

ABSTRACT
Only a small subset of colorectal cancer (CRC) patients benefits from immunotherapies, comprising blocking antibodies (Abs) against checkpoint receptor “programmed-cell-death-1” (PD1) and its ligand (PD-L1), because most cases lack the required mutational burden and neo-antigen load caused by microsatellite instability (MSI) and/or an inflamed, immune cell-infiltrated PD-L1+ tumor microenvironment. Peroxisome proliferator-activated-receptor-gamma (PPARγ), a metabolic transcription factor stimulated by anti-diabetic drugs, has been previously implicated in pre/clinical responses to immunotherapy. We therefore raised the hypothesis that PPARγ induces PD-L1 on microsatellite stable (MSS) tumor cells to enhance Ab-target engagement and responsiveness to PD-L1 blockage. We found that PPARγ-agonists upregulate PD-L1 mRNA/protein expression in human gastrointestinal cancer cell lines and MSS+ patient-derived tumor organoids (PDOs). Mechanistically, PPARγ bound to and activated DNA-motifs similar to cognate PPARγ-responsive-elements (PPREs) in the proximal −2 kb promoter of the human PD-L1 gene. PPARγ-agonist reduced proliferation and viability of tumor cells in co-cultures with PD-L1 blocking Ab and lymphokine-activated killer cells (LAK) derived from the peripheral blood of CRC patients or healthy donors. Thus, metabolic modifiers improved the antitumoral response of immune checkpoint Ab, proposing novel therapeutic strategies for CRC.
KEYWORDS: Immunotherapy, cancer, colorectal, PD-L1, PPAR, MSS
Introduction
Genome-wide sequencing classified gastrointestinal cancers including colorectal cancer (CRC) into subtypes with distinct mutations1 and immune microenvironments.2 However, translation of molecular profiles to the clinic for individualized treatments or response prediction (“Precision Medicine”) remains a challenge.3 The recent success of immune checkpoint inhibitors, as exemplified by therapeutic Abs which block the “programmed-cell-death-1” (PD1/PD-L1) receptor–ligand system (e.g. pembrolizumab, atezolizumab), is limited to a minority of patients. As such, only cases with microsatellite-instable (MSI+) tumors, having a high mutational burden, neoantigen load and immune cell infiltration score, so far benefit from checkpoint inhibitor therapy.4 Thus, increasing eligibility rates is of urgent medical need.
To close the gap between in vitro cancer models and the clinics, patient-derived-tumor-organoids (PDOs) have been recently developed and hold the promise to improve translational research. PDOs are living biobanks for personalized, mechanistic studies recapitulating the clinical performance of patients in hospital’s real-life.5 Since tumor/epithelium-centered therapies fail due to intrinsic unresponsiveness and/or acquired resistance, extension of treatment concepts to other cell types is warranted. Reconstitution of these “avatars” with autologous or allogenic immune cells shall identify novel drugable targets to avoid lack of response, prevent resistance, relieve anergy and empower the full antitumor potential of innate and adaptive immunity.6,7 In this highly individualized contact-dependent co-culture model system, genomic profiles and response performances can be monitored in time and space and correlated with real-time clinical responses of patients in combination with standard of care (e.g. radio/chemotherapy).8,9
Peroxisome-proliferator-activated-receptors (PPARs) belong to the nuclear hormone receptor superfamily and comprise three genes PPAR α, β/δ and γ.10 Beyond its appreciated role as an insulin sensitizer in patients with type-2-diabetes-mellitus, PPARγ promotes differentiation of mucosal epithelial cells and orchestrates the immune response in the intestinal tract.11 Lipids derived from the diet, such as nitrated linoleic acid (LNO2) and eicosanoids released from sites of tissue inflammation, e.g. during colitis, activate PPARγ together with prescription-approved anti-diabetic drug agonists of the thiazolidinedione class: rosiglitazone (rosi) and pioglitazone (pio).12 PPARγ is expressed throughout the gastrointestinal tract with high levels in the colorectum. It inhibits inflammation and, thereby, may prevent cancers associated with chronic inflammation.13 As such, PPARγ increases expression of genes related to mucosal defense, reshapes the intestinal immune response toward polarization of M2 macrophages and mitigates Th1-driven inflammatory responses in preclinical rodent models.11 In this context, PPARγ is also a major driver for regulatory T-cells in the white adipose tissue.14
On the other hand, PPARγ has been shown to exert efficacy against established human hematopoietic malignancies, e.g. in leukemia15–17 and multiple preclinical settings (as reviewed in Peters et al.11). Thus, the involvement of PPARγ may be in part mediated by alterations in bone marrow-derived cell lineages which determine the “host” immune response also in solid tumors.18 For example, PPARγ potentiates the pro-tumor actions of cytotoxic T-lymphocytes and macrophages.19–22
In this context, PPARγ could be a suitable target for immunotherapy. A functional connection between metabolism and the cytotoxic activity of natural killer (NK) and T-cells has been established by demonstrating a role for all three PPAR proteins (α,γ,δ).23–25 Conclusively, mitochondrial fatty acid oxidation seems to be one predominant factor determining immune cell activity.26–28 Moreover, PPARγ is a master transcription factor for adipocyte differentiation and fat deposition, causative for its insulin-sensitizing action. Notably, obesity has also been linked to an improved response to PD1/PD-L1 inhibition in patients with solid tumors (melanoma, lung e.a.).29,30 Intrigued by the fact that PPARγ is drugable by clinically approved agonists and rewires metabolism with NK/T-cell activity, we resorted to this member of the PPAR gene family to assess its antitumor and immune-stimulatory features in CRC.
We raised the hypothesis that the immunogenicity of MSS+ CRC can be increased by pharmacological administration of PPARγ-agonists inducing PD-L1 expression31 on tumor cells, followed by enhanced sensitivity to immune cell attack in presence of therapeutic blocking Abs against PD1/PD-L1.2 Thus, the overall aim was to assess whether Ab-target engagement and antitumoral efficacy can be improved by this approach. To test this idea, we employed a translational program studying molecular aspects of PDL1 promoter regulation in CRC cell lines, organoids and patients.
Materials and methods
Reagents
Chemicals were purchased from Merck (Darmstadt, Germany) if not stated otherwise. Antibodies (Ab) for detection and therapeutic use (functional grade) and conditions for Western blotting (WB), immunofluorescence (IF), immunohistochemistry (IHC) and flow cytometry (FC) are listed in Table S1. Rosiglitazone (rosi), pioglitazone (pio) and GW9662 were from Cayman (Ann Arbor, MI), recombinant human IL2 and IFNγ from PeproTech (Hamburg, Germany).
Cell lines
Human embryonic kidney cells immortalized by the large T-antigen from Simian Virus-40 (HEK293T), leukemia (K562) and gastrointestinal adenocarcinoma cell lines (CRC: HT29, HCT116, SW480; Gastric: AGS) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultivated in complete Roswell Park Memorial Institute (RPMI) 1640 medium or Dulbecco’s Modified Eagle’s Medium (DMEM) according to the guidelines of the distributors. Basal media, herewith termed “complete” media, were supplemented with 10% (v/v) fetal calf serum (FCS), 2 mM l-glutamine and 100 U/ml penicillin/streptomycin (all from Thermofisher, Waltham, MA).
Patients
Written informed consent was provided by all patients. The study followed the principles of the Declaration of Helsinki and was approved by the Medical Ethics Committee II of the Medical Faculty Mannheim, Heidelberg University (2014–633 N-MA; 2016–607 N-MA).32,33 Patients with a fresh diagnosis of primary colon or rectal carcinoma (University Hospital Mannheim, Heidelberg University, Mannheim, Germany) were included prior to any intervention treatment. Cases with active HIV, HBV or HCV infections were excluded. Biopsies from primary tumors were collected by endoscopy and transferred into ice-cold phosphate-buffered saline (PBS) for generation of PDOs as detailed in.32 A prospective anonymized database was established merging clinical parameters and molecular tumor characteristics.32
A subset of seven patients (n = 3 female; n = 4 male) with MSS+ CRC and a mean age of 69 y [median: 70 y] was selected with respect to their KRAS gene mutation profiles32 (Table S2). Viable and expandable PDOs were available from four patients for all assays; no tissue material was archived for two patients.
Software and statistics
Results are displayed as mean ± S.E. from independent experiments, herewith defined as replicates, from different cell passages or individuals (healthy donors or patients). Optical densities (O.D.) of bands in gels from Western blots and PCRs were measured using automated imaging devices and quantified with Image J (imagej.nih.gov/ij). Data were normalized to housekeeping genes or proteins as indicated in the legends to figures and calculated as -fold or % compared to control. Statistical analysis was done with GraphPad Prism software (version 4.0, La Jolla, CA). Therein, data were first tested for non- vs. parametric distribution, followed by the appropriate statistical procedures with Bonferroni posttests for two-way ANOVA and Tukey or Dunn posttests for one-way-ANOVA or Kruskal–Wallis/Friedmann test, respectively. The two group comparisons were done with Mann–Whitney/Wilcoxon for nonparametric or t-tests for parametric data. All tests were unpaired and two-sided if not stated otherwise. p-Values <0.05 were considered significant (*).
Additional information has been deposited under Supplementary Materials and Methods.
Results
PPARγ binds to and activates the proximal −2 kb promoter of the human PDL1 gene
MSI+ and a PD-L1+ inflamed/immune-infiltrated (“hot”) tumor microenvironment predicts favorable clinical response to PD1/PD-L1 blocking Abs, whereas lack of response is associated with MSS+ and absence of PD-L1.2 We therefore asked if PPARγ induces PD-L1 in MSS+ tumor cells to strengthen the physical contact (synapse) between PD-L1+ tumor and PD1+ immune cells, followed by improved antitumoral efficacy of PD1/PD-L1 blocking Abs.
To identify potential PPARγ-binding sites in the human PDL1 promoter, we searched for cognate PPARγ-responsive-elements (PPREs, 5ʹ-AGGTCA-3ʹ) in the primary sequence of the upstream regulatory region of the gene. To this end, we performed an in silico query in the proximal −2 kb promoter using AliBaba2.1. The genomic DNA sequence of the PDL1 promoter with predicted PPARγ-binding motifs is depicted in S1. Three putative PPRE sites (1691, 2172, 2923 bp) upstream of the transcriptional start site were identified in proximity to consensus steroid hormone receptor- and interferon-responsive elements.34 To measure DNA-binding, we amplified the predicted PPREs by genomic PCR upon chromatin immunoprecipitation (ChIP) of the endogenous PDL1 promoter in human CRC cell lines (Figure 1a). MSS+ HT29 and MSI+ HCT116 (as pos. control35) cells were treated with the PPARγ-agonist rosiglitazone (rosi, 1–10 µM) for 48 h, followed by ChIP. PPARγ Ab pulled-down DNA harboring the three PPREs under basal conditions in both cell lines, however, only the distal motif was enriched upon ligand treatment of HT29 cells (by ~2-fold; *p < .05 vs. vehicle or bead control, two-way ANOVA with Bonferroni posttests, n = 3 per cell line) (Figure 1b, S2). These data indicated that PPARγ occupies the PDL1 promoter and is responsive to pharmacological activation.
Figure 1.
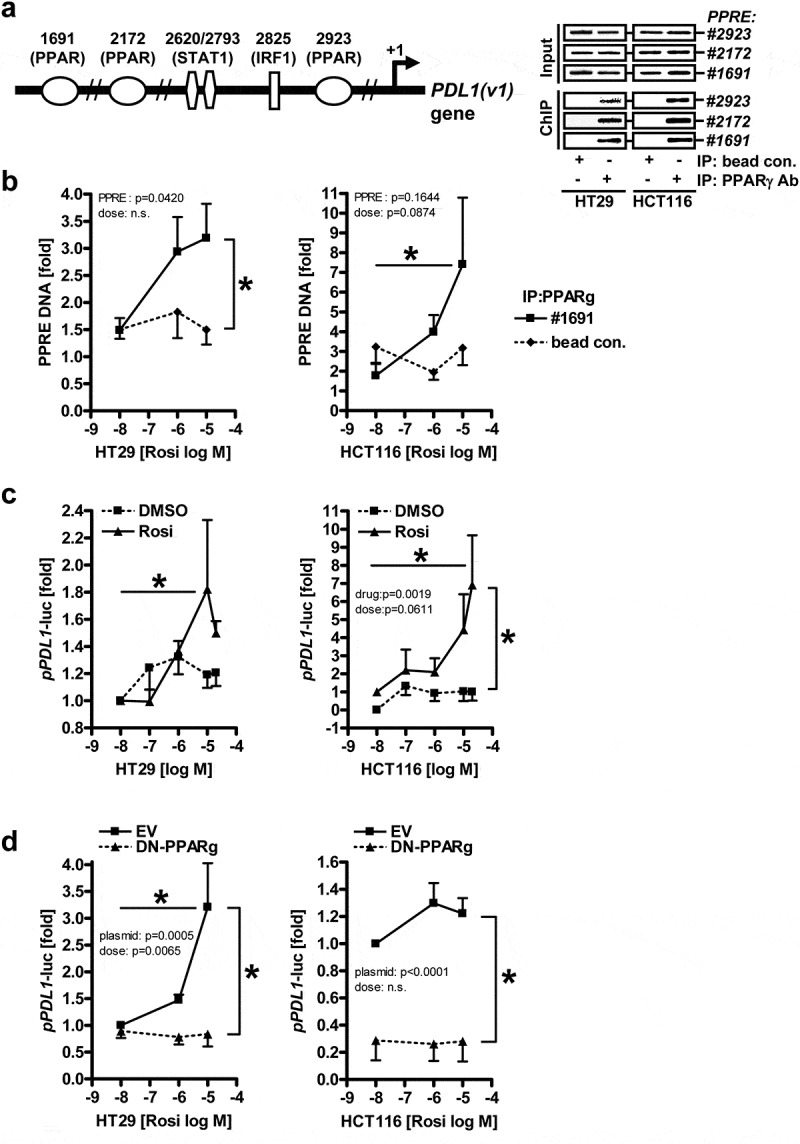
PPARγ binds to and activates the proximal −2 kb promoter of the human PDL1 gene.A-B, PPARγ protein binds to three predicted PPREs (1691, 2172, 2923 bp) in the DNA of the -2 kb human PDL1 promoter. Cells (HT29, HCT116) were treated with vehicle (DMSO) or PPARγ-agonist rosiglitazone (rosi, 1-10 µM) for 48 h, and chromatin-immunoprecipitation (ChIP) was performed using Abs against PPARγ for IP and genomic qPCR for amplification of bound DNA; A, Left: Scheme of the human PDL1 promoter (S1); Right: Representative genomic amplification products on ethidium bromide-stained agarose gels; B, Ct-values for PPRE 1691 were normalized to β2-microglobulin (B2M) (of input DNA) and expressed as -fold ± S.E. (*p<0.05 vs. vehicle or bead control, 2way-ANOVA with Bonferroni post-tests, n=3 per cell line). Ct-values for PPREs 2172 and 2923 are show in S2.; C, PPARγ-agonist activates the proximal -2 kb human PDL1 promoter. Cells (HT29, HCT116) were transfected with luciferase reporter plasmid and incubated with vehicle (DMSO) or rosi (0.1-20 µM) for 48 h. Luciferase activity was normalized to protein content and expressed as -fold ± S.E. (*p<0.05 vs. vehicle, 2way-ANOVA with Bonferroni post-tests, n=3 per cell line); D, Inhibition of PPARγ-activity by a dominant-negative (DN) mutant reduces activation of the PDL1 promoter. Cells (HT29, HCT116) were transfected either with empty vector (EV) or GFP-PPARγΔDbox (abbrev. DN-PPARγ) together with luciferase reporter plasmid followed by incubation with vehicle (DMSO) or rosi (0.1-10 µM) for 48 h. Data are presented as in C (*p<0.05 vs. vehicle or EV, 2way-ANOVA with Bonferroni post-tests, n=3 per cell line)
To explore if PPARγ also transactivates the human PDL1 promoter (Figure 1c), the upstream region of the PDL1 gene (transcript variant 1), covering −2 kb until the start of the protein coding sequence, was inserted into the pGL3 luciferase reporter plasmid. Then, HT29 and HCT116 cells were transfected and incubated with rosi (0.1–20 µM) for 48 h. Rosi increased luciferase activity driven by the −2 kb PDL1 promoter in both cell lines (by ≥2-fold; *p < .05 vs. vehicle, two-way ANOVA with Bonferroni posttests, n = 3 per cell line).
Similar though weaker effects were obtained with the related PPARγ-agonist pioglitazone (pio), but not for the PPARγ-antagonist GW9662 (abbrev. GW) (S3a).
To decide if PD-L1 promoter activation is mediated through the receptor itself or off-target effects of the agonist(s), HT29 and HCT116 cells were transfected with empty vector (EV) or an expression plasmid encoding a dominant-negative (DN) PPARγ mutant and stimulated with rosi as earlier. This mutant was deficient in heterodimerization with retinoid X receptor (RXR) and DNA-binding due to deletion of the “D-box” docking motif in the transition region of the second zinc finger of the DBD and the hinge region (ΔDbox).36 As expected, luciferase activity was abrogated under these conditions (*p < .05 vs. vehicle or EV, two-way ANOVA with Bonferroni posttests, n = 3 per cell line) (Figure 1d). The mutant also attenuated transcription driven by 3xPPREs in the enhancer region of the acyl-CoA oxidase (ACO) gene and of other bona fide PPARγ-target genes (S3b,c). These data indicated that PPARγ binds and transactivates the human PDL1 promoter.
Likewise, knock-down of PPARγ by shRNA diminished activation of both reporters (S4). However, regulation was cell line-dependent, possibly due to different mutations in given lines (e.g. KRAS, MSI e.a.). Conclusively, this data showed that the PDL1 gene can be addressed by pharmacological and genetic modulation of PPARγ.
PPARγ-agonists upregulate PD-L1 mRNA and protein expression
To assess whether PPARγ-ligands also regulate expression of the endogenous PDL1 gene, HT29 cells, as an exemplary line for MSS+ CRC, were treated with vehicle (DMSO), rosi (1–10 µM), pio (10–100 µM) or GW9662 (1–10 µM) for 24–48 h, followed by RNA extraction. RT-qPCR analyses demonstrated that rosi increased PDL1 mRNA (Figure 2a) (by three to fourfold; *p < .05 vs. vehicle, two-way ANOVA with Bonferroni posttests, n = 3 per drug). Pio showed a similar though weaker trend, whereas the antagonist was ineffective.
Figure 2.
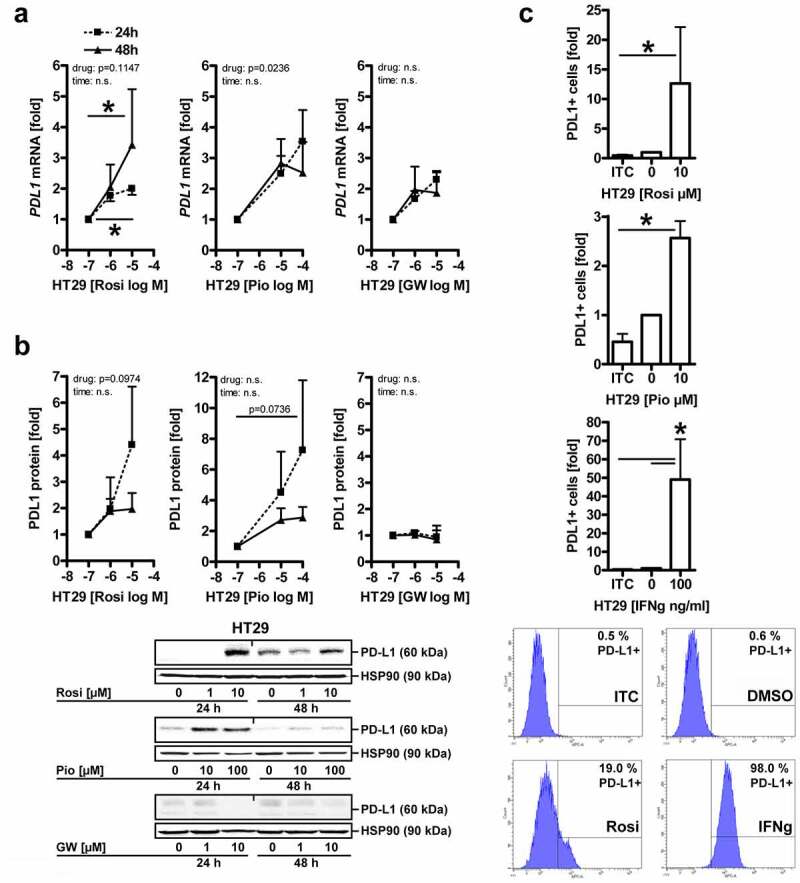
PPARγ-agonists upregulate PD-L1 mRNA and protein expression.A, Agonists, but not antagonists of PPARγ increase PDL1 mRNA. HT29 cells were treated with vehicle (DMSO), rosi (1-10 µM), pio (10-100 µM) or GW9662 (1-10 µM) for 24-48 h, followed by RNA extraction. Ct-values from RT-qPCRs were normalized to B2M and calculated as -fold ± S.E. (*p<0.05 vs. vehicle, 2way-ANOVA with Bonferroni post-tests, n=3 per drug); B, Agonists, but not antagonists of PPARγ increase PD-L1 protein. HT29 cells were treated as in A, followed by extraction as total cell lysates. Quantitative analyses (top) and representative images (bottom) from Western blots. O.D. values from gels were normalized to HSP90 and are -fold ± S.E. (*p<0.05 vs. vehicle, 2way-ANOVA with Bonferroni post-tests, n=3 per drug); C, PPARγ increases surface PD-L1 protein. HT29 cells were treated with vehicle (DMSO), rosi or pio (both 10 µM) or IFNγ (100 ng/ml as pos. control) for 48 h, then dissociated with Accutase™, and single cells were stained with Abs and viability dye (7AAD) as indicated in Table S1 and analysed by flow cytometry (FC). Quantitative analyses (top) and representative intensity plots (bottom). Data were calculated as -fold ± S.E. PD-L1+ cells compared to unstained samples (*p<0.05 vs. isotype control (ITC), Kruskal Wallis test with Dunn post-test, n=5 per drug)
Next, cells were treated as in A, followed by extraction of total cell lysates and Western blotting. Quantitative analysis evinced that PPARγ activation also increased PD-L1 protein (Figure 2b) (by ~2- to 7-fold; *p < .05 vs. vehicle, two-way ANOVA with Bonferroni posttests, n = 3 per drug). As above, similar results were obtained for other PPARγ-ligands.
Results from more gastrointestinal cancer cell lines are presented in S5-6. Active Ras-MEK1/2-ERK1/2 signaling inhibits transcription driven by PPARγ.37 Hence, cell lines with low Ras activity (KRAS: HT29 wt, HCT116 G13D) upregulated PD-L1 to a greater extent upon exposure to PPARγ-ligand than those with high Ras activity (KRAS: SW480 G12V, AGS G12D).
Finally, cell-surface associated PD-L1 protein was determined by flow cytometry (FC) in live HT29 cells. After a 48 h incubation, rosi increased the frequency of PD-L1+ cells by >10-fold and pio by 2- to 3-fold (*p < .05 vs. isotype control (ITC), Kruskal–Wallis test with Dunn posttest, n = 5 per drug) (Figure 2c).
PPARγ-agonists upregulate PD-L1 expression in a subset of PDOs
We then resorted to PDOs with the aim to investigate the role of PPARγ in regulation of PD-L1 in a more clinically relevant setting. Constitutively active Ras signaling is a major oncogenic driver for CRC1 and a negative regulator of PPARγ.37 We therefore selected four patients with MSS+ CRC based on the tumor’s KRAS gene mutation (wt/A146T, n = 2; G12D/C, n = 2) (Table S2), and who had been well characterized in our hospital with regard to their clinical performance and tumor genetics.32
Tumor organoids from these patients were treated with vehicle (DMSO), rosi (1 and 10 µM) or IFNγ (100 ng/ml as pos. control) for 48 h, followed by RNA extraction. RT-qPCR analyses evinced that all PDOs were responsive to IFNγ,34 whereas the PPARγ-agonist increased PDL1 mRNA only in a subset (2 of 4/50%) by more than twofold (Figure 3a) (*p < .05 vs. vehicle, Friedmann test with Dunn posttests, n = 4 patients, n = 3 replicates per patient). As before in cell lines, PPARγ bound to PPREs in the PDL1 promoter, and PPARγ-agonist induced mRNA/protein expression of cognate PPARγ-target genes (ACO, CD36 e.a.) in organoids (S7).
Figure 3.
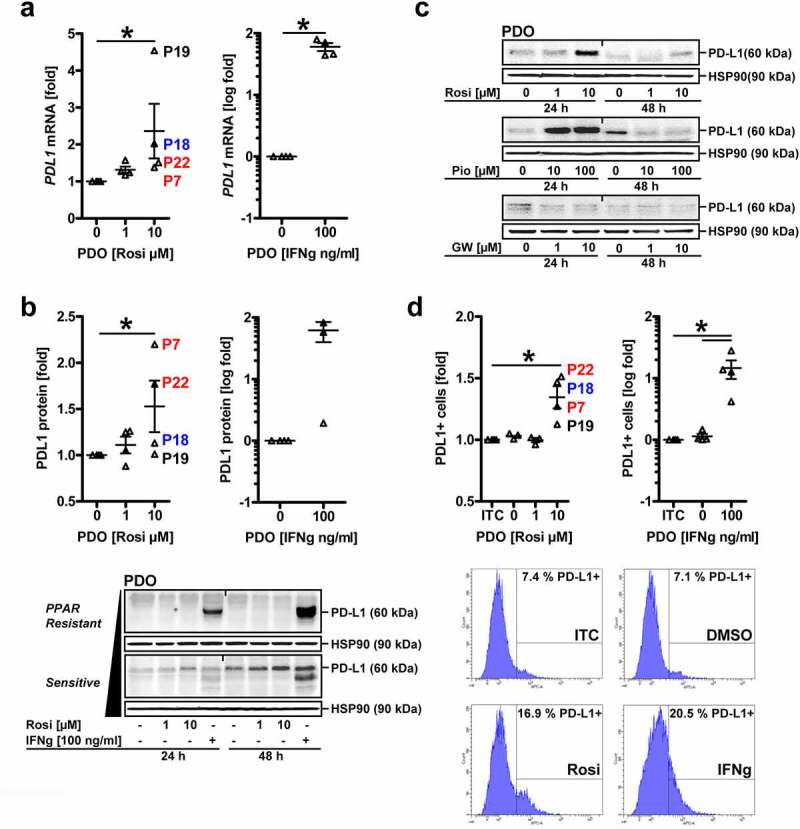
PDO subsets are sensitive to upregulation of PD-L1 by PPARγ-agonist.A, PPARγ-agonist increases PD-L1 mRNA in a subset of PDOs. Tumor organoids from CRC patients (n=4 MSS+ cases: KRAS wt = blue, mutant A146T = black, G12D/C = red) were treated with vehicle (DMSO), rosi (1 and 10 µM) or IFNγ (100 ng/ml) for 48 h, followed by RNA extraction. Ct-values from RT-qPCRs normalized to B2M are -fold ± S.E. (*p<0.05 vs. vehicle, Friedmann test with Dunn post-tests, n=4 patients, n=3 replicates per patient); B, PPARγ-agonist increases total PD-L1 protein in a subset of PDOs. Organoids were treated as in A, followed by extraction as whole cell lysates. Quantitative analyses (top) and representative images (bottom) from Western blots. O.D. values of bands in gels normalized to HSP90 are -fold ± S.E. (t=48 h: *p<0.05 vs. vehicle, Friedmann test with Dunn post-tests, n=4 patients, n≥3 replicates per patient). Arrow (black) marks exemplary PDOs sensitive (lower) vs. resistant (upper) to rosi-mediated PD-L1 up-regulation. IFNγ served as pos. control for all PDOs; C, Agonists, but not the antagonist for PPARγ increase PD-L1 protein in PDOs. Organoids were treated with vehicle (DMSO), rosi (1 and 10 µM), pio (10 and 100 µM) or GW (1 and 10 µM) for 24-48 h. Representative images from Western blot on whole cell lysates are shown; D, PPARγ increases cell surface-associated PD-L1 protein in a subset of PDOs. Organoids were treated as in A (rosi: 10 µM), then dissociated with Accutase™, and single cells were stained with Abs and viability dye (7AAD) as indicated in Table S1 and analysed by FC. Quantitative analyses (top) and representative intensity plots (bottom). Data are calculated from intensity plots as -fold ± S.E. PD-L1+ cells (*p<0.05 vs. isotype control (ITC), Friedmann test with Dunn post-tests, n=4 patients with n≥2 passages per patient)
This upregulation was confirmed by quantitative analyses from Western blots upon extraction of PDOs as total cell lysates. After 24–48 h, IFNγ augmented total cellular PD-L1 protein in all patients tested. Again, rosi increased PD-L1 only in a subset (2 of 4/50%) (Figure 3b) (t = 48 h: *p < .05 vs. vehicle, Friedmann test with Dunn posttests, n = 4 patients, n ≥ 3 replicates per patient). Similar effects were obtained with pio, but not for the PPARγ-antagonist GW9662 (Figure 3c). Due to lack of consistent efficacy in MatriGel®, pio was discontinued for further PDO assays. Nevertheless, cell surface-associated PD-L1 protein as determined by FC after 48 h was elevated in presence of rosi (by ~50%, *p < .05 vs. ITC, Friedmann test with Dunn posttests, n = 4 patients with n ≥ 2 passages per patient) (Figure 3d).
Notably, total cellular PD-L1 levels did not correlate with the KRAS status of a given PDO line. PD-L1 is subjected to glycosylation for proper insertion into and function at the plasma membrane and also exists as secreted forms.38 To distinguish between these PD-L1 pools,39 we compared its in situ expression in PDOs with the corresponding matched primary tumor (abbrev. pTU) tissue of the same patient (Figure 4a). FFPE-sections from PDOs and surgical resection material were stained by immunohistochemistry (IHC). Human lymph node tissue served as pos. control. Patient-wise assessment of baseline PD-L1 positivity in tumor cells and tumor-infiltrating immune cells, quantified as “tumor proportion score” [TPS in %], “inflammatory cell score” [IC in %] and “combined positivity score” [CPS = TPS+IC in a.u.],39 revealed that all MSS+ specimens (n = 4 patients; n = 1 no material available) tested were negative for PD-L1 in tumor cells (TPS = 0%), but some displayed a low intensity and frequency of membrane-accentuated PD-L1 with cytoplasmic staining in cells of the tumor-adjacent stroma (IC<5%) Overall, a positive association of PD-L1 expression in PDOs and the matched primary tumor tissue (CPS>0), as defined by observable immune cell infiltration (IC≥1),39 was evident in only 2 of 4 (50%) cases (Table S5). Tissue biopsies contained a mixture of tumor and stroma (immune) cells. Hence, we could not exclude the possibility that PD-L1 positivity was progressively lost during generation and passaging of PDOs in selection media.32
Figure 4.
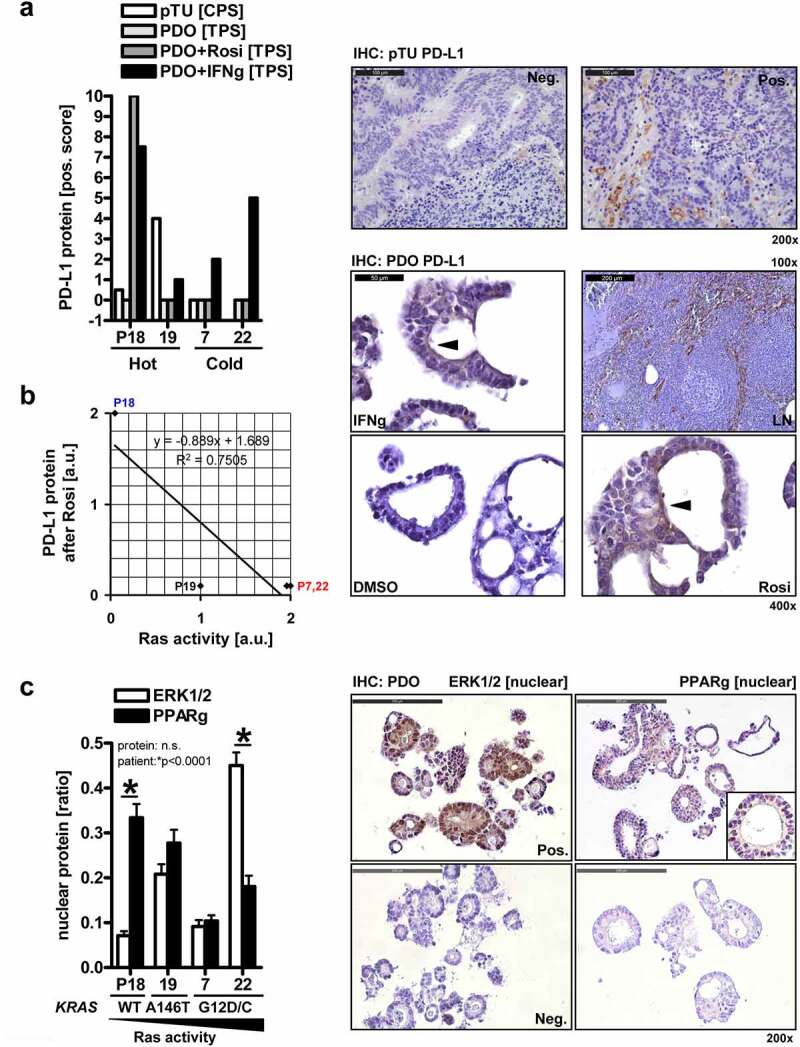
PD-L1 expression in PDO subsets and matched patients’ primary tumor tissue.A, Expression of PD-L1 protein in PDOs compared with the primary tumor tissue of the same patient. PDOs (n=4 MSS+ cases) were treated with vehicle (DMSO), rosi (10 µM) or IFNγ (100 ng/ml as pos. control) for 48 h, followed by processing for FFPE. Sections from PDOs and matched surgery resection material were stained with PD-L1 Ab by immunohistochemistry (IHC). Right: Representative pictures; original magnifications 200x. Left: Data are combined PD-L1 positivity scores derived from tumor/epithelial cells and infiltrated immune cells per patient. Legend: pTU = primary tumor tissue; TPS [%], IC [%], CPS = TPS+IC [a.u.]; “hot” = observable immune cell infiltration; “cold” = no immune cell infiltration; Y-axis: Value <0 = no material available; Value 0 = no staining recorded; B, Correlation plot. Organoids were treated as in A, and FFPE-sections were stained with PD-L1 Abs by IHC. Human lymph node tissue served as pos. control. Right: Representative pictures; original magnifications 100-400x. PD-L1 was in part localized to the cytoplasm. Note the membrane-accentuated (arrow: brown color) PD-L1 staining in this exemplary KRAS wt patient (P18). Left: Values (a.u.) for Ras activity according to the KRAS gene mutation status were aligned to the ability of individual PDOs to up-regulate membrane-bound PD-L1 protein upon treatment with rosi (Pearson correlation coefficient: n=4 patients: r -0.8, p=0.13; n=6 all patients: r -0.8, p=0.06, Table S5). Legend (x/y axis units): 0 = no; 1 = weak; 2 = high; C, Localisation of ERK1/2 and PPARγ in PDOs. FFPE-sections from B were stained by IHC. Right: Representative pictures; original magnifications 200x. Left: Quantitative analyses; White bars: In PDOs with mutant KRAS G12, ERK1/2 localized to the nucleus. Data are means ± S.E. of nEPI (nuclear ERK1/2 positivity index = ratio of nuclear positive cells / total cells); Black bars: High nuclear PPARγ positivity was evident in the KRAS wt PDO with low nuclear ERK1/2. Data are means ± S.E. of nPPI (nuclear PPARγ positivity index = ratio of nuclear positive cells / total cells) (*p<0.05, 2way-ANOVA with Bonferroni post-tests, n=4 cases; n≥3 fields per patient)
We then investigated treatment-induced changes of PD-L1 in the same PDOs. Organoids were treated as detailed in legend to Figure 3a, and FFPE-sections were stained as above. PD-L1 was again in part distributed to the cytoplasm and the plasma membrane. All untreated or vehicle-treated PDOs were PD-L1 negative (TPS = 0%) and, again, INFγ increased the TPS in all PDOs tested (4 of 4/100%), whereas rosi (at 10 µM) augmented staining only in the KRAS wt PDO of patient P18 (S8, Table S5).
To elucidate the underlying mechanisms of resistance in PDOs toward PPARγ-agonist-mediated PD-L1 upregulation, we resorted to the Ras pathway. This signaling cascade inactivates PPARγ by (i) ERK1/2-mediated phosphorylation of serine 84/112 (γ1/γ2) within its N-terminal transcriptional activation domain (AF1) and (ii) nuclear export and cytosolic retention by MEK1/2.37 We hypothesized that this post-translational mechanism also exists in PDOs. To this end, we aligned the KRAS gene mutation status of each PDO line32 with its ability to exhibit membrane-accentuated PD-L1 in situ expression in FFPE-sections after treatment with rosi (Figure 4b). Herein, PDOs with high Ras activity (KRAS mut) were less sensitive than those with weak Ras activity (KRAS wt) (Pearson correlation coefficient: n = 4 patients: r − 0.8, p = .13; n = 6 all patients: r − 0.8, p = .06, Table S5).
To test this further, we detected ERK1/2 in PDOs by monitoring their nuclear translocation as a surrogate for active kinase signaling. FFPE-sections (from Figure 4a) were stained with ERK1/2 Ab by IHC. ERK1/2 were localized to the nucleus in the PDO with the activating mutant KRAS G12C (P22), whereas the lowest level of nuclear ERK1/2 was found in the KRAS wt case (P18) (Figure 4c). Likewise, the highest nuclear accumulation of PPARγ, as an indicator for its transcriptional activity, was observed in the same KRAS wt PDO which had the lowest levels of active nuclear ERK1/2 (Figure 4c).
In sum, the KRAS mutation status in PDOs could be in part correlated with a reciprocal subcellular distribution of PPARγ and ERK1/2 (*p < .05, two-way ANOVA with Bonferroni posttests, n = 4 cases; n ≥ 3 fields per patient). A similar pattern was recorded upon subcellular fractionation (SCF) of PDOs from the same two patients, KRAS wt (P18) and KRAS G12C (P22) (*p < .05, two-way ANOVA with Bonferroni posttests, n = 2 patients, n = 3 replicates per patient) (S9). However, due to the low case numbers assessed, correlation of cellular phenotypes with individual gene mutations (e.g. Her2, PI3KCA) has to be handled with caution.
LAK adhere to, invade and reduce growth and viability of PDOs
The observed induction of PD-L1 by PPARγ-agonists allowed us to assess if the lack of response to PD1/PD-L1 blockage in MSS+ CRC can be alleviated by combination with PD-L1-inducing treatments. To test this hypothesis, we generated co-cultures of PDOs with lymphokine-activated killer cells (abbrev. LAK), a mixture of CD3+ CD16+ CD56+ CD8+ NK/T-like cells.40 PBMCs were isolated from healthy donors, and suspended lymphocytes stimulated with phytohemagglutinin (PHA at 10 µg/ml) and IL2 (100–1.000 IU/ml) for 48–72 h, followed by live FC analysis using fluorescence-labeled PD1 detection Ab. Consistent with general knowledge,40 LAK contained >80% CD8+ T-lymphocytes, and a subpopulation was PD1+ (>10%) at the cell surface, justifying the use of PD1/PD-L1 blocking Abs in this model system (not shown).
First, the localization of LAK was determined by fluorescence microscopy. PMBCs were isolated from healthy donors as above, and suspended lymphocytes were stimulated with IL2 for 24 h, labeled with Qtracker (red) and co-embedded in MatriGel® with intact PDOs (“spheroids”) at an effector:target ratio of 100:1, followed by live cell imaging after 1–3 d. LAK adhered to and partially invaded PDOs (Figure 5a). Again, patient-dependent phenotypes were recorded, as exemplified by the two KRAS mutant PDOs, P7 who allowed adhesion of LAK to a higher extent than P22 (n = 2 patients, n ≥ 2 images per patient & day). Herewith, we defined this differential behavior as “Responder to LAK” (abbrev. R) vs. “Non-Responder to LAK” (abbrev. NR) in the consecutive assays.
Figure 5.
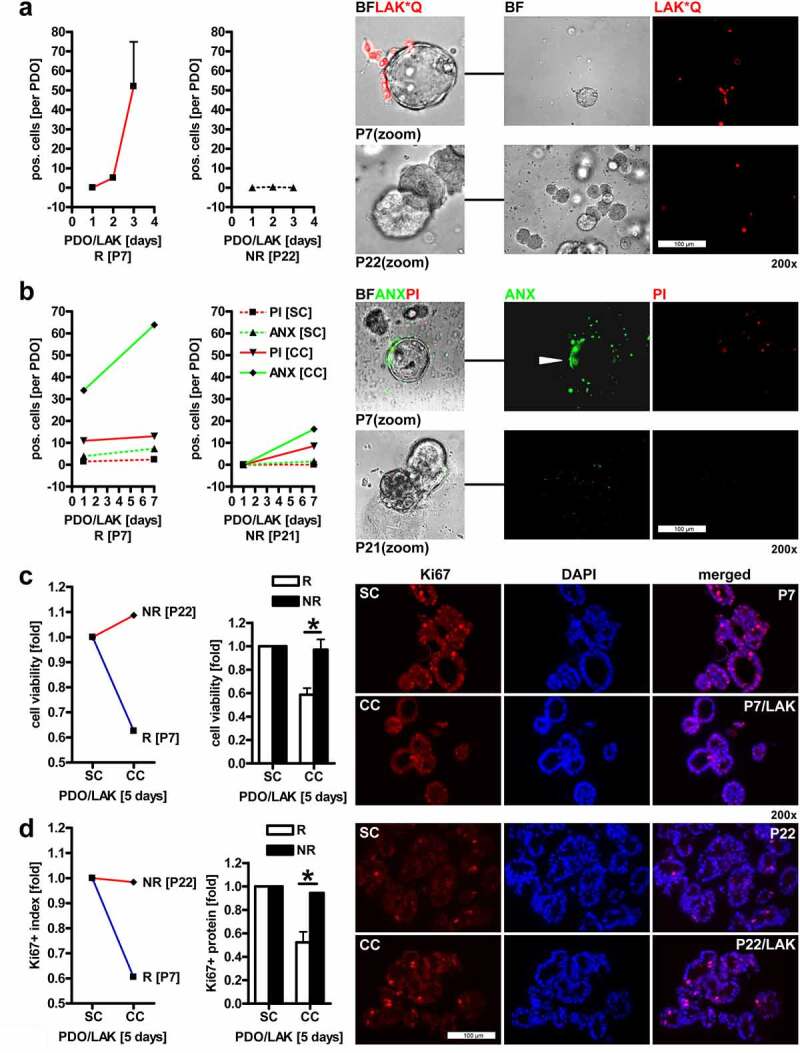
LAK adhere to, invade and reduce growth and viability of CRC PDOs.A, Localisation of LAK in co-cultures with PDOs. IL2-stimulated allogenic LAK (from healthy donors) were labelled with Qtracker (red, abbrev. “*Q”) and co-embedded in MatriGel® with intact PDOs at an effector:target ratio of 100:1, followed by live cell imaging after 1 to 3 days. Quantitative analysis (left) and representative pictures (right). Data are mean numbers of LAK per PDO (as defined by an intact spheroid) ± S.E. (n=2 patients, n≥2 images per patient & day). NR = “non-responder” PDO (P22) with few adherent LAK; R = “responder” PDO (P7) with many adherent LAK. Color code: red = LAK*Q (labelled); BF (bright field) = PDO. Original magnifications 200x; B, Detection of cell death in co-cultures of LAK with PDOs. Organoids were co-cultured as in A, and viability was detected after 1 to 7 days by annexin (ANX) / propidium iodide (PI) staining and fluorescence imaging. Tumor cells and LAK suffered from cell death. Quantitative analysis (left) and representative pictures (right). Data are mean numbers of PI+/ANX+ signals per cell (LAK) or spheroid (PDO) ± S.E. (n=2 patients, n≥2 images per patient & day). NR = “non-responder” PDO (here P21, no signals were recorded for P22) with few dead cells; R = “responder” PDO (P7) with many dead cells in co-cultures. Color code: red = PI (necrosis); green = ANX (apoptosis); BF (bright field) = PDO+LAK. Original magnifications 200x; C, Co-culture of PDOs with LAK reduces overall cell viability. Organoids were co-cultivated as in A, followed by colorimetric MTT assay after 5 days. O.D. values were calculated as -fold ± S.E. PDO+LAK co-culture (CC) vs. PDO single culture (SC) (*p<0.05 R vs. NR, 2way-ANOVA with Bonferroni post-tests, n=6 patients, n=3 replicates per patient). Representative (left panel) and all (right panel) patients are presented. NR = “non-responder” PDO (P22) with many viable cells; R = “responder” PDO (P7) with few viable cells after co-culture with LAK; D, Co-culture of PDOs with LAK reduces CRC cell proliferation. Organoids were co-cultured as in A for 5 days. FFPE-sections were stained with Ki67 Ab for immunofluorescence microscopy. Quantitative analysis (left) and representative pictures (right). The ratio of Ki67+ nuclei vs. total nuclei was calculated as -fold ± S.E. (*p<0.05 R vs. NR, 2way-ANOVA with Bonferroni post-tests, n=6 patients, n=3 replicates per patient). Representative (left) and all (right) patients are presented. NR = “non-responder” PDO (P22) with many proliferating cells; R = “responder” PDO (P7) with few proliferating cells after co-culture with LAK. Color code: red = Ki67 (Ab), blue = DAPI (nuclei). Original magnifications 200x
To detect cell death (Figure 5b), organoids were co-cultured with LAK (as in Figure 5a), and viability was visualized after 1–7 d by annexin (ANX)/propidium iodide (PI) staining and fluorescence imaging. Both, necrotic (PI+) and apoptotic (ANX+) cells were recorded. Note that also LAK suffered from cell death, not only tumor cells. Again, co-cultures with P7 displayed more dead cells (n = 2 patients, n ≥ 2 images per patient & day).
We then asked if co-culture of PDOs with LAK reduces overall cell viability (Figure 5c). Organoids were co-cultivated as in A, followed by colorimetric MTT assay after 5 d. O.D. values were compared between PDO+LAK co-culture (CC) vs. PDO single culture (SC). Cell viability was diminished by 20–50% after co-culture (*p < .05 R vs. NR, two-way ANOVA with Bonferroni posttests, n = 6 patients, n = 3 replicates per patient). Again, co-cultures with P7 were less viable than those with P22. FC allowed the distinction of dead (ANX+ 7AAD+) EpCAM+ PDO cells from CD45+ LAK in single-cell suspensions from Accutase™-dissociated 5-d MatriGel®-embedded co-cultures (not shown).
Moreover, co-cultures of PDOs with LAK also reduced the proliferation of CRC cells (Figure 5d). FFPE-sections were stained with Ki67 Ab for immunofluorescence microscopy. The ratio of Ki67+ nuclei vs. total nuclei was calculated yielding a reduction in cell growth by 30–65% upon co-culture (*p < .05 R vs. NR, two-way ANOVA with Bonferroni posttests, n = 6 patients, n = 3 replicates per patient). Again, co-cultures with P7 were less proliferative than those with P22. Other patients followed this dichotome pattern, partitioning into R (P13, P19) (S10) vs. NR (P21, P30) (not shown).
Conclusively, PDOs differed with regard to their adhesiveness to and growth inhibition by LAK, indicative of individual immunogenic properties for each patient.
PPARγ-agonist plus IFNγ sensitizes PDOs to PD-L1 Ab in co-cultures with LAK
In search of the underlying molecular players why certain PDOs are responder (R) or non-responder (NR) to LAK, we resorted to custom-made PCR arrays. To characterize selected immune receptor–ligand systems which mediate the cross-talk between LAK and tumor cells, expression profiling of 42 genes was conducted (Table S4). Two KRAS mutant PDOs, as again exemplified by P7 (R to LAK) and P22 (NR to LAK), were co-embedded with allogenic (from a healthy donor) or syngenic (autologous, from the matched CRC patient) LAK into MatriGel® and co-cultivated for 5 d in the presence or absence of rosi (10 µM). Total RNA was extracted from PDO+LAK co-cultures (CC) vs. PDO single cultures (SC), and Ct-values from RT-qPCRs (Table S6) were calculated as -fold change according to the ΔΔCt method for further bioinformatics analysis (S11).
Hierarchical clustering of the PCR data confirmed that P22 expressed less activatory (CPA), but more inhibitory (CPI) checkpoint molecules than P7 (S12). For example, non-classical MHC class I genes whose protein products mediate NK/T cell tolerance/anergy (such as HLA-G) were elevated, whereas mRNAs encoding classical MHC class I molecules (HLA-B/C) which are required for efficient recognition by CD8+ T-cells were reduced. Similar results were obtained for other immune receptor–ligands (Table S7). Thus, case-dependent expression of checkpoints beyond PD-L1 may explain the differential sensitivity of PDOs to LAK.
Moreover, PPARγ-agonist altered mRNAs for several immune checkpoints beyond PD-L1 in a patient-dependent manner (Table S8). In cocultures with LAK, rosi increased expression of activatory checkpoints (TNFRSF4/9/18) in P7 and decreased inhibitory checkpoints (e.g. NKG2A, PD1) in P22. However, future experiments have to validate those as potential novel PPARγ-target genes.
After collecting evidence that PDOs differ in their individual immunogenicity profiles, we asked if this feature is translated to differential recognition and attack by the host immune system in presence of PD1/PD-L1 blocking Abs. To test this vulnerability, organoids were co-cultured with LAK in MatriGel® for 5 d in presence or absence of rosi (10–100 µM) or IFNγ (100 ng/ml) supplemented with either the clinically-in-use blocking Abs, anti-PD1 [pembrolizumab, ADCC incompetent, IgG4] or anti-PD-L1 [atezolizumab, ADCC incompetent, IgG1 mutant (mut)], or an experimental anti-PD-L1 Ab [ADCC competent, IgG1 wild type (wt)] and compared with the respective isotype controls (all at 500 µg/ml). Thereafter, overall cell viability was measured by MTT assay: Rosi (at 100 µM) in combination with IFNγ (100 ng/ml) decreased the viability of co-cultures to ~50% compared with single cultures or each agent alone (*p < .05, Kruskal–Wallis test with Dunn posttests; n = 6 patients, n ≥ 2 passages per patient, n ≥ 2 healthy donors per patient) (Figure 6a). However, this treatment failed to augment the cytotoxic/static effect of ADCC incompetent PD1 blocking Ab (Figure 6b). Instead, the same drug combination further reduced cell viabilities to ~40% in presence of ADCC competent PD-L1wt Ab (Figure 6c), possibly supported by enhanced Ab-target engagement upon pharmacological PD-L1 upregulation along the 5 d of co-culture. Again, the clinically-in-use ADCC incompetent PD-L1 Ab was ineffective, indicative of additional cellular mechanisms strengthening the efficacy of epitope blocking Abs.
Figure 6.
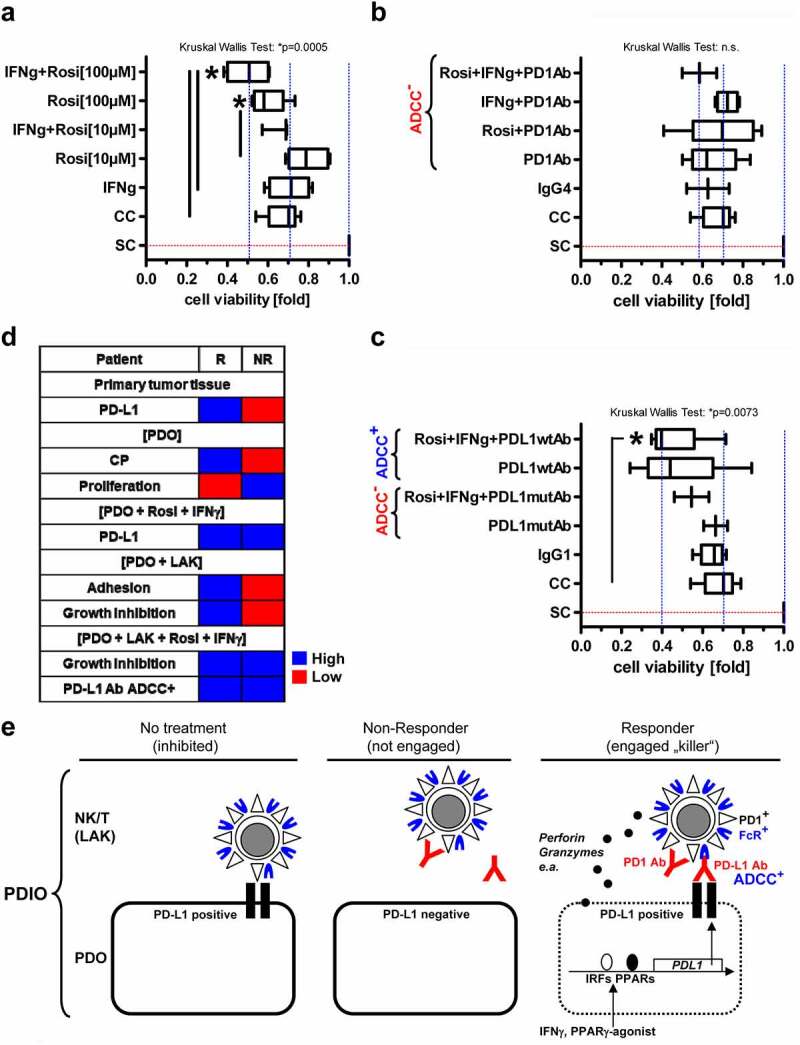
PPARγ-agonist plus IFNγ sensitizes PDOs to PD-L1 Ab in co-cultures with LAK.A-C, Organoids were co-cultured with LAK (like in Fig.5) for 5 days in presence or absence of (A) rosi (10 or 100 µM) or IFNγ (100 ng/ml) and (B) together with PD1 Ab (pembrolizumab, ADCC-incompetent IgG4) or (C) mutant (mut) PD-L1 Ab (atezolizumab, ADCC-incompetent IgG1) or wildtype (wt) PD-L1 Ab (ADCC+ competent IgG1) and compared with the respective isotype controls (all at 500 µg/ml). Cell viability was measured by MTT assay. O.D. values were calculated as -fold ± S.E. PDO+LAK co-culture (CC) vs. PDO single culture (SC) (*p<0.05, Kruskal Wallis test with Dunn post-tests; n=6 patients, n≥2 passages per patient, n≥2 healthy donors per patient); D, Alignment of characteristics determining response (R, exemplified by P7) vs. non-response (NR, exemplified by P22) of PDOs to LAK in patients with KRAS mutant CRC: R exhibited high levels of PD-L1 and other CPs, slow proliferation, good adhesiveness to and growth inhibition by LAK; NR low levels of PD-L1 and other CPs, rapid proliferation, poor adhesiveness to and no growth inhibition by LAK. Both, R and NR to LAK up-regulated PD-L1 upon exposure to PPARγ-agonist and displayed growth inhibition in presence of PD-L1 blocking Ab. Comparisons of primary tumor tissues and functional data from co-cultures are presented. Patient-wise information is listed in (Tables S2&5). Color code: blue = high; red = low expression/response; E, Proposed model of tumor-immune cell cross-talk in CRC. “Responder” PDOs (R, exemplified by P7) were sensitive, whereas “non-responder” (NR, exemplified by P22) PDOs resistant to NK/T-cell-mediated recognition, growth inhibition and cytotoxicity (“killing”), presumably due to differential expression of regulatory immune checkpoints and other immune-ligand-receptor systems as evinced from our experimental data (S7-14, Tables S2&5). We hypothesize that these highly individualized expression profiles may be exploited by precise tailoring of therapeutic blocking Abs and complemented by ADCC. Pharmacological up-regulation of PD-L1 on tumor cells by interferons or metabolic modifiers (as shown here for PPARγ ligands) may facilitate Ab-target engagement (as shown here for anti-PD-L1 Abs)
Stratification of patients’ CRCs according to their KRAS mutations (S13) confirmed that PDOs with weak Ras activity (KRASwt/A146T) were more sensitive to drug-mediated growth inhibition than PDOs with high Ras activity (KRASG12D/C). Nevertheless, all PDOs profited from PD-L1wt Ab treatment (Figure 6d). However, due to limited case numbers, larger prospective studies are necessary to corroborate these associations.
PPARγ-agonist fails to augment degranulation and cytotoxicity of NK cells toward PDOs
To focus on a professional subgroup of killer cells among the LAK, CD56+ NK cells from healthy donors were enriched by magnetic sorting (MACS). We first tested if PBMC-derived LAK contain functional NK cells (S14a). Allogenic isolated CD56+ NK cells were stimulated with IL2 or left untreated, followed by co-incubation (at an 1:1 effector:target ratio) with Accutase™-dissociated single tumor cells from PDOs or K562 target cells (as pos. control, not shown) for 4 h. Thereafter, degranulation of NK cells was measured by staining for CD107a+ (LAMP1+) in FC. The percentage of CD107a+ cells in dot plots was calculated (S14b). Alike K562 cells (not shown), PDOs robustly induced degranulation of IL2-stimulated NK cells (*p < .05, two-way ANOVA with Bonferroni posttests, n = 2 patients, n = 1–3 passages per patient with n = 2 donors per passage).
To confirm that PBMC-derived LAK contain cytotoxic NK cells, cells were co-cultured as above, and death protease release was measured by CytoTox-Fluor™ Cytotoxicity Assay (S14c). Fluorescence intensity in co-cultures was calculated compared with single PDO cultures (*p < .05, two-way ANOVA with Bonferroni posttests, n = 2 patients, n ≥ 2 passages per patient with n = 2 donors per passage). As for LAK, PDOs classified into high (R: P7) and low (NR: P22) stimulators of NK cell degranulation and cytotoxicity, underscoring the medical need for tailored immunotherapeutic strategies in individual MSS+ CRC patients.
We finally intended to see if PD-L1 blockage also boosts recognition of PDOs by NK cells as it did for LAK (S14b). Thus, allogenic CD56+ NK cells were generated, followed by co-incubation (at an 1:1 effector:target ratio) with Accutase™-dissociated single tumor cells from PDOs, which had been pre-treated for 48 h with rosi (10 µM) and IFNγ (100 ng/ml), or K562 target cells (not shown) for 4 h in presence or absence of anti-PD-L1 Ab. The percentage of CD107a+ NK cells was calculated.
As expected, blockage of PD-L1 on PDOs by ADCC competent PD-L1wt Ab augmented NK cell degranulation compared with untreated organoids (*p < .05, two-way ANOVA with Bonferroni posttests, n = 2 patients, n = 1–3 passages per patient with n = 2 donors per passage). However, the combination of IFNγ with rosi failed to further strengthen recognition of PDOs by NK cells compared with PD-L1wt Ab alone. This finding suggested that drug compounds act both in a patient- and immune cell-type dependent manner.
Conclusively, resistance and/or non-response of MSS+ PDOs to PD1/PD-L1 blockage may be overcome by harnessing of the PD-L1 antigen for improved Ab-target engagement and antitumoral efficacy in CD8 + T-and NK cells.
Discussion
Here, we demonstrated that PPARγ upregulates PD-L1 in MSS+ CRC cells and promotes the antitumoral activity of LAK (mainly CD8+ T-lymphocytes) together with IFNγ and ADCC competent PD-L1 blocking Ab. Modifiers of metabolic reprogramming including ligands for PPARs (α/γ) alter the sensitivity to immune checkpoint Abs in animal models and patients. Overall, metabolic dysfunction seems to be a hallmark of CD8+ T-cell exhaustion.41 For example, activation of PPARα by hypoglycemia or hypoxia enhances fatty acid oxidation in tumor-infiltrating CD8+ T-cells followed by increased checkpoint Ab efficacy and attenuation of tumor growth in mice.23,24,26 Mechanistically, bezafibrate, a bona fide agonist of PPARα/PPARγ-coactivator-1-alpha (PGC1α) transcription augments tumoricidal effects of PD1 blockade by boosting mitochondrial oxidative phosphorylation and proliferation, survival and effector functions of cytotoxic T-cells.
Likewise, PPARγ in adipose, tumor or immune cells is associated with an altered response to immunotherapy. Here, obesity exerts apparent paradoxical effect on T-cells.30 In general, obesity evokes low-grade inflammation, immune cell aging/exhaustion, tumor progression and PD1-driven T-cell dysfunction. In contrary, obesity also correlates with increased efficacy of PD1/PD-L1 blockade in mice and humans: A retrospective multicenter study of patients with advanced cancers treated with anti-PD1/PD-L1 Abs was conducted regarding clinical outcomes stratified for body mass index (BMI).29 Notably, overall response rate (ORR) and survival (OS/PFS) were higher in overweight/obese patients. Since, PPARγ is a master transcription factor for adipocyte differentiation,10 overweight may be regarded as a surrogate for PPARγ-activity and a tool to improve functions in adipose- and tumor-associated immune cells. Supporting this conjecture, the PD1/PD-L1 axis is altered in peripheral blood cells from individuals with type-2-diabetes-mellitus.42 Recent phase II/III clinical trials in lung cancer patients revealed that an elevated BMI is a favorable prognostic factor for overall survival (OS) and response to PD-L1 blockage (atezolizumab) in PD-L1+ tumors.43
In addition to cell-intrinsic mechanisms, para/autocrine factors determine outcomes of Ab or cell-based immunotherapies: As such, PPARγ in myeloid cells improves the preclinical response to GM-CSF-secreting-tumor-cell-vaccine.22 Instead, others suggested an unfavorable role for anabolic PPARγ (lipogenesis) vs. catabolic PPARα (fatty acid oxidation): In bladder cancer patients,44 β-catenin, PPARγ and FGFR3 are activated in non-T cell-inflamed (“cold”) tumors, and Wnt5a-β-catenin-PPARγ signaling fosters immune evasion in mouse melanoma.45 Adipocytes from visceral fat derived from obese individuals and CRC patients secrete ω6-polyunsaturated fatty acids to deliver suppressive signals to innate immune cells.46
In NK cells, PPARs confer lipotoxicity to limit antitumor responses in vivo.25 Obesity evokes lipid accumulation causing ”paralysis” of NK cell functions. Herein, PPARα/δ agonists and natural fatty acids mimic obesity, inhibit glycolysis and abolish delivery of cytotoxic factors from the NK/tumor cell synapse. Natural (15d-PGJ2) and synthetic (ciglitazone) PPARγ-agonists compromise IFNγ synthesis and cytotoxic activity of human/murine NK cells.47 Thus, metabolic reprogramming of innate and adaptive immune cells in the systemic or local tumor microenvironments via pharmacological targetable nuclear receptors of the PPAR/PGC1 families may be exploited to improve the efficacy of current clinically-in-use checkpoint Ab therapies.
Consistent with this evidence, we demonstrated that PPARγ binds to the proximal promoter of the human PDL1 gene followed by upregulation of PDL1 mRNA and PD-L1 protein expression. Accordingly, others recorded antagonistic effects of PPARs on the PDL1 promoter, i.e. upregulation by PPARγ vs. downregulation by PPARα. For example, PPARα suppressed PD-L1-driven immune escape in human hepatocellular carcinoma cells.48 In contrast, PPARγ-agonist (rosi) together with IL5-neutralizing Ab prevented chronic rejection of MHC class II-mismatched mouse xenografts31 by increasing PD-L1 on grafts and reducing CD8+ T cell and eosinophil infiltration.
PD-L1 expression rises during adipocyte differentiation.49 Thus, inhibition of adipogenesis by PPARγ-antagonists reduced PD-L1 in fat tissue, e.g. in mouse breast cancer. Likewise, PD1 is regulated by PPARγ in mice to govern host defense against infections and during allergic responses.50 As such, PPARγ-agonists increased, whereas antagonists decreased PD1 on innate lymphoid cells (ILC type 2).
Hence, PPARγ-mediated upregulation of PD-L1, as evident from our study, proposed an enhanced Ab-target engagement for PD-L1 blocking Ab in the synapse of LAK with tumor cells. Several formats of anti-PD-L1 Abs exist that are either competent in mediating ADCC or incompetent due to different isotypes and Fc portions. This concept was supported by our data from MSS+ CRC cases. PDOs were resistant to PD1/PD-L1 blockage by ADCC incompetent clinical Abs (pembrolizumab, atezolizumab), but sensitive to experimental ADCC competent anti-PD-L1 Ab. In addition to lack of ADCC, Ab efficacy may be obliterated by concomitant presence of alternative inhibitory immune checkpoints as exemplified by the TIGIT/PVR system.51 Namely, all tumor cell lines and PDOs had high levels of TIGIT receptors (PVR/CD155; PVRL2/CD112), whereas TIGIT blocking Ab reduced viability of PDOs (not shown).
Further, PD-L1 induction and engagement may fail in KRAS-mutated PDOs due to post-translational inactivation of PPARγ.37 PDOs expressed similar levels of total PPARγ mRNA, as evinced by cDNA array transcription profiling,32 and protein in our hands (not shown). In general, ERK1/2 and PPARγ are more active when localized to the nucleus, whereas inactive when located in the cytoplasm (e.g. ERK1/2 when un-phosphorylated and PPARγ when bound to MEK1/2).37 Consistently, only the wt KRAS PDO had considerable amounts of nuclear PPARγ and was responsive to PPARγ-ligand. In contrast, KRAS mutant PDOs with constitutively active Ras-MEK1/2-ERK1/2 signaling retained PPARγ in the cytoplasm attenuating their ability to activate transcription.
Moreover, tumor heterogeneity and frequent polymorphisms in the PPARG gene locus (e.g. P12A, P115G)52 may alter sensitivity of individual PDOs to ligands, including mutations in distinct PPARγ domains and phosphorylation epitopes.53 As such, high-Ras activity cases may include a higher percentage of PPARγ loss-of-function cell clones. Consistent with PPARγ-inhibition by the Ras-MEK1/2-ERK1/2 pathway,37 KRAS wt PDOs responded better to all treatments (LAK, drugs or Abs) than PDOs with constitutively activating KRAS mutations (S13). However, small case numbers limit this conclusion. Larger prospective patient studies have to corroborate these associations.
Nevertheless, the question remains whether it is clinically desirable to induce PD-L1 on tumor cells. High PD-L1 expression in patients’ tissues per se may be negative prognostic, marking immune exhaustion and/or anergy, but positive predictive regarding PD1/PD-L1 Ab engagement at the tumor-immune cell synapse.2,3 Hence, PD-L1+ MSI+ solid tumors independently of tissue origin have been approved for checkpoint blockage immunotherapy.1,4 In analogy, overexpression of the oncogene Her2 is unfavorable regarding overall survival (OS), but allows efficient Ab-target binding (avidity) in Her2+ patient subgroups, such as in breast and gastric cancer, who then profit from Her2 blocking Abs.
This paradox integrates into the concept to convert a “cold” non-immunogenic MSS+ tumor microenvironment with low immune infiltration into a “hot” inflamed, PD-L1+, MSI+-like environment eligible for checkpoint Ab therapy. As such, PD-L1 positivity in tumor-infiltrating immune cells of MSS+ CRC improved prognosis,54 and patients with MSS+ tumors and high tumor mutational burden under PD1 Ab therapy displayed prolonged progression-free survival (PFS).55 Overall, a high “immunoscore” in CRC seems to be a better predictor of patient survival than the MSI+ status.56,57 Consequently, radiation, chemotherapy or targeted drugs which boost immunogenicity of MSS+ cancers are an emerging theme. In this context, PD-L1 induction by interferons is a promising strategy,58 as evinced by pegylated IFNα with PD1 Ab (pembrolizumab) in a phase Ib/II study in advanced melanoma.59 Likewise, MEK1/2 inhibitor cobimetinib received attention, however, was not confirmed in phase I/Ib and III clinical studies combined with PD-L1 Ab (atezolizumab) (NCT01988896;60 IMblaze37061). Conclusively, high PD-L1 on tumor cells and low PD-L1 on immune cells including antigen-presenting cells (e.g. dendritic cells, myeloid-derived suppressor cells, macrophages) may be optimal to achieve tumor recognition by and antitumor cytotoxic effects of immune cells. Nevertheless, how to reach this critical balance remains to be elaborated in future studies including innate immune cells. Severe immune-related adverse events (IRAE) limit clinical use of current checkpoint blockers, and dose reduction by combinations is warranted.62 In this context, anti-diabetic drugs may be beneficial by counteracting auto-immunity and preserving organ function (e.g. β-cells pancreas) and may complement steroids exerting anti-inflammatory effects.11
As a perspective, alternative immune checkpoints beyond PD1/PD-L1 were identified in our PCR arrays, which may be addressed in future clinical applications. Differential a priori expression profiles of immune receptor–ligand systems correlated with different sensitivities of individual PDOs toward recognition, growth inhibition and killing by LAK, a mixture of T/NK cells, personalized features of immunogenicity which may be translated to individualized immunotherapies. Of note, PDOs with few immune checkpoints, poor adhesiveness and rapid growth were less efficiently recognized and killed by LAK than those with many immune checkpoints, good adhesiveness and slow growth (Figure 6d).
Hence, individual immunogenicity profiles of patients’ PDOs (Tables S5 and S7) could allow stratification of single cases in “responder” (R) vs. “non-responder” (NR) to LAK, and immunogenicity-enhancing drugs may help to convert NR toward R (Figure 6e). As exemplified in our study, resistance/non-response to clinical PD1/PD-L1 Abs was relieved by an experimental ADCC competent PD-L1 Ab in combination with PPARγ-agonist plus IFNγ in all PDOs tested. Both “NR and R to LAK” cases profited from drug-mediated upregulation of PD-L1 (and other immune genes) and antitumoral effects (Table S8).
However, the effects of PPARγ agonists cannot be attributed to PD-L1 upregulation alone, instead a plethora of target genes contributes to growth inhibition complemented by non-genomic receptor-independent effects (e.g. on mitochondria). Overall, pio was the weaker and more transiently active agonist than rosi in our assays. This may be due to different pharmacological stability of the agent in the medium, cells or MatriGel®.32 We also used high concentrations of glitazones (µM) to guarantee diffusion and permeability in 3D matrices; thus, translatability of the observed in vitro data is warranted. In other words, the pleiotropic targets of transcription factors driven by interferons and PPAR ligands, affecting the transcriptomes of both immune and epithelial cells (e.g. cell cycle regulator P21), shall culminate in a common outcome, i.e. reduced viability and/or proliferation of tumor stem cells.11 Since morphological and functional phenotypes of PDOs and LAK are mutually influenced in a bidirectional cross-talk in the cocultures and the real-life tumor microenvironment, future in-depth dissection of cell type-specific PPAR-dependent vs. ligand-mediated mechanisms are necessary.
Unexpectedly, rosi added on the efficacy of ADCC competent PD-L1 Ab only in co-cultures with LAK but not with purified NK cells. This discrepancy could be explained by PPARγ-mediated lipotoxicity evoking NK cell dysfunction.25 Since our LAK cells contained mainly CD8+ cytotoxic T-cells, one may conclude that rosi triggers antitumoral effects predominantly in the cross-talk of PDOs with this lymphoid subset. Since clinical ADCC incompetent PD1/PD-L1 Abs (pembrolizumab, atezolizumab) were ineffective against PDOs in all settings tested, the role of the Fc portion of these therapeutics could be reconsidered despite safety concerns.
In summary, metabolic reprogramming of innate or adaptive immune cells by repurposing of approved anti-diabetic drugs and biologicals (like interferons and metabolic modifiers such as metformin or PPAR-ligands) may reinforce Ab-target engagement by induction of PD-L1 (and other checkpoints), reduce effective doses of therapeutic Abs and thereby prevent adverse effects, lower costs and envision eligibility for non-MSI+ patients (see model in Figure 6e).
Supplementary Material
Funding Statement
EB received funding from German Cancer Aid (Deutsche Krebshilfe 108287, 111086), German Research Foundation (Deutsche Forschungsgemeinschaft DFG, Bu2285) and German Cancer Research Center (Deutsches Krebsforschungszentrum DKFZ-MOST, Ca158). LH, JR, TS and VH were supported by MD fellowships; JB, MEc, PW and TGu by the “Translational Physician Scientist” (TraPS) program; WW by the “Translational Medical Research” (TMR) master program (all from the Medical Faculty Mannheim, University Heidelberg); TZ by the Clinician Scientist program “Interfaces and Interventions in complex chronic conditions” (ICON) of the DFG. BL received an MD fellowship from the Chinese Scholarship Council (CSC). ID held funding from the MD/PhD Masterprogram (University of Strasbourg and Universite Descartes Paris). AC, MB and ME were supported by a grant provided by the MERCK Heidelberg Innovation Call (Darmstadt, Germany). AC received funding from DFG [SFB1366 (394046768-SFB1366; C2 to AC); TRR179 (TP07 to AC); SFB-TRR156 (B10N to AC); RTG2099 (259332240-RTG2099; P9 to AC)], Baden-Württemberg Foundation special program “Angioformatics Single Cell Platform” and a network grant of the European Commission (H2020-MSCA-MC-ITN-765104-NATURE-NK)
| Abbreviations | |
| 7AAD | 7-Aminoactinomycin D |
| Ab | Antibody |
| ACO | Acyl-CoA oxidase |
| ADCC | Antibody-dependent cellular cytotoxicity |
| APC | Antigen-presenting cells |
| CC | Co-culture |
| CD | Cluster of differentiation |
| ChIP | Chromatin-immunoprecipitation |
| CPA/I | Activatory/inhibitory (immune) checkpoint |
| CPS | Combined positivity score |
| CRC | Colorectal cancer |
| EpCAM | Epithelial cell adhesion molecule |
| ERK | Extracellular signal-regulated kinase |
| Fc | Fragment crystallizable (Ab isotype) |
| FC | Flow cytometry |
| FFPE | Formalin-fixed (agarose) paraffin-embedded |
| GW | GW9662 |
| H&E | Hematoxylin and eosin |
| IC | Inflammatory cell (score) |
| IF | Immunofluorescence |
| IFN | Interferon |
| IgG | Immunoglobulin G |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| IRF | Interferon regulatory factor |
| ITC | Isotype control |
| LAK | Lymphokine-activated killer cells |
| LNO2 | 10-Nitrolinoleate |
| MEK | Mitogen-activated protein kinase kinase |
| MSI/MSS | Microsatellite instable/stable |
| NK | Natural killer |
| OD | Optical density |
| PBMCs | Peripheral blood mononuclear cells |
| PD1 | Programmed cell death 1 (PDCD1/CD279) |
| PD-L1 | Programmed cell death 1 ligand 1 (PDCD1LG1/CD274) |
| PDO | Patient-derived (tumor) organoid |
| PDIO | Patient-derived immune organoid |
| pio | Pioglitazone |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PPRE | PPARγ-responsive (DNA binding) element |
| R/NR | Responder/non-responder (to LAK) |
| RAS | Rous sarcoma oncogene |
| rosi | Rosiglitazone |
| SC | Single culture |
| TPS | Tumor proportion score |
| WB | Western blot |
| WT | Wild type |
Author contributions
All authors cooperated and contributed to, critically reviewed and approved the manuscript. EB wrote the paper. AC, EB and ME defined the research theme. BL, EB, FH, ID, JB, JP, JR, KK, LH, MEc, PW, TGu, TS, VH, WW designed methods and carried out the experiments. EB, JP, KK, PW and TGu analyzed the data and interpreted the results. TGa gave patient samples, performed and analysed immunohistochemistry stainings. ME, JB, SB and TZ collected biopsies and conducted clinical studies. CS, JB, TZ and MB provided bioinformatics and sequencing data.
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
Transcript profiling
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE117548.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Guinney J, Dienstmann R, Wang X, De Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–16. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018. Oct 12;362(6411):eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed]
- 3.Becht E, De Reynies A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautès-Fridman C, Laurent-Puig P, Fridman WH, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22(16):4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 4.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuveson D, Clevers H.. Cancer modeling meets human organoid technology. Science. 2019;364(6444):952–955. doi: 10.1126/science.aaw6985. [DOI] [PubMed] [Google Scholar]
- 6.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, Van De Haar J, Fanchi LF, Slagter M, Van Der Velden DL, Kaing S, Kelderman S, et al. Generation of Tumor-Reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174(6):1586–1598 e12. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou S-H, Salahudeen AA, Smith AR, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972–1988 e16. doi: 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359(6378):920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutting T, Burgermeister E, Hartel N, Ebert MP. Checkpoints and beyond - Immunotherapy in colorectal cancer. Semin Cancer Biol. 2018;55:78–89. doi: 10.1016/j.semcancer.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13(1):36–49. doi: 10.1038/nrendo.2016.135. [DOI] [PubMed] [Google Scholar]
- 11.Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181–195. doi: 10.1038/nrc3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollenberg AN. Metabolic health and nuclear-receptor sensitivity. N Engl J Med. 2012;366(14):1345–1347. doi: 10.1056/NEJMcibr1114529. [DOI] [PubMed] [Google Scholar]
- 13.Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol. 2018;15:721–737. doi: 10.1038/s41575-018-0054-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, Mathis D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486(7404):549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, Beressi J-P, Verhoeyen E, Raggueneau V, Maneglier B, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature. 2015;525:380–383. doi: 10.1038/nature15248. [DOI] [PubMed] [Google Scholar]
- 16.Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR-gamma signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med. 2018;24(3):360–367. doi: 10.1038/nm.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z, Nakanishi M, Porras DP, Almakadi M, Campbell CJV, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. 2017;19(11):1336–1347. doi: 10.1038/ncb3625. [DOI] [PubMed] [Google Scholar]
- 18.Korpal M, Puyang X, Jeremy WZ, Seiler R, Furman C, Oo HZ, Seiler M, Irwin S, Subramanian V, Julie Joshi J, et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat Commun. 2017;8(1):103. doi: 10.1038/s41467-017-00147-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen B, Wang Q, Zhao X, Chen J, Cheng N, et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat Commun. 2017;8:766. doi: 10.1038/s41467-017-00523-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, Raes G, De Baetselier P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. 2006;108:525–535. doi: 10.1182/blood-2005-09-3777. [DOI] [PubMed] [Google Scholar]
- 21.Wu L, Yan C, Czader M, Foreman O, Blum JS, Kapur R, Du H. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119:115–126. doi: 10.1182/blood-2011-06-363093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goyal G, Wong K, Nirschl CJ, Souders N, Neuberg D, Anandasabapathy N, Dranoff G. PPARgamma contributes to immunity induced by cancer cell vaccines that secrete GM-CSF. Cancer Immunol Res. 2018;6:723–732. doi: 10.1158/2326-6066.CIR-17-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32(3):377–391 e9. doi: 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates Anti–PD-1 therapy. Cancer Immunol Res. 2018;6(11):1375–1387. doi: 10.1158/2326-6066.CIR-18-0095. [DOI] [PubMed] [Google Scholar]
- 25.Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19(12):1330–1340. doi: 10.1038/s41590-018-0251-7. [DOI] [PubMed] [Google Scholar]
- 26.Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A. 2017;114(5):E761–E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215(4):1091–1100. doi: 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harel M, Ortenberg R, Varanasi SK, Mangalhara KC, Mardamshina M, Markovits E, Baruch EN, Tripple V, Arama-Chayoth M, Greenberg E, et al. Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. Cell. 2019;179(1):236–250 e18. doi: 10.1016/j.cell.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cortellini A, Bersanelli M, Buti S, Cannita K, Santini D, Perrone F, Giusti R, Tiseo M, Michiara M, Di Marino P, et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: when overweight becomes favorable. J Immunother Cancer. 2019;7(1):57. doi: 10.1186/s40425-019-0527-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, Mirsoian A, Minnar CM, Stoffel KM, Sturgill IR, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25(1):141–151. doi: 10.1038/s41591-018-0221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Li D, Tsang JY, Niu N, Peng J, Zhu J, Hui K, Xu A, Lui VCH, Lamb JR, et al. PPAR-γ signaling and IL-5 inhibition together prevent chronic rejection of MHC Class II–mismatched cardiac grafts. J Heart Lung Transplant. 2011;30(6):698–706. doi: 10.1016/j.healun.2011.01.704. [DOI] [PubMed] [Google Scholar]
- 32.Betge JRN, Sauer J, Rauscher B, Dingert C, Gaitantzi H, Herweck F, Miersch T, Valentini E, Hauber V, Gutting T, et al. Multiparametric phenotyping of compound effects on patient derived organoids. Preprint. Cold Spring Harbor Laboratories . 2019 Jun 07; https://www.biorxiv.org/content/10.1101/660993v1; doi: 10.1101/660993. [DOI] [Google Scholar]
- 33.Zhan T, Ambrosi G, Wandmacher AM, Rauscher B, Betge J, Rindtorff N, Häussler RS, Hinsenkamp I, Bamberg L, Hessling B, et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat Commun. 2019;10(1):2197. doi: 10.1038/s41467-019-09898-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017;19(6):1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duldulao MP, Lee W, Le M, Chen Z, Li W, Wang J, Gao H, Li H, Kim J, Garcia-Aguilar J, et al. Gene expression variations in microsatellite stable and unstable colon cancer cells. J Surg Res. 2012;174(1):1–6. doi: 10.1016/j.jss.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgermeister E, Friedrich T, Hitkova I, Regel I, Einwachter H, Zimmermann W, Rocken C, Perren A, Wright MB, Schmid RM, et al. The Ras inhibitors caveolin-1 and docking protein 1 activate peroxisome proliferator-activated receptor through spatial relocalization at helix 7 of its ligand-binding domain. Mol Cell Biol. 2011;31(16):3497–3510. doi: 10.1128/MCB.01421-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burgermeister E, Seger R. PPARγ and MEK interactions in cancer. PPAR Res. 2008;2008:309469. doi: 10.1155/2008/309469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee HH, Wang YN, Xia W, Chen C-H, Rau K-M, Ye L, Wei Y, Chou C-K, Wang S-C, Yan M, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36(2):168–178 e4. doi: 10.1016/j.ccell.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schildhaus HU. [Predictive value of PD-L1 diagnostics]. Pathologe. 2018;39:498–519. doi: 10.1007/s00292-018-0507-x. [DOI] [PubMed] [Google Scholar]
- 40.Linn YC, Hui KM. Cytokine-induced killer cells: NK-like T cells with cytotolytic specificity against leukemia. Leuk Lymphoma. 2003;44(9):1457–1462. doi: 10.3109/10428190309178764. [DOI] [PubMed] [Google Scholar]
- 41.Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8 + T cell exhaustion. Immunity. 2016;45(2):358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi B, Du X, Wang Q, Chen Y, Zhang X. Increased PD-1 on CD4(+)CD28(-) T cell and soluble PD-1 ligand-1 in patients with T2DM: association with atherosclerotic macrovascular diseases. Metabolism. 2013;62(6):778–785. doi: 10.1016/j.metabol.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 43.Kichenadasse G, Miners JO, Mangoni AA, et al. Association between body mass index and overall survival with immune checkpoint inhibitor therapy for advanced non-small cell lung cancer. JAMA Oncol. 2020 Apr 1;6(4):512–518.. doi: 10.1001/jamaoncol.2019.5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF. Molecular drivers of the Non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol Res. 2016;4(7):563–568. doi: 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, Liu J, Liu X, Boczkowski D, Nair S, et al. Paracrine Wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48:147–160 e7. doi: 10.1016/j.immuni.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Corno M, D’Archivio M, Conti L, Scazzocchio B, Varì R, Donninelli G, Varano B, Giammarioli S, De Meo S, Silecchia G, et al. Visceral fat adipocytes from obese and colorectal cancer subjects exhibit distinct secretory and ω6 polyunsaturated fatty acid profiles and deliver immunosuppressive signals to innate immunity cells. Oncotarget. 2016;7(39):63093–63105. doi: 10.18632/oncotarget.10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Rodriguez-Galan MC, Subleski JJ, Ortaldo JR, Hodge DL, Wang J-M, Shimozato O, Reynolds DA, Young HA. Peroxisome proliferator-activated receptor-gamma and its ligands attenuate biologic functions of human natural killer cells. Blood. 2004;104:3276–3284. doi: 10.1182/blood-2004-02-0664. [DOI] [PubMed] [Google Scholar]
- 48.Wang G, Liu Y, Huang J, Liu J, Wang J, Yang J. PPARalpha suppresses PD-L1-mediated immune escape by down-regulating spp1 in human hepatocellular carcinoma. Cancer Res Treat. 2019. doi: 10.4143/crt.2019.111. [DOI] [PubMed] [Google Scholar]
- 49.Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, Chiang H-C, Zhang X, Zhang C, Zhang D, et al. Adipose PD-L1 Modulates PD-1/PD-L1 checkpoint blockade immunotherapy efficacy in breast cancer. Oncoimmunology. 2018;7(11):e1500107. doi: 10.1080/2162402X.2018.1500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Batyrova B, Luwaert F, Maravelia P, Miyabayashi Y, Vashist N, Stark JM, Soori SY, Tibbitt CA, Riese P, Coquet JM, et al. PD-1 expression affects cytokine production by ILC2 and is influenced by peroxisome proliferator-activated receptor-gamma. Immun Inflamm Dis. 2019;8(1):8–23. doi: 10.1002/iid3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. 2017;276(1):112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 52.Liang X, Fan X, Tan K, Zhang L, Jian L, Yu L. Peroxisome proliferators-activated receptor gamma polymorphisms and colorectal cancer risk. J Cancer Res Ther. 2018;14(9):S306–S310. doi: 10.4103/0973-1482.235346. [DOI] [PubMed] [Google Scholar]
- 53.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, De La Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3(6):799–804. doi: 10.1016/S1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 54.Lee KS, Kwak Y, Ahn S, Shin E, Oh H-K, Kim D-W, Kang S-B, Choe G, Kim WH, Lee HS, et al. Prognostic implication of CD274 (PD-L1) protein expression in tumor-infiltrating immune cells for microsatellite unstable and stable colorectal cancer. Cancer Immunol Immunother. 2017;66(7):927–939. doi: 10.1007/s00262-017-1999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7(10):1570–1573. doi: 10.1158/2326-6066.CIR-19-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kikuchi T, Mimura K, Okayama H, Nakayama Y, Saito K, Yamada L, Endo E, Sakamoto W, Fujita S, Endo H, et al. A subset of patients with MSS/MSI-low-colorectal cancer showed increased CD8(+) TILs together with up-regulated IFN-γ. Oncol Lett. 2019;18(6):5977–5985. doi: 10.3892/ol.2019.10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44(3):698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 58.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davar D, Wang H, Chauvin JM, Pagliano O, Fourcade JJ, Ka M, Menna C, Rose A, Sander C, Borhani AA, et al. Phase Ib/II study of pembrolizumab and pegylated-interferon Alfa-2b in advanced melanoma. J Clin Oncol. pp.JCO1800632. 2018. doi: 10.1200/JCO.18.00632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hellmann MD, Kim T-W, Lee CB, Goh B-C, Miller WH, Oh D-Y, Jamal R, Chee C-E, Chow LQM, Gainor JF, et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol. 2019;30(7):1134–1142. doi: 10.1093/annonc/mdz113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, Falcone A, Fakih M, Kozloff M, Segal NH, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20(6):849–861. doi: 10.1016/S1470-2045(19)30027-0. [DOI] [PubMed] [Google Scholar]
- 62.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.