

Abstract
Purpose:
To explore the effects of pelareorep on autophagy in multiple models of CRC, including patient derived PBMC.
Experimental Design:
HCT116 (KRAS-Mut) and Hke3 (KRAS-WT) cells were treated with pelareorep (5MOI) and harvested at 6 and 9 hours. LC3A/B expression was determined by immunofluorescence and flow cytometry; five autophagic proteins were analyzed by western blot (WB). The expression of 88 autophagy-genes were determined by qPCR.
Syngeneic mouse models; CT26/Balb-C (KRAS-mut) and MC38/C57B6 (KRAS-WT) were developed; and treated with pelareorep (10x106 PFU/day) intraperitoneally. Protein and RNA were extracted from harvested tumor tissues. PBMC from 5 experimental and 3 control patients were sampled at 0 (pre) and 48 hours, days 8 and 15. The gene expression normalized to “Pre” was determined using 2−ΔΔCT method.
Results:
Pelareorep induced significant upregulation of LC3A/B in HCT116 as compared to Hke3 cells by immunofluorescence (3.24X and 8.67X); flow cytometry (2.37X and 2.58X); autophagosome formation (2.02X and 1.57X), at 6 and 9 hours, respectively; all p<0.05. WB analysis showed increase in LC3A/B (2.38X and 6.82X), Beclin1 (1.17X and 1.24X) at 6 and 9 hours; ATG5 (2.4X), P-62 (1.52X) at 6 hours; and VPS-34 (1.39X) at 9 hours (all p<0.05). Induction of 13 transcripts in cell lines (>4X; 6 and 9 hours; p<0.05), 12 transcripts in CT26 (qPCR), and 14 transcripts in human PBMC (p<0.05) was observed. LC3A/B, RICTOR and RASD1 expression was upregulated in all 3 model systems.
Conclusion:
Pelareorep hijacks host autophagic machinery in KRAS-Mutant conditions to augment its propagation and preferential oncolysis of the cancer cells.
Keywords: Autophagy, KRAS, colorectal cancer, pelareorep, LC3
INTRODUCTION
Therapeutic assessment of oncolytic reovirus (pelareorep) has been in progress for over a decade. Pelareorep is a non-enveloped dsRNA virus that selectively lyses KRAS-mutated colorectal tumors cells. About 45% of colorectal cancer (CRC) patients harbor KRAS mutation in their tumors and have limited treatment options [1, 2]. At present, there are no therapeutic agents directly targeting the KRAS pathway partially due to the complexity of network and multitude of downstream collateral pathways. Thus, finding adjuncts to the current treatment arsenal is challenging, yet vital. We have previously demonstrated that KRAS mutation promotes pelareorep propagation in human CRC cell lines [3]. However, the detailed molecular pathways involved in this process are yet to be fully deciphered. Our studies indicate that pelareorep induces apoptosis and increased cell death in KRAS-mutated CRC, and the infection augments expression of apoptotic biomarkers such as Caspase 3 and PARP1 [4]. To further understand the mechanism of cell death induced by pelareorep, we evaluated the contribution of autophagic machinery by analyzing the process of infection and propagation of reovirus in in vitro and in vivo models.
Autophagy is a homeostatic mechanism for intracellular recycling and regulation. It plays a vital role in the removal of damaged proteins and organelles, thus limiting cellular damage facilitating sustained viability under adverse conditions [5]. Abnormalities in autophagic machinery are observed in several diseases, including many forms of cancer [6]. It is the dual role of autophagy - cell savior under some, and promotion of cell death in other conditions - that makes it an explorative tool for cancer treatments [7].
Autophagy also serves as a powerful tool that a cell utilizes to defend itself from viral infection [8]. There has been increasing evidence that viruses have acquired the potent ability to takeover and subvert autophagy for their life cycle and cell lysis[8, 9]. Autophagic machinery is influenced by multiple cellular pathways and its close interplay with apoptosis is complex [10]. Many stimuli that ultimately cause cell death also trigger autophagy. In such cases, autophagy is usually manifested well before apoptosis dismantles the cell.
It has been reported that under special circumstances autophagy related proteins help to induce apoptosis particularly when the cytoplasm and its components are degraded excessively [10]. Autophagic induction can either relieve the cell from stress caused by viral infection by degrading waste contents via its pro-survival mechanism, or it can also promote a type of programmed cell death to prevent propagation of infected cells [11]. Due to a higher proliferation rate, cancer cells induce the autophagic machinery more than normal cells as their survival mechanism.[12] The role of autophagy in cancer is fairly complex since it appears to be tumor-suppressive in the initial stages of cancer evolution, but has also been implicated in tumor progression in the later stages.[13, 14].
KRAS activation is known to facilitate pelareorep propagation [4] but hinder autophagy via activating mTOR and other downstream pathways [15]. KRAS mutation and its correlation to autophagic machinery is complex with scientific data indicating both induction and inhibition of autophagic machinery[5, 15-17]. The inter-relation between KRAS activation, pelareorep propagation and their influence on autophagic machinery is the arena we explored in order to increase the efficacy of pelareorep as a therapeutic tool for KRAS mutated CRC [18, 19]. We have employed a variety of model systems, to incorporate various combinations of the KRAS mutational status and pelareorep exposure, to conclusively prove the potentiating effect on autophagy in pelareorep mediated cell death. By utilizing various tools of genomics and proteomics, we have traced out the contribution of autophagic proteins towards pelareorep propagation.
METHODS
REOVIRUS (pelareorep)
Pelareorep (reovirus type 3 Dearing strain) was provided by Oncolytics Biotech Inc. (Calgary, Canada). It was stored long term at −80° C and at +4° C for 4 weeks when in use.
Dosage
Mouse Tumor:
The experimental group of mice were treated with pelareorep intratumorally (i.t.) at a daily dose of 10 million TCID50 in 100 uL PBS throughout the study till the end point was reached.
Patient Samples:
Pelareorep was administered as a 60-minute infusion for 5 consecutive days every 28 days, at a tissue culture infective dose (TCID50) of 3x1010/day.
Cell Culture and Treatment
Two CRC cell lines, HCT116 (KRAS mut) and its isogenic Hke3 (KRAS WT ), were obtained from ATCC and from Prof. Lidija Klampfer, respectively [4]. All cell lines were (authenticated at Genomic Core facility, Albert Einstein College of Medicine on 6/27/2018 by STR profiling and mycoplasma cleared) cultured in MEM, 10% FBS, 2mM L-Glutamine and 1% Pentstrep. Half to 2 X 106 cells (depending on the experiment) were treated with pelareorep at 5MOI (multiplicity of infection). Simultaneously, the same number of cells were cultured without pelareorep as controls. Cell were harvested at 6 or 9 hours.
Syngeneic in vivo models and allograft studies
Male and female BALB/c and C57BL/6 mice (6–8 weeks; Envigo Research Models, Inc) were used in accordance with protocols approved by Albert Einstein College of Medicine’s Institutional Animal Care and Use Committee (IACUC). Two transplantable (syngeneic) models of murine tumor cell lines, CT26 (BALB/C-derived colon adenocarcinoma cells; microsatellite stable and KRAS mutation at codon 12) and MC38 (C57BL6-derived colon adenocarcinoma cells; microsatellite unstable and KRAS WT) were received as kind gift from Dr. Xingxing Zang (Albert Einstein College of Medicine). CT26 was maintained in RPMI 1640 and MC38 in DMEM complete media. BALB/c and C57BL/6 mice were inoculated by subcutaneous injection of CT26 and MC38 (5×105 cells per mouse), respectively, suspended in 100uL of calcium- and magnesium-free PBS (1x PBS, pH 7.4) on the flank region. When the tumors had grown to approximately 100 mm3, mice were randomly divided into two groups (5 animals in each group) and treated with either pelareorep intratumorally (i.t.) at a daily dose of 10 million TCID50 in 100 uL PBS (pelareorep group) or 100 uL of PBS daily i.t. (control group). Tumor volume was measured every three days using calipers and calculated as follows: volume = longest tumor diameter × (shortest tumor diameter)2/2. Animals were euthanized, and tumors were excised upon reaching tumor volume of 2 cm3 size.
Human CRC Patient PBMC Analysis
Following the institutional review board’s (Ethics Committee) approval, blood samples were collected from 8 patients with KRAS-mutated metastatic CRC at 0 (pre) and 48 hours, days 8 and 15 and peripheral blood mononuclear cells were isolated for transcriptome analysis. Five patients had received pelareorep as part of a phase 1 clinical trial (NCT01274624) [19], while 3 patients were not enrolled in this trial but did receive equivalent background chemotherapy (i.e., FOLFIRI and bevacizumab).
Immunofluorescence
Cells were plated on Poly-L lysine coated coverslip (Sigma-Aldrich, P8920). Post pelareorep treatment, the coverslips were rinsed once with 1X PBS and fixed with absolute methanol at 20°C for 20 min. After removing methanol and 3 washes with 1X PBS, the coverslips were incubated overnight with 1:200 LC3 A/B antibody (cell signaling, 3868S). Post incubation coverslips were washed thrice with 1X PBS, then incubated anti-rabbit secondary antibody with Alexa Fluor 488 conjugate (Thermo-Fisher, A27034). Coverslips were incubated for 20 min at room temperature with Nuc-Blue Live Ready Probe (Thermo-Fisher, R37605), washed and mounted using Pro-Long gold antifade reagent (Invitrogen, P36934) on microscope slides (Fisher-brand, 12-550-143), and cured in the dark (24-Hrs; Room temp) and scanned on 3D Histech P250 High Capacity Slide Scanner at Albert Einstein Analytical Imaging Facility (AIF).
Flow cytometry with autophagy assay kit and LC3 A/B antibody
After treatment, cells were stained using either Autophagy Assay Kit (Abcam, ab139484) or LC3 A/B-DyLight 488 antibody (PA5-22938) and prepared for flow cytometry analysis employing the following protocols.
Autophagic flux detection via Autophagy Assay Kit.
Cells were fixed for 15 min at room temperature in 4% paraformaldehyde, stained with Green Detection Reagent and prepared for flow cytometry analysis following the kit’s protocol (Abcam, ab139484).
LC3 A/B detection.
Cells were fixed with IC Fixation buffer for 20 min at room temperature and resuspended in 1X Permeabilization Buffer (Invitrogen, 00-5523-00), then centrifuged at 500 xg for 10 min. Cells were stained LC3 A/B Dy-Light 488 antibody (1:500 ;1 hr/ 0°C), Followed by resuspension in Staining Buffer ( e-Bioscience 00-4222-57). Data was collected in BDFACS-Diva (BD Bioscience) and processed by flow Jo. V9 software.
Cell line protein isolation, estimation, and Western Blot:
Harvested
Cells (1X PBS) were put through 3 freeze-thaw cycles (liquid nitrogen) before 40 minutes incubation on ice with RIPA buffer (25 mM Tri-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium-deoxycholate, 0.1 SDS). The cells were centrifuged at >12,000 rpm for 1 hour at 4°C, and protein supernatant was collected.
Cell line
Protein was estimated by Micro BCA method (Thermo-Fisher, 23235). 50 ugm protein were loaded, separated by SDS-PAGE electrophoresis, transferred to PVDF (polyvinylidene difluoride) membranes, blocked in 5% milk (Bio-Rad, 1706404) and probed with primary antibodies and either anti-mouse or anti-rabbit secondary antibodies (cell signaling,7076, 7074, respectively). The blot was then incubated with HRP-conjugated secondary antibodies (1hr; Room-temp) (Supplementary Table S1) and visualized by chemiluminescence using Clarity Western ECL (Bio-Rad # 170-5061).
Mouse allograft protein isolation, estimation and Western Blot:
The whole tissue lysate was collected by mincing and grinding the tumor tissue with pestle and incubated (40mins; RIPA buffer+ protease inhibitor (SigmaP8340). The lysate was centrifuged at >12,000 rpm (1hour at 4°C). Post protein estimation( Bradford assay) 50 μg of protein was loaded into each lane, resolved by SDS-PAGE electrophoresis and transferred to PVDF membranes and probed with anti-mouse LC3A/B antibody (CST#4108) followed by HRP-conjugated secondary anti-rabbit antibody (CST#7074) and visualized by chemiluminescence (ECL Bio-Rad #170-5061).
Densitometry analysis.
Visualization and densitometry were done using Li-Cor Odyssey Fc imaging system. For densitometry, the band intensity was quantified using Image Studio Lite software (Li-Cor Corporate). The quantified intensity of each sample was normalized to the intensity of its respective housekeeping protein: β-actin or GAPDH.
RNA isolation, cDNA synthesis, and RT-qPCR analysis
Cell line:
RNA isolation and cDNA synthesis.
Cells were harvested and washed in 1X PBS before the isolation of RNA using RNeasy Mini Kit (Qiagen, 74106). One thousand ng of RNA was used to synthesize cDNA for each sample using iScript Reverse Transcription Supermix following the kit’s protocol (Bio-Rad, 1708840).
RT-qPCR Analysis with Autophagy Primer Library.
RT-qPCR reactions were prepared following Power-Up SYBR Green Master Mix’s instruction (Thermo-Fisher, A25777), and probed with Autophagy primer library diluted to the working concentration following the product’s protocol (RealTimePrimers, HATPL-1) composed of 88 autophagy-associated targets (supplementary table-2) and 8 reference genes (ACTB, B2M, GAPDH, GRUSB, HPRT1, GK1, PPIA, and RPL13A). The q-PCR cycle and conditions were set according to the instruction by Power-Up SYBR Green Master Mix. Gene expression data were analyzed by the 2−ΔΔCT method [20] and normalized to the median of the eight housekeeping genes. Two tailed t-test was used to determine statistical significance (p<0.05). Statistics were calculated using Microsoft Office Excel.
Mouse tumor:
RNA was isolated from tumor tissue and purified using Qiagen’s RNeasy Mini kit (Qiagen, cat, no.74106). RNA concentration was estimated using NanoDrop. 500 ng of RNA was used to synthesize cDNA with iScript Reverse Transcription Supermix (Bio-Rad, cat. no. 1001708841). cDNA from the five animals in each of the four study conditions, i.e. CT26- untreated, CT26- pelareorep treated, MC38- untreated and MC38- pelareorep treated, were pooled. Real time gene expression was performed by way of a mouse autophagy PCR array (Real Time Primers, MATPL-I) composed of 88 autophagy-associated targets (Supplementary Table-3) and 8 reference genes (ACTB, B2M, GAPDH, GRUSB, HPRT1, PGK1, PPIA, and RPL13A). The full list of genes is available on http://realtimeprimers.com/moauprli.html [21]. The qPCR (BioRad CFX-97) was conducted (manufacturers recommendation) in triplicate with AzuraQuant™ Green(AZ-2120) Probe Fast qPCR Master Mixes and analyzed as detailed in cell line qPCR.
Gene expression profile by RNA sequencing of patient samples TRANSCRIPTOME Analysis
Total RNA was isolated from the peripheral blood mononuclear cells (PBMC) from 5 patients treated with pelareorep and 3 patients treated with standard of care therapy only at days 0 (pre), 2 (48 hours), 8- and 15-days post pelareorep treatment. Thermo-Fisher Scientific, Clariom-D Pico-Assay, (Catalog Number 902924) single stranded cDNA from total RNA follows with fragmentation and labeling as per manufacturer’s protocol.
For this study we analyzed 100 autophagy related genes (Supplementary Table 4). The genes that were found to be significantly altered (2−ΔΔCT method) in pelareorep receiving patient cohort were confirmed for no such alterations in the control group.
STATISTICAL ANALYSIS
All experiments were performed at least 3 times and the mean values from different treatment groups compared by two way unpaired student’s t test with a p value < 0.05 considered statistically significant. Analysis was performed using Microsoft Excel (MS Office 2013).
RESULTS
KRAS mutant HCT116 has a greater expression of LC3 at 6 and 9 hours post pelareorep treatment
LC3 is one of the most extensively studied protein of the autophagy pathway. The distribution of fluorescence labeled LC3 A/B protein was determined microscopically by the number of puncta in treated and untreated samples of HCT116 and Hke3 cells. There was a significant, and time dependent difference in the increase in expression of LC3 A/B between the two cell lines at 6 hours (3.24 fold; p= 0.008) and 9 hours (8.67 fold; p= 0.015) when treated with pelareorep (Figure 1A-D).
Figure 1. Immunofluorescence analysis of LC3 A/B.


(A & C) Visualization of LC3 A/B showed overall higher LC3 A/B puncta (green) in HCT116 in comparison to Hke3 at 6 and 9 hours respectively. (B&D) Green fluorescence intensity measurement showed higher fold change of LC3 A/B in HCT116 in comparison to Hke3 at both time points (p<0.05). The fold change of each cell line was obtained by normalizing the pelareorep-treated samples to the untreated counterparts (n=4) * p<0.05.
Pelareorep treatment increases autophagosome formation and number of LC3 positive cells in HCT116 culture
To verify our finding, DyLight 488 labeled LC3 A/B and fluorescently labeled autophagosomes (Green Detection Reagent- AbCam) in untreated and pelareorep treated HCT116 and Hke3 cells at 6 and 9 hours were analyzed by flow cytometry (Figure 2Ai-iii & 2B i-iii). We observed significant difference in number of LC3 A/B positive cells normalized to untreated samples between the two cell lines at 6 hours (2.37 fold; p=0.018) and 9 hours (2.58-fold; p=0.045) (Figure 2A). Similarly, the fold difference of autophagosome positive cells in pelareorep treated/untreated samples were also significantly higher in HCT116 when compares to Hke3 by 2.02 fold at 6 hours (p= 0.012), and by 1.57 fold at 9 hours (p=0.016) (Figure 2B).
Figure 2A: Expression-level difference of LC3 A/B in human CRC cell lines.

Flow cytometry analysis (Flo Jo 9.2) detected higher fold change of LC3 A/B in HCT116 in comparison to Hke3 at 6 and 9 hours (p<0.05) i) The dot plot ii) Histogram iii) Graphical representation. Cells were stained for LC3 A/B and detected through FITC channel. The fold change of each cell line was obtained by normalizing the pelareorep treated samples to the untreated counterparts (n=4).
Figure 2B. Flow cytometry analysis detected higher fold change of autophagosome accumulation in HCT116 in comparison to Hke3 at 6 and 9 hours (p<0.05).

Cells were stained for autophagosome using Abcam Autophagy Detection kit and detected through FITC channel. (i) Histogram (ii) dot plot (iii) graphical representation. The fold change (y-axis) of each cell line was obtained by normalizing the pelareorep-treated samples to the untreated counterparts (n=3).
Pelareorep treatment upregulates proteins related to autophagy machinery in in vitro and in vivo models
CRC cell lines:
To observe autophagy at molecular level, expression of five autophagy related proteins were analyzed at 6 and 9 hours in both, pelareorep-treated and untreated samples of HCT116 and Hke3 (Figures 3A-B). We observed significant difference in the protein expression (normalized to untreated samples) between the two cell lines at 6 and 9 hours for LC3 A/B (p=0.021, and 0.036, respectively) and Beclin-1 (p=0.044, and 0.043, respectively). However, we only observed this significant difference in ATG-5 and P-62 at 6 hours (p=0.051 and p=0.004), and in VPS-34 at 9 hours (p=0.039) (Figure 3A). For all significant differences, HCT116 shown higher protein expression when compared to Hke3. At both time points, the expression of LC3A/B was higher in HCT116 by 2.38 and 6.82 folds, respectively, and slightly higher expression of Beclin-1 (1.17 and 1.24 folds, respectively). At 6 hours, the expression of ATG-5 and P-62 was higher by 1.84 and 1.52 folds, and at 9 hours the expression of VPS-34 was higher by 1.39 fold (Figure 3B).
Figure 3A and 3B: Expression of autophagy related proteins in untreated (−) and Pelareorep treated (+) HCT116 and Hke3 samples at 6 and 9 hours.
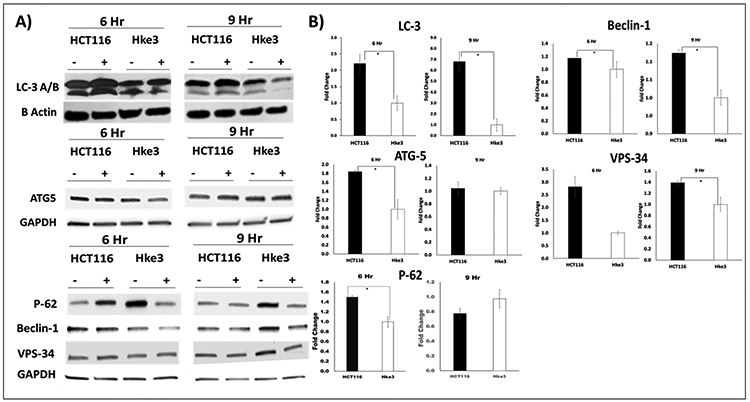
A) The western portrayed the protein expression of autophagy related protein: LC3 A/B, ATG-5, P-62, Beclin-1, and VPS-34 on the left. B) The corresponding densitometry analysis showing fold change of pelareorep treated to untreated are on the right (n= 3).
Syngeneic mouse models:
We tested the overall expression of LC3A/B protein normalized to GAPDH in 20 syngeneic allograft residual tumors - CT26 cells injected into BALB/c and MC38 into C57BL/6 - post-euthanization in mice (5 animals per group of pelareorep-treated and control). MC38 had a baseline higher normalized expression of LC3A/B compared with CT26 (Figure 3C). Both groups of animals showed increase in the expression of LC3, with the magnitude of change being significant only in the KRAS mutant CT26 group (3.8 fold; p=0.005) and not in KRAS WT MC38 syngeneic mice (p=0.11) indicating that pelareorep induces LC3A/B protein expression significantly in KRAS mutant allograft tumors.
Figure 3C: Western blot analysis of total LC3 expression in CT26 and MC38-mice xenograft tumor in the untreated (Control) or pelareorep Treated group. (Left) The graphical depiction of the protein expression of LC3 I and II. (Right).

The corresponding densitometry showing normalized overall LC3 expression in control and Pelareorep treated groups of either CT26 or MC38 (n=5 for each group). For CT26, the control group was statistically significantly different from the pelareorep treated group (p=0.005).
Pelareorep exposure augments autophagy induction in human colorectal cancer cell lines
To further investigate autophagy at molecular level, expression of 88 autophagy marker genes in pelareorep-treated and untreated HCT116 and Hke3 cells at 6 and 9 hours were analyzed by real time quantitative PCR. Expression of pelareorep-treated individual cell type was normalized to respective untreated samples. Genes including AMBRA1, ATG16L2, BCL2, EIF4EBP1, EIF4G1, HSPA5, MAP1LC3 A/B, MCL1, and PIK3R4 showed a 4-fold or higher upregulation in HCT116 compared to Hke3 at 6 hours (p<0.05) (Figure 4A). At this time point, all genes except for MCL-1 were below basal level for Hke3. Similarly, genes such as ATG-4D, GPSM1, SEC23A, and GABARAP were significantly upregulated (p<0.05) in HCT116 at 9 hours treatment (Figure 4B).
Figure 4A and 4B. Alterations in autophagy related gene expression at 6 and 9 hours.
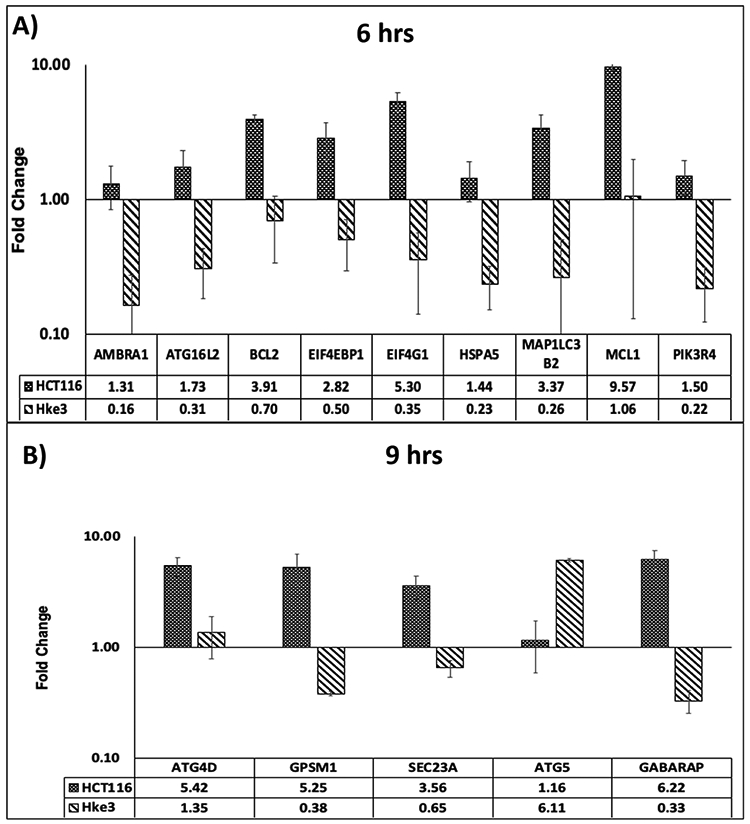
Average fold change of autophagy gene expression in HCT116 and Hke3 at 6 hours (A: Top panel) and 9 hours (B: Bottom panel) (n=4). The genes presented are at least 4-fold difference between HCT116 and Hke3 (p<0.05). The error bars were generated from standard error of mean, and y-axis was plotted in log scale.
Pelareorep exposure triggers autophagy induction in allografts of mouse models
Expression profiling of tumor tissues isolated from the two syngeneic mouse models, CT26 and MC38, revealed a differential regulation of autophagy-associated genes. Of the 88 genes analyzed, 13 genes could not be captured due to very low expression (Cq unreadable) or high Cq values >38 wherein efficiency was questionable and were found to be unreadable across multiple runs of the experiment.
On analyzing the degree of change induced by pelareorep treatment, 12 genes (Figure 5) were identified to have statistically significant difference (p≤0.05), between the two mouse models. It is interesting to note that all 12 genes were induced in CT26, and suppressed in MC38. Of note, the BARKOR gene had an ~30-fold induction in the CT26 model. This gene has been identified as a mammalian autophagy-specific factor, overexpression of which leads to autophagy activation, increased number and enlarged volume of autophagosomes [22].
Figure 5: Gene expression pattern in allografted syngeneic mouse model.
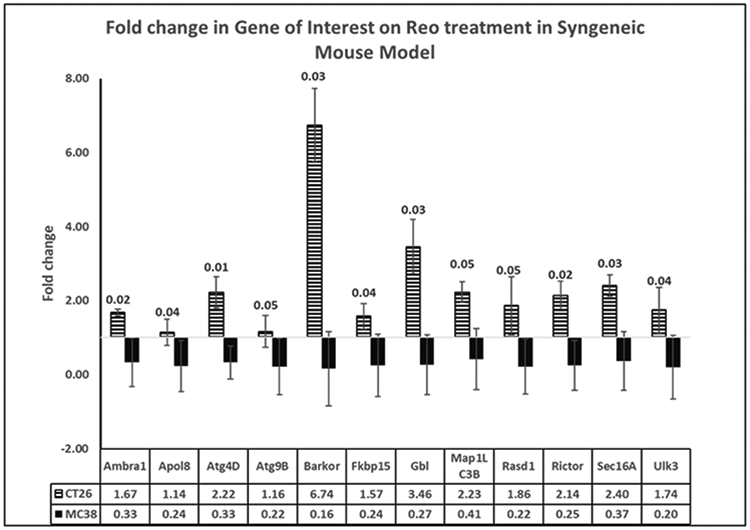
Comparison between fold-change in genes of interest expression between CT26 and MC38 mouse models on treatment with pelareorep, calculated by the 2−ΔΔCT method. Respective p values listed as legend on the bars.
Pelareorep treatment enhances autophagy induction in CRC patients with KRAS mutation
Encouraged by our pre-clinical results, we examined 100 autophagy related genes (Supplementary Table 4) selected from RNA sequencing database generated by transcriptome analysis of the patient PBMC samples for differences in expression. Since the study entailed sampling of blood at three post-treatment time points, genes which showed significant changes (p<0.05) on at least two occasions, compared to pre-treatment levels, were selected for analysis. The time points studied in this analysis allowed for assessment of changes in autophagy markers over a period of days-weeks. Fourteen genes were thus identified in the patients who received pelareorep treatment - APOB, ATG16L1, ATG3, ATG7, DRAM1, GNAI3, MAPLC3, PIK3CA, PPM1K, RICTOR, RASD1, SQSTM1, ULK3, and UVRAG. Figure 6 depicts the changes noted in graphical form, which allows us to visualize the trend over time.
Figure 6:
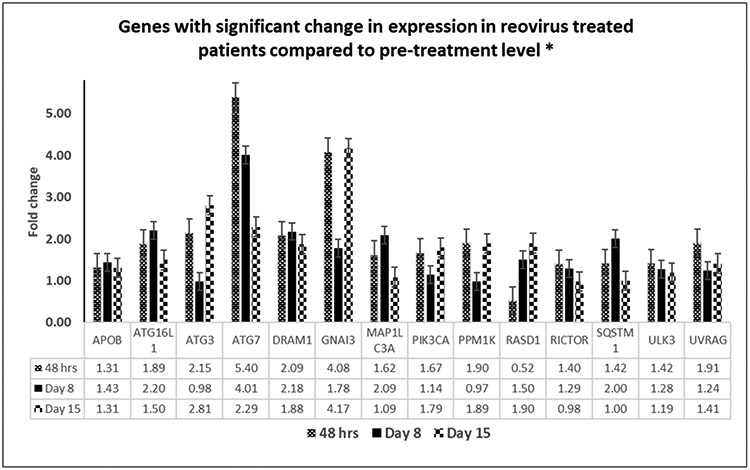
Genes showing significant change in expression (p=<0.05) on exposure to pelareorep in KRAS mutated patients at least in two out of three time points (48 hours, Day8 and Day 15) normalized to pre-pelareorep therapy.
*Genes with signficant change on at least 2/3 time points
Identification of genes with significant changes across all three experimental models
In order to validate the involvement of autophagic machinery, we investigated gene expression across the 3 models – namely, preclinical in vitro (cell lines), pre-clinical in vivo (syngeneic mouse models), and clinical in vivo (human patient samples). We have compiled the genes which showed significant changes in same trend across experiments into a Venn diagram (Figure 7). Three genes, MAP1LC3, RASD1 and RICTOR, were found to be common across all three experimental models. One gene, ULK3 is shared between the mouse model and human patient samples only while three other genes were common between the human cell line model and human patient samples (ATG16L1/2, PPM1K, SQSTM1). Similarly, four genes (AMBRA1, ATG4, GB and SEC16A) were common between mouse model and human cell lines. The 88 genes that were analyzed across samples were assessed by the STRING database [23] cluster analysis to understand the protein-protein interactions and interplay of the network of genes identified to be significant (Supplementary Figure 1).
Figure 7:
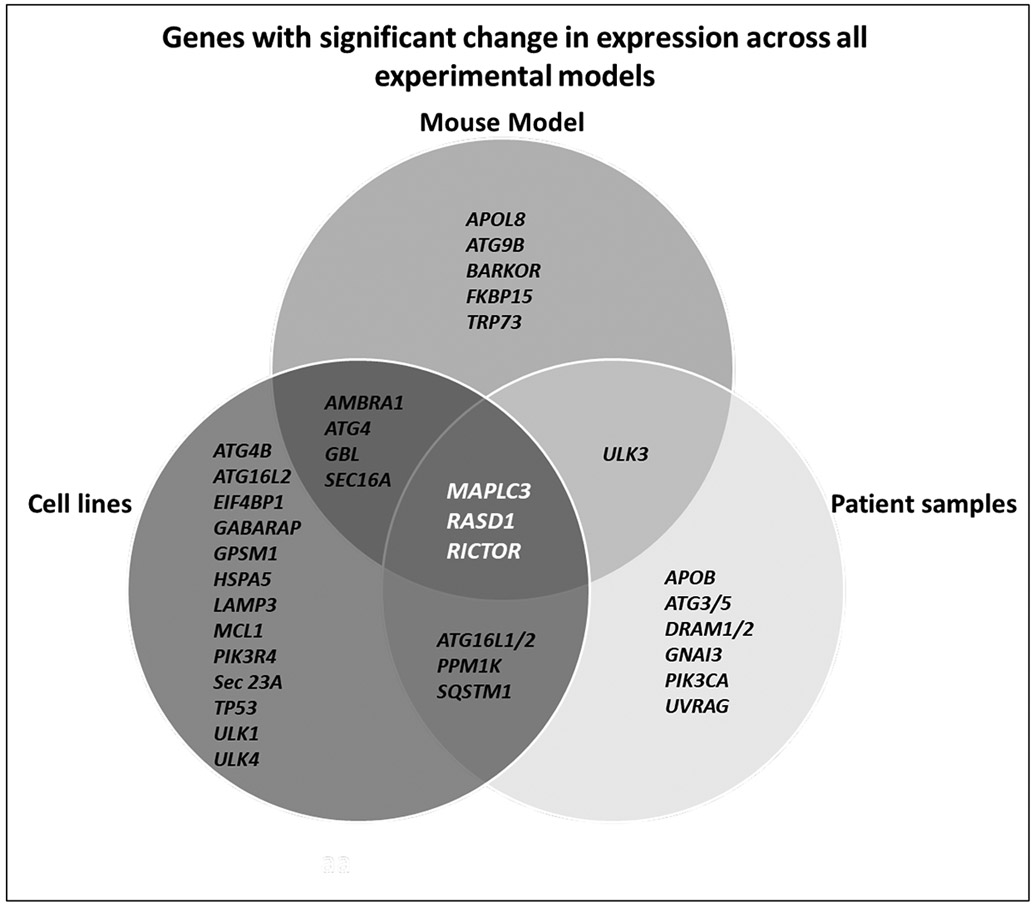
Venn diagram depicting genes with significant change in expression (p<=0.05) across the three models studied.
DISCUSSION
The association between KRAS mutation and autophagy induction in cancer is complex. Several studies have shown that KRAS mutation induces autophagy while others have reported inhibition [5, 15, 17]. We instituted a broad-spectrum study including three different models to assess the contribution of autophagic machinery towards efficient self-propagation and oncolytic activity of pelareorep in KRAS mutated CRC.
In the in vitro cell line experiments, we observed an early upregulation of autophagy related proteins starting at 3 hours and reaching significance between 6 and 9 hours. Within 24 hours post infection the difference in level of the autophagic proteins between the KRAS mutant and WT disappears. We thus hypothesize that autophagy related proteins augment the oncolytic process of pelareorep during the initial steps of viral infection creating a favorable cellular environment for its effective propagation. There is clear evidence that RNA viruses like polio, corona (CoV), dengue, zika, west nile and mouse hepatitis (MHV) exploit autophagy for their replication [8]. It is proposed that LC3 forms a complex with ATG5/ATG12 and P-62 (SQSTM) during the initial steps of the formation of autophagosomes and serves as a platform for viral replication, further supported by the report stating that reovirus utilize ATG5 for its replication [24]. ATG5 is an important protein that plays a critical role not only in autophagic pathway but extends further to activate the innate immune response, an inevitable natural event post virus invasion [25]. ATG5 is indispensable for autophagic vesicle formation. Knocking down ATG5 can result in downregulation or total inhibition of autophagy, proving its central role in autophagy [25]. Recent studies have demonstrated that ATG5 modulates the immune system and enables crosstalk with apoptosis [26-28]. We have previously shown that pelareorep preferentially induces apoptosis in an array of KRAS mutant CRC cells [4] and the effect was most discernable at 48 hours post pelareorep administration. Our cell line data shows significant alterations of autophagic proteins at 6 hours post infection indicating autophagic induction preceded apoptosis. ATG5 is an important protein and one of the early players that critically controls the switch of the autophagic machinery from a pro survival mode to programmed cell death. P-62, a marker for early autophagic events and indicative of accumulation of autophagosome is significantly upregulated in HCT116 at 6 hours (early onset). Another important autophagic protein that networks with lipid kinase VP34 and regulates autophagy and apoptosis is Beclin-1, that can intervene at every major step in autophagic pathways, from autophagosome formation, to autophagosome/endosome maturation [29]. This protein was significantly upregulated in HCT116 at both time points with a significant upregulation of VP34 at 9 hours strongly supporting a preferential induction of autophagic machinery by pelareorep that is furthered to trigger the apoptotic pathway as a downstream event in KRAS mutant cells. [8]. These results are consistent with the previously reported role of autophagy as a “double edged sword” with abilities to salvage the cell as well as induce programmed cell death (apoptosis) [8]; and along the same line of our previously reported study where apoptosis is triggered beyond 24 hours [4]. Upon trigger of the apoptotic machinery the expression of autophagic machinery is no longer significantly upregulated.
The qPCR analysis of the 88 autophagy related genes (supplementary tables 2, 3 and 4) in the cell lines at 6 and 9 hours clearly indicate upregulation of specific autophagy related genes only in pelareorep treated HCT116 cells, but lacking in Hke3 cells. Nine genes showed an early induction within 6 hours of pelareorep treatment and included LC3, RASD1, ATG16L1 among others. A rapid induction of autophagy, reflecting the instinct of the cell to adapt to stress, is followed by the activation of cell death pathways in response to multiple external signals such as invading pathogens [20, 22]. The fact that many signal transduction pathways that are elicited by cell-intrinsic stress regulate both autophagy and apoptosis might explain the sequential activation of both processes.
Our syngeneic mouse model study was an endpoint experiment wherein tumors generated with CT26 (KRAS mutant) were compared to MC38 (KRAS WT) tumors. Expression of LC3 was significantly upregulated in KRAS mutant tumors, which again confirmed that pelareorep efficiently induces autophagy in KRAS mutant tumors. It is well established that pelareorep propagates and oncolyses KRAS mutant CRC cell significantly better than KRAS wild type CRC [4, 30, 31]. The qPCR analysis of mouse tumor tissue shows an upregulation in expression of 12 autophagy related genes in CT26 tumor, which were all downregulated in the MC38 tumor (figure 5). Overexpression of BARCOR leads to autophagy activation and augmentation of autophagosome formation [22]. The most prominent and impressive (30-fold) overexpression of gene BARCOR strongly supports our hypothesis. Furthermore, important genes including LC3, ATG4, RASD1, Sec16A and ULK were also significantly upregulated in CT26 tumors.
In the clinical samples using patient PBMC, the expression of 100 (including the corresponding 88 genes used for the cell line and the mouse model study) genes from the transcriptome assay similarly documented significant upregulation of several genes (Figure 6). These genes were significantly (p<0.05) upregulated in two out of three time points when compared to pre-pelareorep samples, and this was absent in control patients receiving standard of care treatment (Supplementary Figure 2). Our analysis of normalized genes utilizing strict statistical parameters yielded 13 genes that were significantly (p<0.05) upregulated in patients with a KRAS mutation treated with pelareorep in comparison to untreated patients. Three of these genes were common across the models (Figure 7).
Microtubule associated protein light chain 3 (LC3), one of the most well studied markers of autophagy [32], was found to be the centrally upregulated gene across every model and assay in our study, in complete agreement that autophagy was upregulated with pelareorep treatment in a KRAS mutant environment. RASD1 is a member of the RAS superfamily of small G-proteins is known to regulate signal transduction pathways through both G proteins and G protein-coupled receptors[33]. Although RASD1 is a member of the RAS superfamily of small G-proteins, which often promotes cell growth and tumor expansion, it plays an active role in preventing aberrant cell growth [34]. We speculate that pelareorep augments the destruction of KRAS mutant tumor cells by inducing RASD1, a phenomenon not witnessed in KRAS wild type tumors. We also observed an upregulation of RICTOR which is a crucial component of mTORC2, and plays an essential role in regulating and activating AKT phosphorylation [35]. Reports indicate that AKT promotes cell growth, apoptosis and autophagy by triggering the phosphorylation and activation of mTOR [36]. RICTOR is a multifaceted protein that can cross talk and interact not only with the proteins of mTOR2 complexes but also with growth factors like EGFR [37] and cytoskeleton protein actin [38]. RICTOR’s oncogenic role has gained momentum over recent years, with reports uncovering both canonical rate limiting activity on mTORC2-AKT, and the presence of additional mTORC2-independent functionalities [39].
Apart from these three genes that were upregulated across the all three subsets of study, an additional four genes were commonly upregulated in the mouse and cell line studies including AMBRA1, ATG4, GBL and Sec16A. AMBRA1 is known to be phosphorylated by ULK-1 and is upregulated upon autophagy activation, leading to complex formation with Beclin-1 and PI3KIII, followed by translocation to ER where autophagosome formation takes place [40]. ATG4D plays an important role in phagophore expansion by complexing with ATG-7, ATG3, and ATG5-ATG12 [41]. GβL is a subunit of both mTORC1 and mTORC2 complexes that regulate cell growth and survival and an integral component of autophagy machinery [42]. Sec16A is known to control ER-to-Golgi trafficking of specific cargo and is probably one important protein that pelareorep utilizes for synthesizing viral proteins and trafficking virions to the cell surface in a noncanonical manner [43].
Similarly, ULK3 is shared between mouse model and cell line while ATG16- 1 and 2, PPM1K and SQSTM1 are shared between patient samples and cell lines. A string database analysis of mouse ULK3 Protein (https://string-db.org/network/10090.ENSMUSP00000059947) indicates that it is strongly associated with ATG proteins. ULK3, a serine theronine protein kinase primarily phosphorylates ATG proteins which in turn activates autophagy. Autophagy related 16 like 1 and 2 (ATG16L1/2) plays an essential role in autophagy by interacting with ATG12-ATG5 to mediate the conjugation of phosphatidylethanolamine (PE) to LC3 to produce a membrane-bound activated form of LC3 and thereby, controls the elongation of the nascent autophagosomal membrane. Interestingly ATG16L1/2 also regulates mitochondrial antiviral signaling (MAVS)-dependent type I interferon (IFN-I) production. It has also been reported that ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I Interferon [44]. PPM1K encodes mitochondrial targeted 2C-type serine/threonine protein phosphatase (PP2Cm) and is highly conserved among vertebrates. PP2Cm expression is dynamically regulated by the nutrient environment and pathological stresses [45] and significant upregulation in KRAS mutated conditions indicates the viral control on mitochondrial homeostasis. STSQM1 alternatively known as p-62 has been previously discussed as we have also confirmed its expression western blot analysis as well as by qPCR in cell lines. Analysis of the patient sample transcriptome also confirmed the same degree of up-regulation indicating similar effects occurring in actual patients receiving pelareorep. The effectiveness (patient response) pharmacodynamic and immune profiling of our trial patients have been explicitly discussed in our previous publications[19, 46] Apart from the common genes, each study also had a group of other genes transcripts that were also significantly (p<0.05) upregulated which are all indicative of autophagic induction.
CONCLUSION
This is the first study evaluating the alterations in autophagic pathway in colorectal cancer secondary to pelareorep intervention in which we focused on KRAS mutation, as pelareorep is significantly effective under KRAS mutated condition. We have previously shown that apoptosis is triggered upon pelareorep administration in a wide array of CRC cell lines, an effect most prominent at 48 hours [4]. Our current study clearly reveals that several important autophagic proteins are significantly upregulated in KRAS mutation as compared to the KRAS wildtype conditions, especially at early time points. Autophagy can be associated with both cancer progression and tumor suppression [47], and can promote cell survival or activate programmed cell death. We predict that administration of pelareorep in conjunction with autophagy activating drugs [48] will synergistically improve the efficacy of pelareorep and can be used as a dedicated therapy for the KRAS mutated CRC patient population.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE:
Oncolytic reovirus (pelareorep), a non-enveloped double stranded (ds) RNA virus, selectively replicates in and lyses KRAS mutated colorectal cancer cells and is a potential therapy option for patients. Although several mechanisms of this preferential oncolytic activity have been proposed, the involvement of autophagic machinery remains unexplored. Understanding its role in the preferential propagation of pelareorep may help to identify novel biomarkers and potential therapeutic targets in a pursuit to find effective treatment against KRAS mutated colorectal cancer. In this manuscript, we successfully demonstrate in a multitude of CRC models, the role of autophagy in the selective susceptibility of KRAS mutated cancer. Specifically, upregulation of LC3 A/B, a key early autophagic protein is consistent observed. This study may form the foundation for a first in human study of pelareorep with an autophagy inducer to augment its anti-cancer property.
ACKNOWLEDGMENTS
We gratefully acknowledge the genomic facility, Flow cytometry Core facility and the Analytical Imaging Facility of Albert Einstein College of Medicine along with the NCI cancer center support grant (P30CA013330), which partially supports all morphometric work conducted with 3D Histech P250 High Capacity Slide Scanner SIG #1S10OD019961-01 of the shared facilities. We also thankfully acknowledge Prof. Xingxing Zang (Department of Microbiology and Immunology, Albert Einstein College of Medicine) for kind gift of two transplantable (syngeneic) models of murine tumor cell lines, CT26 and MC38.
FUNDING
SG is supported by NIH/AG1R21 NIH AG 1R21AG058027-01
RM is supported by Yeshiva University startup fund 2A4108
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
REFERENCES
- 1.Peeters M, et al. , Prevalence of RAS mutations and individual variation patterns among patients with metastatic colorectal cancer: A pooled analysis of randomised controlled trials. Eur J Cancer, 2015. 51(13): p. 1704–13. [DOI] [PubMed] [Google Scholar]
- 2.Han CB, et al. , Concordant KRAS mutations in primary and metastatic colorectal cancer tissue specimens: a meta-analysis and systematic review. Cancer Invest, 2012. 30(10): p. 741–7. [DOI] [PubMed] [Google Scholar]
- 3.Maitra R, Ghalib MH, and Goel S, Reovirus: a targeted therapeutic--progress and potential. Mol Cancer Res, 2012. 10(12): p. 1514–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maitra R, et al. , Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget, 2014. 5(9): p. 2807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White E and DiPaola RS, The double-edged sword of autophagy modulation in cancer. Clin Cancer Res, 2009. 15(17): p. 5308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beau I, Mehrpour M, and Codogno P, Autophagosomes and human diseases. Int J Biochem Cell Biol, 2011. 43(4): p. 460–4. [DOI] [PubMed] [Google Scholar]
- 7.Apel A, et al. , Autophagy-A double-edged sword in oncology. Int J Cancer, 2009. 125(5): p. 991–5. [DOI] [PubMed] [Google Scholar]
- 8.Choi Y, Bowman JW, and Jung JU, Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol, 2018. 16(6): p. 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmad L, Mostowy S, and Sancho-Shimizu V, Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front Cell Dev Biol, 2018. 6: p. 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marino G, et al. , Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol, 2014. 15(2): p. 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang YP, et al. , Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin, 2013. 34(5): p. 625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amaravadi RK, et al. , Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res, 2011. 17(4): p. 654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rouschop KM and Wouters BG, Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med, 2009. 9(4): p. 417–24. [DOI] [PubMed] [Google Scholar]
- 14.Gozuacik D and Kimchi A, Autophagy and cell death. Curr Top Dev Biol, 2007. 78: p. 217–45. [DOI] [PubMed] [Google Scholar]
- 15.Paquette M, El-Houjeiri L, and Pause A, mTOR Pathways in Cancer and Autophagy. Cancers (Basel), 2018. 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo Re AE, et al. , Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J Biol Chem, 2012. 287(30): p. 25325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitz KJ, et al. , Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J Surg Oncol, 2016. 14(1): p. 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gollamudi R, et al. , Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs, 2010. 28(5): p. 641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goel S, et al. , Elucidation of Reovirus Pharmacodynamics in a Phase I Trial in Patients with Kras Mutated Colorectal Cancer. Mol Cancer Ther, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402–8. [DOI] [PubMed] [Google Scholar]
- 21.Primers RT Mouse Autophagy Primer Library. 2018. [cited 2018 12/27/2018]; Mouse Autophagy Primer Library]. Available from: https://www.realtimeprimers.com/moauprli.html.
- 22.Sun Q, et al. , Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A, 2008. 105(49): p. 19211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szklarczyk D, et al. , STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res, 2019. 47(D1): p. D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemp V, et al. , Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery. Viruses, 2017. 9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye X, Zhou XJ, and Zhang H, Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front Immunol, 2018. 9: p. 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikoletopoulou V, et al. , Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta, 2013. 1833(12): p. 3448–3459. [DOI] [PubMed] [Google Scholar]
- 27.Lee HR, et al. , Modulation of Immune System by Kaposi’s Sarcoma-Associated Herpesvirus: Lessons from Viral Evasion Strategies. Front Microbiol, 2012. 3: p. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills KR, et al. , Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci U S A, 2004. 101(10): p. 3438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang R, et al. , The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ, 2011. 18(4): p. 571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WQ, et al. , Efficacy and safety evaluation of human reovirus type 3 in immunocompetent animals: racine and nonhuman primates. Clin Cancer Res, 2004. 10(24): p. 8561–76. [DOI] [PubMed] [Google Scholar]
- 31.Goel S, et al. , Elucidation of Pelareorep Pharmacodynamics in A Phase I Trial in Patients with KRAS-Mutated Colorectal Cancer. Mol Cancer Ther, 2020. 19(5): p. 1148–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizushima N and Yoshimori T, How to interpret LC3 immunoblotting. Autophagy, 2007. 3(6): p. 542–5. [DOI] [PubMed] [Google Scholar]
- 33.Wie J, et al. , The Roles of Rasd1 small G proteins and leptin in the activation of TRPC4 transient receptor potential channels. Channels (Austin), 2015. 9(4): p. 186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaidyanathan G, et al. , The Ras-related protein AGS1/RASD1 suppresses cell growth. Oncogene, 2004. 23(34): p. 5858–63. [DOI] [PubMed] [Google Scholar]
- 35.Sarbassov DD, et al. , Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science, 2005. 307(5712): p. 1098–101. [DOI] [PubMed] [Google Scholar]
- 36.Wan G, et al. , Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy, 2014. 10(1): p. 70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jebali A and Dumaz N, The role of RICTOR downstream of receptor tyrosine kinase in cancers. Mol Cancer, 2018. 17(1): p. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagan GN, et al. , A Rictor-Myo1c complex participates in dynamic cortical actin events in 3T3-L1 adipocytes. Mol Cell Biol, 2008. 28(13): p. 4215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruder D, et al. , Concomitant targeting of the mTOR/MAPK pathways: novel therapeutic strategy in subsets of RICTOR/KRAS-altered non-small cell lung cancer. Oncotarget, 2018. 9(74): p. 33995–34008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cianfanelli V, et al. , Ambra1 at a glance. J Cell Sci, 2015. 128(11): p. 2003–8. [DOI] [PubMed] [Google Scholar]
- 41.Yin Z, Pascual C, and Klionsky DJ, Autophagy: machinery and regulation. Microb Cell, 2016. 3(12): p. 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DH, et al. , GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell, 2003. 11(4): p. 895–904. [DOI] [PubMed] [Google Scholar]
- 43.Piao H, et al. , Sec16A is critical for both conventional and unconventional secretion of CFTR. Sci Rep, 2017. 7: p. 39887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimm WA, et al. , The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut, 2016. 65(3): p. 456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan BF, et al. , Regulation of PP2Cm expression by miRNA-204/211 and miRNA-22 in mouse and human cells. Acta Pharmacol Sin, 2015. 36(12): p. 1480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parakrama R, et al. , Immune characterization of metastatic colorectal cancer patients post reovirus administration. BMC Cancer, 2020. 20(1): p. 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen N and Debnath J, Autophagy and tumorigenesis. FEBS Lett, 2010. 584(7): p. 1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marinkovic M, et al. , Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid Med Cell Longev, 2018. 2018: p. 8023821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.