

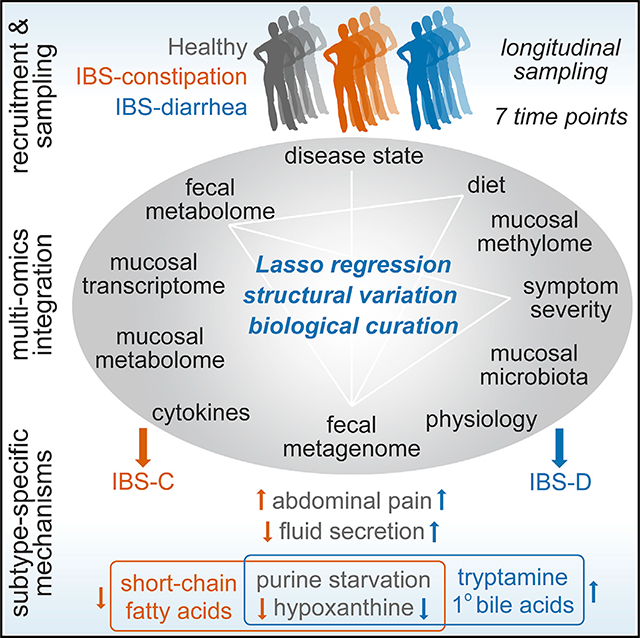
SUMMARY
The gut microbiome has been implicated in multiple human chronic gastrointestinal (GI) disorders. Determining its mechanistic role in disease has been difficult due to apparent disconnects between animal and human studies and lack of an integrated multi-omics view of disease-specific physiological changes. We integrated longitudinal multi-omics data from the gut microbiome, metabolome, host epigenome, and transcriptome in the context of irritable bowel syndrome (IBS) host physiology. We identified IBS subtype-specific and symptom-related variation in microbial composition and function. A subset of identified changes in microbial metabolites correspond to host physiological mechanisms that are relevant to IBS. By integrating multiple data layers, we identified purine metabolism as a novel host-microbial metabolic pathway in IBS with translational potential. Our study highlights the importance of longitudinal sampling and integrating complementary multi-omics data to identify functional mechanisms that can serve as therapeutic targets in a comprehensive treatment strategy for chronic GI diseases.
In Brief
Integrated and longitudinal multiomic analyses of patients with irritable bowel syndrome reveals a role for the gut microbiota in modulating purine metabolism and influencing host gastrointestinal function.
Graphical Abstract
INTRODUCTION
Irritable bowel syndrome (IBS) is a globally prevalent disorder characterized by recurrent abdominal pain or discomfort. IBS is predominantly seen in females and is associated with changes in stool form or frequency and is based on the predominant stool form classified as constipation predominant (IBS-C), diarrhea predominant (IBS-D), or mixed (IBS-M).
IBS pathogenesis involves changes in gastrointestinal motility, intestinal secretion, visceral hypersensitivity, and intestinal permeability, all of which can be modified by the gut microbiome (Bhattarai et al., 2017a). In addition, IBS symptoms are affected by diet, host genetics, and environment, which are also known to modulate the human gut microbiome (Bhattarai et al., 2017a). Experimental evidence supporting a role for the gut microbiome in IBS is based on transplantation experiments, where transit time (De Palma et al., 2017; Touw et al., 2017), pain sensation thresholds (Crouzet et al., 2013), and intestinal permeability (Edogawa et al., 2020) differences associated with IBS-C and IBS-D were replicated in gnotobiotic mice following transplantation of fecal microbiota from patients. However, in the absence of robust animal models mimicking all aspects of IBS pathophysiology, human studies are needed to uncover the interaction of the gut microbiome with relevant human-specific disease pathways.
Human studies in IBS are limited by cross-sectional sampling and lack of subtype stratification, which is reflected in the lack of agreement in findings across the large number of microbiome studies (Duan et al., 2019; Pittayanon et al., 2019). The latter further increases variability given the well-described influence of gastrointestinal transit on the gut microbiome (Kashyap et al., 2013; Roager et al., 2016). In addition, IBS, like other chronic gastrointestinal disorders, is characterized by periods of remission and exacerbation in symptoms, and cross-sectional samples thus fail to account for temporal variability in disease. Finally, inherent differences in host physiology between human and animal studies have been a barrier in advancing our understanding of mechanistic roles for the gut microbiome in IBS.
We performed a longitudinal study in subsets of IBS patients integrating multi-omic measurements including the microbial metagenome, host transcriptome, and methylome with assessment of host functions and identified IBS subtype-specific mechanisms driven by altered microbial metabolism, which corresponded with concurrent changes in host physiology.
RESULTS
This is a prospective observational study using longitudinal multi-omics sampling of microbiome and host samples with the goal of identifying microbial mediators driving subtype-specific phenotypes in IBS. This design allowed us to compare healthy controls (HCs) to patients with IBS-C or IBS-D (Rome III criteria). We matched study participants for gender, age, and body mass index (BMI); further inclusion and exclusion criteria are outlined in STAR Methods. A total of 77 participants provided stool samples for at least one time point, and 42 participants agreed to undergo flexible sigmoidoscopy allowing us to obtain colonic biopsies longitudinally. Sample types and demographics of study subjects, as well as extensive metadata including medication use, hospital anxiety and depression score, and IBS symptom severity score (SSS), are outlined in Figures 1A–1C and S1A and Table S1. Study subjects provided dietary recall (Table S1) and symptom severity at each visit. IBS patients were given the option to provide an additional stool sample and complete symptom severity questionnaire between regular visits at the time of a self-identified flare, which was defined as a substantial worsening of symptoms between visits. A total of 12 subjects provided the additional stool sample at the time of flare (6 each for IBS-C and IBS-D).
Figure 1. Gut Microbiota Composition of IBS-C Patients Is More Distinct and Variable.
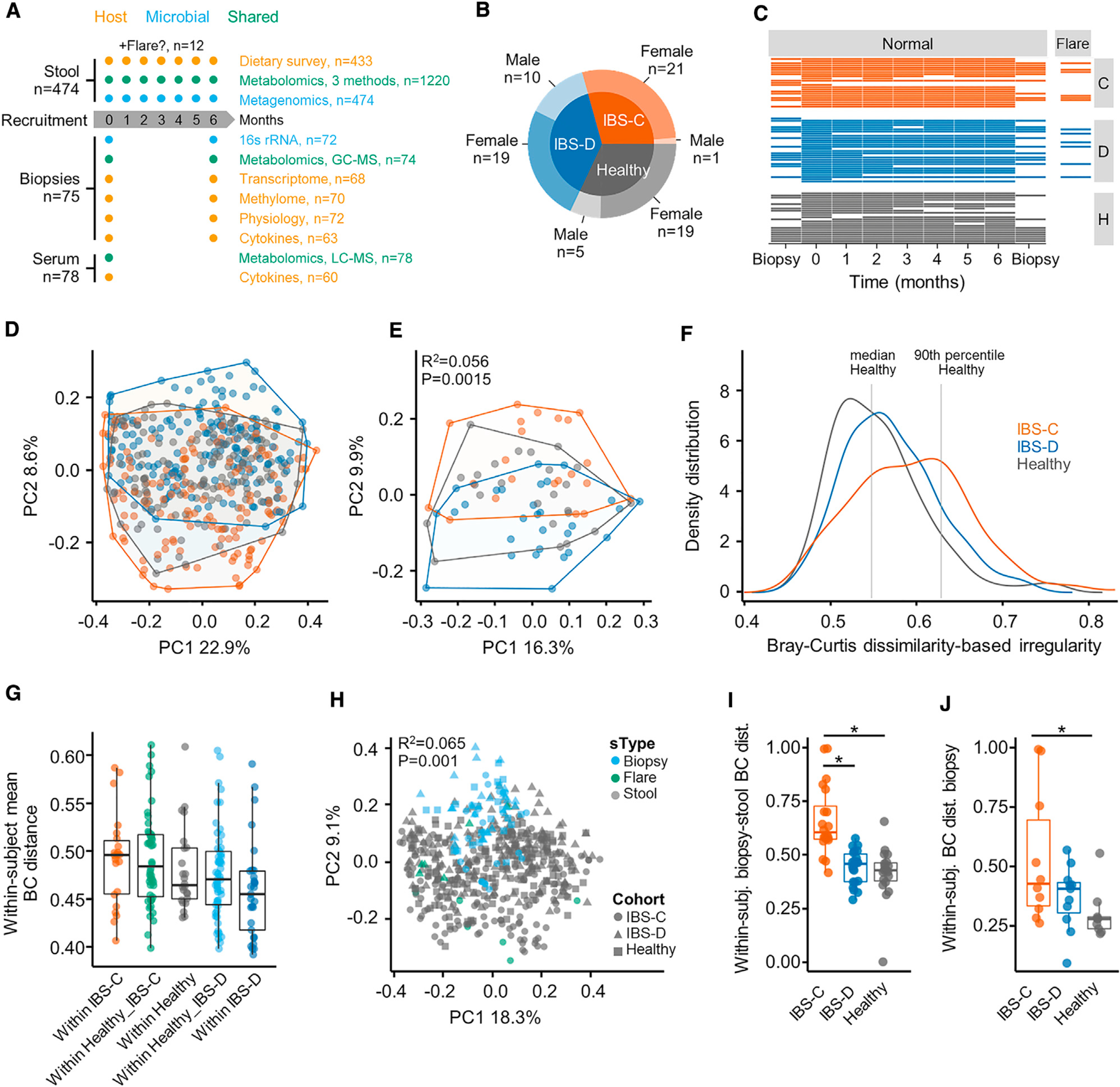
(A) Outline of sample collection.
(B) Number of subjects and distribution of biological sex by cohort.
(C) Total number of samples per subject collected longitudinally.
(D) Bray Curtis β-diversity ordination of samples from IBS-C, IBS-D, and HC considering all samples from all subjects (n = 474 stool samples, no. of samples per subject 1–7).
(E) Same as (D) considering by-subject averaged data (statistics in inset from PERMANOVA on group membership). IBS-C and IBS-D versus HC, p = < 0.05, IBS-C versus IBS-D, p value = 0.001, dispersion around centroids via pairwise PERMANOVA (n = 22, 29, and 24 averaged gut microbiome profiles for IBS-C, IBS-D, and HC, respectively).
(F) Bray-Curtis dissimilarity (BCD)-based irregularity (BCDI) showing distribution of the three groups linear mixed-effect model correcting for subject HC versus IBS-C p = < 0.011 (n = 142, 170, and 143 stool samples for IBS-C, IBS-D, and HC, respectively).
(G) Community variability determined by the mean within-subject Bray Curtis distance (within-IBS-D versus within-IBS-C, p = < 0.005, ANOVA Tukey, n = 22, 46, 24, 53, and 29, Bray-Curtis distances between stool samples of the same subject for within-IBS-C, Healthy versus IBS-C, within-Healthy, Healthy versus IBS-D, and within-IBS-D, respectively).
(H) Bray Curtis β-diversity ordination of biopsy and stool samples (statistics in inset from PERMANOVA on group membership. n = 72, 12, and 462 stool samples for biopsy, flare, stool, respectively).
(I) Difference in mucosa associated and luminal microbiota composition based on Bray-Curtis distance (HC versus IBS-C, IBS-D versus IBS-C p = < 0.001, ANOVA Tukey HSD; n = 20, 22, and 19 paired mucosal-stool microbiome samples for IBS-C, IBS-D, and HC, respectively).
(J) Community variability within each group based on mean Bray Curtis Distance (HC versus IBS-C, p value = 0.02, ANOVA Tukey HSD, n = 10, 11, and 9 mucosal microbiome samples for IBS-C, IBS-D, and HC, respectively).
Boxplot center represents median and box interquartile range (IQR). Whiskers extend to most extreme data point <1.5 × IQR. C: IBS-C, D: IBS-D, H: HC. Symbols indicate significance (*p = < 0.05).
See also Figures S1 and S2 and Tables S1 and S2.
To identify microbial drivers of subtype-specific symptoms in IBS, we performed shotgun metagenomic sequencing and metabolomics on stool samples, metabolomics and cytokine measurements on serum samples, and 16S rRNA gene sequencing, metabolome, transcriptome, and methylome analyses on biopsy samples (Figure 1A; STAR Methods).
Longitudinal Sampling Overcomes Heterogeneity Seen in Cross-Sectional Microbiome Studies
A cross-sectional study of the gut microbiome in chronic GI conditions provides a snapshot of a highly dynamic ecosystem. In addition to the effects of diet, medication use, lifestyle, and other environmental factors, the variability in microbiome seen over time may also reflect changes in disease activity. We assessed the effect of longitudinal sampling on the identification of compositional changes compared to cross-sectional sampling by subsampling our longitudinal data, testing for significant taxa, and comparing the results at single time points with results obtained on data that were averaged by subject across all time points.
Differences in taxa abundances between HC and disease groups observed in individual time-points were highly inconsistent when comparing the different time points and did not overlap with changes observed in the averaged data (Figure S1B). When using averaged data, but not the single-time point data, we found a significantly higher abundance of multiple Streptococcus spp. individually in IBS-C and IBS-D as well as in the composite IBS group, compared to HC (log2(FC) ~1, at Mann-Whitney U test false discovery rate [FDR] <0.25) (Figure S1C; Table S2). In addition, we found a significantly lower abundance of the recently identified phylum Synergistetes in IBS-D compared to HC (log2(FC) −2.1, FDR 0.017; Figure S1D; Table S2). We also found that inter-individual variation dominates over intra-individual variation, which supports our approach of averaging the longitudinal data from each individual (STAR Methods, t test p < 0.0001). These findings highlight the need for longitudinal sampling in chronic diseases to reliably identify microbiota changes that may be missed using cross-sectional sampling. Hence, we primarily report findings from time-averaged data. This is further supported by a recent study showing that commonly used “omics” methods are more accurate when using averages over multiple sampling time points (Poyet et al., 2019).
PCoA-based on Bray-Curtis β diversity showed that stool microbiota composition in IBS clustered by subtype, and IBS-D and IBS-C displayed significantly different dispersion from HC samples as well as from each other (Figures 1D and 1E). To further confirm differences in β diversity, we calculated a Bray-Curtis dissimilarity (BCD)-based irregularity (BCDI) score (STAR Methods). BCDI scores for IBS-C were significantly elevated (linear mixed-effect model correcting for subject; Figure 1F). When considering a sample to be irregular beyond the 90th percentile of the HC distribution, we found that more IBS-C samples are irregular than IBS-D (31.7% for IBS-C, 14.1% for IBS-D, equality-of-proportions test p < 0.001).
Longitudinal Sampling Reveals Greater Variability in IBS-C Microbiota over Time
Stool microbiota composition exhibited greater variability over time in patients with IBS-C compared to HC and IBS-D subjects (Figure 1G). In addition, there was higher Shannon α diversity in averaged IBS-C stool samples compared to IBS-D samples (ANOVA with Tukey HSD p value 0.016).
We then tested for differences in luminal and mucosa-associated microbiota. These differences are relevant in IBS because disease subtypes, defined by differences in stool form, are partly the result of alteration in epithelial fluid secretion into the lumen. The microbial composition of the colonic mucosa was significantly different from the luminal microbiota in stool samples (Figure 1H). The mucosa-associated microbiota in IBS patients were characterized by significantly higher levels of Proteobacteria when compared to HC (Figure S1G). The mucosa-associated microbiota in patients with IBS-C were less similar to their respective luminal microbiota than those of IBS-D or HC (Figure 1I). This potentially reflects the longer transit time in IBS-C subjects, where there is more time for the communities to diverge. In addition, there was greater intra-individual variability in mucosa-associated microbiota in patients with IBS-C across time, similar to what we observed in the luminal microbiota (Figure 1J).
IBS Symptom Severity Is Associated with Functional Changes in the Gut Microbiota
The severity of IBS at a particular sampling point was reported using the IBS SSS (0–500), which is a cumulative metric of abdominal pain intensity, frequency, distension, dissatisfaction with bowel habits, and influence of IBS on life in general. We observed a higher relative abundance of more than 20 Lactobacillus spp. in severe IBS-D (SSS >300) compared to mild-moderate IBS-D (SSS <300; >10-fold, Mann-Whitney FDR <0.1, Table S2). This was not related to probiotic or dairy consumption by the subjects (Figures S2A–S2C). When considering functional variation through Kyoto Encyclopedia of Genes and Genomes (KEGG) ontology (KO) term abundance in the stool metagenomics data, we found that 74 KO terms were associated with severe IBS-C, and 44 with severe IBS-D at an FDR <0.1 (STAR Methods; Table S3). KO terms for alcohol dehydrogenase (ADH) were found in both severe IBS-C and IBS-D compared to mild-moderate IBS (~0.6 log2(FC) higher in severe IBS). These ADH KO terms are positively correlated to Bifidobacterium sp. and Streptococcus sp. (Table S4). The considerable genetic diversity among ADH enzymes complicates constructing a link between this abundance and specific metabolic products. However, these data suggest that ADH activity may be related to abdominal pain, which is the primary symptom common to both IBS-C and IBS-D. In addition, we found that stool form recorded as Bristol stool scale and abdominal pain preceding bowel movements were associated with specific bacteria and metabolites (Table S4).
Metabolomics Integrated with Physiologic Measurements Provides Mechanistic Insight into the Effect of Gut Microbiota Metabolism on Gastrointestinal Function
To better understand the mechanisms by which the gut microbiota could drive symptom pathophysiology, we quantified the metabolic output of the microbiome reflected in the biochemical profiles of the luminal and mucosa-associated samples. We first focused on microbiota-derived metabolites previously reported to drive changes in gastrointestinal physiology. H1-nuclear magnetic resonance (NMR) spectroscopy showed the short-chain fatty acids (SCFAs) propionate, butyrate, and acetate to be significantly lower in the stool samples of patients with IBS-C compared to HC (Figure 2A, Figure S3A for averaged data). Consistent with the luminal metabolites, acetate (measured by gas chromatography-mass spectrometry [GC-MS]) was also significantly reduced in the colonic mucosal biopsy samples from the IBS-C group compared to the HC group (Figure 2B). Notably, these differences in SCFAs were independent of the overall intake of dietary fiber as this was not significantly different between the groups (Figure S2C).
Figure 2. Metabolomics Integrated with Physiologic Measurements Provides Mechanistic Insight into the Effect of Gut Microbiota Metabolism on Gastrointestinal Function.
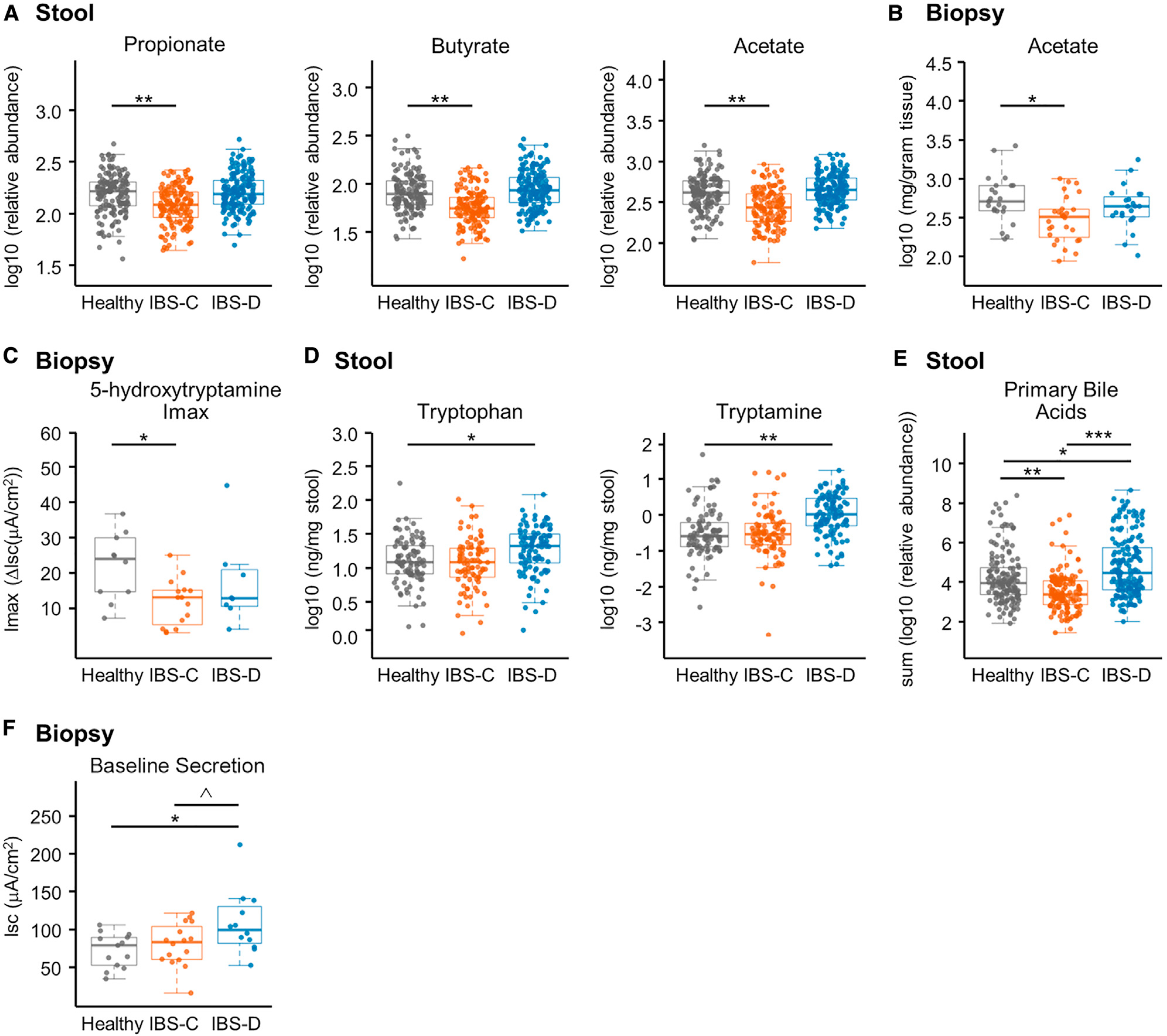
(A) Relative abundance of propionate, butyrate, and acetate in stool samples determined with 1H NMR (linear mixed-effect models on log10-transformed data correcting for subject, FDR corrected, n = 136, 170, and 146 metabolite profiles for IBS-C, IBS-D, and HC, respectively).
(B) Absolute abundance of acetate in colonic biopsies determined with GC-MS (linear mixed-effect models on log10-transformed abundance correcting for subject, FDR corrected, n = 28, 23, and 23 averaged metabolomes for IBS-C, IBS-D, and HC, respectively).
(C) Maximal ΔIsc (Imax) following application of increasing concentrations of serotonin (5-HT) basolaterally in colonic biopsies from time-point 1 (ANOVA Tukey HSD, n = 13, 12, and 10 colonic biopsies for IBS-C, IBS-D, and HC, respectively).
(D) Absolute abundance of tryptophan and tryptamine in a subset of the stool samples determined with LC-MS/MS (ng/mg stool) (linear mixed-effect models on log10-transformed data correcting for subject, FDR adjusted, n = 84, 91, and 103 metabolite profiles for IBS-C, IBS-D, and HC, respectively).
(E) Relative abundance of primary unconjugated bile acids in stool samples determined with LC-MS/MS. Data shown are the sum of cholic acid and chenodeoxycholic acid relative abundances (linear mixed-effect models on log10-transformed data correcting for subject, n = 136, 170, and 146 metabolite profiles for IBS-C, IBS-D, and HC, respectively).
(F) Baseline Isc (ANOVA Tukey HSD, n = 16, 12, and 13 colonic biopsies for IBS-C, IBS-D, and HC, respectively).
Boxplot center represents median and box IQR. Whiskers extend to most extreme data point <1.5 × IQR. Symbols indicate significance (***p = < 0.001, **p = < 0.01, *p = < 0.05, ^p = < 0.10).
We have previously described the role of SCFAs in modulating the serotonergic pathway in host GI tissue (Bhattarai et al., 2017b; Reigstad et al., 2015). To determine the physiologic relevance of lower SCFAs in stool and biopsies seen in IBS-C patients, we investigated the change in short circuit current (Isc; a measure of ionic flux across the epithelium reflecting intestinal secretion) in colonic epithelium in response to serotonin (5-HT) using an Ussing chamber (STAR Methods). Water secretion accompanies ionic flux, and decreased secretion thus results in lower water content of stool as seen in constipation. Conversely, increased ionic flux can result in increased water secretion resulting in diarrhea. The secretory response of colonic biopsies from IBS-C patients to 5-HT was significantly lower than HC (Figure 2C), which is consistent with the stool form seen in IBS-C patients.
We recently found that the bacterially derived monoamine tryptamine (a tryptophan metabolite similar to serotonin) activates serotonin receptor-4 (5-HT4R) resulting in increased fluid secretion and decreased transit time in gnotobiotic mice (Bhattarai et al., 2018), but its physiologic role in human disease has not yet been determined. Hence, we investigated changes in tryptamine and other tryptophan metabolites in stool samples using a targeted liquid chromatography-mass spectrometry (LC-MS) method. We found that both tryptophan and tryptamine were significantly increased in stool samples from IBS-D patients (Figure 2D; Figure S3B) and could thus in part be responsible for the increased water content of stools in IBS-D. We verified that these metabolite changes were not associated with dietary differences in protein intake (Figure S2).
To establish the capacity of tryptamine to stimulate fluid secretion in human tissue, we investigated the tryptamine-induced change in Isc in colonic biopsies from IBS subjects and HC, again using an Ussing chamber. We found that tryptamine significantly increased colonic secretion in all three groups, but there were no significant differences among the groups (Figure S3C). The lack of such differences indicates that the colonic epithelium of IBS patients and HCs is capable of tryptamine-induced fluid secretion, and observed changes could thus be due to changes in tryptamine abundance.
In humans, the primary bile acids (BAs) cholic acid (CA) and chenodeoxycholic acid (CDCA) are deconjugated from their glycine or taurine conjugate by microbial bile salt hydrolases (BSHs). These deconjugated primary BAs then serve as substrates for a diverse range of microbial modifications, including conversion to the secondary BAs deoxycholic acid (DCA) and lithocholic acid (LCA) and desulfation of DCA-S to DCA. Certain forms of BAs such as hydroxylated BAs have been found to increase intestinal fluid secretion in humans (Camilleri, 2014). We therefore examined whether there are differences in microbial biotransformation of BAs in IBS, which may contribute to altered intestinal secretion. We identified variation in BA signatures associated with IBS (measured with LC-MS/MS), with significantly higher amounts of unconjugated primary BAs in stool samples from patients with IBS-D and significantly lower amounts of unconjugated primary BAs in stool samples from IBS-C patients (Figure 2E) compared to HC. We also found higher amounts of individual primary conjugated and unconjugated BAs and DCA-S in IBS-D compared to HC and IBS-C subjects (Figures S3D–S3F). As hydroxylated primary BAs like CDCA may increase colonic secretion, we tested the effect of CDCA in colon mucosa-submucosa preparations from germ-free mice in an Ussing chamber. Indeed, we observed a significant increase in Isc in response to CDCA (Figure S3G), which supported a physiological role for the elevated CDCA levels in increasing the water content in stools from IBS-D patients.
To determine the physiologic relevance of differences in tryptamine and primary BA levels, we measured differences in Isc in colonic biopsies obtained from the three groups. As expected from our findings in gnotobiotic mice (Bhattarai et al., 2018), we found colonic biopsies from IBS-D patients also exhibited significantly higher baseline Isc (Figure 2F), which is consistent with the secretory effect of tryptamine and primary BA that we have described above.
Integrated Microbiome-Metabolome Analysis Identifies a Novel Microbial Metabolic Pathway in IBS
In addition to the above targeted approach, we employed an untargeted metabolomics approach to identify novel microbial pathways that may be driving pathophysiologic changes in IBS. A projection to latent structures discriminant analysis (PLS-DA) model based on untargeted 1H-NMR spectral profiles identified metabolic variation between the IBS subgroups and HC stool samples (Figures S4A and S4B). Lysine, uracil, and hypoxanthine were all found to be significantly lower in stool samples from IBS-C patients compared to HC (Figures 3A–3C; Figures S4C–S4F). Hypoxanthine was also lower in IBS-D patients albeit not at the same significance as in IBS-C. Hypoxanthine can serve as an energy source for intestinal epithelial cells and promotes intestinal cellular barrier development and recovery following injury or hypoxia (Lee et al., 2018; Lee et al., 2020). Lower fecal hypoxanthine levels could reflect decreased production or elevated breakdown of hypoxanthine by the microbiome in the gut of IBS patients.
Figure 3. Integrated Microbiome-Metabolome Analysis Identifies a Novel Microbial Metabolic Pathway in IBS.
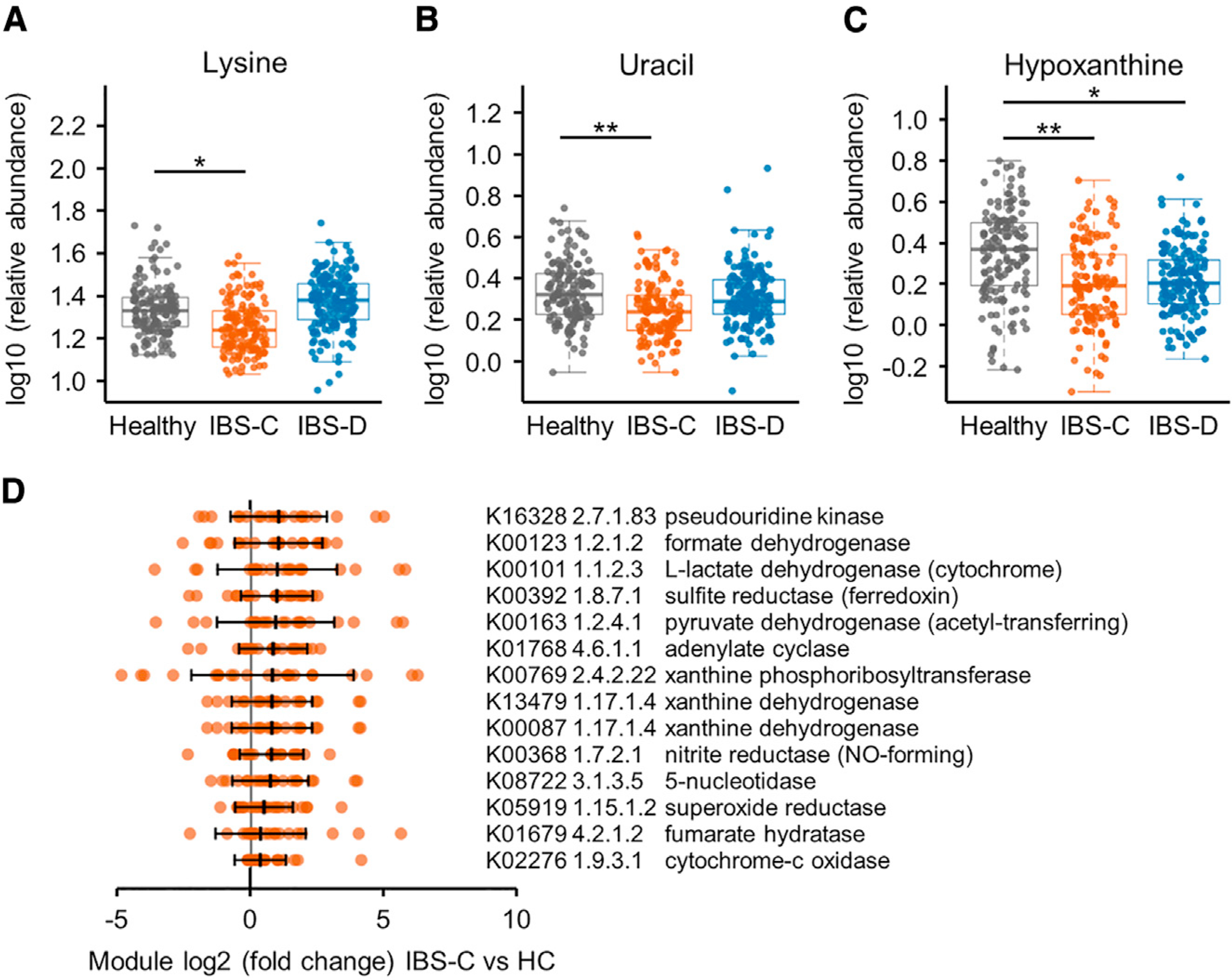
(A–C) Relative abundance of (A) lysine, (B) uracil, and (C) hypoxanthine in stool samples determined with 1H NMR (linear mixed-effect models on log10-transformed data correcting for subject, FDR adjusted, n = 136, 170, and 146 metabolite profiles for IBS-C, IBS-D, and HC, respectively). Boxplot center represents median and box IQR. Whiskers extend to most extreme data point <1.5 × IQR. Symbols indicate significance (***p = < 0.001, **p = < 0.01, *p = < 0.05).
(D) Selected hypoxanthine-related gut metagenome KO term abundance in stools from IBS-C subjects compared to the median abundance of the healthy control (HC) subjects. By-subject averaged data (FDR <0.1, Mann-Whitney test; except for K00769, which had q value 0.12). The maximal log2(FC) of the either of the xanthine dehydrogenase (XDH)/oxidase modules is 0.73, p = < 0.005, q value 0.09 for IBS-C, and log2(FC) 0.49, p = < 0.07 for IBS-D. Error bars show SD and middle line indicates median (IBS-C n = 22 averaged microbiome compositions). All KO term associations can be found in Table S3.
See also Figures S3 and S4 and Table S3.
To gain insights into possible microbial contributions to the fecal hypoxanthine pool, we interrogated metagenomics functional modules related to hypoxanthine in stool samples from patients with IBS and HC (Table S3). Among the KO terms, we found that xanthine dehydrogenase/oxidase (XO; 1.17.1.4) and xanthine phosphoribosyltransferase (XPRT; 2.4.2.22) modules were elevated in IBS-C relative to HC (Figure 3D). XPRT liberates xanthine from xanthosine-monophosphate as an early step in purine salvage. Downstream, XO is an enzyme with low substrate specificity that acts on xanthine or hypoxanthine to produce uric acid. Higher levels of these XPRT and XO modules suggest increased purine breakdown by gut microbiota in IBS patients.
We inspected the metagenomic KO terms (Table S3) further to explore two aspects of hypoxanthine metabolism, namely, its role in modulating the epithelial energy state (Lee et al., 2020) and generation of H2O2 and superoxide anions given the putative role of oxidants in IBS (Mete et al., 2013). Four modules from the TCA cycle (L-lactate-, pyruvate-, and formate dehydrogenase, and fumarate hydratase) related to energy metabolism and 4 terms for alternative forms of respiration (sulfite reductase/ferredoxin, sulfite-, nitrite reductase, and cytochrome-C oxidase) were present at significantly higher abundance in IBS-C stools compared to HC (q < 0.1) (Figure 3D). Interestingly, the superoxide reductase (1.15.1.2) term was elevated in IBS-C, which could reflect increased capacity to deal with oxidative stress in the IBS-C gut microbiome. This might be necessary in situations of high XO activity. Together this suggests that the microbiome in IBS patients exhibits an increased capacity for hypoxanthine utilization and breakdown, which is congruent with the lower hypoxanthine levels in IBS-C stools.
Microbial Gene-Region Analysis Corroborates Associations with Bile Acids, Butyrate, and Hypoxanthine Metabolism
To further elucidate the microbial contribution to differential metabolite abundances identified in IBS, we first performed direct multivariate correlation analysis based on linear models (Maaslin; http://huttenhower.sph.harvard.edumaaslin). This identified 60 significant metabolite-species correlations for HC samples, 28 for IBS-C, and 46 for IBS-D (Table S4). No correlations were present in all groups; 12 were present in HC and IBS-C or IBS-D. Two correlations were present in both IBS-C and IBS-D subgroups (Table S4).
While the above correlational approach allows us to identify potential microbial drivers of differences in fecal metabolites, it is unable to identify specific microbial genes that might be relevant for the differences in detected metabolites. We therefore tested for specific bacterial genomic regions that may be responsible for the variation in metabolic output between the groups using the recently described method that associates structurally variable genomic regions to metabolite abundances (SV association) (Zeevi et al., 2019). This analysis allows identification of microbial genes involved in the production or consumption of metabolites by either identifying deletion regions (DRs) that are completely missing from some microbiomes or variable regions (VRs), which display variable abundance in some microbiomes. We identified 16 DRs and 20 VRs that correlate with 9 and 8 metabolites, respectively, at q < 0.1 (Table S4). All DRs were from a single bacterium, Blautia wexlerae DSM19850. CDCA was the most frequently associated metabolite, as it covaried with 4 DRs and 7 VRs. This was followed by CA with 3 DRs and 7 VRs. The multitude of associations of microbial genomic regions with BAs could reflect presently unknown genes that are involved in modification of primary BAs.
Two regions from Blautia obeum ATCC 29174 were present at significantly lower levels in IBS-C samples, and these regions positively correlated with butyrate (Figures S4G–S4I). This is consistent with Blautia spp. being butyrate producers and the lower butyrate levels in IBS-C. These regions are 2676–2677, which contains a tetricoat peptide and 2704–2705, which is annotated as a type III ribonuclease.
The strongest observed correlation for the VRs was for a single region from Lachnospiraceae sp. 3_1_46FAA with hypoxanthine (Figures 4A and 4B). pBLAST analysis (Camacho et al., 2009) perfectly matched the coding sequence (CDS) with topoisomerase III (E = 0). As hypoxanthine is a precursor for energy metabolites and topoisomerase III is linked to DNA replication, this may indicate increased utilization of hypoxanthine for growth. These examples underscore the increased functional resolution that can be achieved using SV association analysis in guiding targeted experiments focused on specific microbial gene regions.
Figure 4. Hypoxanthine Consumption by Specific Gut Microbiome Members as Suggested by Microbial Gene Region Associations.
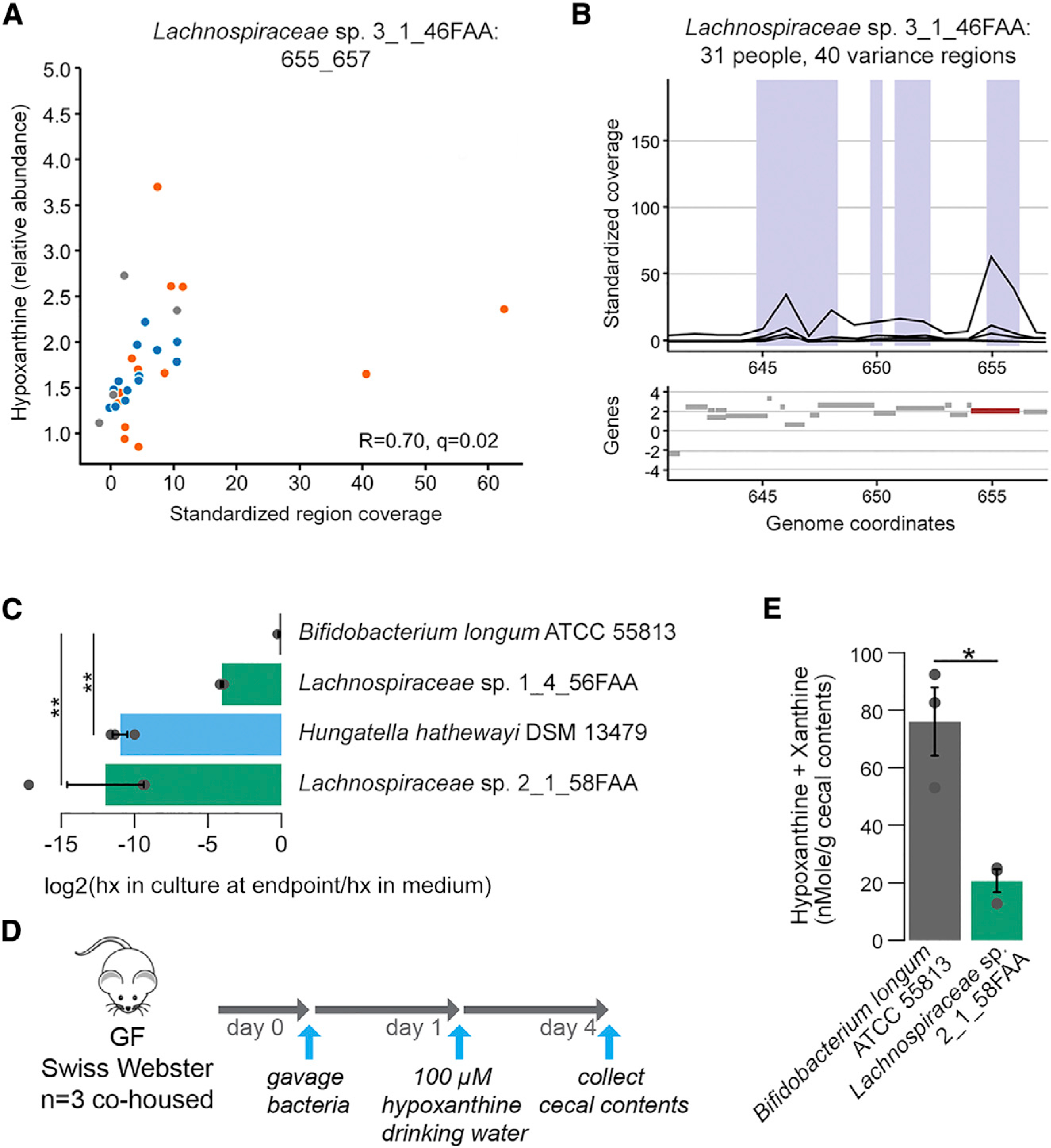
(A) Scatterplot of metabolite intensities and standardized region coverage for SV association result for Lachnospiraceae sp. 3_1_46FAA genomic region positively correlated to hypoxanthine (Spearman correlation inset, n = 13, 13, and 5 averaged microbiome abundances with Lachnospiraceae bacterium 3–146FAA present above threshold for IBS-C, IBS-D, and HC, respectively).
(B) Genomic context of region from (A) with relevant gene highlighted in red.
(C) 3 Clostridiales strains and B. longum were grown in Mega medium. Hypoxanthine levels in the culture supernatant after overnight growth were determined with LC-MS (ANOVA Tukey HSD on log2(FC), n = 3 cultures per strain). hx: hypoxanthine
(D) Outline of monocolonization mouse experiment verifying in vivo hypoxanthine consumption. 3 female GF Swiss Webster mice were oral gavaged with ~2*106 colony-forming units (CFUs) of either B. longum or Lachnospiraceae sp. 2_1_58FAA and co-housed for the duration of the experiment. Hypoxanthine was supplied in drinking water to mimic exogenous production by the microbiome. On day 4 after, gavage mice were sacrificed and cecal contents were collected.
(E) Hypoxanthine and xanthine pool size was determined in cecal contents using enzyme assays. Samples were corrected for baseline levels of H2O2 in the sample based on parallel reactions without XO enzyme (Welch t test on averaged duplicate samples, n = 3 cecal contents from 3 mice per colonization status).
Error bars indicate standard error of the mean (SEM). Symbols indicate significance (**p = < 0.01, *p = < 0.05).
Microbial Metabolism Contributes to Luminal Hypoxanthine Levels
To get more insight into the role of the microbiome in reducing hypoxanthine levels, we selected 2 Lachnospiraceae strains based on genomic similarity to Lachnospiraceae sp. 3_1_46FAA identified in the gene-region analysis presented above (Figures 4A and 4B). We confirmed the presence of the XO gene in these genomes using pBLAST. We included Hungatella hathewayi as a positive control as it was highly correlated with the XO KEGG term (Table S4, K00087; Spearman rho 0.43, q < 0.005). All these strains belong to the Clostridiales order. Bifidobacterium longum ATCC 55813 was included as a negative control since it does not encode XO (as evidenced by pBLAST analysis). We found significant reductions in hypoxanthine levels in the growth medium from the 2 Lachnospiracea strains and H. hathewayi when compared with B. longum using LC-MS (Figure 4C).
To determine whether Lachnospiraceae also consume hypoxanthine in vivo, we mono-colonized germ-free mice with either Lachnospiracea sp. 2_1_58FAA or B. longum and supplemented hypoxanthine in drinking water (Figure 4D). We found significantly lower hypoxanthine levels in cecal contents of mice colonized with Lachnospiraceae sp. 2_1_58FAA as compared to B. longum colonized mice (Figure 4E). Since hypoxanthine levels increase upon conventionalization (Matsumoto et al., 2017), this shows that microbially determined hypoxanthine levels are a consequence of the balance between microbial production and consumption.
Alteration in Gut Microbiome and Microbial Metabolites Underlie Flares in IBS Patients
IBS is a chronic disease with temporal variability in symptom severity, where most patients will experience transient worsening of symptoms. Our longitudinal analysis above identified a potential link between the gut microbiome and symptom severity in IBS patients. To further confirm that there is a microbial basis for potential exacerbation in symptoms, we examined the additional stool samples collected at the time of self-reported worsening of symptoms (flare) in the subset of patients who provided the additional sample. The flare samples exhibited a higher BCDI compared to the non-flare baseline combined IBS samples (Figures 5A and 5B), and lower Shannon α-diversity when compared to the averaged samples from the respective IBS subgroup (Figure 5C). Specific bacterial taxa were significantly associated with flares, both when considering IBS patients as one group, as well as within IBS-D and IBS-C patients (168 species for combined IBS, 40 for IBS-C, and 7 species for IBS-D at q <0.1 from Mann-Whitney U test compared to respective averaged baseline samples; Table S2). These significant species decreased in abundance almost universally during flare episodes. However, one species of Archaea, Halobiforma nitratireducens, was consistently elevated in the flare samples for both subtypes (Figure 5D). This Archaeon is capable of nitrate reduction, an alternative form of respiration that was identified above to be among the energy metabolism-related KO terms that were present at higher abundance in IBS-C.
Figure 5. Alteration in Gut Microbiome and Microbial Metabolites Underlie Flares in IBS Patients.
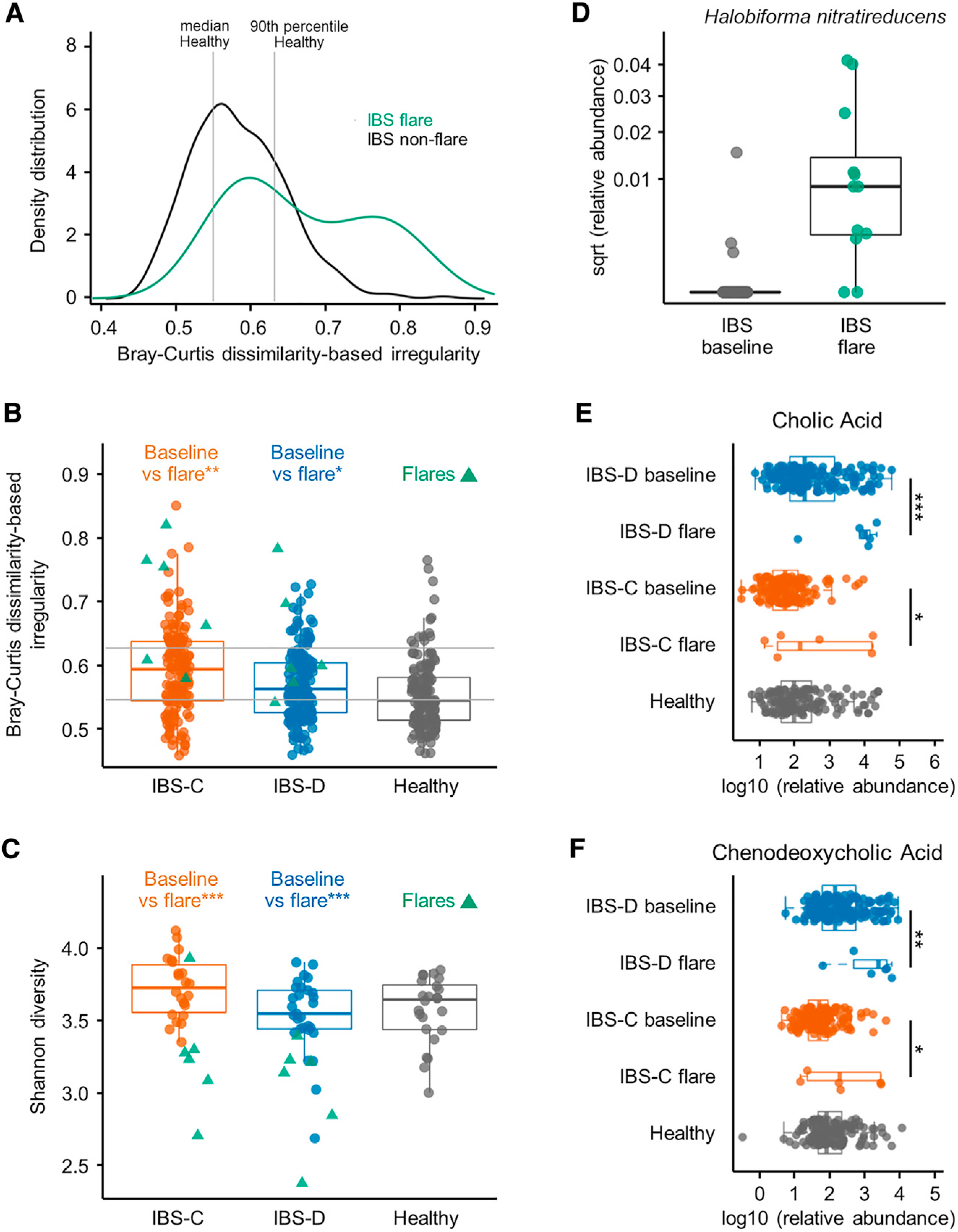
(A) BCDI showing distribution of IBS flare and all non-flare IBS samples (linear mixed-effect model correcting for subject, IBS non-flare versus IBS flare p = < 0.01, n = 312, 12 gut microbiome profiles for IBS non-flare, IBS flare, respectively).
(B) Within-disease comparisons of BCDI score (p values from linear mixed-effect model correcting for subject, n = 142, 6, 170, and 6 gut microbiome profiles for IBS-C non-flare, IBS-C flare, IBS-D non-flare, and IBS-D flare, respectively).
(C) Within-disease comparisons of α-diversity in flare samples compared to by-subject averaged baseline data (Shannon diversity at species level, p values from Mann-Whitney U test, n = 22, 6, 29, and 6 averaged gut microbiome profiles for IBS-C non-flare, IBS-C flare, IBS-D non-flare, and IBS-D flare, respectively).
(D) Relative abundance of Halobiforma nitratireducens in flare and non-flare IBS samples (q < 0.001, Mann-Whitney U test, n = 51, 12 averaged gut microbiome profiles for IBS non-flare, IBS flare, respectively).
(E) Relative abundance of cholic acid in stool samples determined with LC-MS/MS (linear mixed-effect models on log10-transformed data correcting for subject, FDR adjusted, n = 136, 6, 170, and 6 metabolite profiles for IBS-C non-flare, IBS-C flare, IBS-D non-flare, and IBS-D flare, respectively).
(F) Same as in (E) for chenodeoxycholic acid.
Boxplot center represents median and box IQR. Whiskers extend to most extreme data point <1.5 × IQR. Symbols indicate significance (***p = < 0.001, **p = < 0.01, *p = < 0.05).
Primary BAs were significantly elevated in flare samples of both IBS-C and IBS-D patients (Figures 5E and 5F) inviting speculation on their potential contribution to abdominal pain, which is common to both subtypes. We also investigated functional metagenomic KO modules that were associated with flares, with a focus on modules that were previously identified to be associated with symptom severity as well as the newly implicated hypoxanthine metabolic pathway. Of these, an alcohol dehydrogenase and XO module were found at higher abundance in IBS-D flares (log2(FC) 0.78, q value 0.147, p < 0.02; XO; log2(FC) 1.36, q value 0.147, p < 0.02), which again coincided with increases in TCA and respiration terms identified above (Table S3). These observations are also consistent with the findings described above linking IBS symptom severity with the gut microbiome.
The heterogeneous nature of IBS suggests there are likely distinct changes that underlie symptoms in individual patients. Hence, we next performed time-course analysis of the microbiome in individual patients to identify individual-specific patterns underlying symptom flares. In a patient with IBS-C, time-course permutation analysis revealed an increase in BCDI over time with this trajectory culminating in a flare episode (Figure S5A). Several Streptococcus spp. were positively correlated with BCDI scores in a subset of subjects (3/6 subjects; Table S5). At the functional level, again we found that secretory metabolites including tryptamine, CA, and CDCA were elevated in subsets of IBS patients (6/11 for BA and 4/11 for tryptamine) at the time of flare. These observations highlight that unique microbial and metabolic features may underlie worsening symptoms in different patients.
Microbiome and Metabolome Data Integrated with Transcriptomic and Epigenetic Differences Reveal Novel Host-Microbiome Interactions in IBS
As for most chronic conditions, the pathophysiology of IBS is multifactorial with contributions from host pathways, microbial pathways, and host-microbial co-metabolism. To determine the effect of microbial metabolism on host function, we first compared transcriptional and epigenetic changes observed in colonic biopsy tissue (Figure S6; Table S6). We also used these data to identify putative host-microbial-metabolite interactions in an untargeted way by constructing cross-omics correlation networks that integrate transcriptome data with metabolite and microbiota abundances (Figure S7; Table S7).
We identified 82 and 78 differentially expressed (DE) genes when comparing IBS-C and HC or IBS-D and HC (>1 absolute log2(FC) change and p < 0.05), respectively, with 17 genes overlapping in both comparisons (Figures S6A–S6D; Table S6). A KEGG pathway enrichment analysis revealed that immune and inflammation-related pathways were enriched in IBS patients (Figure S6F). Enriched categories for IBS-C contained genes for prostaglandin D2 synthesis, which is involved in regulating smooth muscle contraction (PTGDS), B cell responses to antigen challenges (CD19 and CD22), and antigen presentation via the HLA class II molecule (HLA-DQA1 and HLA-DQB1). Notably, we found no significant changes in any of the pro-inflammatory cytokines that were previously implicated in IBS in either blood or colonic biopsy samples (Mendeley data VI, https://doi.org/10.17632/29n2z5r5ph.3).
For epigenome analysis, we focused on differentially methylated regions (DMRs). We detected 54 DMRs when comparing IBS-C and HC, 75 DMRs when comparing IBS-D versus HC, and 39 DMRs comparing IBS-C and IBS-D (Figure S6E). We next inspected DE genes that belong to a DMR and identified two genes important for intestinal secretion, KCNE4 and AQP1, which were expressed at lower levels in IBS-D (Table S6). KCNE4 is a voltage-gated potassium (Kv) channel that is involved in neuronal excitability, epithelial electrolyte transport, and smooth muscle contraction (Jepps et al., 2009). AQP1 encodes an aquaporin, which are integral membrane proteins that facilitate the transport of water across biological membranes.
KEGG pathway enrichment analysis on the DMR genes showed that the antigen processing and presentation pathway was enriched in both IBS subtypes (Figure S6F; Table S6). The same pathway was also enriched in DE genes from IBS-C samples. This enrichment is due to human leukocyte antigen (HLA) class II genes. HLA genes are particularly interesting for IBS as the presence of celiac disease-related HLA-DQ-2/8 variants were found to be predictive of a favorable response to gluten-free diet in patients with IBS (Vazquez-Roque et al., 2012).
To better understand the implications of the HLA pathway in IBS-C, we inspected the underlying HLA class II genes in more detail and looked for correlations of these genes with the other omics. HLA class II complexes are present on antigen-presenting cells (APCs) but also on epithelial cells of the gastrointestinal tract and play a central role in the immune system by presenting peptides derived from extracellular proteins. We found a 4-fold higher expression of HLA-DQA1 and HLA-DQB1, which encode the α and β chains from the HLA class II molecule, respectively, in the IBS-C biopsy tissue compared to HC biopsies (Table S6). In addition, HLA-DQB1 is preceded by a DMR suggesting that its expression could be due to differential methylation in IBS-C. We found a correlation between HLA-DQA1 and Bacteroides vulgatus in our luminal cross-dataset correlation network (Figure S7B; Table S7) inviting speculation on the potential role of (bacterial) antigens in IBS-C.
In addition to the correlation networks highlighted above, we employed a powerful machine learning-based integration approach by applying a Lasso penalized regression model and used this to identify additional gene-microbe and gene-metabolite associations (Figure 6; Table S7). In this network, a hub is present around acetate and the PGLYRP1 and KIFC3 genes (Figure 6B). This could be relevant because acetate is a metabolite that is present at lower abundance both in stool and biopsy samples for IBS-C. PGLYRP1 is pattern receptor that binds to murein peptidoglycans (PGN) of Gram-positive bacteria and can result in bactericidal activity through interference with peptidoglycan biosynthesis. Indeed, the gene is negatively correlated with the broad family of Gram-positive bacteria Peptostreptococcaceae. KIFC3 is a minus-end microtubule-dependent motor protein required for zonula adherens maintenance and thus implicated in barrier function. This illustrates that the use of various omics integration approaches generates a wealth of hypotheses that can be used to contextualize the observed changes across omics data layers (Figure 6; Figure S7; Table S7).
Figure 6. Multi-omics Integration Results from Lasso Penalized Regression.
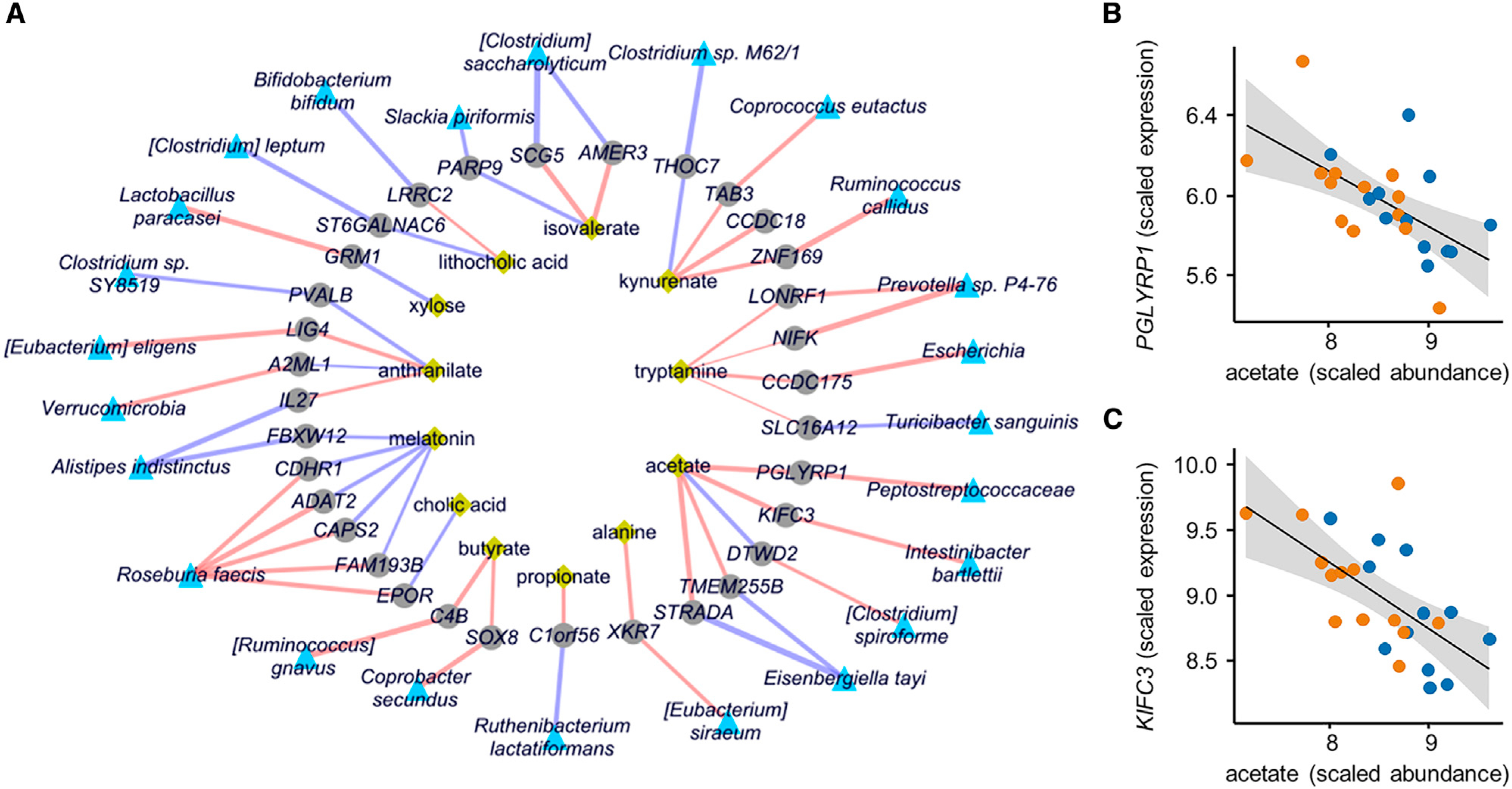
(A) Network representing significant and stability-selected correlations of host genes (gray nodes) with fecal microbial taxa (blue nodes) and fecal metabolites (yellow nodes) at FDR <0.25. Purple edges indicate positive correlation and red edges indicate negative correlation, and edge width indicates the strength of correlation (Lasso regression using 25 IBS patients and 13 matched HC datasets).
(B) Lasso correlation plots between acetate with PGLYPR1 (FDR <0.001) and acetate (FDR <0.05) from network shown in A). Orange and blue points represent IBS-C and IBS-D subjects, respectively.
(C) Same as (B) for KIFC3.
Multi-omics Integration Identifies Purine Starvation in Colonic Epithelium as a Potential Novel Mechanism Underlying IBS
Above we identified significantly lower fecal hypoxanthine abundance in IBS-C and IBS-D, established that microbial hypoxanthine degradation leads to lower intestinal hypoxanthine levels, and identified functional changes that point to increased purine degradation by the microbiome in IBS-C stools (Figures 3 and 7). However, since hypoxanthine is a host-microbial co-metabolite, its pool can be affected by both microbial and host metabolism. To determine the host response to the reduced hypoxanthine pool, we examined changes in purine metabolism gene expression in colonic biopsies. Gene expression of human xanthine oxidase (encoded by the XDH gene) was elevated in colonic biopsies of both IBS subtypes when compared to HC and this was consistent in both time points with a log2 (FC) ranging between 0.26 and 0.65 (Figure 7B; Table S6). This suggests that depletion of the hypoxanthine pool may be the result of increased XO activity from both the microbiome and the host.
Figure 7. An Integrated Multi-omics View of IBS Points to Microbiome-Host Interactions in the Purine Salvage Pathway.
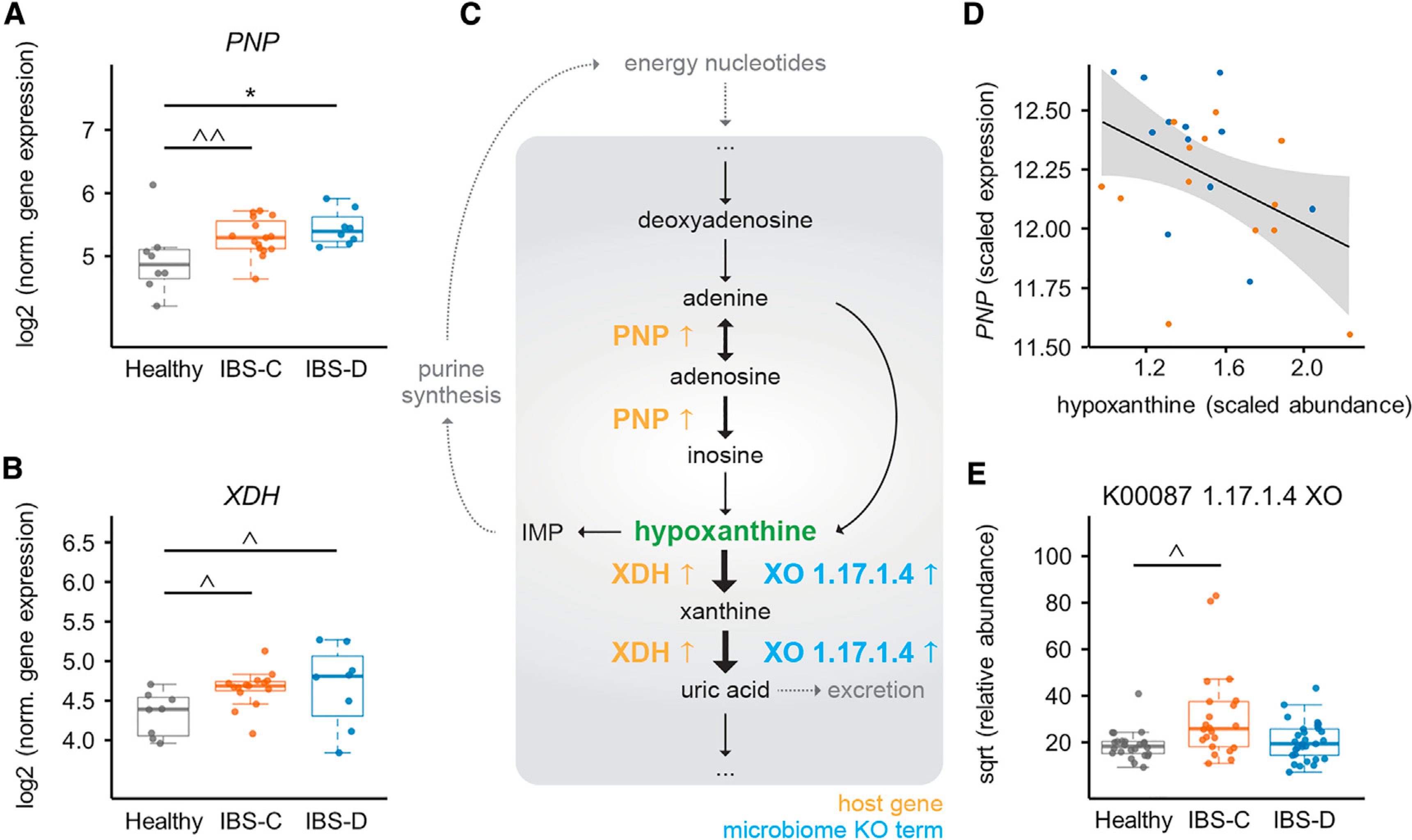
(A) Purine nucleoside phosphorylase (PNP) expression in colonic biopsy tissue (for A and B, ANOVA Tukey HSD, n = 15, 8, and 8 time-point-averaged female biopsy transcriptomes for IBS-C, IBS-D, and HC, respectively). For full statistical results split by time point, see Table S6 (p = < 0.001 for IBS-C and IBS-D versus HC for biopsies from first time point (IBS-D versus HC FDR 0.018), from generalized binomial test).
(B) Gene expression of human XDH in colonic biopsy tissue. For full statistical results split by time point, see Table S6 (p value 0.022 for IBS-C and 0.101 for IBS-D in time point 1 and <0.005 for time point 2 (with IBS-C versus HC FDR <0.05), from generalized binomial test).
(C) Simplified human-microbiome purine nucleotides degradation pathway with identified IBS-relevant changes indicated. Black arrows indicate metabolic steps, and yellow and blue up arrows indicate elevated expression or abundance in IBS.
(D) Lasso correlation plot between hypoxanthine and PNP (FDR <0.001). Orange and blue points represent IBS-C and IBS-D subjects, respectively.
(E) Metagenomic xanthine oxidase module abundance for all groups (also shown in Figure 3D for IBS-C; IBS-C versus HC FDR <0.1, Mann-Whitney U test. n = 22, 29, and 24 averaged gut microbiome profiles for IBS-C, IBS-D, and HC, respectively).
Boxplot center represents median and box IQR. Whiskers extend to most extreme data point <1.5 × IQR. Symbols indicate significance (*p = < 0.05, ^p = < 0.1, ^p = < 0.2).
Intestinal epithelial cells have limited capacity for de novo synthesis of purines and instead predominantly rely on salvage pathways for adenylate biosynthesis (Figure 7C) (Biaggioni et al., 2015; Grimble, 1994). Hence, to identify secondary effects in the host resulting from depletion of the hypoxanthine pool, we inspected possible transcriptional changes in the purine salvage pathway. Purine nucleoside phosphorylase (PNP), the first gene in the purine salvage pathway, was expressed ~2-fold higher in both IBS-C and IBS-D (Figure 7A; Table S6), and within IBS patients PNP expression displayed a negative correlation with hypoxanthine levels (Figure 7D). Importantly, variation in host genetics is not responsible for these differences in gene expression as Illumina global screening arrays revealed that single nucleotide polymorphisms (SNPs) in XDH and PNP were not differentially distributed between IBS subgroups and HCs. Together, these findings suggest a model where elevated degradation of purine nucleotides by the microbiota and the host induces metabolic stress in colonic tissue. In turn, this may lead to a compensatory response by increasing purine salvage. Using this multiomics view, we suggest that low levels of purine nucleotides may result in lower epithelial energy state and capacity for mucosal repair, which may in part underlie the pathophysiology of IBS.
DISCUSSION
In this study, we describe findings from an integrated longitudinal multi-omics analysis of the gut microbiome, metabolome, host epigenome, and transcriptome in the context of host physiology in patients with different subtypes of IBS. Several multiomics integration tools enhanced our ability to focus on a specific set of pathways with potential biological significance in IBS.
Baseline colonic secretion was increased in IBS-D patient biopsies, which would suggest either an inherent change in epithelial transport or an increase in metabolites that promote fluid secretion. The observed increases in secretagogues such as the primary BA CDCA and bacterial metabolite tryptamine suggest that higher levels of microbiota-related secretory compounds likely drives increased secretion in IBS-D. This is further supported by the lack of significant differences in secretory response to tryptamine among colonic biopsies from the three groups, which would be expected if there were inherent defects in the colonic epithelium.
Previous studies have suggested that BA malabsorption drives increased intestinal secretion in IBS-D, but the lack of a concurrent increase in secondary BA along with primary BAs in our study, suggests decreased microbial biotransformation of primary BA may at least in part be driving this effect.
Through further targeted integration of multiple host and microbiome data layers, we identified the host-microbial pathway of purine metabolism, which may play an important role in the pathophysiology of IBS. This is the first time that hypoxanthine has been implicated in IBS pathogenesis, including prior gnotobiotic studies on animal models. This illustrates the relevance of employing multi-omics measurements in humans to identify potential disease mechanisms that may depend on human-specific responses in gene expression. Hypoxanthine is an appealing drug target given the availability of the xanthine oxidase inhibitor allopurinol, which is used to treat gout (Becker et al., 2009), and thiopurines, which are used to optimize inflammatory bowel disease therapy (Moreau et al., 2017). There are also multiple drugs in development with more specific activity against xanthine oxidase, which may be relevant in the future.
Our study provides multiple new therapeutic targets for future investigation. The apparent decreased microbial biotransformation of BA in IBS-D patients may be treated using defined microbial consortia with a high capacity of conversion of CDCA to LCA. Similarly, in IBS-C patients, increased production of bacterial SCFAs and/or tryptamine may be viable therapeutic strategies. Finally, stimulating microbial hypoxanthine production or inhibition of xanthine oxidase locally in the gut would be a novel approach to increase the amount of luminal hypoxanthine without systemic effects and may be beneficial in IBS independent of disease subtype. Another interesting focus is the reported effect of liver BA signaling on XO activity, as it is unclear whether this relationship also exists in the digestive tract (Kanemitsu et al., 2017).
In summary, our integrated longitudinal multi-omics study highlights how we can leverage human studies to advance our understanding of diseases with both host and microbial components to identify targets for improved treatment.
Limitations of Study
We acknowledge that our study has some limitations. We realize that our findings are not a proof of causation even though we primarily focused on biologically plausible mechanisms. In order to increase rigor and confidence in our findings, we integrated multiple data types and primarily reported pathways that were identified across different data types. We focused primarily on the colonic microbiome, but we are aware that the small intestine likely plays an important role in generation of IBS symptoms (Saffouri et al., 2019). Longitudinal studies dedicated to the small intestinal microbiome are needed to complement our findings and advance our understanding of IBS. In addition, longitudinal multi-omics data like presented in this study could help in moving away from phenotype-based to mechanism-based IBS subtyping.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Purna Kashyap, MBBS (kashyap.purna@mayo.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Processed data for metagenomics, metabolomics, and cytokine measurements is available as a Mendeley dataset (https://doi.org/10.17632/29n2z5r5ph.3). RNaseq and methylome data was deposited at the Gene Expression Omnibus (GEO) (GSE146853). Microbiome sequencing reads are deposited at the European Nucleotide Archive (ENA) (PRJEB37924). Raw data from metabolomics measurements are deposited at the MetaboLights repository (MTBLS1396). Code used for metagenome and metabolome data analysis is available at https://gitlab.com/rubenmars/ibs-multi-omics-code. Code used for correlation networks can be found at https://github.com/mhoutti/ibs_network.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Recruitment and exclusion criteria
Mayo Clinic Rochester was the only medical center participating in the study. Participants were recruited through Mayo Clinic Institutional Review Board (IRB)-approved advertisements (newspaper advertisement, Mayo Clinic classifieds, “Volunteers Sought” flyers posted on campus, pamphlets given to IBS patients, letters mailed to home, word of mouth, clinicaltrials.gov, and Mayo research website). Healthy subjects and IBS-C and IBS-D patients between 18–65 years old who expressed interest were invited to undergo screening to assess eligibility. Participants were given the option of undergoing two flexible sigmoidoscopies. All IBS-C and IBS-D subjects fulfilled Rome III criteria. Recruitment of healthy subjects was matched with IBS subjects for age, sex and BMI.
Volunteers with prior history of abdominal surgeries (except appendectomy and cholecystectomy), diagnosis of inflammatory bowel disease, microscopic colitis, celiac disease, or other inflammatory conditions, antibiotic use within the past 4 weeks, bleeding risk or taking medication that increases bleeding risk (only for those who chose to undergo the flexible sigmoidoscopies), bowel prep for colonoscopy in the past week, pregnancy, plan to become pregnant during study, being a vulnerable adult, and age below 18 or over 65 were excluded. In addition, people were excluded with other diseases, conditions, or habits that would interfere with study completion, increased risks with flexible sigmoidoscopies (if chosen), or that in judgment of the investigator would potentially interfere with compliance to study or adversely affect outcomes.
Regulatory compliance
The study was approved and reviewed yearly through the IRB of Mayo Clinic Rochester. All participants provided written informed consent before any study-related activities were completed.
Participant and sample metadata
Additional info on study subjects was collected at the first visit after study consent for IBS and healthy volunteers. This included recording of medical history and a limited exam by the study physician where height, weight, BMI and vital signs were noted. Furthermore, study subjects underwent a dietitian consult where explanation on Food Frequency Questionnaires (FFQ) and 24-hour dietary recall questionnaire training was given. Additional questionnaires at the first visit were Rome III criteria for IBS diagnosis, IBS symptom severity (also completed monthly for IBS participants), microbiome health, bowel disease questionnaire (BDQ-6), Hospital Anxiety and Depression, IBS Quality of Life, and a 7-day Bowel Diary (also completed monthly for all participants).
Animal experiments
Mouse experiments were performed on female mice born and maintained in the Mayo Clinic Germ-Free Mouse Facility as previously described (Reigstad et al., 2015). Experiments complied with Institutional Animal Care and Use Committee guidelines (IACUC protocol no. A00003920–18).
METHOD DETAILS
Specimen collection and data generation
Stool specimens were completed via home collection kits at the earliest convenience after the initial visit and then monthly for six months. Sample tubes were returned on frozen gel packs overnight using FedEx or dropped off at the clinical core facility of Mayo Clinic Center for Cell Signaling where samples were stored at −80°C.
Blood samples (plasma, serum, whole blood) were collected at the initial visit only and stored at −80°C upon further processing.
Biopsies were obtained through flexible sigmoidoscopy from the sigmoid colon 20–30 cm from the anal verge essentially as described previously (Bhattarai et al., 2018; Peters et al., 2017). Up to two tap water enemas were given to cleanse colon for each procedure. All endoscopic procedures were performed by a single endoscopist (P.C.K.) and up to twelve colonic biopsies were collected using a large-capacity (2.8 mm) biopsy forceps without pin. Depending on downstream processing the biopsies were placed in RNAlater stabilization solution (Invitrogen), directly frozen in liquid nitrogen, or placed in glucose Krebs solution on ice (composition in mM: 11.5 D-glucose, 120.3 NaCl, 15.5 NaHCO3, 5.9 KCl, 1.2 NaH2PO4, 2.5 CaCl2.2H2O, and 1.2 MgCl2; pH 7.3–7.4) and immediately transported to the laboratory for experiments.
Ussing chamber experiments
Colonic mucosal secretory responses were assessed using Ussing chamber setups. Biopsies were mounted within 45 min of collection in 4 mL Ussing chambers (Physiologic instruments, San Diego, USA) with an aperture of 0.31 cm2. The basolateral side of the chamber was bathed with 4 mL of glucose Krebs solution while the apical side was bathed with 4 mL of Krebs Mannitol solution (composition in mM: 11.5 D-mannitol, 120.3 NaCl, 15.5 NaHCO3, 5.9 KCl, 1.2 NaH2PO4, 2.5 CaCl2.2H2O, and 1.2 MgCl2; pH 7.3–7.4). The chamber was bubbled with a 97% O2 and 3% CO2 gas mixture. Tissue viability was confirmed by using concentration response measurements to acetylcholine (1 mM–3 mM) added on the submucosal side prior to the start of experiments. Short circuit current (Isc) was continuously recorded using Acquire and Analyze software (Physiologic instruments, San Diego, USA). ΔIsc values were calculated using Isc measurements before and after application of compounds to the basolateral side and normalized to the tissue area. Tryptamine and serotonin were added at 11 cumulatively increasing concentrations from 0.003 μM to 300 μM. Imax is the maximal Isc value achieved at any of the concentrations.
Microbiome DNA sequencing and alignment
DNA extraction and sequencing was performed at the University of Minnesota Genomics Center (UMGC). DNA was extracted from stool and biopsy sections using the QIAGEN PowerSoil kit (QIAGEN, Germantown, MD, USA) and was quantified using a NanoDrop8000 UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and PicoGreen assays. Shotgun metagenomic sequencing library preparation for stool samples was completed using a modified NexteraXT protocol followed by sequencing on a HiSeq 2500 (Rapid Mode) with 100 bp single-end reads (1×100) or on a NextSeq with 150 bp single-end reads (1×150). To minimize sequencing batch effects samples from the three cohorts were assigned randomly to two sequencing runs. Shotgun reads were trimmed to a maximum of 100 bp prior to alignment. Shotgun sequences were aligned to the RefSeq representative prokaryotic genome collection (release 86) at 97% identity with BURST using default settings (Al-Ghalith et al., https://zenodo.org/record/806850). The generated alignment table was filtered by dropping samples with low depth (< 10,000 reads per sample). Functional profiling of the shotgun sequencing data was completed using the KEGG Orthology group annotations for RefSeq-derived genes from direct alignment. KEGG Orthology profiles were also predicted from reference genomes and the predicted profiles were augmented to improve the estimates of low-abundance genes using SHOGUN (https://github.com/knights-lab/SHOGUN).
Biopsy samples were sequenced via amplification of the V4 region of the 16S ribosomal RNA gene (Gohl et al., 2016), followed by paired-end 2×250 bp sequencing on an Illumina MiSeq. Adapters were trimmed and low-quality reads (< 25 Q-score) were dropped using Shi7 (Al-Ghalith et al., 2018). Amplicon reads were stitched also using Shi7 (Al-Ghalith et al., 2018). Amplicon sequences were aligned to the 16S rRNA genes from the same bacterial genomes in the shotgun sequencing approach using BURST (Al-Ghalith et al., 2018, https://zenodo.org/record/806850) with the same setting as above.
Microbiome data analysis
Downstream analysis of taxa and KEGG Orthology tables was performed in R (R Foundation for Statistical Computing, Vienna, Austria). Computing PERMANOVA, Shannon diversity, and Bray Curtis dissimilarity was done using adonis, diversity(x, index = “shannon”), and vegdist (x, method = “bray”) functions from the vegan package. Before testing for taxa differences between the subgroups, taxa were removed that were absent in 90% of the subjects (averaged data excluding flares). To identify differentially abundant features an FDR cutoff of < 0.25 was used. In specified cases this was cutoff was made more rigorous post hoc to display only top features due to the great number of significant changes at FDR < 0.25.
Inter-individual variation was compared to intra-individual variation by comparing the mean of microbiome distances between samples from different individuals with the mean of the distances between samples from each individual.
Bray-Curtis dissimilarity (BCD)-based irregularity (BCDI) was computed by extracting the pairwise dissimilarities between all healthy control (HC) and HC or IBS samples, and the median of these dissimilarities was stored. The 90th percentile of the HC values was used as a cutoff for identifying microbiome samples that were different compared to those of HC. For this analysis samples from one HC subject (10007557) were removed since the median of these samples was above the 90th percentile of the HC BCDI scores. A sensitivity analysis of the 90th percentile cutoff values was performed by randomly drawing one sample per HC subject and identifying the BCDI within these samples 500 times. In addition, the 90th percentile cutoffs from averaged HC microbiome abundances were computed. Using the average did not change the 90th percentile cutoff (0.63).
Time course permutation analysis of BCDI scores was performed by fitting 3rd order polynomial splines to BCDI scores for every subject. Sum of squares (SS) from the original fit were compared to an ensemble of SS from 999 time-point perturbations.
Metabolomics
1H NMR metabolome profiling of serum and stool samples
Aliquoted stool samples (~100 mg) were randomized in order and transferred to a screw-cap tube containing 50 mg 1.0 mm Zirconia beads (BioSpec). Metabolites were extracted by addition of 400 μL of acetonitrile:H2O (approximate volumetric ratio of 1:3) and homogenized for 30 s in a Biospec beat beater at maximum speed. The homogenized samples were then centrifuged for 20 minutes at 16000 × g, after which the supernatant was transferred to Spin-X 0.22μm spin filter tubes (COSTAR®) and centrifuged for 30 minutes at 16000 × g. 80 μL of the filtered samples was aliquoted into 96 well plates, and 10 μL was kept separately for downstream quality control purposes. Samples were dried under nitrogen flow before reconstituting in 540 μL of D2O and 60 μL of NMR buffer, all in 96 well deep well plates (COSTAR®). The plate was then placed on an Eppendorf MixMate plate shaker at 1300 rpm for 5 minutes. The reconstituted fecal water and buffer mixture was transferred to 5 mm NMR tubes. Plasma buffer with 1.5 M KH2PO4 was prepared by dissolving 20.4g of KH2PO4 in 80mL of D2O. 6 mL of D2O containing 100 mg of 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid sodium salt (TSP) (Millipore-Sigma) and 13 mg of NaN3 was added and mixed by shaking and sonication. The pH was adjusted to pH 7.4 with NaOH pellets. Total volume was adjusted with D2O.
Serum samples were thawed and centrifuged at 4°C at 12000 × g for 5 minutes. All samples were kept at −40°C or colder until analyzed. 90 μL of the supernatant was mixed with 90 μL of plasma buffer before being transferred to a 3 mm NMR tube. Plasma buffer with 0.075 M NaH2PO4 was prepared by dissolving 1.064 g of NaH2PO4 in 80 mL of D2O. 4mL of D2O containing 80 mg of 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid sodium salt (TSP) (Millipore-Sigma) and 40 mg of NaN3 was added and mixed by shaking and sonication. The pH was adjusted to pH 7.4 with NaOH pellets. Total volume was adjusted with D2O.
Metabolic profiles were recorded essentially as described previously (Dona et al., 2014) on a Bruker 600 MHz spectrometer (Bruker Biospin) set at a constant temperature of 300K for fecal samples and 310K for plasma samples. A 1D nuclear Overhauser enhancement spectroscopy (NOSEY) experiment and a 2D J-resolved experiment was performed for each fecal and serum sample. A total of 32 scans were acquired with an acquisition time of 4 minutes and 3 s per fecal sample following 4 dummy scans and the spectral data was collected into 64K data points. Automatic phasing, baseline correction and spectral calibration to TSP (0 ppm) was performed in Topspin 3.1 (Bruker Biospin).
The pre-processed spectral data was imported into MATLAB (Version 8.3.0.532 R2014a, Mathworks Inc, Natick, MA, USA). A series of in-house scripts were used for the following executions. The spectra were manually aligned to correct for subtle alterations in the chemical shifts of the peaks due to variation in pH. To account for the difference in sample concentration, probabilistic quotient normalization (PQN) was applied to the spectral data. A projection to latent structures-discriminant analysis (PLS-DA) model based on the Monte Carlo cross-validation (MCCV) method was constructed on the complete spectral profiles to identify discriminatory features in relevant comparisons (Garcia-Perez et al., 2017; Posma et al., 2018). A total of 1,000 MCCV models with 25 bootstrap rounds in each model was used to assess model robustness and to calculate the mean prediction (Tpred) of each sample. Discriminatory spectral features were annotated using statistical total correlation spectroscopy (STOCSY) (Cloarec et al., 2005) and a combination of in-house and online databases (https://hmdb.ca/). An in-house developed peak integration script was applied to calculate the integral of spectral peaks of interest.
Bile acid profiling through LC-MS/MS
Metabolites were extracted as detailed above for 1H NMR. Samples were analyzed on an ACQUITY ultraperformance liquid-chromatography (UPLC) system (Waters Ltd., UK) coupled to a Xevo G2-S quadrupole time of flight (Q-TOF) mass spectrometer (Waters Ltd.). A reversed-phase column ACQUITY BEH C8 column (1.7 μm, 100 mm × 2.1 mm) was used at an operating temperature of 60°C. The aqueous part of the mobile phase consisted of 1 mM ammonia acetate in ultrapure water, pH 4.15. The organic mobile phase was 1:1 isopropanol acetonitrile. For detailed description of the experimental methods see Sarafian et al. (2015).
Data files were imported into MassLynx (Waters Ltd.) where peaks were automatically integrated. Manual inspection on each processed sample file was carried out to ensure that the spectra had been correctly integrated. The extracted peak integral was then normalized to the total ion current (TIC) of each sample.
SCFA quantification with GC-MS/MS
Colonic biopsies were stored below −40°C until extraction. Biopsy tissues were transferred to a screw-cap tube and weighed, after which 5 1.0 mm Zirconia beads and 100 μL of ultrapure water were added. The tissue was homogenized in a Biospec bead-beater using two 30 s cycles at max speed. An eleven-point calibration curve and a pooled QC sample was constructed using genuine SCFA standards. Metabolites were then extracted using methyl tert-butyl ether (MTBE) (Millipore-Sigma) and derivatized with N-tert-butyl-dimethylsilyl-N-methyltrifluoroacetamide with 1% tert-butyldimethylchloro-silane (MTBSTF + 1% TBDMSCI). Samples were analyzed on a 7000D Triple-Quadrupole Gas chromatography-mass spectrometer (GC-MS) (Agilent Technologies Ltd.).
Data files were imported and analyzed in MassHunter Workstation Software Quantitation Analysis for QQQ version B.07.01 (Agilent Technologies Ltd.). The resulting SCFA concentration were corrected for dilution factor and normalized by sample weight.
Tryptophan quantification with LC-MS/MS
Stool was weighed on an analytical balance (sample weights ~50 mg) after which 1 mL of −20°C extraction solvent with internal standards was added to each sample, and sample mixed by vortexing at max speed for 3–5 s. Extraction solvent contained 200 ng/mL tryptamine-d4, 500 ng/mL L-tryptophan-d3, 1000 ng/mL 3-methylindole-d3, 200 ng/mL indole-3-acetic acid-d5, 200 ng/mL serotonin-d4 in 80% methanol. Samples were sonicated in a sonication bath at RT for 10 minutes and vortexed. Samples were placed at −80°C for 1 hour to facilitate protein precipitation. Extracts were cleared of debris via centrifugation at 18,000 × g, for 20 minutes at 4°C, and the resulting supernatant was transferred to a new microfuge tube. A quality control sample was prepared by pooling 10 μL of every sample. 100 μL of the sample was transferred to a glass autosampler vial and remaining extracts were stored at −80°C. Standard curves were prepared in 80% methanol in a dilution series from 1000 ng/mL to 0.1 ng/mL.
LC-MS/MS was performed on a Waters Acquity UPLC with T3 C18 stationary phase (1 × 50 mm, 1.7 μM) column coupled to a Waters Xevo TQ-S triple quadrupole mass spectrometer. Mobile phases were 100% methanol (B) and water with 0.1% formic acid (A). The analytical gradient was: 0 min, 5% B; 0.5 min, 5% B; 2.5 min, 95% B; 3.5 min, 95% B; 3.55 min, 5% B; 5 min, 5% B. Flow rate was 350 μL/min with an injection volume of 2.5 μL. Samples were held at 4°C in the autosampler, and the column was operated at 45°C. The MS was operated in selected reaction monitoring (SRM) mode. Product ions, collision energies, and cone voltages were optimized for each analyte by direct injection of individual synthetic standards. Inter-channel delay was set to 3 ms. The MS was operated in positive ionization mode with capillary voltage set to 3.2 kV. Source temperature was 150°C and desolvation temperature at 550°C. Desolvation gas flow was 1000 L/h, cone gas flow was 150 L/h, and argon collision gas flow was 0.2 mL/min. Nebulizer pressure (nitrogen) was set to 7 Bar.
Raw data files were imported into Skyline software (MacLean et al., 2010). Each target analyte was manually inspected for retention time and peak area integration. Peak areas were extracted for target compounds detected in biological samples and normalized to the peak area of the appropriate internal standard or surrogate in each sample. Normalized peak areas were exported to Microsoft excel where concentrations were obtained using the linear regression formulas generated from the calibration curve. Limits of detection (LOD) and limits of quantification (LOQ) were calculated as 3× or 10× the standard deviation of the blank divided by the slope of the calibration curve respectively (Broccardo et al., 2013; Shrivastava and Gupta, 2011). One compound, 3-methyl-indole did not produce a linear response and only raw peak areas are reported for this compound (relative quantification only).
Metabolomics data analysis
Metabolomics data was log10 transformed (with pseudo count 1 for zero values) and tested for significance in R with linear mixed-effect models correcting for subject using the lmer function from the lmerTest package with formula lmer(data ~cohort + (1|subject_id). Comparison contrasts were extracted using the get_contrasts function from the psycho package version 0.4–91, and nominal p values were adjusted per contrast table using the p.adjust(, method = “fdr”) function. Significance of by subject-averaged data was tested using Mann-Whitney tests using the wilcox.test function.
Time-series analysis on NMR data was carried out using the santaR version 1.0 package in R. The analysis was based on 1,000 bootstrap rounds (95% confidence interval), 1,000 permutation rounds and 4 degrees of freedom.
Cytokine measurements
Multiplexed Luminex according to manufacturer’s instructions was used for quantification of IL-8, IFNγ, IL-10, Il-18, IL-22, Leptin, VEGF, MIG, IL-1β, IL-17A, IL-1RA, IL-6 & TNFα. The beads were recorded on a Bioplex 200 Luminex instrument. Samples were tested in duplicate and values were quantified by interpolation from a 5 point standard curves. TGFβ−1 was quantified using enzyme-linked immunosorbent assay (ELISA) according to manufacturer’s instructions (R&D Systems, Minneapolis MN). Absorbance was measured on a Bio-Rad 550 microtiter plate reader. Samples were assayed in duplicate and values were interpolated from log-log fitted standard curves.
RNA sequencing and analysis
mRNA was extracted from biopsy samples and used for RNA-Seq library preparation following instructions in the Illumina TruSeq RNA Library Prep Kit v2. Sequencing was run on an Illumina High Seq-2000 in the Mayo Clinic Sequencing Core with 101bp paired end reads. Gene expression counts were obtained using the MAP-RSeq v.2.0.0 workflow (Kalari et al., 2014) which is part of the Mayo Bioinformatics Core pipeline. MAP-RSeq consists of alignment with TopHat 2.0.12 (Kim et al., 2013) against the human hg19 genome build and gene counts with the Subread package 1.4.4 (Liao et al., 2019). Gene annotation files were obtained from Ensemble version 75. Gene counts were normalized using RPKM (Reads Per Kilobase per million Mapped reads). Differential expression analysis was performed using edgeR 2.6.2 (Robinson et al., 2010). Pathway enrichment analyses were performed using R package RITAN (Rapid Integration of Term Annotation and Network resources, https://www.bioconductor.org/packages/release/bioc/html/RITAN.html).
Methylome sequencing and analysis
Illumina Infinium MethylationEPIC BeadChips with ~850K CpG sites were used to assess genome-wide methylation in genomic DNA isolated from biopsy samples. For data pre-processing the raw data (.idat) files were loaded into R package ChAMP version 2.9.10 (Tian et al., 2017). Probes that had detection p value > 0.01, bead count < 3, overlapped with SNP sites, or with multiple alignments in the human genome were removed, which resulted in 773,789 CpG sites for downstream analyses. Potential batch effects were corrected by the Combat method (Johnson et al., 2007; Sun et al., 2011). Differentially methylated CpG sites were detected using the limma function with Benjamini-Hochberg (BH) multiple testing correction. CpG sites with between-group differential p value < 0.01 and methylation difference greater than 5% were considered as differentially methylated CpGs (DMCs). Clusters within these DMCs (differentially methylated regions (DMRs)) were identified using the Bumphunter algorithm (Jaffe et al., 2012) and defined as a minimum of 4 probes in the region with adjusted DMR p value < 0.05 through permutation test. Genes associated with DMCs or DMRs were used for pathway enrichment analysis with the R package RITAN.
To correct for differences in gender and small numbers of male samples only biopsy samples from female subjects were included for interpretation. We focused our analysis on samples from the first biopsy time point and used time point 2 samples as a verification cohort.
Multi-omics data integration
Association between stool microbial features and stool metabolites was investigated using the Maaslin2 package in R (http://huttenhower.sph.harvard.edumaaslin). Maaslin2 was ran using minimum abundance and minimum prevalence for microbial features were set at 0.0001 and 0.5 respectively. Threshold for FDR corrected q-value was set at 0.25. Linear mixed effects models were applied to the association with subject set as random-effect.
Identification of structural deletion and variable regions and subsequent association with significant metabolite features was performed as reported in the methods paper (Zeevi et al., 2019). Due to low number of male samples only data from females was included.
Before running correlations, all taxa abundance data was adjusted for compositionality with centered log ratio (CLR) transformation from the robCompositions package in R. For correlation networks, species were removed that had a mean per-sample abundance under 0.001%, data was adjusted for compositionality, and each set of significant Spearman correlations were assembled into a network using igraph and plotted using ggraph in R.
Lasso penalized regression machine learning was performed using a model for regularization and feature selection to integrate host gene expression with microbiome and metabolomics data. Host biopsy gene expression from time point 1, collapsed fecal microbiome abundance and collapsed fecal metabolite data were subject-matched resulting in a subset of 25 IBS patients and 13 healthy controls. The biomaRt R package was used to remove non-protein-coding genes, lowly expressed genes (expressed in less than half of the samples), and genes with low variance, resulting in 12132 unique genes. A variance stabilizing transformation was performed on the filtered gene expression data using the DESeq2 R package. For the microbiome data, the counts taxa matrix was summarized at species, genus, family, and phylum taxonomic levels, and only taxa found at 0.01% relative abundance in at least 20% of the samples were kept. This filtered taxon matrix was CLR transformed. The fecal metabolomic data, NMR metabolites, bile acids, and tryptophan panel data was concatenated, and log2 transformed.
The Lasso regression model was fit separately in order to identify gene-microbiome and gene-metabolite associations. The gene-wise model uses gene expression for each gene as response and microbiome abundance or metabolite concentrations as predictors. The effect of gender and IBS-subtype was controlled for by including them as binary covariates in our predictor matrix. Leave-one-out cross validation was used for tuning the penalty parameters in the Lasso model fits using the R package glmnet. Inference for Lasso models was performed using regularized projections to obtain significance and confidence interval for each variable associated with a given gene. Multiple hypothesis testing was corrected for using the Benjamini-Hochberg method. Since the Lasso model is sensitive to small variations of the predictor, we used stability selection to select robust variables associated with the host genes. Intersects of outputs from Lasso and stability selection models were inspected and filtered at FDR < 0.25. Host gene-gender and host gene-IBS subtype associations were removed.
In vitro and in vivo hypoxanthine consumption experiments
Bacteria were cultivated in Mega medium (Romano et al., 2015). After overnight growth, hypoxanthine levels in the culture supernatant were determined using LC-MS. Hypoxanthine identity was verified through matching of accurate mass and retention time of an authentic standard (Sigma-Aldrich).
Monocolonization experiments were performed on 6-week old germ-free Swiss Webster mice. Three female mice were orally gav-aged with ~2*106 CFU of overnight bacterial culture grown in BHI medium + 0.01% cysteine and co-housed for the duration of the experiment. Hypoxanthine (Sigma-Aldrich) was supplied at 100 μM in filter sterilized NANOpure water. Water consumption was monitored and not different between the groups. On day 4 after gavage mice were sacrificed and cecal contents were collected.
The combined hypoxanthine and xanthine concentration was determined in cecal contents using Amplex Red Xanthine/Xanthine Oxidase Assay Kit (Thermo Fisher). Sample weights were normalized by addition of H2O and samples were homogenized by beat beating. Metabolites were extracted by addition of 19 volumes of 80°C ultrapure H2O and incubation at 80°C for 3 minutes while shaking at 800 rpm in a heating block. The supernatant after centrifugation for 10 minutes at 4°C served as the metabolite sample. Samples were analyzed in duplicate for 30 minutes and normalized based on parallel reactions without XO enzyme to correct for baseline levels of H2O2 in the sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details on statistical tests, n numbers, and significance cutoffs can be found in the figure legends. When relevant, further details are found in the method details for the specific measurement.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Bifidobacterium longum | ATCC | ATCC 55813 |
| Lachnospiraceae sp. 2_1_58FAA | BEI Resources | HM-161, HMP ID 0991 |
| Lachnospiraceae sp. 1_4_56FAA | BEI Resources | HM-154, HMP ID 0988 |
| Hungatella hathewayi | DSM | DSM 13479 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNAlater stabilization solution | Invitrogen | AM7020 |
| Tryptamine hydrochloride | Millipore Sigma | CAS# 343–94-2 |
| Hypoxanthine | Millipore Sigma | CAS# 68–94-0 |
| Critical Commercial Assays | ||
| Amplex Red Xanthine/Xanthine Oxidase Assay Kit | Thermo Fisher | A22182 |
| Deposited Data | ||
| Mendeley dataset, supplemental data I-VI | This paper | https://doi.org/10.17632/29n2z5r5ph.3 |
| Microbiome data | This paper | European Nucleotide Archive, PRJEB37924 |
| Metabolomics data | This paper | MetaboLights repository, MTBLS1396 |
| Transcriptome and methylome data | This paper | Gene Expression Omnibus, GSE146853 |
| Experimental Models: Organisms/Strains | ||
| Germ-free mice | Taconic Farms | Swiss Webster |
| Software and Algorithms | ||
| Maaslin2 | Huttenhower lab | http://huttenhower.sph.harvard.edumaaslin |
| BURST | Knights lab | https://zenodo.org/record/806850 |
| SHOGUN | Knights lab | https://github.com/knights-lab/SHOGUN |
| Other | ||
| Code used for metagenome and metabolome data analysis | This study | https://gitlab.com/rubenmars/ibs-multi-omics-code |
| Code used for correlation networks | This study | https://github.com/mhoutti/ibs_network |
Highlights.
Longitudinal sampling limits heterogeneity seen in cross-sectional microbiome studies
Alteration in the gut microbiome and microbial metabolites underlie IBS and symptom flares
Data integration reveals effect of microbial metabolites on gastrointestinal function
Purine starvation is identified as a possible therapeutic target in IBS
ACKNOWLEDGMENTS
The authors would like to thank Lyndsay Busby for secretarial assistance and J.M. Nielsen for help with figures. This work was supported by NIH grant DK114007 (P.C.K.); the Center for Individualized Medicine, Mayo Clinic, Rochester, MN (P.C.K.); the Minnesota Partnership for Biotechnology and Medical Genomics (P.C.K. and D.K.); the STRATiGRAD PhD training program of Imperial College London (https://www.imperial.ac.uk/stratigrad/) (Y.Y.); Société des Produits Nestle (J.R.S.); Imperial College NIHR BRC (J.R.S.); and NIH grant R35-GM128716 (R.B.).
Footnotes
DECLARATION OF INTERESTS
P.C.K. is on the Advisory Board of Novome Biotechnologies and is an ad hoc consultant for Pendulum Therapeutics, IP group, and Otsuka Pharmaceuticals. P.C.K. holds patent US20170042860A1 for use of tryptamine producing bacteria (“Methods and materials for using Ruminococcus gnavus or Clostridium sporogenes to treat gastrointestinal disorders”), and P.C.K. and Mayo Clinic have a financial interest related to this research. These interests have been reviewed and managed in accordance with Mayo Clinic Conflict-of-Interest policies. D.B.K. serves as CEO of CoreBiome, a company involved in the commercialization of microbiome analysis and a wholly owned subsidiary of OraSure Technologies. These interests have been reviewed and managed by the University of Minnesota in accordance with its Conflict-of-Interest policies.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.007.
A video abstract is available at https://doi.org/10.1016/j.cell.2020.08.007#mmc8.
REFERENCES
- Al-Ghalith GA, Hillmann B, Ang K, Shields-Cutler R, and Knights D (2018). SHI7 Is a Self-Learning Pipeline for Multipurpose Short-Read DNA Quality Control. mSystems 3. Published online April 2018. 10.1128/mSystems.00202-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MA, Schumacher HR, MacDonald PA, Lloyd E, and Lademacher C (2009). Clinical efficacy and safety of successful longterm urate lowering with febuxostat or allopurinol in subjects with gout. J. Rheumatol 36, 1273–1282. [DOI] [PubMed] [Google Scholar]
- Bhattarai Y, Muniz Pedrogo DA, and Kashyap PC (2017a). Irritable bowel syndrome: a gut microbiota-related disorder? Am. J. Physiol. Gastrointest. Liver Physiol 312, G52–G62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai Y, Schmidt BA, Linden DR, Larson ED, Grover M, Beyder A, Farrugia G, and Kashyap PC (2017b). Human-derived gut microbiota modulates colonic secretion in mice by regulating 5-HT3 receptor expression via acetate production. Am. J. Physiol. Gastrointest. Liver Physiol 313, G80–G87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai Y, Williams BB, Battaglioli EJ, Whitaker WR, Till L, Grover M, Linden DR, Akiba Y, Kandimalla KK, Zachos NC, et al. (2018). Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 23, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biaggioni I, Freeman R, Mathias CJ, Low P, Hewitt LA, and Kaufmann H; Droxidopa 302 Investigators (2015). Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension 65, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccardo CJ, Schauer KL, Kohrt WM, Schwartz RS, Murphy JP, and Prenni JE (2013). Multiplexed analysis of steroid hormones in human serum using novel microflow tile technology and LC-MS/MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 934, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilleri M (2014). Advances in understanding of bile acid diarrhea. Expert Rev. Gastroenterol. Hepatol 8, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloarec O, Dumas ME, Craig A, Barton RH, Trygg J, Hudson J, Blancher C, Gauguier D, Lindon JC, Holmes E, and Nicholson J (2005). Statistical total correlation spectroscopy: an exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal. Chem 77, 1282–1289. [DOI] [PubMed] [Google Scholar]
- Crouzet L, Gaultier E, Del’Homme C, Cartier C, Delmas E, Dapoigny M, Fioramonti J, and Bernalier-Donadille A (2013). The hypersensitivity to colonic distension of IBS patients can be transferred to rats through their fecal microbiota. Neurogastroenterol. Motil 25, e272–e282. [DOI] [PubMed] [Google Scholar]
- De Palma G, Lynch MD, Lu J, Dang VT, Deng Y, Jury J, Umeh G, Miranda PM, Pigrau Pastor M, Sidani S, et al. (2017). Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci. Transl. Med 9, eaaf6397. [DOI] [PubMed] [Google Scholar]
- Dona AC, Jiménez B, Schäfer H, Humpfer E, Spraul M, Lewis MR, Pearce JT, Holmes E, Lindon JC, and Nicholson JK (2014). Precision high-throughput proton NMR spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Anal. Chem 86, 9887–9894. [DOI] [PubMed] [Google Scholar]
- Duan R, Zhu S, Wang B, and Duan L (2019). Alterations of Gut Microbiota in Patients With Irritable Bowel Syndrome Based on 16S rRNA-Targeted Sequencing: A Systematic Review. Clin. Transl. Gastroenterol 10, e00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edogawa S, Edwinson AL, Peters SA, Chikkamenahalli LL, Sundt W, Graves S, Gurunathan SV, Breen-Lyles M, Johnson S, Dyer R, et al. (2020). Serine proteases as luminal mediators of intestinal barrier dysfunction and symptom severity in IBS. Gut 69, 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez I, Posma JM, Gibson R, Chambers ES, Hansen TH, Vestergaard H, Hansen T, Beckmann M, Pedersen O, Elliott P, et al. (2017). Objective assessment of dietary patterns by use of metabolic phenotyping: a randomised, controlled, crossover trial. Lancet Diabetes Endocrinol 5, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R, et al. (2016). Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol 34, 942–949. [DOI] [PubMed] [Google Scholar]
- Grimble GK (1994). Dietary nucleotides and gut mucosal defence. Gut 35 (1, Suppl), S46–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, and Irizarry RA (2012). Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int. J. Epidemiol 41, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps TA, Greenwood IA, Moffatt JD, Sanders KM, and Ohya S (2009). Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver Physiol 297, G107–G115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, et al. (2014). MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics 15, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemitsu T, Tsurudome Y, Kusunose N, Oda M, Matsunaga N, Koyanagi S, and Ohdo S (2017). Periodic variation in bile acids controls circadian changes in uric acid via regulation of xanthine oxidase by the orphan nuclear receptor PPARα. J. Biol. Chem 292, 21397–21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap PC, Marcobal A, Ursell LK, Larauche M, Duboc H, Earle KA, Sonnenburg ED, Ferreyra JA, Higginbottom SK, Million M, et al. (2013). Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Wang RX, Alexeev EE, Lanis JM, Battista KD, Glover LE, and Colgan SP (2018). Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J. Biol. Chem 293, 6039–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Wang RX, Goldberg MS, Clifford GP, Kao DJ, and Colgan SP (2020). Microbiota-Sourced Purines Support Wound Healing and Mucous Barrier Function. iScience 23, 101226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Ooga T, Kibe R, Aiba Y, Koga Y, and Benno Y (2017). Colonic Absorption of Low-Molecular-Weight Metabolites Influenced by the Intestinal Microbiome: A Pilot Study. PLoS ONE 12, e0169207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mete R, Tulubas F, Oran M, Yılmaz A, Avci BA, Yildiz K, Turan CB, and Gurel A (2013). The role of oxidants and reactive nitrogen species in irritable bowel syndrome: a potential etiological explanation. Med. Sci. Monit 19, 762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau B, Clement P, Theoret Y, and Seidman EG (2017). Allopurinol in combination with thiopurine induces mucosal healing and improves clinical and metabolic outcomes in IBD. Therap. Adv. Gastroenterol 10, 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters SA, Edogawa S, Sundt WJ, Dyer RB, Dalenberg DA, Mazzone A, Singh RJ, Moses N, Smyrk TC, Weber C, et al. (2017). ConstipationPredominant Irritable Bowel Syndrome Females Have Normal Colonic Barrier and Secretory Function. Am. J. Gastroenterol 112, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittayanon R, Lau JT, Yuan Y, Leontiadis GI, Tse F, Surette M, and Moayyedi P (2019). Gut Microbiota in Patients With Irritable Bowel Syndrome-A Systematic Review. Gastroenterology 157, 97–108. [DOI] [PubMed] [Google Scholar]
- Posma JM, Garcia-Perez I, Ebbels TMD, Lindon JC, Stamler J, Elliott P, Holmes E, and Nicholson JK (2018). Optimized Phenotypic Biomarker Discovery and Confounder Elimination via Covariate-Adjusted Projection to Latent Structures from Metabolic Spectroscopy Data. J. Proteome Res 17, 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M, Groussin M, Gibbons SM, Avila-Pacheco J, Jiang X, Kearney SM, Perrotta AR, Berdy B, Zhao S, Lieberman TD, et al. (2019). A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med 25, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Reigstad CS, Salmonson CE, Rainey JF 3rd, Szurszewski JH, Linden DR, Sonnenburg JL, Farrugia G, and Kashyap PC (2015). Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 29, 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roager HM, Hansen LBS, Bahl MI, Frandsen HL, Carvalho V, Gøbel RJ, Dalgaard MD, Plichta DR, Sparholt MH, Vestergaard H, et al. (2016). Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 1, 16093. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador-Noguez D, and Rey FE (2015). Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio 6, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffouri GB, Shields-Cutler RR, Chen J, Yang Y, Lekatz HR, Hale VL, Cho JM, Battaglioli EJ, Bhattarai Y, Thompson KJ, et al. (2019). Small intestinal microbial dysbiosis underlies symptoms associated with functional gastrointestinal disorders. Nat. Commun 10, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian MH, Lewis MR, Pechlivanis A, Ralphs S, McPhail MJ, Patel VC, Dumas ME, Holmes E, and Nicholson JK (2015). Bile acid profiling and quantification in biofluids using ultra-performance liquid chromatography tandem mass spectrometry. Anal. Chem 87, 9662–9670. [DOI] [PubMed] [Google Scholar]
- Shrivastava A, and Gupta V (2011). Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chronicles of Young Scientists 2, 21–25. [Google Scholar]
- Sun Z, Chai HS, Wu Y, White WM, Donkena KV, Klein CJ, Garovic VD, Therneau TM, and Kocher JP (2011). Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med. Genomics 4, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, and Teschendorff AE (2017). ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touw K, Ringus DL, Hubert N, Wang Y, Leone VA, Nadimpalli A, Theriault BR, Huang YE, Tune JD, Herring PB, et al. (2017). Mutual reinforcement of pathophysiological host-microbe interactions in intestinal stasis models. Physiol. Rep 5, e13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Roque MI, Camilleri M, Smyrk T, Murray JA, O’Neill J, Carlson P, Lamsam J, Eckert D, Janzow D, Burton D, et al. (2012). Association of HLA-DQ gene with bowel transit, barrier function, and inflammation in irritable bowel syndrome with diarrhea. Am. J. Physiol. Gastrointest. Liver Physiol 303, G1262–G1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Godneva A, Bar N, Kurilshikov A, Lotan-Pompan M, Weinberger A, Fu J, Wijmenga C, Zhernakova A, and Segal E (2019). Structural variation in the gut microbiome associates with host health. Nature 568, 43–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data for metagenomics, metabolomics, and cytokine measurements is available as a Mendeley dataset (https://doi.org/10.17632/29n2z5r5ph.3). RNaseq and methylome data was deposited at the Gene Expression Omnibus (GEO) (GSE146853). Microbiome sequencing reads are deposited at the European Nucleotide Archive (ENA) (PRJEB37924). Raw data from metabolomics measurements are deposited at the MetaboLights repository (MTBLS1396). Code used for metagenome and metabolome data analysis is available at https://gitlab.com/rubenmars/ibs-multi-omics-code. Code used for correlation networks can be found at https://github.com/mhoutti/ibs_network.