

Abstract
Molecular profiling technologies, such as genome sequencing and proteomics, have transformed biomedical research, but most such technologies require tissue dissociation, which leads to loss of tissue morphology and spatial information. Recent developments in spatial molecular profiling technologies have enabled the comprehensive molecular characterization of cells while keeping their spatial and morphological contexts intact. Molecular profiling data generate deep characterizations of the genetic, transcriptional and proteomic events of cells, while tissue images capture the spatial locations, organizations and interactions of the cells together with their morphology features. These data, together with cell and tissue imaging data, provide unprecedented opportunities to study tissue heterogeneity and cell spatial organization. This review aims to provide an overview of these recent developments in spatial molecular profiling technologies and the corresponding computational methods developed for analyzing such data.
Keywords: spatial molecular profiling, spatial transcriptomic data, mass spectrometry, super-resolution microscopy, FISH, scRNA-seq, spatial organization, cell morphology
Introduction
Understanding the spatial organization of cells, together with their mRNA and protein abundances, is essential to understanding how cells from different origins form tissues with distinctive structures and functions. Such information can bridge the gap between biological functions and morphological/genomic features and advance our understanding of important biological activity, such as tumorigenesis, embryonic development and tissue morphogenesis. However, for a long time, information gathered from molecular profiling and tissue imaging was analyzed separately with little or no crosstalk and was limited either by the low throughput of measuring one target at a time [1–3] or by the difficulties involved with manually collecting samples from multiple tissue locations [4, 5]. This was due to the technical difficulties involved: most of the current molecular profiling technologies require tissue dissociation, which leads to the loss of tissue morphology and spatial information, while current microscopes can only detect a limited number of fluorescent channels, which constrains the number of markers available for visualization over tissue slides. To overcome the problem of missing information, scientists have come up with different strategies, such as cell type deconvolution [6–10] from RNA-seq data or spatial reconstruction of cell positions through unsupervised [11] or supervised learning [12, 13]. However, such algorithms rely on statistical assumptions that might not hold in real data and can only recover limited information, hampering their applications in downstream analysis. Only recently, researchers have developed spatial molecular profiling technologies that can quantify and map gene expression and protein abundance simultaneously. Specifically, molecular profiling technologies provide high-throughput quantification of gene products (mostly RNA transcripts with a few being able to measure protein abundance), while imaging technologies provide the positions of individual cells and their morphological features. Together, these techniques provide a comprehensive characterization of cells and their spatial organizations.
Spatial molecular profiling technologies
To map and measure gene expression or protein abundance simultaneously, either of the following technical challenges needs to be addressed: how to quantify transcript and protein abundance in situ in a multiplexed manner or how to retain spatial information during sequencing. This leads to two main approaches for developing spatial molecular profiling technologies: imaging-based and sequencing-based.
Imaging-based spatial molecular profiling technologies
On the imaging side, single-molecule fluorescence in situ hybridization (smFISH) [1, 2] enables scientists to visualize the locations of individual molecules within a cell. By counting the fluorescent signals of a gene product, one can directly deduce its expression value. In 2014, Lubeck et al. [14] developed a sequential barcoding technique to uniquely identify a variety of RNA species by florescent sequence readouts through multiple rounds of smFISH, which greatly expanded the set of RNA molecules that could be measured at the same time. In one round of hybridization, probes labeled with one of the four fluorophores were introduced to immobilized samples, imaged and then stripped by DNase treatment. In the next round of hybridization, the same probes were used but labeled with different dyes. This procedure was repeated for N rounds, and the number of unique barcodes to represent different transcripts could scale quickly as 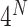 . The authors named their technology seqFISH. In this first paper, the authors barcoded 12 genes in single yeast cells with 4 dyes and 2 rounds of hybridization for demonstration. In 2019, the same group presented an improved version, seqFISH+ [15], in which they used 60 pseudocolors, 3 fluorescent channels and 4 rounds of pseudocolor imaging to achieve transcriptome-wide profiling (theoretically, 24 000 genes). Multiplexed error-robust FISH (MERFISH), developed by Chen et al. [16], was another sequential barcoding FISH technology that shared a largely similar strategy with seqFISH. Compared with seqFISH, MERFISH employed fewer fluorescent channels (only 0 and 1) and more hybridization rounds. As a consequence, MERFISH was less efficient in terms of multiplexing, but because it conducted more hybridization rounds than the theoretical requirement, it was able to distance target transcript barcodes from each other to prevent potential misidentification due to one-bit color error, and that was why the authors named their method ‘error-robust’. They measured the expression of 1001 genes with 14 rounds of hybridization using 14 bits Hamming-distance-2 (MHD2) code. osmFISH developed by Codeluppi et al. [17] was also based on smFISH, but instead of applying sequential barcoding, like seqFISH and MERFISH, osmFISH only involved one round of hybridization per transcript. Therefore, the number of profiled targets only scaled linearly with the number of fluorescence channels and the number of hybridization cycles. There are also non-FISH-based methods that use the florescent sequence as readout. Spatially resolved transcript amplicon readout mapping (STARmap) [18] first labeled cellular RNAs by pairs of DNA probes followed by enzymatic amplification to form a DNA amplicon. The amplicon contained a five-base unique barcode that later could be used as the identifier of its target and amine-modified nucleotides that could be conjugated into a polymer network. Following polymerization fixed the amplicons in their native spatial coordinates in a hydrogel polymer network. Proteins and lipids were digested to enhance transparency of the hydrogel polymer. The identities of the probes were later determined by decoding five-base DNA barcodes in multicolor fluorescence. STARmap achieved 3D structure restoration of cellular RNAs, compared with FISH methods, which are all in 2D. Also, it had a better signal-to-noise ratio compared with smFISH by removing unwanted substances. GeoMx Digital Spatial Profiler (DSP) [19] is a commercially available platform developed by NanoString for spatial protein or RNA detection. Formalin-fixed paraffin-embedded samples are hybridized with photo-cleavable oligonucleotide-tagged antibodies or probes. Specific regions of interest are then subjected to UV light ablation, causing the detachment of oligonucleotide tags from their targets. The florescent sequences of the oligo-tags are then scanned in the microscope and quantified. Instead of using fluorescence, Giesen et al. [20] and Angelo et al. [21] employed mass spectrometry (MS) for multiplexing. Unique metal isotopes were conjugated to antibodies specific to targets and were later liberated using a UV laser or duoplasmatron ion beam and further visualized and quantified as a readout. Fluidigm Hyperion Imaging System [22] is a commercially available multiplexed imaging mass cytometry platform that is capable of detecting dozens of protein markers simultaneously. Note that theoretically, all these hybridization-based strategies can detect both RNAs and proteins based on the probes selected (DNA or antibodies) such as DSP, while FISH methods are developed more for RNAs as proteins usually do not allow multiple probes binding and MS methods are more for proteins.
. The authors named their technology seqFISH. In this first paper, the authors barcoded 12 genes in single yeast cells with 4 dyes and 2 rounds of hybridization for demonstration. In 2019, the same group presented an improved version, seqFISH+ [15], in which they used 60 pseudocolors, 3 fluorescent channels and 4 rounds of pseudocolor imaging to achieve transcriptome-wide profiling (theoretically, 24 000 genes). Multiplexed error-robust FISH (MERFISH), developed by Chen et al. [16], was another sequential barcoding FISH technology that shared a largely similar strategy with seqFISH. Compared with seqFISH, MERFISH employed fewer fluorescent channels (only 0 and 1) and more hybridization rounds. As a consequence, MERFISH was less efficient in terms of multiplexing, but because it conducted more hybridization rounds than the theoretical requirement, it was able to distance target transcript barcodes from each other to prevent potential misidentification due to one-bit color error, and that was why the authors named their method ‘error-robust’. They measured the expression of 1001 genes with 14 rounds of hybridization using 14 bits Hamming-distance-2 (MHD2) code. osmFISH developed by Codeluppi et al. [17] was also based on smFISH, but instead of applying sequential barcoding, like seqFISH and MERFISH, osmFISH only involved one round of hybridization per transcript. Therefore, the number of profiled targets only scaled linearly with the number of fluorescence channels and the number of hybridization cycles. There are also non-FISH-based methods that use the florescent sequence as readout. Spatially resolved transcript amplicon readout mapping (STARmap) [18] first labeled cellular RNAs by pairs of DNA probes followed by enzymatic amplification to form a DNA amplicon. The amplicon contained a five-base unique barcode that later could be used as the identifier of its target and amine-modified nucleotides that could be conjugated into a polymer network. Following polymerization fixed the amplicons in their native spatial coordinates in a hydrogel polymer network. Proteins and lipids were digested to enhance transparency of the hydrogel polymer. The identities of the probes were later determined by decoding five-base DNA barcodes in multicolor fluorescence. STARmap achieved 3D structure restoration of cellular RNAs, compared with FISH methods, which are all in 2D. Also, it had a better signal-to-noise ratio compared with smFISH by removing unwanted substances. GeoMx Digital Spatial Profiler (DSP) [19] is a commercially available platform developed by NanoString for spatial protein or RNA detection. Formalin-fixed paraffin-embedded samples are hybridized with photo-cleavable oligonucleotide-tagged antibodies or probes. Specific regions of interest are then subjected to UV light ablation, causing the detachment of oligonucleotide tags from their targets. The florescent sequences of the oligo-tags are then scanned in the microscope and quantified. Instead of using fluorescence, Giesen et al. [20] and Angelo et al. [21] employed mass spectrometry (MS) for multiplexing. Unique metal isotopes were conjugated to antibodies specific to targets and were later liberated using a UV laser or duoplasmatron ion beam and further visualized and quantified as a readout. Fluidigm Hyperion Imaging System [22] is a commercially available multiplexed imaging mass cytometry platform that is capable of detecting dozens of protein markers simultaneously. Note that theoretically, all these hybridization-based strategies can detect both RNAs and proteins based on the probes selected (DNA or antibodies) such as DSP, while FISH methods are developed more for RNAs as proteins usually do not allow multiple probes binding and MS methods are more for proteins.
Sequencing-based spatial molecular profiling technologies
Sequencing-based spatial molecular profiling technologies mainly focus on measuring spatially mapped cell transcriptomic activities. Its major challenge is to trace back the original location of RNA molecules, since transcriptome-wide transcript quantification is well established. To achieve this, additional barcodes need to be incorporated into the sequences before collecting and pooling RNA samples. This gave rise to the development of several methods for adopting such strategies. In 2016, Ståhl et al. [23] first brought up this idea and developed spatial transcriptomics (ST) technology. Histological sections were positioned on glass slides and deposited with reverse transcription primers containing spatial barcodes. Complementary DNA molecules were then synthesized and sequenced to capture both expression and spatial information. In 2018, Spatial Transcriptomics, the original Swedish company that invented the technology, was acquired by 10x Genomics [24]. Slide-seq [25] is a recently developed spatial sequencing method that also borrowed the idea of using drop-seq [26] for single-cell RNA sequencing (scRNA-seq) to introduce unique DNA barcodes onto 10 μm microparticles (‘beads’). In this method, they then transferred frozen tissue sections to the arrayed beads’ surfaces to prepare for the barcoded RNA-seq library. It was able to reveal the fine single cell layer features in a mouse hippocampus coronal section experiment. High-definition spatial transcriptomics [27] is an upgraded version of ST that produced barcoded beads with an even smaller size than Slide-seq. It increased the spatial resolution from 100 μm in the original ST to 2 μm.
Figure 1 demonstrates the workflows of imaging- and sequencing-oriented spatial transcriptomic technologies, and Table 1 provides a summary of current spatial molecular profiling technologies. There have been successful applications of both of these two branches of methods to the profiling embryonic development [28], cancer tissue [27] and complex structure of neural layers [25, 29]; the differences between the technologies and strategies behind them give them their unique features. The advantages of the probe hybridization-based approach are that (i) it is capable of quantifying both RNA transcript and protein abundance while sequencing is only suitable for measuring RNA; (ii) in measuring RNA, it avoids the reverse transcription and amplification required by sequencing, which may introduce bias; and (iii) for FISH-based technologies that use super-resolution microscopy, the resolution is at the single-molecule level, allowing further subcellular analysis such as RNA compartmentalization [30]. The advantages of the sequencing-based approach are that it is a mature technology and relatively easy to operate and that, because the actual nucleotide sequences are obtained, traditional mutation calling and copy number analysis are also suitable to detect genomic variations.
Figure 1.
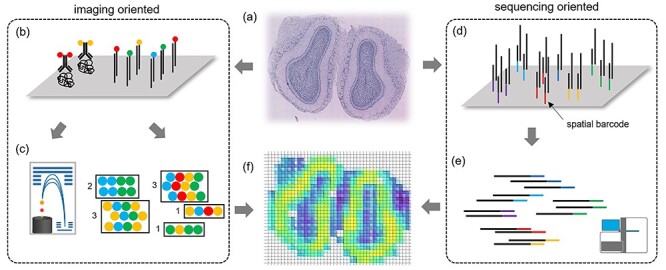
Overview of the workflows of imaging- and sequencing-oriented spatial molecular profiling technologies. Because each method differs in technical detail, the figure is intended to give only a demonstrative idea. (A) Prepared tissue slides. (B) Hybridization phase. Proteins or RNAs are hybridized with metal conjugated antibodies or fluorescent probes. (C) Quantification phase. Metal isotopes or fluorescent sequences are quantified as readout by MS or microscopy. (D) Barcoding phase. RNA molecules are captured by barcoded surface probes. (E) Sequencing phase. cDNA library is synthesized and sequenced. (F) Visualization of spatial transcriptomic data as a heatmap.
Table 1.
Summary of spatial molecular profiling technologies
| Name | Spatial information | Expression quantification | Target | Target size | Year | Reference |
|---|---|---|---|---|---|---|
| CyTOF-ICC/IHC | image | MS | protein | ~100 | 2014 | [18] |
| MIBI | protein | ~100 | 2014 | [19] | ||
| Hyperion | protein | ~30 | 2017 | [20] | ||
| seqFISH | fluorescent probe | mRNA | ~16 | 2014 | [12] | |
| seqFISH+ | mRNA | ~10 000 | 2019 | [13] | ||
| MERFISH | mRNA | ~1000 | 2015 | [14] | ||
| osmFISH | mRNA | ~50 | 2018 | [15] | ||
| STARmap | mRNA | ~1000 | 2018 | [16] | ||
| DSP | mRNA, protein | ~1000 | 2019 | [17] | ||
| ST | spatial barcode | sequencing | mRNA | Transcriptome | 2016 | [21] |
| HDST | mRNA | Transcriptome | 2019 | [25] | ||
| Slide-seq | mRNA | Transcriptome | 2019 | [23] |
Analyze spatial molecular profiling data
These new developments in spatial molecular profiling have enabled the comprehensive molecular characterization of cells while keeping their spatial and morphological contexts intact. This opens up new possibilities for scientists to look into the heterogeneity of mRNA expression, protein abundance, gene regulation and cell interaction in space. Recently, different analysis methods have been proposed to utilize spatial molecular profiling datasets in studying novel biological questions. In this section, we summarize the newly proposed methodologies for analyzing spatial molecular profiling data grouped by their application scopes (demonstrated in Figure 2). Table 2 (at the end of this section) provides a brief summary of existing analysis methods. Most of the existing analysis methods use spatial transcription profiling data, but they are also applicable to other types of spatial molecular profiling data. We also include methods that have been developed for pure spatial coordinate data without expression values, as they aim to address the same question of understanding cell spatial organizations and interactions.
Figure 2.
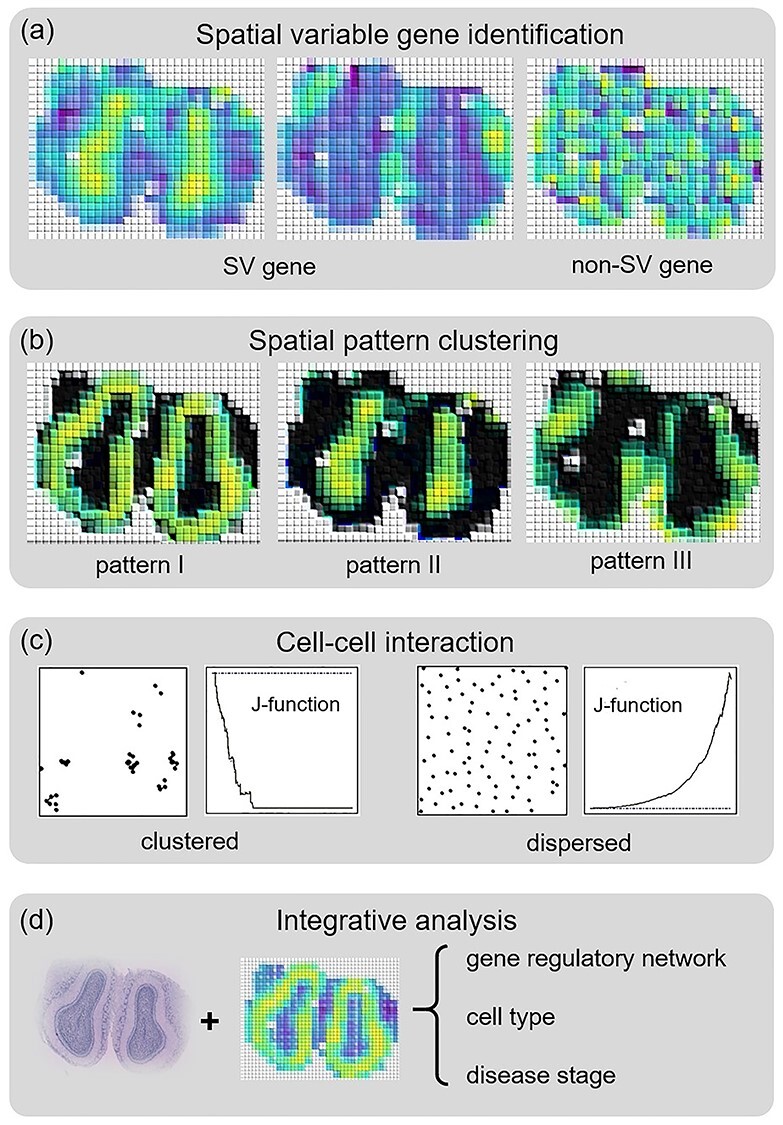
Summary of the applications of spatial transcriptomic data. (A) Identify SV genes. (B) Cluster SV genes into patterns. (C) Spatial cell–cell interaction analysis. (D) Integrate image data with spatial transcriptomic data for downstream functional analysis.
Table 2.
Summary of methods for analyzing spatial molecular profiling data
| Method | Framework | Data | Implementation | Link |
|---|---|---|---|---|
| SpatialDE | GP | spatial gene expression profile | Python | https://github.com/Teichlab/SpatialDE |
| SPARK | GP | R | https://xzhoulab.github.io/SPARK/ | |
| trendsceek | marked point process | R | https://github.com/edsgard/trendsceek | |
| staNMF | matrix factorization | Python | https://github.com/greenelab/staNMF | |
| SVCA | GP | Python | https://github.com/damienArnol/svca | |
| Moran’s I | spatial autocorrelation | R | https://cran.r-project.org/web/packages/lctools/index.html | |
| K,G,F,J,L function | point process | spatial coordinates | R | https://cran.r-project.org/web/packages/spatstat/index.html |
| BayesHiddenPottsMixture | Potts model | spatial coordinates, cell type annotation | R | https://github.com/liqiwei2000/BayesHiddenPottsMixture |
| BayesMarkInteractionModel | marked point process | R | https://github.com/liqiwei2000/BayesMarkInteractionModel | |
| histoCAT | NA | image | Matlab | http://www.bodenmillerlab.com/research-2/histocat/ |
| GripDL | neural network | spatial gene expression profile, gene regulatory network | Python | https://github.com/2010511951/GripDL |
| SpaCell | neural network | spatial gene expression profile, image | Python | https://github.com/BiomedicalMachineLearning/Spacell |
Spatial differential gene expression or protein abundance analysis
Traditional differential gene analysis methods, such as ANOVA, significance analysis of microarrays [31], DEseq [32] and EdgeR [33], focused on comparative analysis between groups of samples with different phenotypes or between cells manually collected from distinct locations, in order to test the significance of the correlation between a grouping variable and gene expression. In spatially resolved expression profiles, a new statistical problem is how to test the association between gene expression levels and their spatial coordinates and reject the null hypothesis when spatial inhomogeneity is exhibited. The Gaussian process (GP) model, which was adopted by both SpatialDE [34] and SPARK [35], can serve as a natural fit for this problem because of its ability to model temporal [36] or spatial [37] dependence. These two methods share a common methodology framework, with SPARK being more explicit in modeling count data, sample normalization and P-value calibration. The general framework can be formulated as a multivariate normal distribution of the following form:
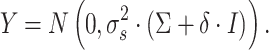 |
Here, 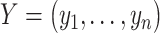 represents the normalized expression values of a given gene across
represents the normalized expression values of a given gene across 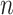 spatial coordinates,
spatial coordinates, 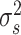 is a scaling factor,
is a scaling factor, 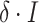 is the independent nonspatial variance and
is the independent nonspatial variance and 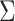 is the spatial covariance matrix defined by a covariance function
is the spatial covariance matrix defined by a covariance function 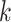 for expression levels in every pair of locations (e.g. cells)
for expression levels in every pair of locations (e.g. cells) 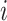 and
and 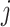 :
:
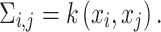 |
The advantage of GP is that it is versatile for different pattern identification based on the covariance function 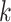 selected using prior knowledge. SpatialDE tested three types of kernels—square exponential, linear and periodic—to search for focal correlation, linear trend and spatial oscillation, respectively. SPARK devised a total of 10 kernels (5 periodic and 5 square-exponential) with different hyper-parameters to capture spatial patterns.
selected using prior knowledge. SpatialDE tested three types of kernels—square exponential, linear and periodic—to search for focal correlation, linear trend and spatial oscillation, respectively. SPARK devised a total of 10 kernels (5 periodic and 5 square-exponential) with different hyper-parameters to capture spatial patterns.
Trendsceek [38] took another approach. It modeled the spatial distribution of cells as a realization of the point process and the gene expression value of cells as their attached marks. It calculated four summary statistics, Stoyan’s mark-correlation, mean-mark function, variance-mark function and mark-variogram for all pairs of points in the space and examined whether the distributions of the four statistics are independent of the pairwise distance of points to identify explainable spatial variability.
We performed a benchmark comparison of the three methods in simulated datasets (Supplementary S1). Our result was consistent with Sun et al. [35] that SPARK had the best performance, followed by SpatialDE in most settings (Figures S2–S10). The four statistics of Trendsceek have less statistical power in identifying spatial variable (SV) genes, while also suffering from a long computation time because of the permutations needed to generate null distributions. In real dataset, Sun et al. [35] reported that SPARK detected 772 SV genes, SpatialDE detected 67 and Trendsceek detected none in mouse olfactory bulb data. In a human breast cancer dataset, they showed that the three methods identified 290, 115 and 15 SV genes, respectively.
Spatial pattern identification
An immediate task following spatial differential gene detection is to group those genes into distinctive clusters based on their spatial gene expression pattern, thus summarizing high-dimensional spatial data into a number of histological patterns. This can help reveal the underlying causal effect of spatial variability and explicate how genes are spatially regulated. Both SpatialDE and SPARK implement this function. SpatialDE extends its GP model to a GP mixture model and clusters SV genes identified beforehand into pre-specified K patterns. SPARK, on the contrary, performs an ad hoc hierarchical agglomerative clustering on spatial genes independent of the GP framework, thus ignoring spatial information. Stability-driven nonnegative matrix factorization (staNMF) [39] is another spatial decomposition algorithm that was applied to the Drosophila embryonic spatial expression dataset and decomposed the gene expression into concise spatial representations that corresponded to biologically meaningful regions in the Drosophila embryo.
Spatial cell–cell interaction and neighborhood analysis
Cell–cell interaction and cell community analyses are longstanding interests in scientific domains like the tumor microenvironment and brain regional functionality studies. The task is usually to assess whether any attractive or repulsive effect exists between cells or different types of cells, statistically speaking, in order to determine how much the spatial distribution of cells deviates from spatial randomness. Schapiro et al. [40] and Enfield et al. [41] performed a simple calculation of the proportions of different types of cells adjacent to a given cell type. Xia et al. [30] used Moran’s I in spatial autocorrelation to evaluate the expression spatial heterogeneity. de Back et al. [42] introduced the usage of the K function in point processes, which is similar to Moran’s I, to detect any clustering or dispersion that occurs in the distribution of spatial points (Figure 2). There are also other distance-based measurements, like G-, F-, J- and L-functions and their bivariate versions, that accomplish the same goal [43, 44].
Beyond descriptive statistics, there are more complicated model-based approaches for analyzing spatial patterns and interactions, especially when cell marks can be decoded as a categorical variable. Bayesian hidden Potts mixture [45] is a Bayesian hierarchical model that incorporates a hidden Potts model and a Markov random field model. It projects the irregularly distributed cells onto a square lattice and quantifies the interactions between different regions (small squares defined by the grid). The Bayesian mark interaction model [46] aims to directly model the interaction of spatial points rather than grid regions through a geostatistical marking model under the Bayesian framework. The advantage of these two methods is that they can output scalar parameters indicating interaction strengths, which can be later used to correlate with outcome variables for downstream analysis. The authors investigated the associations between inferred interaction parameters among tumor, stromal and lymphocyte cells in non-small-cell lung cancer (NSCLC) patients’ pathology images and their survivals and found that the interaction between tumor and stromal cells can be a predictor for patients’ prognoses after adjusting for clinical information.
Spatial variance component analysis (SCVA) [47] is a recently published method that incorporates both spatial coordinates and transcriptomic data for spatial interaction inference. Similar to SpatialDE, SCVA also borrows the framework of GP, while extending the covariance matrix to contain three terms: intrinsic effect 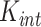 , environmental effect
, environmental effect 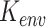 and cell–cell interaction effect
and cell–cell interaction effect 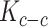 . This gives
. This gives
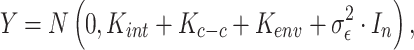 |
where 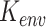 is the same as the square exponential kernels of SpatialDE,
is the same as the square exponential kernels of SpatialDE, 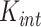 measures the similarity of cells in terms of their intrinsic state and
measures the similarity of cells in terms of their intrinsic state and 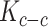 quantifies the similarity between neighboring cells. Unlike the previous methods in this section, which were purely based on the spatial distribution of cells, SCVA also utilizes their expression profiles to generate a quantitative measurement of the fraction of the variability from cell–cell interaction for each cell rather than a single coefficient over a whole image slide.
quantifies the similarity between neighboring cells. Unlike the previous methods in this section, which were purely based on the spatial distribution of cells, SCVA also utilizes their expression profiles to generate a quantitative measurement of the fraction of the variability from cell–cell interaction for each cell rather than a single coefficient over a whole image slide.
Integrative analysis and spatial prediction
The aforementioned methods focused mainly on mining the spatial gene profile to uncover spatial patterns and potential explanatory factors; however, a few studies took a step further to explore the possibility of using features extracted from spatial context as predictors for downstream functional analysis. GripDL [48] is a supervised deep learning model for reconstructing gene regulatory networks using spatial gene expression images. SpaCell [49] is also a deep learning framework that incorporates pixel information from Hematoxylin and Eosin staining images with matched gene expression measurements for cell-type and disease-stage classification. Jackson et al. [50] quantified 35 biomarkers in 720 breast cancer pathology images and identified survival-associated tumor microenvironment and subgroups.
With both the spatial coordinates and expression profiles of cells available, Battich et al. [51] examined the interplay of these two sources of information and found that transcript abundance was predictable by 183 predefined features capturing the intrinsic and microenvironmental properties of cells, including cell crowding, molecular profiles, nuclear morphology and neighborhood activity. Goltsev et al. [52] conducted a similar study and discovered that some surface-marker expressions in immune cells were highly correlated with the neighborhood cell type composition. While these two studies elucidated some aspects of the dependency of RNA/protein expression on spatial context, the spatial abundance of transcripts will likely be more readily known in the future, obviating the need to predict them.
Other potential analyses
Besides the above-mentioned methods, there are still many interesting potential applications worth exploring. Note that the sequencing-based spatial profiling technology is just an extension of regular scRNA-seq; analysis performed with scRNA-seq is also suitable for spatial molecular profiling data with an additional layer of spatial information. There are tools that have been developed to identify mutation and copy number alteration events in single-cell sequencing data [53, 54]. Lu et al. [55] and Zhou et al. [56] leveraged genetic variations to perform single-cell lineage tracing and phylogenetic tree reconstruction. Joshi et al. [57] examined the spatial heterogeneity of the T-cell receptor repertoire in NSCLC. Adding spatial information can potentially further enhance the analysis of scRNA-seq data and generate more insights.
Table 2 provides a summary of current analysis methods for spatial molecular profiling datasets.
Conclusion and outlook
With the rapid emergence of spatial molecular profiling technologies and platforms, the prevalence of high-throughput spatial gene expression profiling with high resolution is foreseeable in the near future. It will not only enable scientists to see the real geographical landscape of gene expression in cellular resolution but also spark new opportunities to investigate novel scientific questions that could not be addressed otherwise. Currently, various exciting applications of spatial molecular profiling technologies indicate that this will be the trend for future molecular profiling analysis.
However, there are several potential caveats and challenges we need to be aware of when investigating spatial data. Currently, most spatial gene identification and clustering algorithms lack meaningful biological assumptions. It is not of primary interest to simply match spatial inhomogeneity patterns with tissue morphology. In fact, this can already be done by using morphological features to partially predict spatial transcript abundance, according to [51] and [52]. Although the authors of SCVA tried to decompose variance even further, their definitions of cell–cell interaction and environmental effect are still based on the same rule; exponentially decaying correlation with distance and other artificially designed kernels do not generate obviously meaningful patterns. It would be a critical step to properly incorporate spatial molecular profiling with genetic information and morphological features, and it would be even more interesting to have spatial-temporal data for more systematic modeling. Moreover, a comparative analysis of how the spatial pattern of a particular gene or a group of genes varies among different experimental conditions can be an important topic in fields like tumor immune cell infiltrating. A statistical model that is extended to take multiple conditions into consideration is desired. In multi-condition setting, researchers need to be aware of batch effects. For an imaging approach, involving negative control (background noise) probes and positive control (stable expressed gene) probes in experiments like DSP is suggested. For a sequencing approach, applying typical RNA-seq or scRNA-seq normalization methods, such as DEseq [32] and Seurat [58], to the spatial expression matrix is recommended. In summary, the development and further evolution of spatial molecular profiling technology together with new analysis methods is an important breakthrough in the field and will greatly facilitate biomedical research.
Key Points
This article is by far to our knowledge the first paper that summarizes the development of spatial molecular profiling technologies and the corresponding statistical analysis methods.
We introduced both the imaging- and sequencing-based spatial molecular profiling technologies, compared their different strategies to obtain gene expression profile with spatial resolution and discussed their advantages and disadvantages.
We did a comprehensive overview of the statistical methods for spatial molecular data analysis, including spatial gene identification, spatial pattern clustering, spatial interaction and neighborhood analysis, with more focus on the GP-based model.
We also discussed the future outlook of integrative analysis and challenge for spatial molecular data analysis.
Supplementary Material
Minzhe Zhang is a PhD student in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. His research focuses on bioinformatics and data sciences.
Thomas Sheffield is a postdoctoral researcher in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. He received his PhD degree in applied math. His research focuses on data sciences.
Xiaowei Zhan is an assistant professor in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. His research focuses on statistical genetics, bioinformatics and data sciences.
Qiwei Li is an assistant professor in the Department of Mathematics Sciences at University of Texas at Dallas. His research focuses on Bayesian methodologies, and high-dimensional and large-scale data modeling.
Donghan M. Yang is a bioinformatics project manager in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. His research focuses on clinical informatics and data sciences.
Yunguan Wang is a data scientist in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. His research focuses on image analysis, bioinformatics and data sciences.
Shidan Wang is a data scientist in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. Her research focuses on deep learning, machine learning and image analysis.
Yang Xie is a professor and director of the Quantitative Biomedical Research Center at the University of Texas Southwestern Medical Center. Her research interests include predictive modeling, cancer biomarkers, and medical informatics.
Tao Wang is an assistant professor in the Department of Population and Data Sciences at University of Texas Southwestern Medical Center. Dr. Wang’s research revolves around using state-of-the-art bioinformatics and biostatistics approaches to study the implications of tumor immunology for tumorigenesis, metastasis, prognosis, and treatment response in a variety of cancers.
Guanghua Xiao is a professor in the Department of Population and Data Sciences at the University of Texas Southwestern Medical Center. His research interests include deep learning, spatial statistics, and imaging analysis.
Funding
This work was partially supported by the National Institutes of Health [R35 GM136375, P30 CA142543 and P50CA70907], and the Cancer Prevention and Research Institute of Texas [RP190107 and RP180805].
Conflict of interest
None declared.
Reference
- 1. Femino AM, Fay FS, Fogarty K, et al. . Visualization of single RNA transcripts in situ. Science 1998;280(5363):585–90. [DOI] [PubMed] [Google Scholar]
- 2. Raj A, Van Den Bogaard P, Rifkin SA, et al. . Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 2008;5(10):877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frise E, Hammonds AS, Celniker SE. Systematic image-driven analysis of the spatial Drosophila embryonic expression landscape. Mol Syst Biol 2010;6(1):345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Junker JP, Noël ES, Guryev V, et al. . Genome-wide RNA tomography in the zebrafish embryo. Cell 2014;159(3):662–75. [DOI] [PubMed] [Google Scholar]
- 5. Lovatt D, Ruble BK, Lee J, et al. . Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 2014;11(2):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du R, Carey V, Weiss S. deconvSeq: deconvolution of cell mixture distribution in sequencing data. Bioinformatics 2019;35(24):5095–5102. [DOI] [PubMed] [Google Scholar]
- 7. Wang X, Park J, Susztak K, et al. . Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun 2019;10(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao Y, Lin Y, Ormerod JT, et al. . scDC: single cell differential composition analysis. BMC Bioinformatics 2019;20(19):721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang T, Lu R, Kapur P, et al. . An empirical approach leveraging tumorgrafts to dissect the tumor microenvironment in renal cell carcinoma identifies missing link to prognostic inflammatory factors. Cancer Discov 2018;8(9):1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Z, Luo D, Zhong X, et al. . SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes 2019;10(7):531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nitzan M, Karaiskos N, Friedman N, et al. . Gene expression cartography. Nature 2019;576(7785):132–7. [DOI] [PubMed] [Google Scholar]
- 12. Satija R, Farrell JA, Gennert D, et al. . Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 2015;33(5):495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Achim K, Pettit JB, Saraiva LR, et al. . High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol 2015;33(5):503. [DOI] [PubMed] [Google Scholar]
- 14. Lubeck E, Coskun AF, Zhiyentayev T, et al. . Single-cell in situ RNA profiling by sequential hybridization. Nat Methods 2014;11(4):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eng CHL, Lawson M, Zhu Q, et al. . Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019;568(7751):235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen KH, Boettiger AN, Moffitt JR, et al. . Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015;348(6233):aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Codeluppi S, Borm LE, Zeisel A, et al. . Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 2018;15(11):932–5. [DOI] [PubMed] [Google Scholar]
- 18. Wang X, Allen WE, Wright MA, et al. . Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018;361(6400):eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Merritt CR, Ong GT, Church SE, et al. . Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nature Biotechnology 2020;38(5):586–599. [DOI] [PubMed] [Google Scholar]
- 20. Giesen C, Wang HA, Schapiro D, et al. . Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 2014;11(4):417–22. [DOI] [PubMed] [Google Scholar]
- 21. Angelo M, Bendall SC, Finck R, et al. . Multiplexed ion beam imaging of human breast tumors. Nat Med 2014;20(4):436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fluidigm. Hyperion Imaging System: A comprehensive system for highly multiplexed imaging. https://fluidigm.com/products/hyperion-imaging-system (access date: 21 June 2020).
- 23. Ståhl PL, Salmén F, Vickovic S, et al. . Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 24. 10x Genomics. 10x Genomics Acquires Spatial Transcriptomics. https://www.10xgenomics.com/news/10x-genomics-acquires-spatial-transcriptomics/ (access date: 21 June 2020).
- 25. Rodriques SG, Stickels RR, Goeva A, et al. . Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Macosko EZ, Basu A, Satija R, et al. . Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161(5):1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vickovic S, Eraslan G, Salmén F, et al. . High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods 2019;16(10):987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frieda KL, Linton JM, Hormoz S, et al. . Synthetic recording and in situ readout of lineage information in single cells. Nature 2017;541(7635):107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lignell A, Kerosuo L, Streichan SJ, -et al. Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nat Commun 2017;8(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xia C, Fan J, Emanuel G, et al. . Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci 2019;116(39):19490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 2001;98(9):5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Svensson V, Teichmann SA, Stegle O. SpatialDE: identification of spatially variable genes. Nat Methods 2018;15(5):343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun S, Zhu J, Zhou X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat Methods 2020;17(2):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roberts S, Osborne M, Ebden M, et al. . Gaussian processes for time-series modelling. Philos Trans A Math Phys Eng Sci 2013;371(1984):20110550. [DOI] [PubMed] [Google Scholar]
- 37. Diggle PJ, Tawn JA, Moyeed RA. Model-based geostatistics. J R Stat Soc Ser C Appl Stat 1998;47(3):299–350. [Google Scholar]
- 38. Edsgärd D, Johnsson P, Sandberg R. Identification of spatial expression trends in single-cell gene expression data. Nat Methods 2018;15(5):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu S, Joseph A, Hammonds AS, et al. . Stability-driven nonnegative matrix factorization to interpret spatial gene expression and build local gene networks. Proc Natl Acad Sci 2016;113(16):4290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schapiro D, Jackson HW, Raghuraman S, et al. . histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat Methods 2017;14(9):873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Enfield KS, Martin SD, Marshall EA, et al. . Hyperspectral cell sociology reveals spatial tumor-immune cell interactions associated with lung cancer recurrence. J Immunother Cancer 2019;7(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. de Back W, Zerjatke T, Roeder I. Statistical and mathematical modeling of spatiotemporal dynamics of stem cells. In: Stem Cell Mobilization. New York, NY: Humana, 2019, 219–43. [DOI] [PubMed] [Google Scholar]
- 43. Illian J, Penttinen A, Stoyan H, et al. . Statistical Analysis and Modelling of Spatial Point Patterns, Vol. 70. John Wiley & Sons: Hoboken, New Jersey, 2008. [Google Scholar]
- 44. Baddeley A, Turner R. spatstat: an R package for analyzing spatial point patterns. J Stat Softw 2005;(i06):12. [Google Scholar]
- 45. Li Q, Wang X, Liang F, et al. . A Bayesian hidden Potts mixture model for analyzing lung cancer pathology images. Biostatistics 2018; 20(4):565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Q, Wang X, Liang F, et al. . A Bayesian mark interaction model for analysis of tumor pathology images. Ann Appl Statistics 2019;13(3):1708–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arnol D, Schapiro D, Bodenmiller B, et al. . Modeling cell-cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep 2019;29(1):202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang Y, Fang Q, Shen HB. Predicting gene regulatory interactions based on spatial gene expression data and deep learning. PLoS Comput Biol 2019;15(9):e1007324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tan X, Su A, Tran M, et al. . SpaCell: integrating tissue morphology and spatial gene expression to predict disease cells. Bioinformatics 2019;36(7):2293–2294. [DOI] [PubMed] [Google Scholar]
- 50. Jackson HW, Fischer JR, Zanotelli VR, et al. . The single-cell pathology landscape of breast cancer. Nature 2020;1–6. [DOI] [PubMed] [Google Scholar]
- 51. Battich N, Stoeger T, Pelkmans L. Control of transcript variability in single mammalian cells. Cell 2015;163(7):1596–610. [DOI] [PubMed] [Google Scholar]
- 52. Goltsev Y, Samusik N, Kennedy-Darling J, et al. . Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 2018;174(4):968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vu TN, Nguyen HN, Calza S, et al. . Cell-level somatic mutation detection from single-cell RNA sequencing. Bioinformatics 2019;35(22):4679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petti AA, Williams SR, Miller CA, et al. . A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun 2019;10(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu T, Park S, Zhu J, et al. . Overcoming genetic drop-outs in variants-based lineage tracing from single-cell RNA sequencing data. bioRxiv 2020. [Google Scholar]
- 56. Zhou Z, Xu B, Minn A, et al. . DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing. Genome Biol 2020;21(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Joshi K, de Massy MR, Ismail M, et al. . Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med 2019;25(10):1549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stuart T, Butler A, Hoffman P, et al. . Comprehensive integration of single-cell data. Cell 2019;177(7):1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.