

Abstract
Pancreatic ductal adenocarcinoma is a deadly disease with a 5-year survival rate of ~9%. An improved understanding of PDAC initiation and progression is paramount for discovering strategies to better detect and combat this disease. While transcriptomic analyses have uncovered distinct molecular subtypes of human PDAC, the factors that influence subtype development remain unclear. Here, we interrogate the impact of cell-of-origin and different Trp53 alleles on tumor evolution, using a panel of tractable genetically-engineered mouse models. Oncogenic KRAS expression, coupled with Trp53 deletion or point mutation, drives PDAC from both acinar and ductal cells. Gene expression analysis reveals further that ductal cell-derived and acinar cell-derived tumor signatures are enriched in basal-like and classical subtypes of human PDAC, respectively. These findings highlight cell-of-origin as one factor that influences PDAC molecular subtypes and provide insight into the fundamental impact that the very earliest events in carcinogenesis can have on cancer evolution.
Keywords: Pancreatic ductal adenocarcinoma, cell-of-origin, mouse models, p53, molecular subtype
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a deadly cancer that is projected to be the second leading cause of cancer-related deaths in the United States by 2030 (1). The 5-year survival rate for PDAC patients is a mere 9% (2) and is attributable to late-stage diagnosis – when patients are rarely eligible for surgical resection – and therapeutic strategies being largely ineffective (3). A better understanding of how PDAC arises is vital for improving both early detection and treatment.
The genetics underlying PDAC development are well-described. Human PDAC genome sequencing has identified several recurrent genomic alterations in pancreatic tumors, including activating mutations in KRAS in >90% of PDACs (4,5) and mutations in the TP53, CDKN2A, and SMAD4 tumor suppressor genes in 72%, 30%, and 32% of cases, respectively (6). Analysis of human pancreas samples through DNA and immunohistochemical analyses has led to a proposed progression model for PDAC, with oncogenic KRAS mutations serving as the initiating event and specific tumor suppressor gene mutations occurring at defined stages thereafter to promote cancer progression (7). In this model, cancers arise when oncogenic KRAS promotes the formation of preinvasive Pancreatic Intraepithelial Neoplasias (PanINs) that progress through increasingly more dysplastic stages, culminating in frank carcinomas and metastasis. Studies in genetically engineered mouse models in which oncogenic KRAS is expressed and tumor suppressor genes are inactivated in the pancreas have been instrumental for demonstrating the importance of these combined mutations in driving PDAC development (8–12). Importantly, these models faithfully reproduce the entire progression of human PDAC, from neoplastic lesions to metastatic cancer.
As a complementary approach to genomic analyses, transcriptomic analyses have been utilized to define molecular subtypes of human PDAC, with the goal of identifying distinct classes that could guide clinical decision-making. Three seminal studies established a framework for PDAC classification. Depending on the methodology, transcriptomics analysis of resected PDAC samples from untreated patients revealed between 2 and 4 distinct molecular subtypes (13–15). While differences in sample cellularity and technology used for transcriptomic analysis may underlie the differences in specific subtypes defined by each study, there is a consensus from these initial studies as well as subsequent ones that two broad pancreatic cancer subtypes exist: the classical and the basal-like subtypes, with the latter being associated with poorer prognosis (16–24). The cellular and molecular factors that drive each molecular subtype, however, remain unknown. Unequivocally establishing the factors critical for dictating the subtypes will refine our understanding of the underlying biology of these classes and ultimately improve risk stratification and therapeutic development. Such an understanding, however, requires tractable genetic experiments in model systems to allow interrogation of such factors as specific gene mutations and cell type-of-origin on the development of different subtypes.
Correlative analyses have suggested particular genetic associations with specific subtypes, such as mutations in the TP53 gene, which encodes the p53 transcription factor, with the basal-like subtype (13,20,21,23,24). TP53 commonly sustains missense mutations in the DNA binding domain that not only induce loss of wild-type p53 function, but may also promote gain-of-function properties through the production of a mutant p53 protein (25). Missense mutations comprise two major classes: contact mutations that alter residues required by p53 to contact DNA and structural mutations that affect the three-dimensional structure of the protein (26). The most frequently observed TP53 mutations in PDAC are at codons 175 and 273 (corresponding to mouse codons 172 and 270, respectively), which represent structural and contact residue mutations (26,27). While the majority of mouse PDAC studies have relied on combined oncogenic KRAS and p53R172H expression to drive metastatic PDAC, oncogenic KRAS and Trp53 deletion also promote metastatic PDAC (9,12,28). How Trp53 deletion and different classes of p53 point mutants impact the development of different subtypes of PDAC, however, remains unexplored.
It also remains unclear how cell-of-origin influences pancreatic cancer subtype. Acinar and ductal cells are the major epithelial cell types in the exocrine pancreas, but which of these serves as the cell-of-origin in PDAC has been debated. Early studies in mouse PDAC models failed to reveal the cell-of-origin because they relied on strains in which Cre recombinase is expressed in multipotent pancreas progenitor cells during embryogenesis (8). While PanINs resemble ducts and were originally thought to arise from normal ductal cells, more recent evidence using tamoxifen-regulatable Cre to induce genetic mutations in adult mice has suggested that acinar cells can give rise to PanINs and PDAC through a cellular reprogramming process termed Acinar-to-Ductal Metaplasia (ADM) (29–37). Some evidence from adult mouse PDAC models also supports the notion that PDAC can arise from ductal cells, in the context of oncogenic KRAS expression and homozygous Trp53 or Fbw7 deletion (30,33,36). Thus, human PDAC likely originates from pancreatic acinar or ductal cells.
Here, we seek to understand the factors that influence the development of different transcriptional subtypes of PDAC using a comprehensive panel of tractable genetically engineered mouse models. To better recapitulate the process of PDAC development in humans, we have investigated PDAC development in adult mice, from pancreatic acinar or ductal cells, and in the contexts of intact p53, p53 inactivation, or expression of the p53 mutants, p53R172H or p53R270H. We have performed transcriptomic analysis of tumors arising from acinar and ductal cells, then utilized these profiles to illuminate a connection between cell-of-origin and human PDAC subtype. Our studies reveal that different cells-of-origin can influence the ultimate molecular subtype of PDAC, providing critical new insight into the basis for intertumoral PDAC heterogeneity.
RESULTS
Oncogenic KRAS activation can induce metastatic PDAC from adult pancreatic acinar cells
To deconstruct the genetic requirements for pancreatic cancer development from adult mouse pancreatic acinar cells, we established a system to activate oncogenic KrasG12D and modulate Trp53 in mouse pancreatic acinar cells through the use a tamoxifen-inducible knock-in Ptf1aCreER allele. Importantly, to induce genetic alterations in cells of the fully developed adult pancreas, mice were aged to adulthood (8 – 10 weeks) before tamoxifen treatment (Figure 1A). Tamoxifen treatment induced efficient genomic recombination in the majority of acinar cells, but not other pancreatic cell types, as indicated by the selective expression of the tdTomato reporter allele in amylase-positive acinar cells (Figure 1B). To initially assess tumor development in the presence and absence of p53, we generated, tamoxifen treated, aged, and examined cohorts of KrasLSL-G12D/+;Rosa26LSL-tdTomato/LSL-tdTomato;Ptf1aCreER;Trp53+/+ (KrasLSL-G12D/+;Rosa26LSL-tdTomato/LSL-tdTomato heretofore denoted as KT), KT;Ptf1aCreER;Trp53fl/+, and KT;Ptf1aCreER;Trp53fl/fl mice for pancreatic cancer-free survival upon morbidity.
Figure 1. Pancreatic cancer can arise from adult mouse acinar cells.
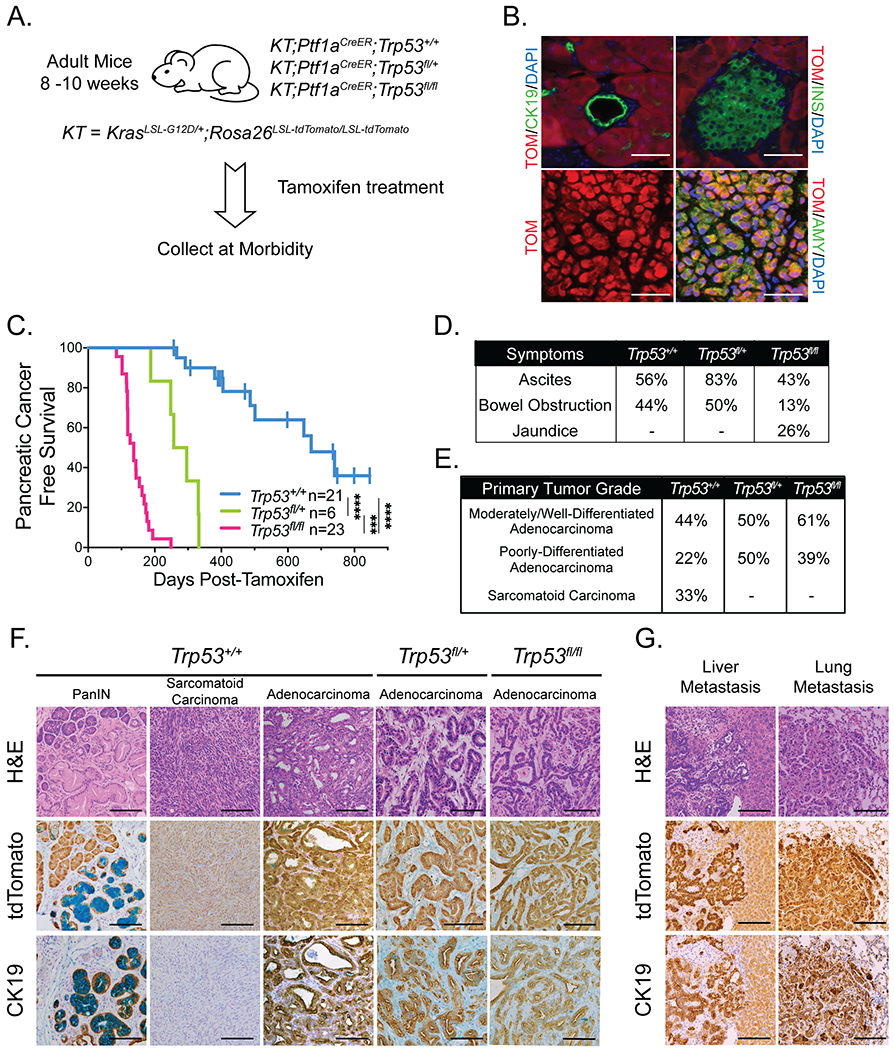
(A) Schematic for acinar cell-derived pancreatic cancer study of KT;Ptf1aCreER;Trp53+/+, KT;Ptf1aCreER;Trp53fl/+, and KT;Ptf1aCreER;Trp53fl/fl mouse cohorts. Mice were treated with tamoxifen for 3 consecutive days at 8-10 weeks of age, then aged to analyze pancreatic cancer-free survival.
(B) Representative co-immunofluorescence staining for tdTomato (TOM), amylase (AMY; acinar cell marker), cytokeratin 19 (CK19; ductal cell marker), and insulin (INS; islet marker), in a Rosa26LSL-tdTomato/LSL-tdTomato;Ptf1aCreER;Trp53+/+ mouse pancreas 3 days after the last dose of tamoxifen (n = 5). DAPI stains nuclei. Scale Bar = 50 μm.
(C) Kaplan-Meier analysis of pancreatic cancer-free survival of cohorts listed in (A). Labels indicate the Trp53 status of each cohort. Pancreatic cancer-free survival in KT;Ptf1aCreER;Trp53fl/fl mice (n = 23) and KT;Ptf1aCreER;Trp53fl/+ mice (n = 6) is significantly shorter than that in KT;Ptf1aCreER;Trp53+/+ mice (n = 21), based on the log-rank test. Pancreatic cancer-free survival in KT;Ptf1aCreER;Trp53fl/fl mice (n = 23) is significantly shorter than that in KT;Ptf1aCreER;Trp53fl/+ mice (n = 6), based on the log-rank test. ***p-value < 0.001, ****p-value < 0.0001.
(D) Table summarizing the percentages of tumor-bearing mice in KT;Ptf1aCreER;Trp53+/+ (n = 9), KT;Ptf1aCreER;Trp53fl/+ (n = 6), and KT;Ptf1aCreER;Trp53fl/fl (n = 23) cohorts presenting with clinical symptoms of pancreatic cancer (ascites, bowel obstruction, and jaundice) at morbidity.
(E) Table summarizing the percentages of tumor-bearing KT;Ptf1aCreER;Trp53+/+ (n = 9), KT;Ptf1aCreER;Trp53fl/+ (n = 6), and KT;Ptf1aCreER;Trp53fl/fl (n = 23) mice with primary tumor grade (comprising > 50% of the tumor) called as moderately/well-differentiated adenocarcinoma, poorly-differentiated adenocarcinoma, or sarcomatoid carcinoma. The primary tumor grades of KT;Ptf1aCreER;Trp53+/+ mice are significantly different from KT;Ptf1aCreER;Trp53fl/fl mice by the Fisher’s Exact Test (p-value < 0.05).
(F) Representative histological images of lesions found in each cohort analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Alcian blue staining marks PanINs. Scale Bar = 100 μm.
(G) Representative histological images of metastases to the liver and lungs in KT;Ptf1aCreER;Trp53fl/fl mice analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Scale Bar = 100 μm.
Kaplan-Meier analysis revealed first that loss of p53 in KT;Ptf1aCreER;Trp53fl/fl mice led to rapid, fully penetrant development of PDAC, with a median pancreatic cancer-free and overall survival of 137 days after tamoxifen treatment (Figures 1C, S1A; Table S1). As expected, KT;Ptf1aCreER;Trp53fl/+ mice with heterozygous expression of wild-type Trp53 displayed a longer tumor latency with a median pancreatic cancer-free survival of 276.5 days after tamoxifen treatment. Interestingly, KT;Ptf1aCreER;Trp53+/+ mice also succumbed to pancreatic cancer, albeit with a significantly longer latency of 670 days. Approximately half of this latter cohort developed pancreatic cancer, with the rest displaying widespread premalignant PanIN lesions at morbidity. Of note, male KT;Ptf1aCreER;Trp53+/+ mice displayed significantly shorter pancreatic cancer-free and overall survival than female mice, in contrast to KT;Ptf1aCreER;Trp53fl/fl mice, where survival was similar between sexes (Figures S1B, S1C). At morbidity, some mice in all cohorts exhibited common human clinical symptoms of PDAC such as ascites, jaundice, and bowel obstruction (Figures 1D, S1D).
Histopathological analysis provided additional insight into PDAC development. We assigned a primary and secondary histological grade for each tumor (Table S1) and found that KT;Ptf1aCreER;Trp53fl/+ and KT;Ptf1aCreER;Trp53fl/fl mice consistently developed moderately/well-differentiated or poorly-differentiated adenocarcinomas characterized by Cytokeratin 19 (CK19) positivity (Figures 1E, 1F). Of tumor-bearing KT;Ptf1aCreER;Trp53+/+ mice, 44% developed moderately/well-differentiated adenocarcinomas, 22% developed poorly-differentiated adenocarcinomas, and 33% developed sarcomatoid carcinomas, a statistically significant shift in spectrum relative to KT;Ptf1aCreER;Trp53fl/fl mice. These tumors were either CK19-positive adenocarcinomas or sarcomatoid carcinomas with a notable positivity for vimentin and absence of CK19 immunostaining (Figures 1F, S1E). The majority of tumors in mice of all genotypes had tumor-adjacent PanIN lesions, as indicated by alcian blue positivity (Figure 1F, Table S1). Interestingly, one KT;Ptf1aCreER;Trp53+/+ mouse had evidence of a rare intraductal papillary mucinous neoplasm (IPMN; Figure S1F). Lineage tracing using the tdTomato allele verified that all PanINs, IPMNs, tumors, and metastases originated from Ptf1a-expressing cells, suggesting an acinar cell origin (Figures 1F, 1G, S1F, S1G, S1H). In addition, metastatic lesions were observed in all cohorts of mice at both the gross and histological levels. Liver metastases were the most common, with rare peritoneum, diaphragm, and lung metastases observed upon dissection in some animals (Figures 1G, S1H). Quantification of liver metastases by histological analysis revealed similar rates of metastasis, with 33% and 32% of tumors metastasizing in p53-expressing and p53-deficient mice, respectively (Table S1). Collectively, these findings demonstrate that oncogenic KRAS expression can drive pancreatic cancer development from adult mouse acinar cells. Although PDAC can develop in mice initially expressing wild-type p53, the latency of tumor development is significantly reduced with targeted p53 inactivation, underscoring the importance of p53 as a barrier to pancreatic cancer development in adults.
Expression of p53 missense mutants does not reduce the latency of acinar cell-derived PDAC development
To assess how the expression of mutant p53, given its potential gain-of-function effects, compares to complete p53 deficiency in adult acinar cell-derived pancreatic cancer development, we next generated mouse cohorts with oncogenic KRASG12D expression and expression of the p53 structural mutant p53R172H (KT;Ptf1aCreER;Trp53fl/LSL-R172H), the p53 contact mutant p53R270H (KT;Ptf1aCreER;Trp53fl/LSL-R270H) or p53 deficiency (KT;Ptf1aCreER;Trp53fl/−). We generated hemizygous cohorts in which expression of a Trp53 point mutant allele and a Trp53 null allele in the pancreas allows a clear assessment of potential gain-of-function activity rather than dominant-negative activity observed when the mutants are combined with a wild-type Trp53 allele. Importantly, by using a combination of a Trp53 floxed allele and conditional Lox-stop-lox activatable alleles for the Trp53 mutants (which are Trp53 null until Cre is active), we generated mice with a uniform Trp53 heterozygous genetic background in stromal tissues, for a controlled comparison of pancreatic cancer-free survival across all genotypes (Figure 2A).
Figure 2. Analysis of p53 missense mutants in acinar cell-derived pancreatic cancer development.
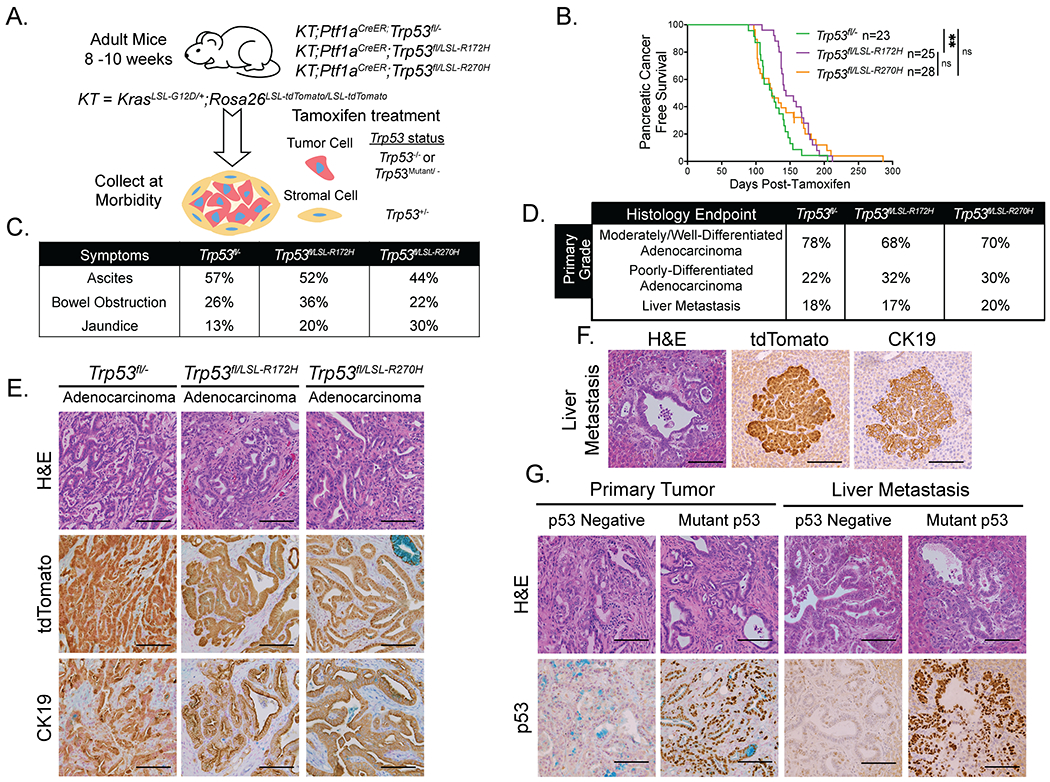
(A) Schematic for mutant p53 acinar cell-derived pancreatic cancer study of KT;Ptf1aCreER;Trp53fl/−, KT;Ptf1aCreER;Trp53fl/LSL-R172H, and KT;Ptf1aCreER;Trp53fl/LSL-R270H mouse cohorts. Mice were treated with tamoxifen for 3 consecutive days at 8-10 weeks of age, then aged to analyze pancreatic cancer-free survival. The genotypes of tumor cells and stromal cells after Cre action are indicated.
(B) Kaplan-Meier analysis of pancreatic cancer-free survival of cohorts listed in (A). Labels indicate the Trp53 status of each cohort. Pancreatic cancer-free survival of KT;Ptf1aCreER;Trp53fl/− mice (n = 23) is significantly shorter than that of KT;Ptf1aCreER;Trp53fl/LSL-R172H mice (n = 25) but similar to that of KT;Ptf1aCreER;Trp53fl/LSL-R270H mice (n = 28), based on the log-rank test. Pancreatic cancer-free survival is similar in KT;Ptf1aCreER;Trp53fl/LSL-R172H and KT;Ptf1aCreER;Trp53fl/LSL-R270H mice, based on the log-rank test. **p-value < 0.01. Not significant = ns.
(C) Table summarizing the percentages of KT;Ptf1aCreER;Trp53fl/− (n = 23), KT;Ptf1aCreER;Trp53fl/LSL-R172H (n = 25), and KT;Ptf1aCreER;Trp53fl/LSL-R270H (n = 27) mice presenting with clinical symptoms of pancreatic cancer (ascites, bowel obstruction, and jaundice) at morbidity.
(D) Table summarizing the percentages of tumor-bearing KT;Ptf1aCreER;Trp53fl/− (n = 23), KT;Ptf1aCreER;Trp53fl/LSL-R172H (n = 25), and KT;Ptf1aCreER;Trp53fl/LSL-R270H (n = 27) mice with primary tumor grade (comprising > 50% of the tumor) called as moderately/well-differentiated adenocarcinoma or poorly-differentiated adenocarcinoma as well as the frequencies of metastasis to the liver. All cohorts have similar primary tumor grades and metastasis frequencies, by Fisher’s Exact Test. *With the exception with of one mouse that developed a thymic lymphoma, necessitating early sacrifice, all of the mice in the KT;Ptf1aCreER;Trp53fl/LSL-R270H cohort had evidence of PDAC.
(E) Representative histological images of adenocarcinomas found in each cohort, analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Scale Bar = 100 μm.
(F) Representative histological image of a liver metastasis in a KT;Ptf1aCreER;Trp53fl/− mouse analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Scale Bar = 100 μm
(G) Examples of p53-negative and mutant p53-positive primary pancreatic tumors in a KT;Ptf1aCreER;Trp53fl/LSL-R172H mouse as well as p53-negative and mutant p53-positive liver metastases in KT;Ptf1aCreER;Trp53fl/LSL-R172H and KT;Ptf1aCreER;Trp53fl/LSL-R270H mice, respectively, analyzed by H&E staining and p53 immunohistochemistry. Scale Bar = 100 μm.
Analysis of these cohorts revealed that KT;Ptf1aCreER;Trp53fl/− mice deficient for p53 rapidly developed cancer with a median pancreatic cancer-free survival of 123 days after tamoxifen treatment (Figure 2B; Table S2). This latency is similar to that seen in KT;Ptf1aCreER;Trp53fl/fl mice, suggesting that the Trp53 heterozygous stroma does not significantly change tumor latency (Figure S2A). Furthermore, KT;Ptf1aCreER;Trp53fl/LSL-R270H mice expressing the contact mutant p53R270H had a similar pancreatic cancer-free survival of 123.5 days to KT;Ptf1aCreER;Trp53fl/− mice (Figure 2B), indicating that there is no significant gain-of-function effect driven by p53R270H expression in terms of tumor latency. Interestingly, analysis of the p53R172H structural mutant revealed a slightly enhanced pancreatic cancer-free survival of 144 days in the KT;Ptf1aCreER;Trp53fl/LSL-R172H mice relative to KT;Ptf1aCreER;Trp53fl/− mice with Trp53 deficiency, again indicating no clear gain-of-function effects with this mutant in terms of tumor latency (Figure 2B). As with KT;Ptf1aCreER;Trp53fl/fl mice, there was no significant difference in pancreatic cancer-free survival of female and male mice in each cohort (Figure S2B). Some mice in all cohorts exhibited common human clinical symptoms of PDAC such as ascites, jaundice, and bowel obstruction at morbidity at roughly the same frequencies (Figures 2C, S2C). Histopathological analysis revealed that p53 deficiency or expression of either p53 mutant consistently led to the development of adenocarcinomas, with 68-78% of tumors manifesting a moderately/well-differentiated primary tumor grade (Figure 2D, Table S2). These tumors were characterized by CK19 positivity and frequently had tumor-adjacent PanINs (Figure 2E, Table S2). Histological analysis of livers, the most common site of metastasis, revealed similar rates of metastasis across cohorts (Figures 2D, 2F, S2D, S2E). Each cohort also had evidence of rare metastases to the peritoneum, diaphragm and lung observed upon dissection (Figure S2F). Lineage tracing using the tdTomato allele confirmed that all tumors and metastases originated from Ptf1a-expressing cells, suggesting an acinar cell origin (Figures 2E, 2F, S2D, S2E, S2F).
That mutant p53 expression did not obviously change metastatic rates suggests that there is no clear gain-of-function effect of p53 point mutant expression in enhancing metastasis. Accordingly, ~25% of KT;Ptf1aCreER;Trp53fl/LSL-R172H and KT;Ptf1aCreER;Trp53fl/LSL-R270H mice had at least one primary tumor negative for mutant p53 expression, often accompanied by a metastasis negative for p53 expression (Figure 2G). These observations suggest that there is not an absolute selection for cells expressing mutant p53. Collectively, these findings demonstrate that, like p53 deficiency, mutant p53 promotes PDAC development, but does not exhibit a clear gain-of-function effect in terms of tumor latency and rates of metastasis in acinar cell-derived pancreatic cancer development.
Trp53 mutation is required for PDAC development from adult pancreatic ductal cells
We next sought to define the genetic requirements for PDAC development from adult pancreatic ductal cells. Toward this end, we generated mouse cohorts in which we activated oncogenic KrasG12D and modulated Trp53 using the tamoxifen-inducible Sox9CreER transgene (Figure 3A). Tamoxifen treatment of adult mice resulted in efficient genomic recombination in ductal cells, as indicated by expression of the tdTomato reporter allele in CK19-positive ductal cells (Figure 3B). We first assessed tumor development in mice expressing or lacking p53 by generating, tamoxifen treating, aging, and examining cohorts of KT;Sox9CreER;Trp53fl/fl and KT;Sox9CreER;Trp53+/+ mice for pancreatic cancer-free survival upon morbidity.
Figure 3. Pancreatic cancer can develop from adult mouse ductal cells.
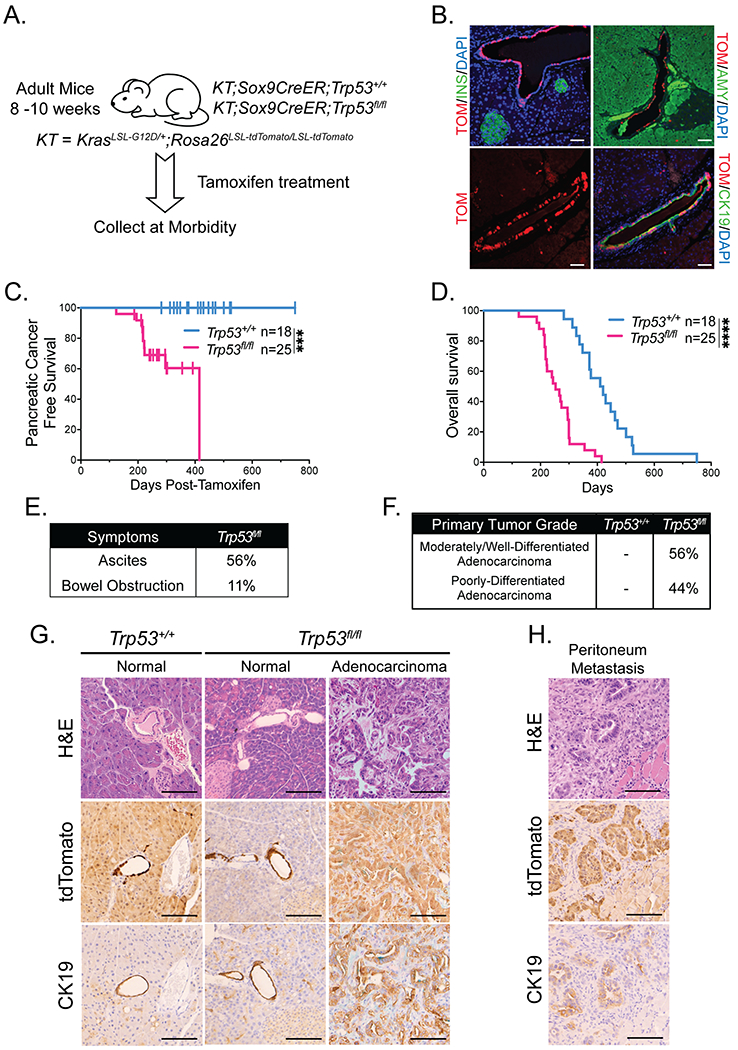
(A) Schematic for ductal cell-derived pancreatic cancer study of KT;Sox9CreER;Trp53+/+and KT;Sox9CreER;Trp53fl/fl mouse cohorts. Mice were treated with tamoxifen for 3 consecutive days at 8-10 weeks of age, then aged to analyze survival.
(B) Representative co-immunofluorescence staining for tdTomato (TOM) and amylase (AMY; acinar cell marker), cytokeratin 19 (CK19; ductal cell marker), and insulin (INS; islet marker), in a Rosa26LSL-tdTomato/LSL-tdTomato;Sox9CreER;Trp53+/+ mouse pancreas 3 days after the last dose of tamoxifen (n = 4). DAPI stains nuclei. Scale Bar = 50 μm.
(C) Kaplan-Meier analysis of pancreatic cancer-free survival of cohorts listed in (A). Labels indicate the Trp53 status of each cohort. Pancreatic cancer-free survival in KT;Sox9CreER;Trp53fl/fl mice (n = 25) is significantly different from that of KT;Sox9CreER;Trp53+/+ mice (n = 18), based on the log-rank test. ***p-value < 0.001.
(D) Kaplan-Meier analysis of overall survival of cohorts listed in (A). Labels indicate the Trp53 status of each cohort. Overall survival in KT;Sox9CreER;Trp53fl/fl mice (n = 25) is significantly different from that of KT;Sox9CreER;Trp53+/+ mice (n = 18), based on the log-rank test. ****p-value < 0.0001.
(E) Table summarizing the percentages of pancreatic tumor-bearing KT;Sox9CreER;Trp53fl/fl mice presenting with clinical symptoms of pancreatic cancer (ascites and bowel obstruction) at morbidity (n = 9).
(F) Table summarizing the percentages of tumor-bearing KT;Sox9CreER;Trp53fl/fl mice (n = 9) with primary tumor grade (comprising > 50% of the tumor) of moderately/well-differentiated or poorly-differentiated adenocarcinomas. KT;Sox9CreER;Trp53+/+ mice (n = 18) had no evidence of pancreatic tumors.
(G) Representative histological images of typical pancreas morphology found in each cohort, analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Scale Bar = 100 μm.
(H) Representative image of a peritoneum metastasis in a KT;Sox9CreER;Trp53fl/fl mouse analyzed by H&E staining and immunohistochemistry for tdTomato and CK19. Scale Bar = 100 μm.
Kaplan Meier analysis revealed that 32% of KT;Sox9CreER;Trp53fl/fl mice developed pancreatic cancer by ~300 days post-tamoxifen treatment (Figure 3C; Table S3). In contrast, none of the 18 KT;Sox9CreER;Trp53+/+ mice developed pancreatic cancer, suggesting that Trp53 loss is necessary for ductal cell-derived pancreatic cancer development. However, the ductal model is complicated by the expression of Sox9 in adult tissues beyond the pancreas, leading to the development of non-pancreatic tumors, such as Harderian gland adenocarcinomas, that necessitates early sacrifice with a median overall survival of 415 days in KT;Sox9CreER;Trp53+/+ mice and 253 days in KT;Sox9CreER;Trp53fl/fl mice (Figure 3D). Pancreata were therefore analyzed when mice reached morbidity due either to pancreatic or non-pancreatic tumors. At morbidity, some KT;Sox9CreER;Trp53fl/fl mice succumbing to PDAC exhibited common human clinical symptoms of PDAC such as ascites and bowel obstruction (Figure 3E). We observed both moderately/well-differentiated and poorly-differentiated adenocarcinomas in pancreatic tumor-bearing KT;Sox9CreER;Trp53fl/fl mice, at similar frequencies to KT;Ptf1aCreER;Trp53fl/fl mice (44% versus 39% poorly-differentiated tumors, respectively; Figures 1E, 3F). The adenocarcinomas were characterized by CK19-positivity and importantly, lineage tracing with tdTomato suggested a Sox9-expressing pancreatic cell-of-origin for these tumors (Figures 3G, S3A). Histological analysis and alcian blue staining failed to reveal PanINs associated with tumors in KT;Sox9CreER;Trp53fl/fl mice (Figure 3G; Table S3). In terms of establishing the incidence of metastasis, neither tumor histology nor tdTomato positivity could be used to distinguish PDAC metastases from primary cholangiocarcinomas resulting from Sox9CreER activity in the liver where it can promote neoplasia. However, visual inspection upon dissection, coupled with histological analysis, identified small clusters of tdTomato-positive malignant cells in the peritoneum and lungs of mice with clear PDACs, suggesting that metastasis had occurred (Figure 3H).
Next, to assess the gain-of-function phenotypes of mutant p53 expression in adult ductal cell-derived pancreatic cancer development, we generated mouse cohorts with oncogenic KRASG12D expression and expression of the structural mutant p53R172H (KT;Sox9CreER;Trp53fl/LSL-R172H), expression of the contact mutant p53R270H (KT;Sox9CreER;Trp53fl/LSL-R270H), or conditional Trp53 knockout (KT;Sox9CreER;Trp53fl/−). As with the acinar model, these mice were generated to create uniform Trp53 heterozygous stroma. Mice in all three cohorts displayed similar pancreatic cancer-free and overall survivals (Figures S3B, S3C) and exhibited clinical symptoms of pancreatic cancer development (Figure S3D). However, it was unclear if there was a gain-of-function phenotype as the incidence of pancreatic cancer development was low. Histopathological analysis revealed the development of poorly to moderately/well-differentiated adenocarcinomas in all cohorts and a sarcomatoid carcinoma in one KT;Sox9CreER;Trp53fl/− mouse (Figures S3E, S3F). Again, the adenocarcinomas were characterized by CK19 expression and tdTomato expression, confirming a Sox9-expressing pancreatic cell origin for these tumors (Figure S3F).
To investigate a potential precursor lesion for the ductal cell-derived tumors, we examined the pancreata of mice prior to cancer development. Analysis of KT;Sox9CreER;Trp53fl/fl mice at ~120 days – the midpoint of median overall survival in the ductal model – consistently failed to reveal evidence of potential precursor lesions, including ADMs or PanINs (Figure 4A). In stark contrast, KT;Ptf1aCreER;Trp53fl/fl mice showed early evidence of ADM and PanIN development at ~70 days, the midpoint of median overall survival in the acinar model (Figure 4B). When ductal cell-derived tumors developed, tumors were often were surrounded by uninvolved pancreas (Figure 4C), whereas acinar cell-derived tumors were typically surrounded by ADMs and PanINs (Figure 4D). Notably, some ducts in the pancreata of KT;Sox9CreER;Trp53fl/fl mice displayed phospho-ERK1/2 staining, as did ADMs and PanINs of KT;Ptf1aCreER;Trp53fl/fl mice, indicative of KRAS activation in these cells (Figure 4E). This observation, coupled with the lack other precursor lesions, suggests that ductal cell-derived tumors might originate directly from ducts. Collectively, these findings demonstrate that oncogenic KRAS expression and Trp53 inactivation or mutation can drive pancreatic cancer development directly from adult mouse ductal cells, highlighting a role for p53 in blocking pancreatic cancer development from ductal cells in adult mice.
Figure 4. Analysis of the precursors in PDAC development from acinar and ductal cells.
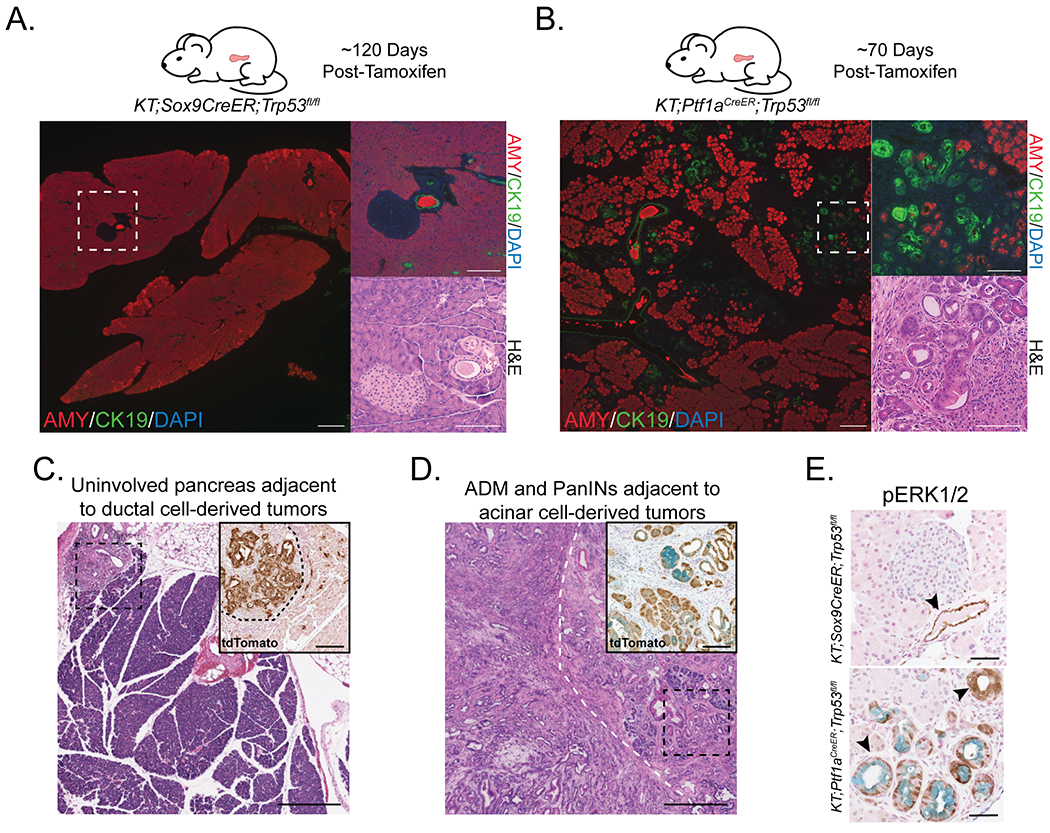
(A) (Left) Representative histological image of pancreas in KT;Sox9CreER;Trp53fl/fl mouse at ~120 days post-tamoxifen analyzed by co-immunofluorescence for AMY (acinar cell marker) and CK19 (ductal cell marker; n = 6). DAPI stains nuclei. Scale Bar = 200 μm. (Right) High magnification image of area within dashed box by co-immunofluorescence and H&E staining. Scale Bar = 100 μm.
(B) Representative histological image of pancreas in KT;Ptf1aCreER;Trp53fl/fl mouse at ~70 days post-tamoxifen analyzed by co-immunofluorescence for AMY (acinar cell marker) and CK19 (ductal cell marker; n = 7). DAPI stains nuclei. Scale Bar = 200 μm. (Right) High magnification image of area within dashed box by co-immunofluorescence and H&E staining. Scale Bar = 100 μm.
(C) Representative histological image of a tumor found in a KT;Sox9CreER;Trp53fl/fl mouse analyzed by H&E staining. Scale Bar = 400 μm. (Inset) High magnification of area within dashed box analyzed by immunohistochemistry for tdTomato. Black dashed curved line separates tumor from non-tumor area. Scale Bar = 100 μm.
(D) Representative histological image of a tumor found in a KT;Ptf1aCreER;Trp53fl/fl mouse, analyzed by H&E staining. White dashed curved line roughly separates tumor from non-tumor area. Scale Bar = 400 μm. (Inset) High magnification of area within black dashed box analyzed by immunohistochemistry for tdTomato. Alcian blue staining indicates PanINs. Scale Bar = 100 μm.
(E) Representative phospho-ERK1/2 immunostaining of the pancreas in KT;Sox9CreER;Trp53fl/fl (n = 6) and KT;Ptf1aCreER;Trp53fl/fl (n = 7) mice at approximately 120 and 70 days, respectively, the midpoint of median survival. Alcian blue staining marks PanINs. Arrowheads denote an example of a phospho-ERK1/2 expressing duct, ADM, or PanIN. Scale Bar = 50 μm.
Acinar and ductal cell-derived tumors are transcriptionally distinct
Our studies have demonstrated that pancreatic cancer can originate from either PTF1A- or SOX9-expressing pancreatic cells, which we posit represent acinar and ductal cells, respectively. To gain insight into whether the cell-of-origin affects the molecular subtype of PDAC, we performed transcriptomic analysis of bulk acinar cell-derived and ductal cell-derived tumors by RNA-sequencing (RNA-seq; Figure S4A). We focused on tumors with Trp53 deletion, rather than Trp53 point mutation, given the robust phenotypes observed with p53 deficiency in both tumor models. Notably, the set of acinar cell-derived and ductal-cell derived tumors we analyzed had similar differentiation profiles (Figures S4A, S4B). PCA analysis of transcriptomes revealed that acinar cell-derived and ductal cell-derived tumors cluster distinctly, suggestive of different molecular profiles (Figure 5A). Indeed, analysis of differentially-expressed genes by DESeq2 revealed that 1,075 genes are more highly expressed in acinar cell-derived tumors than in ductal cell-derived tumors and that 417 genes are more highly expressed in ductal cell-derived tumors than in acinar cell-derived tumors (log2 fold change > 1.0, p-adjusted value < 0.05; Figure 5B, Table S4). Immunohistochemical analysis confirmed the differential expression of proteins encoded by several of these genes, including CLDN18, TFF1, and MUC5AC, all of which showed higher expression in acinar cell-derived tumors than in ductal cell-derived tumors (Figure 5C).
Figure 5. Transcriptome analysis of mouse acinar and ductal cell-derived tumors.
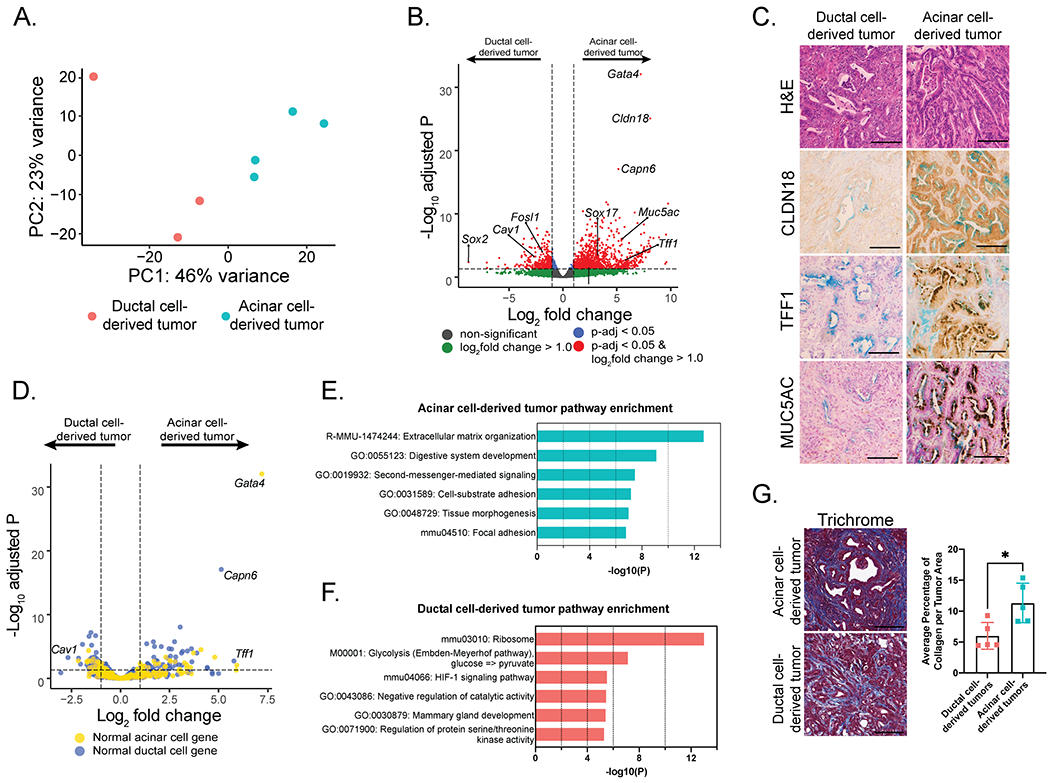
(A) Principal component analysis of gene expression profiles of PDACs from KT;Ptf1aCreER;Trp53fl/fl and KT;Sox9CreER;Trp53fl/fl mice.
(B) Volcano plot of differentially-expressed genes in acinar and ductal cell-derived tumors. A horizontal dashed line indicates an adjusted p-value of 0.05. Vertical dashed lines indicate an absolute log2 fold change of 1.0. Genes are color-coded based on adjusted p-value and absolute log2 fold change cut-offs.
(C) Representative histological images of acinar cell-derived (n = 5) or ductal cell-derived (n = 5) PDAC analyzed by H&E staining and immunohistochemistry for CLDN18, TFF1, or MUC5AC. Alcian blue staining indicates mucinous gland structures within tumors. Scale Bar = 100 μm.
(D) Volcano plot of normal pancreatic acinar cell genes highlighted in yellow and normal pancreatic ductal genes highlighted in blue. Genes are plotted based on their expression levels in acinar and ductal cell-derived tumors. A horizontal dashed line indicates an adjusted p-value of 0.05. Vertical dashed lines indicate an absolute log2 fold change of 1.0.
(E) The top 6 expression signatures enriched in acinar cell-derived tumors relative to ductal cell-derived tumors, as identified by Metascape analysis.
(F) The top 6 expression signatures enriched in ductal cell-derived tumors relative to acinar cell-derived tumors, as identified by Metascape analysis.
(G) (Left) Representative histological images of PDACs in KT;Sox9CreER;Trp53fl/fl (n = 5) and KT;Ptf1aCreER;Trp53fl/fl (n = 5) mice analyzed by Masson’s Trichrome staining. Scale Bar = 100 μm. (Right) Average percentage of collagen per tumor area +/− standard deviation. Each dot indicates a mouse. *p-value < 0.05 assessed by a two-tailed Student’s t-test.
We sought to understand the significance of the gene expression profiles specific to acinar-derived and ductal-derived tumors. We first asked whether each tumor type resembled the cell-of-origin. Comparison of the acinar and ductal cell-derived tumor signatures to single cell RNA-seq data for normal pancreatic acinar and ductal cells (38) failed to reveal a significant likeness of tumors to the cell-of-origin (Figures 5D, S4C). We next used functional annotation to reveal pathways specific to tumors derived from each cell-of-origin (Figures 5E, 5F). Gene Ontology analysis of the top pathways enriched in acinar cell-derived tumors revealed categories linked to extracellular matrix (ECM) organization, cell adhesion and digestive system development (Figure 5E). Consistent with the enhanced ECM organization, Masson’s Trichrome staining revealed increased collagen staining in acinar cell-derived tumors compared to ductal cell-derived tumors (Figure 5G). In contrast to the acinar cell-derived tumors, the ductal cell derived-tumors displayed enrichment for GO terms such as ribosome function and glycolysis (Figure 5F). These findings demonstrate that the molecular profile of pancreatic tumors is dependent on the cell-of-origin.
Acinar and ductal cell-derived tumor signatures correlate with distinct molecular subtypes of human PDAC
As described above, human PDAC has been classified into discrete molecular subtypes by transcriptomic analyses (13–15). We hypothesized that the emergence of these tumor subtypes could be influenced by cell-of-origin. We therefore sought to determine whether the acinar and ductal cell-derived tumor signatures are enriched in different molecular subtypes of human pancreatic cancer. We defined acinar cell-derived and ductal cell-derived tumor signatures based on the highest-confidence differentially-expressed mouse genes, using a log2 fold change > 1.5 and p-adjusted value < 0.01. We thus identified 640 genes as differentially-expressed between acinar cell-derived and ductal cell-derived tumors, 573 of which had human orthologs (Figure 6A). Of these, 496 genes were more highly expressed in acinar cell-derived tumors than in ductal cell-derived tumors and 77 genes were more highly expressed in ductal cell-derived tumors than in acinar cell-derived tumors.
Figure 6. Comparison of acinar and ductal cell-derived tumor signatures to molecular subtypes of human PDAC.
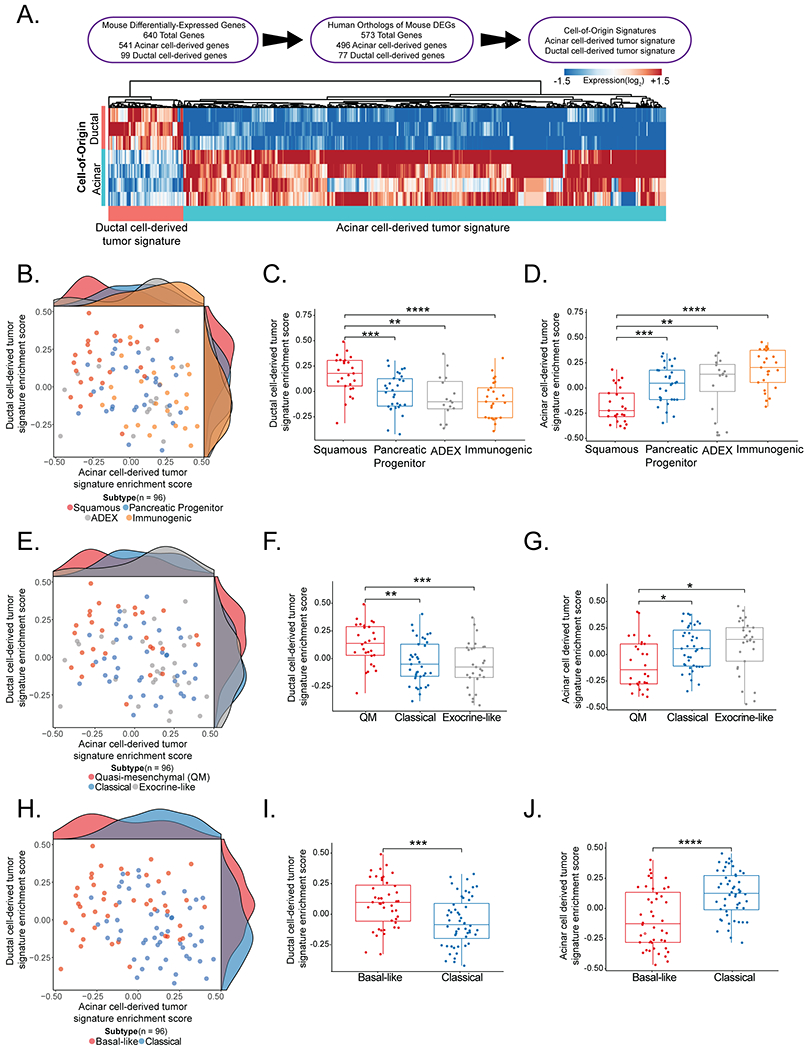
(A) (Top) Scheme summarizing how acinar and ductal cell-derived tumor signatures were derived. (Bottom) A heat map of the genes defining the signatures.
(B) Scatter plot of acinar cell-derived tumor and ductal cell-derived tumor signature enrichment scores in primary PDAC samples (ICGC - AU, n = 96) classified as squamous, pancreatic progenitor, immunogenic, or ADEX molecular subtypes. Each dot represents a tumor sample, color coded by molecular subtype.
(C) Box plot of the ductal cell-derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as squamous, pancreatic progenitor, immunogenic, or ADEX molecular subtypes. The ductal cell-derived signature enrichment score is significantly lower in tumors classified as the pancreatic progenitor (n = 30), ADEX (n = 16), or immunogenic (n = 25) subtypes than the squamous subtype (n = 25), based on a two-tailed Student’s t-test. ** p-value < 0.01 ***p-value < 0.001, ****p-value < 0.0001.
(D) Box plot of the acinar cell-derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as squamous, pancreatic progenitor, immunogenic, or ADEX molecular subtypes. The acinar cell-derived signature enrichment score is significantly higher in tumors classified as the pancreatic progenitor (n = 30), ADEX (n = 16), or immunogenic (n = 25) subtype than the squamous subtype (n = 25), based on a two-tailed Student’s t-test. **p-value < 0.01 ***p-value < 0.001, ****p-value < 0.0001.
(E) Scatter plot of acinar cell-derived tumor and ductal cell-derived tumor signature enrichment scores in primary PDAC samples (ICGC - AU, n = 96) classified as quasi-mesenchymal, classical, or exocrine-like molecular subtypes. Each dot represents a tumor sample, color coded by molecular subtype.
(F) Box plot of the ductal cell-derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as quasi-mesenchymal, classical, or exocrine-like molecular subtypes. The ductal cell-derived signature enrichment score is significantly lower in tumors classified as the classical (n = 39) or exocrine-like (n = 29) subtype than the quasi-mesenchymal subtype (n = 28), based on a two-tailed Student’s t-test. ** p-value < 0.01 ***p-value < 0.001.
(G) Box plot of the acinar cell-derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as quasi-mesenchymal, classical, or exocrine-like molecular subtypes. The acinar cell-derived signature enrichment score is significantly higher in tumors classified as the classical (n = 39) or exocrine-like (n = 29) subtype than the quasi-mesenchymal subtype (n = 28), based on a two-tailed Student’s t-test. *p-value < 0.05.
(H) Scatter plot acinar cell-derived tumor and ductal cell-derived tumor signature enrichment scores in primary PDAC samples (ICGC - AU, n = 96) classified as basal-like or classical molecular subtypes. Each dot represents a tumor sample, color coded by molecular subtype.
(I) Box plot of the ductal cell derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as basal-like or classical molecular subtypes. The ductal cell-derived signature enrichment score is significantly higher in tumors classified as the basal-like (n = 44) than the classical (n = 52) subtype, based on a two-tailed Student’s t-test. ***p-value < 0.001.
(J) Box plot of the acinar cell derived tumor signature enrichment score in primary PDAC samples (ICGC - AU, n = 96) classified as basal-like or classical molecular subtypes. The acinar cell-derived signature enrichment score is significantly lower in tumors classified as the basal-like (n = 44) than the classical (n = 52) subtype, based on a two-tailed Student’s t-test. ****p-value < 0.0001.
To investigate whether these defined signatures were associated with particular human PDAC subtypes, we interrogated whether there is an enrichment of either the acinar cell-derived tumor signature or ductal cell-derived tumor signature in human PDACs classified using published datasets. We began with the 4 molecular subtype classification scheme – squamous, pancreatic progenitor, immunogenic, and abnormally differentiated endocrine exocrine (ADEX) – defined through analysis of the Australian International Cancer Genome Initiative (ICGC) patient cohort using RNA-seq of untreated bulk primary PDACs of high cellularity (18). Notably, generation of this data set through RNA-seq analysis of bulk primary tumors mirrors the input material and methodology of our mouse gene expression analysis. Strikingly, gene set variation analysis (GSVA) of these data revealed a significant enrichment of the ductal cell-derived tumor signature in tumors of the squamous subtype (Figure 6B, 6C). In contrast, the acinar cell-derived tumor signature was significantly enriched in the pancreatic progenitor, ADEX, and immunogenic subtypes – which are now thought to be within a single broad subtype known as classical – relative to the squamous subtype (19) (Figure 6B, 6D). To confirm subtype enrichment of the cell-of-origin signatures in an independent cohort, we leveraged an RNA-seq data set of bulk primary PDACs from The Cancer Genome Atlas (TCGA) Research Network (6), classified according to the four subtype classification scheme. Again, GSVA uncovered a significant enrichment of the ductal cell-derived tumor signature in the squamous subtype relative to the pancreatic progenitor, immunogenic and ADEX subtypes (Figure S5A, S5B). In contrast, the acinar cell-derived tumor signature was significantly enriched in the pancreatic progenitor, ADEX, and immunogenic subtypes relative to the squamous subtype (Figure S5A, S5C).
We then compared the patterns of enrichment of our signatures in the Australian ICGC PDAC patient cohort processed using the other major classification schemes (14,15). The ICGC data set classified using the classical, quasi-mesenchymal, and exocrine-like molecular subtype designations, which were derived from microarray analysis of microdissected epithelium of untreated, primary PDACs (15), revealed a significant enrichment of the ductal cell-derived tumor signature in tumors classified as the quasi-mesenchymal subtype relative to the other subtypes (Figures 6E, 6F). In contrast, the acinar cell-derived tumor signature was significantly enriched in the classical and exocrine-like subtypes relative to the quasi-mesenchymal subtype (Figures 6E, 6G). The Australian ICGC PDAC patient data was then classified using the basal-like and classical designations originally derived from analysis of bulk, resected untreated primary PDACs and metastases by both microarrays and RNA-seq, followed by the exclusion of transcripts expressed in the normal pancreas (14). The ductal cell-derived tumor signature was significantly enriched in basal-like tumors (Figures 6H, 6I) and the acinar cell-derived tumor signature was significantly enriched in classical tumors (Figures 6H, 6J). Subsequent studies have led to the consensus that there are two broad subtypes – the basal-like (including the squamous or quasi-mesenchymal subtypes) and classical subtype (including the pancreatic progenitor, immunogenic, ADEX, and exocrine-like subtypes) (19,20,22), and our findings suggest that the acinar cell-derived tumor signature is associated with the classical subtype and the ductal cell-derived tumor signature correlates with the basal-like subtype.
The cell-of-origin signatures are associated with patient survival outcomes
We next evaluated the clinical significance of our cell-of-origin signatures by examining associations with PDAC patient overall survival in the Australian ICGC cohort. Interestingly, patients whose tumors showed low expression of the acinar cell-derived signature had significantly worse overall survival than those with high expression of the acinar signature (Figure 7A). In contrast, patients whose tumors showed high expression of the ductal cell-derived signature had significantly worse overall survival than those with low expression of the ductal signature (Figure 7B). Cox proportional hazard analysis demonstrated that high expression of the ductal cell-derived signature remained significantly associated with decreased overall survival in a model that incorporated key clinical covariates such as age, tumor stage and grade and subtype classification, while high expression of the acinar cell derived signature was independently associated with improved overall survival (Figure 7C). These independent survival associations were confirmed in a meta-analysis of three PDAC patient cohorts, including the Australian and Canadian ICGC cohorts and the TCGA cohort (Figure 7D). Similar to the Australian ICGC cohort, the Canadian ICGC cohort again demonstrated significant associations of the acinar cell-derived signature with improved survival and of the ductal cell-derived signature with decreased survival (Figure S6A). It is unclear why the TCGA cohort alone does not reflect the same outcome associations as the ICGC cohorts (Figure S6B), but this could be related to differences in epithelial purity of these samples (13,20). Collectively, our findings indicate that the acinar and ductal cell-derived tumor signatures are independently associated with survival of human PDAC patients. Moreover, these findings demonstrate that the very earliest events in carcinogenesis can impart on tumors a fundamental program that drives cancer subtype and phenotype.
Figure 7. The cell-of-origin signature is associated with survival outcomes.
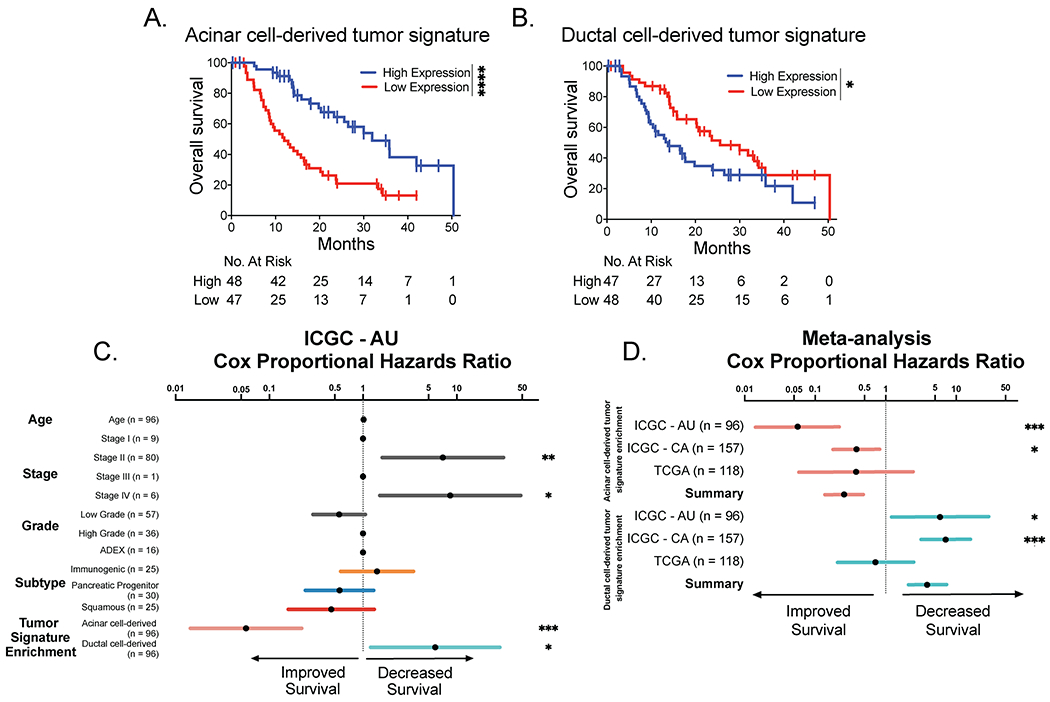
(A) Kaplan-Meier analysis of the Australian ICGC PDAC patient cohort overall survival stratified by expression of the acinar cell-derived tumor signature. Patients with high expression of the acinar cell-derived tumor signature (n = 48) exhibit significantly better survival than in patients with low expression of the acinar cell-derived signature (n = 47) based on the log-rank test. ****p-value < 0.0001.
(B) Kaplan-Meier analysis of the Australian ICGC PDAC patient cohort overall survival stratified by the ductal cell-derived tumor signature. Patients with high expression of the ductal cell-derived tumor signature (n = 47) exhibit significantly worse survival than patients with low expression of the ductal cell-derived signature (n = 49) based on the log-rank test. ****p-value < 0.0001.
(C) Cox proportional hazards ratios of the association between age, tumor, stage or grade, 4 subtype PDAC classification (ADEX, immunogenic, pancreatic progenitor, or squamous) or the acinar or ductal cell-derived tumor signature enrichment (where high expression is reflective of enrichment) and overall survival of the Australian ICGC patient cohort (ICGC – AU; n = 96). Asterisks indicate the factors that are significantly associated with overall survival. *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001.
(D) Meta-analysis of the cox proportional hazards ratios of the acinar or ductal cell-derived tumor signature enrichment (where high expression is reflective of enrichment) in the Australian ICGC (ICGC – AU), Canadian ICGC (ICGC – CA), and TCGA patient cohorts, shown individually and combined. Asterisks indicate invidual cohorts where the cell-of-origin signature enrichment is significantly associated with overall survival. *p-value < 0.05, ***p-value < 0.001.
DISCUSSION
Here, we use genetically engineered mouse models to better understand the cellular and molecular origins of different subtypes of pancreatic cancer. To best recapitulate human cancer development, we induced mutations in different cellular compartments of the pancreas in adult mice and aged large cohorts until morbidity. Our aging studies revealed first that oncogenic KRAS expression, coupled with Trp53 deletion, in adult acinar cells efficiently drives metastatic PDAC, supporting the idea that p53 imposes an important barrier to PDAC development from acinar cells. Interestingly, by aging our acinar cell-derived tumor cohorts extensively, we observed that oncogenic KRAS expression can also drive metastatic PDAC development in the context of wild-type p53. In addition, KRASG12D expression in acinar cells, coupled with Trp53 point mutant alleles, promotes tumorigenesis. Upon interrogating adult ductal cells as a cell-of-origin, we found that expression of activated KRAS can promote PDAC, but only in the context of Trp53 inactivation or mutation, and that tumor latency is significantly longer than for acinar cell-derived PDAC development. Transcriptomic analysis of tumors arising from each compartment upon KRAS activation and Trp53 inactivation revealed that the ductal cell-derived tumor signature is enriched in the basal-like subtype, while the acinar cell-derived tumor signature is enriched in the classical subtype. Our studies thus illuminate cell-of-origin as one factor that determines subtypes of PDAC.
The cell-of-origin of human pancreatic cancer has been controversial. Historically, pancreatic ductal cells were presumed to be the PDAC cell-of-origin based on the histological similarity of PDACs to normal pancreatic ducts (39,40). However, this notion was revised when experiments in mouse models demonstrated that KRAS expression and Trp53 mutation in acinar cells can trigger PDAC, suggesting that PDAC can originate from acinar cells that undergo ADM, which then become PanINs and ultimately PDAC (29–31,34–36). Subsequent studies of ductal cells – one using 3-4 week old Sox9CreER;KrasLSL-G12D;Trp53fl/fl mice and the other using 7-9 week old Hnf1bCreER;KrasLSL-G12D;Trp53LSL-R172H/LSL-R172H;R26mTmG mice – suggested that ductal cells can also give rise to PDAC in specific contexts (30,33). Whereas the former model showed PDAC development accompanied by high-grade PanINs, possibly due to the age of tumor initiation when the pancreas was still rapidly growing (41), our studies in adult mice and the latter model demonstrated PDAC without clear evidence of PanINs, suggesting another trajectory for these tumors. This path contrasts dramatically with that of acinar cell-derived tumors, which are associated with ADM and PanINs, the presumed precursor for PDACs. Alternatively, it is possible that PanINs arise in the ductal model but universally progress into tumors, thus obliterating evidence of the initial lesion. Indeed, earlier studies observed the development of PanIN-like lesions after orthotopic transplantation of genetically modified primary human ductal cells (42). We did not, however, observe PanINs, hyperplasia, or dysplasia in numerous mice, even when pancreata were collected at earlier timepoints. The phospho-ERK1/2 positive ducts seen in these mice, in conjunction with the morphology of early tumors, suggests that tumors may therefore arise from ductal cells. While the tumors in the Sox9CreER mice most likely originated from ductal cells, it is also formally possible that they originated from other SOX9-expressing cells in the pancreas such as centroacinar cells or rare SOX9-expressing acinar cells (43–46). Regardless of the exact identity of the SOX9-expressing cell that gives rise to PDAC, our results clearly suggest that different cells-of-origin targeted by Sox9CreER and Ptf1aCreER give rise to fundamentally different pancreatic cancers.
Interestingly, pancreatic tumor histology was significantly influenced by p53 status. We found that Trp53 loss or mutation led to the development of primarily poor to moderately/well-differentiated adenocarcinomas, similar to reports in Pdx1-Cre;KrasLSL-G12D/+;Trp53LSL-R172H/+ mice (9,12). In contrast, KT;Ptf1aCreER;Trp53+/+ mice developed both adenocarcinomas and sarcomatoid carcinomas tumors. This latter observation is reminiscent of previous reports of sarcomatoid tumors developing in Pdx1-Cre;KrasLSL-G12D/+ and Pdx1-Cre;KrasLSL-G12D/+;p16−/− mice, cohorts with intact p53 (12). Notably, an IPMN, which have been associated with Smad4 deficiency (11,47), was also observed in a KT;Ptf1aCreER;Trp53+/+ mouse. Future studies will delineate how loss of different tumor suppressor genes affects the histological type of pancreatic cancer arising from different cells-of-origin.
Notably, we did not observe a gain-of-function effect of mutant p53 expression, with either the p53R270H contact mutant or the p53R172H structural mutant, in terms of tumor latency and rates of metastasis. Previous characterization of these point mutants in mouse cancer models has similarly shown that tumor latency is not altered by expression of the point mutants, although gain-of-function phenotypes relating to the appearance of new tumor types and enhanced rates of metastasis were reported (48–50). We did not observe enhanced metastasis with expression of the point mutants relative to p53 deficiency, which suggests some context-dependency for this phenotype. Indeed, this behavior is not unprecedented, as expression of p53R172H in a hepatocellular carcinoma model also did not enhance development and progression (51). Interestingly, we noted that acinar cell-derived PDAC was in fact significantly delayed in the presence of p53R172H, an observation potentially explained by the ability of this protein to fold into the proper conformation in certain settings (52). Overall, at the level of resolution of our studies, it appears that loss of wild-type p53 function, rather than gain-of-function p53 activities, is the predominant driver of PDAC development.
In characterizing PDAC molecular subtypes by transcriptomic and genomic analyses, associations with specific genetic events have been described (13–15,23). The basal-like subtype has been correlated with certain mutations, such as those in TP53 and KDM6A, along with downregulation of genes of endodermal identity (13–15,19–21,23,24). The classical subtype is characterized by expression of genes related to an endodermal cell fate such as GATA transcription factors (13–15,19,20,24). While these molecular subtypes have been linked to certain genetic lesions post facto, our transcriptome analysis directly interrogates the connection between the cell-of-origin and molecular subtype. We show not only that acinar cells can give rise to tumors of the classical subtype and ductal cells can give rise to tumors of the basal-like subtype, but also that both subtypes can originate from cells deficient for Trp53. In future, single cell RNA-seq analysis of malignant and adjacent pre-malignant pancreas tissue in our mouse PDAC models will enhance our understanding of PDAC evolution. In particular, it is now feasible to computationally order cellular trajectories from such data (53), which would provide insight into the dynamics of tumor development from acinar and ductal cells. Single cell resolution will help not only to identify intermediate states during tumor progression but also to decipher the significance of recent observations of human PDACs with intratumoral heterogeneity, where tumors are comprised of cells of both the classical and basal-like subtypes (21,24).
Elucidating the cell-of-origin of specific cancer types not only provides key insight into the biological basis for tumor evolution, but also illuminates possible routes to improved clinical intervention. Genetically engineered mouse models have been fundamental for identifying the cell-of-origin of diverse cancers, such as intestinal, breast, and lung cancers, and for defining cell-of-origin as a critical parameter influencing intertumoral heterogeneity (54,55). Our study underscores the importance of both acinar and ductal cells as tumor-initiating cells for PDAC, as well as illuminating the distinct molecular subtypes of PDAC arising from these cells. Although different molecular classes of PDAC have been previously defined, the existing subtype signatures are not currently used to guide therapeutic decision-making. Understanding the underlying biological basis for these distinct subtypes may help to refine signatures and thus improve strategies for early diagnosis, prognostication, and therapy. As new PDAC therapies are pursued, it will be instructive to stratify patients using our cell-of-origin signatures, which have a clear biological basis, when assessing the efficiency of such therapies to determine if they preferentially act in tumors of a particular subtype. Indeed, we have identified distinct functional annotation signatures for acinar and ductal-cell tumors, and these may provide insight into therapeutic vulnerabilities specific to each subtype. For example, our ductal cell-derived signature displays a striking enrichment of genes involved in glycolysis, and, accordingly, some patient-derived cell lines of the squamous subtype are sensitive to glycolysis inhibition (56). In addition, although our study suggests that cell-of-origin is one factor that influences PDAC molecular subtype, other factors such as genetic and epigenetic changes during tumor progression likely also influence tumor subtype (24). Continued investigation will further reveal which additional factors, such as genetic and epigenetic changes, influence molecular subtype in PDAC and will enable this information to be utilized for improved clinical management of PDAC.
METHODS
Mouse work
All mouse work was approved and performed in compliance with the Stanford University Institutional Animal Care and Use Committee known as the Administrative Panel on Laboratory Animal Care (APLAC). Mice were maintained on a mixed 129/Sv-C57BL/6 background. Previously described mouse strains carrying the following alleles were used: Ptf1aCreER (RRID:IMSR_JAX:019378), Sox9CreER (RRID:IMSR_JAX:018829), Trp53fl (RRID:IMSR_JAX:008462), Trp53null (RRID:IMSR_JAX:002101), Trp53LSL-R172H (RRID:IMSR_JAX:008652), Trp53LSL-R270H (RRID:IMSR_JAX:008651), KrasLSL−12D (RRID:IMSR_JAX:008179), and Rosa26LSL-tdTomato (RRID:IMSR_JAX:007909). Mice were genotyped by PCR. Cohorts with similar male and female representation were generated to account for sex as a biological variable. At 8-10 weeks of age, mice were administered 5 mg tamoxifen dissolved in 2% ethanol/corn oil via oral gavage once per day for 3 consecutive days. Unless indicated otherwise, mice were aged until morbidity. At morbidity, tumor tissue was fixed for histopathology or flash-frozen in liquid nitrogen for additional analyses.
Histology
Hematoxylin and eosin (H&E) staining and Masson’s trichrome staining were performed on paraffin-embedded tissues using standard protocols. All tumors were graded under blind review by a pathologist. Masson’s trichrome staining was quantified using the FIJI Software (RRID:SCR_002285). Liver metastases were assessed through analysis of one H&E section per liver. Whole mount brightfield and fluorescent images were captured from freshly dissected tissue using a Leica stereo microscope and Nightsea stereo microscope fluorescent adapter.
Immunostaining
Immunohistochemistry and immunofluorescence were performed using standard protocols. For unmasking, sections were incubated in 0.01 M citrate buffer (pH 6.0) or Tris-EDTA buffer (10 mM Tris, 1 mM EDTA, 0.05% Tween 20, pH 9.0) in a pressure cooker for 13 minutes. Primary antibodies directed against the following proteins were used: Amylase (Santa Cruz Biotechnology Cat# sc-46657, RRID:AB_626668), CK19 (DSHB Cat# TROMA-III, RRID:AB_2133570), CLDN18 (Thermo Fisher Scientific Cat# 700178, RRID:AB_2532290), Insulin (Cell Marque Cat# 273A-14, RRID:AB_1158520), MUC5AC (Lab Vision Cat# MS-145-P1, RRID:AB_62736), phospho-ERK1/2 (Cell Signaling Technology Cat# 4370, RRID:AB_2315112), p53 (Leica Biosystems Cat# P53-CM5P, RRID:AB_2744683), RFP/tdTomato (Rockland Cat# 600-401-379, RRID:AB_2209751), TFF1 (ABclonal Cat# A1789, RRID:AB_2763830) and Vimentin (Cell Signaling Technology Cat# 5741, RRID:AB_10695459). For immunohistochemistry, the following biotinylated secondary antibodies were used: anti-rabbit (Vector Laboratories Cat# BA-1000, RRID:AB_2313606), anti-rat (Vector Laboratories Cat# BA-9401, RRID:AB_2336208), and anti-mouse (Vector Laboratories Cat# BA-2000, RRID:AB_2313581). Following biotinylated antibody incubation, the VECTASTAIN Elite ABC HRP Kit (Vector Laboratories Cat# PK-6100, RRID:AB_2336819) was used. Subsequent chromogenic staining was performed with the DAB peroxidase substrate kit (Vector Laboratories Cat# SK-4100, RRID:AB_2336382) and sections were counterstained using Gill’s formula hematoxylin (Vector Laboratories Cat# H-3401, RRID:AB_2336842) or the NovaUltra Alcian Blue Stain kit (IHC World, Cat# IW-3000). For immunofluorescence, the following secondary antibodies were used: Alexa Fluor 488 anti-rat (Molecular Probes Cat# A-21208, RRID:AB_141709), Alexa Fluor 546 anti-mouse (Molecular Probes Cat# A-11030, RRID:AB_144695), fluorescein anti-rabbit (Vector Laboratories Cat# FI-1000, RRID:AB_2336197) and fluorescein anti-mouse (Vector Laboratories Cat# FI-2000, RRID:AB_2336176). Sections mounted with DAPI mounting media (ProLong Fluoromount G Mounting Medium, RRID:SCR_015961). All images were taken using a Leica DM6000B microscope (RRID:SCR_018713), an Aperio AT2 slide scanner (Leica Biosytems), and/or a NanoZoomer 2.0-RS slide scanner (Hamamatsu).
RNA-seq Library Preparation and Data Analysis
RNA was extracted from bulk tumors using the RNeasy midi kit (Qiagen, Cat#75144). RNA quality was assessed using a BioAnalyzer (2100 Bioanalyzer Instrument, RRID:SCR_018043), and only samples with an RNA integrity number (RIN) greater than 8.0 were included for library preparation. Novogene generated and sequenced RNA-seq libraries using the Illumina TruSeq RNA Sample Preparation kit (Cat# RS-122-2001) and Illumina HiSeq4000 (RRID:SCR_016386). Reads were aligned to the GRCm38 mouse genome using HISAT2 (v 2.0.4; RRID:SCR_015530). Sorted BAM files were generated using Samtools (v 1.3.1; RRID:SCR_002105). The number of reads mapping to each gene in the mouse Ensembl database (v 87; RRID:SCR_002344) was counted using HTSeq-count (v 0.6.1; RRID:SCR_011867). Differential gene expression analysis was performed using DESeq2 (v 1.24.0; RRID:SCR_015687). Pathway analysis was performed using Metascape (v 3.5; RRID:SCR_016620).
Acinar and Ductal Cell-Derived Tumor Signature Enrichment Analysis
The acinar or ductal-cell derived tumor signatures comprise genes with an absolute log2 fold change >1.5 and an adjusted p-value <0.01 in the differential gene expression analysis described above. Human orthologs of the derived mouse genes were obtained using R package BioMart (v 2.40.5; RRID:SCR_002987). We used publicly available gene expression and clinical data for the ICGC Australian (13), ICGC Canadian (PACA-CA, downloaded from the ICGC data portal – https://dcc.icgc.org/) and TCGA (20) PDAC cohorts. R package Gene Set Variation Analysis (GSVA) v 1.32.0 (57) was used to estimate the enrichment of the acinar or ductal-cell derived tumor signatures in human PDAC cohorts.
PDAC Molecular Subtype Assignment
Human PDAC samples from the ICGC Australian and TCGA cohorts were assigned to molecular subtypes of squamous, pancreatic progenitor, ADEX, and immunogenic based on published subtype designations (13,20). Subtype designations of quasi-mesenchymal, classical, exocrine-like, and basal-like were not published for the ICGC Australian cohort. Therefore, GSVA was used to calculate a signature enrichment score using the published gene sets for each subtype classification (14,15). Tumors were then clustered using the k-means algorithm for all signatures and the subtype was assigned to the cluster with the highest score.
Statistical Analyses
GraphPad Prism (v 8.3.1; RRID:SCR_002798) and R (v 3.6.1; RRID:SCR_001905) were used for analysis and graphical representations. The n number is included in the figure legends and represents the number of mice analyzed per genotype for biological replication. GraphPad Prism was used to perform the log-rank test for Kaplan-Meier analysis. The Survival package (v 2.44-1.1) was used for the cox proportional hazards regression model. The SurvComp package (v 1.32; RRID:SCR_003054) version 1.32 was used to compute meta-analysis statistics. Power analysis was not used in this study because sample sizes depended on available datasets.
Data Availability
Next-generation sequencing data for the RNA-seq experiments have been deposited at the Gene Expression Omnibus (RRID:SCR_005012) under accession number GSE164180.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Although human PDAC has been classified into different molecular subtypes, the etiology of these distinct subtypes remains unclear. Using mouse genetics, we reveal that cell-of-origin is an important determinant of PDAC molecular subtype. Deciphering the biology underlying pancreatic cancer subtypes may reveal meaningful distinctions that could improve clinical intervention.
ACKNOWLEDGEMENTS
We thank Tyler Jacks and Monte Winslow for mouse strains. We thank the staff at the Stanford Functional Genomics Facility for their technical support. We thank Pauline Chu for the processing and sectioning of all paraffin-embedded mouse tissue samples. We thank Steven Artandi for the Insulin antibody. We thank Monte Winslow, Anthony Boutelle, and Alyssa Kaiser for critical reading of the manuscript. This work was supported by a National Cancer Institute R35 grant CA197591 to L.D. Attardi, a Blavatnik Family Fellowship to B.M. Flowers, and a National Institutes of Health Director’s Pioneer Award DP1-CA238296 to C. Curtis.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
REFERENCES
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74(11):2913–21 doi 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Oberstein PE, Olive KP. Pancreatic cancer: why is it so hard to treat? Therap Adv Gastroenterol 2013;6(4):321–37 doi 10.1177/1756283X13478680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2016;30(4):355–85 doi 10.1101/gad.275776.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 2018;8(9) doi 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguirre AJ, Hruban RH, Raphael BJ, Canc Genome Atlas Res N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32(2):185-+ doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res 2000;6(8):2969–72. [PubMed] [Google Scholar]
- 8.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4(6):437–50 doi 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 9.Hingorani SR, Wang LF, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53(R172H) and KraS(G12D) cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7(5):469–83 doi 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 10.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17(24):3112–26 doi 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006;20(22):3130–46 doi 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A 2006;103(15):5947–52 doi 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 14.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genet 2015;47(10):1168–78 doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu SD, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17(4):500–3 doi 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu SD, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17(4):500–U140 doi 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genet 2015;47(10):1168-+ doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47-+ doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 19.Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2019;16(4):207–20 doi 10.1038/s41575-019-0109-y. [DOI] [PubMed] [Google Scholar]
- 20.Aguirre AJ, Hruban RH, Raphael BJ, Canc Genome Atlas Res N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32(2):185–203 doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi A, Fan J, Chen R, Ho Y-j, Makohon-Moore AP, Lecomte N, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer 2020;1(1):59–74 doi 10.1038/s43018-019-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurer C, Holmstrom SR, He J, Laise P, Su T, Ahmed A, et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019;68(6):1034–43 doi 10.1136/gutjnl-2018-317706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018;155(6):1999–2013 e3 doi 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 24.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020;52(2):231–40 doi 10.1038/s41588-019-0566-9. [DOI] [PubMed] [Google Scholar]
- 25.Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ 2018;25(1):161–8 doi 10.1038/cdd.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mello SS, Attardi LD. Not all p53 gain-of-function mutants are created equal. Cell Death Differ 2013;20(7):855–7 doi 10.1038/cdd.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat 2016;37(9):865–76 doi 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 28.Mello SS, Valente LJ, Raj N, Seoane JA, Flowers BM, McClendon J, et al. A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 2017;32(4):460–73 e6 doi 10.1016/j.ccell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Habbe N, Shi GL, Meguid RA, Fendrich V, Esni F, Chen HP, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 2008;105(48):18913–8 doi 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee AYL, Dubois CL, Sarai K, Zarei S, Schaeffer DF, Sander M, et al. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2019;68(3):487–98 doi 10.1136/gutjnl-2017-314426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedlander SYG, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009;16(5):379–89 doi 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinorna by k-Ras Oncogenes in adult mice. Cancer Cell 2007;11(3):291–302 doi 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Bailey JM, Hendley AM, Lafaro KJ, Pruski MA, Jones NC, Alsina J, et al. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016;35(32):4282–8 doi 10.1038/onc.2015.441. [DOI] [PubMed] [Google Scholar]
- 34.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JP, et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012;22(6):737–50 doi 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011;19(6):728–39 doi 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferreira RMM, Sancho R, Messal HA, Nye E, Spencer-Dene B, Stone RK, et al. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Reports 2017;21(4):966–78 doi 10.1016/j.celrep.2017.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology 2009;137(3):1072–82, 82, e1–6 doi 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muraro MJ, Dharmadhikari G, Grun D, Groen N, Dielen T, Jansen E, et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst 2016;3(4):385–94 e3 doi 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cubilla AL, Fitzgerald PJ. MORPHOLOGICAL LESIONS ASSOCIATED WITH HUMAN PRIMARY INVASIVE NONENDOCRINE PANCREAS CANCER. Cancer Res 1976;36(7):2690–8. [PubMed] [Google Scholar]
- 40.Gidekel Friedlander SYG, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009;16(5):379–89 doi 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonner-Weir S, Aguayo-Mazzucato C, Weir GC. Dynamic development of the pancreas from birth to adulthood. Ups J Med Sci 2016;121(2):155–8 doi 10.3109/03009734.2016.1154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J, Snyder ER, Liu Y, Gu X, Wang J, Flowers BM, et al. Reconstituting development of pancreatic intraepithelial neoplasia from primary human pancreas duct cells. Nat Commun 2017;8:14686 doi 10.1038/ncomms14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A 2007;104(6):1865–70 doi 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabula Muris C, Overall c, Logistical c, Organ c, processing, Library p, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562(7727):367–72 doi 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 2005;8(3):185–95 doi 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 46.Wollny D, Zhao S, Everlien I, Lun X, Brunken J, Brune D, et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev Cell 2016;39(3):289–301 doi 10.1016/j.devcel.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007;11(3):229–43 doi 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 48.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004;119(6):847–60 doi 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004;119(6):861–72 doi 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A 2010;107(1):246–51 doi 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahronian LG, Driscoll DR, Klimstra DS, Lewis BC. The p53R172H mutant does not enhance hepatocellular carcinoma development and progression. PLoS One 2015;10(4):e0123816 doi 10.1371/journal.pone.0123816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 2007;26(15):2226–42 doi 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- 53.Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019;37(5):547–54 doi 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- 54.Blanpain C Tracing the cellular origin of cancer. Nat Cell Biol 2013;15(2):126–34 doi 10.1038/ncb2657. [DOI] [PubMed] [Google Scholar]
- 55.Visvader JE. Cells of origin in cancer. Nature 2011;469(7330):314–22 doi 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 56.Brunton H, Caligiuri G, Cunningham R, Upstill-Goddard R, Bailey UM, Garner IM, et al. HNF4A and GATA6 Loss Reveals Therapeutically Actionable Subtypes in Pancreatic Cancer. Cell Rep 2020;31(6):107625 doi 10.1016/j.celrep.2020.107625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data for the RNA-seq experiments have been deposited at the Gene Expression Omnibus (RRID:SCR_005012) under accession number GSE164180.