

Abstract
Age‐related macular degeneration (AMD) in its various forms is a leading cause of blindness in industrialized countries. Here, we provide evidence that ligands for neuropilin‐1 (NRP1), such as Semaphorin 3A and VEGF‐A, are elevated in the vitreous of patients with AMD at times of active choroidal neovascularization (CNV). We further demonstrate that NRP1‐expressing myeloid cells promote and maintain CNV. Expression of NRP1 on cells of myeloid lineage is critical for mitigating production of inflammatory factors such as IL6 and IL1β. Therapeutically trapping ligands of NRP1 with an NRP1‐derived trap reduces CNV. Collectively, our findings identify a role for NRP1‐expressing myeloid cells in promoting pathological angiogenesis during CNV and introduce a therapeutic approach to counter neovascular AMD.
Keywords: age‐related macular degeneration, angiogenesis, inflammation, mononuclear phagocytes, neuropilin‐1
Subject Categories: Vascular Biology & Angiogenesis
A population of innate immune myeloid cells expressing NRP1 receptor invades the retina where it drives and maintains pathological neovascularization during age‐related macular degeneration (AMD). A recombinant NRP1‐derived trap prevents choroidal neovascularization‐associated pathological angiogenesis.

The paper explained.
Problem
Age‐related macular degeneration (AMD) is a slowly progressing condition of the aging eye and the leading cause of central vision loss in industrialized countries. Advanced AMD is often classified into “dry” atrophic AMD or “wet” neovascular (NV) AMD. Wet AMD typically occurs when neovascularization from the choroid (choroidal neovascularization; CNV), sprouts into the subretinal space and neuro‐retina, hemorrhages, leaks and ultimately provokes photoreceptor death, fibrovascular scarring, and retinal detachment of the macular region. This can rapidly compromise the central visual field.
Results
In the current study, we show that several ligands of Neuropilin1 (NRP1, a transmembrane receptor that binds several growth factors and guidance cues and potentiates their signaling) are induced in the vitreous of patients with active NV AMD. We observed that myeloid cells expressing NRP1 contribute to pathological angiogenesis in later stages of CNV in mice. Moreover, we demonstrate that while mononuclear phagocyte‐resident NRP1 is not essential for recruitment of immune cells to sites of CNV, it is critical for mitigating myeloid cell inflammation and favors alternative activation. Using a NRP1‐derived trap, we significantly reduced CNV.
Impact
Our study ultimately shows that therapeutic targeting of NRP1 ligands or NRP1‐expressing myeloid cells hinders CNV.
Introduction
Age‐related macular degeneration (AMD) is a slowly progressing condition of the aging eye and the leading cause of central vision loss in industrialized countries (Friedman et al, 2004; Rein et al, 2006; Jonas et al, 2014; Wong et al, 2014). Central vision loss from AMD poses a significant burden on health care systems and profoundly impacts well‐being and mental health (Taylor et al, 2016). In the early asymptomatic stages of disease, insoluble extracellular lipid aggregates termed drusen accumulate in the subretinal space. Inadequate clearance of these deposits can trigger a pathologic inflammatory response and ensuing tissue damage (Guillonneau et al, 2017; Handa et al, 2017). Advanced AMD is often classified into “dry” atrophic AMD or “wet” neovascular (NV) AMD. Visual impairment in the late stages of atrophic AMD is characterized by areas with progressive degeneration of the retinal pigment epithelium (RPE) and the photoreceptors that rely on RPE for support (Ambati & Fowler, 2012; Lim et al, 2012). While vision loss resulting from the dry form of AMD is typically gradual and protracted, wet AMD can rapidly compromise the central visual field. This typically occurs when neovascularization from the choroid (choroidal neovascularization; CNV) sprouts into the subretinal space and neuro‐retina, hemorrhages, leaks and ultimately provokes photoreceptor death, fibrovascular scarring and retinal detachment of the macular region (Lim et al, 2012).
To date, the mechanisms that precipitate NV AMD remain only partially defined, with vascular endothelial growth factor A (VEGF‐A) playing a cardinal role in CNV (Ambati & Fowler, 2012). Current standards of care for wet AMD such as Aflibercept, Ranibizumab, and off‐label Bevacizumab target VEGF‐A and have revolutionized treatment of NV AMD. Unfortunately, long‐term use of anti‐VEGF‐A therapies may have limited efficacy (Comparison of Age‐related Macular Degeneration Treatments Trials Research Group et al, 2016), possible neuronal side‐effects (Robinson et al, 2001), and have been shown to cause degeneration of the RPE‐choriocapillaris complex in mouse models (Saint‐Geniez et al, 2008; Kurihara et al, 2012; Berber et al, 2017). In addition, approximately 1 out of 10 treated patients does not respond to anti‐VEGF‐A therapy (Krebs et al, 2013) with NV network complexes persisting despite monthly intravitreal injections. Therefore, alternative treatments that block retinal neovascularization in AMD are required. A pharmacogenomic link has been suggested with the variability in treatment response (Lazzeri et al, 2013; Lores‐Motta et al, 2018), and it has recently been proposed that a single‐nucleotide polymorphism (SNP) in Neuropilin‐1 (Nrp1; rs2070296) is associated with decreased anti‐VEGF‐A therapy response (Lores‐Motta et al, 2016).
NRP1 is a single‐pass transmembrane receptor with a large ~860 amino acid extracellular domain subdivided into 3 sub‐domains: a large extracellular domain with two CUB motifs (A1, A2), two domains with similarity to coagulation factor V/VIII (B1, B2), a MAM domain (C), and a single transmembrane domain (TM) followed by a short cytoplasmic domain (CD; Geretti et al, 2008). The A1, A2 and B1 domains bind Semaphorin 3A (SEMA3A) while the B1 and B2 domains bind VEGF‐A, transforming growth factor beta (TGF‐β), placental growth factor 2 (PGF) (Mamluk et al, 2002), and platelet‐derived growth factor (PDGF; Antipenko et al, 2003; Raimondi et al, 2016; Muhl et al, 2017; Miyauchi et al, 2018). NRP1 can collaborate with several receptors and their ligands such as VEGFR2 and VEGF‐A (Soker et al, 1998; Soker et al, 2002), Plexin A1 and SEMA3A (Takahashi et al, 1999), TGF‐βR and TGF‐β (Glinka et al, 2011), and PDGF‐R and PDGF‐BB (Ball et al, 2010) and hence has the potential to modulate multiple receptor signaling pathways (Nakamura & Goshima, 2002; Prud'homme & Glinka, 2012; Rizzolio, 2017). Of note, all above ligands are known to regulate angiogenesis, suggesting that NRP1‐mediated signaling could be of interest for NV AMD (Battegay et al, 1994; Massague et al, 2000; Hoeben et al, 2004; Acevedo et al, 2008; Funasaka et al, 2014).
In addition to vessels and neurons, NRP1 is highly expressed on cells of both the innate and adaptive immune system (Roy et al, 2017) where it plays an important role in homing and modulating myeloid cell function (Takamatsu & Kumanogoh, 2012; Casazza et al, 2013; Roy et al, 2017; Wilson et al, 2018) and in T‐cell migration (Lepelletier et al, 2007) and differentiation (Renand et al, 2013). Here, we sought to elucidate the contribution of myeloid‐resident NRP1 in NV AMD.
Results
NRP1 ligands are elevated in patients with NV AMD and in a mouse model of CNV
NRP1 has been implicated in diseases characterized by deregulated vasculature such as cancer (Ellis, 2006; Rizzolio & Tamagnone, 2011; Jubb et al, 2012; Prud'homme & Glinka, 2012; Chaudhary et al, 2014), and in diseases of the retina such as proliferative diabetic retinopathy (Dejda et al, 2014), diabetic macular edema (Cerani et al, 2013; Sodhi et al, 2019) and retinopathy of prematurity (Joyal et al, 2011; Dejda et al, 2014). We therefore sought to determine whether NRP1 ligands were present in NV AMD. We obtained vitreous from patients diagnosed with active proliferating NV AMD as determined by fundus imaging and optical coherence tomography (OCT) and from age‐ and sex‐matched control patients with non‐vascular retinal pathologies such as macular hole or epiretinal membranes. Representative horizontal B‐scan and thickness maps of an AMD patient with active CNV lesions and a control patient with a non‐vascular retinal pathology (macular hole) are shown in Fig 1A and B. Detailed characteristics of patients are included in Table 1. We used ELISA‐based detection for NRP1 ligands and found a significant increase in VEGF‐A, from 129 to 513 ng/ml and SEMA3A, from 0.001 to 0.33 ng/ml (Fig 1C and D). TGF‐β showed a trend toward increase from 1 ng/ml in control patients to 1.3 ng/ml in NV AMD patients (Fig 1E), while PDGF‐BB and PGF did not change (Fig 1F and G) as previously reported (Mimura et al, 2019).
Figure 1. NRP1 ligands are elevated in patients with NV AMD and in a mouse model of CNV.
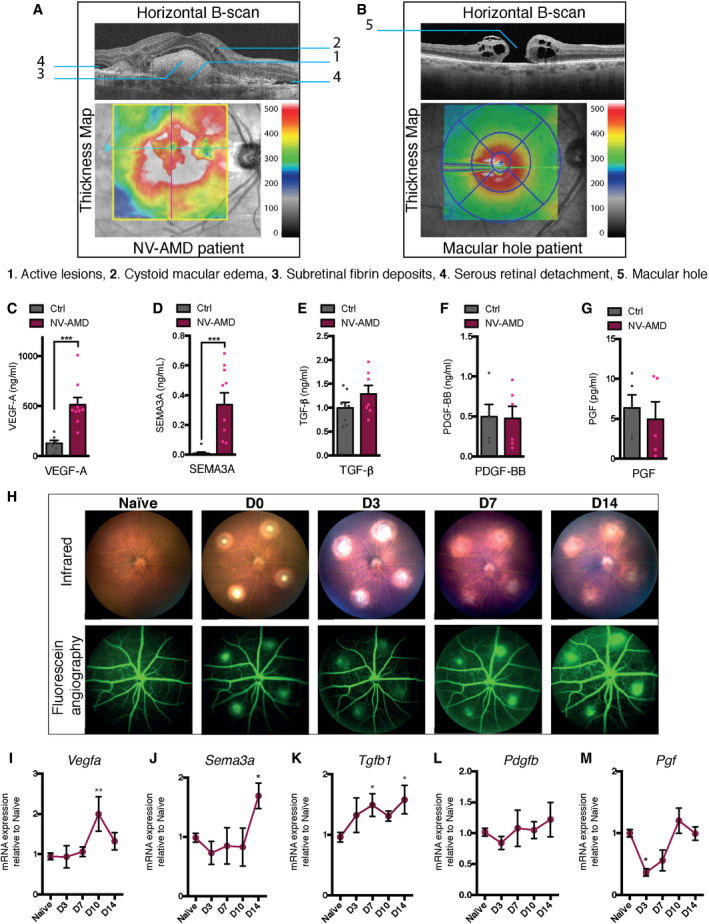
-
AOptical coherence tomography (OCT) horizontal B‐scan and thickness map of neovascular age‐related macular degeneration (NV AMD) patient with active lesions (1), cystoid macular edema (2), subretinal fibrin deposits (3), and serous retinal detachment (4).
-
BOptical coherence tomography (OCT) horizontal B‐scan and thickness map of control patient with a medium sized, stage 3, full thickness macular hole (5).
-
C–GVitreous humor analyzed by ELISA for VEGF‐A (C); n = 7 (Ctrl), 9 (NV AMD), SEMA3A (D); n = 10 (Ctrl), 10 (NV AMD), TGFβ (E); n = 8 (Ctrl), 7 (NV AMD), PDGF‐BB (F); n = 5 (Ctrl),6 (NV AMD), PGF (G): n = 5 (Ctrl), 5 (NV AMD). Dots represent concentrations of individual patient samples.
-
HMicron IV infrared and fluorescein in vivo imaging of naïve mouse fundus and following laser‐induced CNV at D0, D3, D7, D14.
-
I–MTime course of mRNA expression of NRP1 ligands in mouse RPE‐choroid‐sclera complexes relative to naïve (no burn), 3 (D3), 7 (D7), 10 (D10), and 14 (D14) days after burn for Vegfa (I); n = 11 (No burn), 6 (D3), 7 (D7), 4 (D10 and D14), Sema3a (J); n = 17 (No burn), 6 (D3), 4 (D7), 3 (D10 and D14), Tgfb1 (K); n = 14 (No burn), 3 (D3), 7 (D7), 3 (D10), 6 (D14), Pdgfb (L); n = 9 (No burn), 5 (D3 and D7), 4 (D10 and D14), Pgf (M); n = 6 (No burn), 4 (D3), 3 (D7), 6 (D10), 3 (D14).
Data information: All comparisons between groups were analyzed using a Student’s unpaired t‐test (C‐G) or a one‐way analysis of variance (ANOVA) and Dunnett’s multiple comparisons test (I‐M); *P < 0.05, **P < 0.01, ***P < 0.001; error bars represent mean ± SEM; exact P‐values listed in Appendix Table S1.
Table 1.
Characteristics of donors of vitreous samples used in Fig 1C–G
| Pathology | ELISA | Age |
|---|---|---|
| Control Patients | ||
| MH | VEGF, S3A, TGF, PDGF | – |
| ERM | VEGF, S3A, TGF, PDGF | 72 |
| MH | VEGF, S3A, TGF, PGF | 82 |
| ERM | VEGF, S3A, TGF | 57 |
| ERM | VEGF, S3A, TGF | 93 |
| ERM | VEGF, S3A | 76 |
| ERM | VEGF, PGF | 82 |
| MH | S3A, TGF, PDGF | 66 |
| ERM | TGF, PDGF | 54 |
| MH | TGF, PDGF | 65 |
| MH | S3A | 71 |
| ERM | S3A | 71 |
| ERM | S3A, PGF | 64 |
| ERM | PGF | 81 |
| ERM | PGF | 80 |
| Age Mean ± SEM; VEGF: 77 ± 4.9; S3A: 72 ± 3.5; TGF: 69 ± 5.2; PDGF: 64 ± 3.7; PGF: 78 ± 3.5 | ||
| NV AMD Patients | ||
| NV AMD | VEGF, S3A, TGF, PDGF | 96 |
| NV AMD | VEGF, S3A, TGF, PDGF | 79 |
| NV AMD | VEGF, S3A, TGF, PDGF | 86 |
| NV AMD | VEGF, S3A, TGF, PDGF | 75 |
| NV AMD | VEGF, S3A, TGF, PDGF | 76 |
| NV AMD | VEGF, TGF, PDGF | 80 |
| NV AMD | VEGF, S3A | 84 |
| NV AMD | VEGF | 80 |
| NV AMD | VEGF | 74 |
| NV AMD | S3A, TGF, PDGF, PGF | 79 |
| NV AMD | S3A, PGF | 91 |
| NV AMD | S3A | 84 |
| NV AMD | S3A, PGF | 80 |
| NV AMD | PGF | 71 |
| NV AMD | PGF | 85 |
| Age Mean ± SEM; VEGF: 81 ± 2.3; S3A: 83 ± 2.1; TGF: 82 ± 2.7; PDGF: 82 ± 2.7.; PGF: 81 ± 3.3 | ||
MH, Macular hole; ERM, Epiretinal membrane; NV AMD, Neovascular AMD.
Table 2.
Table of P‐values.
| Graph | Data | P‐value | Test used | Summary |
|---|---|---|---|---|
| Figure 1 | ||||
| C | ELISA | 0.0007 | T‐test | *** |
| D | ELISA | 0.0007 | T‐test | *** |
| I | qPCR | 0.0034 | ANOVA + Dunnett's | ** |
| J | qPCR | 0.0361 | ANOVA + Dunnett's | * |
| K | qPCR | 0.0338, 0.0167 | ANOVA + Dunnett's | *,* |
| M | qPCR | 0.0189 | ANOVA + Dunnett's | * |
| Figure 2 | ||||
| B | FACS | 0.0303 | T‐test | * |
| C | FACS | 0.0301 | T‐test | * |
| D | qPCR | 0.0001 | T‐test | *** |
| E | WB | < 0.0001 | T‐test | **** |
| L | CNV | 0.0089 | T‐test | ** |
| N | CNV | 0.0069 | T‐test | ** |
| Figure 3 | ||||
| A | qPCR | 0.0463 | T‐test | * |
| B | qPCR | 0.0063 | T‐test | ** |
| E | qPCR | 0.0409 | T‐test | * |
| J | WB | 0.0416 | T‐test | * |
| K | qPCR | 0.0335 | T‐test | * |
| L | qPCR | 0.0332 | T‐test | * |
| N | FACS | 0.0028 | T‐test | ** |
| O | FACS | 0.0025 | T‐test | ** |
| Figure 4 | ||||
| F | CNV | 0.0074 | T‐test | ** |
| H | CNV | 0.0005 | T‐test | *** |
| J | CNV | < 0.0001, 0.0197 | ANOVA + Tukey's | ****,* |
| L | CNV | < 0.0001, 0.0238 | ANOVA + Tukey's | ****,* |
| Expanded View Figure 1 | ||||
| D | FACS | 0.0002 | T‐test | *** |
| E | FACS | 0.0075 | T‐test | ** |
| Expanded View Figure 3 | ||||
| A | qPCR | 0.041 | T‐test | * |
T‐test = Student’s unpaired t‐test, ANOVA + Dunnett's = Ordinary one‐way ANOVA with Dunnett's multiple comparisons test, ANOVA + Tukey's = Ordinary one‐way ANOVA with Tukey's multiple comparisons test.
We next modeled NV AMD in mice by subjecting them to the laser‐induced photocoagulation model of CNV, where disruption of the Bruch’s membrane triggers sprouting of subretinal vessels from the choroid (Lambert et al, 2013) as depicted with fundus photography and fluorescein angiography (Fig 1H). Although this model does not mimic chronic aspects of human disease, it is relatively reproducible and widely used to model CNV in NV AMD. Following induction of CNV, we sacrificed mice and collected RPE‐choroid‐sclera complexes over the 2‐week period of active CNV (3, 7, 10, or 14 days after laser burn). We assessed transcript levels of Nrp1 ligand by real‐time quantitative PCR (RT‐qPCR) and Vegfa levels were found to rise significantly compared with naïve choroids at day 10 (Fig 1I), while Sema3a levels rose twofold by day 14 (Fig 1J). Tgfb1 transcripts increased as early as 3 days after laser and were elevated throughout the course until day 14 (Fig 1K). Consistent with PDGF‐BB levels in human vitreous (Fig 1F), levels of Pdgfb did not significantly vary (Fig 1L). Pgf expression dropped in the first week after burn and rose slightly around day 10 (Fig 1M) as reported by others (Crespo‐Garcia et al, 2017). Collectively, these data obtained in both humans and mice suggest that various ligands of NRP1 are elevated in NV AMD.
NRP1‐expressing mononuclear phagocytes rise in the retina upon injury and promote CNV
Under physiological conditions, the subretinal space and photoreceptor cell layer are devoid of mononuclear phagocytes (Guillonneau et al, 2017). In late AMD, the immunosuppressive subretinal environment is disturbed and mononuclear phagocytes accumulate and contribute to pathogenesis (Langmann, 2007; Ambati et al, 2013; Guillonneau et al, 2017; Rashid et al, 2019). Secondary to laser‐induced CNV, mononuclear phagocytes including microglia and circulating monocytes are recruited to sites of neovascularization (Yu et al, 2020). NRP1 is highly expressed on retinal mononuclear phagocytes (Dejda et al, 2016), and we have previously demonstrated a role for NRP1‐expressing myeloid cells in mediating pathological angiogenesis in oxygen‐induced retinopathy (Dejda et al, 2014; Dejda et al, 2016). Hence, we sought to determine the contribution of NRP1‐expressing mononuclear phagocytes to CNV.
Analysis by FACS of whole retinas and RPE‐choroid‐sclera complexes at day 3 (D3) of CNV revealed a rise in Ly6G−, F4/80+, CD11b+ mononuclear phagocytes in laser‐burned eyes when compared to controls (Fig 2A and B). Importantly, we observed a proportional threefold increase in NRP1+ mononuclear phagocytes at D3 when compared to non‐laser eyes (Fig 2C) (gating scheme and internal controls in Fig EV1, EV2, EV3A–C). In order to establish the role of mononuclear phagocyte‐resident NRP1 on CNV, we generated a myeloid‐specific knockout of Nrp1 by crossing Nrp1‐floxed mice with LysM‐Cre (LysM‐Cre/Nrp1+/+) mice, yielding LysM‐Cre/Nrp1fl/fl offspring. The resulting mice showed a robust reduction in Nrp1 transcript (Fig 2D) and protein (Fig 2E and F) expression in bone marrow‐derived macrophages (BMDM) when compared to LysM‐Cre/Nrp1+/+ littermate controls. FACS analysis revealed a ~30% reduction in NRP1+ microglia in the retina and RPE‐choroid‐sclera complexes before and 3 days after laser (Fig EV1, EV2, EV3D and E). LysM‐Cre/Nrp1fl/fl mice on regular diets did not show any difference in body weight, size, or open‐field activity when compared to littermates. Immunofluorescence of lesion sites on flat‐mounted RPE‐choroid‐sclera complexes at D3 post‐laser burn reveal that NRP1‐positive mononuclear phagocytes (labeled with IBA1) are recruited to sites of injury (Fig 2G) while, as expected, LysM‐Cre/Nrp1fl/fl mice did not show accretion of NRP1‐expressing mononuclear phagocytes.
Figure 2. NRP1‐expressing mononuclear phagocytes increase in the retina upon injury and promote CNV.
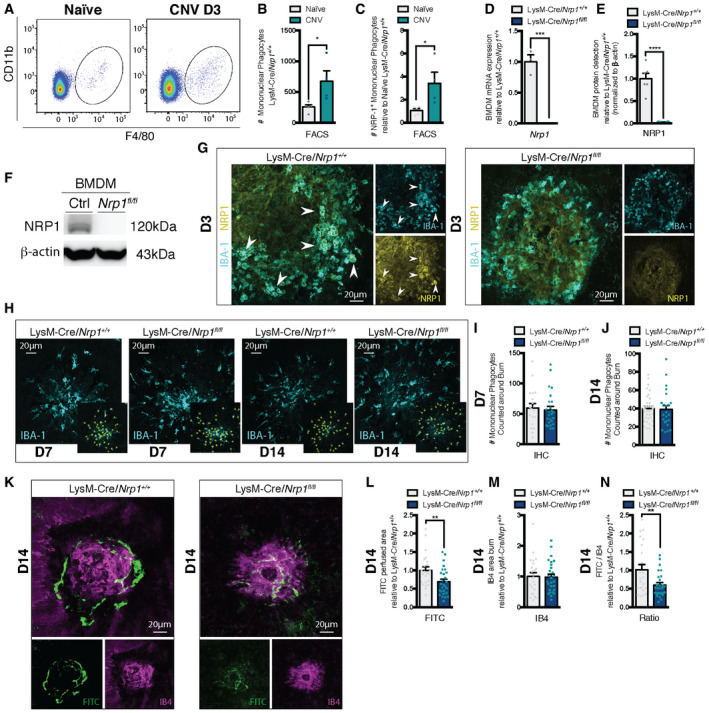
-
ARepresentative FACS plots of retinas and sclera‐choroid‐RPE cell complexes from naïve and burned mice 3 days (D3) after laser burn.
-
BQuantification of mononuclear phagocytes (Ly6G−, F4/80+, CD11b+) in retinas and sclera‐choroid‐RPE cell complexes at D3 relative to naïve; n = 5 (Naïve), 4 (CNV).
-
CQuantification of NRP1+ mononuclear phagocytes (Ly6G−, F4/80+, CD11b+, NRP1+) in retinas and sclera‐choroid‐RPE cell complexes at D3 relative to naïve; n = 5 (Naïve), 4 (CNV).
-
DmRNA expression of Nrp1 in BMDM relative to LysM‐Cre/Nrp1+/+; n = 3 (LysM‐Cre/Nrp1+/+), n = 4 (LysM‐Cre/Nrp1fl/fl).
-
EQuantification of NRP1 protein expression in LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl BMDM; n = 6.
-
FRepresentative Western blot showing NRP1 expression in LysM‐Cre/Nrp1+/+ (Ctrl) and LysM‐Cre/Nrp1fl/fl (Nrp1fl/fl).
-
GRepresentative confocal images of NRP1 and IBA1‐stained mononuclear phagocytes on choroidal flat mounts from LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D3. Arrowheads indicate NRP1‐positive mononuclear phagocytes. Scale bar: 20 μm.
-
HRepresentative confocal images of IBA‐1‐stained mononuclear phagocytes on choroidal flat mounts from LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D7 and D14. Examples of macrophage quantification (yellow stars) are presented in side panels. Scale bar: 20 μm.
-
I, JTotal number of IBA‐1‐positive mononuclear phagocytes counted around laser impact area on confocal images of choroidal flat mounts at D7 (I) and D14 (J); n = 19 burns (D7 LysM‐Cre/Nrp1+/+), n = 25 burns (D7 LysM‐Cre/Nrp1fl/fl), n = 37 burns (D7 LysM‐Cre/Nrp1+/+), n = 23 burns (D7 LysM‐Cre/Nrp1fl/fl), 3–5 mice with ~4 burns per eye.
-
KCompilation of representative compressed Z‐stack confocal images of FITC–dextran‐labeled CNV and IB4‐stained laser impact area from LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D14. Scale bar: 20 μm.
-
L–NQuantification of area of FITC–dextran‐labeled CNV (L), isolectin B4 (IB4)‐stained laser impact area (M) and the ratio of FITC/IB4 per laser burn (N) relative to LysM‐Cre/Nrp1+/+ at D14; n = 23 burns (LysM‐Cre/Nrp1+/+), n = 27 burns ( LysM‐Cre/Nrp1fl/fl).
Data information: All comparisons between groups were analyzed using a Student’s unpaired t‐test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; error bars represent mean ± SEM; exact P‐values listed in Appendix Table S1.
Figure EV1. Mononuclear phagocyte‐resident NRP1 expression in LysM‐Cre/Nrp1fl/fl .
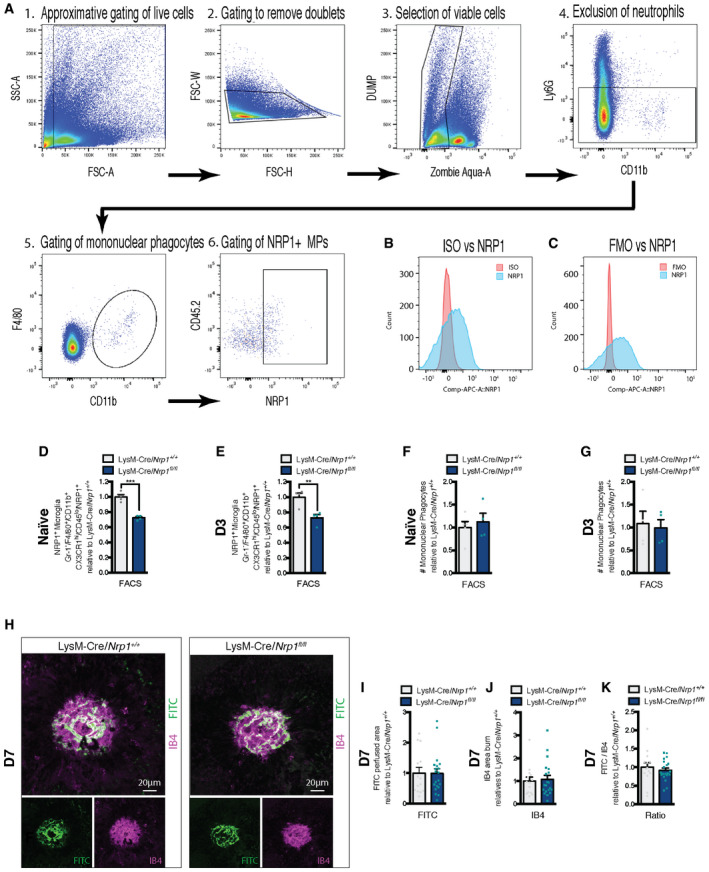
-
AGating scheme explaining the identification of the Ly6G−, F4/80+, CD11b+, and NRP1+ mononuclear phagocytes in retinas and sclera‐choroid‐RPE cell complexes. 1. Gating of live cells, 2. Removal of doublets, 3. Selection of viable cells, 4. Exclusion of neutrophils, 5. Gating of mononuclear phagocytes, 6. Gating of NRP1+ mononuclear macrophages.
-
BFACS histogram of APC‐conjugated rat IgG2A isotype control (red) versus anti‐mNRP1 APC‐conjugated rat IgG2A (R&D systems) (blue).
-
CFACS histogram of FMO (fluorescence minus one) versus anti‐mNRP1 APC‐conjugated rat IgG2A.
-
D, EQuantification of NRP1‐positive microglia (Ly6G−, F4/80+, CD11b+, CX3CR1hi, CD45lo, NRP1+) in retinas and sclera‐choroid‐RPE cell complexes in Naïve (non‐burned) mice (D); n = 4 and at D3 (E); n = 4.
-
F, GQuantification of mononuclear phagocytes (Ly6G−, F4/80+, CD11b+) in retinas and sclera‐choroid‐RPE cell complexes in Naïve (non‐burned) mice (F); n = 5 (LysM‐Cre/Nrp1+/+), 4 (LysM‐Cre/Nrp1fl/fl) and at D3 (G); n = 4.
-
HCompilation of representative compressed Z‐stack confocal images of FITC–dextran‐labeled CNV and IB4‐stained laser impact area from LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D7. Scale bar: 20 μm.
-
I–KQuantification of area of FITC–dextran‐labeled CNV (I), isolectin B4 (IB4)‐stained laser impact area (J) and the ratio of FITC/IB4 per laser burn (K) relative to LysM‐Cre/Nrp1+/+ at D7; n = 14 burns (LysM‐Cre/Nrp1+/+), n = 20 burns (LysM‐Cre/Nrp1fl/fl).
Data information: All comparisons between groups were analyzed using a Student’s unpaired t‐test; **P < 0.01, ***P < 0.001; error bars represent mean ± SEM; exact P‐values listed in Table 2.
Figure EV2. Gating scheme explaining the identification of the Ly6G−, F4/80+, CD11b+, CD11c+, CD206− and Ly6G−, F4/80+, CD11b+, CD11c−, CD206+ BMDMs.
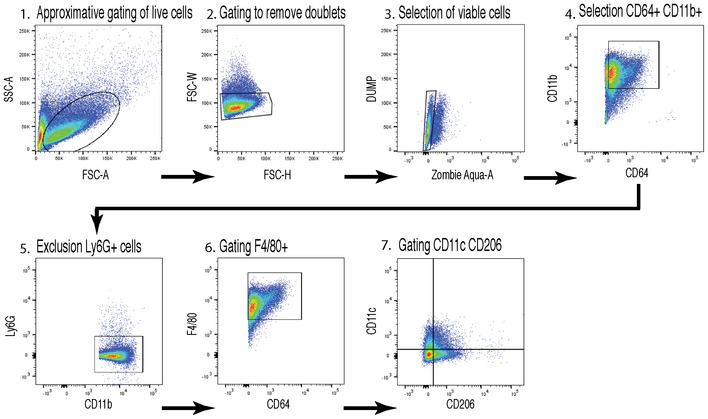
1. gating of live cells, 2. removal of doublets, 3. selection of viable cells, 4. Selection of myeloid cells (CD64+) 5. exclusion of neutrophils, 6. gating of mononuclear macrophages, 7. Gating on CD11c and CD206.
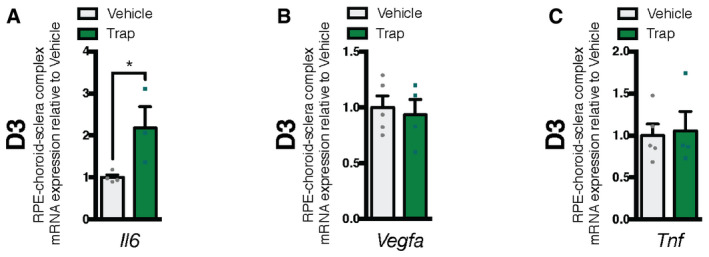
Figure EV3. Inflammatory state of RPE‐choroid‐sclera complexes following treatment with trap.
-
A–CmRNA expression of inflammation markers relative to vehicle in mouse RPE‐choroid‐sclera complexes at D3 for Il6 (A); n = 4 (vehicle), n = 4 (trap), Vegfa (B); n = 5 (vehicle), n = 4 (trap), Tnf (C); n = 5 (vehicle), n = 4 (trap).
Data information: All comparisons between groups were analyzed using a Student’s unpaired t‐test; *P < 0.05; error bars represent mean ± SEM; exact P‐values listed in Table 2.
Throughout the course of CNV, prior to laser burn until D14 post‐laser burn, the number of mononuclear phagocytes present in RPE‐choroid‐sclera complexes of either LysM‐Cre/Nrp1fl/fl or control LysM‐Cre/Nrp1+/+ mice followed similar trends. Analysis by FACS or immunofluorescence revealed that mononuclear phagocytes in either LysM‐Cre/Nrp1fl/fl or control LysM‐Cre/Nrp1+/+ mice remained similar over time and did not show significant difference either prior to laser burn or at D3, D7, or D14 (Figs 2H–J and EV1, EV2, EV3F and G).
Two weeks after laser burn (D14), quantification of compressed Z‐stack confocal images of FITC‐dextran‐perfused neovessels revealed a ~30–40% decrease in CNV in LysM‐Cre/Nrp1fl/fl mice compared with controls (Fig 2K–N). The average size of isolectin B4 (IB4)‐labeled impact areas did not differ between groups (Fig 2M) suggesting that the observed effect is directly on nascent vasculature. Interestingly, the extent of CNV did not vary between groups in the first week post‐laser burn (Fig EV1, EV2, EV3H–K) implying that NRP1‐expressing mononuclear phagocytes partake in later stages of disease. Together, these data suggest that while levels of mononuclear phagocytes are similar between LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl during CNV, NRP1‐expressing mononuclear phagocytes promote and maintain neovascularization in the later stages of CNV.
NRP1‐expressing mononuclear phagocytes display a pro‐angiogenic phenotype
Given that equal numbers of mononuclear phagocytes are present in the back of the eye of LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice following laser burn, yet NRP1+ mononuclear phagocytes promote CNV (Fig 2), we sought to determine the impact of loss of myeloid‐resident NRP1 on choroidal inflammation during CNV. Three days after laser burn, we observed a significant rise in mRNA transcripts for interleukin 1 β (Il1b) and interleukin 6 (Il6) (Fig 3A and B) in RPE‐choroid complexes of LysM‐Cre/Nrp1fl/fl mice when compared to controls, with a ~twofold increase in Il1b and ~sevenfold increase in Il6. No significant change was detected in Vegfa or tumor necrosis factor (Tnf) (Fig 3C and D). A similar pattern of expression was observed seven days post‐laser burn (Fig 3E–H).
Figure 3. NRP1‐expressing mononuclear phagocytes display a pro‐angiogenic alternatively activated phenotype.
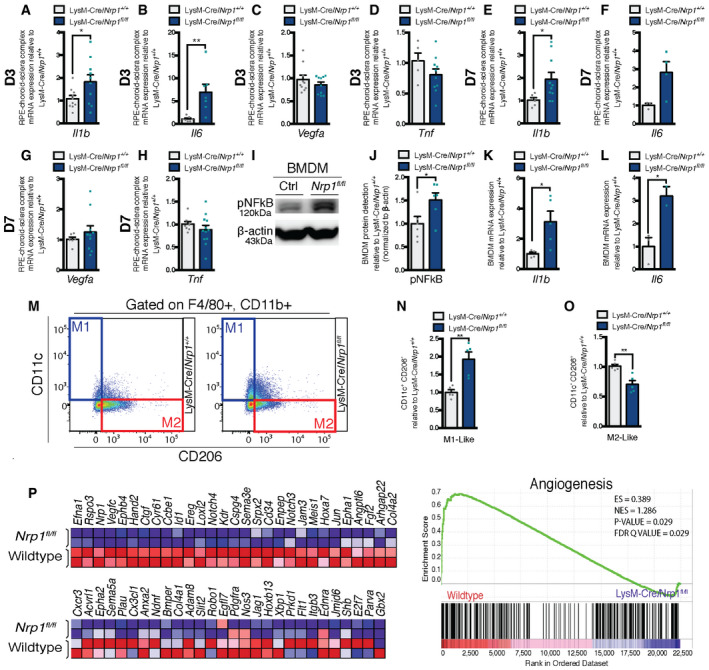
-
A–DmRNA expression of inflammation markers relative to LysM‐Cre/Nrp1+/+ in mouse RPE‐choroid‐sclera complexes at D3 for Il1b (A); n = 10 (LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl), Il6 (B); n = 7 (LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl), Vegfa (C); n = 10 (LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl), Tnf (D); n = 5 (LysM‐Cre/Nrp1+/+), n = 10 (LysM‐Cre/Nrp1fl/fl).
-
E–HmRNA expression of inflammation markers relative to LysM‐Cre/Nrp1+/+ in mouse RPE‐choroid‐sclera complexes at D7 for Il1b (E); n = 6 (LysM‐Cre/Nrp1+/+), n = 10 (LysM‐Cre/Nrp1fl/fl), Il6 (F); n = 3 (LysM‐Cre/Nrp1+/+), n = 4 (LysM‐Cre/Nrp1fl/fl), Vegfa (G); n = 6 (LysM‐Cre/Nrp1+/+), n = 10 (LysM‐Cre/Nrp1fl/fl), Tnf (H); n = 8 (LysM‐Cre/Nrp1+/+), n = 12 (LysM‐Cre/Nrp1fl/fl).
-
IRepresentative Western blot showing pNF‐κB expression in LysM‐Cre/Nrp1+/+ (Ctrl) and LysM‐Cre/Nrp1fl/fl (Nrp1fl/fl).
-
JQuantification of pNF‐κB expression in LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl BMDM; n = 6.
-
K, LmRNA expression relative to LysM‐Cre/Nrp1+/+ of inflammation markers in mouse BMDMs for Il1b (K); n = 4 (LysM‐Cre/Nrp1+/+), n = 5 (LysM‐Cre/Nrp1fl/fl) and Il6 (L); n = 3 (LysM‐Cre/Nrp1+/+), n = 2 (LysM‐Cre/Nrp1fl/fl).
-
MRepresentative FACS plots of M1 and M2‐Like macrophages in LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl BMDMs.
-
N, OQuantification of M1‐Like macrophages (F4/80+, CD11b+, CD11c+, CD206−)(N), M2‐Like macrophages (F4/80+, CD11b+, CD11c−, CD206+) (O) in LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl BMDMs relative to LysM‐Cre/Nrp1+/+; n = 5.
-
PHeatmap (left) and enrichment plot (right) of GO Angiogenesis gene set enrichment analysis (GSEA) of wild‐type and LysM‐Cre/Nrp1fl/fl peritoneal macrophages; n = 2. NES, normalized enrichment score; FDR, false discovery rate.
Data information: All comparisons between groups were analyzed using a Student’s unpaired t‐test; *P < 0.05, **P < 0.01; error bars represent mean ± SEM; exact P‐values listed in Appendix Table S1.
We next investigated immune activation in bone marrow‐derived macrophages (BMDM) from LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice. BMDMs were derived from bone marrow cells using macrophage colony‐stimulating factor (M‐CSF) which can induce a predominantly anti‐inflammatory M2‐like cell population (Fleetwood et al, 2007; Lacey et al, 2012). However, we observed a significant increase in NF‐B p65 phosphorylation in NRP1‐deficient BMDMs (Fig 3I and J), accompanied by increased levels of Il1b and Il6 (Fig 3K and L) indicative of classical activation of mononuclear phagocytes and suggesting that the absence of NRP1 in myeloid cells led to increased levels of these cytokines in RPE‐choroid‐sclera complexes (Fig 3A and B). In line with these findings, FACS analysis revealed a ~twofold increase in M1‐like (F4/80+, CD11b+, CD11c+, CD206−) BMDMs in LysM‐Cre/Nrp1fl/fl mice (Fig 3M and N) (gating scheme in Fig EV2), accompanied by a ~25% decrease in M2‐like (F4/80+, CD11b+, CD11c−, CD206+) cells (Fig 3M and O).
Moreover, transcriptomic analysis by RNA sequencing (RNA‐seq) and gene set enrichment analysis (GSEA) of NRP1‐deficient peritoneal macrophages revealed a significant decrease in transcripts from the GO Angiogenesis gene set (Fig 3P) in LysM‐Cre/Nrp1fl/fl macrophages when compared to wild‐type controls. These data support the notion that the absence of NRP1 on mononuclear phagocytes renders them less pro‐angiogenic and more pro‐inflammatory. These findings are consistent with the lower levels of CNV observed in LysM‐Cre/Nrp1fl/f mice (Fig 2K–N) and other studies suggesting that heightened inflammation may reduce CNV in the laser‐induced mouse model (Zandi et al, 2015).
Therapeutic intravitreal administration of soluble NRP1 reduces CNV in mice
Based on the above data, we sought to determine the therapeutic value of interfering with NRP1 ligands on the outcome of CNV. We generated a recombinant NRP1‐derived trap consisting of the extracellular domain of NRP1 (Fig 4A). This trap binds and neutralizes NRP1 ligands (Cerani et al, 2013; Dejda et al, 2014). We initially confirmed the binding of the NRP1 ligands SEMA3A and VEGF‐A to the trap by surface plasmon resonance (SPR). The trap was covalently immobilized by standard amine coupling on a carboxymethyl dextran sensor chip with lower charge (CM4) and ligands were injected at various concentrations to evaluate binding kinetics in real‐time. Sensorgrams revealed fast association and low dissociation for both ligands, which followed a simple 1:1 stoichiometry model. Equilibrium dissociation constants (Kd) of 0.67 nM and 4.32 nM were, respectively, extracted for SEMA3A and VEGF‐A (Fig 4B–D), indicative of high and physiologically relevant binding affinities. Interestingly, while the association rate constants were somewhat similar for both ligands, SEMA3A showed a much slower dissociation to the immobilized trap compared with VEGF‐A, suggesting that the SEMA3A‐trap complex is very stable.
Figure 4. Therapeutic intravitreal administration of soluble NRP1 reduces CNV in mice.
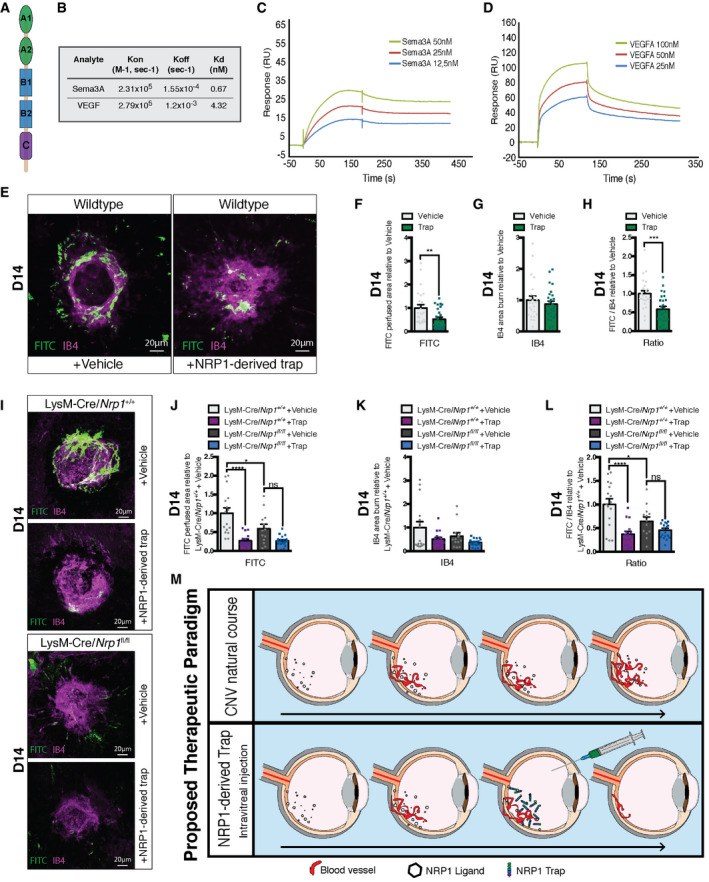
-
ASchematic representation of a soluble receptor Neuropilin‐1, consisting of five domains: two CUB motifs (A1, A2), two coagulation factor domains (B1, B2), and the MAM domain (C).
-
BRate constant and binding affinities of Sema3A and VEGF to immobilized trap obtained using a one‐site Langmuir binding model.
-
C, DRepresentative SPR sensorgrams for various concentrations of Sema3A (C) and VEGF (D) binding to immobilized Trap.
-
ECompilation of representative compressed Z‐stack confocal images of FITC–dextran‐labeled CNV and isolectin B4 (IB4)‐stained laser impact area from vehicle and NRP1‐derived trap treated wild‐type mice. Scale bar: 20 μm.
-
F–HQuantification of area of FITC–dextran‐labeled CNV (F), IB4‐stained laser impact area (G) and the ratio of FITC/IB4 per laser burn (H) relative to vehicle at D14; n = 24 burns (vehicle), n = 26 burns (NRP1‐derived trap).
-
ICompilation of representative compressed Z‐stack confocal images of FITC–dextran‐labeled CNV and IB4‐stained laser impact area from vehicle and NRP1‐derived trap‐treated LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D14. Scale bar: 20 μm.
-
J–LQuantification of area of FITC–dextran‐labeled CNV (J), isolectin B4 (IB4)‐stained laser impact area (K), and the ratio of FITC/IB4 per laser burn (L) relative to LysM‐Cre/Nrp1+/+ + vehicle in vehicle and NRP1‐derived trap‐treated LysM‐Cre/Nrp1+/+ and LysM‐Cre/Nrp1fl/fl mice at D14; n = 16 burns (LysM‐Cre/Nrp1+/+ + vehicle), n = 16 burns (LysM‐Cre/Nrp1+/+ + trap), n = 13 burns (LysM‐Cre/Nrp1fl/fl + vehicle), n = 19 burns (LysM‐Cre/Nrp1fl/fl + trap).
-
MProposed therapeutic paradigm. From a therapeutic perspective, intravitreal injection of NRP1‐derived traps reduces pathological angiogenesis associated with CNV.
Data information: Comparisons between groups were analyzed using a Student’s unpaired t‐test; (E–G) or one‐way ANOVA with Tukey's multiple comparisons test (I–K); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; error bars represent mean ± SEM; exact P‐values listed in Appendix Table S1.
Intravitreal injections of NRP1‐derived trap in C57BL/6J wild‐type mice at D0 led to a robust ~47% decrease in Dextran‐FITC‐perfused neovessels compared with vehicle controls at day 14 post‐laser burn (Fig 4E and F). The average size of IB4‐labeled impact area was not significantly affected by the treatment (Fig 4E and G) and ratios of FITC‐labeled neovessels to the size of IB4‐labeled post‐laser burn scarring were reduced by 41% after trap treatment (Fig 4E and H). Transcripts for Vegfa and Tnf did not vary following treatment with trap while Il6 rose (Fig EV3A–C).
In order to determine whether the beneficial effects of NRP1‐derived traps on CNV were mediated by influencing NRP1+ mononuclear phagocytes, we injected traps into the vitreous of LysM‐Cre/Nrp1fl/fl and LysM‐Cre/Nrp1+/+ mice following laser burn. Similar to wild‐type mice, trap treatment in LysM‐Cre/Nrp1+/+ led to a significant decrease in FITC‐perfused vessels and FITC/IB4‐ratios (Fig 4I, J and L). Treatment of LysM‐Cre/Nrp1fl/fl mice reduced CNV to similar levels as seen in controls; however, the magnitude of effect was diminished (Fig 4I–L). NRP1 is expressed by several other cells in the retina and sclera‐choroid‐RPE complex such as endothelial cells and neurons. Intravitreal injection of the NRP1‐derived traps will influence ligands that signal in all NRP1‐expressing cells. The reduced therapeutic effect of the trap in LysM‐Cre/Nrp1fl/fl mice suggests that a portion of the therapeutic effect of the NRP1 trap is mediated through myeloid cells. Together, these data suggest that sequestering NRP1 ligands is an effective strategy to reduce pathological subretinal neovascularization.
Discussion
Here, we show that several ligands of NRP1 are induced in the vitreous of patients with active NV AMD and that myeloid‐resident NRP1 contributes to pathological angiogenesis in later stages of CNV in mice. Moreover, we demonstrate that while mononuclear phagocyte‐resident NRP1 is not essential for cellular recruitment to sites of CNV, it is critical for mitigating myeloid cell inflammation and skews myeloid cells toward a pro‐angiogenic phenotype. The absence of NRP1 leads to enhanced production of pro‐inflammatory factors as we have previously suggested for dendritic cells (Oussa et al, 2016) and adipose tissue macrophages (Wilson et al, 2018). These data add to the notion that less inflammatory and more M2‐like mononuclear phagocytes are enriched with age, and exacerbate CNV in the laser‐induced mouse model (Nakamura et al, 2015; Zandi et al, 2015).
Accumulation of mononuclear phagocytes in AMD secondary to the disruption of the physiologically immunosuppressive subretinal environment is central to the etiology of both atrophic and wet forms of disease (Sennlaub et al, 2013; Guillonneau et al, 2017; Rashid et al, 2019). Our data suggest that NRP1 keeps mononuclear phagocytes in a less inflammatory and more reparative state, toward the M2 portion of the spectrum. This is consistent with findings demonstrating that NRP1‐deficient myeloid cells are more pro‐inflammatory, classically activated cells in models of obesity (Wilson et al, 2018), tumor growth (Casazza et al, 2013; Roy et al, 2017; Miyauchi et al, 2018; Chen et al, 2019), and sepsis (Dai et al, 2017). With respect to being pro‐angiogenic, NRP1‐expressing mononuclear phagocytes have been described as dispensable for physiological angiogenesis in the retina and elsewhere (Fantin et al, 2013; Dejda et al, 2016), yet important for vessel growth during weight gain (Wilson et al, 2018). They have also been reported to normalize tumor blood vessels (Carrer et al, 2012) and promote pathological angiogenesis in the retina (Dejda et al, 2014) and tumors (Casazza et al, 2013).
Based on the above findings, we sought to determine the therapeutic potential of neutralizing NRP1 ligands. A single intravitreal injection of a recombinant NRP1‐derived trap was effective at preventing CNV, highlighting the therapeutic potential of NRP1 for exudative AMD. In addition, others (Sodhi et al, 2019) and us (Cerani et al, 2013; Dejda et al, 2014) have demonstrated efficacy for recombinant NRP1 for retinal vasculopathies characterized by preretinal neovascularization or vasogenic edema. Potentially, non‐responders to anti‐VEGF therapy could benefit from this treatment paradigm, since a genetic variation of Nrp1 is an indicator of reduced treatment response to anti‐VEGF therapeutics like Ranibizumab in patients with NV AMD (Lores‐Motta et al, 2016; Lores‐Motta et al, 2018). NRP1‐derived traps have a considerably lower affinity for VEGF‐A compared with current anti‐VEGF therapeutics such as Ranibizumab, Bevacizumab, and Aflibercept (García‐Quintanilla et al, 2019) and hence may limit toxicity associated with sustained VEGF‐A deprivation. Of note, the NRP1‐derived trap was less effective in retinas of LysM‐Cre/Nrp1fl/fl suggesting a mechanism of action in part redundant with deletion of NRP1 in myeloid cells.
In sum, while a role for endothelial‐resident NRP1 has been demonstrated in choroidal and retinal neovascularization (Fernandez‐Robredo et al, 2017), we provide evidence that NRP1‐expressing immune cells contribute to CNV. Collectively, we provide rationale for therapeutic targeting of NRP1 ligands or NRP1‐expressing myeloid cells for exudative AMD.
Materials and Methods
Vitrectomy
All patients were previously diagnosed with NV AMD and were followed and treated by a single vitreoretinal surgeon (F.A. Rezende). Control patients underwent surgical treatment for non‐vascular pathology (epiretinal membrane or macular hole) by the same surgeon. Patients underwent surgery under local retro/peribulbar anesthesia. Three‐port 25‐gage transconjunctival pars plana vitrectomy was performed through 25‐gage valved cannulas (Alcon). Under microscope visualization using a wide‐angle viewing system (Resight, Zeiss), undiluted vitreous at the macular area was collected with a 25‐gage vitrector. Vitreous samples were frozen on dry ice immediately after biopsy and stored at −80°C. We obtained approval of human clinical protocols from the Hôpital Maisonneuve‐Rosemont ethics committee (Ref. CER: 10059). Written informed consent was obtained from all subjects and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Quantification of ligands of NRP1 in human vitreous by ELISA
Samples were centrifuged at 15,000 g for 5 min at 4°C prior to analysis. NRP1 ligands were quantified in supernatants using ELISAs following manufacturer’s instructions; VEGF‐A (DVE00,. R&D Systems), SEMA3A (LS‐F29822, LSBio), TGF‐β (BMS‐249‐4, Thermo Fisher Scientific Inc.), PGF (EHPGF, Thermo Fisher Scientific Inc.), and PDGF‐BB (BMS2071, Thermo Fisher Scientific Inc.)
Animals
All studies were performed in accordance with the Association for Research in Visions and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. All animal procedures were validated by the Animal Care committee of the University of Montreal and Hôpital Maisonneuve‐Rosemont in agreement with the guidelines established by the Canadian Council on Animal Care.
C57BL/6J wild‐type (WT), LysM‐Cre (Lyz2tm1(cre)Ifo/J; no. 004781), and Neuropilin1‐floxed (Nrp1tm2Ddg/J; no. 005247) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred in house. We generated a line of myeloid‐specific transgenic mice by breeding LysM‐Cre mice (Cre‐recombinase expressed in the myeloid lineage) with NRP1‐floxed mice, resulting in a mouse with attenuated Nrp1 in myeloid cells (LysM‐Cre/Nrp1fl/fl). Mice were raised under sterile barrier conditions and housed under a 12‐h light cycle with water and food ad libitum. Only male mice were used in this study.
In vivo imaging following laser‐induced choroidal neovascularization (CNV)
In vivo imaging was performed using a scanning laser ophthalmoscope (Micron IV; Phoenix Laboratories, Pleasanton, CA, USA). Mice of 9–11 weeks of age were subjected to pupil dilation (Mydriacyl; Alcon, Mississauga, ON, Canada) and anesthetized with a mix of 10% ketamine and 4% xylazine (10 µl/g body weight). Fluorescein (Alcon, 1 unit/g body weight of a 5% fluorescein dilution in 0.9% sodium chloride) was injected subcutaneously and corneas were lubricated with Optixcare ophthalmic gel (Aventix Animal Health, Burlington, ON, Canada). After a fluorescein circulation of 5 min, retinas were imaged before and after inducing choroidal neovascularization with 4 distinct laser burns (50 µm, 300 mW, 0.05 s). Animals were followed‐up 3, 7, and 14 days after laser burn.
Surface plasmon resonance
Surface plasmon resonance analyses were performed using a Biacore T200 instrument (GE Healthcare). Purified recombinant trap was immobilized by standard amine‐coupling chemistry on a Biacore CM4 carboxymethylated dextran sensor chip, which was pre‐activated with 100 mM N‐hydroxysuccinimide (NHS) and 100 mM of 3‐(N,N‐dimethylamino) propyl‐N‐ethylcarbodiimide (EDC). Surfaces were blocked by injecting 1 M ethanolamine. An immobilization abundance of 100–150 RU of Trap was reached. SEMA3A and VEGF‐A were, respectively, injected over the sample and reference flow cells at increasing concentrations (12.5–100 nM) at a flow rate of 40 μl/min in PBS buffer supplemented with 0.025% (v/v) Tween‐20. Binding sensorgrams were obtained by subtracting the reference flow cell. Response curves were analyzed using BIAevaluation software (GE Healthcare), and data from all concentrations were globally fit to a one‐site Langmuir binding model.
Real‐time quantitative PCR analysis
Immediately after enucleation, eyes were dissected to isolate the sclera–choroid–RPE cell complex. BMDMs were washed 3× in PBS and collected in TRIzol. RNA was isolated using TRIzol and digested with DNase I to prevent amplification of genomic DNA contaminants. All‐In‐One RT MasterMix (ABM) was used for the reverse transcription, and BrightGreen qPCR MasterMix (ABM) to determine gene expression in an ABI Biosystems Real‐Time PCR machine with β‐actin (Actb) as a reference gene. We used the following primers: Mouse Actb = F: 5’‐GAC GGC CAG GTC ATC ACT ATT G‐3’, R: 5’‐CCA CAG GAT TCC ATA CCC AAG A‐3’; Mouse Vegfa = F: 5’‐GCC CTG AGT CAA GAG GAC AG‐3’, R: 5’‐CTC CTA GGC CCC TCA GAA GT‐3’; Mouse Sema3a = F: 5’‐GGG ACT TCG CTA TCT TCA GAA‐3’, R: 5’‐GGC GTG CTT TTA GGA ATG TTG‐3’; Mouse Tgfb1 = F: 5’‐ACG CCT GAG TGG CTG TCT TTT GAC‐3’, R: 5’‐GGG CTG ATC CCG TTG ATT TCC ACG‐3’; Mouse Pdgfb = F: 5’‐GAA GTT GGC ATT GGT GCG AT‐3’, R: 5’‐TGG AGT CGA GTC GGA AAG CT‐3’; Mouse Nrp1 = F: 5’‐ ACC CAC ATT TCG ATT TGG AG‐3’, R: 5’‐TTC ATA GCG GAT GGA AAA CC‐3’; Mouse Il1b = F: 5’‐CTG GTA CAT CAG CAC CTC ACA‐3’, R: 5’‐GAG CTC CTT AAC ATG CCC TG‐3’; Mouse Il6 = F: 5’‐AGA CAA AGC CAG AGT CCT TCA GAG A‐3’, R: 5’‐GCC ACT CCT TCT GTG ACT CCA GC‐3’; Mouse Tnf = F: 5’‐CCC TCA CAC TCA GAT CAT CTT CT‐3’, R: 5’‐GCT ACG ACG TGG GCT ACA G‐3’; Mouse iNos = F: 5’‐CGG CAA ACA TGA CTT CAG GC‐3’, R: 5’‐GCA CAT CAA AGC GGC CAT AG‐3’; Mouse Cd163 = F: 5’‐ATG CTT CCA TCC AGT GCC TC‐3’, R: 5’‐CAC AAA CCA AGA GTG CCG TG‐3’; Mouse Cd206 = F: 5’‐GTT CAC CTG GAG TGA TGG TTC TC‐3’, R: 5’‐AGG ACA TGC CAG GGT CAC CTT T‐3’; Mouse Arg1 = F: 5’‐CAF CAC TGA GGA AAG CTG GT‐3’, R: 5’‐CAG ACC GTG GGT TCT TCA CA‐3’; Mouse Pgf = F: 5’‐CAG TTG CTT CTT ACA GGT CC‐3’, R: 5’‐CAC CTC ATC AGG GTA TTC AT‐3’.
Laser‐induced CNV
At the age of 6 weeks, mice were anesthetized by intraperitoneal injection with 10 μl/g body weight of a 10% ketamine and 4% xylazine solution. Using an argon laser, we ruptured their Bruch’s membrane, as described previously (Lambert et al, 2013). Mice were sacrificed at 3, 7, 10, or 14 days after we induced 4 burns per eye for the immunohistochemistry analysis and 6 burns per eye for the RT‐qPCR and FACS analyses.
Immunohistochemistry
7 or 14 days after CNV induction, mice were sedated with isoflurane gas and cardiacally perfused with 0.5 ml of 15 mg/ml of fluorescein isothiocyanate (FITC)‐dextran (average mol wt 2,000 kDa) and euthanized. Eyes were enucleated and fixed for 30 min in 4% PFA at room temperature, before dissection of the sclera‐choroid‐RPE cell complex. After a secondary fixation of 15 min in 4% PFA at room temperature, the choroids were stained with rhodamine‐labeled Griffonia (bandeiraea) Simplicifolia Isolectin I (RL‐1102‐2, Vector Laboratories Inc.) (1:100), NRP1 (AF566, goat polyclonal; R&D Systems) (1:250), and IBA‐1 (019‐19741, rabbit polyclonal; Wako) (1:350) overnight. After 1‐h incubation with secondary antibodies, the sclera‐choroid‐RPE cell complex was mounted onto a slide, and the burns and macrophages were captured in a Z‐stack with an Olympus FV1000 microscope. The Z‐stacks were compressed into one image and quantified in ImageJ.
FACS on retina and sclera‐choroid‐RPE cell complexes
Retinas and sclera‐choroid‐RPE cell complexes of non‐burned (D0) and burned mice at D3 were cut into small pieces and homogenized in a solution of 750 U/ml DNAse I (Sigma‐Aldrich Corp.) and 0.5 mg/ml of collagenase (Roche) for 20 min at 37°C. Homogenates were filtered through a 70 μm cell strainer and washed in PBS. Viability of the cells was checked by Zombie Aqua (423101: BioLegend) staining for 15 min at room temperature. After incubation with LEAF‐purified anti‐mouse CD16/32 (101310; BioLegend) for 10 min at 4°C to block Fc receptors, cells were incubated for 25 min at 4°C with the following antibodies: BV711 anti‐mouse/human CD11b (101242; BioLegend), PE anti‐mouse F4/80 (123110; BioLegend), APC anti‐mouse CD64 (139305; BioLegend), FITC anti‐mouse CD38 (102705; BioLegend), APC/Cy7 anti‐mouse Ly‐6G (127624; BioLegend), BV785 anti‐mouse CD11c (117335; BioLegend), and PE/Cy7 anti‐mouse CD206 (141719; BioLegend). Antibody dilution was determined with titration by lot. Fluorescence‐activated cell sorting (FACS) was performed on a BD LSRFortessaTM X‐20 cell analyzer, and data were analyzed using FlowJo software (FlowJo version 10.2).
Generation of BMDM
Bone marrow from both femurs and tibiae was harvested through bone flushing with PBS supplemented with 10% FBS. After red blood cell (RBC) lysis, cells were seeded in complete medium (Dulbecco’s modified Eagle’s medium (DMEM) plus 10% FBS and 1% streptomycin/penicillin) and stimulated with macrophage colony‐stimulating factor (M‐CSF) (Mouse M‐CSF Recombinant Protein, eBioscience™; Invitrogen) 1:5,000. After 3 days of incubation at 37°C with 5% CO2, fresh medium containing M‐CSF was added. Cells were allowed to differentiate for a total of 6 days, before their medium was replaced by complete medium without M‐CSF. LPS stimulated cells were stimulated for 24 h with 250 ng/ml LPS (Escherichia coli O55:B5 lipopolysaccharide; Sigma‐Aldrich). The cells were not tested for mycoplasma contamination. As evaluated by flow cytometry, the purity was usually around 99%.
Western blot analysis
For assessment of BMDM protein levels, we collected BMDM by scraping the cells in 1× RIPA on ice. Protein concentration was assessed by bicinchoninic acid (BCA) assay (Sigma‐Aldrich), and 30 μg protein was analyzed for each condition by standard SDS–PAGE technique. Anti‐NRP1 antibody (ab81321) (1:2,000) and Anti‐NF‐κB p65 (ab16502) (1:500) antibody were purchased from Abcam.
FACS on BMDM for extracellular staining
BMDMs were collected in PBS through scraping. Viability of the cells was checked by Zombie Aqua (423101: BioLegend) staining for 15 min at room temperature. After incubation with LEAF‐purified anti‐mouse CD16/32 (101310; BioLegend) for 10 min at 4°C to block Fc receptors, cells were incubated for 25 min at 4°C with the following antibodies: BV711 anti‐mouse/human CD11b (101242; BioLegend), PE anti‐mouse F4/80 (123110; BioLegend), APC anti‐mouse CD64 (139305; BioLegend), FITC anti‐mouse CD38 (102705; BioLegend), APC/Cy7 anti‐mouse Ly‐6G (127624; BioLegend), BV785 anti‐mouse CD11c (117335; BioLegend), and PE/Cy7 anti‐mouse CD206 (141719; BioLegend). Antibody dilution was determined with titration by lot. Fluorescence‐activated cell sorting (FACS) was performed on a BD LSRFortessaTM X‐20 cell analyzer, and data were analyzed using FlowJo software (FlowJo version 10.2).
RNA‐seq sample preparation, sequencing, and analysis
RNA‐seq data are available from a previous study (Wilson et al, 2018) in Gene Expression Omnibus (GEO) under the entry GSE110447. RNA‐seq was performed as described previously (Wilson et al, 2018). Gene set enrichment analysis GSEA was conducted using GSEA v2.2.1 software provided by Broad Institute of Massachusetts Institute of Technology and Harvard University. We used the ANGIOGENESIS gene set contributed by the Gene Ontology Consortium from the Molecular Signature Database of the Broad Institute, Inc.
Intravitreal injections
Wild‐type, LysM‐Cre/Nrp1fl/fl, and LysM‐Cre/Nrp1+/+ mice subjected to laser burn were intravitreally injected with NRP1‐derived trap or vehicle (Saline 0.9%) on the day of the burn and sacrificed at D14.
Statistical analysis
Data are presented as mean ± SEM Student’s t‐test was used to compare two different groups or, when indicated, a one‐way analysis of variance (ANOVA) and Dunnett’s multiple comparisons test. A P < 0.05 was considered statistically different. N was indicated for each experiment.
Author contributions
Research design and study: E.A., P.S.; Experimental work: E.A., F.B., F.F., F.P., M.H., A.D., G.M., A.W., N.B., K.B., S.C‐G., S.B.; All retinal surgeries: F.A.R.; Data analysis: E.A., F.B., A.W., M.B.; Valuable conceptual insight on research design: JS.D., G.C., V.B.; Manuscript writing with valuable input from authors: E.A., A.W., P.S.
Conflict of interest
P.S. is the founder of and a consultant for SemaThera Inc. G.C. and V.B. are consultants for SemaThera Inc. F.B., N.B., and K.B. are employees of SemaThera Inc. The rest of the authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgments
We dedicate this study to the memory of our colleague and friend Normand Beaulieu who made invaluable contributions to the understanding of Neuropilin biology and the translational merit of targeting its ligands. P.S. holds the Wolfe Professorship in Translational Research and a Canada Research Chair in Retinal Cell Biology. This work was supported by operating grants to P.S from The Foundation Fighting Blindness Canada and SemaThera Inc. Additional funding was provided by the Canadian Institutes of Health Research (Foundation grant #148460), the Diabetes Canada (DI‐3‐18‐5444‐PS), Heart and Stroke Foundation of Canada (G‐16‐00014658), and Natural Sciences and Engineering Research Council of Canada (418637), the Fonds de Recherche en Ophtalmologie de l’Université de Montréal (FROUM), and the Réseau en Recherche en Santé de la Vision. M.H. holds the Banting Fellowship from the CIHR. S.C‐G. holds a Fonds de Recherche Santé du Québec (FRQS) scholarship.
EMBO Mol Med (2021) 13: e11754.
Data availability
This study includes no data deposited in external repositories.
References
- Acevedo LM, Barillas S, Weis SM, Gothert JR, Cheresh DA (2008) Semaphorin 3A suppresses VEGF‐mediated angiogenesis yet acts as a vascular permeability factor. Blood 111: 2674–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati J, Atkinson J, Gelfand B (2013) Immunology of age‐related macular degeneration. Nat Rev Immunol 13: 438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati J, Fowler BJ (2012) Mechanisms of age‐related macular degeneration. Neuron 75: 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipenko A, Himanen JP, van Leyen K, Nardi‐Dei V, Lesniak J, Barton WA, Rajashankar KR, Lu M, Hoemme C, Puschel AW et al (2003) Structure of the semaphorin‐3A receptor binding module. Neuron 39: 589–598 [DOI] [PubMed] [Google Scholar]
- Ball SG, Bayley C, Shuttleworth CA, Kielty CM (2010) Neuropilin‐1 regulates platelet‐derived growth factor receptor signalling in mesenchymal stem cells. Biochem J 427: 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay EJ, Rupp J, Iruela‐Arispe L, Sage EH, Pech M (1994) PDGF‐BB modulates endothelial proliferation and angiogenesis in vitro via PDGF beta‐receptors. J Cell Biol 125: 917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berber P, Grassmann F, Kiel C, Weber BH (2017) An eye on age‐related macular degeneration: the role of MicroRNAs in disease pathology. Mol Diagn Ther 21: 31–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrer A, Moimas S, Zacchigna S, Pattarini L, Zentilin L, Ruozi G, Mano M, Sinigaglia M, Maione F, Serini G et al (2012) Neuropilin‐1 identifies a subset of bone marrow Gr1‐ monocytes that can induce tumor vessel normalization and inhibit tumor growth. Cancer Res 72: 6371–6381 [DOI] [PubMed] [Google Scholar]
- Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24: 695–709 [DOI] [PubMed] [Google Scholar]
- Cerani A, Tetreault N, Menard C, Lapalme E, Patel C, Sitaras N, Beaudoin F, Leboeuf D, De Guire V, Binet F et al (2013) Neuron‐derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin‐1. Cell Metab 18: 505–518 [DOI] [PubMed] [Google Scholar]
- Chaudhary B, Khaled YS, Ammori BJ, Elkord E (2014) Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother 63: 81–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Wu S, Yan RM, Fan LS, Yu L, Zhang YM, Wei WF, Zhou CF, Wu XG, Zhong M et al (2019) The role of the hypoxia‐Nrp‐1 axis in the activation of M2‐like tumor‐associated macrophages in the tumor microenvironment of cervical cancer. Mol Carcinog 58: 388–397 [DOI] [PubMed] [Google Scholar]
- Comparison of Age‐related Macular Degeneration Treatments Trials Research Group , Maguire MG, Martin DF, Ying GS, Jaffe GJ, Daniel E, Grunwald JE, Toth CA, Ferris FL III, Fine SL (2016) Five‐year outcomes with anti‐vascular endothelial growth factor treatment of neovascular age‐related macular degeneration: the comparison of age‐related macular degeneration treatments trials. Ophthalmology 123: 1751–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo‐Garcia S, Corkhill C, Roubeix C, Davids AM, Kociok N, Strauss O, Joussen AM, Reichhart N (2017) Inhibition of placenta growth factor reduces subretinal mononuclear phagocyte accumulation in choroidal neovascularization. Invest Ophthalmol Vis Sci 58: 4997–5006 [DOI] [PubMed] [Google Scholar]
- Dai X, Okon I, Liu Z, Wu Y, Zhu H, Song P, Zou MH (2017) A novel role for myeloid cell‐specific neuropilin 1 in mitigating sepsis. FASEB J 31: 2881–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejda A, Mawambo G, Cerani A, Miloudi K, Shao Z, Daudelin JF, Boulet S, Oubaha M, Beaudoin F, Akla N et al (2014) Neuropilin‐1 mediates myeloid cell chemoattraction and influences retinal neuroimmune crosstalk. J Clin Invest 124: 4807–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejda A, Mawambo G, Daudelin JF, Miloudi K, Akla N, Patel C, Andriessen EM, Labrecque N, Sennlaub F, Sapieha P (2016) Neuropilin‐1‐expressing microglia are associated with nascent retinal vasculature yet dispensable for developmental angiogenesis. Invest Ophthalmol Vis Sci 57: 1530–1536 [DOI] [PubMed] [Google Scholar]
- Ellis LM (2006) The role of neuropilins in cancer. Mol Cancer Ther 5: 1099–1107 [DOI] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Plein A, Denti L, Fruttiger M, Pollard JW, Ruhrberg C (2013) NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood 121: 2352–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Robredo P, Selvam S, Powner MB, Sim DA, Fruttiger M (2017) Neuropilin 1 involvement in choroidal and retinal neovascularisation. PLoS One 12: e0169865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD (2007) Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 178: 5245–5252. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O'Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J, Eye Diseases Prevalence Research G (2004) Prevalence of age‐related macular degeneration in the United States. Arch Ophthalmol 122: 564–572 [DOI] [PubMed] [Google Scholar]
- Funasaka T, Raz A, Nangia‐Makker P (2014) Galectin‐3 in angiogenesis and metastasis. Glycobiology 24: 886–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Quintanilla L, Luaces‐Rodríguez A, Gil‐Martínez M, Mondelo‐García C, Maroñas O, Mangas‐Sanjuan V, González‐Barcia M, Zarra‐Ferro I, Aguiar P, Otero‐Espinar FJ et al (2019) Pharmacokinetics of intravitreal anti‐VEGF drugs in age‐related macular degeneration. Pharmaceutics 11: 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geretti E, Shimizu A, Klagsbrun M (2008) Neuropilin structure governs VEGF and semaphorin binding and regulates angiogenesis. Angiogenesis 11: 31–39 [DOI] [PubMed] [Google Scholar]
- Glinka Y, Stoilova S, Mohammed N, Prud'homme GJ (2011) Neuropilin‐1 exerts co‐receptor function for TGF‐beta‐1 on the membrane of cancer cells and enhances responses to both latent and active TGF‐beta. Carcinogenesis 32: 613–621 [DOI] [PubMed] [Google Scholar]
- Guillonneau X, Eandi CM, Paques M, Sahel JA, Sapieha P, Sennlaub F (2017) On phagocytes and macular degeneration. Prog Retin Eye Res 61: 98–128 [DOI] [PubMed] [Google Scholar]
- Handa JT, Cano M, Wang L, Datta S, Liu T (2017) Lipids, oxidized lipids, oxidation‐specific epitopes, and Age‐related Macular Degeneration. Biochim Biophys Acta 1862: 430–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA (2004) Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56: 549–580 [DOI] [PubMed] [Google Scholar]
- Jonas JB, Bourne RR, White RA, Flaxman SR, Keeffe J, Leasher J, Naidoo K, Pesudovs K, Price H, Wong TY et al (2014) Visual impairment and blindness due to macular diseases globally: a systematic review and meta‐analysis. Am J Ophthalmol 158: 808–815 [DOI] [PubMed] [Google Scholar]
- Joyal J‐S, Sitaras N, Binet F, Rivera JC, Stahl A, Zaniolo K, Shao Z, Polosa A, Zhu T, Hamel D et al (2011) Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood 117: 6024–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubb AM, Strickland LA, Liu SD, Mak J, Schmidt M, Koeppen H (2012) Neuropilin‐1 expression in cancer and development. J Pathol 226: 50–60 [DOI] [PubMed] [Google Scholar]
- Krebs I, Glittenberg C, Ansari‐Shahrezaei S, Hagen S, Steiner I, Binder S (2013) Non‐responders to treatment with antagonists of vascular endothelial growth factor in age‐related macular degeneration. Br J Ophthalmol 97: 1443–1446 [DOI] [PubMed] [Google Scholar]
- Kurihara T, Westenskow PD, Bravo S, Aguilar E, Friedlander M (2012) Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest 122: 4213–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA (2012) Defining GM‐CSF– and macrophage‐CSF–dependent macrophage responses by in vitro models. J Immunol 188: 5752–5765 [DOI] [PubMed] [Google Scholar]
- Lambert V, Lecomte J, Hansen S, Blacher S, Gonzalez M‐L, Struman I, Sounni N, Rozet E, de Tullio P, Foidart J et al (2013) Laser‐induced choroidal neovascularization model to study age‐related macular degeneration in mice. Nat Protoc 8: 2197–2211 [DOI] [PubMed] [Google Scholar]
- Langmann T (2007) Microglia activation in retinal degeneration. J Leukoc Biol 81: 1345–1351 [DOI] [PubMed] [Google Scholar]
- Lazzeri S, Figus M, Orlandi P, Fioravanti A, Di Desidero T, Agosta E, Sartini MS, Posarelli C, Nardi M, Danesi R et al (2013) VEGF‐A polymorphisms predict short‐term functional response to intravitreal ranibizumab in exudative age‐related macular degeneration. Pharmacogenomics 14: 623–630 [DOI] [PubMed] [Google Scholar]
- Lepelletier Y, Smaniotto S, Hadj‐Slimane R, Villa‐Verde DM, Nogueira AC, Dardenne M, Hermine O, Savino W (2007) Control of human thymocyte migration by Neuropilin‐1/Semaphorin‐3A‐mediated interactions. Proc Natl Acad Sci USA 104: 5545–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY (2012) Age‐related macular degeneration. Lancet 379: 1728–1738 [DOI] [PubMed] [Google Scholar]
- Lores‐Motta L, van Asten F, Muether PS, Smailhodzic D, Groenewoud JM, Omar A, Chen J, Koenekoop RK, Fauser S, Hoyng CB et al (2016) A genetic variant in NRP1 is associated with worse response to ranibizumab treatment in neovascular age‐related macular degeneration. Pharmacogenet Genomics 26: 20–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lores‐Motta L, Riaz M, Grunin M, Corominas J, van Asten F, Pauper M, Leenders M, Richardson AJ, Muether P, Cree AJ et al (2018) Association of genetic variants with response to anti‐vascular endothelial growth factor therapy in age‐related macular degeneration. JAMA Ophthalmol 136: 875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, Klagsbrun M (2002) Neuropilin‐1 binds vascular endothelial growth factor 165, placenta growth factor‐2, and heparin via its b1b2 domain. J Biol Chem 277: 24818–24825 [DOI] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103: 295–309 [DOI] [PubMed] [Google Scholar]
- Mimura T, Funatsu H, Noma H, Shimura M, Kamei Y, Yoshida M, Kondo A, Watanabe E, Mizota A (2019) Aqueous humor levels of cytokines in patients with age‐related macular degeneration. Ophthalmologica 241: 81–89 [DOI] [PubMed] [Google Scholar]
- Miyauchi JT, Caponegro MD, Chen D, Choi MK, Li M, Tsirka SE (2018) Deletion of Neuropilin 1 from microglia or bone marrow‐derived macrophages slows Glioma progression. Cancer Res 78: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl L, Folestad EB, Gladh H, Wang Y, Moessinger C, Jakobsson L, Eriksson U (2017) Neuropilin 1 binds PDGF‐D and is a co‐receptor in PDGF‐D‐PDGFRbeta signaling. J Cell Sci 130: 1365–1378 [DOI] [PubMed] [Google Scholar]
- Nakamura F, Goshima Y (2002) Structural and functional relation of neuropilins. Adv Exp Med Biol 515: 55–69 [DOI] [PubMed] [Google Scholar]
- Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N, Apte RS (2015) IL10‐driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun 6: 7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oussa NA, Dahmani A, Gomis M, Richaud M, Andreev E, Navab‐Daneshmand AR, Taillefer J, Carli C, Boulet S, Sabbagh L et al (2016) VEGF requires the receptor NRP‐1 to inhibit lipopolysaccharide‐dependent dendritic cell maturation. J Immunol 197: 3927–3935 [DOI] [PubMed] [Google Scholar]
- Prud'homme GJ, Glinka Y (2012) Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget 3: 921–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi C, Brash JT, Fantin A, Ruhrberg C (2016) NRP1 function and targeting in neurovascular development and eye disease. Prog Retin Eye Res 52: 64–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid K, Akhtar‐Schaefer I, Langmann T (2019) Microglia in retinal degeneration. Front Immunol 10: 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rein D, Zhang P, Wirth K, Lee P, Hoerger T, McCall N, Klein R, Tielsch J, Vijan S, Saaddine J (2006) The economic burden of major adult visual disorders in the United States. Arch Ophthalmol 124: 1754–1760 [DOI] [PubMed] [Google Scholar]
- Renand A, Milpied P, Rossignol J, Bruneau J, Lemonnier F, Dussiot M, Coulon S, Hermine O (2013) Neuropilin‐1 expression characterizes T follicular helper (Tfh) cells activated during B cell differentiation in human secondary lymphoid organs. PLoS One 8: e85589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolio STL (2017) Neuropilins as signaling hubs, controlling tyrosine kinases and other cell surface receptors. In The neuropilins: role and function in health and disease, Neufeld G (ed.), pp 23–39. New York: Springer International Publishing; [Google Scholar]
- Rizzolio S, Tamagnone L (2011) Multifaceted role of neuropilins in cancer. Curr Med Chem 18: 3563–3575 [DOI] [PubMed] [Google Scholar]
- Robinson GS, Ju M, Shih SC, Xu X, McMahon G, Caldwell RB, Smith LE (2001) Nonvascular role for VEGF: VEGFR‐1, 2 activity is critical for neural retinal development. FASEB J 15: 1215–1217 [DOI] [PubMed] [Google Scholar]
- Roy S, Bag AK, Singh RK, Talmadge JE, Batra SK, Datta K (2017) Multifaceted role of neuropilins in the immune system: potential targets for immunotherapy. Front Immunol 8: 1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint‐Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, Darland DC, Young MJ, D'Amore PA (2008) Endogenous VEGF is required for visual function: evidence for a survival role on müller cells and photoreceptors. PLoS One 3: e3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E et al (2013) CCR2(+) monocytes infiltrate atrophic lesions in age‐related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med 5: 1775–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodhi A, Ma T, Menon D, Deshpande M, Jee K, Dinabandhu A, Vancel J, Lu D, Montaner S (2019) Angiopoietin‐like 4 binds neuropilins and cooperates with VEGF to induce diabetic macular edema. J Clin Invest 129: 4593–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M (2002) VEGF165 mediates formation of complexes containing VEGFR‐2 and neuropilin‐1 that enhance VEGF165‐receptor binding. J Cell Biochem 85: 357–368 [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin‐1 is expressed by endothelial and tumor cells as an isoform‐specific receptor for vascular endothelial growth factor. Cell 92: 735–745 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM (1999) Plexin‐neuropilin‐1 complexes form functional semaphorin‐3A receptors. Cell 99: 59–69 [DOI] [PubMed] [Google Scholar]
- Takamatsu H, Kumanogoh A (2012) Diverse roles for semaphorin‐plexin signaling in the immune system. Trends Immunol 33: 127–135 [DOI] [PubMed] [Google Scholar]
- Taylor DJ, Hobby AE, Binns AM, Crabb DP (2016) How does age‐related macular degeneration affect real‐world visual ability and quality of life? A systematic review. BMJ Open 6: e011504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AM, Shao Z, Grenier V, Mawambo G, Daudelin JF, Dejda A, Pilon F, Popovic N, Boulet S, Parinot C et al (2018) Neuropilin‐1 expression in adipose tissue macrophages protects against obesity and metabolic syndrome. Sci Immunol 3: eaan4626 [DOI] [PubMed] [Google Scholar]
- Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY (2014) Global prevalence of age‐related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta‐analysis. Lancet Glob Health 2: e106–e116 [DOI] [PubMed] [Google Scholar]
- Yu C, Roubeix C, Sennlaub F, Saban DR (2020) Microglia versus monocytes: distinct roles in degenerative diseases of the retina. Trends Neurosci 43: 433–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi S, Nakao S, Chun KH, Fiorina P, Sun D, Arita R, Zhao M, Kim E, Schueller O, Campbell S et al (2015) ROCK‐isoform‐specific polarization of macrophages associated with age‐related macular degeneration. Cell Rep 10: 1173–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.