BRAFi. The combo (SMAD3 depletion and BRAFi (5 µM)) efficiently reduced the number of resistant/persister cells in these two cell lines, suggesting that SMAD3 is an interesting target to limit resistance to BRAFi. Similar results were obtained by targeting BIRC3, EGFR, IL6, or AQP1 (Fig EV3, EV4, EV5).
To transfer this strategy (SMAD3 inhibition + BRAFi) into clinic, we looked for an efficient inhibitor of SMAD3. We identified the chemical inhibitor SIS3 (SMAD3 inhibitor, SMAD3i; Jinnin et al, 2006; Wu et al, 2020). Firstly, we validated the inhibitory efficiency of SMAD3i in melanoma cells since it decreased the levels of phospho‐SMAD3 Ser423/425 induced by TGFβ (Fig 5J) in accordance with previous studies (Jinnin et al, 2006; Chihara et al, 2017). Since the SMAD3 regulation by phosphorylation is not fully understood and the phospho‐SMAD3 Ser423/425 status is not strictly correlated with SMAD3 transcriptional activity (Ooshima et al, 2019), we evaluated the effect of SMAD3i using a reporter assay. We demonstrated that SMAD3i reduced the transcriptional activity of SMAD3 in response to TGFβ exposure in 4 melanoma cell lines (Fig 5K). To know if the combination (SMAD3i + BRAFi) could be broadly used to eradicate persister cells emerging in response to BRAFi treatment, we selected three melanoma cell lines in function of SMAD3 expression levels (Fig 5L). The differentiated cells (SMAD3low) are highly sensitive to BRAFi (decrease of cell density: > 80% at 5 µM BRAFi, Fig 5M)) in contrast to dedifferentiated cells (SMAD3high), which are highly resistant to BRAFi (decrease of cell density: ~50% at 5 µM BRAFi, Fig 5M).
We showed that SMAD3i (SIS3) reduced the cell density of all melanoma cell lines (Fig 5N). These results are in agreement with the SMAD3 knock‐down results (Fig 5G and H). Our experiments also indicated that melanocyte survival is weakly affected by SMAD3 inhibition (up to 20 µM), in agreement with the non‐toxicity of this inhibitor observed in vivo (Tang et al, 2017; Wu et al, 2020). The inhibitory effect of SMAD3i on melanoma cells seems to be associated with the total SMAD3 expression levels (Fig 5L). Our results might suggest that these melanoma cells could be “addicted” to SMAD3 activity.
We finally investigated the interest to combine BRAFi (Vem, alone or in combination with MEKi (Cobi)) and SMAD3i (SIS3) to eradicate the BRAFi‐resistant cell lines (SKMel28R, M229R, and M238R) (Figs 5O and EV3K). We showed that SMAD3 inhibitor alone or in combination with BRAFi (Vem 5 µM) or BRAFi + MEKi (Cobi 1 µM) might be a promising treatment to reduce the amount of persister cells (melanoma). Together, these results identified SMAD3 as an amenable target to limit resistance to BRAFi and tumor growth.
The transcription factor AhR drives SMAD3 expression
Having shown that SMAD3 expression mediates BRAFi‐resistance and tumor growth, we explored the transcriptional program promoting its expression in BRAFi‐resistant melanoma cells. We recently reported that the Aryl hydrocarbon Receptor (AhR), a ligand‐dependent transcription factor is an upstream central node regulating the expression of BRAFi‐resistance genes and melanoma dedifferentiation (Corre et al, 2018; Leclerc et al, 2021; Paris et al, 2021).
We postulated that AhR may govern SMAD3 expression in BRAFi‐resistant cells. We identified three putative canonical binding sites for AhR (XRE for xenobiotic responsive element) in the proximal promoter of SMAD3 (Fig 6A) in accordance with chromatin immunoprecipitation coupled to massively parallel DNA sequencing data (AhR ChIP‐Seq) showing AhR binding on SMAD3 promoter (Lo & Matthews, 2012; Yang et al, 2018). To demonstrate the SMAD3 induction by AhR, 501Mel cells were exposed to the most potent and well‐known AhR ligands (TCDD for 2,3,5,7‐tetrachlorodibenzodioxin; ITE for 2‐(1H‐Indol‐3‐ylcarbonyl)‐4‐thiazolecarboxylic acid methyl ester). These AhR ligands increased SMAD3 expression, in an AhR‐dependent manner (Fig 6B). Comparable results were obtained with a canonical target gene of AhR; the TCCD‐induced poly(ADP ribose) polymerase gene (TIPARP), supporting the role of AhR in regulating SMAD3 expression (Fig 6C). The need of an activated AhR‐promoting SMAD3 expression was further confirmed by the use of an AhR antagonist (CH‐223191) in SKMel28 cells (Fig 6D). Long‐term chemical inhibition of AhR activity reduced SMAD3 and TIPARP expression levels. In accordance with these results, SMAD3 expression levels decreased in AhR KO SKMel28 cells (Fig 6E).
Figure 6. The Transcription Factor AhR Drives SMAD3 Expression.
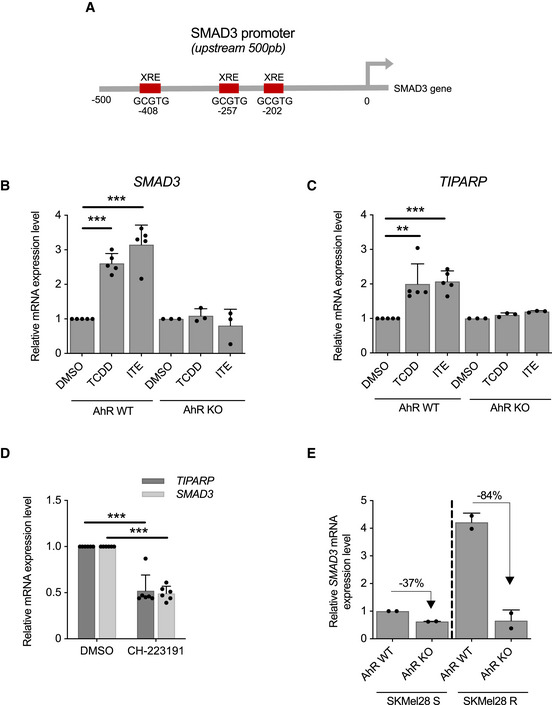
- AhR binding sites (xenobiotic responsive element (XRE); GCGTG) in human SMAD3 proximal promoter.
- AhR activation by exogenous and endogenous ligands promotes SMAD3 induction. 501Mel cells AhR wild‐type or knockout have been exposed to exogenous and endogenous AhR ligands; TCDD (5 nM) or ITE (10 µM) or the solvent (DMSO) during 10 days. n = 5 biologically independent experiments for AhR WT cells and n = 3 for AhR KO cells. Each histogram represents the mean ± s.d.; multiple comparisons have been done using ordinary one‐way ANOVA, ***P < 0.001.
- AhR activation by exogenous and endogenous AhR ligands promotes TIPARP induction. 501Mel cells have been treated as described in B. n = 5 biologically independent experiments for AhR WT cells and n = 3 for AhR KO cells. Each histogram represents the mean ± s.d.; multiple comparisons have been done using ordinary one‐way ANOVA, **P < 0.01, ***P < 0.001.
- AhR antagonist (CH‐223191) reduces SMAD3 and TIPARP expression levels. SKMel28 cells (AhR wild type) have been exposed to CH‐223191 (5 µM) or the solvent (DMSO) during 7 days. n = 6 biologically independent experiments. Each histogram represents the mean ± s.d.; Bilateral Student test (with non‐equivalent variances): ***P < 0.001.
- Loss of AhR reduces SMAD3 expression levels. SMAD3 expression has been investigated in SKMel28 cells AhR wild‐type (WT) or knockout (KO). SKMel28R has been obtained from SKMel28S by chronic exposure to non‐lethal doses of BRAFi (Hugo et al, 2015b). R for BRAFi‐resistant SKMel28 cells and S for sensitive. n = 2 biologically independent experiments. Each histogram represents the mean ± s.d.
Source data are available online for this figure.
Together, our results support the hypothesis that AhR activity drives SMAD3 expression along with the acquisition of BRAFi resistance.
SMAD3 drives phenotype switching and resistance to melanoma therapies
To explore the mechanism underlying therapy sensitization upon SMAD3 inhibition, we examined the transcriptional program regulated by SMAD3 (Fig 7). We hypothesized that the transcription factor SMAD3 may regulate the expression levels of several resistant genes and thereby induce a multifactorial effect, in which multiple drug resistance pathways are activated. We compared the SMAD3 ChIP‐Seq (Ramachandran et al, 2018) data to BRAFi‐resistance gene sets established from three different sources, namely the enclosed screen, the screen performed in A375 (Konermann et al, 2015), and other established BRAFi‐resistance genes such as AXL or NRP1 (Fig 7A). We established a list of SMAD3‐regulated genes, which includes SLIT2, RUNX2, NRP1, MMP2, JUNB, ITGB5, AXL, AFAP1, and EGFR (SMAD3‐signature). We next showed that basal SMAD3‐signature is higher in the three BRAFi‐resistant cell lines when compared to parental cell lines (SKMel28S, M229S, and M238S) (Fig 7B). The stimulation of the TGFβ‐SMAD3 pathway by the recombinant TGFβ promoted the SMAD3 signature in these melanoma cell lines (Fig 7C). The inducibility is higher in parental cell lines (S) since the basal expression level of genes forming the SMAD3 signature is weak in parental cells (Fig 7B). Next, we examined the SMAD3 signature in a large panel of melanoma cell lines representative of the distinct differentiation states (U ‐ NC ‐T ‐ M) (n = 53) (Fig 7D) and cutaneous melanoma (n = 118, TCGA cohort, tumors not exposed to targeted therapy; Fig 7E). The SMAD3 signature correlated with a dedifferentiation status as suggested above (Figs 2G and 5B, and EV4A). Importantly, a subset of BRAF(V600E) melanoma patients (~20%) expressing the SMAD3‐signature was identified (Fig 7E), suggesting that these tumors contained dedifferentiated melanoma cells with potential intrinsic resistance to BRAFi. In addition, SMAD3 signature could be useful to clinicians to propose immune checkpoint immunotherapy to their patients since SMAD3 signature identified anti‐PD1 non‐responders (prior the selection of the treatment) (Fig 7F). Therefore, these results strongly suggest that the SMAD3‐signature may be useful to identify a population of pre‐existing drug‐resistant cells within drug‐naive lesions. To further illustrate the clinical relevance of our results, we assessed the expression levels of the SMAD3 signature in drug‐naive and BRAFi‐resistant patients using publicly available dataset. SMAD3 signature increased in 14/16 patients exposed to BRAFi (baseline vs. relapse, Fig 7G). Altogether, these results indicated that the SMAD3 signature is associated with resistance to both current melanoma therapies.
Figure 7. SMAD3 drives phenotype switching and resistance to melanoma therapies.
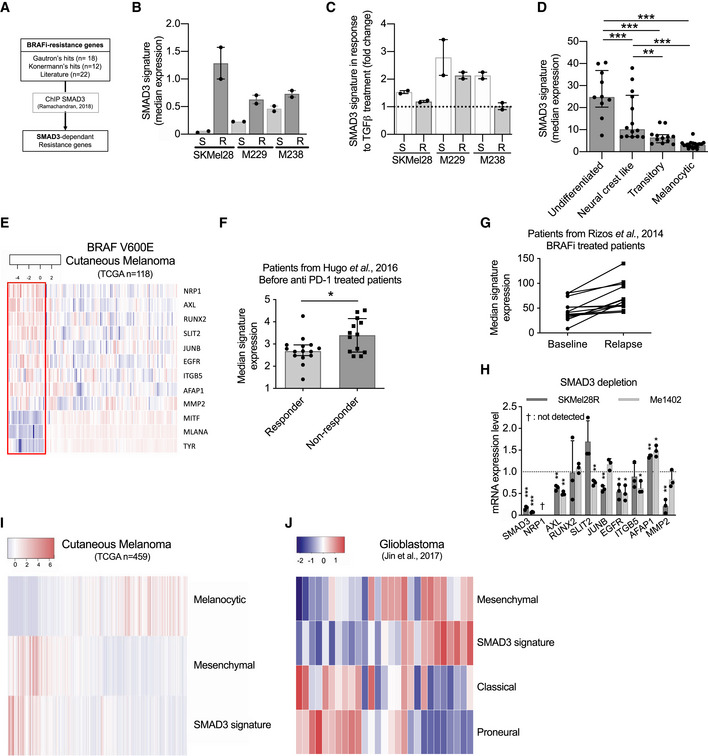
- SMAD3 signature has been established by comparing BRAFi‐resistance genes and SMAD3‐regulated genes identified by chromatin immunoprecipitation followed by DNA sequencing (ChIP‐Seq) (Ramachandran et al, 2018).
- Basal SMAD3 signature in six melanoma cell lines (S for sensitive to BRAFi and R for Resistant). n = 2 biologically independent experiments. Each histogram represents the median with interquartile range.
- Inducibility of SMAD3‐signature in six melanoma cell lines exposed to TGF‐β (10 ng/ml, 48 h). Data were normalized to cell lines exposed to solvent (4 mM HCl + 1 mg/ml human BSA). Values obtained with the TGF‐β stimulated M238R cell line have been set to 1. n = 2 biologically independent experiments. Each histogram represents the median with interquartile range.
- SMAD3 signature discriminates differentiation states of 53 melanoma cell lines (Tsoi et al, 2018). The SMAD3 signature in four subgroups of melanoma cell lines. Each point represents a cell line (n = 17, 12, 14, and 10, respectively, for melanocytic, transitory, neural crest‐like, and undifferentiated cell lines); each histogram represents the median with interquartile range; multiple comparisons have been done using ordinary one‐way ANOVA; **P < 0.01, ***P < 0.001.
- Heat map depicting mRNA levels of SMAD3 signature in BRAF (V600E) non‐treated melanoma patients (dataset from TCGA; SKCM, BRAF(V600E) mutated: n = 118). Three pigmentation genes (MITF, MLANA, and TYR) have been added to highlight the differentiation states of tumors. ~20% of tumors are considered as dedifferentiated tumors (red box) with a high SMAD3 signature. Scale corresponds to Z scores.
- SMAD3 signature in pre‐treatment biopsies from responders and non‐responders to anti‐PD‐1 treatment (from Hugo’s cutaneous melanoma cohort, n = 15 and 13, respectively) (Hugo et al, 2016). Each histogram represents the median with interquartile range, one‐tailed Mann–Whitney test; *P = 0.0324.
- SMAD3 signature in two groups (before BRAFi treatment or during relapse) of V600E patients from Rizos’s cohort (Rizos et al, 2014) (14 on 16 patients displayed an upregulation of SMAD3 signature during relapse).
- SMAD3 depletion decreases expression of SMAD3‐regulated genes. SMAD3 knock‐down by siRNA decreased mRNA expression of BRAFi‐resistance genes in SKMel28R and Me1402. NRP1 mRNA was not detected in our experimental conditions. n = 3 biologically independent experiments. Each histogram represents the mean ± s.d.; Bilateral Student test (with non‐equivalent variances) *P < 0.05, **P < 0.01, *** P < 0.001. Dotted line highlights the value of 1.
- SMAD3 signature overlapped with mesenchymal signature in melanoma tumors (TCGA, n = 459) (Akbani et al, 2015). Melanocytic signature highlights the differentiation states of tumors (Corre et al, 2018). SMAD3 signature and mesenchymal signature correlate in melanoma tumors (TCGA, n = 459) (Mak et al, 2016). Scale corresponds to Z scores.
- The SMAD3 signature overlaps with the glioblastoma mesenchymal subtype (Jin et al, 2017). Scale corresponds to Z scores.
Source data are available online for this figure.
The analyses of the SMAD3 signature in tumor samples highlighted the tumor heterogeneity (mRNA expression level of genes forming the SMAD3 signature is variable between patients, and all genes forming the SMAD3 signature are not high in each tumor.). As expected, this heterogeneity has been retrieved in melanoma cell lines (Fig 7H). In response to SMAD3 depletion, AXL and EGFR decreased in the two models in contrast to other genes such as MMP2, which decreased in only one model. Altogether, our results indicate that the SMAD3 signature assessment is probably more appropriate than quantification of specific mRNAs such as EGFR or SLIT2 to track pre‐existing drug‐resistant cells within drug‐naive lesions.
As the shift toward the mesenchymal‐like state confers broad resistance to therapies (Redfern et al, 2018), we postulated that the SMAD3 signature may be associated with this particular dedifferentiated phenotype. Comparing the SMAD3 signature with the classical mesenchymal‐like signature (Mak et al, 2016) of melanoma (TCGA cohort) highlighted a significant correlation (Figs 7I and EV4A). To further confirm the link between the SMAD3 signature and a mesenchymal state, we searched for similarities with other mesenchymal states identified in two other cancers (glioblastoma [GBM] and hepatoma). As described for the cutaneous melanoma, different differentiation states have been characterized for GBM (proneural, classical, and mesenchymal GBM) (Jin et al, 2017). We found that the SMAD3 signature overlaps with the mesenchymal GBM signature (Fig 7J). Mesenchymal GBM is the most aggressive GBM usually associated with poor overall survival (Patel et al, 2014; Jin et al, 2017). The SMAD3 signature was also associated with epithelial–mesenchymal transition (EMT) in hepatoma (Fig EV4B and C).
Altogether, these results indicate that the transcription factor SMAD3 and its downstream target genes confer resistance to targeted therapies by promoting a mesenchymal‐like phenotype. Our work identifies AhR‐SMAD3 axis as a target to overcome therapy resistance of melanoma.
Discussion
Targeted therapy and immunotherapy have greatly improved the prognosis of patients with cancer, but resistance to these treatments restricts the overall survival of patients. Increasing evidence indicates that transcriptomic reprogramming is associated with persister cell emergence (Puisieux et al, 2014; Bai et al, 2019) but the mechanism underlying resistance from this pool of cells remains elusive. Targeting tumor‐promoting genes leading reprogramming could therefore constitute an attractive approach to prevent relapse, at least in some specific contexts (Bailey et al, 2018). Here, using a whole genome approach, we searched for pathways that trigger the transcriptional reprogramming of persister cells into drug‐resistant cells. Based on CRISPR screen, data mining, and in vivo experiments, we identified and validated three genes (SMAD3, BIRC3, and SLC9A5) able to promote both BRAFi‐resistance and tumor growth. Our work expands our understanding of the biology of persister cells and highlights novel drug vulnerabilities that can be exploited to develop long‐lasting antimelanoma therapies.
Even if CRISPR‐SAM screen is a leading‐edge genetic tool, several concerns must be considered. As observed for all screening approaches, false positives and false negatives are engendered rendering the validation experiments a crucial step. In this study, we clearly showed that the number of sgRNAs per target is an important parameter. For our best hit, EGFR, only two sgRNAs were enriched in BRAFi‐treated cells. Thus, it is highly likely that we missed interesting BRAFi‐resistance genes (false negatives) due to the number of sgRNA/gene (at least 3 sgRNAs/gene in this library). A recent publication confirmed that sgRNAs are not all functional in CRISPRa libraries and it could be interesting to increase the number of sgRNAs per target and to cover more TSS per gene (Sanson et al, 2018). Interestingly, the sgRNA library used in our study displays for several genes up to 27 sgRNAs. These sgRNAs target different isoforms (or TSS) of these genes. By examining the sgRNAs targeting SMAD3, we found that 2 sgRNA are enriched (BRAFi resistance) (Fig EV5). These two sgRNAs promote the expression of the longest SMAD3 isoform; the SMAD3 mRNA expressed in our model (501Mel cells) (Fig EV5A–E). It is important to note that the nine other sgRNAs targeting SMAD3 are not able to confer the BRAFi resistance since they target other SMAD3 isoforms. Based on these observations, we believe that mRNA isoforms identified by RNA sequencing should be considered during the sgRNAs selection for each model. By this way, it would be easy to disqualify a part of the sgRNAs of the library targeting weakly expressed isoforms. This attitude could increase the score per isoform. In other words, the use of the z‐score calculated per gene may discard interesting candidates. So, an analysis based on RNA isoforms could reduce the false negatives.
The second lesson of this CRISPR screen is the weak sgRNAs enrichment in BRAFi‐exposed cells. Except for EGFR, the sgRNAs enrichment is about 2. Despite these values, we showed that these candidates are robust as for SMAD3 (in vitro, in vivo, and in patients). It is tempting to explain this fact by the duration of treatment (BRAFi exposure) and the dose (2 µM). By increasing the dose (i.e., 5 µM), we showed that BRAFi killed more than 90% of 501Mel cells in four days (Fig 5M). This protocol is not achievable because it would induce too many false negatives. The other option consists to increase the duration of BRAFi treatment (and keep a low BRAFi concentration, i.e., 2 µM). However, a recent publication demonstrated that a long‐term BRAFi exposure promotes a dedifferentiation process conferring BRAFi resistance (Tsoi et al, 2018; Bai et al, 2019). This alternative protocol could be perilous by inducing cell resistance to BRAFi independently of the sgRNA expression. Here, we selected a melanoma cell model exhibiting a differentiated profile as the vast majority of metastatic melanoma tumors (89% in the TCGA cohort), a short period of treatment (14 days) and an intermediate dose of BRAFi (2 µM). In fact, we followed, except the cell line, the protocol established by Feng Zhang’s laboratory, who developed the CRISPR‐SAM library (Konermann et al, 2015). The differentiation status of the cell line could be important since the transactivation mediated by CRISPR‐SAM is possible only for active promoter in basal condition and the magnitude of transactivation relies on the basal expression level. Here, we showed that 501Mel cells express low level of SMAD3 in basal condition and the transactivation obtained by CRISPR‐SAM is substantial (Fig 1I). Moreover, SMAD3 is a transcription factor which promotes the expression of various genes including potent BRAFi‐resistance genes (AXL and EGFR). The robust transactivation of genes encoding a transcription factor, a transporter, or an enzyme is more inclined to be enriched with our protocol, especially if the basal gene expression is low. Here, we identified and validated in vitro and in vivo the transcription factor SMAD3 and the transporter SLC9A5, validated in vitro and in vivo.
BRAFi resistance relies, at least in part, on the phenotypic plasticity of melanoma cells (Tsoi et al, 2018; Rambow et al, 2018; Corre et al, 2018). These cells may escape the deleterious effect of drug combinations such as BRAFi + MEKi. Among these cells, those harboring a mesenchymal‐like phenotype (usually named invasive cells) display high intrinsic resistance to MAPK therapeutics (Konieczkowski et al, 2014; Müller et al, 2014; Verfaillie et al, 2015; Shaffer et al, 2017). Enrichment in AXLhigh subpopulation (considered as invasive and mesenchymal‐like cells) is a common feature of drug‐resistant melanomas (Müller et al, 2014; Konieczkowski et al, 2014). Targeting mesenchymal‐like cells using an antibody‐drug conjugate, AXL‐107‐MMAE, showed promising effects in a preclinical model of melanoma (Boshuizen et al, 2018). The emergence of AXLhigh cells is currently explained by the decrease in MITF activity, but the mechanism of resistance to MAPK therapeutics remains unclear. Here, we demonstrate that the AhR‐SMAD3 axis governs the expression levels of potent BRAFi‐resistance genes, including AXL, EGFR, and MMP2.
The dedifferentiation process, conferring BRAFi resistance, requires transcriptomic reprogramming by transcription factors. Retinoic acid receptor gamma (RXRγ) was identified as a crucial transcription factor that promotes the emergence of the drug‐tolerant subpopulation of NCSCs (Rambow et al, 2018). An increase in AXLhigh‐positive cell population was reported following MAPK inhibition in the presence of an RXRγ antagonist. This increase may explain why this co‐treatment only delays but does not completely prevent relapse in PDXs (preprint: Marin‐Bejar et al, 2020). This confirms the need to develop strategies that prevent melanoma dedifferentiation during BRAFi treatment. Thus, our data strongly suggest that SMAD3 is a key transcriptional factor involved in the emergence of drug‐resistant mesenchymal‐like cells in response to MAPK and identify a clinically compatible approach (SMAD3i) that might abrogate such a trajectory. Other transcription factors have been associated to BRAFi resistance such as JUN (Titz et al, 2016) and AhR (Liu et al, 2017; Corre et al, 2018). We recently showed that a sustained AhR activation promotes the dedifferentiation of melanoma cells and the expression of BRAFi‐resistance genes (Corre et al, 2018). As proof‐of concept, we demonstrated that differentiated and BRAFi‐sensitive cells can be directed toward an AhR‐dependent resistant program using AhR agonists. To abrogate the deleterious AhR sustained‐activation, we identified Resveratrol, a clinically compatible AhR antagonist. Combined with BRAFi, Resveratrol reduces the number of BRAFi‐resistant cells and delays relapse. Recently, an independent team confirmed that AhR inhibition is reachable in vivo using other AhR antagonists (Kyn 101, Ikena Oncology, or the CH‐223191; Campesato et al, 2020) to improve the melanoma therapy. AhR antagonists validated in mice are currently evaluated in clinical trials (NCT04200963, Ikena Oncology) and (NCT04069026, Bayer). In addition, a water‐soluble SMAD3 inhibitor has been recently published for the in vivo treatment (Wu et al, 2020), suggesting that clinical trials should start soon. Another option overcoming the potential pharmacological caveat would be to use antisense oligonucleotides (ASO) targeting AhR or SMAD3 (Leclerc et al, 2021). We and others demonstrated in melanoma that ASO strategy is feasible in vivo by targeting SAMMSON mRNA and the lncRNA TYRP1 (Leucci et al, 2016; Gilot et al, 2017).
In this study, we propose an AhR‐SMAD3 impairment as a strategy to overcome melanoma resistance. Recently, conditional deletion of Smad7, a negative regulator of TGF‐β/SMAD pathway, led to sustained melanoma growth and at the same time promoted metastasis formation (Tuncer et al, 2019), confirming that TGF‐β/SMAD pathway is a promising target for melanoma (Javelaud et al, 2007). In addition, Rizos’s team further illustrated the link between the TGF‐β and melanoma therapy resistance. They showed that TGF‐β promotes a dedifferentiation phenotype, which is a common mechanism of resistance to PD‐1 inhibitors (Lee et al, 2020).
Several questions remain unsolved. We previously reported that activated AhR reprograms the transcriptome of melanoma cells mediating BRAFi resistance. In this study, we demonstrate that a SMAD3‐regulated gene expression program promotes therapy resistance in cutaneous melanoma and EMT. Importantly, SMAD3 expression levels during resistance acquisition are dependent, at least in part, on AhR. Thus, it would be interesting to precisely define the role of AhR and SMAD3 in the induction of each BRAFi‐resistance gene. To date, no physical interaction between AhR and SMAD3 proteins has been reported, suggesting that AhR acts as an upstream regulator of SMAD3 axis. It is noteworthy that the increased expression levels of SMAD3 by AhR expands the possibility of fine tuning gene expression since SMAD3 interacts with SMAD2 but also with JUN, TEADs, and YAP1 (Zhang et al, 1998; Fujii et al, 2012; Piersma et al, 2015). Because these three transcription factors have also been associated with therapies resistance in melanoma (Nallet‐Staub et al, 2014; Ramsdale et al, 2015; Verfaillie et al, 2015; Hugo et al, 2015b), our results suggest that regulation of BRAFi‐resistance genes expression is multiparametric and probably more sophisticated than initially though. Nonetheless, the elucidation of these transcriptional programs and networks governing BRAFi‐resistance genes and relapse is important for optimal target selection and the development of rationale and effective combination strategies.
BRAFi resistance may be achieved through the exposure of melanoma cells to TGF‐β, demonstrating that transcriptome reprogramming may confer resistance without the need for pre‐existing or de novo mutations (Viswanathan et al, 2017). The TGF‐β pathway promotes a shift toward the mesenchymal state (Antony et al, 2019). The resulting dedifferentiation modifies the expression of the adhesion molecules in the cell, supporting a migratory and invasive behavior. Together, our results strongly indicate that the SMAD3‐regulated genes are critical players in melanoma resistance to therapies by promoting an EMT‐like process. EMT reversal represents a powerful approach, as it may reduce the invasive behavior of cancer cells and favor re‐differentiation, synonymous of a decrease in BRAFi‐resistance gene expression (Giannelli et al, 2014; Rodón et al, 2015). By combining anti‐EMT drug and targeted therapy such as SMAD3i and BRAFi, we should efficiently reduce amount of persister cells. We anticipate that SMAD3 inhibition should limit the risk of resistance to therapies, since a decrease of expression levels of several BRAFi‐resistance genes is obtained with SMAD3i (SIS3). SMAD3 inhibition is expected to be more efficient than inhibitors targeting single downstream targets such as AXL or EGFR. In conclusion, our work highlights novel drug vulnerabilities that can be exploited to develop long‐lasting antimelanoma therapies.
Given the plasticity of melanoma cells and the capability of tumor microenvironment to produce TGF‐β (Chakravarthy et al, 2018), our work also warrants further investigation of the source of TGF‐β as another approach to prevent acquisition of the therapy‐resistant mesenchymal phenotype.
Materials and Methods
Reagents
DMSO—Sigma‐Aldrich (D8418)
BRAF inhibitors: Vemurafenib (PLX4032)—Selleckchem (RG7204); Paradox Breaker (PLX8394)—MedChemExpress (HY‐18972)
SMAD3 inhibitor: SIS3—Santa Cruz Biotechnology (sc‐222318)
MEK inhibitor: Cobimetinib—Selleckchem (GDC‐0973)
2,3,7,8‐TCDD (TCDD)—Sigma‐Aldrich (48599)
2‐(1H‐Indol‐3‐ylcarbonyl)‐4‐thiazolecarboxylic acid methyl ester (ITE)—MedChemExpress (HY‐19317)
CH‐223191—Selleckchem (S7711)
TGF‐β recombinant—Santa Cruz Biotechnology (240‐B‐010)
BIRC inhibitor: Birinapant—Selleckchem (S7015)
Cell lines and culture conditions
501Mel, Me1402, and HEK293T cell lines were obtained from ATCC. SKMel28 S & R cell lines were obtained from J.C Marine’s laboratory at VIB Center for Cancer Biology, VIB, Leuven, Belgium. 501Mel and SKMel28 AhR knockout cell lines have been established as previously described (Corre et al, 2018). M229S, M229R, M238S, M238R, and M249 were obtained from Thomas Graeber’s laboratory at department of Molecular and Medical Pharmacology, University of California, Los Angeles, USA. All melanoma cell lines were grown in humidified air (37°C, 5% CO2) in RPMI‐1640 medium (Gibco BRL, Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum (FBS) (PAA cell culture company) and 1% penicillin–streptomycin (PS) antibiotics (Gibco, Invitrogen). HEK293T was grown in DMEM (Gibco BRL, Invitrogen, Paisley, UK) supplemented as melanoma cell lines media. SKMel28R, M229R, and M238R are cultivated in presence of 0.1 µM vemurafenib. All cell lines have been routinely tested for mycoplasma contamination (Mycoplasma contamination detection kit; rep‐pt1; InvivoGen—San Diego—CA).
CRISPR‐SAM screens
A detailed protocol is available as supplementary Methods. Briefly, lentiviral productions have been performed as recommended (http://tronolab.epfl.ch), using HEK293T cells, psPAX2, pVSVG, and vectors required for CRISPR‐SAM according to Zhang lab (Joung et al, 2017). Infections were performed overnight in presence of 4 or 8 μg of polybrene per ml. All vectors have been provided by Addgene. SgRNA Library was amplified and prepared as described by Zhang Lab (Joung et al, 2017). 501Mel cells were transduced to stably express dCAS‐VP64 (cat. no. 61425) and MS2‐P65‐HSF1 (cat. no. 61426), before to transduce them with SAM sgRNA library (lentiSAMv2, 3‐plasmid system, cat. no. 1000000057) at a MOI of 0.2. Infected cells have been selected using antibiotics: Blasticidin (2 µg/ml, 5 days), Hygromycin B (200 µg/ml, 5 days), and Zeocine (600 µg/ml, 5 days). In vitro CRISPR‐SAM screens (Fig 2) were conducted as described by Zhang laboratory (Joung et al, 2017). The 501Mel cells expressing the sgRNA library were split into three groups: DMSO (solvent for BRAFi), vemurafenib (PLX4032, 2 µM, Selleckchem), and Paradox Breaker (PLX8394, 2 µM, MedChemExpress). After 14 days, resistant cell populations have been amplified. A minimum of 36 × 106 cells has been pellet and further sgRNA enrichment analysis. Cell libraries have been cryopreserved at −80°C for further in vivo experiments. The results presented are pooled data from two independent screens. To generate 501Mel cells overexpressing individually SMAD3, BIRC3, or SLC9A5, 501Mel expressing dCAS‐VP64 and MS2‐P65‐HSF1 were transduced to stably express specific sgRNAs (Table EV8). Infected cells have been selected using zeocin (600 µg/ml, 5 days). Manipulations of lentivirus were performed in the biosafety level 3 containment laboratory core facility of the Biology and Health Federative Research Structure of Rennes (Biosit).
Xenograft
Nude mice (4 weeks) were purchased from Janvier Labs and maintained under specific pathogen‐free conditions in our accredited animal house (A 35_238_40). The animal study follows the 3R (replace_reduce_refine) framework and has been filed with and approved by the French Government Board (No. 04386.03). Animal welfare is a constant priority: animals were thus euthanized under anesthesia. No blinding was done for animal studies. The animals were randomly allocated into different groups.
To identify tumor‐promoting genes in vivo (Fig 1D), two cell populations were subcutaneously xenografted on female NMRI nude mice flanks (3 × 106 cells per mouse); 501Mel (6 mice) and 501Mel CRISPR‐SAM cell library (10 mice, Table EV2) for Fig 1. For xenograft of other libraries (Fig 4): Vem‐persister cells (n = 11 mice) and (PLX8394)‐persister cells (n = 12 mice) (Table EV7). Tumor volume was assessed according to the formula V = (W(2) × L)/2, during 20 weeks. After mice sacrifice, tumors were sampled and conserved at −80°C for further sgRNA enrichment analysis.
For individual validation of tumor‐promoting genes (Fig 1H–K): 3 × 106 of 501Mel cells overexpressing either SMAD3, BIRC3, or SLC9A5 were subcutaneously xenografted on female NMRI nude mice flanks (6 mice per group). In each group, mice were injected on both flanks: sgRNA 1 on right flank and sgRNA 2 on left flank (see Table EV8 for sgRNA sequences). Tumor growth was assessed as previously described (Gilot et al, 2017) until the endpoint (600 mm3).
In order to identify BRAFi‐resistance and tumor growth genes in vivo (Fig 4), three cell populations were subcutaneously xenografted on female NMRI nude mice flanks (3 × 106 cells per mice); 501Mel (6 mice), 501Mel CRISPR‐SAM vemurafenib resistant (10 mice), and 501Mel CRISPR‐SAM Paradox Breaker resistant (12 mice). Tumor growth was assessed as previously described (Gilot et al, 2017), during 20 weeks. After mice sacrifice, tumors were sampled and conserved at −80°C for further sgRNA enrichment analysis.
These experiments are compliant with all relevant ethical regulations regarding animal research.
sgRNA enrichment analysis
Genomic DNAs from cell pellets (>36.106 cells) and tumors (> 400 mg) were extracted using the Zymo Research Quick‐gDNA MidiPrep according to the manufacturer’s protocol. PCR amplifications and quality controls have been done as described by Zhang laboratory (Joung et al, 2017).
sgRNA sequencing
Sequencing was performed by the Human & Environmental Genomics core facility of Rennes on a HiSeq 1500 (Rapid SBS kit v2 1x100 cycles, Illumina). Base Calling was performed with Illumina's CASAVA pipeline (Version 1.8).
Bioinformatic analysis of sgRNA and gene hits
Data processing was conducted using the MAGeCK v0.5.6 software (Li et al, 2014). Briefly, read counts from different samples are first median‐normalized to adjust for the effect of library sizes and read count distributions (mageck count with option: norm‐method median). Then, in an approach similar to those used for differential RNA‐Seq analysis, the variance of read counts is estimated by sharing information across features and a negative binomial model is used to test whether sgRNA abundance differs significantly between the treatment conditions and the DMSO control. Positively or negatively selected sgRNA are ranked according to adjusted P‐values (false discovery rate) and gene log fold changes computed with the modified robust ranking aggregation algorithm implemented in MAGeCK (mageck test with options: norm‐method median, gene‐lfc‐method alphamedian, andadjust method fdr).
RNA interference
All siRNAs were transfected at 66 nM using Lipofectamine RNAiMAX (Invitrogen). For survival assay, 4,000 cells were seeded in 96‐well plates, in quadruplicates. The following day, cells have been FBS‐starved (1% FBS overnight). Thirty‐six hours after transfection, cells were exposed to DMSO or BRAFi (vemurafenib (PLX4032); Selleckchem; 1 µM). After 84 h of treatment, cell density was measured by methylene blue assay, as previously described (Gilot et al, 2011). For RNA analysis, 50,000 cells were seeded in 12‐well plates. Cells were harvested 48 h after transfection. All siRNAs were purchased from IDT DNA (Table EV8).
Treatment experiments
8,000 (SKMel28R) or 12,000 (Me1402) cells were seeded in 96‐well plates, in quadruplicates. For SMAD3i + vemurafenib combination treatment: 5,000 (501Mel) or 8,000 (M249, SKMel28R) cells were seeded in 96‐well plates, in triplicates. Cells were exposed, 6 h after seeding, to either DMSO, BRAFi (vemurafenib; 5 µM), or MEKi (Cobi, 1 µM) or combination BRAFi (5 µM) + SMAD3i (0, 3, 10, 15, or 20 µM or solvent) ± MEKi (1 µM) for 4 days. 8,000 (SKMel28R) or 12,000 (Me1402) cells were seeded in 96‐well plates, in quadruplicates. For Birinapant treatment (96 h): cells were exposed, 6 h after seeding, to either DMSO or Birinapant (100 nM). Cell density was evaluated using methylene blue assay, as previously described (Gilot et al, 2011).
Half maximal inhibitory concentrations (IC50 values)
Cell sensitivity to vemurafenib or SMAD3i (SIS3) has been established by cell density measurement and calculation of the IC50 using GraphPad (PRISM8.0®) as previously described (Corre et al, 2018). 5,000 (501Mel) or 8,000 cells (other cell lines) were plated and exposed to inhibitor(s) at indicated concentrations for 4 days. Cells have been exposed to inhibitors 6 h after plating.
Melanoma spheroids
Spheroids were prepared using the liquid overlay method. Briefly, 500 μl of melanoma cells (10,000 cell/ml) were added to a 24‐well plate coated with 1.5% agar (Invitrogen). Plates were left to incubate for 72 h; by this time, cells had organized into 3‐dimensional (3D) spheroids. Spheroids were then harvested and added into 1 ml of a solution of collagen I (2 mg/ml – Corning) with MEM 1X (Gibco), acetic acid 0.02N and neutralization buffer (HEPES 200 mM pH 7.4; sodium bicarbonate 2.2%; NaOH 0.2 N). The suspension was then added to a 12‐well plate coated with 1.5% agar. Normal medium was overlaid on top of the solidified collagen after 2 h of incubation. After 48 h, medium was renewed. Pictures of the invading spheroids were monitored at different times using a Zeiss microscope.
RNA extraction and RT–qPCR expression
Experiments have been done as previously described (Gilot et al, 2017). Primers used for RT–qPCR experiments are available in Table EV8.
Western blot experiments
Experiments were performed as previously described (Gilot et al, 2017). Membranes were probed with suitable antibodies and signals were detected using the LAS‐3000 Imager (Fuji Photo Film). The primary and secondary antibodies are described in Table EV8.
SMAD‐luciferase assay
Experiments are based on the SMAD‐responsive element: Cignal Lenti SMAD Reporter (luc) (CLS‐017L from Qiagen) and as control the Cignal Lenti Negative Control (luc). The SMAD‐responsive luciferase vector encodes the Firefly luciferase reporter gene under the control of a minimal (m)CMV promoter and tandem repeats of the SMAD Binding Element (SBE). Melanoma cells line were infected as previously described (Gilot et al, 2017), and infected cells were selected using antibiotic selection (puromycin 2 µg/ml, 7 days). Cells were exposed to TGFβ ± SMAD3i as detailed in Fig 5K legend. Luciferase assays were then performed with a Promega kit according to the manufacturer’s instructions. Data were expressed in arbitrary units, relative to the value of luciferase activity levels found in TGFβ‐exposed cells, arbitrarily set at 100%. Firefly luciferase activity was normalized to protein content using Bicinchoninic Acid Kit from Sigma‐Aldrich® and measured with using a luminometer (Centro XS3 LB960, Berthold Technologies).
Immunohistochemistry
Tissue samples from representative lesions (preprint: Marin‐Bejar et al, 2020) were collected and fixed in 4% paraformaldehyde for 24 h and then processed for paraffin embedding (HistoStar™ Embedding Workstation). Sections of 5 µm of thickness obtained from the paraffin‐embedded tissues (Thermo Scientific Microm HM355S microtome) were mounted on SuperFrost™ Plus Adhesion slides (Thermo Scientific).
Immunofluorescence with and without tyramide signal amplification
Antibody targeted SMAD3 was used for detecting the following protein: (rabbit, 1:100, Cell Signaling Technologies, #9513, incubation for 30 min at RT).
Furthermore, the Akoya Opal Polaris 7 Color Automation IHC Detection Kit (Akoya, NEL871001KT) was used for the tyramide signal amplification according to the manufacturer’s protocol. All dewaxing and staining steps were performed using the Leica Bond RX slide stainer with the standard settings of the Opal 7‐color (v5.2 plus) IHC protocol provided by the manufacturer. The steps specific for the staining is as follows: Antigen retrieval was performed for 20 min at 100°C with Bond TM Epitope Retrieval 1 solution (Leica, AR9961). Tissue was blocked for 15 min using a blocking buffer (TBS with 1% BSA (VWR, 22013, bovine serum albumin (BSA), fraction V, biotium (50 g))) with 10% normal goat serum (Invitrogen, 10000C). For introduction of the secondary‐HRP, the Envision+/HRP goat anti‐Rabbit (Dako Envision + Single Reagents, HRP, Rabbit, Code K4003) was used for the antibody raised in rabbit (SMAD3). The protein SMAD3 was detected using the OPAL 690 reagent.
After the staining in the Leica Bond RX, slides were washed twice for 2 min in TBS‐Tween‐20 (0.5%) and were then mounted using ProLong Diamond Antifade Mountant (Thermo Fisher, P36961). Numbers of count cells are indicated in Appendix Fig S2 source data file.
In silico analyses
Heat maps were generated with R‐packages heatmap3 (Zhao et al, 2014).
Gene Set Enrichment Analysis was performed using the Broad Institute software.
SKCM TCGA expression data were obtained using OncoLNC portal (http://www.oncolnc.org; (Anaya, 2016)). Cell state categorization into four differentiation states (Undifferentiated, Neural crest‐like, Transitory, Melanocytic) of SKCM TCGA tumors and were performed using expression data of previously established gene sets (Tsoi et al, 2018).
Genomic alterations of SKCM TCGA tumors were analyzed using cBioPortal (http://www.cbioportal.org).
CCLE cell lines expression data and IC50 were obtained from GSE36139 and from the original publication (Barretina et al, 2012), respectively.
Expression data of 501Mel were obtained from our previously published RNA‐seq (GSE95589) (Gilot et al, 2017).
Expression data of M229, M238, and SKMel28 at different resistance steps were obtained from GSE75313 (Song et al, 2017).
Patient’s median expression of our 18 hits in baseline vs relapse (BRAFi) is based on expression data from GSE65186 (Hugo et al, 2015b) and GSE50509 (Rizos et al, 2014).
Expression data of invasive vs proliferative cell lines were obtained from GSE60666 (Verfaillie et al, 2015).
Analysis of our 18 hits expression in EGFR‐positive sorted cells was realized based on GSE97682 (Shaffer et al, 2017).
Analysis of SMAD3, BIRC3, and EGFR expression among 52 cell lines previously categorized as Undifferentiated, Neural crest‐like, Transitory, or Melanocytic were performed using http://systems.crump.ucla.edu/dediff and GSE80829.
SMAD3 ChIP‐Seq data have been obtained from GSE92443 (Ramachandran et al, 2018).
The comparison of the median SMAD3 signature in anti‐PD‐1 responsive vs resistant patients has been performed via expression data from GSE78220 (Hugo et al, 2016).
SMAD3 signature median expression was compared to melanocytic one (MITF, OCA2, MLANA, TYR, DCT) and mesenchymal one (52 genes; (Mak et al, 2016)) via expression data from SKCM TCGA obtained from OncoLNC.
The comparison between median expression of SMAD3 signature with classical, proneural, and mesenchymal ones in a cohort of glioblastoma patients was based on expression data from GSE103366 (Jin et al, 2017).
Gene Set Enrichment Analysis on Huh28 ± TGF‐β was realized based on GSE102109 (Merdrignac et al, 2018). Gene Set Enrichment Analysis on 3sp cells (mesenchymal) versus 3p cells (epithelial) was realized based on GSE26391 (van Zijl et al, 2011).
Single‐cell RNA‐seq data of a BRAF‐mutant PDX model undergoing BRAF and MEK inhibition were retrieved (GSE116237). The 674 single cells were projected into a two‐dimensional space using t‐distributed stochastic neighbor embedding (tSNE) and colored according to their drug‐tolerant state (DTC) identity (Rambow et al, 2018). In a second step, the activity of the resistance gene expression signature (18 genes) was quantified per cell using the AUCell algorithm (Aibar et al, 2017) resulting into an AUCell score (0 < range<0.4), which was used to gradient‐color the tSNE plot. Finally, the activity of the resistance gene signature was quantified per DTC population using GraphPad (****P < 0.0001, Mann–Whitney test).
Statistical analyses
Data are presented as mean ± s.d. unless otherwise specified, and differences were considered significant at a P‐value of less than 0.05. In the box plots, the line within the box is the median, the bottom and top of the box are, respectively, the first and the third quartiles, and the whiskers represent the minimum and maximum of all the data points. Comparisons between groups normalized to a control were carried out using bilateral Student’s test (with non‐equivalent variances). When at least two factors were compared between two groups, a two‐way ANOVA (without adjustment) was used as specified in figure legends. The statistical significance between two independent groups of patient samples was examined using the Mann–Whitney test. All statistical analyses were performed using Prism 8 software (GraphPad, La Jolla, CA, USA) or Microsoft Excel software. All experiments were performed three or more times independently under similar conditions, unless otherwise specified in the figure legends and statistics table containing the raw data (Appendix Fig S2–S3 source data) and Appendix Table S1 (P‐values).
Author contributions
Conceptualization: AG and DG. Methodology: AG, LB, AMQ, MA, and DG. Software: AG, MA, SC, CC, and FR. Formal analysis: AG, LB, AMQ, MA, DL, SC, OM‐B, CC, and FR. Investigation: AG, LB, AMQ, AP, MA, DL, SC, AP, HML, OM‐B, NT, and DG. Writing‐original draft: AG and DG. Writing‐review and editing: AG, DG, LB, AMQ, SC, M‐DG, FR, MA, CC, and J‐CM. Visualization: AG, LB, AMQ, MA, SC, FR, CC, and DG. Supervision: DG. Project Administration: DG and M‐DG. Funding: DG, SC, and M‐DG.
Conflict of interest
The authors declare that they have no conflict of interest.
For more information.
The Graeber laboratory provides a webtool to investigate the expression profile of your favorite RNAs in 53 melanoma cell lines, classified in four categories based on the differentiation status.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The authors thank the Gene Expression and Oncogenesis team from the CNRS UMR6290 especially M. Migault, A. Forestier, and L. Boussemart; the Rennes FHU CAMIn team; and E. Watrin and C. Hitte for providing scientific expertise. The authors acknowledge the SFR Biosit core facilities of Rennes 1 University with the ARCHE animal housing facility, the cell culture L3 facility, and the Human and Environmental Genomics platform for their help and support. This study received financial support from the following: Fondation ARC pour la Recherche (ARC labellized team); Ligue Nationale Contre le Cancer (LNCC); Départements du Grand‐Ouest; Région Bretagne; University of Rennes 1; CNRS; SFR Biosit, Association Vaincre le Cancer. Further support was provided by a “Ligue Nationale Contre le Cancer” (LNCC) Grand Ouest fellowship (AG) and from the Région Bretagne (AG), and Fondation ARC pour la Recherche (AG) and the LNCC (AQ) and from French Ministry of Research (NT and DL) and from Institut National contre le Cancer (INCa) (AP). The authors are grateful to Feng Zhang for providing the Human CRISPR 3‐plasmid activation pooled library (SAM) (Addgene).
EMBO Mol Med (2021) 13: e13466.
Contributor Information
Marie‐Dominique Galibert, Email: mgaliber@univ-rennes1.fr.
David Gilot, Email: david.gilot@univ-rennes1.fr.
Data availability
The datasets generated during current study are available: https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8595/.
References
- Aibar S, González‐Blas CB, Moerman T, Huynh‐Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J‐C, Geurts P, Aerts J et al (2017) SCENIC: single‐cell regulatory network inference and clustering. Nat Methods 14: 1083–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B et al (2015) Genomic classification of cutaneous melanoma. Cell 161: 1681–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaya J (2016) OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput Sci 2: e67 [Google Scholar]
- Antony J, Thiery JP, Huang RY‐J (2019) Epithelial‐to‐mesenchymal transition: Lessons from development, insights into cancer and the potential of EMT‐subtype based therapeutic intervention. Phys Biol 16(4): 041004 [DOI] [PubMed] [Google Scholar]
- Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L et al (2016) Cobimetinib combined with vemurafenib in advanced BRAFV600‐mutant melanoma (coBRIM): updated efficacy results from a randomised, double‐blind, phase 3 trial. Lancet Oncol 17: 1248–1260 [DOI] [PubMed] [Google Scholar]
- Bai X, Fisher DE, Flaherty KT (2019) Cell‐state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat Rev Clin Oncol 16: 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta‐Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B et al (2018) Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 174: 1034–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al (2012) The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M et al (2019) Prioritization of cancer therapeutic targets using CRISPR‐Cas9 screens. Nature 568: 511–516 [DOI] [PubMed] [Google Scholar]
- Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P (2001) Transforming growth Factor‐b1 increases survival of human melanoma through stroma remodeling. Cancer Res 61: 8306–8316 [PubMed] [Google Scholar]
- Boshuizen J, Koopman LA, Krijgsman O, Shahrabi A, Van Den Heuvel EG, Ligtenberg MA, Vredevoogd DW, Kemper K, Kuilman T, Song JY et al (2018) Cooperative targeting of melanoma heterogeneity with an AXL antibody‐drug conjugate and BRAF/MEK inhibitors. Nat Med 24: 203–212 [DOI] [PubMed] [Google Scholar]
- Campesato LF, Budhu S, Tchaicha J, Weng C‐H, Gigoux M, Cohen IJ, Redmond D, Mangarin L, Pourpe S, Liu C et al (2020) Blockade of the AHR restricts a Treg‐macrophage suppressive axis induced by L‐Kynurenine. Nat Commun 11: 4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD (2018) TGF‐β‐associated extracellular matrix genes link cancer‐associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 9: 4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chihara Y, Shimoda M, Hori A, Ohara A, Naoi Y, Ikeda J, Kagara N, Tanei T, Shimomura A, Shimazu K et al (2017) A small‐molecule inhibitor of SMAD3 attenuates resistance to anti‐HER2 drugs in HER2‐positive breast cancer cells. Breast Cancer Res Treat 166: 55–68 [DOI] [PubMed] [Google Scholar]
- Corre S, Tardif N, Mouchet N, Leclair HM, Boussemart L, Gautron A, Bachelot L, Perrot A, Soshilov A, Rogiers A et al (2018) Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF‐inhibitors in melanoma. Nat Commun 9: 4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, Ito H, Murakami H, Kondo Y, Kondo E et al (2012) TGF‐β synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J Exp Med 209: 479–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannelli G, Villa E, Lahn M (2014) Transforming growth factor‐ as a therapeutic target in hepatocellular carcinoma. Cancer Res 74: 1890–1894 [DOI] [PubMed] [Google Scholar]
- Gilot D, Le Meur N, Giudicelli F, Le Vée M, Lagadic‐Gossmann D, Théret N, Fardel O (2011) RNAi‐based screening identifies kinases interfering with dioxin‐mediated up‐regulation of CYP1A1 activity. PLoS One 6: e18261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilot D, Migault M, Bachelot L, Journé F, Rogiers A, Donnou‐Fournet E, Mogha A, Mouchet N, Pinel‐Marie ML, Mari B et al (2017) A non‐coding function of TYRP1 mRNA promotes melanoma growth. Nat Cell Biol 19: 1348–1357 [DOI] [PubMed] [Google Scholar]
- Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, Krauthammer M, McCusker JP, Kluger Y, Sznol M (2010) PLX4032, a selective BRAFV600Ekinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAFWTmelanoma cells. Pigment Cell Melanoma Res 23: 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, Goding CR (2010) Cancer stem cells versus phenotype‐switching in melanoma. Pigment Cell Melanoma Res 23: 746–759 [DOI] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB et al (2015a) Non‐genomic and immune evolution of melanoma acquiring MAPKi resistance HHS public access. Cell 162: 1271–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB et al (2015b) Non‐genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162: 1271–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S, Berent‐Maoz B, Pang J, Chmielowski B, Cherry G et al (2016) Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 165: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javelaud D, Mohammad KS, McKenna CR, Fournier P, Luciani F, Niewolna M, André J, Delmas V, Larue L, Guise TA et al (2007) Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res 67: 2317–2324 [DOI] [PubMed] [Google Scholar]
- Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T, Gimple RC, Wang X, Mack SC, Miller TE et al (2017) Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med 23: 1352–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Tamaki K (2006) Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor‐beta1‐induced extracellular matrix expression. Mol Pharmacol 69: 597–607 [DOI] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F (2017) Genome‐scale CRISPR‐Cas9 knockout and transcriptional activation screening. Nat Protoc 12: 828–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H et al (2015) Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 517: 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT, Barzily‐Rokni M, Straussman R, Haq R et al (2014) A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4: 816–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Chunduru SK, Halloran MB, He X, Xiao M, Vultur A, Villanueva J, Mitsuuchi Y, Neiman EM, Benetatos C et al (2013) The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin Cancer Res 19: 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamar JM, Stern P, Liu H, Schindler JW, Jiang Z‐G, Hynes RO (2012) The Hippo pathway target, YAP, promotes metastasis through its TEAD‐interaction domain. Proc Natl Acad Sci 109: E2441–E2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc D, Staats Pires AC, Guillemin GJ, Gilot D (2021) Detrimental activation of AhR pathway in cancer: an overview of therapeutic strategies. Curr Opin Immunol 70: 15–26 [DOI] [PubMed] [Google Scholar]
- Lee JH, Shklovskaya E, Lim SY, Carlino MS, Menzies AM, Stewart A, Pedersen B, Irvine M, Alavi S, Yang JYH et al (2020) Transcriptional downregulation of MHC class I and melanoma de‐ differentiation in resistance to PD‐1 inhibition. Nat Commun 11: 1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, Radaelli E, Eyckerman S, Leonelli C, Vanderheyden K et al (2016) Melanoma addiction to the long non‐coding RNA SAMMSON. Nature 531: 518–522 [DOI] [PubMed] [Google Scholar]
- Levy C, Khaled M, Fisher DE (2006) MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12: 406–414 [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS (2014) MAGeCK enables robust identification of essential genes from genome‐scale CRISPR/Cas9 knockout screens. Genome Biol 15: 554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, Ji T, Zhang H, Dong W, Jin X et al (2017) Blockade of IDO‐kynurenine‐AhR metabolic circuitry abrogates IFN‐γ‐induced immunologic dormancy of tumor‐repopulating cells. Nat Commun 8: 15207 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lo R, Matthews J (2012) High‐resolution genome‐wide mapping of AHR and ARNT binding sites by ChIP‐Seq. Toxicol Sci 130: 349–361 [DOI] [PubMed] [Google Scholar]
- Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, Skoulidis F, Parra ER, Rodriguez‐Canales J, Wistuba II et al (2016) A patient‐derived, pan‐cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial‐to‐mesenchymal transition. Clin Cancer Res 22: 609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Bejar O, Rogiers A, Dewaele M, Femel J, Karras P, Pozniak J, Bervoets G, Van Raemdonck N, Pedri D, Swings T et al (2020) A neural crest stem cell‐like state drives nongenetic resistance to targeted therapy in melanoma. bioRxiv 10.1101/2020.12.15.422929 [PREPRINT] [DOI] [Google Scholar]
- Merdrignac A, Angenard G, Allain C, Petitjean K, Bergeat D, Bellaud P, Fautrel A, Turlin B, Clément B, Dooley S et al (2018) A novel transforming growth factor beta‐induced long noncoding RNA promotes an inflammatory microenvironment in human intrahepatic cholangiocarcinoma. Hepatol Commun 2: 254–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S et al (2017) Computational correction of copy number effect improves specificity of CRISPR‐Cas9 essentiality screens in cancer cells. Nat Genet 49: 1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, Koya RC, Samatar AA, Khanlou N, Braun J et al (2015) Tunable‐combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 27: 240–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, Kong X, Possik PA, Cornelissen‐Steijger PDM, Foppen MHG et al (2014) Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5: 5712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallet‐Staub F, Marsaud V, Li L, Gilbert C, Dodier S, Bataille V, Sudol M, Herlyn M, Mauviel A (2014) Pro‐invasive activity of the hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol 134: 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar D, Blanpain C (2016) Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol Mech Dis 11: 47–76 [DOI] [PubMed] [Google Scholar]
- Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac‐de Paillerets B, Hofman P et al (2011) Senescent cells develop a PARP‐1 and nuclear factor‐ B‐associated secretome (PNAS). Genes Dev 25: 1245–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooshima A, Park J, Kim SJ (2019) Phosphorylation status at Smad3 linker region modulates transforming growth factor‐β‐induced epithelial‐mesenchymal transition and cancer progression. Cancer Sci 110: 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris A, Tardif N, Galibert MD, Corre S (2021) AhR and cancer: from gene profiling to targeted therapy. Int J Mol Sci 22: 752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL et al (2014) Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piersma B, Bank RA, Boersema M (2015) Signaling in fibrosis: TGF‐β, WNT, and YAP/TAZ converge. Front Med 2: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483: 100–103 [DOI] [PubMed] [Google Scholar]
- Puisieux A, Brabletz T, Caramel J (2014) Oncogenic roles of EMT‐inducing transcription factors. Nat Cell Biol 16: 488–494 [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Vizán P, Das D, Chakravarty P, Vogt J, Rogers KW, Müller P, Hinck AP, Sapkota GP, Hill CS (2018) TGF‐β uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial‐to‐mesenchymal transition. Elife 7: 1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin‐Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec‐Rychter M et al (2018) Toward minimal residual disease‐directed therapy in melanoma. Cell 174: 843–855 [DOI] [PubMed] [Google Scholar]
- Ramsdale R, Jorissen RN, Li FZ, Al‐Obaidi S, Ward T, Sheppard KE, Bukczynska PE, Young RJ, Boyle SE, Shackleton M et al (2015) The transcription cofactor c‐JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 8: ra82 [DOI] [PubMed] [Google Scholar]
- Rapino F, Delaunay S, Rambow F, Zhou Z, Tharun L, De Tullio P, Sin O, Shostak K, Schmitz S, Piepers J et al (2018) Codon‐specific translation reprogramming promotes resistance to targeted therapy. Nature 558: 605–609 [DOI] [PubMed] [Google Scholar]
- Redfern AD, Spalding LJ, Thompson EW (2018) The Kraken Wakes: induced EMT as a driver of tumour aggression and poor outcome. Clin Exp Metastasis 35: 285–308 [DOI] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC et al (2014) BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 20: 1965–1977 [DOI] [PubMed] [Google Scholar]
- Rizzolio S, Cagnoni G, Battistini C, Bonelli S, Isella C, Van Ginderachter JA, Bernards R, Di Nicolantonio F, Giordano S, Tamagnone L (2018) Neuropilin‐1 upregulation elicits adaptive resistance to oncogene‐targeted therapies. J Clin Invest 128: 3976–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodón J, Carducci M, Sepulveda‐Sánchez JM, Azaro A, Calvo E, Seoane J, Braña I, Sicart E, Gueorguieva I, Cleverly A et al (2015) Pharmacokinetic, pharmacodynamic and biomarker evaluation of transforming growth factor‐β receptor I kinase inhibitor, galunisertib, in phase 1 study in patients with advanced cancer. Invest New Drugs 33: 357–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F et al (2018) Optimized libraries for CRISPR‐Cas9 genetic screens with multiple modalities. Nat Commun 9: 5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M et al (2017) Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature 546: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F (2015) High‐throughput functional genomics using CRISPR–Cas9. Nat Rev Genet 16: 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MP, Brunton H, Rowling EJ, Ferguson J, Arozarena I, Miskolczi Z, Lee JL, Girotti MR, Marais R, Levesque MP et al (2016) Inhibiting drivers of non‐mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 29: 270–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Piva M, Sun L, Hong A, Moriceau G, Kong X, Zhang H, Lomeli S, Qian J, Yu CC et al (2017) Recurrent tumor cell‐intrinsic and ‐extrinsic alterations during MAPKi‐induced melanoma regression and early adaptation. Cancer Discov 7: 1248–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RJ, Flaherty KT (2013) Resistance to BRAF‐targeted therapy in melanoma. Eur J Cancer 49: 1297–1304 [DOI] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM et al (2014) Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508: 118–122 [DOI] [PubMed] [Google Scholar]
- Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R, Vanderhoydonc F, Duarte JAG, Bosisio F, Van den Eynde K et al (2018) Sustained SREBP‐1‐dependent lipogenesis as a key mediator of resistance to BRAF‐targeted therapy. Nat Commun 9: 2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang PM‐K, Zhou S, Meng X‐M, Wang Q‐M, Li C‐J, Lian G‐Y, Huang X‐R, Tang Y‐J, Guan X‐Y, Yan BP‐Y et al (2017) Smad3 promotes cancer progression by inhibiting E4BP4‐mediated NK cell development. Nat Commun 8: 14677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 352: 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titz B, Lomova A, Le A, Hugo W, Kong X, ten Hoeve J, Friedman M, Shi H, Moriceau G, Song C et al (2016) JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. Cell Discov 2: 16028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X et al (2018) Multi‐stage differentiation defines melanoma subtypes with differential vulnerability to drug‐induced iron‐dependent oxidative stress. Cancer Cell 33: 890–904.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncer E, Calçada RR, Zingg D, Varum S, Cheng P, Freiberger SN, Deng C‐X, Kleiter I, Levesque MP, Dummer R et al (2019) SMAD signaling promotes melanoma metastasis independently of phenotype switching. J Clin Invest 129: 2702–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L et al (2015) Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun 6: 6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetma V, Rožanc J, Charles EM, Hellwig CT, Alexopoulos LG, Rehm M (2017) Examining the in vitro efficacy of the IAP antagonist birinapant as a single agent or in combination with dacarbazine to induce melanoma cell death. Oncol Res Featur Preclin Clin Cancer Ther 25: 1489–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore‐Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ et al (2017) Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature 547: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC et al (2011) Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29: 3085–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh SJ, Rizos H, Scolyer RA, Long GV (2016) Resistance to combination BRAF and MEK inhibition in metastatic melanoma: where to next? Eur J Cancer 62: 76–85 [DOI] [PubMed] [Google Scholar]
- Wu N, Lian G, Sheng J, Wu D, Yu X, Lan H, Hu W, Yang Z (2020) Discovery of a novel selective water‐soluble SMAD3 inhibitor as an antitumor agent. Bioorg Med Chem Lett 30: 127396 [DOI] [PubMed] [Google Scholar]
- Yang SY, Ahmed S, Satheesh SV, Matthews J (2018) Genome‐wide mapping and analysis of aryl hydrocarbon receptor (AHR)‐ and aryl hydrocarbon receptor repressor (AHRR)‐binding sites in human breast cancer cells. Arch Toxicol 92: 225–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng XH, Derynck R (1998) Smad3 and Smad4 cooperate with c‐Jun/c‐Fos to mediate TGF‐beta‐induced transcription. Nature 394: 909–913 [DOI] [PubMed] [Google Scholar]
- Zhao S, Guo Y, Sheng Q, Shyr Y (2014) Advanced heat map and clustering analysis using heatmap3. Biomed Res Int 2014: 986048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zijl F, Mall S, Machat G, Pirker C, Zeillinger R, Weinhaeusel A, Bilban M, Berger W, Mikulits W (2011) A human model of epithelial to mesenchymal transition to monitor drug efficacy in hepatocellular carcinoma progression. Mol Cancer Ther 10: 850–860 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The datasets generated during current study are available: https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8595/.