

Abstract
The N-acylsulfonamide functional group is a feature of the pharmacophore of several biologically active molecules, including marketed drugs. Although this acidic moiety presents multiple point of attachments that could be exploited to introduce structural diversification, depending on the circumstances, the replacement of the functional group itself with a suitable surrogate, or bioisostere, may be desirable. A number of N-acylsulfonamide bioisosteres have been developed over the years that provide opportunities to modulate both structure and physicochemical properties of this important structural motif. To enable an assessment of the relative impact on physicochemical properties that these replacements may have compared to the N-acylsulfonamide group, we conducted a structure-property relationship study based on matched molecular pairs, in which the N-acylsulfonamide moiety of common template reference structures is replaced with a series of bioisosteres. The data presented, which include an assessment of relative changes in acidity, permeability, lipophilicity and intrinsic solubility, provides a basis for informed decisions when deploying N-acylsulfonamides, or surrogates thereof, in analog design.
Keywords: N-Acylsulfonamide isostere, bioisostere, isosteric replacement, physicochemical properties, structure property relationship (SPR), oxetane, thietane
Graphical Abstract
Introduction.
Acidic moieties are of great importance in drug design as they can significantly impact, through ionic and electrostatic interactions, both physicochemical properties (e.g., lipophilicity), ADME-PK parameters, as well as on target/off target interactions (e.g., with proteins) of biologically active compounds.1 As a result, the modulation of one or more properties of the acidic moiety of candidate compounds can have important ramifications, especially during the hit/lead optimization process.
The N-acylsulfonamide group is an acidic moiety that is frequently employed in medicinal chemistry as a carboxylic acid bioisostere,2–6 with several reported examples in which such a replacement led to improved derivatives.7–10 This structural motif, however, can also be in itself a constituent of the pharmacophore as exemplified in many different classes of biologically active compounds,11 including antivirals,12 antibacterial,13, 14 and antiproliferative agents.15–17 The importance of N-acylsulfonamides in the field of drug design and medicinal chemistry is evident by the fact that as many as nine novel N-acylsulfonamide drugs (Figure 1) have been marketed in the USA between 2015 and 2020.18 Moreover, a review of the literature revealed that between 2019 and 2020, >50 research articles have been published in medicinal chemistry journals in which N-acylsulfonamides were studied for a variety of indications.
Figure 1:
Examples of N-acylsulfonamides that are either marketed or are in advanced stages of clinical development.
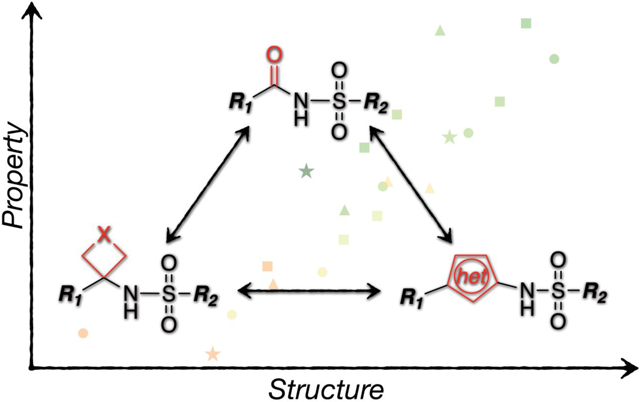
Although the N-acylsulfonamide structural motif presents alternative points of attachment that provide opportunities for structural diversification and modulation of physicochemical properties, on occasions, the replacement of this functional group itself with a suitable bioisostere may be desirable. The success of this strategy, however, like any isosteric replacement strategy, is highly context dependent and ultimately relies upon the availability of alternative structures that could be considered as potential surrogates. The concept of isosteric replacement has evolved significantly since it was first introduced. The current conception, which presents bioisosteres more as molecular metaphors rather than close structural equivalents of what they are replacing, is considerably broader than originally conceived and leads to the evaluation of a relatively wide range of alternative structures that could potentially emulate the desired biological activity.19–21 In the case of the N-acylsulfonamide, over the years, a select number of surrogates have been introduced in which the carbonyl moiety is replaced with an oxetane or a 5-membered ring heterocycle (Figure 2A). These structures likely originate from earlier observations that 5-membered ring heterocycles, such as triazoles, isoxazoles and others, have found broad applications as possible mimetics of different carboxylic acid derived functional groups.22–25 Likewise, the oxetane ring is known to be a potentially effective replacement of the carbonyl moiety of ketones, aldehydes, carboxylic acids, esters and amides,26–31 and this presumably led to its incorporation in N-acylsulfonamides.32, 33 Compared to the parent N-acylsulfonamide structure, these replacements can be expected to introduce some changes in terms of properties (e.g., acidity), geometry and/or electrostatic potential (Figure 2B). However, interestingly, different examples have been reported in which incorporation of such structural motifs resulted in biologically active analogs in a variety of contexts (e.g., see 1–3, 5, 7 and 8, Figure 2C).33–40 Among these examples, the match paired molecules, 4 and 5, as well as 6–8, are especially notable as they reveal a comparable biological activity between the N-acylsulfonamide and the corresponding surrogate.35, 36
Figure 2:
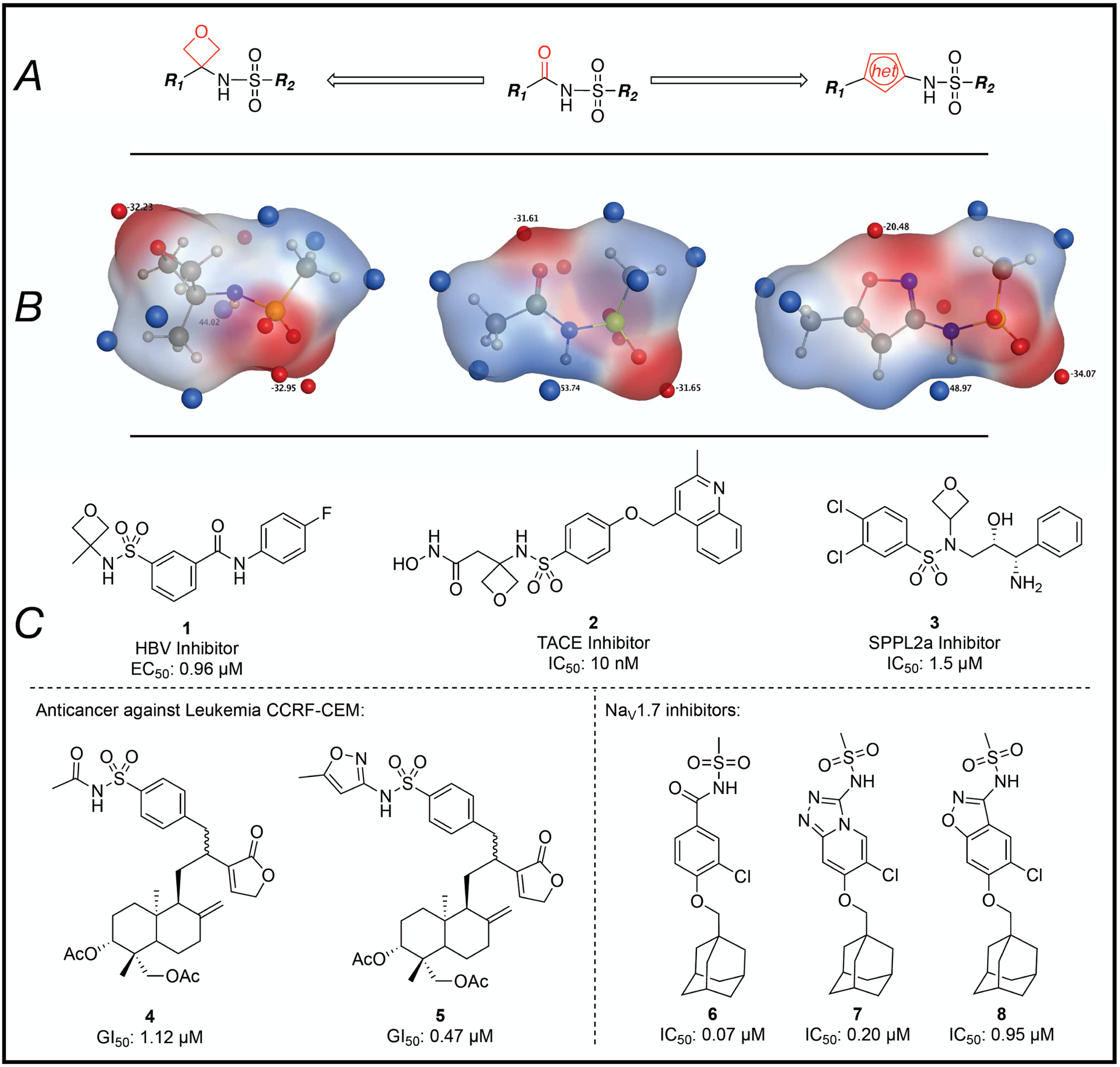
(A) General outline of N-acylsulfonamide bioisosteres that are based on replacement of the carbonyl moiety with either an oxetane or a 5-membered ring heteroaromatic ring. (B) comparison of Gaussian-optimized geometry and electrostatic potential maps of the N-acylsulfonamide moiety (center) and the corresponding oxetane (left) and isoxazole (right) derivatives. The areas colored in red and blue represent respectively negative and positive regions of the electrostatic potential; the corresponding surface minima and maxima are indicated as red and blue spheres. (C) Representative literature examples,37–39 including match paired comparisons, 4 and 5, as well as 6–8.35, 36
Given the importance of N-acylsulfonamides in medicinal chemistry and the potential utility of N-acylsulfonamide bioisosteres in analog design, an assessment of the relative impact on physicochemical properties that these replacements can have, compared to the parent N-acylsulfonamide group, may ultimately facilitate informed decisions in the hit/lead optimization stage. As a result, to enable an informative and meaningful comparison of the properties of these structures, we constructed a focused set of matched molecular pairs (MMP) comprised of different N-acylsulfonamides and relative derivatives, as well as a series of corresponding surrogates, and evaluated how these replacements can affect key physicochemical properties, such as: (a) acidity (pKa); (b) lipophilicity (logD7.4); (c) intrinsic solubility; and (d) passive diffusion in a parallel artificial membrane permeability assay (PAMPA). Finally, an assessment of the hydrogen bonding ability of selected examples was also conducted.
Results:
Library Design and Synthesis.
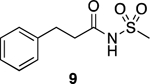
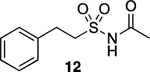
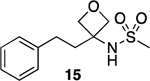
The compound library was designed following a general strategy utilized previously in structure property relationship (SPR) studies of carboxylic acid bioisosteres,41 in which the phenylpropionic acid fragment was used as structural template for analog design. The N-acylsulfonamide derivative 9 and related sulfuric diamide derivative 10 (Table 1), the physicochemical properties of which had been determined in these earlier studies,41 were chosen here as reference N-acylsulfonamide molecules. In addition, to better capture the differences in physicochemical properties that may arise from different substitutions and alternative orientations of the N-acylsulfonamide group, two additional control compounds, 11 and 12 (Table 1), were also included. For each of the four reference compounds, a series of related derivatives bearing different surrogates of the N-acylsulfonamide group were synthesized leading to a total of 24 entries (9–32, Table 1). Among the different classes of bioisosteres evaluated in the study are structures that have already been exemplified in analog design that are based on N-acyl moiety replacement with either a 5-membered ring heteroaromatic, such as isoxazole and 1,2,3-triazole, or an oxetane ring. In addition, since previous studies from our laboratories suggested that appropriately substituted thietanes may also serve as effective surrogates of the acyl group,26 a series of examples of N-acylsulfonamide derivatives bearing these 4-membered ring heterocycles were also chosen so as to expand the scope of the study beyond the examples of N-acylsulfonamide bioisosteres described in the literature. A comparison of the geometry and electrostatic potential of these structures relative to the corresponding N-acylsulfonamides is presented in Supporting Information.
Table 1.
Calculated and Experimental Properties of Test Compounds
| Cmpd | Structure | mp (°C)a | % Stability aq. buffer pH 7.4–7.6b | Intrinsic solubility (mol/L)c | logPd | logD7.4e | logP calcf | logD7.4 calcf | PAMPA | pKaj | pKa calcf | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pe (cm/s)g | % retentionh | logPappi | |||||||||||
| 9 | 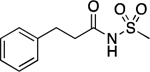 |
105.5–106.5 | 99.6 | 3.89E–2 | 1.10±0.01 | −0.64 (−1.02) | 0.86 | −0.27 | 3.45E–7 | 1.35E–2 | −6.46 | 4.75±0.01 (4.94) | 4.08 |
| 10 | 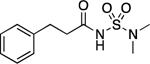 |
103.0–104.0 | 94.3 | 1.14E–2 | 1.66±0.02 | 0.12 (0.17) | 0.99 | −0.15 | 1.53E–6 | 2.15E–2 | −5.81 | 5.79±0.01 (5.86) | 4.12 |
| 11 | 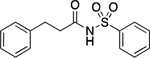 |
107.8–108.5 | 100.0 | 9.97E–4 | 2.67±0.01 | −0.09 | 2.92 | 1.78 | 1.64E–6 | −3.05E–2 | −5.79 | 4.49±0.04 | 4.24 |
| 12 | 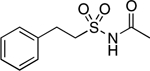 |
76.2–78.4 | 91.4 | 8.65E–2 | 1.04±0.01 | −0.87 | 0.78 | −0.36 | 1.00E–06 | −5.00E–2 | −6.00 | 4.91±0.01 | 4.09 |
| 13 | 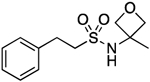 |
43.1–44.0 | 100.0 | 5.43E–2 | 1.58±0.06 | 1.58 | 0.83 | 0.83 | 6.79E–6 | −3.20E–2 | −5.17 | 10.45±0.01 | 9.59 |
| 14 | 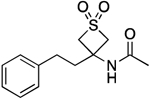 |
135.3–136.8 | 94.5 | 1.62E–3 | ‡ | 1.18† | 0.10 | 0.10 | 2.11E–6 | −1.20E–2 | −5.68 | >12 | 13.53 |
| 15 | 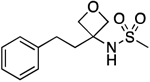 |
71.4–71.9 | 100.0 | 2.65E–2 | 1.61±0.03 | 1.61 | 0.76 | 0.76 | 8.44E–6 | 0.157 | −5.1 | 10.05±0.01 | 9.59 |
| 16 | 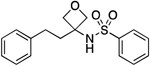 |
105.8–107.2 | 94.1 | 6.38E–4 | ‡ | 2.76† | 2.81 | 2.81 | 7.06E–6 | 0.260 | −5.2 | 10.29±0.12 | 10.15 |
| 17 | 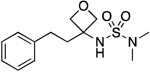 |
ND | 93.7 | 3.47E–2 | ‡ | 1.96† | 0.89 | 0.89 | 1.02E–5 | 5.89E–2 | −4.99 | 11.02±0.01 | 9.78 |
| 18 | 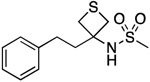 |
115.2–116.0 | 91.5 | 1.43E–3 | ‡ | 2.44† | 1.34 | 1.34 | 1.46E–05 | 0.439 | −4.8 | 10.45±0.02 | 10.70 |
| 19 | 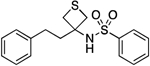 |
74.2–75.8 | 92.8 | 1.10E–3 | ‡ | 2.96† | 3.40 | 3.40 | ND | ND | ND | >12 | 10.16 |
| 20 | 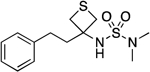 |
56.6–58.9 | 93.5 | 2.20E–3 | ‡ | 2.83† | 1.47 | 1.47 | 1.14E–5 | 0.373 | −4.9 | 11.93±0.05 | 10.89 |
| 21 | 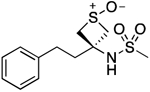 |
151.2–154.2 | 100.0 | 2.20E–2 | 0.88±0.02 | 0.87 | −0.43 | −0.43 | ND | ND | ND | 9.87±0.01 | 10.16 |
| 22 | 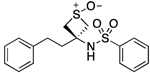 |
135.3–136.8 | 90.3 | 4.268E–2 | ‡ | 0.76† | 1.62 | 1.62 | 2.91E–6 | 5.07E–2 | −5.54 | 9.80±0.06 | 10.16 |
| 23 | 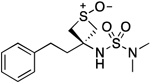 |
127.1–127.8 | 93.4 | 1.06E–2 | ‡ | 1.45† | −0.30 | −0.31 | 3.41E–6 | 7.13E–2 | −5.47 | 10.33±0.02 | 10.42 |
| 24 | 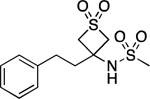 |
108.3–108.7 | 100.0 | 4.32E–2 | 1.03±0.01 | 1.03 | −0.24 | −0.25 | 3.66E–06 | −3.00E–3 | −5.4 | 9.44±0.02 | 9.07 |
| 25 | 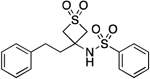 |
182.5–184.2 | 98.3 | 1.06E–3 | ‡ | 1.89† | 1.81 | 1.81 | ND | ND | ND | 10.22±0.15 | 10.16 |
| 26 | 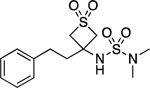 |
100.2–101.5 | 100.0 | 8.16E–3 | 1.83±0.05 | 1.82 | −0.11 | −0.12 | 4.43E–6 | 0.105 | −5.35 | 9.58±0.01 | 9.35 |
| 27 | 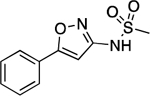 |
158.0–158.8 | 97.7 | 3.85E–2 | 0.58±0.02 | −0.42 | 1.21 | 0.42 | 1.16E–6 | 3.42E–2 | −5.94 | 5.22±0.01 | 6.44 |
| 28 | 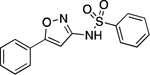 |
164.8–166.0 | 95.5 | 2.17E–3 | 1.76±0.03 | 0.99 | 3.26 | 2.24 | 5.79E–6 | −4.81E–2 | −5.24 | 6.62±0.02 | 5.82 |
| 29 | 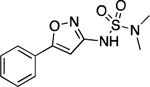 |
152.5–152.9 | 97.3 | 2.36E–2 | 0.85±0.01 | 0.42 | 1.33 | 0.95 | 2.87E–6 | 0.131 | −5.54 | 5.62±0.02 | 7.17 |
| 30 | 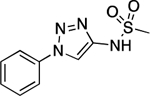 |
156.0–157.5 | 93.5 | 1.05E–2 | 1.16±0.03 | 0.06 | 0.57 | −0.19 | 2.42E–6 | −4.07E–2 | −5.62 | 6.34±0.01 | 6.48 |
| 31 | 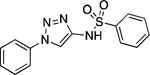 |
163.7–164.5 | 93.3 | 3.62E–4 | 2.55±0.04 | 1.06 | 2.63 | 1.59 | 4.09E–6 | 2.76E–4 | −5.39 | 5.93±0.05 | 5.72 |
| 32 | 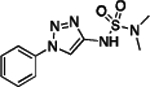 |
158.5–159.7 | 93.9 | 2.75E–3 | 1.72±0.01 | 0.69 | 0.70 | 0.34 | 3.77E–6 | −4.43E–2 | −5.42 | 6.42±0.01 | 7.20 |
Melting point of crystalline material;
Test compound (%) remaining after 5 h of incubation at rt in aqueous buffer (BICCA® Simulated intestinal fluid, without pancreatin; pH 7.40–7.60) as determined by relative changes in peak area of LC/MS chromatograms over time;
Intrinsic solubility determined from the general solubility equation (GSE) by using experimentally determined logP and mp values;
Log of the partition coefficient between n-octanol and water (unless otherwise noted, logP determinations were conducted via potentiometric titrations using a Sirius T3, Pion);
Log of the distribution coefficient between n-octanol and aqueous buffer at pH 7.4 (unless otherwise noted, logD7.4 determinations were conducted via potentiometric titrations using a Sirius T3, Pion);
Calculated values using ChemAxon47;
Effective permeability (PAMPA assay run by Analiza);
Membrane retention;
Log of the apparent permeability coefficient (Papp);
pKa values determined by potentiometric titrations using a Sirius T3, Pion (values in brackets are from41);
Test compound was an oil;
logD7.4 value determined via shake-flask assay (experiment run by Analiza);
logP value is considered equal to the logD7.4 as these compounds exhibit pKa values >9.4; ND = not determined.
Reference N-acylsulfonamide-derived compounds, 9–11, were either already available (9 and 10) or prepared (11) from phenylpropionic acid as described previously,41 while reference compound 12 was obtained via aminolysis of phenethylsulfonyl chloride, followed by N-acylation with acetic anhydride. The synthesis of all other test compounds is highlighted in Schemes 1 and 2. Sulfonamide, 13, was prepared by sulfonylating commercially available 3-methyloxetan-3-amine (33) under basic conditions (Scheme 1).
Scheme 1.
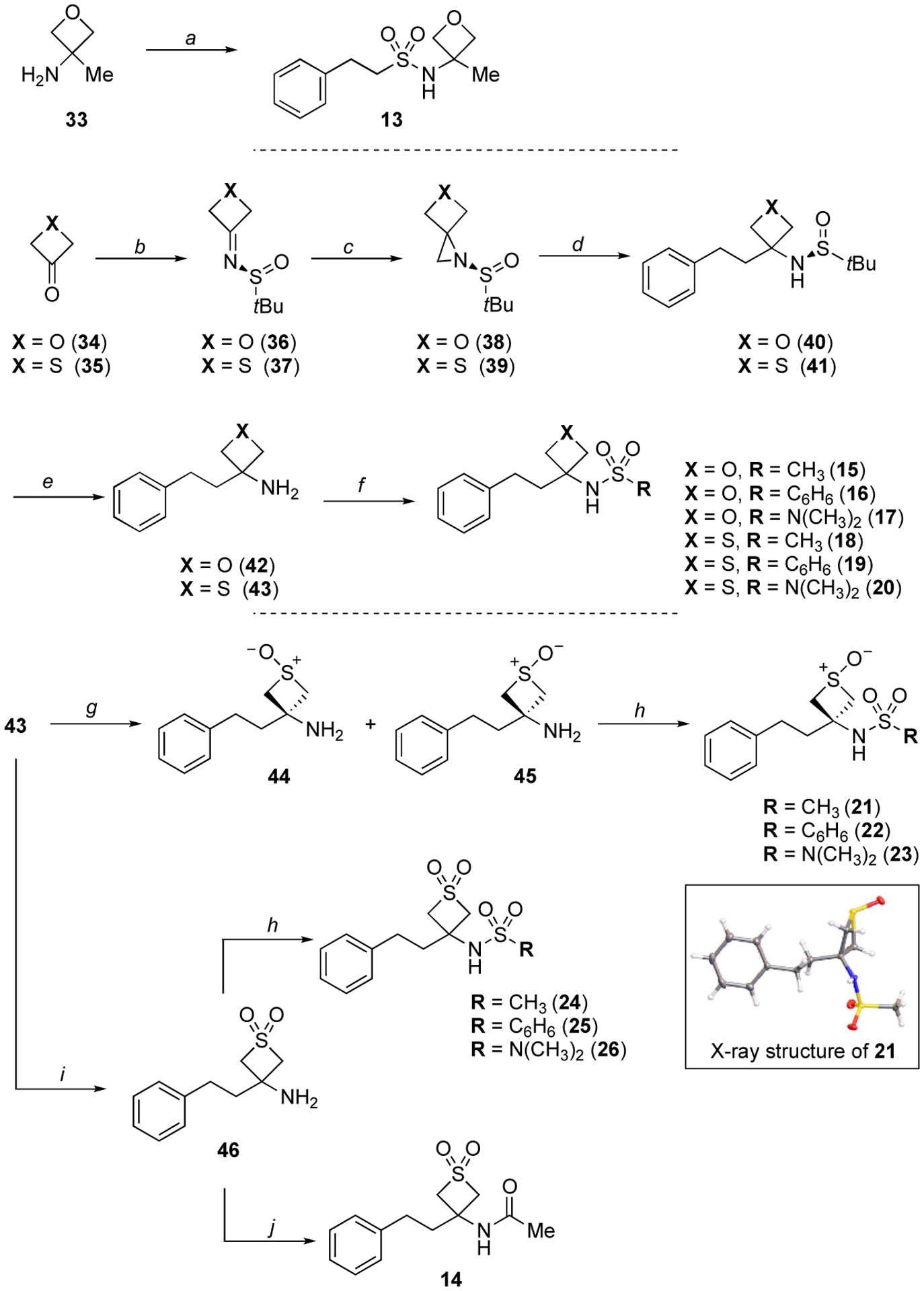
Reagents and conditions: (a) Et3N, 2-phenylethane-1-sulfonyl chloride, CH2Cl2, 0 °C to rt, 16 h, (34%); (b) (S)-(−)-2-methyl-2-propanesulfinamide, Ti(iPrO)4, CH2Cl2, reflux, 16 h, (77%); (c) TMSOI, NaH, DMSO, rt, 2 h, (39–49%); (d) benzylmagnesium chloride, CuI, THF, −30 to 0 °C, 1 h, (48–90%); (e) HCl, CH3OH, 0 °C to rt, 16 h, (62%); (f) Et3N, appropriate sulfonyl chloride or sulfamoyl chloride, CH2Cl2, 0 °C to rt, 16 h, (24–68%); (g) m-CPBA, CH2Cl2, −78 °C, 2 h, (70%); (h) Et3N, appropriate sulfonyl or sulfamoyl chloride, CH2Cl2, 0 °C to rt, 16 h, (15–78%); (i) oxone, acetone/H2O (1:1), 0 °C to rt, 16 h, (47%); (g) acetic anhydride, AgOTf, CH2Cl2, 0 °C to rt, 2 h, (67%).
Scheme 2.
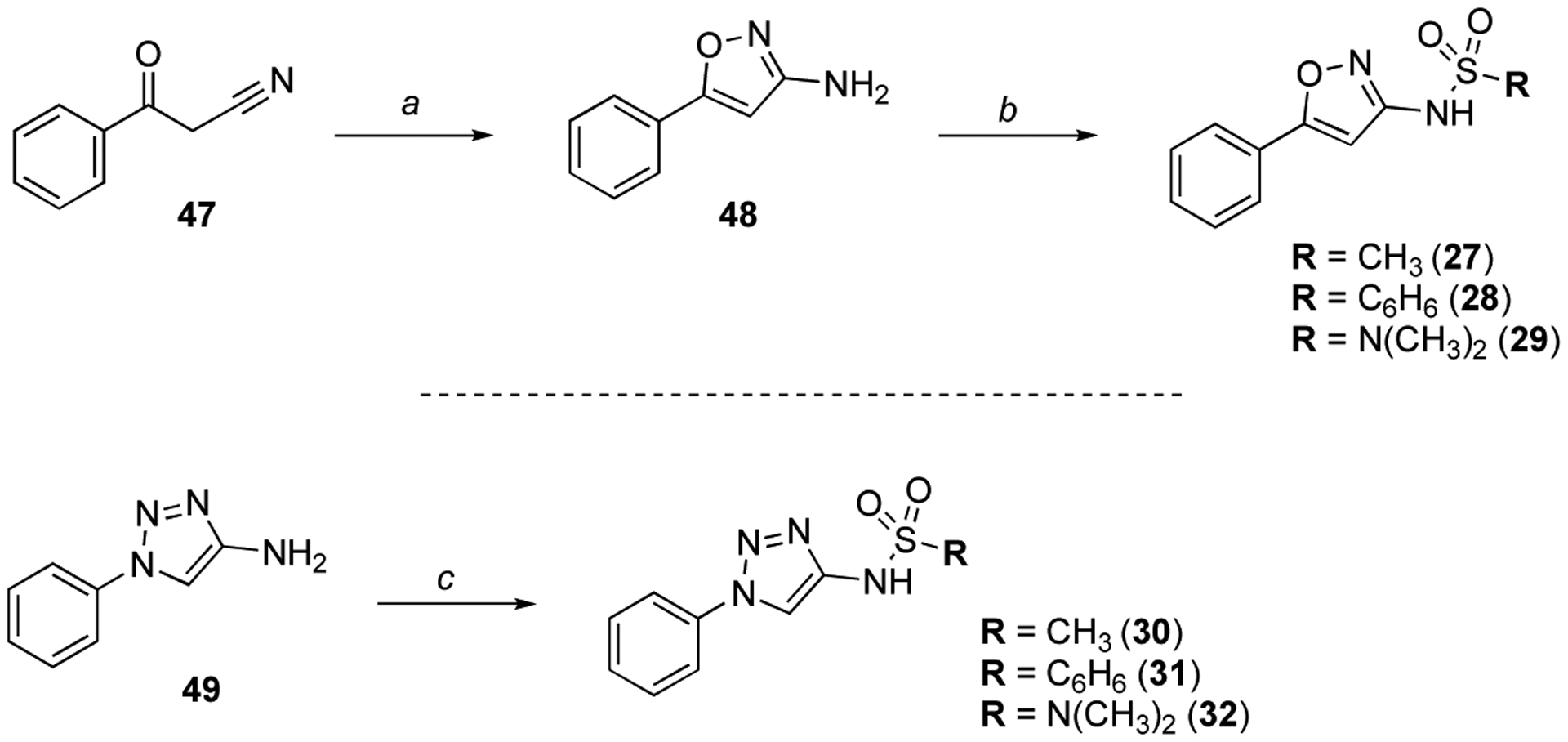
Reagents and conditions: (a) NH2OH·HCl, NaOH, H2O/EtOH (1:1), 80 °C, 2 h, (36%); (b) appropriate sulfonyl chloride or sulfamoyl chloride, pyridine, 0 °C to rt, 16 h, (18–75%); (c) appropriate sulfonyl chloride or sulfamoyl chloride, Et3N, CH2Cl2, 0 °C to rt, 16 h, (21–27%).
Oxetane and thietane derivatives, 15–20, were prepared in five steps starting from commercially available oxetan-3-one (34) and thietan-3-one (35). Condensation of the appropriate ketone and (S)-(−)-2-methyl-2-propanesulfinamide to the corresponding imines (36 and 37) followed by treatment with trimethylsulfoxonium iodide (TMSOI), led to aziridines 38 and 39, which were then subjected to ring opening using benzylmagnesium chloride to provide N-protected amines 40 and 41.42–44 Finally, deprotection under acidic conditions to the amine 42 and 43, followed by sulfonylation, furnished the desired sulfonamides 15–20 (Scheme 1).
Thietane-1-oxide (21–23) and −1,1-dioxide compounds (24–26) were obtained from thietane 43. Thus, oxidation of 43 with m-CPBA furnished a separable mixture of trans- and cis- 1-oxido-3-phenethylthietan-3-amines, 44 and 45. The major cis-isomer, 45, was then used for the synthesis of sulfonamides 21–23. Alternatively, oxone mediated conversion of 43 to the corresponding 1,1-dioxide, 46, followed by N-sulfonylation, yielded sulfonamides 24–26 (Scheme 1), while acylation of 46 with acetic anhydride provided acetamide 14.45
Isoxazole derivatives 27–29 were obtained via cyclocondensation of α-ketonitrile 47 in the presence of hydroxylamine, followed by sulfonylation of the resulting amino-isoxazole 48 with the appropriate sulfonyl chloride or sulfamoyl chloride46 (Scheme 2). Likewise, triazole compounds 30–32 were prepared via sulfonylation of commercially available 1-phenyl-1H-1,2,3-triazol-4-amine 49 (Scheme 2).
In addition to the structures of final product being established by 1H- and 13C-NMR, as well as IR, and HRMS, X-ray structures of selected compounds (15, 16, 18, 19, 21) were also obtained (see Supporting Information).
Determination of Physicochemical Properties.
All test compounds were initially evaluated for chemical stability in aqueous buffer (pH 7.4) by determining the percentage of remaining compound after 5 h of incubation. The results of this initial screening confirmed that the test compounds were generally stable (i.e., >90% unchanged) suggesting that spontaneous hydrolysis under typical assay buffer conditions would not be a limiting factor. Next, evaluation of physicochemical properties of test compounds included the determination of: (a) acidity (pKa); (b) lipophilicity (logD7.4); (c) intrinsic solubility; and (d) passive diffusion in a PAMPA assay. Determination of pKa values was conducted via potentiometric titrations using a Sirius T3 (Pion, Inc.). Likewise, the logP and logD7.4 values of compounds 9–13, 15, 21, 24, 26–32 were obtained via potentiometric titrations, while for selected compounds (14, 16–20, 22, 23, 25) the logD7.4 determinations were conducted via shake-flask method. Calculated logP, logD7.4, and pKa values were also obtained for comparison employing ChemAxon.47 For all solid compounds, the melting point (mp) of crystalline material was obtained and from the knowledge of mp and logP values, the intrinsic solubility was estimated via the general solubility equation.48, 49 Finally, an estimation of hydrogen bonding ability of selected compounds (i.e., 9, 12, 13, and 15) was conducted using experimental ΔlogP values in various solvents via potentiometric titrations.50–54 The results from these studies are summarized in Tables 1 and 2.
Table 2.
The ΔlogP values of selected compounds.a
 |
 |
 |
 |
|
|---|---|---|---|---|
| ΔlogPcyclohexane | 2.13 ± 0.04 | 1.63 ± 0.05 | 1.87 ± 0.04 | 2.01 ± 0.09 |
| ΔlogPheptane | 1.63 ± 0.05 | 1.48 ± 0.04 | 1.60 ± 0.05 | 1.72 ± 0.13 |
| ΔlogPtoluene | 0.89 ± 0.03 | 0.40 ± 0.06 | 0.43 ± 0.04 | 0.52 ± 0.07 |
ΔlogP = logPoctanol-water − logPhydrocarbon-water; the partition coefficients in different solvents were experimentally determined via potentiometric titrations (see Supporting Information).
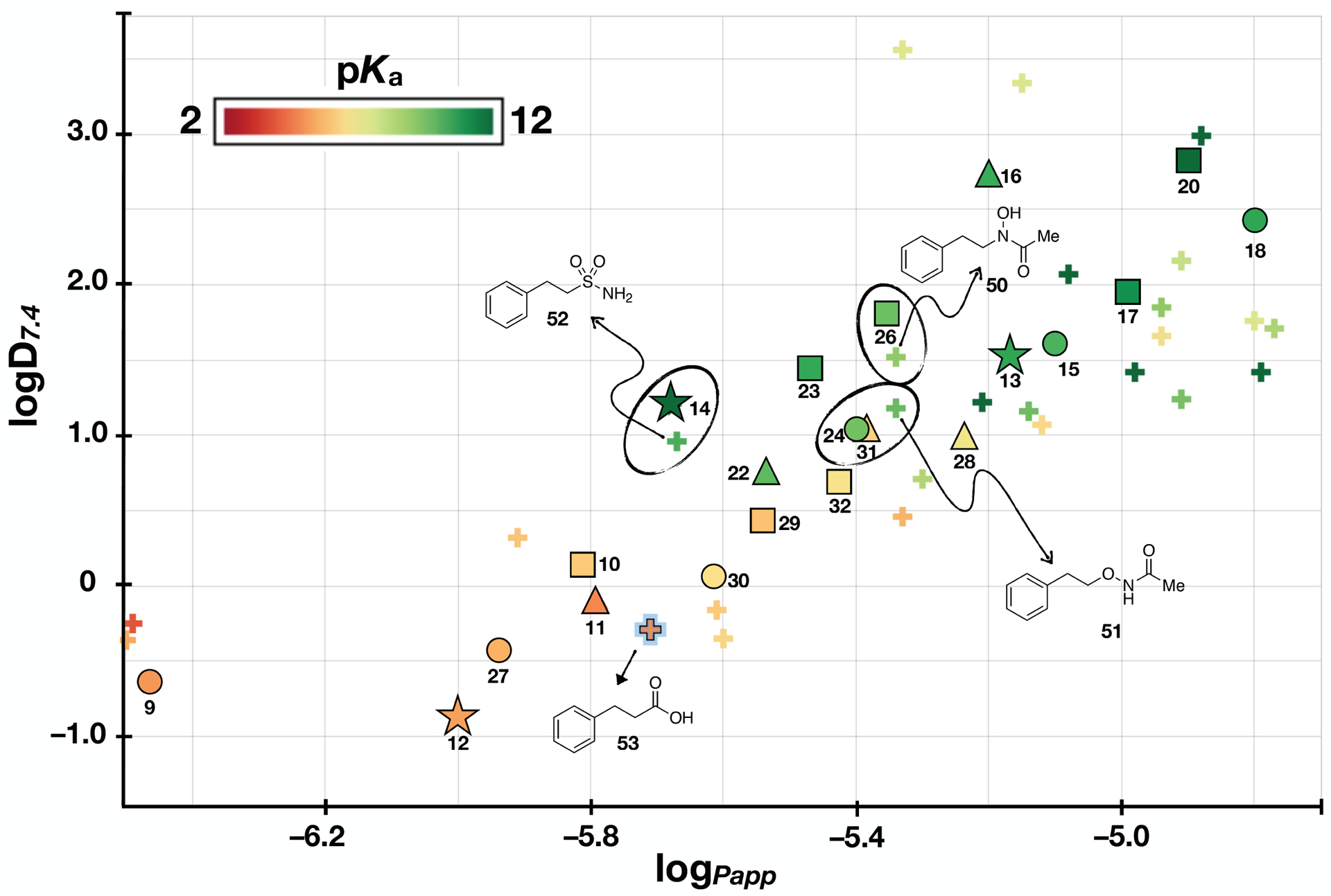
From the data accumulated in this study, different elements of SPRs can be identified. First, within each series of compounds (denoted with different symbols in Figure 3), the reference N-acylsulfonamides, 9–12, were found to be generally the most acidic, as well as the least permeable and lipophilic molecules, with 10 and 11 being comparatively more lipophilic and permeable than the two other controls, 9 and 12. Although a good overall correlation linking the logD7.4 values and the apparent permeability coefficients (Papp) was observed across the entire data set (r2 = 0.890, Figure 3), some interesting discrepancies were noted. For example, the results from the PAMPA assay indicate a possible difference in permeability between 9 and 12, with the latter being more permeable than the former. This result may be somewhat unexpected considering that these two compounds are structural isomers characterized by very similar pKa and logD7.4 values.
Figure 3.
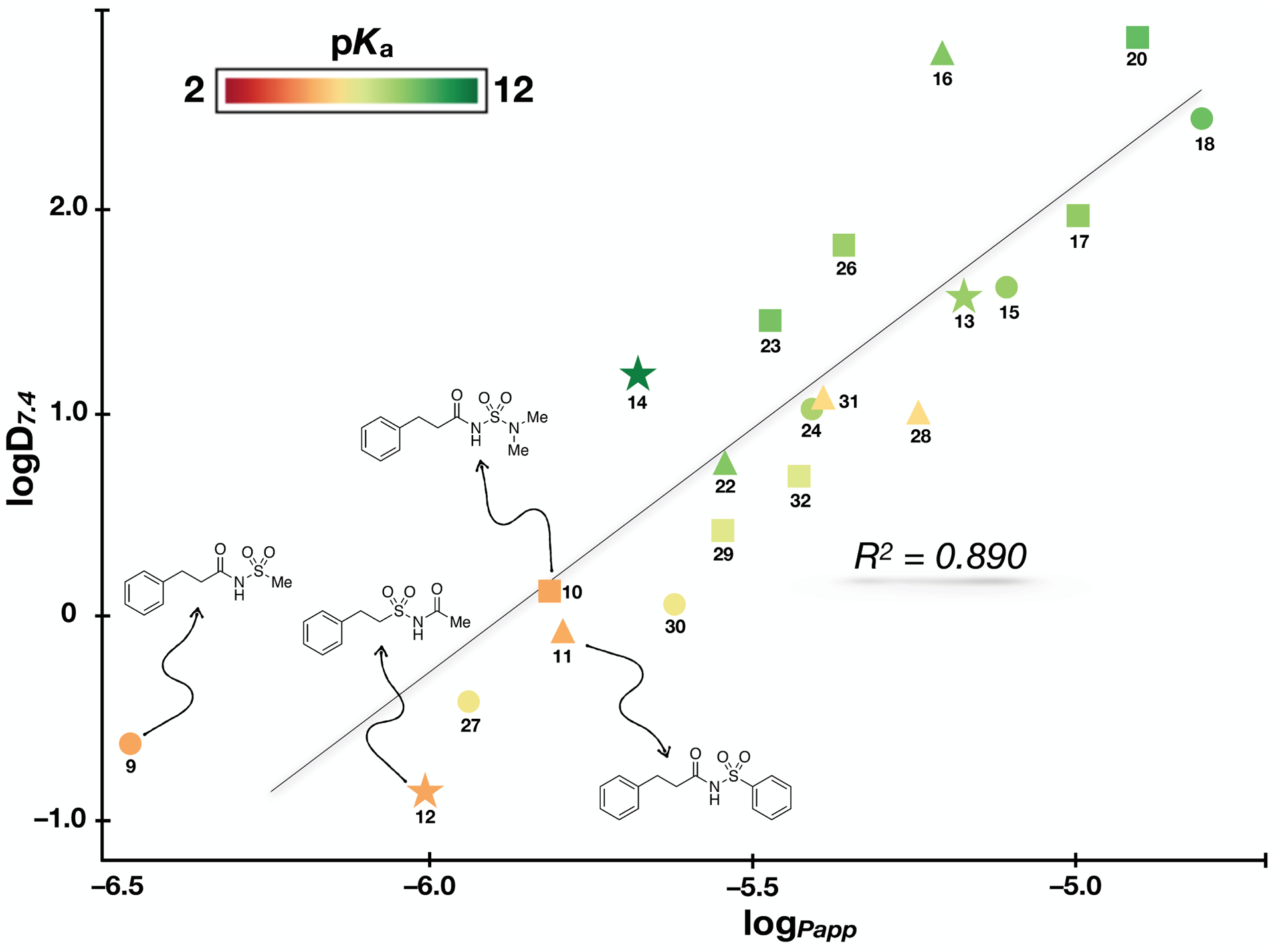
Plot showing lipophilicity (i.e., logD7.4), acidity (i.e., pKa), and permeability (i.e., logPapp) of test compounds. Each of the four reference compounds (structures shown) and their corresponding analogs are identified with different symbols.
We have previously observed that differences in PAMPA permeability values of compounds having similar acidity and lipophilicity may be ascribed to the differential degree of hydrogen bonding and solvation.41 However, in the particular case of 9 and 12, these two compounds are structural isomers that share identical polar surface area (tPSA = 63.24 Å2) suggesting that the ability to hydrogen bond of these two isomers may be closely comparable. Thus, to assess experimentally possible differences in hydrogen bonding between 9 and 12, we evaluated the difference in the partition coefficients (ΔlogP) of each of the two test compounds when the partition solvent is changed from the protic solvent, n-octanol, to a non-polar aprotic hydrocarbon, such as cyclohexane, heptane or toluene (Table 2). The ΔlogP values have been used before to estimate the propensity of compounds to hydrogen bond,50, 55 and this parameter has been found to correlate well with passive diffusion.51, 53, 56, 57 Interestingly, although 9 and 12 have identical tPSA, which would suggest a near identical hydrogen bonding ability, the ΔlogP values of isomer 9 were found to be consistently higher (0.15 – 0.50) than the corresponding values of 12 regardless of the aprotic solvent used in the experiment. By comparison, the relative difference in ΔlogP values in the case for the oxetane derivatives, 13 and 15, which are also structural isomers but with more closely comparable PAMPA permeability values, appeared to be generally narrower (0.09 – 0.14, Table 2). These results suggest that the N-acylsulfonamide, 9, has greater propensity to hydrogen bond than the structural isomer, 12, and this likely results in comparatively tighter solvation and lower permeability in the PAMPA assay.
Among the different series of model compounds, the derivatives of acylsulfonamide 9 (shown as circles in Figure 3) exhibit a wider and more evenly distributed change in properties compared to other series, with relative increases in lipophilicity, permeability and acidity that ranged respectively: 0.2 – 3.1 (logD7.4); 0.5 – 1.7 (logPapp); 0.5 – 5.7 (pKa). Notably, the oxetane/thietane derivatives of 9 (e.g., 15, 18, 21 and 24) are significantly less acidic with pKa values that are >5 units higher than the pKa value of 9. The reduced acidic character of these compounds, however, may not be always associated with a reduction in hydrogen bonding ability as suggested again by ΔlogP values (Table 2) that in the case of oxetane derivative, 15, appear to be within the range of values registered for N-acylsulfonamide 9 and 12. This is in agreement with other reports of the oxetane ring being an excellent HB acceptor, often better than carbonyls.58, 59
Further comparison of the properties of different series of model compounds suggests that in the case of 9 and 10 (circles and squares in Figure 3) the impact that each of the 4- and 5-membered ring carbonyl replacements have on compound lipophilicity and permeability follows a similar trend. Within each series of analogs and for both parameters (i.e., logD7.4 and logPapp) the isoxazole derivatives (27, 29) registered the smallest increase relative to the corresponding N-acylsulfonamides, while the oxetane and thietane derivatives (15, 17, 18, 20) produced the largest differential in lipophilicity and permeability. This trend is less evident in the case of compounds derived from 11 (triangles in Figure 3). Although the oxetane (16) and thietane (19) derivatives are still the most lipophilic members within this series, the corresponding thietane-1-oxide (22), isoxazole (28) and triazole (31) compounds appear to fall within a relatively narrow range of logD7.4 values. Some series-specific differences can also be seen when plotting the relative changes in intrinsic solubility values (Figure 4). However, in the majority of cases, carbonyl replacement with a 1,2,3-triazole (i.e., 30, 31, and 32) or a thietane (e.g., 18 and 20) appears to result in a relative decrease in intrinsic solubility, whereas replacement with an isoxazole (i.e., 27, 28, and 29) or a thietane-1-oxide (e.g., 22) produces solubility values that are either similar or above the intrinsic solubility of the corresponding control compound.
Figure 4.
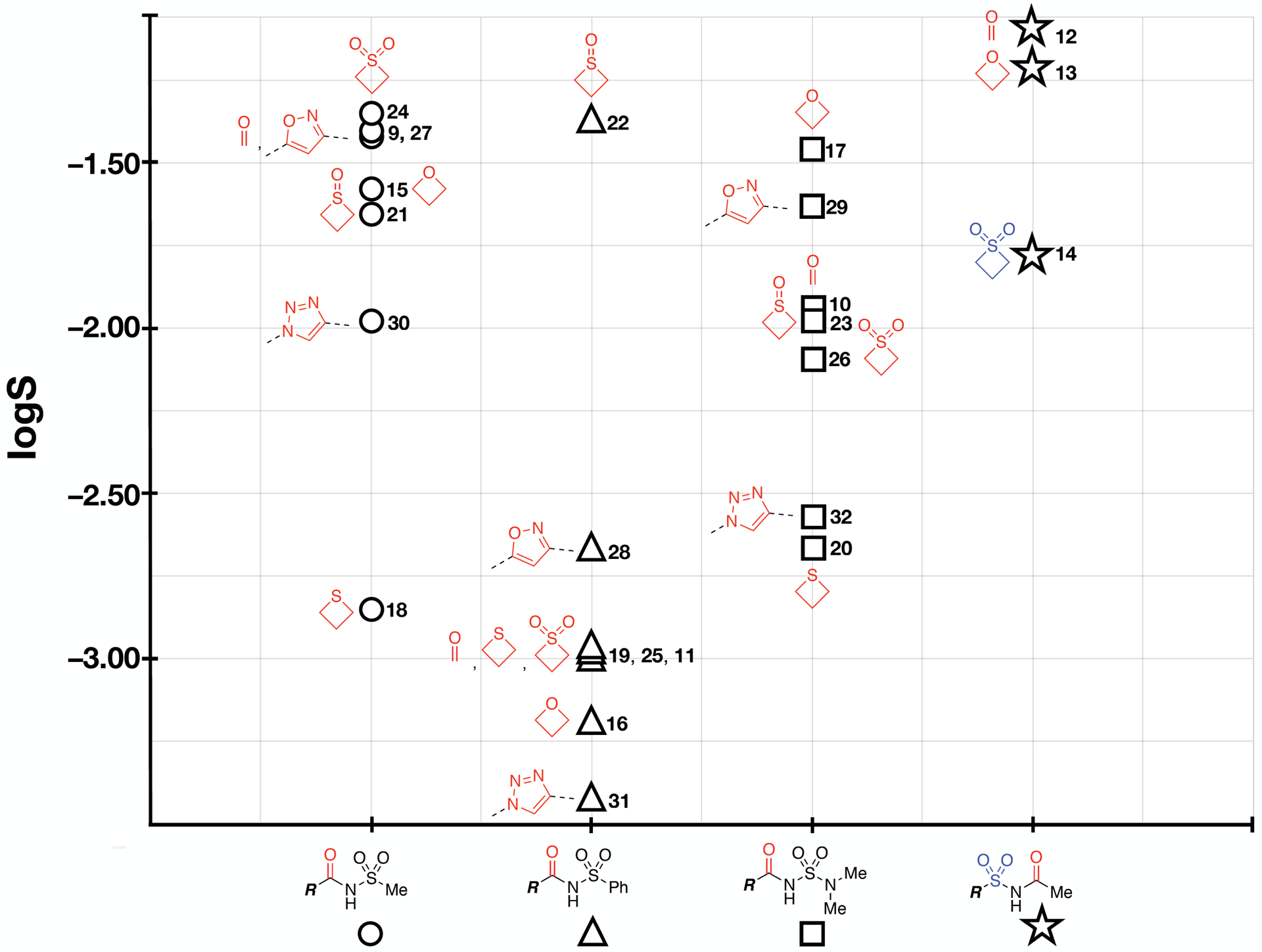
Plot of the log of the intrinsic solubility (S) of the different series of test compounds; logS values are obtained from the general solubility equation [logS = −logP – 0.01 × (mp – 25) + 0.5] by using experimentally determined mp and logP values.
Finally, comparison of calculated and experimental values, showed that the pKa predictions (ChemAxon47) are generally accurate across the entire data set (r2 = 0.95, Figure 5A). However, the r2 values are significantly lower if the linear regression analysis is performed separately within the more and less acidic compound clusters (Figure 5A). A relatively modest correlation was also noted between calculated and experimental logD7.4 values (r2 = 0.57, Figure 5B). These observations suggest that although the rapid, inexpensive availability of predicted pKa and lipophilicity values remains undeniably a great resource to medicinal chemistry, especially in programs requiring the rapid assessment of large number of compounds, experimental determinations are clearly necessary in SPR studies.
Figure 5.
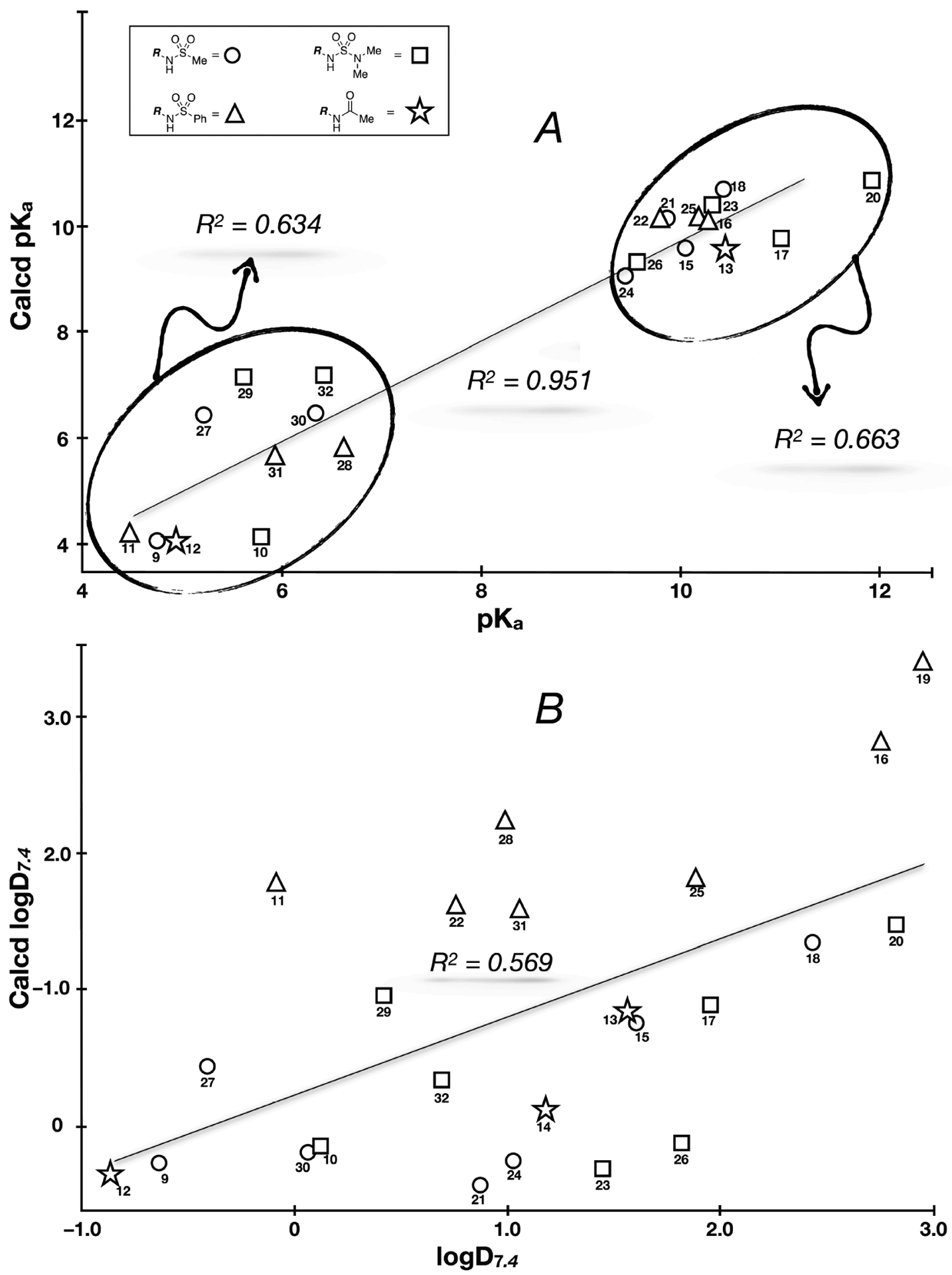
Comparison between experimental and calculated pKa (A) and logD7.4 values (B).
Discussion
The isosteric replacement of specific atoms, functional groups, or fragments with appropriate surrogate structures is a validated strategy in medicinal chemistry that can be utilized to modulate the intrinsic physicochemical properties of compounds of interest, resulting in derivatives with potentially improved pharmacokinetics and/or pharmacodynamics.20 Considering that the outcome of any isosteric replacement can vary considerably depending on the particular context in which it is applied, a screening and evaluation of alternative isosteres is almost invariably needed. Under these circumstances, knowledge of the relative ranking of different isosteres based on experimentally determined physicochemical properties can be helpful to enable informed decisions in the analog design process. Structure property relationship studies that are based on comparisons between MMPs comprise an effective strategy to reveal relative differences and trends associated with specific chemical transformations. As such, this strategy is especially suited to investigate the property space of isosteric replacements.41 The present study was aimed at assessing the properties of different representative N-acylsulfonamides, as well as different classes of related surrogates. With respect to the four N-acylsulfonamides examined in these studies, the data collected illustrate that, as expected, relatively simple manipulations of the substituent at the sulfur center (e.g., 9 – 12) can be exploited to modulate the physicochemical properties of these compounds. Perhaps of particular interest is the observation that the alternative arrangement of the N-acylsulfonamide group, as in 9 and 12, produces structural isomers that exhibit different hydrogen bonding ability, as determined by ΔlogP values, that in turn correlates with different apparent permeability coefficients in the PAMPA assay.
With respect to the N-acylsulfonamide surrogates examined, these included examples where the carbonyl moiety is replaced with either a 5-membered ring heteroaromatic, or an oxetane ring, as these structural motifs already found applications in drug design as N-acylsulfonamide bioisosteres. Moreover, a series of thietane derivatives were also included and depending on the particular oxidation state in the thietane sulfur atom, these compounds could significantly expand the range of properties of their respective series. Although the N-(thietan-3-yl)sulfonamide substructure is still relatively rare and unexplored and the possible bioisosteric relationships of these derivatives have not yet been investigated, a comparison of geometry and electrostatic potential of the N-acylsulfonamide 9, and the corresponding oxetane/thietane derivatives (Figure 6 and Supporting Information) would suggest that these types of structures may potentially serve as candidate replacements. In addition, considering that the model compounds studied here were derived from the same phenylpropionyl structural template used in earlier SPR studies of carboxylic acid bioisosteres,26, 41 the physicochemical properties of these novel structures could be directly compared against different classes of acidic moieties, gaining additional insight.
Figure 6.
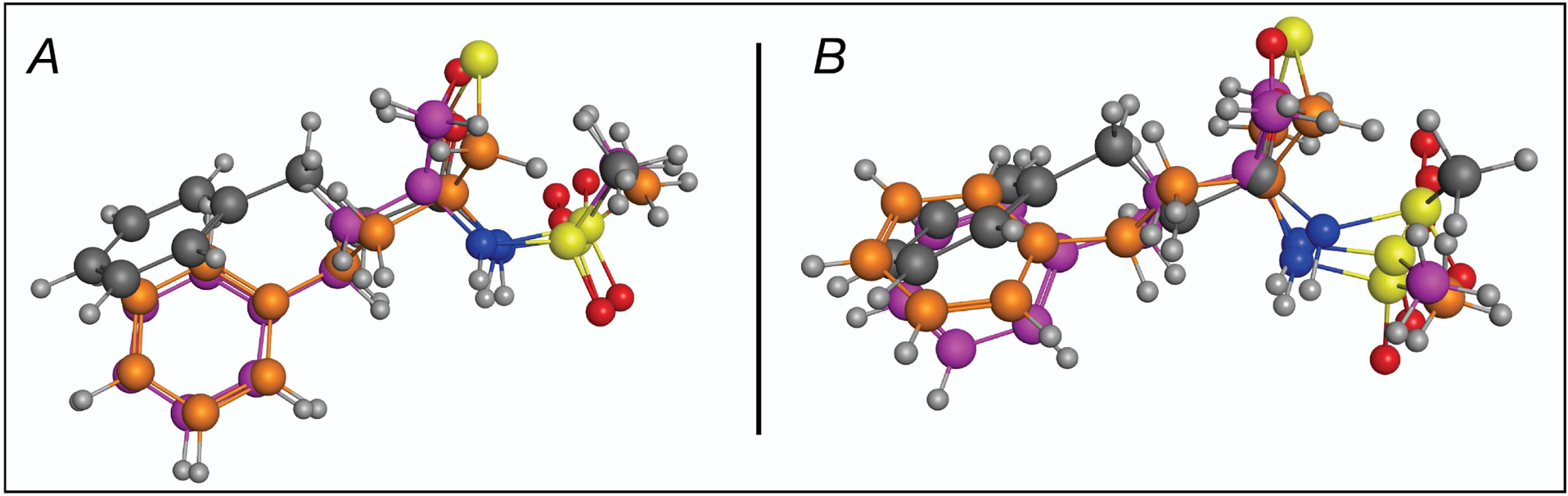
Overlay of the X-ray structures (A) and structures obtained via Gaussian geometry optimization (B) of compound 9 (grey), 15 (purple), and 18 (orange).
Indeed, comparison of the pKa, logD7.4 and PAMPA permeability data of the different sets of compounds (Figure 7 and Supporting Information) shows that many of the thietane derivatives fall within the property space of acidic moieties, with selected structures being near isometric with other acidic groups. For example, the acidity, lipophilicity and permeability of 24 and 26 appear to be closely comparable respectively to the hydroxamic ester 50 and acid 51 derivatives. Likewise, interestingly, the physicochemical properties of amide 14 are similar to the properties of the corresponding phenethylsulfonamide 52. Although these similarities in physicochemical properties cannot be used to infer bioisosteric relationships, these results help in further contextualizing the properties of the novel structures and in providing a rationale for their possible use in medicinal chemistry programs.
Figure 7.
Comparison of lipophilicity (i.e., logD7.4), acidity (i.e., pKa), and permeability (i.e., logPapp) of test compounds vs other classes of carboxylic acid bioisosteres from41 (crosses). Structures and data for all compounds are in the Supporting Information.
Thus, taken together, these similarities as well as the general trends of SPRs identified in this study may facilitate informed decisions when deploying N-acylsulfonamides, or surrogates thereof, in analog design.
Conclusions.
The results from this study provide an assessment of the physicochemical properties of different N-acylsulfonamides, as well as a series of corresponding surrogate structures. This data set may be helpful in drug design efforts that involve the incorporation or replacement of N-acylsulfonamides. In addition, by complementing and expanding previous SPR studies of carboxylic acid bioisosteres, the data presented may contribute in further defining the property space of acidic moieties and their surrogates.
MATERIALS AND METHODS
All solvents and reagents were reagent grade. The hexane solvent was a mixture of isomers. All reagents were purchased from reputable vendors and used as received. Thin layer chromatography (TLC) was performed with 200 μM MilliporeSigma precoated silica gel aluminum sheets. TLC spots were visualized under UV light or using KMnO4 stain. Flash chromatography was performed with SiliaFlash P60 (particle size 40–63 μM) supplied by Silicycle. Melting points were taken from Mel-Temp II by Barnstead Thermolyne, using an Omega digital thermometer. Infrared (IR) spectra were recorded on a PerkinElemer FT-IR spectrometer. Proton and carbon NMR spectra were recorded on a 600 MHz NMR spectrometer. Chemical shifts were reported relative to residual solvent’s peak. High-resolution mass spectra were measured at the University of California San Diego Molecular Mass Spectrometry Facility. Single crystal X-ray structure determinations were performed at the University of California San Diego Crystallography Facility. Analytical reverse phase high-performance liquid chromatography (HPLC) was performed using a SunFire C18 (4.6 × 50 mm, 5 mL) analytical column, while preparative reverse phase HPLC purifications were performed using a SunFire preparative C18 OBD column (5 μm 19 × 50 mm) on a Gilson instrument. Samples were analyzed with analytical HPLC and employed 10% to 90% of CH3CN in H2O over 6–12 min and flow rate of 2 mL/min. Samples purified by preparative HPLC employed 10% to 90% of CH3CN in H2O over 6–20 min and flow rate of 20 mL/min. All final compounds were found to be >95% pure by HPLC/UV.
3-Phenyl-N-(phenylsulfonyl)propanamide (11).
To a solution of phenylpropionic acid (0.500 g, 3.33 mmol, 1.00 eq), EDC·HCl (0.766 g, 4.00 mmol, 1.20 eq) and DMAP (0.488 g, 4.00 mmol, 1.20 eq) in anh. CH2Cl2 (20.0 mL), benzenesulfonamide (0.523 g, 3.33 mmol, 1.00 eq) was added at rt under N2. The resulting mixture was stirred at reflux for 8 h and then at rt for an additional 48 h. The reaction was quenched with H2O, then extracted with CH2Cl2 (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 15% of EtOAc in hexane) provided the title compound as a white solid (0.641 g, 2.22 mmol, 67%). 1H NMR (600 MHz, CDCl3) δ 8.03 – 7.96 (m, 2H), 7.66 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.9 Hz, 2H), 7.24 – 7.13 (m, 3H), 7.09 – 6.97 (m, 2H), 2.87 (t, J = 7.7 Hz, 2H), 2.56 (t, J = 7.7 Hz, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 170.33, 139.48, 138.31, 133.91, 128.93, 128.52, 128.13, 126.32, 37.84, 30.18 ppm. HRMS (ES+) calculated for C15H15NO3S [M + H]+ 290.0845, found 290.0846. IR (neat) ν 3107.25, 2886.93, 1682.94, 1460.06, 1452.37, 1344.62, 1170.12, 1089.75, 1072.22 cm−1.
N-(Phenethylsulfonyl)acetamide (12).
Synthesis of 12 followed previously described procedures with some modifications.60 To a solution of 2-phenylethane-1-sulfonyl chloride (0.500 g, 2.44 mmol, 1.00 eq) in anh. CH2Cl2 (6.0 mL), NH3 (7.0 M in methanol, 1.74 mL, 5.00 eq) was added dropwise at – 78 °C under N2. The reaction mixture was slowly warmed to rt and stirred at this temperature for 3 h. The resulting mixture was neutralized with 1.0 M HCl and extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The resulting material was used in the subsequent step without further purification (0.452 g, 2.44 mmol, quantitative yield). 1H NMR (600 MHz, CDCl3) δ 7.33 (t, J = 7.7 Hz, 2H), 7.27 (d, J = 7.2 Hz, 1H), 7.22 (d, J = 7.2 Hz, 2H), 3.88 (s, 2H), 3.40 – 3.34 (m, 2H), 3.19 – 3.13 (m, 2H) ppm. To a solution of 2-phenylethane-1-sulfonamide (0.100 g, 0.540 mmol, 1.00 eq), DMAP (0.007 g, 0.055 mmol, 0.10 eq), and Et3N (0.164 g, 1.62 mmol, 3.00 eq) in anh. CH2Cl2 (5.0 mL), acetic anhydride (0.138 g, 1.35 mmol, 2.50 eq) was added at −78 °C under N2. The mixture was slowly warmed to rt and stirred for 24 h. The resulting mixture was diluted with H2O and extracted with EtOAc (×3). The combined organic extracts were dried over Mg2SO4, filtered, and concentrated in vacuo. Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound as an off-white solid (0.018 g, 0.079 mmol, 15%). 1H NMR (600 MHz, CDCl3) δ 7.33 (t, J = 7.6 Hz, 2H), 7.27 (d, J = 7.9 Hz, 1H), 7.23 (d, J = 7.5 Hz, 2H), 3.75 (t, J = 7.7 Hz, 2H), 3.15 (t, J = 7.7 Hz, 2H), 1.94 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 169.05, 136.93, 129.12, 128.67, 127.34, 53.95, 29.65, 23.60 ppm. HRMS (ES+) calculated for C10H14NO3S [M + Na]+ 250.0508, found 250.0509. IR (neat) ν 3238.70, 2925.23, 1722.07, 1404.68, 1326.17, 1146.20, 1135.20 cm−1.
N-(3-Methyloxetan-3-yl)-2-phenylethane-1-sulfonamide (13).
To a solution of 3-methyloxetan-3-amine (0.050 g, 0.570 mmol, 1.00 eq) in anh. CH2Cl2 (5.0 mL), Et3N (0.170 g, 1.70 mmol, 3.00 eq) was added at 0 °C under N2, followed by 2-phenylethane-1-sulfonyl chloride (0.350 g, 1.70 mmol, 3.00 eq). The resulting mixture was warmed to rt and stirred overnight. After 16 h, H2O was added, and the crude was extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 30% of EtOAc in hexane) provided the title compound (0.050 g, 0.200 mmol, 34%). 1H NMR (600 MHz, CDCl3) δ 7.33 (t, J = 7.6 Hz, 2H), 7.27 (s, 1H), 7.22 (d, J = 7.5 Hz, 2H), 4.70 (d, J = 6.8 Hz, 2H), 4.53 (s, 1H), 4.41 (d, J = 6.8 Hz, 2H), 3.38 – 3.30 (m, 2H), 3.19 – 3.13 (m, 2H), 1.72 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 137.80, 129.11, 128.57, 127.23, 83.13, 57.76, 56.19, 30.23, 24.71 ppm. HRMS (ES+) calculated for C12H18NO3S [M + Na]+ 278.0821, found 278.0822. IR (neat) ν 3265.69, 2965.91, 2880.01, 1455.03, 1312.55, 1128.64, 1007.16 cm−1.
N-(1,1-Dioxido-3-phenethylthietan-3-yl)acetamide (14).
To a solution of 3-amino-3-phenethylthietane 1,1-dioxide 46 (0.060 g, 0.270 mmol, 1.00 eq) in anh. CH2Cl2, acetic anhydride (0.054 g, 0.530 mmol, 2.00 eq) was added at 0 °C under N2, followed by addition of silver triflate (0.001 g, 0.004 mmol, 0.01 eq). The mixture was stirred at this temperature for 30 min, then stirred at rt for 2 h. The solvent was evaporated in vacuo. Purification via silica gel column chromatograph (up to 50% EtOAc in hexane) provided the title compound (0.048 g, 0.180 mmol, 67%). 1H NMR (600 MHz, CDCl3) δ 7.30 (t, J = 7.5 Hz, 2H), 7.24 – 7.20 (m, 1H), 7.18 – 7.14 (m, 2H), 5.81 (s, 1H), 4.24 – 4.18 (m, 2H), 4.11 – 4.06 (m, 2H), 2.64 (dd, J = 8.8, 6.4 Hz, 2H), 2.49 (dd, J = 8.7, 6.5 Hz, 2H), 1.91 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 170.36, 139.86, 129.01, 128.45, 126.79, 74.30, 45.26, 38.77, 31.26, 23.54 ppm. HRMS (ES+) calculated for C13H17NO3S [M + Na]+ 290.0821, found 290.0821. IR (neat) ν 3298.61, 1647.68, 1540.27, 1455.85, 1358.84, 1315.32, 1301.07, 1286.78, 1194.79, 1137.89, 1072.13, 1033.51 cm−1.
3-Phenethyloxetan-3-amine (42) and N-(3-phenethyloxetan-3-yl)methanesulfonamide (15).
To (S)-2-methyl-N-(3-phenethyloxetan-3-yl)propane-2-sulfinamide 40 (0.150 g, 0.533 mmol, 1.00 eq) in anh. CH3OH (4.0 mL), a solution of HCl (2.13 mL, 1.25 M solution in CH3OH, 2.67 mmol, 5.00 eq) was added dropwise at 0 °C under N2. The mixture was stirred at rt overnight. The mixture was concentrated in vacuo, and the resulting material was used in the next step without purification. To 3-phenethyloxetan-3-amine hydrochloride salt in anh. CH2Cl2 (4.0 mL), Et3N (0.150 mL, 1.07 mmol, 2.00 eq) was added at 0 °C under N2, followed by the addition of methanesulfonyl chloride (0.083 mL, 1.07 mmol, 2.00 eq) dropwise at 0 °C. The reaction mixture was stirred at rt overnight. The reaction was then quenched with H2O, and the resulting mixture was extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 30% of EtOAc in hexane) provided the title compound as a white solid (0.051 g, 0.197 mmol, 37% over two steps). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.22 (d, J = 7.9 Hz, 3H), 4.73 (d, J = 7.0 Hz, 3H), 4.48 (d, J = 7.0 Hz, 2H), 3.09 (s, 3H), 2.81 – 2.72 (m, 2H), 2.44 – 2.38 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.53, 128.90, 128.44, 126.61, 81.29, 59.60, 44.58, 38.54, 30.16, 24.91 ppm. HRMS (ES+) calculated for C12H17NO3S [M + Na]+ 278.0821, found 278.0821. IR (neat) ν 3142.48, 3029.94, 2954.18, 2931.24, 2886.46, 1682.06, 1601.77, 1469.16, 1453.11, 1348.08, 1308.44, 1154.59 cm−1.
N-(3-Phenethyloxetan-3-yl)benzenesulfonamide (16).
Following the same procedure described for the synthesis of 15, using (S)-2-methyl-N-(3-phenethyloxetan-3-yl)propane-2-sulfinamide 40 (0.173 g, 1.95 mmol, 1.00 eq), benzenesulfonyl chloride (0.345 g, 1.95 mmol, 2.00 eq), and Et3N (0.198 g, 1.95 mmol, 2.00 eq). Purification by silica gel column chromatography (up to 20% EtOAc in hexane) provided the title compound (0.210 g, 0.662 mmol, 68%). 1H NMR (600 MHz, CDCl3) δ 7.93 (d, J = 8.1 Hz, 2H), 7.67 – 7.60 (m, 1H), 7.55 (t, J = 7.8 Hz, 2H), 7.24 (t, J = 7.3 Hz, 2H), 7.21 – 7.15 (m, 1H), 7.00 – 6.91 (m, 2H), 5.01 (s, 1H), 4.61 (d, J = 7.0 Hz, 2H), 4.35 (d, J = 7.0 Hz, 2H), 2.47 – 2.40 (m, 2H), 2.36 – 2.29 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 142.02, 140.70, 133.13, 129.51, 129.48, 128.38, 127.01, 126.28, 81.28, 59.25, 38.37, 29.85 ppm. HRMS (ES+) calculated for C17H19NO3S [M + Na]+ 340.0978, found 340.0975. IR (neat) ν 3115.92, 2981.87, 1444.04, 1320.06, 1147.53, 1092.66 cm−1.
N,N-Dimethyl-[3-(2-phenylethyl)oxetane-3-yl]sulfamoyl-amine (17).
Following the same procedure described for the synthesis of 15, using (S)-2-methyl-N-(3-phenethyloxetan-3-yl)propane-2-sulfinamide 40 (0.173 g, 0.976 mmol, 1.00 eq), dimethylsulfamoyl chloride (0.280 g, 1.95 mmol, 2.00 eq), and Et3N (0.198 g, 1.95 mmol, 2.00 eq). Purification by silica gel column chromatography (up to 20% EtOAc in hexane) provided the title compound (0.098 g, 0.340 mmol, 35%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.7 Hz, 2H), 7.22 (d, J = 6.8 Hz, 3H), 4.74 (d, J = 7.0 Hz, 2H), 4.45 (d, J = 6.8 Hz, 2H), 4.38 (s, 1H), 2.86 (s, 6H), 2.75 – 2.70 (m, 2H), 2.43 – 2.35 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.98, 128.78, 128.46, 126.41, 81.08, 59.23, 38.43, 38.01, 30.01 ppm. HRMS (ES+) calculated for C13H20N2O3S [M + H]+ 285.1267, found 285.1268. IR (neat) ν 2923.70, 1455.67, 1326.43, 1144.23, 1050.98, 1033.09 cm−1.
N-(3-Phenethylthietan-3-yl)methanesulfonamide (18).
To a solution of 3-phenethylthietan-3-amine 43 (0.245 g, 1.27 mmol, 1.00 eq) in anh. CH2Cl2 (5.0 mL), Et3N (0.256 g, 2.53 mmol, 2.00 eq) was added, followed by methanesulfonyl chloride (0.290 g, 2.53 mmol, 2.00 eq) at 0 °C under N2. The reaction mixture was stirred at rt overnight. H2O was added, and the crude was extracted with EtOAc (×3). The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 30% of EtOAc in hexane) provided the title compound (0.118 g, 0.435 mmol, 34%). 1H NMR (600 MHz, CDCl3) δ 7.32 (t, J = 7.6 Hz, 2H), 7.24 (d, J = 7.2 Hz, 3H), 4.66 (s, 1H), 3.66 (d, J = 9.9 Hz, 2H), 3.14 (d, J = 10.1 Hz, 2H), 3.08 (s, 3H), 2.76 (dd, J = 10.4, 6.5 Hz, 2H), 2.49 (dd, J = 10.3, 6.6 Hz, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.75, 128.86, 128.48, 126.49, 63.28, 44.79, 39.95, 38.16, 29.85 ppm. HRMS (ES+) calculated for C12H17NO2S2 [M + Na]+ 294.0593, found 294.0592. IR (neat) ν 3234.79, 2967.55, 1732.48, 1442.63, 1307.46, 1153.47, 1130.11 cm−1.
N-(3-Phenethylthietan-3-yl)benzenesulfonamide (19).
Following the same procedure described for the synthesis of 18, using of 3-phenethylthietan-3-amine 43 (0.200 g, 1.03 mmol, 1.00 eq), Et3N (0.209 g, 2.07 mmol, 2.00 eq) and benzenesulfonyl chloride (0.365 g, 2.07 mmol, 2.00 eq). Purification by silica gel column chromatography (up to 20% EtOAc in hexane) provided the title compound (0.270 g, 0.810 mmol, 78%). 1H NMR (600 MHz, CDCl3) δ 7.96 – 7.90 (m, 2H), 7.64 – 7.58 (m, 1H), 7.57 – 7.49 (m, 2H), 7.27 (d, J = 2.0 Hz, 1H), 7.24 (d, J = 2.0 Hz, 1H), 7.22 – 7.15 (m, 1H), 7.06 – 6.99 (m, 2H), 4.89 (s, 1H), 3.54 – 3.48 (m, 2H), 3.02 – 2.95 (m, 2H), 2.54 – 2.48 (m, 2H), 2.45 – 2.40 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 142.48, 140.93, 133.02, 129.43, 128.56, 126.98, 126.16, 63.00, 39.63, 38.08, 29.60 ppm. HRMS (ES+) calculated for C17H19NO2S2 [M + Na]+ 356.0749, found 356.0749. IR (neat) ν 3276.99, 2948.31, 1448.03, 1320.15, 1154.08, 1090.87, 1063.51 cm−1.
N,N-Dimethyl-[3-(2-phenylethyl)thietan-3-yl]sulfamoyl-amine (20).
Following the same procedure described for the synthesis of 18, using of 3-phenethylthietan-3-amine 43 (0.050 g, 0.260 mmol, 1.00 eq), Et3N (0.052 g, 0.520 mmol, 2.00 eq) and dimethylsulfamoyl chloride (0.074 g, 0.520 mmol, 2.00 eq). Purification by silica gel column chromatography (up to 20% EtOAc in hexane) provided the title compound (0.024 g, 0.080 mmol, 24%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.25 – 7.20 (m, 3H), 4.38 (s, 1H), 3.69 (d, J = 9.9 Hz, 2H), 3.09 (d, J = 10.3 Hz, 2H), 2.84 (s, 6H), 2.74 – 2.72 (m, 2H), 2.47 – 2.45 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 141.15, 128.73, 128.51, 126.31, 62.92, 39.76, 38.00, 37.68, 29.70 ppm. HRMS (ES+) calculated for C13H20N2O2S2 [M + Na]+ 323.0858, found 323.0861. IR (neat) ν 2923.28, 2853.49, 1453.49, 1323.19, 1143.97, 1033.10, 1054.35 cm−1.
cis-N-(1-Oxido-3-phenethylthietan-3-yl)methanesulfonamide (21).
Following the same procedure described for the synthesis of 18, using of cis-3-amino-3-phenethylthietane 1-oxide 38 (0.030 g, 0.140 mmol, 1.00 eq), Et3N (0.04 g, 0.430 mmol, 3.00 eq) and methanesulfonyl chloride (0.049 g, 0.043 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.028 g, 0.097 mmol, 68%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.3 Hz, 1H), 7.19 (d, J = 7.5 Hz, 2H), 5.44 (s, 1H), 3.97 – 3.88 (m, 2H), 3.70 – 3.59 (m, 2H), 3.08 (s, 3H), 2.77 – 2.68 (m, 2H), 2.07 (dd, J = 10.1, 6.4 Hz, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 139.77, 128.97, 128.39, 126.78, 62.78, 51.54, 44.75, 42.53, 29.97 ppm. HRMS (ES+) calculated for C12H17NO3S2 [M + Na]+ 310.0542, found 310.0542. IR (neat) ν 3125.00, 2862.28, 1465.89, 1310.97, 1033.39, 1022.67 cm−1.
cis-N-(1-Oxido-3-phenethylthietan-3-yl)benzenesulfonamide (22).
Following the same procedure described for the synthesis of 18, using of cis-3-amino-3-phenethylthietane 1-oxide 38 (0.030 g, 0.133 mmol, 1.00 eq), Et3N (0.040 g, 0.400 mmol, 3.00 eq) and benzenesulfonyl chloride (0.071 g, 0.400 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.034 g, 0.093 mmol, 70%). 1H NMR (600 MHz, CDCl3) δ 7.93 (d, J = 7.9 Hz, 2H), 7.64 (t, J = 7.5, 1H), 7.56 (t, J = 7.7 Hz, 2H), 7.28 (s, 1H), 7.20 (t, J = 7.4 Hz, 2H), 7.02 (d, J = 7.7 Hz, 2H), 5.02 (s, 1H), 3.78 – 3.70 (m, 2H), 3.39 – 3.33 (m, 2H), 2.60 – 2.54 (m, 2H), 2.00 – 1.94 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 141.73, 139.79, 133.53, 129.71, 128.85, 128.39, 127.22, 126.67, 62.90, 51.09, 42.57, 29.78 ppm. HRMS (ES–) calculated for C17H19NO3S2 [M + Na]+ 372.0699, found 372.0700.
cis-3-[(Dimethylsulfamoyl)amino]-3-(2-phenylethyl)-1lambda4-thietan-1-one (23).
Following the same procedure described for the synthesis of 18, using of cis-3-amino-3-phenethylthietane 1-oxide 38 (0.060 g, 0.290 mmol, 1.00 eq), Et3N (0.087 g, 0.860 mmol, 3.00 eq) and dimethylsulfamoyl chloride (0.120 g, 0.860 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.014 g, 0.44 mmol, 15%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.5 Hz, 2H), 7.22 (td, J = 7.2, 1.5 Hz, 1H), 7.21 – 7.17 (m, 2H), 4.65 (s, 1H), 3.90 – 3.82 (m, 2H), 3.64 – 3.57 (m, 2H), 2.86 (s, 6H), 2.75 – 2.67 (m, 2H), 2.06 – 1.99 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) 140.13, 128.89, 128.44, 126.63, 62.43, 50.98, 42.53, 38.02, 29.83 ppm. HRMS (ES+) calculated for C13H20N2O3S2 [M + Na]+ 339.0808, found 339.0801.
N-(1,1-Dioxido-3-phenethylthietan-3-yl)methanesulfonamide (24).
Following the same procedure described for the synthesis of 18, using of 3-amino-3-phenethylthietane 1,1-dioxide 46 (0.050 g, 0.220 mmol, 1.00 eq), Et3N (0.067 g, 0.670 mmol, 3.00 eq) and methanesulfonyl chloride (0.076 g, 0.670 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.043 g, 0.014 mmol, 64%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.22 (dd, J = 27.3, 7.3 Hz, 3H), 5.50 (s, 1H), 4.36 – 4.28 (m, 2H), 4.13 – 4.07 (m, 2H), 3.13 (s, 3H), 2.84 – 2.73 (m, 2H), 2.49 – 2.37 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 139.29, 129.06, 128.45, 126.94, 74.73, 48.35, 44.75, 41.64, 30.86 ppm. HRMS (ES–) calculated for C12H17NO4S2 [M − H]– 302.0526, found 302.0526. IR (neat) ν 3275.93, 3028.34, 2924.35, 1455.59, 1315.13, 1232.45, 1143.38, 1090.29 cm−1.
N-(1,1-Dioxido-3-phenethylthietan-3-yl)benzenesulfonamide (25).
Following the same procedure described for the synthesis of 18, using of 3-amino-3-phenethylthietane 1,1-dioxide 46 (0.030 g, 0.133 mmol, 1.00 eq), Et3N (0.040 g, 0.399 mmol, 3.00 eq) and benzenesulfonyl chloride (0.071 g, 0.399 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.034 g, 0.093 mmol, 70%). 1H NMR (600 MHz, CDCl3 δ 8.71 (s, 1H), 7.85 – 7.80 (m, 2H), 7.65 – 7.61 (m, 1H), 7.57 (td, J = 8.0, 7.5, 2.1 Hz, 2H), 7.13 – 7.09 (m, 2H), 7.05 (dd, J = 8.5, 6.3 Hz, 1H), 6.74 – 6.70 (m, 2H), 4.34 – 4.29 (m, 2H), 4.23 (d, J = 14.8 Hz, 2H), 2.21 – 2.15 (m, 2H), 1.98 – 1.92 (m, 2H) ppm. 13C NMR (150 MHz, DMSO-d6) δ 142.50, 140.49, 132.99, 129.63, 128.29, 128.19, 126.50, 126.00, 74.13, 46.86, 40.97, 29.75 ppm. HRMS (ES–) calculated for C17H19NO4S2 [M − H]– 364.0683, found 364.0685. IR (neat) ν 3235.00, 2919.23, 1449.08, 1342.17, 1302.06, 1243.77, 1161.36 cm−1.
3-[(Dimethylsulfamoyl)amino]-3-(2-phenylethyl)-1lambda6-thietane-1,1-dione (26).
Following the same procedure described for the synthesis of 18, using of 3-amino-3-phenethylthietane 1,1-dioxide 46 (0.080 g, 0.355 mmol, 1.00 eq), Et3N (0.108 g, 1.07 mmol, 3.00 eq) and dimethylsulfamoyl chloride (0.153 g, 1.07 mmol, 3.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the title compound (0.018 g, 0.054 mmol, 15%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.25 – 7.18 (m, 3H), 4.68 (s, 1H), 4.36 – 4.29 (m, 2H), 4.07 – 4.02 (m, 2H), 2.88 (s, 6H), 2.78 (dd, J = 6.8, 4.0 Hz, 2H), 2.46 – 2.40 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 139.54, 129.02, 128.53, 126.87, 74.27, 48.00, 41.35, 38.06, 30.86 ppm. HRMS (ES+) calculated for C12H17NO4S2 [M + Na]+ 355.0747, found 355.0760. IR (neat) ν 3258.71, 1453.15, 1341.72, 1310.12, 1249.77, 1153.23 cm−1.
N-(5-Phenylisoxazol-3-yl)methanesulfonamide (27).
To a solution of 5-phenylisoxazol-3-amine 48 (0.030 g, 0.190 mmol, 1.00 eq) in anh. CH2Cl2 (1.8 mL), Et3N (0.038 g, 0.370 mmol, 2.00 eq) and methanesulfonyl chloride (0.026 g, 0.220 mmol, 1.20 eq) were added at 0 °C under N2. The reaction mixture was slowly warmed to rt and stirred at this temperature overnight. After 16 h, solvent was removed in vacuo. Purification by silica gel column chromatography (up to 30% EtOAc in hexane) provided the desired product (0.025 g, 0.10 mmol, 56%). 1H NMR (600 MHz, CD3OD) δ 7.84 (dd, J = 7.7, 2.0 Hz, 2H), 7.55 – 7.49 (m, 3H), 6.74 (s, 1H), 3.21 (s, 3H) ppm. 13C NMR (150 MHz, CD3OD) δ 171.63, 159.90, 131.62, 130.08, 126.60, 93.83, 49.00, 40.70 ppm. HRMS (ES+) calculated for C10H10N2O3S [M + Na]+ 261.0310, found 261.0305. IR (neat) ν 2919.22, 2850.47, 1623.00, 1578.33, 1470.58, 1332.46, 1152.41, 1042.94 cm−1.
N-(5-Phenylisoxazol-3-yl)benzenesulfonamide (28).
Synthesis of 28 was previously described.61 To a solution of 5-phenylisoxazol-3-amine 48 (0.030 g, 0.190 mmol, 1.00 eq) in anh. pyridine (1.8 mL), benzenesulfonyl chloride (0.043 g, 0.240 mmol, 1.30 eq) was added at 0 °C under N2. The reaction mixture was slowly warmed to rt and stirred at this temperature overnight. The solvent was then removed in vacuo. Purification by silica gel column chromatography (up to 30% EtOAc in hexane) provided the desired product (0.042 g, 0.140 mmol, 75%). 1H NMR (600 MHz, CDCl3) δ 7.89 (dd, J = 8.5, 2.3 Hz, 2H), 7.76 (dd, J = 6.7, 3.2 Hz, 2H), 7.62 – 7.56 (m, 1H), 7.52 – 7.45 (m, 5H), 6.80 (s, 1H) ppm. 13C NMR (150 MHz, CDCl3) δ 171.25, 157.96, 139.01, 133.83, 130.96, 129.55, 129.20, 127.19, 126.98, 125.99, 93.27 ppm. HRMS (ES+) calculated for C15H12N2O3S [M + H]+ 301.0639, found 301.0639. IR (neat) ν 2963.47, 1616.6, 1578.49, 1470.68, 1398.46, 1317.67, 1165.18, 1082.67 cm−1.
N-(5-Phenylisoxazol-3-yl)-N,N-dimethylaminesulfonamide (29).
Following the same procedure described for the synthesis of 28, using 5-phenylisoxazol-3-amine 48 (0.030 g, 0.190 mmol, 1.00 eq) and dimethylsulfamoyl chloride (0.035 g, 0.240 mmol, 1.30 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the desired product (0.009 g, 0.034 mmol, 18%). 1H NMR (600 MHz, CDCl3) δ 8.49 (s, 1H), 7.77 (dd, J = 6.6, 3.3 Hz, 2H), 7.52 – 7.45 (m, 3H), 6.73 (d, J = 3.3 Hz, 1H), 2.93 (s, 6H) ppm. 13C NMR (150 MHz, CDCl3) δ 170.88, 159.01, 130.89, 129.18, 127.05, 125.97, 93.24, 38.48 ppm. HRMS (ES+) calculated for C11H13N3O3S [M + H]+ 268.0750, found 268.0753. IR (neat) ν 2918.50, 1621.68, 1596.55, 1577.84, 1470.33, 1417.05, 1399.21, 1355.58, 1166.61, 1035.13 cm−1.
N-(1-Phenyl-1H-1,2,3-triazol-4-yl)methanesulfonamide (30).
Following the same procedure described for the synthesis of 18, 1-phenyl-1H-1,2,3-triazol-4-amine (0.030 g, 0.190 mmol, 1.00 eq), methanesulfonyl chloride (0.024 g, 0.210 mmol, 1.10 eq), and Et3N (0.021 g, 0.210 mmol, 1.10 eq). Purification by silica gel column chromatography (up to 30% EtOAc in hexane) provided the desired product (0.012 g, 0.050 mmol, 27%). 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 2.4 Hz, 1H), 7.77 – 7.73 (m, 2H), 7.56 (td, J = 7.8, 2.2 Hz, 2H), 7.48 (dd, J = 8.6, 6.4 Hz, 1H), 6.74 (s, 1H), 3.12 (s, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 142.83, 136.91, 130.04, 129.46, 120.54, 114.88, 40.30 ppm. HRMS (ES+) calculated for C9H11N4O2S [M + H]+ 239.0597, found 239.0599. IR (neat) ν 2919.65, 1597.66, 1573.98, 1498.02, 1411.81, 1345.55, 1330.76, 1215.67, 1153.87, 1053.08 cm−1.
N-(1-Phenyl-1H-1,2,3-triazol-4-yl)benzenesulfonamide (31).
Following the same procedure described for the synthesis of 18, using 1-phenyl-1H-1,2,3-triazol-4-amine (0.030 g, 0.190 mmol, 1.00 eq), benzenesulfonyl chloride (0.033 g, 0.19 mmol, 1.00 eq), and Et3N (0.019 g, 0.190 mmol, 1.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the desired compound (0.012 g, 0.040 mmol, 21%). 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 2.4 Hz, 1H), 7.83 (d, J = 7.7 Hz, 2H), 7.73 (dd, J = 6.3, 4.3 Hz, 2H), 7.57 – 7.51 (m, 3H), 7.49 – 7.44 (m, 3H) ppm. 13C NMR (150 MHz, CDCl3) δ 143.19, 139.07, 136.95, 133.57, 130.00, 129.40, 129.32, 127.21, 120.43, 113.40 ppm. HRMS (ES+) calculated for C12H13N4O2S [M + H]+ 301.0754, found 301.0755. IR (neat) ν 2922.24, 1567.23, 1492.90, 1449.21, 1414.92, 1353.95, 1333.27, 1164.36, 1093.35, 1053.00 cm−1.
N-(1-Phenyl-1H-1,2,3-triazol-4-yl)-N,N-dimethylaminesulfonamide (32).
Following the same procedure described for the synthesis of 15, using 1-phenyl-1H-1,2,3-triazol-4-amine (0.030 g, 0.190 mmol, 1.00 eq), dimethylsulfamoyl chloride (0.027 g, 0.19 mmol, 1.00 eq), and Et3N (0.019 g, 0.190 mmol, 1.00 eq). Purification by reverse phase HPLC (10% to 90% CH3CN in H2O) provided the desired compound (0.013 g, 0.049 mmol, 26%). 1H NMR (600 MHz, CDCl3) δ 7.99 (s, 1H), 7.77 – 7.72 (m, 2H), 7.54 (dd, J = 8.8, 7.3 Hz, 2H), 7.48 – 7.45 (m, 1H), 2.89 (s, 6H) ppm. 13C NMR (150 MHz, CDCl3) δ 143.99, 137.02, 129.99, 129.28, 120.46, 113.65, 38.60 ppm. HRMS (ES+) calculated for C10H14N5O2S [M + H]+ 268.0863, found 268.0864. IR (neat) ν 2917.46, 1597.33, 1576.48, 1494.69, 1426.58, 1364.30, 1232.86, 1164.11, 1148.06 cm−1.
(S)-2-Methyl-N-(oxetan-3-ylidene)propane-2-sulfinamide (36).
Synthesis of 36 closely followed previously described procedures.42, 43 1H NMR (600 MHz, CDCl3) δ 5.80 (ddd, J = 15.4, 4.5, 2.1 Hz, 1H), 5.67 (ddd, J = 15.4, 4.4, 2.0 Hz, 1H), 5.53 – 5.41 (m, 2H), 1.27 (s, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 175.98, 85.45, 85.34, 57.40, 21.77 ppm. HRMS (ES+) calculated for C7H14NO2S [M + H]+ 176.0740, found 176.0741. IR (neat) ν 2961.88, 2926.46, 1696.95, 1626.13, 1456.15, 1362.66, 1264.92, 1131.77, 1079.36 cm−1.
(S)-2-Methyl-N-(thietan-3-ylidene)propane-2-sulfinamide (37).
Synthesis of 37 closely followed previously described procedures,42, 43 using 3-thietanone (5.00 g, 56.7 mmol, 1.00 eq), (S)-(−)-2-methylpropane-2-sulfinamide (8.25 g, 68.1 mmol, 1.20 eq), and titanium (IV) isopropoxide (32.3 g, 113 mmol, 2.00 eq). Purification by silica gel column chromatography (up to 10% of EtOAc in hexane) provided the title compound as a light brown liquid (8.40 g, 44.0 mmol, 77%). 1H NMR (600 MHz, CDCl3) δ 4.74 (dd, J = 17.2, 4.5 Hz, 1H), 4.50 (dd, J = 17.1, 4.2 Hz, 1H), 4.24 (dd, J = 16.1, 4.5 Hz, 1H), 4.14 (dd, J = 15.9, 4.3 Hz, 1H), 1.20 (s, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 175.05, 57.87, 46.48, 45.10, 22.39 ppm. HRMS (ES+) calculated for C7H14NOS2 [M + H]+ 192.0511, found 192.0513. IR (neat) ν 2959.04, 2944.88, 2869.80, 1784.77, 1657.29, 1456.15, 1395.24, 1362.66, 1264.92, 1164.35, 1077.95 cm−1.
(S)-1-(tert-Butylsulfinyl)-5-oxa-1-azaspiro[2.3]hexane (38).
Synthesis of 38 closely followed previously described procedure.43 1H NMR (600 MHz, CDCl3) δ 5.04 (d, J = 7.9 Hz, 1H), 5.00 – 4.88 (m, 1H), 4.82 (d, J = 7.9 Hz, 1H), 4.78 (d, J = 7.2 Hz, 1H), 2.67 (s, 1H), 1.98 (s, 1H), 1.22 (s, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 78.35, 75.66, 56.94, 40.92, 22.51 ppm. HRMS (ES+) calculated for C8H16NO2S [M + H]+ 190.0896, found 190.0897. IR (neat) ν 2954.79, 2872.64, 1456.15, 1393.82, 1362.66, 1301.72, 1150.19, 1066.62 cm−1.
(S)-1-(tert-Butylsulfinyl)-5-thia-1-azaspiro[2.3]hexane (39).
Synthesis of 39 closely followed previously described procedure,43 using of NaH (1.84 g, 46.0 mmol, 1.10 eq), trimethylsulfoxonium iodide (10.1 g, 46.0 mmol, 1.10 eq), and (S)-2-methyl-N-(thietan-3-ylidene)propane-2-sulfinamide 37 (8.00 g, 41.8 mmol, 1.00 eq). Purification by silica gel column chromatography (up to 10% of EtOAc in hexane) provided the title compound (3.35 g, 16.3 mmol, 39%). 1H NMR (600 MHz, CDCl3) δ 3.98 (d, J = 10.1 Hz, 1H), 3.80 (d, J = 9.7 Hz, 1H), 3.23 (d, J = 10.1 Hz, 1H), 3.17 (d, J = 9.7 Hz, 1H), 2.71 (s, 1H), 2.03 (s, 1H), 1.21 (s, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 57.17, 42.89, 36.33, 33.72, 22.65 ppm. HRMS (ES+) calculated for C8H16NOS2 [M + H]+ 206.0668, found 206.0668. IR (neat) ν 2853.38, 2927.88, 1514.22, 1454.73, 1357.00, 1263.51, 1170.03, 1124.69, 1055.28 cm−1.
(S)-2-Methyl-N-(3-phenethyloxetan-3-yl)propane-2-sulfinamide (40).
To a solution of copper (I) iodide (0.101 g, 0.528 mmol, 0.10 eq) in anh. THF (35 mL), benzylmagnesium chloride (11.0 mL, 1.40 M solution in THF-Me, 15.9 mmol, 3.00 eq) was added at rt under N2. The mixture was cooled to −30 °C using an CH3CN/dry ice bath. 1-(tert-Butylsulfinyl)-5-oxa-1-azaspiro[2.3]hexane 38 (1.00 g, 5.28 mmol, 1.00 eq) in anh. THF (5.0 mL) was added dropwise at −30 °C. The reaction was stirred at this temperature for 10 min, after which it was warmed to 0 °C and stirred for 1 h. Satd. aq. NaCl was added and the reaction was warmed to rt. The resulting crude was extracted with EtOAc (×3), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 50% EtOAc in hexane) provided the title compound (1.34 g, 4.75 mmol, 90%). 1H NMR (600 MHz, CDCl3) δ 7.29 (t, J = 7.6 Hz, 2H), 7.20 (dd, J = 8.1, 5.9 Hz, 3H), 4.74 (d, J = 6.8 Hz, 1H), 4.69 (d, J = 7.0 Hz, 1H), 4.51 (d, J = 6.8 Hz, 1H), 4.47 (d, J = 6.6 Hz, 1H), 3.72 (s, 1H), 2.68 (t, J = 8.1 Hz, 2H), 2.45 – 2.30 (m, 2H), 1.25 (d, J = 2.2 Hz, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 141.04, 128.76, 128.49, 126.38, 82.52, 82.33, 59.99, 56.11, 38.96, 29.91, 22.63 ppm. HRMS (ES+) calculated for C15H24NO2S [M + H]+ 282.1522, found 282.1520. IR (neat) ν 3413.74, 3138.94, 2959.96, 2875.47, 1492.98, 1356.15, 1368.33, 1338.58, 1263.51, 1164.35, 1114.14, 1083.61, 1004.29 cm−1.
(S)-2-Methyl-N-(3-phenethylthietan-3-yl)propane-2-sulfinamide (41).
Following the same procedure described for the synthesis of 40 using 1-(tert-butylsulfinyl)-5-thia-1-azaspiro[2.3]hexane 39 (1.00 g, 4.87 mmol, 1.00 eq), benzylmagnesium chloride (14.6 mL, 1.0 M solution in THF-Me, 15.9 mmol, 3.00 eq), and copper (I) iodide (0.093 g, 0.487 mmol, 0.10 eq). Purification by silica gel column chromatography (up to 25% EtOAc in hexane) provided the title compound (0.696 g, 2.34 mmol, 48%). 1H NMR (600 MHz, CDCl3) δ 7.33 – 7.29 (m, 2H), 7.24 – 7.19 (m, 3H), 3.67 (s, 1H), 3.60 (d, J = 9.7 Hz, 1H), 3.54 (d, J = 10.1 Hz, 1H), 3.19 (dd, J = 9.9, 1.5 Hz, 1H), 3.13 (dd, J = 9.8, 1.4 Hz, 1H), 2.68 (ddd, J = 9.0, 6.5, 1.9 Hz, 2H), 2.49 – 2.43 (m, 1H), 2.42 – 2.35 (m, 1H), 1.24 (s, 9H) ppm. 13C NMR (150 MHz, CDCl3) δ 141.26, 128.75, 128.52, 126.31, 63.68, 56.19, 40.82, 39.25, 29.50, 22.66 ppm. HRMS (ES+) calculated for C15H24NOS2 [M + H]+ 298.1294, found 298.1296. IR (neat) ν 3199.85, 2943.46, 1495.81, 1453.32, 1362.66, 1263.51, 1218.18, 1162.94, 1051.03 cm−1.
3-Phenethylthietan-3-amine (43).
To a solution of (S)-2-methyl-N-(3-phenethylthietan-3-yl)propane-2-sulfinamide 41 (0.670 g, 2.25 mmol, 1.00 eq) in anh. CH3OH (11.0 mL), a solution of HCl (9.01 mL, 1.25 M solution in CH3OH, 2.67 mmol, 5.00 eq) was added dropwise at 0 °C under N2. The mixture was stirred at rt overnight. The mixture was concentrated in vacuo, and the resulting salt was dissolved in minimal H2O and the pH of the solution was brought to 9. The mixture was then extracted with EtOAc (×3), and the combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 20% EtOAc in hexane) provided the title compound (0.270 g, 1.40 mmol, 62%). 1H NMR (600 MHz, CDCl3) δ 7.30 (t, J = 7.7 Hz, 2H), 7.25 – 7.17 (m, 3H), 3.23 – 3.10 (m, 4H), 2.74 – 2.66 (m, 2H), 2.15 – 2.08 (m, 2H), 1.98 (d, J = 14.3 Hz, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 141.85, 128.64, 128.49, 60.55, 42.45, 41.50, 29.89 ppm. HRMS (ES+) calculated for C11H15NS [M + H]+ 194.0998, found 194.1002. IR (neat) ν 3349.61, 2972.83, 2924.02, 2843.43, 1602.51, 1490.12, 1448.10 cm−1.
trans-3-Amino-3-phenethylthietane 1-oxide (44) and cis-3-amino-3-phenethylthietane 1-oxide (45).
To a solution of 3-phenethylthietan-3-amine 43 (0.100 g, 0.517 mmol, 1.00 eq) in anh. CH2Cl2 (2.5 mL), a solution of m-CPBA (0.136 g, 0.569 mmol, 1.10 eq) in anh. CH2Cl2 (1.5 mL) was added dropwise at −78 °C under N2. The reaction was stirred at this temperature for 2 h, and then the reaction was quenched with H2O and satd. NaHCO3. The mixture was extracted with CH2Cl2 (×3) and washed with brine. The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 30% of EtOAc in hexane) led to the separation of product 44 (0.011 g, 0.053 mmol, 10%) and product 45 (0.076 g, 0.360 mmol, 70%). trans-3-Amino-3-phenethylthietane 1-oxide (44): 1H NMR (600 MHz, CDCl3) δ 7.30 (t, J = 7.7 Hz, 2H), 7.20 (dd, J = 26.5, 7.5 Hz, 3H), 3.47 (dd, J = 8.8, 3.1 Hz, 2H), 3.03 (dd, J = 8.8, 3.1 Hz, 2H), 2.71 – 2.61 (m, 2H), 2.03 – 1.93 (m, 2H), 1.25 (s, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.52, 128.83, 128.40, 63.91, 51.44, 47.21, 29.86 ppm. HRMS (ES+) calculated for C11H15NOS [M + Na]+ 232.0767, found 232.0768. IR (neat) ν 3362.74, 2923.99, 1602.64, 1373.96, 1224.97, 1051.38, 1033.11 cm−1. cis-3-Amino-3-phenethylthietane 1-oxide (45). 1H NMR (600 MHz, CDCl3) δ 77.30 (t, J = 7.6 Hz, 2H), 7.24 – 7.12 (m, 3H), 3.92 – 3.79 (m, 2H), 3.21 – 3.02 (m, 2H), 2.75 – 2.58 (m, 2H), 1.87 (s, 2H), 1.83 – 1.72 (m, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.74, 128.78, 128.34, 65.78, 48.82, 43.89, 30.03 ppm. HRMS (ES+) calculated for C11H15NOS [M + Na]+ 232.0767, found 232.0767. IR (neat) ν 3372.63, 2913.66, 1602.52, 1492.51, 1453.07, 1044.94, 1028.83, 1018.41 cm−1.
3-Amino-3-phenethylthietane 1,1-dioxide (46).
To a solution of 3-phenethylthietan-3-amine 43 (0.100 g, 0.517 mmol, 1.00 eq) in acetone (2.5 mL), a solution of Oxone® (0.477 g, 1.55 mmol, 3.00 eq) in H2O (2.5 mL) was added dropwise at 0 °C. The mixture was stirred at this temperature for 10 min, then warmed to rt and stirred overnight. The reaction mixture was then cooled to 0 °C and a solution of Na2SO3 (1.0 M, 5.0 mL) was added. The resulting mixture was extracted with EtOAc (×3) and washed with brine. The combined organic extracts were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by silica gel column chromatography (up to 25% of EtOAc in hexane) provided the title compound (0.055 g, 0.240 mmol, 47%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J = 7.6 Hz, 2H), 7.25 – 7.17 (m, 3H), 4.09 – 4.03 (m, 2H), 3.85 – 3.80 (m, 2H), 2.75 (dd, J = 9.6, 6.6 Hz, 2H), 2.19 – 2.11 (m, 2H), 1.80 (s, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 140.17, 128.90, 128.46, 76.55, 44.58, 43.71, 31.18, 24.25, 24.05 ppm. HRMS (ES+) calculated for C11H16NO2S [M + H]+ 226.0896, found 226.0897. IR (neat) ν 3385.41, 3313.17, 3017.12, 2920.80, 2852.81, 1494.39, 1389.57, 1303.17, 1229.51, 1179.94, 1041.69, 1077.95 cm−1.
5-Phenylisoxazol-3-amine (48).
Synthesis of 48 closely followed previously described procedures.46, 62 1H NMR (600 MHz, CDCl3) δ 7.72 (d, J = 7.7 Hz, 2H), 7.43 (dt, J = 8.8, 6.4 Hz, 3H), 6.09 (s, 1H), 3.99 (s, 2H) ppm. 13C NMR (150 MHz, CDCl3) δ 169.64, 163.84, 130.17, 128.96, 127.77, 125.76, 92.04 ppm. HRMS (ES+) calculated for C6H9N2O [M + H]+ 161.0709, found 161.0710. IR (neat) ν 3444.90, 3324.50, 1621.88, 1517.06, 1488.73, 1342.83, 1264.92, 1051.03 cm−1.
Stability Studies.
Stock solutions (5 mM) were prepared in DMSO. From the stock solution, test compounds solutions (100 μM final concentration) were prepared and stirred in BICCA® Simulated intestinal fluid (without pancreatin, pH 7.40–7.60) at rt. Percentages of compound left after 5 h of incubation were determined by assessing relative change in peak area of the analytical LC/MS chromatograms. LC/MS conditions employed gradients of CH3CN in H2O (from 10% to 90% CH3CN) over 4 min and flow rate of 2 mL/min.
Determination of Physicochemical Properties.
Determinations of pKa values were obtained via automated potentiometric titrations using a Sirius T3 instrument (Pion, Inc) according to manufacturer instructions. Three or more titrations were performed from pH 1.8 to pH 12.2 using ionic strength adjusted water (0.15 M KCl), acid (0.5 M HCl) and base (0.5 M KOH). For compounds with relatively low aqueous solubility (16 and 25), the pKa determinations were conducted using a cosolvent (methanol) protocol; Yasuda-Shedlovsky extrapolation method was used to extrapolate the pKa at 0% cosolvent. LogP measurements of compounds with known experimental pKa were obtained via potentiometric titrations using a Sirius T3 instrument (Pion, Inc). LogD7.4 values were extrapolated from the measured logP. LogD7.4 of selected compounds with pKa values > 10 (i.e., 14, 16 – 20, 22, 23, 25) were measured via shake-flask method (experiments carried out by Analyza, Inc). Effective permeability values (logPapp) were measured by Parallel Artificial Membrane Permeability Assay (PAMPA) using the Corning GentestTM pre-coated PAMPA plate system with quantitation by HPLC-UV (experiments carried out by Analyza, Inc).
Computational Studies.
All molecular modelling experiments were performed on Asus WS X299 PRO Intel® i9–10980XE CPU @ 3.00GHz × 36 running Ubuntu 18.04. Molecular Operating Environment (MOE),63 Gaussian 0964 and Multiwfn65 were used as molecular modelling software. The molecular structures were initially prepared by MOE QuikPrep tool generating possible ionization states at pH 7 ± 2, followed by a molecular superposition using the Refined flexible alignment. Successively, all the geometries were optimized in the vacuum-phase using B3LYP/ 6–311++ G(d,p) Pople basis set of density functional theory (DFT) by Gaussian 09 software. Quantitative calculation of the electrostatic potential (ESP) distribution of the molecule was performed using a constant electronic density of 0.002 au. The ESP surface minima and maxima values were generated by Multiwfn using the same optimized structures at which the final electron densities and electrostatic potentials were calculated.
Supplementary Material
Highlights:
Design and syntheses of matched molecular pairs of N-acylsulfonamides and related bioisosteres are described.
Structure property relationship studies of N-acylsulfonamides and bioisosteres – including acidity, permeability, and lipophilicity – show that these structures cover a wide range of physicochemical properties.
Despite difference in ionization states of N-acylsulfonamides and a subset of isosteres, comparison of Gaussian-optimized geometry and electrostatic potential maps show similar electrostatic potentials.
Novel thietane and oxidized thietane derivatives show potential as isosteric replacements of the carbonyl of the N-acylsulfonamides.
Acknowledgments.
Financial support for this work has been provided in part by the NIH/NIA (AG061173 for KRF, CV, AB and CB).
List of Nonstandard Abbreviations:
- m-CPBA
meta-chloroperbenzoic acid
- TMSOI
trimethylsulfoxonium iodide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available: Potentiometric acidity (pKa) reports of compounds 9–13, 15–18, 20–32; potentiometric lipophilicity (logPoctanol and logD7.4) reports of compounds 9–13, 15, 21, 24, 26, 27–32; logPhydrocarbon (cyclohexane, heptane, toluene) of compounds 9, 12, 13, 15; NMR spectra of test compounds; HPLC traces of representative compounds; X-ray crystal structures of compounds 15 (CCDC 1998391), 16 (CCDC 1995771), 18 (CCDC 1995773), 19 (CCDC 1995772), 21 (CCDC 1995769), Authors will release the atomic coordinates and experimental data upon article publication; experimental details for the permeability assay (PAMPA), and logD7.4 determinations; the SMILES string structures along the full data set in tabular form (csv file format).
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Iain G; Obach RS; Dennis AS; Zhuang M; Alexander AA; Kevin B; Amit K; Don W; Deepak D; Chandra P; Alf V Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET. RSC: 2010; p P001–P530. [Google Scholar]
- 2.Borhade SR; Svensson R; Brandt P; Artursson P; Arvidsson PI; Sandström A Preclinical Characterization of Acyl Sulfonimidamides: Potential Carboxylic Acid Bioisosteres with Tunable Properties. ChemMedChem 2015, 10, 455–460. [DOI] [PubMed] [Google Scholar]
- 3.Pinter T; Jana S; Courtemanche RJM; Hof F Recognition Properties of Carboxylic Acid Bioisosteres: Anion Binding by Tetrazoles, Aryl Sulfonamides, and Acyl Sulfonamides on a Calix[4]arene Scaffold. J. Org. Chem 2011, 76, 3733–3741. [DOI] [PubMed] [Google Scholar]
- 4.Uehling DE; Donaldson KH; Deaton DN; Hyman CE; Sugg EE; Barrett DG; Hughes RG; Reitter B; Adkison KK; Lancaster ME; Lee F; Hart R; Paulik MA; Sherman BW; True T; Cowan C Synthesis and Evaluation of Potent and Selective β3 Adrenergic Receptor Agonists Containing Acylsulfonamide, Sulfonylsulfonamide, and Sulfonylurea Carboxylic Acid Isosteres. J. Med. Chem 2002, 45, 567–583. [DOI] [PubMed] [Google Scholar]
- 5.Winters MP; Crysler C; Subasinghe N; Ryan D; Leong L; Zhao S; Donatelli R; Yurkow E; Mazzulla M; Boczon L; Manthey CL; Molloy C; Raymond H; Murray L; McAlonan L; Tomczuk B Carboxylic acid bioisosteres acylsulfonamides, acylsulfamides, and sulfonylureas as novel antagonists of the CXCR2 receptor. Bioorg. Med. Chem 2008, 18, 1926–1930. [DOI] [PubMed] [Google Scholar]
- 6.Pelz NF; Bian Z; Zhao B; Shaw S; Tarr JC; Belmar J; Gregg C; Camper DV; Goodwin CM; Arnold AL; Sensintaffar JL; Friberg A; Rossanese OW; Lee T; Olejniczak ET; Fesik SW Discovery of 2-Indole-acylsulfonamide Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods. J. Med. Chem 2016, 59, 2054–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yee YK; Bernstein PR; Adams EJ; Brown FJ; Cronk LA; Hebbel KC; Vacek EP; Krell RD; Snyder DW A novel series of selective leukotriene antagonists: exploration and optimization of the acidic region in 1,6-disubstituted indoles and indazoles. J. Med. Chem 1990, 33, 2437–2451. [DOI] [PubMed] [Google Scholar]
- 8.Winton VJ; Aldrich C; Kiessling LL Carboxylate Surrogates Enhance the Antimycobacterial Activity of UDP-Galactopyranose Mutase Probes. ACS Infect. Dis 2016, 2, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scola PM; Wang AX; Good AC; Sun L-Q; Combrink KD; Campbell JA; Chen J; Tu Y; Sin N; Venables BL; Sit S-Y; Chen Y; Cocuzza A; Bilder DM; D’Andrea S; Zheng B; Hewawasam P; Ding M; Thuring J; Li J; Hernandez D; Yu F; Falk P; Zhai G; Sheaffer AK; Chen C; Lee MS; Barry D; Knipe JO; Li W; Han Y-H; Jenkins S; Gesenberg C; Gao Q; Sinz MW; Santone KS; Zvyaga T; Rajamani R; Klei HE; Colonno RJ; Grasela DM; Hughes E; Chien C; Adams S; Levesque PC; Li D; Zhu J; Meanwell NA; McPhee F Discovery and Early Clinical Evaluation of BMS-605339, a Potent and Orally Efficacious Tripeptidic Acylsulfonamide NS3 Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. J. Med. Chem 2014, 57, 1708–1729. [DOI] [PubMed] [Google Scholar]
- 10.Rikimaru K; Wakabayashi T; Abe H; Imoto H; Maekawa T; Ujikawa O; Murase K; Matsuo T; Matsumoto M; Nomura C; Tsuge H; , M.; Mol CD; Snell GP; Bragstad KA; Sang B-C; Dougan DR; Tanaka T; Katayama N; Horiguchi Y; Momose Y A new class of non-thiazolidinedione, non-carboxylic-acid-based highly selective peroxisome proliferator-activated receptor (PPAR) γ agonists: Design and synthesis of benzylpyrazole acylsulfonamides. Bioorg. Med. Chem 2012, 20, 714–733. [DOI] [PubMed] [Google Scholar]
- 11.Ammazzalorso A; De Filippis B; Giampietro L; Amoroso R N-acylsulfonamides: Synthetic routes and biological potential in medicinal chemistry. Chem. Biol. Drug Des 2017, 90, 1094–1105. [DOI] [PubMed] [Google Scholar]
- 12.Pilot-Matias T; Tripathi R; Cohen D; Gaultier I; Dekhtyar T; Lu L; Reisch T; Irvin M; Hopkins T; Pithawalla R; Middleton T; Ng T; McDaniel K; Or YS; Menon R; Kempf D; Molla A; Collins C In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob. Agents Chemother 2015, 59, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehmann DE; Demeritt JE; Hull KG; Fisher SL Biochemical characterization of an inhibitor of Escherichia coli UDP-N-acetylmuramyl-l-alanine ligase. Biochim. Biophys. Acta 2004, 1698, 167–173. [DOI] [PubMed] [Google Scholar]
- 14.Patil V; Kale M; Raichurkar A; Bhaskar B; Prahlad D; Balganesh M; Nandan S; Shahul Hameed P Design and synthesis of triazolopyrimidine acylsulfonamides as novel anti-mycobacterial leads acting through inhibition of acetohydroxyacid synthase. Bioorg. Med. Chem. Lett 2014, 24, 2222–2225. [DOI] [PubMed] [Google Scholar]
- 15.Meier T; Uhlik M; Chintharlapalli S; Dowless M; Van Horn R; Stewart J; Blosser W; Cook J; Young D; Ye X; Evans G; Credille K; Ballard D; Huber L; Capen A; Chedid M; Ilaria R; Smith MC; Stancato L Tasisulam Sodium, an Antitumor Agent That Inhibits Mitotic Progression and Induces Vascular Normalization. Mol. Cancer Ther 2011, 10, 2168–2178. [DOI] [PubMed] [Google Scholar]
- 16.Oltersdorf T; Elmore SW; Shoemaker AR; Armstrong RC; Augeri DJ; Belli BA; Bruncko M; Deckwerth TL; Dinges J; Hajduk PJ; Joseph MK; Kitada S; Korsmeyer SJ; Kunzer AR; Letai A; Li C; Mitten MJ; Nettesheim DG; Ng S; Nimmer PM; O’Connor JM; Oleksijew A; Petros AM; Reed JC; Shen W; Tahir SK; Thompson CB; Tomaselli KJ; Wang B; Wendt MD; Zhang H; Fesik SW; Rosenberg SH An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [DOI] [PubMed] [Google Scholar]
- 17.Souers AJ; Leverson JD; Boghaert ER; Ackler SL; Catron ND; Chen J; Dayton BD; Ding H; Enschede SH; Fairbrother WJ; Huang DCS; Hymowitz SG; Jin S; Khaw SL; Kovar PJ; Lam LT; Lee J; Maecker HL; Marsh KC; Mason KD; Mitten MJ; Nimmer PM; Oleksijew A; Park CH; Park C-M; Phillips DC; Roberts AW; Sampath D; Seymour JF; Smith ML; Sullivan GM; Tahir SK; Tse C; Wendt MD; Xiao Y; Xue JC; Zhang H; Humerickhouse RA; Rosenberg SH; Elmore SW ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 2013, 19, 202–213. [DOI] [PubMed] [Google Scholar]
- 18.United States Food and Drug Administration (http://www.fda.gov accessed on 12/22/2020).
- 19.Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 20.Meanwell N Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- 21.Brown N Bioisosteres in Medicinal Chemistry. Wiley: Wiley-VCH, 2012; Vol. 54. [Google Scholar]
- 22.Bonandi E; Christodoulou MS; Fumagalli G; Perdicchia D; Rastelli G; Passarella D The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [DOI] [PubMed] [Google Scholar]
- 23.Giraudo A; Krall J; Nielsen B; Sørensen TE; Kongstad KT; Rolando B; Boschi D; Frølund B; Lolli ML 4-Hydroxy-1,2,3-triazole moiety as bioisostere of the carboxylic acid function: a novel scaffold to probe the orthosteric γ-aminobutyric acid receptor binding site. Eur. J. Med. Chem 2018, 158, 311–321. [DOI] [PubMed] [Google Scholar]
- 24.Sainas S; Temperini P; Farnsworth JC; Yi F; Møllerud S; Jensen AA; Nielsen B; Passoni A; Kastrup JS; Hansen KB; Boschi D; Pickering DS; Clausen RP; Lolli ML Use of the 4-Hydroxytriazole Moiety as a Bioisosteric Tool in the Development of Ionotropic Glutamate Receptor Ligands. J. Med. Chem 2019, 62, 4467–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammed I; Kummetha IR; Singh G; Sharova N; Lichinchi G; Dang J; Stevenson M; Rana TM 1,2,3-Triazoles as Amide Bioisosteres: Discovery of a New Class of Potent HIV-1 Vif Antagonists. J. Med. Chem 2016, 59, 7677–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassalas P; Oukoloff K; Makani V; James M; Tran V; Yao Y; Huang L; Vijayendran K; Monti L; Trojanowski JQ; Lee VMY; Kozlowski MC; Smith AB; Brunden KR; Ballatore C Evaluation of Oxetan-3-ol, Thietan-3-ol, and Derivatives Thereof as Bioisosteres of the Carboxylic Acid Functional Group. ACS Med. Chem. Lett 2017, 8, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell NH; Clarkson GJ; Notman R; Raubo P; Martin NG; Shipman M Synthesis and structure of oxetane containing tripeptide motifs. Chem. Commun 2014, 50, 8797–8800. [DOI] [PubMed] [Google Scholar]
- 28.Beadle JD; Knuhtsen A; Hoose A; Raubo P; Jamieson AG; Shipman M Solid-Phase Synthesis of Oxetane Modified Peptides. Org. Lett 2017, 19, 3303–3306. [DOI] [PubMed] [Google Scholar]
- 29.Möller GP; Müller S; Wolfstädter BT; Wolfrum S; Schepmann D; Wünsch B; Carreira EM Oxetanyl Amino Acids for Peptidomimetics. Org. Lett 2017, 19, 2510–2513. [DOI] [PubMed] [Google Scholar]
- 30.McLaughlin M; Yazaki R; Fessard TC; Carreira EM Oxetanyl Peptides: Novel Peptidomimetic Modules for Medicinal Chemistry. Org. Lett 2014, 16, 4070–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumari S; Carmona AV; Tiwari AK; Trippier PC Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem 2020, 63, 12290–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varnes JG; Gero T; Huang S; Diebold RB; Ogoe C; Grover PT; Su M; Mukherjee P; Saeh JC; Macintyre T; Repik G; Dillman K; Byth K; Russell DJ; Ioannidis S Towards the next generation of dual Bcl-2/Bcl-xL inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 3026–3033. [DOI] [PubMed] [Google Scholar]
- 33.Yang C-C; Wi S-C; Zhong N; Lin M-Y; Tu C-C Benzenesulfonamide Derivatives and Method For Treating Cancer. U. S. Patent US20180311190A1, 2018. [Google Scholar]
- 34.Touré BB; Miller-Moslin K; Yusuff N; Perez L; Doré M; Joud C; Michael W; DiPietro L; van der Plas S; McEwan M; Lenoir F; Hoe M; Karki R; Springer C; Sullivan J; Levine K; Fiorilla C; Xie X; Kulathila R; Herlihy K; Porter D; Visser M The Role of the Acidity of N-Heteroaryl Sulfonamides as Inhibitors of Bcl-2 Family Protein–Protein Interactions. ACS Med. Chem. Lett 2013, 4, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandanur SGS; Nanduri S; Golakoti NR Synthesis and biological evaluation of new C-12(α/β)-(N-) sulfamoyl-phenylamino-14-deoxy-andrographolide derivatives as potent anti-cancer agents. Bioorg. Med. Chem. Lett 2017, 27, 2854–2862. [DOI] [PubMed] [Google Scholar]
- 36.Focken T; Chowdhury S; Zenova A; Grimwood ME; Chabot C; Sheng T; Hemeon I; Decker SM; Wilson M; Bichler P; Jia Q; Sun S; Young C; Lin S; Goodchild SJ; Shuart NG; Chang E; Xie Z; Li B; Khakh K; Bankar G; Waldbrook M; Kwan R; Nelkenbrecher K; Karimi Tari P; Chahal N; Sojo L; Robinette CL; White AD; Chen C-A; Zhang Y; Pang J; Chang JH; Hackos DH; Johnson JP; Cohen CJ; Ortwine DF; Sutherlin DP; Dehnhardt CM; Safina BS Design of Conformationally Constrained Acyl Sulfonamide Isosteres: Identification of N-([1,2,4]Triazolo[4,3-a]pyridin-3-yl)methane-sulfonamides as Potent and Selective hNaV1.7 Inhibitors for the Treatment of Pain. J. Med. Chem 2018, 61, 4810–4831. [DOI] [PubMed] [Google Scholar]
- 37.Boiteau J-G; Ouvry G; Arlabosse J-M; Astri S; Beillard A; Bhurruth-Alcor Y; Bonnary L; Bouix-Peter C; Bouquet K; Bourotte M; Cardinaud I; Comino C; Deprez B; Duvert D; Féret A; Hacini-Rachinel F; Harris CS; Luzy A-P; Mathieu A; Millois C; Orsini N; Pascau J; Pinto A; Piwnica D; Polge G; Reitz A; Reversé K; Rodeville N; Rossio P; Spiesse D; Tabet S; Taquet N; Tomas L; Vial E; Hennequin LF Discovery and process development of a novel TACE inhibitor for the topical treatment of psoriasis. Bioorg. Med. Chem 2018, 26, 945–956. [DOI] [PubMed] [Google Scholar]
- 38.Vandyck K; Rombouts G; Stoops B; Tahri A; Vos A; Verschueren W; Wu Y; Yang J; Hou F; Huang B; Vergauwen K; Dehertogh P; Berke JM; Raboisson P Synthesis and Evaluation of N-Phenyl-3-sulfamoyl-benzamide Derivatives as Capsid Assembly Modulators Inhibiting Hepatitis B Virus (HBV). J. Med. Chem 2018, 61, 6247–6260. [DOI] [PubMed] [Google Scholar]
- 39.Velcicky J; Mathison CJN; Nikulin V; Pflieger D; Epple R; Azimioara M; Cow C; Michellys P-Y; Rigollier P; Beisner DR; Bodendorf U; Guerini D; Liu B; Wen B; Zaharevitz S; Brandl T Discovery of Orally Active Hydroxyethylamine Based SPPL2a Inhibitors. ACS Med. Chem. Lett 2019, 10, 887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Humphries PS; Bersot R; Kincaid J; Mabery E; McCluskie K; Park T; Renner T; Riegler E; Steinfeld T; Turtle ED; Wei Z-L; Willis E Carbazole-containing sulfonamides and sulfamides: Discovery of cryptochrome modulators as antidiabetic agents. Bioorg. Med. Chem. Lett 2016, 26, 757–760. [DOI] [PubMed] [Google Scholar]
- 41.Lassalas P; Gay B; Lasfargeas C; James MJ; Van T; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB III; Huryn DM; Ballatore C Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu G; Cogan DA; Owens TD; Tang TP; Ellman JA Synthesis of Enantiomerically Pure N-tert-Butanesulfinyl Imines (tert-Butanesulfinimines) by the Direct Condensation of tert-Butanesulfinamide with Aldehydes and Ketones. J. Org. Chem 1999, 64, 1278–1284. [Google Scholar]
- 43.Hamzik PJ; Brubaker JD Reactions of Oxetan-3-tert-butylsulfinimine for the Preparation of Substituted 3-Aminooxetanes. Org. Lett 2010, 12, 1116–1119. [DOI] [PubMed] [Google Scholar]
- 44.Hodgson DM; Hughes SP; Thompson AL; Heightman TD Terminal Aziridines by α-Deprotonation/Electrophile Trapping of N-Protected Aziridine. Org. Lett 2008, 10, 3453–3456. [DOI] [PubMed] [Google Scholar]
- 45.Das R; Chakraborty D Silver Triflate Catalyzed Acetylation of Alcohols, Thiols, Phenols, and Amines. Synthesis-Stuttgart 2011, 1621–1625. [Google Scholar]
- 46.Johnson L; Powers J; Ma F; Jendza K; Wang B; Meredith E; Mainolfi N A Reliable Synthesis of 3-Amino-5-Alkyl and 5-Amino-3-Alkyl Isoxazoles. Synthesis-Stuttgart 2013, 45, 171–173. [Google Scholar]
- 47.https://chemaxon.com/products/calculators-and-predictors (accessed on 12/22/2020).
- 48.Hill AP; Young RJ Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discov. Today 2010, 15, 648–655. [DOI] [PubMed] [Google Scholar]
- 49.Jain N; Yalkowsky SH Estimation of the aqueous solubility I: application to organic nonelectrolytes. J. Pharm. Sci 2001, 90, 234–52. [DOI] [PubMed] [Google Scholar]
- 50.Abraham MH; Chadha HS; Whiting GS; Mitchell RC Hydrogen bonding. 32. An analysis of water-octanol and water-alkane partitioning and the delta log P parameter of seiler. J. Pharm. Sci 1994, 83, 1085–100. [DOI] [PubMed] [Google Scholar]
- 51.Conradi RA; Hilgers AR; Ho NF; Burton PS The influence of peptide structure on transport across Caco-2 cells. Pharm. Res 1991, 8, 1453–60. [DOI] [PubMed] [Google Scholar]
- 52.el Tayar N; Testa B; Carrupt P Polar intermolecular interactions encoded in partition-coefficients - an indirect estimation of hydrogen-bond parameters of polyfunctional solutes. J. Phys. Chem 1992, 96, 1455–1459. [Google Scholar]
- 53.Toulmin A; Wood JM; Kenny PW Toward prediction of alkane/water partition coefficients. J. Med. Chem 2008, 51, 3720–30. [DOI] [PubMed] [Google Scholar]
- 54.Shalaeva M; Caron G; Abramov Y; O’Connell T; Plummer M; Yalamanchi G; Farley K; Goetz G; Philippe L; Shapiro M Integrating Intramolecular Hydrogen Bonding (IMHB) Considerations in Drug Discovery Using Delta logP As a Tool. J. Med. Chem 2013, 56, 4870–4879. [DOI] [PubMed] [Google Scholar]
- 55.el Tayar N; Tsai RS; Testa B; Carrupt PA; Leo A Partitioning of solutes in different solvent systems: the contribution of hydrogen-bonding capacity and polarity. J .Pharm. Sci 1991, 80, 590–8. [DOI] [PubMed] [Google Scholar]
- 56.Goodwin JT; Conradi RA; Ho NF; Burton PS Physicochemical determinants of passive membrane permeability: role of solute hydrogen-bonding potential and volume. J. Med. Chem 2001, 44, 3721–9. [DOI] [PubMed] [Google Scholar]
- 57.Huque FT; Box K; Platts JA; Comer J Permeability through DOPC/dodecane membranes: measurement and LFER modelling. Eur. J. Pharm. Sci 2004, 23, 223–32. [DOI] [PubMed] [Google Scholar]
- 58.Berthelot M; Besseau F; Laurence C The Hydrogen-Bond Basicity pKHB Scale of Peroxides and Ethers. Eur. J. Org. Chem 1998, 1998, 925–931. [Google Scholar]
- 59.Besseau F; Lucon M; Laurence C; Berthelot M Hydrogen-bond basicity pK(HB) scale of aldehydes and ketones. J. Chem. Soc., Perkin Trans 1998, 101–107. [Google Scholar]
- 60.Zinczuk J; Sorokin IH; Orazi OO; Corral RA Intramolecular Sulphonamidomethylation. Part II [1,2] Fused Heterocycles from 2-Phenylethanesulphonamides. J. Heterocycl. Chem 1991, 29, 859–866. [Google Scholar]
- 61.Stupple P; Lagiakos H; Foitzik R; Camerino M; Nikolakopoulos G; Bozikis T; Kersen W; Walker S; Bert J A compound of formula (I), or a pharmaceutically acceptable salt thereof. W. I. P. O. Patent WO 2020/002587 A1, 2020.
- 62.Rowbottom M; Faraoni R; Chao Q; Campbell B; Lai A; Setti E; Ezawa M; Sprankle K; Abraham S; Tran L; Struss B; Gibney M; Armstrong R; Gunawardane R; Nepomuceno R; Valenta I; Hua H; Gardner M; Cramer M; Gitnick D; Insko D; Apuy J; Jones-Bolin S; Ghose A; Herbertz T; Ator M; Dorsey B; Ruggeri B; Williams M; Bhagwat S; James J; Holladay M Identification of 1-(3-(6,7-Dimethoxyquinazolin-4-yloxy)phenyl)-3-(5-(1,1,1-trifluoro-2-methylpropan-2-yl)isoxazol-3-yl)urea Hydrochloride (CEP-32496), a Highly Potent and Orally Efficacious Inhibitor of V-RAF Murine Sarcoma Viral Oncogene Homologue B1 (BRAF) V600E. J. Med. Chem 2012, 55, 1082–1105. [DOI] [PubMed] [Google Scholar]
- 63.Molecular Operating Environment (MOE), Montreal, QC, Canada, H3A 2R7, 2019. http://www.Chemcomp.com (accessed on 12/22/2020).
- 64.Gaussian 98 (Gaussian, Inc., Pittsburgh, PA, 1998) http://www.gaussian.com/ (accessed on 12/22/2020). [Google Scholar]
- 65.Lu T; Chen F Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem 2012, 33, 580–592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.