

Abstract
HIV reverse transcriptase (RT) is an enzyme that plays a major role in the replication cycle of HIV and has been a key target of anti-HIV drug development efforts. Because of the high genetic diversity of the virus, mutations in RT can impart resistance to various RT inhibitors. As the prevalence of drug resistance mutations is on the rise, it is necessary to design strategies that will lead to drugs less susceptible to resistance. Here we provide an in-depth review of HIV reverse transcriptase, current RT inhibitors, novel RT inhibitors, and mechanisms of drug resistance. We also present novel strategies that can be useful to overcome RT’s ability to escape therapies through drug resistance. While resistance may not be completely avoidable, designing drugs based on the strategies and principles discussed in this review could decrease the prevalence of drug resistance.
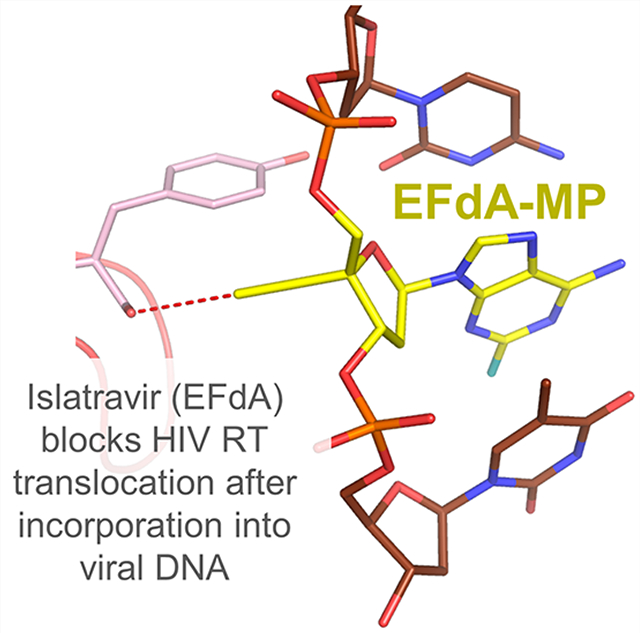
Graphical Abstract
1. INTRODUCTION
Human immunodeficiency virus (HIV) is the cause of acquired immunodeficiency syndrome (AIDS), which is responsible for the deaths of over 38 million people.1 Because there is no cure for HIV, patients are subjected to life-long therapy. The standard treatment for HIV is known as highly active antiretroviral therapy (HAART), which is capable of suppressing viral replication below the limit of detection and reducing viral transmission.2–4 HAART normally consists of three different drugs to keep HIV below detectable levels. There are six main classes of anti-HIV drugs: nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors, integrase inhibitors, fusion inhibitors, and entry inhibitors. In these classes, there are more than 27 different approved drugs.5 Fourteen of the approved drugs target the key viral enzyme reverse transcriptase (RT). The standard of care for most treatment naïve patients is two nucleoside reverse transcriptase inhibitors and one non-nucleoside reverse transcriptase inhibitor or integrase inhibitor.2,4
1.1. HIV Replication Cycle
HIV infection is initiated by the envelope protein binding to a host cell CD4 receptor, followed by binding to CXCR4 or CCR5 coreceptors. In turn, these interactions result in structural changes of the envelope protein which lead to fusion of the viral membrane with the host cell membrane. The HIV virion contains two copies of single-stranded positive sense RNA (+ssRNA) genome, about 50 copies of RT, other viral enzymes such as integrase and protease and nucleocapsid chaperones.6,7 RT has a polymerase domain able to use RNA or DNA as a template and make a DNA copy; it also has a ribonuclease H (RNase H) domain capable of degrading RNA in an RNA:DNA duplex. Using these enzymatic activities RT creates a linear double-stranded (dsDNA) product from the genomic + ssRNA. The viral dsDNA (vDNA) product is then used as substrate for the viral enzyme integrase for integration into the host genome.6 The integrated viral DNA or provirus serves as the template for viral genomic and viral mRNA, which is made by the host RNA polymerase and exported from the nucleus to the cytoplasm. The translation of viral RNAs (vRNA) produce the Gag polyprotein precursor, Pr55gag, which comprises the matrix, capsid, and nucleocapsid structural proteins, the Gag-Pol polyprotein precursor, Pr160gag–pol, with Pol comprising HIV protease, RT, and integrase enzymes. Envelope glycoprotein (Env) and regulatory and accessory viral proteins (Vif, Vpr, Vpu, Nef) are also translated. The Gag, Gag-Pol precursors, and Env are trafficked to the plasma membrane, where the vRNA is encapsidated and this structure buds off from infected cells and the virion contents mature through the action of HIV protease.6,7 Part of the maturation process involves the conversion of the p66/p66 RT homodimer to the p66/p51 heterodimer.8 The p66 subunit is 560 amino acids long and consists of two active enzymatic domains: polymerase, and RNase H. The p51 subunit contains the first 440 amino acids of p66 and it lacks an RNase H domain (Figure 1).
Figure 1.

Structure of HIV-1 RT in complex with dsDNA and dTTP substrates and binding locations of FDA-approved drugs targeting RT. HIV-1 RT (PDB 1RTD)9 is comprised of two subunits, p66 (multicolored cartoon) and p51 (gray cartoon). The enzymatically active p66 subunit has both RNA-dependent and DNA-dependent DNA polymerization activities and contains four subdomains: fingers (blue), palm (red), thumb (green), and connection (yellow), as well as an RNase H domain (purple). Template DNA is shown in light brown, primer DNA is shown in dark brown, and dTTP is shown as gray sticks at the polymerase active site (black dashed circle). Two Mg2+ ions in the polymerase active site are shown as spheres. The polymerase active site is also the location where NRTIs bind. The NNRTI binding pocket (NNIBP) is located in the p66 palm subdomain, near the base of the p66 thumb subdomain and adjacent to the polymerase active site (black dashed rectangle).
1.2. Structural Components of the Polymerase Active Site of HIV Reverse Transcriptase
The p66 polymerase domain of RT is divided into four subdomains: the fingers (residues: 1−84, 119−154), palm (residues: 85−118, 155−241), thumb (residues: 242−313), and connection (residues: 314−426)10–12 (Figure 1). The p51 subunit lacks an RNase H domain; although its structural domains have the same sequence as those in p66, they are organized differently,10,11 and as a result p51 has no enzymatic activity and simply provides structural support for p66.13
The polymerase and RNase H active sites of RT are situated at opposing parts of the nucleic acid binding cleft, which accommodates ~17 base-pairs (bp) of DNA/DNA and ~18 bp of RNA/DNA.14 Near the polymerase active site DNA/DNA substrates assume an A-form,11 whereas RNA/DNA substrates assume a similar H-form.15 The nucleic acid interacts with two α-helices of the p66 thumb subdomain (αH and αI) (Figure 2). The polymerase active site has three carboxylates in the palm subdomain of p66: D110, D185, and D186, which bind two Mg2+ in vivo (Mn2+ can be supported in vitro) and are essential for polymerization. D185 and D186 are part of a highly conserved YXDD motif in retroviral RTs; in HIV RT X is Met (Figure 2).16–19 In addition to the thumb subdomain, another important RT structural element is the primer grip (β12 and β13 hairpin), which also helps position the 3′-OH of the primer at the polymerase catalytic site.11,20,21 Important polymerase active site residues include R7222 and K65 that bind the β- and γ-phosphates of the incoming dNTP, and Y115, which contributes to the binding of the deoxyribose ring of the incoming dNTP and has been termed the “stericgate”, responsible for discriminating between deoxy- and ribonucleoside triphosphates (Figure 2).9,23–25
Figure 2.

Structure of the HIV-1 RT polymerase active site. dTTP (gray sticks) is bound at the polymerase active site (PDB 1RTD).9 Conserved active site residues D110, V111, D185, and D186 chelate two Mg2+ ions (light-green spheres), which also bind the alpha, beta, and gamma phosphates of the dTTP. Residue Y115 is located at the bottom of the active site. dTTP sits above Y115 and is additionally stabilized through hydrogen bond interactions between the alpha and beta phosphates and R72 in the β4 fingers, and the gamma phosphate and K65 in the β3−β4 fingers loop. The conserved YMDD loop is between β9 and β10, and the primer grip is located between β12 and β13 in the p66 palm subdomain. αH and αI of the p66 thumb subdomain interact with the nucleic acid substrate to help position it correctly in the polymerase active site.
1.3. Enzymology of RT
The mechanism of DNA synthesis begins with RT bound to a template/primer. The 3′-end of the primer is positioned at the polymerization site. RT incorporates dNMPs based on complementarity to the template strand.9,11,26 The incoming dNTP initially binds at the nucleotide site (N- site or pretranslocation site), which is followed by a nucleophilic attack to form a phosphodiester bond and release of the pyrophosphate product. Following primer extension, the 3′-end of the elongated DNA moves to the post-translocation or P-site, thus vacating the N-site for the next incoming dNTP.27,28 Insights into the molecular details of these translocation events have been provided by crystal structures of strategically designed covalent RT-nucleic acid intermediates.27
The RNase H domain of RT cleaves RNA in RNA/DNA duplexes. The RNase H has a highly conserved DEDD motif (D443, E478, D498, D549), which binds the two divalent cations required for enzymatic activity.9,29–34
1.4. HIV Reverse Transcription
While there are several viral and cellular factors that aid in reverse transcription, here we only discuss RT. Like any DNA polymerase, RT requires both a primer and a template. RT initiates DNA synthesis from a host tRNA primer: tRNALys335,36 that is reported to be packaged in virions at a 4-fold excess with respect to the viral genomes.37 The tRNALys3 hybridizes to an 18-nucleotide complementary region, known as the primer binding site (PBS), which is located near the 5′ end of the viral genome.38 Polymerization begins from hybridized tRNALys3, which synthesizes a minus-strand DNA in complex with the plus-strand viral RNA. In vitro data have shown the addition of the first five to six nucleotides after the primer is very slow but then DNA synthesis speeds up.39,40 As RT synthesizes DNA, it creates an RNA/DNA hybrid, which is concomitantly degraded by the RNase H enzymatic domain, leaving a single minus-strand DNA (Figure 3). The sequences of the 5′ and 3′ ends of the viral RNA genome are repeat regions.41–44 The RNA-free minus-strand DNA can hybridize to the repeat region of the 3′ end of either of the vRNA genomes, which is known as the first jump or minus-strand transfer and is one of the reasons for viral recombination that may affect drug resistance, as discussed below.45–48 After the minus-strand DNA hybridizes to the 3′ end of the RNA genome, synthesis of the DNA continues as the RNase H function of RT continues to degrade RNA. Additionally, there is a purine-rich sequence near the 3′ end of the vRNA, known as the 3′-polypurine tract (3′-PPT) that is resistant to cleavage by RNase H, which serves as primer for plus-strand DNA synthesis (Figure 3). There is also a central PPT (cPPT), which is known to increase plus-strand DNA synthesis but unlike the 3′-PPT is not essential.49–57 The 3′-PPT primed DNA synthesis continues and copies the first 18 ribonucleotides of the tRNA.51,58,59 RNase H removes the tRNA primer from the tRNA/DNA complex, leaving a ribo-A on the 3′ end of the minus-strand DNA; this sets the stage for the second jump, also known as plus-strand transfer. Once the tRNA is removed, it exposes the 3′ end of a single plus-strand DNA with PBS, at the same time, the minus strand DNA is also exposed (Figure 3).60 The exposed minus- and plus-strands anneal to each other and create double-stranded DNA (dsDNA) that has the same sequence on both ends also known as long terminal repeats (LTRs).26,28,36,61,62
Figure 3.

Reverse transcription of HIV-1. An HIV virion has two copies of the ssRNA genome. Once a host tRNALys3 binds to the primer binding site (PBS), DNA synthesis starts forming the minus-strand strong-stop DNA. During DNA synthesis, RNase H digests RNA, exposing the minus-strand DNA that hybridizes to the R region of the 3′-end of either viral genome copy. The elongation of the minus strand continues until the polypurine tract (PPT), which is RNase H resistant and serves as a primer for plus-strand DNA synthesis. After plus- and minus-strand DNA syntheses are completed, the RNase H removes the tRNA primer and releases the PBS, thus facilitating the “second jump”. Strand displacement activity of RT to the PBS and PPT ends and/or DNA repair and ligation lead to dsDNA or a circular intermediate with two long terminal repeats (LTRs). Figure adapted with permission from ref 63. Copyright 2020 Elsevier. License number: 4943870547699.
2. ROLE OF RT IN HIV GENETIC DIVERSITY AND DRUG RESISTANCE
HIV patients are infected by a highly heterogeneous pool of genetic variants known as quasispecies.64–67 The high genetic variability of HIV is thought to be the result of several factors, including a high mutation rate, high rate of viral replication that results in high viral load, the lack of 3′ to 5′ exonuclease repair function of its replicative enzyme, the RT,68 the introduction of random mutations by the host RNA polymerase II during transcription of the nascent viral RNA from the integrated proviral DNA,69 and by the process of viral recombination. Some of these processes are discussed below.
2.1. The Fidelity and Mutations
RT copies the ~9700 nucleotide long viral RNA genome into dsDNA through RNA- and DNA-dependent polymerization steps, totaling ~19 400 nucleotide incorporation events.5,70,71 The in vitro misincorporation rate for RT has been reported to be ~1 × 10−4, and it is affected by factors such as type of divalent metal used.72,73 The in vivo mutation rate of the viral replication is ~2 × 10−5.55,74–76 The high mutation rate of HIV is critical for its survival during drug therapies. The virus can obtain drug resistance mutations, which is one of the major challenges for effective HAART.77–86 The resistance mutations emerge as a result of poor patient adherence, which permits viral replication and mutation, increasing the chances that drug resistance mutations will occur and spread throughout the patient due to a selective growth advantage in spite of ongoing treatment. Some drug-resistant viruses are also capable of being transmitted, and consequently these strains can increase in prevalence as described below.77,84,87–93
2.2. Viral Recombination
Viral recombination is a process whereby RT engages into template switching between the two viral RNA templates (~3−4 times per replication cycle) that are copackaged into the same virion.94 Recombination of HIV increases the genetic diversity of the viral population affecting resistance to antivirals.47,48,95–101 The rapid turnover of 108 to 109 virions per day, combined with the high error rate of RT and the high rates of viral recombination, contribute significantly to the generation of a very large number of mutated HIV genomes per day.28,45,75,102–106
3. RT-TARGETING ANTIVIRALS
Currently there are two different types of approved RT inhibitors: nucleos(t)ide reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) (Figure 4). NRTIs approved and in use are azidothymidine (AZT), lamivudine (3TC), emtricitabine (FTC), didanosine (ddI), abacavir (ABC), and tenofovir (TFV). Two NRTIs have been discontinued due to toxic side effects: stavudine (d4T) and 2′−3-dideoxycytidine (ddC).26 As mentioned above, two of these NRTIs are typically used in the current HAART regimen. In the US, treatment regimen includes a combination of the NRTIs TFV and FTC.4 NRTIs are also typically used in pre-exposure prophylaxis therapy (PrEP).107–109 NNRTIs that have been approved for the treatment of HIV-1 infection: nevirapine (NVP), efavirenz (EFV), etravirine (ETR), rilpivirine (RPV), and doravirine (DOR). One NNRTI that has been discontinued is delavirdine (DLV). NNRTIs are also used in the HAART regimen, but less frequently.5,26,110
Figure 4.

Chemical structures of approved NRTIs and NNRTIs.
3.1. Current Nucleoside RT Inhibitors
NRTIs are analogues of the natural substrate deoxynucleotide triphosphates (dNTPs). All current approved HIV NRTIs lack a 3′-OH and act as chain terminators when incorporated into the primer strand of the viral DNA and thus prevent the addition of an incoming nucleotide. NRTIs inhibit reverse transcription by mimicking dNTPs and can be incorporated by RT. Because NRTIs lack a 3′-OH, once they are incorporated into the primer, chain termination occurs, ultimately preventing further DNA synthesis. The first anti-HIV drug approved was AZT, a thymidine analogue that has an azido group in the 3′-OH position.111 In contrast, ddC and ddI have no azido group but lack a 3′-OH,112,113 whereas d4T has a double bond between the 2′ and 3′ positions of the ribose and ABC has a cyclopentene ring also without a 3′-OH group.114,115 TFV is an acyclic nucleotide phosphonate116 delivered as a diester prodrug, earlier as a tenofovir disoproxil fumarate (TDF) and more recently (in 2015) as a tenofovir alafenamide (TAF).117 TAF is superior to TDF in terms of efficacy, stability, and safety.118–122
Three factors play a major role in NRTI development: (1) efficient activation to the triphosphate form, (2) stability of the NRTI, and (3) NRTIs barrier to resistance. For NRTIs to be effective, they must be phosphorylated by host cell kinases, which convert them to their active triphosphate form.123 Once activated, they must compete with the natural substrate for incorporation by RT into the elongating primer. All NRTIs must be phosphorylated three times, with the exception of TFV. Because TFV has a phosphonate, it needs an addition of only a β- and γ-phosphate, skipping the addition of the α-phosphate, the first and rate-limiting phosphorylation event for most NRTIs. However, one exception is AZT, where the rate limiting step is the addition of the second and third phosphates; due to these reasons, the cellular levels of AZT-TP are relatively low.124 After an NRTI is converted to its triphosphate form, RT incorporates it as an NRTI monophosphate at the 3′ end of the primer end and a pyrophosphate product is released.
3.2. Non-nucleoside RT Inhibitors
Unlike NRTIs, NNRTIs do not compete for the natural substrate; instead, they block RT as allosteric inhibitors. NNRTIs bind in a hydrophobic pocket ~10 Å away from the polymerase active site known as the NNRTI binding pocket (NNIBP). The NNIBP consists of the following residues: L100, K101, K103, V106, T107, V108, V179, Y181, Y188, V189, G190, F227, W229, L234, and Y318 of p66 and E138 of p51 (Figure 5).10,12
Figure 5.

Structure of the HIV-1 RT NNIBP. Select residues of the NNIBP of HIV-1 RT (PDB 1VRT)125 are shown as sticks, with NVP shown as transparent orange sticks. These include L100, K101, K103, V106, V179, Y181, Y188, F227, W229, L234, and P236 of the p66 palm subdomain (pink sticks), Y318 of the p66 thumb subdomain (green sticks), and E138 of the p51 subunit (gray sticks). Additional residues in the NNIBP include T107, V108, V189, and G190, which are not shown for clarity.
Crystallographic studies have shown that the NNIBP does not exist in the absence of NNRTIs.5,10,125–131 For an NNRTI to bind in the NNIBP, structural changes must occur, particularly in residues Y181 and Y188 at the “floor” and W229 at the “ceiling” of NNIBP. The latter is formed in part by the “primer grip”, a structural element of RT that positions the nucleic acid substrate at the polymerase active site. These changes perturb the “primer grip”, and as a result, NNRTI binding affects the alignment of the primer terminus in the active site (Figure 6), which ultimately inhibits chemical synthesis.125,132–136 Of note, because the primer directly interacts with the base of the thumb (Figure 6), NNRTI binding and primer grip repositioning also affect the position of the thumb, which ends up in an overextended conformation (pale green vs green in Figure 6).26,137
Figure 6.

Structural changes occur in HIV-1 RT upon NNRTI binding, which affect the position of nucleic acid binding. HIV-1 RT covalently cross-linked to dsDNA (p66 palm is shown in red cartoon, p66 thumb is shown in green cartoon, primer is shown as dark-gray sticks; template has been removed for clarity) with NVP (purple sticks) bound at the NNRTI binding pocket and AZT-MP (dark gray sticks) at the 3′-end of the DNA primer strand in the P-site (PDB 3V81).138 The crystal structure of HIV-1 RT (p66 palm and p66 thumb in pink and light-green cartoon, respectively) covalently cross-linked to dsDNA (primer in light gray sticks; template not shown for clarity) with AZT-MP at the P-site and AZT-TP in the N-site (PDB 3V4I)138 was aligned to structure 3V81 by p66 residues 100−210. Binding of NVP at the NNIBP causes Y181 and Y188 in the NNIBP to flip 180° and also a shift in the position of W229 (pink vs red sticks before and after NVP binding). These changes lead to a cascade of structural rearrangements that include repositioning of the conserved YMDD loop at the polymerase active site, the primer grip, and the p66 thumb subdomain, which are important for nucleic acid binding (movements noted with black dashed arrows). The nucleic acid repositioning is noted by the black dashed arrow between the two AZT-MP molecules at the P-site.
NNRTIs can potently inhibit the replication of WT HIV-1; however, because the RT enzymatic activity does not directly involve the NNIBP, this pocket can be relatively easily mutated to impart NNRTI resistance (low barrier to resistance) without significantly affecting the ability of the virus to efficiently replicate, known as viral fitness. The early NNRTIs NVP, EFV, and DLV bind at the NNIBP in a rigid “butterfly-like conformation”.126,128,139,140 A major drawback of early NNRTIs is that, typically, a single NNIBP resistance mutation (such as L100I, K103N, V106A, E138K, Y181C, Y188L, G190A, or H221Y) can result in significant loss of their efficacy.126 In contrast, the newer NNRTIs, ETR and RPV, were developed to have higher conformational flexibility (Figure 7) and inhibit the common NNRTI resistance mutants.129,141,142 Furthermore, this flexibility trait helped maintain the efficacy of ETR and RPV in the presence of K103N and Y181C.141 RPV was designed to efficiently bind in the NNIBP and inhibit effectively using the following interactions: (1) hydrophobic interactions within the NNIBP, (2) hydrogen bonding with the main chain of the K101 residue,143 and (3) water-mediated hydrogen bonds.5,144 Structural evidence demonstrated that RPV could adapt multiple conformations in the presence of various NNRTI resistance mutations. This trait is commonly known as “wiggling” and “jiggling”.143 As of 2018, DOR has been approved and was also shown to be effective against common NNRTI resistance mutations, including K103N, Y181C, and G190A.110,145–147
Figure 7.

Second-generation NNRTIs have more intrinsic flexibility that enables binding and inhibition in the presence of several NNRTI resistance mutations. (A) Chemical structure of second-generation NNRTI rilpivirine (RPV), with labeled torsion angles demonstrating the flexibility of this molecule. (B) The terms “wiggling” and “jiggling” are used to describe how newer NNRTIs position themselves to adopt multiple conformations and adjust for the changing side chains in mutated NNIBPs. (C) Although structurally similar, diarylpyrimidine (DAPY) analogues R120393 (a), TMC120-R147681 (dapivirine) (b), TMC125-R165335 (etravirine) (c), and R185545 (d) are able to bind at the NNIBP assuming diverse conformations, thus avoiding mutated residues in the NNIBP. (B,C) Reproduced unmodified from ref 141. Copyright 2004 American Chemical Society.
3.3. Novel RT Inhibitors
4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA, MK-8591, or islatravir) is an exceptionally potent and promising long-acting RT inhibitor in phase III clinical trials.148–150 EFdA has the following three structural attributes that contribute to the stability, long-acting potential, and potency: (1) 2-fluoro, (2) 4′-ethynyl (4′-E), and (3) 3′-OH.151–153 The 2-fluoro group contributes to the remarkable stability of EFdA by making EFdA resistant to adenosine deaminase, a key metabolizing enzyme of adenosine and adenosine-based antivirals.149,154,155 Unlike any other approved HIV NRTI, EFdA retains a 3′-OH; it is also efficiently phosphorylated to EFdA-MP by the cellular deoxycytidine kinase.149,154 The 4′-E group stabilizes EFdA into a conserved hydrophobic pocket at the polymerization active site.156 EFdA inhibits reverse transcription primarily by blocking translocation of RT on the template-primer. For this reason, EFdA is known as a nucleoside reverse transcriptase translocation inhibitor (NRTTI). EFdA can block translocation through two mechanisms: (1) immediate chain termination (ICT) and (2) delayed chain termination (DCT) (Figure 8).150 During ICT, RT is arrested immediately following incorporation of EFdA-monophosphate (EFdA-MP), and the 3′-end of the terminated primer remains bound at the pretranslocation or N-site. DCT occurs when EFdA-MP is incorporated into the 3′-end of the primer and translocates to the primer- or P-site, allowing the addition of a single nucleotide before chain termination. Crystallographic studies of various EFdA-inhibition intermediates of RT provide glimpses on how EFdA affects RT translocation through two mechanisms.156 Because of the high potency and stability of EFdA, it is being tested as a once-yearly therapy instead of the current once daily dosing regimens (see below).157,158
Figure 8.

Immediate and delayed chain termination mechanisms of EFdA inhibition. Initial binding of EFdA-TP in the N-site (N, orange box), prior to incorporation (left). Once EFdA-MP is incorporated into the 3′-end of the primer, it may cause immediate chain termination (ICT) (middle) or delayed chain termination (DCT) (right). During DCT, after EFdA-MP incorporation at the 3′-end of the primer and translocation, which moves the EFdA-MP at the P-site (P, blue box), RT adds a single additional nucleotide before inhibition of further DNA synthesis. Figure based on ref 156.
Elsulfavirine or VM-1500 is an another potent NNRTI (EC50=1.2 nM) that is active against a broad range of NNRTI resistant viruses (VM-1500 is the name of the prodrug and VM-1500A is the active compound).146,159 Elsulfavirine has been approved for HIV-1 treatment in Russia.160
4. MECHANISMS OF DRUG RESISTANCE
4.1. NRTI Resistance
Under selective drug pressure, drug resistant mutants can emerge and gain a competitive advantage over NRTI-treated WT virus, thus becoming the dominant quasispecies. In principle, mutations should affect drug binding or function but should still allow viral replication. HIV-1 resistance to NRTIs usually involves two general mechanisms: NRTI discrimination and NRTI excision.
4.1.1. (a) NRTI Discrimination.
NRTI discrimination occurs when RT reduces the NRTI incorporation efficiency compared to the natural dNTP. This tends to appear along the dNTP binding pocket from the β3-β4 fingers and the conserved YMDD loop (Figure 2). K65R emerges in response to treatments with TFV, ABC, ddI, and d4T.161–163 Structural studies have revealed that K65R becomes resistant to NRTIs, because R65 together with R72 forms a platform that helps discriminate TFV-DP from dATP (Figure 9).164 The resistance mutation Y115F emerged in response to treatment with ABC; as the Y115 residue supports dNTP binding, mutation to phenylalanine maintains most structural interactions but increases the chance of NRTI discrimination.165 Together, K65R and Y115F further decrease TFV susceptibility.166 The L74V mutation has been shown to confer ddI resistance; the L74 residue supports base-pair formation with the incoming dNTP, and the L74V mutation weakens the template interaction with the dNTP substrate or reduces the template support.167,168 M184V/I RT mutations cause resistance to FTC and 3TC by directly interfering with the binding of the triphosphate form of the drugs at the polymerase active site.9,169–171 The mutated residues at the 184 position, isoleucine or valine, are β-branched amino acids, and sterically clash with the L-oxathiolane ring of 3TC-TP/FTC-TP.9,169–171 The Q151M mutation causes resistance to dideoxynucloside analogues, especially in combination of other associated mutations that are known as the Q151M complex (Q151M/A62V/V75I/F116Y). These mutations emerged in response to combination therapy of AZT with dideoxynucleoside drugs.172,173 The Q151M complex causes resistance to many NRTIs, but not to 3TC/FTC and TFV.
Figure 9.

Mutation K65R in HIV-1 RT imparts resistance to NRTIs through discrimination. Structural studies revealed that K65R becomes resistant to NRTIs because 65R together with R72 form a platform (light-blue sticks and spheres) that helps discriminate dATP (A, magenta sticks and spheres, PDB 3JYT) from TFV-DP (B, gold sticks and spheres, PDB 3JSM) at the polymerase active site (red cartoon and pink sticks and spheres). Figure based on ref 164.
4.1.2. (b) NRTI Excision.
NRTI excision was initially discovered as a resistance mechanism during treatment with AZT.174 Hence, the NRTI resistance mutations were termed as thymidine analogue resistance mutations (TAMs) (Table 1). These mutant RTs are capable of incorporating AZT as efficiently as WT RT. However, RTs containing TAMs can unblock NRTI-terminated primers allowing DNA synthesis to resume.175,176 For an NRTI to be excised, it must be located in the N-site; as the metal binding machinery is required for excision, RT cannot excise from the P-site. AZT-MP can be excised more frequently and better than other NRTIs because of the long azido group that interferes with the binding an incoming dNTP, thus keeping AZT-MP in the N-site more frequently.27 Also, excision occurs because two main mutants K70R and T215Y create an ATP-binding pocket next to the dNTP binding cleft, where the aromatic ring of the tyrosine (T215Y) stacks with the ATP base, and the arginine (K70R) forms a polar interaction with the 3′-OH and the α-phosphate of ATP.177,178 The ATP binding pocket allows binding of ATP and positioning its β- and γ-phosphates in a manner similar to a pyrophosphate group. In turn, this enabling chelation of the two Mg2+ ions, and ultimately promoting pyrophosphorolysis (Figure 10), so RT can remove the AZT-MP from the end of the DNA primer.179 The product of the excision reaction when AZT-terminated primers are unblocked by ATP is an AZTppppA tetraphosphate and an unblocked primer.177,178 The most common mutations responsible for excision are K70R and T215Y, but other mutations are known to enhance excision and the virus’ replication capabilities.177,180,181 D67N and K219Q are found to be associated with K70R,177,182–184 while M41L and L210W are associated with T215Y.185,186 The deletion of codon 67 also increases the excision efficiency of AZT.187,188 Interestingly, a T69 insertion into the β3- and β4-loops of RT was found to help excise other NRTIs.189–192
Table 1.
NRTI Resistance Mutations
| NRTI resistance mutation(s) | mechanism of resistance | drug(s) affected by resistance mutation(s) |
|---|---|---|
| K65R | discrimination | TFV, ABC, ddI, and d4T |
| Y115F | discrimination | ABC |
| L74V | discrimination | ddI |
| M184V/I | discrimination | FTC and 3TC |
| Q151M | discrimination | ddI, AZT, d4T, and ABC |
| Q151M/A62V/V75I/F116Y | discrimination | ddI, AZT, d4T, and ABC |
| Type 1: M41L/L210W/T215Y | excision | AZT and d4T |
| Type 2: D67N/K70R/T215Y/K219Q | excision | AZT and d4T |
Figure 10.

Structural basis for excision-based AZT resistance: interactions of excision product with TAMs RT. HIV-1 RT is shown as cartoon (colored as in Figure 1) bound to dsDNA (template in light brown, primer in dark brown) with key residues shown as pink sticks. The product of AZT-MP excision, AZTppppA, is shown as cyan sticks at the N-site. AZT-resistance mutations (M41L, D67N, K70R, T215Y, K219Q; thymidine-associated mutations or TAMs; shown as magenta sticks) facilitate excision by forming the ATP substrate binding site (PDB 3KLE). Figure based on ref 177.
4.2. NNRTI Resistance
As mentioned in section 3.2, NNIBP residues are not directly involved in the polymerization mechanism of RT, and as such they can mutate without a significant cost in replication fitness (Table 2). Thus, the barrier to NNRTI resistance is low and NNRTI resistance mutations appear relatively fast. There are three main mechanisms by which NNIBP mutations impart NNRTI resistance: (1) loss of key hydrophobic interactions (V106A, V179D, Y181C, Y188L, F227C/L), (2) steric hindrance (L100I or G190A/S), and (3) pocket entrance mutations (K101E/P, K103N, and E138K (of p51)).
Table 2.
Fold-Changes of NNRTI Resistance compared to WT EC50s
| resistance mutation | NVP | ETR | EFV | RPV | DOR | ref |
|---|---|---|---|---|---|---|
| L100I | 7.3 | 1.3 | 20.3 | 0.9 | <3 | 218–220 |
| K101E | NA | 2.7 | 3.8 | 2.4 | 4.5 | 110,219 |
| K103N | 81 | 0.9 | 32.5 | 0.9 | 1 | 218–220 |
| V106A | >42 | 0.5 | 2.0 | 0.6 | ∼45 | 110,219,220 |
| V179D | 4.6 | 1.9 | 2.7 | 1.7 | NA | 219 |
| Y181C | 317 | 4.0 | 2.1 | 2.7 | 1.1 | 218–220 |
| Y188L | >42 | 1.1 | 42.6 | 2.8 | >100 | 110,219,220 |
| G190A | 128 | 1.1 | 8.1 | 1.1 | <5 | 218–220 |
| F227C | 17 | 3.6 | 5.1 | 4.1 | >10 | 110,220,221 |
| E138K (of p51) | 1.1 | 2.6 | 2.0 | 2.8 | 1 | 219,220 |
The amino acid residues that are located in the hydrophobic core of the NNIBP are Y181, Y188, and F227 (Figure 5).10,125–127 Mutations in these residues can cause high resistance through loss of hydrophobic interactions with the incoming NNRTIs.131,193–196 Resistance mutation Y181C is primarily selected with treatment to NVP (>50-fold reduced susceptibility).197,198 Because of their conformational flexibility, the newer generation NNRTIs, ETR and RPV, are able to maintain antiviral activity better than the first-generation NVP, enduring antiviral potency losses of only 5-fold and 4-fold, respectively.198–201 Resistance mutation Y188L confers high-level resistance to both NVP and EFV.197,202–204 In contrast, RPV has only a 5-fold reduced activity against Y188L.205–207 F227C is frequently selected during treatment with DOR or with ETR and RPV.201 It is also selected during NVP or EFV-based therapies, although in these cases it usually appears in combination with V106A, conferring high-levels of resistance.203,204,208
L100I and G190A mutations cause steric hindrance at the center of the NNIBP, leading to resistance by changing the shape of the pocket. For instance, L100I mutates from a γ-branched amino acid to a β-branched amino acid.196 L100I has high-level resistance to EFV, ETR, and RPV. G190A introduces a bulge, mutating from a small residue, glycine, to an alanine.209 Other mutations can also occur at G190 such as a serine, glutamic acid, or a glutamine, which also introduce a bulge and distort the NNIBP. DOR remains effective at inhibiting G190A and has mildly reduced efficacy against the other mutants at this position.210 RPV and ETR are effective at inhibiting G190A/S (unless it is in combination with other NNRTI-resistance mutations) and have reduced susceptibility to G190E/Q.206
Pocket entrance mutations interfere with an NNRTI entering the NNIBP. Mutant K103N does not specifically interact with an NNRTI but rather restricts access to the NNIBP.135,211 However, ETR, RPV, and DOR remain effective against K103N, while NVP and EFV do not.203,206,212 Other pocket entrance mutations K101E/P and E138K (of p51) seem to act similarly to K103N, with their side chains protruding out of the pocket and preventing NNRTI entry. K101P causes resistance to all current NNRTIs, except DOR, unless in combination with other mutations.199,213 K101E causes resistance to RPV and NVP.206,214 In addition, residue 138 can mutate to glycine, glutamine, arginine, or most commonly to lysine (E138K) that imparts RPV resistance.201 E138K is clinically relevant because it was selected together with M184V/I in patients that experienced virological failure while being treated with RPV/TDF/FTC. Virological assays showed that E138K/Q/R mutations can compensate for M184I in both enzymatic fitness and viral replication capacity assays.215–217
5. STRATEGIES TO AVOID DRUG RESISTANCE
HIV RT has been a key target for anti-HIV drugs since 1986.111 HAART has been successful in suppressing HIV. However, due to the high genetic diversity of HIV, prolonged treatments lead to the emergence of drug resistance mutations. Thus, there is a need for novel antivirals with excellent affinity, strong potency, high barrier to resistance, and able to overcome the known mechanisms of resistance using a combination of strategies discussed below.
5.1. Using the “Substrate Envelope” Strategy to Design Inhibitors with Improved Resistance Profile
The substrate envelope is based on the premise that inhibitors that fit into the substrate envelope have a better resistance profile, as they stay protected from binding site mutations that cannot reach into the substrate envelope without significantly affecting the binding of the native substrate.222–224 Hence, inhibitors that protrude from the substrate envelope are more likely to elicit resistance.
The substrate envelope in the case of RT comprises the binding site of dNTP (N-site, Figure 2) and nucleic acid. The resistance profiles of TFV, 3TC, and FTC, which are major NRTIs used in today’s HAART, are relevant to this principle. Specifically, unlike 3TC/FTC that protrude outside the substrate envelope and sterically clash with V/I184 leading to >100-fold resistance, the acyclic TFV is fully active against M184V/I because it binds within the substrate envelope (Figure 11).9,169–171,225 However, RT eventually develops resistance to TFV, by acquiring the K65R mutation, which does not cause a steric clash (Figure 9). Instead, K65R decreases the rate of TFV incorporation and causes mild resistance (3−5-fold).164 Importantly, this mutation also affects the viral fitness by decreasing the efficiency of dNMP incorporation.226,227 In that respect, TFV is an example of a successful design that avoids the steric clash mechanism of drug resistance and leads to a mutation (K65R) that forces escape through a costly decrease in viral replication.
Figure 11.

TFV avoids resistance relationship between NRTI resistance and the substrate envelope. The natural substrate dATP (gray sticks; PDB 5TXL)228 defines the substrate envelope. FTC-TP (blue sticks; PDB 6UIR)229 protrudes from the substrate envelope, thus causing a steric clash with M184V (red dashed lines and explosion cartoon), resulting in >100-fold resistance. TFV-DP (light-cyan sticks; PDB 3JSM)164 binds within the substrate envelope and thus remains active against M184V/I. Structural alignments were based on RT residues 50−200.
5.2. Targeting Evolutionarily Conserved Regions in RT
Targeting conserved regions in a highly genetically diverse virus is essential to drug design because such regions are much less likely to mutate. Evolutionary conservation is expected to have functional significance that is indispensable for virus survival. Ideally, a drug should interact with key residues that are involved in the mechanism of reverse transcription. Thus, mutations of such residues should result in loss of function for RT and decreased viral fitness.
Because NRTIs mimic natural dNTPs and bind at the dNTP or N-site they may interact, at least in part, with highly conserved residues in the polymerase active site. For example, K65 and R72 are highly conserved residues that interact with the γ- and β-phosphates of the incoming dNTP or NRTItriphosphates.9,24 As mentioned above, the K65R mutation may allow narrow escape of TFV with a modest drug resistance, however, even the slight rearrangement of the 65/72 residues observed in the structure of K65R RT (Figure 9) results in a fitness cost that is direct result of mutation of the highly conserved function of precise alignment of the triphosphate component of incoming nucleotide or NRTI triphosphates.164
Another example of targeting conserved residues is the case of novel NRTTI EFdA. This compound has a 4′-E that binds into a conserved hydrophobic pocket defined by these residues: A114, Y115, F160, and M184 and the aliphatic part of D185156 (Figure 12). After incorporation of EFdA-MP into the viral DNA, the 4′-E of the elongated primer is stabilized by the interactions with residues of the conserved pocket leading to inhibition of the DNA synthesis (Figure 12). Three (A114, Y115, F160) out of the four residues that define the 4′-E binding pocket are highly conserved, leading to a high barrier to resistance for EFdA.
Figure 12.

Interactions of EFdA-TP at the N-site of the HIV-1 RT polymerase active site. HIV-1 RT (PDB 5J2M) is colored as in Figures 1 and 2. The α-, β-, and γ-phosphates of EFdA-TP (gold sticks) participate in chelation of the Mg2+ ion along with conserved active site residues D110, the main chain of V111, and D185 (interactions shown as blue dashed lines). The 4′-ethynyl (4′-E) group of EFdA-TP sits in a hydrophobic pocket at the base of the polymerase active site composed of residues A114, Y115, F160, M184, and D185 (interactions shown as black dashed lines). The 3′-OH participates in hydrogen bond interactions with its β-phosphate (interaction shown as green dashed line). For clarity, we do not show additional interactions between the 3′-OH and the main chain of Y115 and water-mediated hydrogen bond interactions with A114 and F116. The 2-fluoro (2-F) group of EFdA-TP participates in water-mediated interactions with Y115 and the main chain nitrogen of G152 (interactions shown as red dashed lines).
Another example of an RT-targeting antiviral that interacts with conserved residues to block translocation of RT is foscarnet or phosphonoformic acid, which is an analogue of the pyrophosphate product of DNA synthesis (Figure 13). Foscarnet has not been approved for the treatment of HIV infection because of its side effects. However, it is an RT inhibitor that stabilizes the pretranslocation state of RT by interacting with conserved residue K65. Of note, RT escapes foscarnet inhibition through the E89G mutation that affects primer translocation, but also viral fitness.230
Figure 13.

Chemical structures of pyrophosphate (PPi) and foscarnet (phosphonoformic acid, PFA).
Finally, another strategy of targeting evolutionarily conserved regions is the design of compounds that affect protein−protein interactions (PPI). Such compounds have been useful in the design of anti-HIV inhibitors that block interactions of LEDGF75 with HIV integrase231 as well gp120 interactions with CD4.232,233 Moreover, inhibitors can also interfere with dimerization of the HIV-1 protease dimerization,234,235 interactions between integrase subunits leading to aggregation,236,237 and misdirection of viral maturation, as well as antivirals that target the interface of the p66/p51 RT heterodimer. There are hydrophobic interactions at p66/p51 interface involving a cluster of tryptophans (W398, W401, W402, W406, W410, and W414), which are fundamental for RT dimerization as demonstrated by mutational studies.238,239 An example of this type of inhibitors that target RT dimerization is MAS0.240,241 Also, as the NNIBP comprises not only residues of the p66 subunit but also of p51 (E138), some NNRTIs, such as EFV, ETR, and dapivirine, have been reported to affect RT dimerization242 and intracellular processing of Gag and Gag-Pol polyproteins.243 Therefore, targeting various conserved regions of RT has been a main strategy of drug design for RT inhibitors.
5.3. Targeting the Conserved Divalent Metal Binding Sites
RT uses divalent metals for both its polymerase and RNase H enzymatic functions. The polymerase site YMDD loop that includes the three highly conserved catalytic carboxylates (D110, D185, and D186) are critical for positioning the two Mg2+ ions that are required for DNA synthesis (Figure 2).16–19 Disrupting their divalent metal binding function would be expected to be an efficient strategy for designing antivirals with high barrier to resistance. As many cellular polymerases have related divalent metal binding motifs, it would be important that such designed inhibitors avoid binding to related motifs of cellular polymerases or kinases and other phosphotransferases that often use divalent metal-binding mechanism for their catalytic functions. A similar approach has been successful in the design of compounds that bind the active site metals of the integrase enzyme of HIV.244,245 In such cases, the inhibitor would be composed of one part that binds the two divalent metals bound to the conserved catalytic carboxylates, and another part of the inhibitor would impart specificity for the viral active site, rather than the cellular two-metal binding enzymes.
Another example of this principle is the design of antivirals that bind the RNase H domain of RT. Various RNase H inhibitors that have been previously explored in the context of HIV-1 have chelating groups, which interact with the two divalent metals of the RNase H domain and inhibit the RNase H activity of RT (Figure 14).246–256
Figure 14.

Interactions of RNase H inhibitors (RNHIs) at the RNase H active site of HIV-1 RT. (A) A 2-hydroxyisoquinoline-1,3-dione RNHI, YLC2–155 (orange sticks), chelates two Mn2+ ions (light-blue spheres) that are bound by conserved RNase H active site residues D443, E478, E498, and D549. YLC2–155 also forms hydrogen bond interactions with Q500 and H539 (PDB 5UV5). YLC2–155 can also bind to the RNase H active site in a different conformation, with the furan ring pointing toward H539 (not shown). Based on ref 252. (B) A hydroxypyridonecarboxylic acid RNHI, 10y (yellow sticks), chelates two Mg2+ ions (light-green spheres) that are bound by the conserved RNase H active site residues. 10y also forms hydrogen bond interactions with H539 and K540 (PDB 5J1E). Based on ref 256.
5.4. Designing Drugs That Interact with Main Chain Residues of RT
Designing antivirals that target main chain polyamide backbone atoms is based on the premise that regardless of the residue mutation, the inhibitor will still maintain its interaction with the main chain of the residue. This approach can be particularly valuable when targeting a virus that has a high mutation rate like HIV. Some examples of such interactions of drugs that target RT are discussed below.
For example, the 2-fluoro substituent of EFdA-TP at the N-site is an important contributor to the success of EFdA because it interacts with the G152 main chain amide (Figure 12A).156 Maintaining these main chain residue interactions helps minimize the potential for drug resistance to occur. Similarly, the inhibitory effect of EFdA during delayed chain termination (DCT) is based on a negative interaction of the main chain of Y183 and the 4′-E of EFdA-MP after its incorporation into the primer strand and elongation by a single nucleotide (DNAEFdA-MPP•dTMPN) (Figure 15).156 This disruption of the interactions of RT with the nucleic acid prevents further DNA synthesis resulting in the DCT mechanism of inhibition. Any mutation at the 183 position would not enable escape and drug resistance, as the steric interactions would be present regardless the type of amino acid present at this position.
Figure 15.

RT interactions during the delayed chain termination (DCT) inhibition mechanism. Superposition of two RT structures with EFdA variants before and after translocation. Transparent light-brown sticks show the position of the DNA primer with ddG at the post-translocation P-site and EFdA-TP at the pretranslocation N-site (PDB 5J2M). After EFdA-MP is incorporated into the 3′-end of the primer and further elongated by a single nucleotide (RT/DNAEFdA-MPP• dTMPN), EFdA-MP is located at the P-site (PDB 5J2N). In this position, the 4′-ethynyl (4′-E) sterically clashes with the main chain carbonyl of Y183, and a large shift occurs only at the P-site of this primer to alleviate this negative interaction, as shown by the distance between the 4′ carbon atoms and the dihedral angles of the two structures. Based on ref 156.
There are several examples of NNRTIs that interact with main chain residues of RT. EFV forms hydrogen bonds with the main chain C=O and N−H groups of residue K101 (Figure 16A).130 Similarly, NVP forms water-mediated hydrogen bonds with the main chain amino and carboxyl groups of the K101 residue (Figure 16B).125 Also, hydrogen bonds between a nitrogen from RPV and the main chain C=O and N−H of K101, and water-mediated hydrogen bonds with between an additional nitrogen and the main chain of E138 in p51 contribute to binding of RPV (Figure 16C).143,257 Finally, DOR interacts with V106 and L100 and the methyl triazolone stacks with P236 and interacts with the main chain atoms of K103 (Figure 16D).145
Figure 16.

Several NNRTIs interact with residues in the NNIBP through main chain interactions. (A) EFV (orange sticks, PDB 1FK9)130 forms hydrogen bond interactions with the main chain C=O and N−H groups of K101. (B) NVP (green sticks, PDB 1VRT)125 interacts with the main chain C=O and N−H groups of K101 through water-mediated hydrogen bonds. (C) RPV (yellow sticks, PDB 2ZD1)143 forms hydrogen bonds with the C=O and N−H groups on the main chain of K101 and interacts with E138 in p51 through water-mediated hydrogen bonds with the main chain C=O group. (D) DOR (blue sticks, PDB 4NCG)145 has hydrophobic interactions with L100 and V106 and forms hydrogen bond interactions with the C=O and N−H groups on the main chain of K103.
Therefore, interactions between hydrogen bond donors or acceptors from a potential RT inhibitor could significantly contribute to its binding potential and decrease its susceptibility to drug resistance.
5.5. Designing Covalent Inhibitors of RT
Covalent inhibitor drugs are typically small molecules that bind irreversibly their target. They have functional groups that can react and bind covalently to an amino acid side chain of the target protein, leading to its inactivation.258–261 They include drugs that have been used for many decades; aspirin and penicillin are some of the early and well-known members of this class of drugs. Among the advantages of covalent inhibitors is that their potency is usually higher than that of the noncovalent version of the inhibitor, as by definition, the koff of an irreversible covalent inhibitor is zero. Also, covalent inhibitors offer long duration of action if the target protein has slow turnover.262 Covalent inhibitors bind a specific region of the target protein and have a reactive functional group known as “warhead” that covalently links to a specific amino acid, leading to a loss of function. Designing covalent inhibitors begins with identifying a conserved nucleophilic amino acid (a cysteine, lysine, serine, threonine, tyrosine, methionine, glutamate, or aspartate), at a site where the inhibitor binding can affect the functionality of the enzyme.263–266 The residue selection is important because it should be near the active site or at a pocket that is important for the function of the target protein.259 Targeting conserved residues is also important, as it should result in compounds that have relatively high barrier to resistance. Furthermore, the binding site needs to be unique or rare, to minimize the chances of nonspecific binding and off-target effects. There are now computational tools that can be used for the design of covalent inhibitors, including AutoDock.267,268
There have been increasing efforts toward the discovery of covalent inhibitors that target HIV. A recent study focused on covalent RT inhibitors (CRTIs) (Figure 17) that specifically target RT that carries the Y181C mutation that renders several NNRTIs ineffective.269,270 The compounds used in this study were based on an originally noncovalent inhibitor that was modified by adding an electrophilic warhead on the carbonchloride bond (Figure 17); this change enabled covalent binding at the C181 residue after the specific initial binding at the NNIBP (Figure 17). Such compounds could be useful for patients that have failed some NNRTI-based therapies that lead to the Y181C mutation. Hence, inhibitors that have the potential to specifically inactivate wild-type or mutant RTs can be a useful strategy in combating HIV RT drug resistance. In this specific case, a compound that specifically targets the sulfydryl group of Y181C NNRTI-resistant viruses could be used in combination with NNRTIs that target the WT version of HIV.
Figure 17.

Covalent inhibitors targeting NNRTI-resistant HIV-1 RT. (A) Chemical structure of compound 1, which served as the basis for the design of covalent RT inhibitors (CRTIs). A chloride bound to a carbon (circled in green) in this compound was replaced with an electrophilic warhead to create various CRTIs that were designed to inhibit Y181C HIV-1 RT. (B) Crystal structure of compound 3 covalently bound to 181C in the NNIBP of Y181C HIV-1 RT (PDB 5VQX).269 Compound 3 (beige sticks) forms additional interactions with the main chain amide of K103.
5.6. Using Conformational Flexibility to Design Inhibitors That Assume Multiple Conformations and Successfully Target Ever-Changing Binding Pockets
Drug molecules that are rigid may be susceptible to multiple drug resistance mutations. This is because they may not adjust efficiently to minor perturbations introduced by mutations that otherwise do not affect the target protein’s function. This may be highly relevant for HIV, a virus with high genetic diversity that can acquire resistance mutations.
It has been proposed that first-generation NNRTIs NVP and DLV have a relatively low barrier to resistance due to their rigid structure that causes loss of interactions and potency through even a single resistance mutation.198 In contrast, the newer generation NNRTI RPV was developed through a strategic approach to have more intrinsic flexibility that enables binding and inhibition even in the presence of NVP and DLV resistance mutations. This approach, known as “wiggling” and “jiggling”, explains the success of newer generation of drugs that orient and position themselves to adopt multiple conformations and adjust for the changing side chains of the NNIBP (Figure 7B).143 Thus, the intrinsic flexibility of these compounds (Figure 7A) allows them to retain their potency in the presence of several common NNRTI resistance mutations. RPV and related diarylpyrimidine (DAPY) analogues are able to bind at the NNIBP using multiple conformations (Figure 7C) and thus angle away from changes at residue I100 and move toward residue 103.143 Hence, designing more flexible inhibitors should result to drugs that adapt multiple conformations and have a better chance to overcome drug resistance.
5.7. Design Inhibitors That Allosterically Interact at Nonactive Site Binding Pockets
Allosteric inhibitors bind to a nonactive site of the enzyme, which alters the confirmation of the active site and ultimately disrupts the substrate bound to the enzyme. An example of HIV allosteric inhibitors are NNRTIs; however, NNRTIs have a relatively low barrier to resistance. The NNRTIs bind in the NNIBP, where this pocket is not directly essential for the function of RT and resistance mutations are more likely to occur. However, work from the Arnold and Tachedjian laboratories have used a strategy known as fragment screening by X-ray crystallography to identify novel potential sites in RT that could provide an allosteric site with a higher barrier to resistance (Figure 18).271–274
Figure 18.

Fragment screening to identify potential binding sites for novel antivirals targeting HIV-1 RT. Fragment screening can be used to identify new binding sites for allosteric inhibitors. Shown are fragment binding sites on HIV-1 RT (orange spheres). Reproduced with permission from ref 273. Copyright 2013 American Chemical Society.
5.8. Designing Combinations of Drugs That Act Synergistically
On several occasions, drugs that belong to the same family, such as NRTIs, are not cross-resistant to each other’s escape mutations. In fact, there are reported cases where resistance to one drug renders RT hypersensitive to the other. Combinations of such drugs exhibit synergy and can be powerful tools in antiviral therapies.
For example, 3TC (or FTC)/AZT (combivir) was one of the relatively successful early combination therapies, because not only FTC (and 3TC) maintains potency against the AZT-resistant TAMs, but also FTC (and 3TC)-resistant M184V/I RT becomes hypersensitive to AZT. This is because M184V/I RT is unable to unblock AZT-terminated primers, thus leading to increased susceptibility to AZT and synergistic action.9,169,170,177–179,275 Another example of successful combination is TFV and FTC (truvada). In this case, the FTC-resistant M184V/I mutant RT is hypersensitive to TFV, while K65R reduces the susceptibility of TFV and FTC. K65R alone is not usually sufficient to suppress the activity of FTC in vivo.9,169,170,276–279 There are also examples of synergy in combinations of NRTIs and NNRTIs that use different mechanisms of RT inhibition. For example, RPV and TFV have been shown to inhibit RT synergistically because RPV stabilized complexes of TFV-terminated DNA primer/templates and RT.280 Similarly, it has also been reported that combination of RPV and EFdA have synergistic effects.281 Finally, another study showed that NNRTIs DOR and RPV had nonoverlapping resistance profiles and it was thus proposed that they could be used in combination.282 Hence, understanding in depth the molecular, biochemical, and virological basis of synergy between drugs is necessary for designing optimal combinations that will maximize suppression of a genetically diverse virus such as HIV.
5.9. Designing Drugs That Block Specific Resistance Mechanisms
In section 5.1, we discussed strategies to avoid the discrimination mechanism of RT resistance to FTC/3TC. Here, we discuss strategies to avoid the excision mechanism of RT. Excision based resistance mutations M41L/D67N/K70R/L210W/T215Y/K219Q form a novel pocket that enables binding of ATP that is used for a phosphorolytic reaction to unblock the chain terminated primer.177 The products of the excision reaction is an unblocked DNA primer that can now continue to be extended by the polymerase action of RT as well as a tetraphosphate (AZTppppA). The structure of this tetraphosphate product in complex with RT carrying TAMs has been solved and the specific molecular interactions are known177 (Figure 10). Hence, it is possible that an antiviral simultaneously targeting these sites can be made. Such a compound would serve both as a polymerase inhibitor, as it will occupy the dNTP binding site, but also as an inhibitor of the excision reaction, as it will also occupy the binding site of the ATP excision substrate. In designing such dual inhibitors, it is important to consider that the excision reaction is to a certain extent reversible, and the AZTppppA can serve as RT substrate, resulting into an AZT-terminated primer and ATP. Thus, nonhydrolyzable versions of the tetraphosphate moiety were made283,284 (AZTpSpCX2ppSA in Figure 19), designed to block the reverse reaction. Therefore, compounds that block both the polymerase and the excision reaction that causes NRTI resistance can be rationally designed.
Figure 19.

Chemical structures of dinucleoside tetraphosphate analogues. ATP-based excision reaction of AZT-chain terminated template/primers leads to the production of AZTppppA dinucleoside tetraphosphate products. Analogues of such products are shown with substitutions of oxygen with sulfur atoms in the tetraphosphate moiety. These modifications ensure that the compounds cannot support DNA synthesis by RT. The design strategy aims for compounds that can be used as inhibitors of the polymerase and excision activities of RTs, especially those carrying excision mutations (TAMs). This is accomplished by binding at the N- and the ATP binding sites. Modified from 284. Copyright 2007 American Chemical Society.
5.10. Targeting DNA Synthesis by Multiple Mechanisms of Action
EFdA is a nucleoside analogue that blocks reverse transcription by multiple mechanism of action: (a) as discussed in section 5.3, it can block DNA synthesis either as an immediate or delayed chain terminator (ICT or DCT) (Figure 8).150 Although the nontranslocated EFdA-MP-terminated primers in ICT can be unblocked, they are often converted back to the EFdA-MP-terminated form. However, when a primer with EFdA-MP at its 3′-end is further extended by a single nucleotide during the DCT mode of inhibition, the EFdA-MP-terminated primers are protected from excision. Thus, compounds that can act by delayed chain termination are less likely to be unblocked by the excision mechanism of drug resistance.
Another strength of EFdA is that it works by an additional mechanism of action; it can be efficiently misincorporated by RT, leading to mismatched primers that are extremely hard to extend and are also protected from excision.
The use of substituent that has an ethynyl (−C≡CH) or cyano (−C≡N) group added to the ribose ring of an NRTI has been used in compounds other than EFdA (Figure 19): a similar approach inspired the design of HBV NRTIs that have a 4′-cyano and are expected to act by a similar mechanism of inhibition (Figure 20).285,286 In addition, remdesivir, a related compound with a 1-ethynyl substitution on the ribose ring, has been introduced as a potent RNA-dependent RNA polymerase inhibitor of Ebola and recently approved as the first antiviral against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, COVID-19) (Figure 20).287–291 Hence, compounds that act by unique and multiple mechanisms of action may be able to maintain antiviral activity if only a single of their mechanism of action is reversed.
Figure 20.

Chemical structures of EFdA, CdG, and remdesivir.
5.11. Targeting the RNase H Function of RT
The RNase H function of RT degrades RNA in an RNA/DNA duplex. It is essential for HIV replication, and as such it could be a target for the development of antivirals that work by a different mechanism of action. Because the RNase H active site is distinct from the polymerase (Figure 1 and 13), it is expected that NRTI- and NNRTI-resistant RTs would still be susceptible to RNase H inhibitors. The topic of RNase H inhibitors has been studied extensively.247,249–256,292–296 Although there are currently no approved RNase H inhibitors, introduction of antivirals that targets this activity and their use as combinations with NRTIs and/or NNRTIs should be an efficient method to combat drug resistance, as there would be a low likelihood for cross-resistance among the various inhibitors that bind different active sites.
5.12. Long-Acting Regimens
Since the introduction of the first anti-HIV drugs, patient adherence has been a key challenge. Patients’ noncompliance leads to resistance mutations that in turn can lead to viral breakthrough and therapy failure. Daily regimens have dramatically improved over the years, and current HAART is based primarily on once-daily dosing regimens. Newer inhibitors that are more potent and pharmacologically stable could decrease the dosing frequency for patients. Consequently, patient compliance would increase, thus decreasing the incidents of drug resistance. Long-acting regimens are also important in pre-exposure prophylaxis (PrEP), as adherence to PrEP can decrease the overall prevalence and transmission of HIV (Table 3).
Table 3.
Half-Lives of Potential Long-Acting Drugs
| median terminal half-lives | ref | |
|---|---|---|
| rilpivirine (RPV) | 50 h | 302 |
| https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/202022s008lbledt.pdf | ||
| tenofovir alafenamide (TAF) | 92 min* | *in dogs |
| 119 | ||
| TFV-DP (active metabolite of TAF) | 50−180 h | 303,304 |
| https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/207561s002lbl.pdf | ||
| islatravir (EFdA) | 78.5−128 h | 299 |
The most promising potential long-acting RT-targeting nucleoside analogue is EFdA, an NRTTI. Studies have shown that a single-injection extended-release form of EFdA maintained efficient inhibition activity for more than 180 days.297 With such promising results, EFdA is being considered for a variety of long-dosing regimens.297–299 Currently, EFdA is in phase 2a trials for once-monthly doses (ClinicalTrials.gov identifier: NCT04003103). Also, EFdA is being considered for a once-yearly implant for PrEP regimens.157,158
Another potential long-acting RT inhibitor is TAF, which has better potency than its predecessor, TDF, and is used at approximately 1/10th lower dose than TDF. A recent study found that a subdermal polyvinyl alcohol implant maintained inhibitory TFV concentration for more than six weeks.118,300
NNRTIs such as RPV and elsulfavirine are also being studied for their long-acting potential. The HPTN 076 clinical trial evaluated the safety and efficacy of a long-acting form of RPV. The trial administered intramuscular injections every eight weeks and found that it was well-tolerated and effective and RPV could be used as a potential drug during PrEP.301 An elsulfavirine preclinical study showed plasma levels of the active form to be above 50 ng/mL for a minimum of 4 weeks, thus providing promise for use as a long-acting drug.160 Hence, introduction of long-acting RT-targeting antivirals will likely help overcome the challenge of patient compliance that leads to drug resistance.
5.13. Leading to a Dead-End Reaction Intermediate
INDOPY-1 is a non-nucleoside analogue RT inhibitor that acts by a distinct mechanism of action: it binds at the N- or dNTP-binding site of the polymerase active site and is thus known as nucleotide-competing RT inhibitor (NcRTI). When bound to RT, INDOPY-1 stacks with the terminal DNA base pair while intercalating with the first overhang template base (Figure 21). In doing so, the inhibitor forms a dead-end reaction intermediate that blocks further DNA synthesis. Notably, interactions of INDOPY-1 at the active site involve some conserved residues, including K65, R72, D110, D185, as well as the main chain N of G152.274,305,306 Resistance to INDOPY-1 has been reported through the FTC/3TC resistance mutation M184V and through Y115F that is known to cause resistance to multiple NRTIs.307 The TFV resistance mutation K65R confers hypersusceptibility to INDOPY-1.308 Hence, compounds that block RT by leading to dead-end reaction intermediates while engaging in interactions with conserved residues and main chain atoms may offer another strategy to minimize drug resistance mutations.
Figure 21.

Structural basis of HIV-1 RT inhibition by nucleotide competing RT inhibitor (NcRTI) INDOPY-1. (A) Chemical structure of INDOPY-1. (B) Structure of INDOPY-1 (orange sticks) in complex with HIV-1 RT (yellow sticks) and template/primer (pink sticks). (B) Reproduced modified from ref 305. Copyright 2019 American Chemical Society.
6. CONCLUSIONS
HIV RT is a key enzyme in the viral replication cycle, and it still remains a major drug target for the development of anti-HIV drugs. Great efforts have been made since the discovery of the first RT inhibitor. However, a major challenge and the reason for therapy failure is the emergence of drug resistance mutations. Developing new inhibitors based on the various strategies mentioned in this review may help improve the ability of compounds to perform better in terms of drug resistance. While it is important to note that fully avoiding drug resistance is likely not possible, using these strategies may help the field move in the right direction. Furthermore, these strategies can be applied to not just RT inhibitors but other viral enzymes as well.
ACKNOWLEDGMENTS
We thank Dr. Kalyan Das for useful discussions and help in the early stages of preparation. This work was funded in part by National Institutes of Health grants R37 AI076119, R01 GM118012, U54 AI150472, and R01 AI121315 (to S.G.S.). M.E.C. received support from NIH grant T32 GM008367. S.G.S. acknowledges funding from the Nahmias-Schinazi Distinguished Chair in Research.
Biographies
Maria E. Cilento is currently a Ph.D. candidate under the supervision of Dr. Stefan Sarafianos in the Division of Laboratory of Biochemical Pharmacology in the Department of Pediatrics at Emory University School of Medicine. She received her B.S. from the Department of Biology at Stockton University, and her Master of Science in Microbiology & Immunology at Thomas Jefferson University, where she conducted research on HIV entry. Her current research focuses on studying the mechanism of inhibition and resistance of HIV from various subtypes to 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA/Islatravir).
Dr. Karen A. Kirby is an Assistant Professor in the Division of Laboratory of Biochemical Pharmacology in the Department of Pediatrics at Emory University School of Medicine. She received her B.S. in Chemistry from the University of Minnesota and her Ph.D. in Chemistry from the University of Missouri, studying small molecule crystallography. She was trained in structural biology related to HIV and other viral proteins, biochemistry, and biophysics (Dr. Stefan Sarafianos, University of Missouri). Her current research focuses on structural mechanisms of inhibition and resistance to HIV and other viral pathogens.
Dr. Stefan G. Sarafianos is a Professor and Associate Director of the Division of Laboratory of Biochemical Pharmacology in the Department of Pediatrics at Emory University School of Medicine. He received his Ph.D. at the Department of Chemistry at Georgetown University. He was trained in molecular biology of HIV (Dr. Mukund Modak, UMDNJ), in structural biology and virology by Dr. Eddy Arnold (Center for Advanced Biotechnology and Medicine, Rutgers University,) and Dr. Stephen Hughes (NCI, NIH), working on structural mechanisms of inhibition and resistance in HIV reverse transcriptase. He is currently the Nahmias Schinazi Distinguished Chair in Research, and his research focuses on HIV, HBV, SARSCoV-2, and other pathogens.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Maria E. Cilento, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, Georgia 30322, United States; Children’s Healthcare of Atlanta, Atlanta, Georgia 30307, United States.
Karen A. Kirby, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, Georgia 30322, United States; Children’s Healthcare of Atlanta, Atlanta, Georgia 30307, United States.
Stefan G. Sarafianos, Laboratory of Biochemical Pharmacology, Department of Pediatrics, Emory University School of Medicine, Atlanta, Georgia 30322, United States; Children’s Healthcare of Atlanta, Atlanta, Georgia 30307, United States.
REFERENCES
- (1).Lucas S; Nelson AM HIV and the spectrum of human disease. J. Pathol. 2015, 235, 229–241. [DOI] [PubMed] [Google Scholar]
- (2).Shafer RW; Vuitton DA Highly active antiretroviral therapy (HAART) for the treatment of infection with human immunodeficiency virus type 1. Biomed. Pharmacother. 1999, 53, 73–86. [DOI] [PubMed] [Google Scholar]
- (3).Pinto AN; Grey P; Shaik A; Cooper DA; Kelleher AD; Petoumenos K Early treatment of primary HIV infection is associated with decreased mortality. AIDS Res. Hum. Retroviruses 2018, 34, 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Eggleton JS; Nagalli S Highly Active Antiretroviral Therapy (HAART); StatPearls: Treasure Island, FL, 2020. [PubMed] [Google Scholar]
- (5).Das K; Arnold E HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vogt V In Retroviruses; Coffin JM, Hughes SH, Varmus, Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 1997. [Google Scholar]
- (7).Freed EO HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jacks T; Power MD; Masiarz FR; Luciw PA; Barr PJ; Varmus HE Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283. [DOI] [PubMed] [Google Scholar]
- (9).Huang H; Chopra R; Verdine GL; Harrison SC Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 1998, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- (10).Kohlstaedt LA; Wang J; Friedman JM; Rice PA; Steitz TA Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 1992, 256, 1783–1790. [DOI] [PubMed] [Google Scholar]
- (11).Jacobo-Molina A; Ding J; Nanni RG; Clark AD Jr.; Lu X; Tantillo C; Williams RL; Kamer G; Ferris AL; Clark P; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ding J; Das K; Hsiou Y; Sarafianos SG; Clark AD Jr.; Jacobo-Molina A; Tantillo C; Hughes SH; Arnold E Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 A resolution. J. Mol. Biol. 1998, 284, 1095–1111. [DOI] [PubMed] [Google Scholar]
- (13).Schuckmann MM; Marchand B; Hachiya A; Kodama EN; Kirby KA; Singh K; Sarafianos SG The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase decreases binding to nevirapine. J. Biol. Chem. 2010, 285, 38700–38709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gotte M; Maier G; Gross HJ; Heumann H Localization of the active site of HIV-1 reverse transcriptase-associated RNase H domain on a DNA template using site-specific generated hydroxyl radicals. J. Biol. Chem. 1998, 273, 10139–10146. [DOI] [PubMed] [Google Scholar]
- (15).Sarafianos SG; Das K; Tantillo C; Clark AD Jr.; Ding J; Whitcomb JM; Boyer PL; Hughes SH; Arnold E Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001, 20, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Larder BA; Purifoy DJ; Powell KL; Darby G Site-specific mutagenesis of AIDS virus reverse transcriptase. Nature 1987, 327, 716–717. [DOI] [PubMed] [Google Scholar]
- (17).Johnson MS; McClure MA; Feng DF; Gray J; Doolittle RF Computer analysis of retroviral pol genes: assignment of enzymatic functions to specific sequences and homologies with nonviral enzymes. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 7648–7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Poch O; Sauvaget I; Delarue M; Tordo N Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Delarue M; Poch O; Tordo N; Moras D; Argos P An attempt to unify the structure of polymerases. Protein Eng., Des. Sel. 1990, 3, 461–467. [DOI] [PubMed] [Google Scholar]
- (20).Jacques PS; Wohrl BM; Ottmann M; Darlix JL; Le Grice SF Mutating the ″primer grip″ of p66 HIV-1 reverse transcriptase implicates tryptophan-229 in template-primer utilization. J. Biol. Chem. 1994, 269, 26472–26478. [PubMed] [Google Scholar]
- (21).Powell MD; Ghosh M; Jacques PS; Howard KJ; Le Grice SF; Levin JG Alanine-scanning mutations in the ″primer grip″ of p66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J. Biol. Chem. 1997, 272, 13262–13269. [DOI] [PubMed] [Google Scholar]
- (22).Sarafianos SG; Pandey VN; Kaushik N; Modak MJ Site-directed mutagenesis of arginine 72 of HIV-1 reverse transcriptase. catalytic role and inhibitor sensitivity. J. Biol. Chem. 1995, 270, 19729–19735. [DOI] [PubMed] [Google Scholar]
- (23).Martin-Hernandez AM; Domingo E; Menendez-Arias L Human immunodeficiency virus type 1 reverse transcriptase: role of Tyr115 in deoxynucleotide binding and misinsertion fidelity of DNA synthesis. EMBO J. 1996, 15, 4434–4442. [PMC free article] [PubMed] [Google Scholar]
- (24).Gao G; Orlova M; Georgiadis MM; Hendrickson WA; Goff SP Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boyer PL; Sarafianos SG; Arnold E; Hughes SH Analysis of mutations at positions 115 and 116 in the dNTP binding site of HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 3056–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sarafianos SG; Marchand B; Das K; Himmel DM; Parniak MA; Hughes SH; Arnold E Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sarafianos SG; Clark AD Jr.; Das K; Tuske S; Birktoft JJ; Ilankumaran P; Ramesha AR; Sayer JM; Jerina DM; Boyer PL; et al. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 2002, 21, 6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hu WS; Hughes SH HIV-1 reverse transcription. Cold Spring Harb. Perspect. Cold Spring Harbor Perspect. Med. 2012, 2, a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Davies JF 2nd; Hostomska Z; Hostomsky Z; Jordan SR; Matthews DA Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 1991, 252, 88–95. [DOI] [PubMed] [Google Scholar]
- (30).Cowan JA; Ohyama T; Howard K; Rausch JW; Cowan SM; Le Grice SF Metal-ion stoichiometry of the HIV-1 RT ribonuclease H domain: evidence for two mutually exclusive sites leads to new mechanistic insights on metal-mediated hydrolysis in nucleic acid biochemistry. JBIC, J. Biol. Inorg. Chem. 2000, 5, 67–74. [DOI] [PubMed] [Google Scholar]
- (31).Cristofaro JV; Rausch JW; Le Grice SF; DeStefano JJ Mutations in the ribonuclease H active site of HIV-RT reveal a role for this site in stabilizing enzyme-primer-template binding. Biochemistry 2002, 41, 10968–10975. [DOI] [PubMed] [Google Scholar]
- (32).Pari K; Mueller GA; DeRose EF; Kirby TW; London RE Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry 2003, 42, 639–650. [DOI] [PubMed] [Google Scholar]
- (33).Nowotny M; Gaidamakov SA; Crouch RJ; Yang W Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 2005, 121, 1005–1016. [DOI] [PubMed] [Google Scholar]
- (34).Nowotny M; Yang W Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006, 25, 1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mak J; Kleiman L Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Telesnitsky A; Goff SP In Retroviruses; Coffin JM, Hughes SH, Varmus HE, Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 1997. [PubMed] [Google Scholar]
- (37).Mak J; Jiang M; Wainberg MA; Hammarskjold ML; Rekosh D; Kleiman L Role of Pr160gag-pol in mediating the selective incorporation of tRNA(Lys) into human immunodeficiency virus type 1 particles. J. Virol. 1994, 68, 2065–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yu Q; Morrow CD Complementarity between 3′ terminal nucleotides of tRNA and primer binding site is a major determinant for selection of the tRNA primer used for initiation of HIV-1 reverse transcription. Virology 1999, 254, 160–168. [DOI] [PubMed] [Google Scholar]
- (39).Isel C; Lanchy JM; Le Grice SF; Ehresmann C; Ehresmann B; Marquet R Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 1996, 15, 917–924. [PMC free article] [PubMed] [Google Scholar]
- (40).Lanchy JM; Keith G; Le Grice SF; Ehresmann B; Ehresmann C; Marquet R Contacts between reverse transcriptase and the primer strand govern the transition from initiation to elongation of HIV-1 reverse transcription. J. Biol. Chem. 1998, 273, 24425–24432. [DOI] [PubMed] [Google Scholar]
- (41).Coffin JM; Haseltine WA Terminal redundancy and the origin of replication of Rous sarcoma virus RNA. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 1908–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schwartz DE; Zamecnik PC; Weith HL Rous sarcoma virus genome is terminally redundant: the 3′ sequence. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Stoll E; Billeter MA; Palmenberg A; Weissmann C Avian myeloblastosis virus RNA is terminally redundant: implications for the mechanism of retrovirus replication. Cell 1977, 12, 57–72. [DOI] [PubMed] [Google Scholar]
- (44).Coffin JM; Hageman TC; Maxam AM; Haseltine WA Structure of the genome of Moloney murine leukemia virus: a terminally redundant sequence. Cell 1978, 13, 761–773. [DOI] [PubMed] [Google Scholar]
- (45).Coffin JM Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 1979, 42, 1–26. [DOI] [PubMed] [Google Scholar]
- (46).van Wamel JL; Berkhout B The first strand transfer during HIV-1 reverse transcription can occur either intramolecularly or intermolecularly. Virology 1998, 244, 245–251. [DOI] [PubMed] [Google Scholar]
- (47).Hu WS; Temin HM Retroviral recombination and reverse transcription. Science 1990, 250, 1227–1233. [DOI] [PubMed] [Google Scholar]
- (48).Yu H; Jetzt AE; Ron Y; Preston BD; Dougherty JP The nature of human immunodeficiency virus type 1 strand transfers. J. Biol. Chem. 1998, 273, 28384–28391. [DOI] [PubMed] [Google Scholar]
- (49).Charneau P; Alizon M; Clavel F A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 1992, 66, 2814–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hungnesi O; Tjotta E; Grinde B Mutations in the central polypurine tract of HIV-1 result in delayed replication. Virology 1992, 190, 440–442. [DOI] [PubMed] [Google Scholar]
- (51).Pullen KA; Ishimoto LK; Champoux JJ Incomplete removal of the RNA primer for minus-strand DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 1992, 66, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zennou V; Serguera C; Sarkis C; Colin P; Perret E; Mallet J; Charneau P The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat. Biotechnol. 2001, 19, 446–450. [DOI] [PubMed] [Google Scholar]
- (53).Schultz SJ; Champoux JJ RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 2008, 134, 86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Beilhartz GL; Gotte M HIV-1 Ribonuclease H: structure, catalytic mechanism and inhibitors. Viruses 2010, 2, 900–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Abram ME; Ferris AL; Shao W; Alvord WG; Hughes SH Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hu C; Saenz DT; Fadel HJ; Walker W; Peretz M; Poeschla EM The HIV-1 central polypurine tract functions as a second line of defense against APOBEC3G/F. J. Virol. 2010, 84, 11981–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Iglesias C; Ringeard M; Di Nunzio F; Fernandez J; Gaudin R; Souque P; Charneau P; Arhel N Residual HIV-1 DNA Flap-independent nuclear import of cPPT/CTS double mutant viruses does not support spreading infection. Retrovirology 2011, 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Smith JS; Roth MJ Specificity of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H in removal of the minus-strand primer, tRNA(Lys3). J. Biol. Chem. 1992, 267, 15071–15079. [PubMed] [Google Scholar]
- (59).Whitcomb JM; Kumar R; Hughes SH Sequence of the circle junction of human immunodeficiency virus type 1: implications for reverse transcription and integration. J. Virol. 1990, 64, 4903–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Levin JG; Guo J; Rouzina I; Musier-Forsyth K Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 217–286. [DOI] [PubMed] [Google Scholar]
- (61).Jacobo-Molina A; Arnold E HIV reverse transcriptase structure-function relationships. Biochemistry 1991, 30, 6351–6356. [DOI] [PubMed] [Google Scholar]
- (62).Fanales-Belasio E; Raimondo M; Suligoi B; Butto S HIV virology and pathogenetic mechanisms of infection: a brief overview. Ann. Ist. Super. Sanita 2010, 46, 5–14. [DOI] [PubMed] [Google Scholar]
- (63).Cilento MKKA; Tedbury PR; Sarafianos SG In Reference Module in Life Sciences; Elsevier: Science Direct, 2020. [Google Scholar]
- (64).Eigen M On the nature of virus quasispecies. Trends Microbiol. 1996, 4, 216–218. [DOI] [PubMed] [Google Scholar]
- (65).Domingo E; Holland JJ RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [DOI] [PubMed] [Google Scholar]
- (66).Keele BF; Giorgi EE; Salazar-Gonzalez JF; Decker JM; Pham KT; Salazar MG; Sun C; Grayson T; Wang S; Li H; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Yu F; Wen Y; Wang J; Gong Y; Feng K; Ye R; Jiang Y; Zhao Q; Pan P; Wu H; et al. The transmission and evolution of HIV-1 quasispecies within one couple: a follow-up study based on next-generation sequencing. Sci. Rep. 2018, 8, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Roberts JD; Bebenek K; Kunkel TA The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [DOI] [PubMed] [Google Scholar]
- (69).Zhang J Host RNA polymerase II makes minimal contributions to retroviral frame-shift mutations. J. Gen. Virol. 2004, 85, 2389–2395. [DOI] [PubMed] [Google Scholar]
- (70).Ratner L; Haseltine W; Patarca R; Livak KJ; Starcich B; Josephs SF; Doran ER; Rafalski JA; Whitehorn EA; Baumeister K; et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 1985, 313, 277–284. [DOI] [PubMed] [Google Scholar]
- (71).Wain-Hobson S; Sonigo P; Danos O; Cole S; Alizon M Nucleotide sequence of the AIDS virus. Cell 1985, 40, 9–17. [DOI] [PubMed] [Google Scholar]
- (72).Achuthan V; Keith BJ; Connolly BA; DeStefano JJ Human immunodeficiency virus reverse transcriptase displays dramatically higher fidelity under physiological magnesium conditions in vitro. J. Virol. 2014, 88, 8514–8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Achuthan V; DeStefano JJ Alternative divalent cations (Zn(2)(+), Co(2)(+), and Mn(2)(+)) are not mutagenic at conditions optimal for HIV-1 reverse transcriptase activity. BMC. Biochem. 2015, 16, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Pathak VK; Temin HM Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 6019–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Mansky LM; Temin HM Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Julias JG; Pathak VK Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 1998, 72, 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Brenner B; Wainberg MA; Salomon H; Rouleau D; Dascal A; Spira B; Sekaly RP; Conway B; Routy JP Resistance to antiretroviral drugs in patients with primary HIV-1 infection. Investigators of the Quebec Primary Infection Study. Int. J. Antimicrob. Agents 2000, 16, 429–434. [DOI] [PubMed] [Google Scholar]
- (78).Blower SM; Aschenbach AN; Gershengorn HB; Kahn JO Predicting the unpredictable: transmission of drug-resistant HIV. Nat. Med. 2001, 7, 1016–1020. [DOI] [PubMed] [Google Scholar]
- (79).Bangsberg DR; Perry S; Charlebois ED; Clark RA; Roberston M; Zolopa AR; Moss A Non-adherence to highly active antiretroviral therapy predicts progression to AIDS. AIDS 2001, 15, 1181–1183. [DOI] [PubMed] [Google Scholar]
- (80).Opravil M; Hirschel B; Lazzarin A; Furrer H; Chave JP; Yerly S; Bisset LR; Fischer M; Vernazza P; Bernasconi E; et al. A randomized trial of simplified maintenance therapy with abacavir, lamivudine, and zidovudine in human immunodeficiency virus infection. J. Infect. Dis. 2002, 185, 1251–1260. [DOI] [PubMed] [Google Scholar]
- (81).Kirk O; Pedersen C; Law M; Gulick RM; Moyle G; Montaner J; Eron JJ Jr.; Phillips AN; Lundgren JD Analysis of virological efficacy in trials of antiretroviral regimens: drawbacks of not including viral load measurements after premature discontinuation of therapy. Antivir. Ther. 2002, 7, 271–281. [PubMed] [Google Scholar]
- (82).Volberding P Adherence, resistance, and timing: current issues in the use of new therapies. AIDS Read. 2002, 12, 349–350 347–356, 368. [PubMed] [Google Scholar]
- (83).Hogg RS; Heath K; Bangsberg D; Yip B; Press N; O’Shaughnessy MV; Montaner JS Intermittent use of triple-combination therapy is predictive of mortality at baseline and after 1 year of follow-up. AIDS 2002, 16, 1051–1058. [DOI] [PubMed] [Google Scholar]
- (84).Walsh JC; Pozniak AL; Nelson MR; Mandalia S; Gazzard BG Virologic rebound on HAART in the context of low treatment adherence is associated with a low prevalence of antiretroviral drug resistance. JAIDS, J. Acquired Immune Defic. Syndr. 2002, 30, 278–287. [DOI] [PubMed] [Google Scholar]
- (85).D’Aquila RT; Schapiro JM; Brun-Vezinet F; Clotet B; Conway B; Demeter LM; Grant RM; Johnson VA; Kuritzkes DR; Loveday C; et al. Drug resistance mutations in HIV-1. Top HIV Med. 2003, 11, 92–96. [PubMed] [Google Scholar]
- (86).Louie M; Hogan C; Di Mascio M; Hurley A; Simon V; Rooney J; Ruiz N; Brun S; Sun E; Perelson AS; et al. Determining the relative efficacy of highly active antiretroviral therapy. J. Infect. Dis. 2003, 187, 896–900. [DOI] [PubMed] [Google Scholar]
- (87).Coffin JM HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [DOI] [PubMed] [Google Scholar]
- (88).Wegner SA; Brodine SK; Mascola JR; Tasker SA; Shaffer RA; Starkey MJ; Barile A; Martin GJ; Aronson N; Emmons WW; et al. Prevalence of genotypic and phenotypic resistance to anti-retroviral drugs in a cohort of therapy-naive HIV-1 infected US military personnel. AIDS 2000, 14, 1009–1015. [DOI] [PubMed] [Google Scholar]
- (89).Duwe S; Brunn M; Altmann D; Hamouda O; Schmidt B; Walter H; Pauli G; Kucherer C Frequency of genotypic and phenotypic drug-resistant HIV-1 among therapy-naive patients of the German Seroconverter Study. J. Acquir. Immune Defic. Syndr. 2001, 26, 266–273. [DOI] [PubMed] [Google Scholar]
- (90).Grant RM; Hecht FM; Warmerdam M; Liu L; Liegler T; Petropoulos CJ; Hellmann NS; Chesney M; Busch MP; Kahn JO Time trends in primary HIV-1 drug resistance among recently infected persons. JAMA 2002, 288, 181–188. [DOI] [PubMed] [Google Scholar]
- (91).Ammaranond P; Cunningham P; Oelrichs R; Suzuki K; Harris C; Leas L; Grulich A; Cooper DA; Kelleher AD Rates of transmission of antiretroviral drug resistant strains of HIV-1. J. Clin. Virol. 2003, 26, 153–161. [DOI] [PubMed] [Google Scholar]
- (92).Taylor S; Cane P; Hue S; Xu L; Wrin T; Lie Y; Hellmann N; Petropoulos C; Workman J; Ratcliffe D; et al. Identification of a transmission chain of HIV type 1 containing drug resistance-associated mutations. AIDS Res. Hum. Retroviruses 2003, 19, 353–361. [DOI] [PubMed] [Google Scholar]
- (93).Barbour JD; Hecht FM; Wrin T; Liegler TJ; Ramstead CA; Busch MP; Segal MR; Petropoulos CJ; Grant RM Persistence of primary drug resistance among recently HIV-1 infected adults. AIDS 2004, 18, 1683–1689. [DOI] [PubMed] [Google Scholar]
- (94).Hu WS; Temin HM Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 1556–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Robertson DL; Sharp PM; McCutchan FE; Hahn BH Recombination in HIV-1. Nature 1995, 374, 124–126. [DOI] [PubMed] [Google Scholar]
- (96).Jetzt AE; Yu H; Klarmann GJ; Ron Y; Preston BD; Dougherty JP High rate of recombination throughout the human immunodeficiency virus type 1 genome. J. Virol. 2000, 74, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Zhang J; Tang LY; Li T; Ma Y; Sapp CM Most retroviral recombinations occur during minus-strand DNA synthesis. J. Virol. 2000, 74, 2313–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Zhuang J; Jetzt AE; Sun G; Yu H; Klarmann G; Ron Y; Preston BD; Dougherty JP Human immunodeficiency virus type 1 recombination: rate, fidelity, and putative hot spots. J. Virol. 2002, 76, 11273–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Dykes C; Balakrishnan M; Planelles V; Zhu Y; Bambara RA; Demeter LM Identification of a preferred region for recombination and mutation in HIV-1 gag. Virology 2004, 326, 262–279. [DOI] [PubMed] [Google Scholar]
- (100).Levy DN; Aldrovandi GM; Kutsch O; Shaw GM Dynamics of HIV-1 recombination in its natural target cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Chin MP; Chen J; Nikolaitchik OA; Hu WS Molecular determinants of HIV-1 intersubtype recombination potential. Virology 2007, 363, 437–446. [DOI] [PubMed] [Google Scholar]
- (102).Wei X; Ghosh SK; Taylor ME; Johnson VA; Emini EA; Deutsch P; Lifson JD; Bonhoeffer S; Nowak MA; Hahn BH; et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 1995, 373, 117–122. [DOI] [PubMed] [Google Scholar]
- (103).Ho DD; Neumann AU; Perelson AS; Chen W; Leonard JM; Markowitz M Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 1995, 373, 123–126. [DOI] [PubMed] [Google Scholar]
- (104).Perelson AS; Neumann AU; Markowitz M; Leonard JM; Ho DD HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [DOI] [PubMed] [Google Scholar]
- (105).Jung A; Maier R; Vartanian JP; Bocharov G; Jung V; Fischer U; Meese E; Wain-Hobson S; Meyerhans A Recombination: multiply infected spleen cells in HIV patients. Nature 2002, 418, 144. [DOI] [PubMed] [Google Scholar]
- (106).Abram ME; Ferris AL; Das K; Quinones O; Shao W; Tuske S; Alvord WG; Arnold E; Hughes SH Mutations in HIV1 reverse transcriptase affect the errors made in a single cycle of viral replication. J. Virol. 2014, 88, 7589–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Nikolopoulos GK; Christaki E; Paraskevis D; Bonovas S Pre-exposure prophylaxis for HIV: evidence and perspectives. Curr. Pharm. Des. 2017, 23, 2579–2591. [DOI] [PubMed] [Google Scholar]
- (108).Castel AD; Magnus M; Greenberg AE Pre-exposure prophylaxis for human immunodeficiency virus: the past, present, and future. Infect. Dis. Clin. North Am. 2014, 28, 563–583. [DOI] [PubMed] [Google Scholar]
- (109).Krakower DS; Mayer KH Pre-exposure prophylaxis to prevent HIV infection: current status, future opportunities and challenges. Drugs 2015, 75, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Pham HT; Xiao MA; Principe MA; Wong A; Mesplede T Pharmaceutical, clinical, and resistance information on doravirine, a novel non-nucleoside reverse transcriptase inhibitor for the treatment of HIV-1 infection. Drugs Context 2020, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Nakashima H; Matsui T; Harada S; Kobayashi N; Matsuda A; Ueda T; Yamamoto N Inhibition of replication and cytopathic effect of human T cell lymphotropic virus type III/lymphadenopathy-associated virus by 3′-azido-3′-deoxythymidine in vitro. Antimicrob. Agents Chemother. 1986, 30, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Kim CH; Marquez VE; Broder S; Mitsuya H; Driscoll JS Potential anti-AIDS drugs. 2’,3′-Dideoxycytidine analogues. J. Med. Chem. 1987, 30, 862–866. [DOI] [PubMed] [Google Scholar]
- (113).Russell JW; Klunk LJ Comparative pharmacokinetics of new anti-HIV agents: 2’,3′-dideoxyadenosine and 2’,3′-dideoxyinosine. Biochem. Pharmacol. 1989, 38, 1385–1388. [DOI] [PubMed] [Google Scholar]
- (114).August EM; Marongiu ME; Lin TS; Prusoff WH Initial studies on the cellular pharmacology of 3′-deoxythymidin-2’ene (d4T): a potent and selective inhibitor of human immunodeficiency virus. Biochem. Pharmacol. 1988, 37, 4419–4422. [DOI] [PubMed] [Google Scholar]
- (115).Daluge SM; Good SS; Faletto MB; Miller WH; St Clair MH; Boone LR; Tisdale M; Parry NR; Reardon JE; Dornsife RE; et al. 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob. Agents Chemother. 1997, 41, 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Balzarini J; Holy A; Jindrich J; Naesens L; Snoeck R; Schols D; De Clercq E Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 1993, 37, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Sax PE; Wohl D; Yin MT; Post F; DeJesus E; Saag M; Pozniak A; Thompson M; Podzamczer D; Molina JM; et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet 2015, 385, 2606–2615. [DOI] [PubMed] [Google Scholar]
- (118).Ruane PJ; DeJesus E; Berger D; Markowitz M; Bredeek UF; Callebaut C; Zhong L; Ramanathan S; Rhee MS; Fordyce MW; Yale K Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of tenofovir alafenamide as 10-day monotherapy in HIV-1-positive adults. JAIDS, J. Acquired Immune Defic. Syndr. 2013, 63, 449–455. [DOI] [PubMed] [Google Scholar]
- (119).Babusis D; Phan TK; Lee WA; Watkins WJ; Ray AS Mechanism for effective lymphoid cell and tissue loading following oral administration of nucleotide prodrug GS-7340. Mol. Pharmaceutics 2013, 10, 459–466. [DOI] [PubMed] [Google Scholar]
- (120).Sax PE; Zolopa A; Brar I; Elion R; Ortiz R; Post F; Wang H; Callebaut C; Martin H; Fordyce MW; et al. Tenofovir alafenamide vs. tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. JAIDS, J. Acquired Immune Defic. Syndr. 2014, 67, 52–58. [DOI] [PubMed] [Google Scholar]
- (121).Markowitz M; Zolopa A; Squires K; Ruane P; Coakley D; Kearney B; Zhong L; Wulfsohn M; Miller MD; Lee WA Phase I/II study of the pharmacokinetics, safety and antiretroviral activity of tenofovir alafenamide, a new prodrug of the HIV reverse transcriptase inhibitor tenofovir, in HIV-infected adults. J. Antimicrob. Chemother. 2014, 69, 1362–1369. [DOI] [PubMed] [Google Scholar]
- (122).Wassner C; Bradley N; Lee Y A review and clinical understanding of tenofovir:: tenofovir disoproxil fumarate versus tenofovir alafenamide. J. Int. Assoc. Provid. AIDS Care 2020, 19, 2325958220919231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Perno CF; Yarchoan R; Cooney DA; Hartman NR; Gartner S; Popovic M; Hao Z; Gerrard TL; Wilson YA; Johns DG; et al. Inhibition of human immunodeficiency virus (HIV-1/HTLV-IIIBa-L) replication in fresh and cultured human peripheral blood monocytes/macrophages by azidothymidine and related 2’,3′dideoxynucleosides. J. Exp. Med. 1988, 168, 1111–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Furman PA; Fyfe JA; St Clair MH; Weinhold K; Rideout JL; Freeman GA; Lehrman SN; Bolognesi DP; Broder S; Mitsuya H; et al. Phosphorylation of 3′-azido-3′deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 8333–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Ren J; Esnouf R; Garman E; Somers D; Ross C; Kirby I; Keeling J; Darby G; Jones Y; Stuart D; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Mol. Biol. 1995, 2, 293–302. [DOI] [PubMed] [Google Scholar]
- (126).Ding J; Das K; Moereels H; Koymans L; Andries K; Janssen PA; Hughes SH; Arnold E Structure of HIV-1 RT/TIBO R 86183 complex reveals similarity in the binding of diverse nonnucleoside inhibitors. Nat. Struct. Mol. Biol. 1995, 2, 407–415. [DOI] [PubMed] [Google Scholar]
- (127).Hsiou Y; Das K; Ding J; Clark AD Jr.; Kleim JP; Rosner M; Winkler I; Riess G; Hughes SH; Arnold E Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design feature for reducing drug resistance. J. Mol. Biol. 1998, 284, 313–323. [DOI] [PubMed] [Google Scholar]
- (128).Zhuang C; Pannecouque C; De Clercq E; Chen F Development of non-nucleoside reverse transcriptase inhibitors (NNRTIs): our past twenty years. Acta Pharm. Sin. B 2020, 10, 961–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).de Bethune MP Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res. 2010, 85, 75–90. [DOI] [PubMed] [Google Scholar]
- (130).Ren J; Milton J; Weaver KL; Short SA; Stuart DI; Stammers DK Structural basis for the resilience of efavirenz (DMP266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure 2000, 8, 1089–1094. [DOI] [PubMed] [Google Scholar]
- (131).Ren J; Nichols C; Bird L; Chamberlain P; Weaver K; Short S; Stuart DI; Stammers DK Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation nonnucleoside inhibitors. J. Mol. Biol. 2001, 312, 795–805. [DOI] [PubMed] [Google Scholar]
- (132).Kati WM; Johnson KA; Jerva LF; Anderson KS Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992, 267, 25988–25997. [PubMed] [Google Scholar]
- (133).Reardon JE Human immunodeficiency virus reverse transcriptase: steady-state and pre-steady-state kinetics of nucleotide incorporation. Biochemistry 1992, 31, 4473–4479. [DOI] [PubMed] [Google Scholar]
- (134).Rodgers DW; Gamblin SJ; Harris BA; Ray S; Culp JS; Hellmig B; Woolf DJ; Debouck C; Harrison SC The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Hsiou Y; Ding J; Das K; Clark AD Jr; Hughes SH; Arnold E Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4, 853–860. [DOI] [PubMed] [Google Scholar]
- (136).Schauer GD; Huber KD; Leuba SH; Sluis-Cremer N Mechanism of allosteric inhibition of HIV-1 reverse transcriptase revealed by single-molecule and ensemble fluorescence. Nucleic Acids Res. 2014, 42, 11687–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Xavier Ruiz F; Arnold E Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr. Opin. Struct. Biol. 2020, 61, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Das K; Martinez SE; Bauman JD; Arnold E HIV-1 reverse transcriptase complex with DNA and nevirapine reveals nonnucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Usach I; Melis V; Peris JE Non-nucleoside reverse transcriptase inhibitors: a review on pharmacokinetics, pharmacodynamics, safety and tolerability. J. Int. AIDS Soc. 2013, 16, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Wang Y; De Clercq E; Li G Current and emerging nonnucleoside reverse transcriptase inhibitors (NNRTIs) for HIV-1 treatment. Expert Opin. Drug Metab. Toxicol. 2019, 15, 813–829. [DOI] [PubMed] [Google Scholar]
- (141).Das K; Clark AD Jr.; Lewi PJ; Heeres J; De Jonge MR; Koymans LM; Vinkers HM; Daeyaert F; Ludovici DW; Kukla MJ; et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [DOI] [PubMed] [Google Scholar]
- (142).Janssen PA; Lewi PJ; Arnold E; Daeyaert F; de Jonge M; Heeres J; Koymans L; Vinkers M; Guillemont J; Pasquier E; et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J. Med. Chem. 2005, 48, 1901–1909. [DOI] [PubMed] [Google Scholar]
- (143).Das K; Bauman JD; Clark AD Jr.; Frenkel YV; Lewi PJ; Shatkin AJ; Hughes SH; Arnold E High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Chong P; Sebahar P; Youngman M; Garrido D; Zhang H; Stewart EL; Nolte RT; Wang L; Ferris RG; Edelstein M; et al. Rational design of potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Med. Chem. 2012, 55, 10601–10609. [DOI] [PubMed] [Google Scholar]
- (145).Cote B; Burch JD; Asante-Appiah E; Bayly C; Bedard L; Blouin M; Campeau LC; Cauchon E; Chan M; Chefson A; et al. Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses. Bioorg. Med. Chem. Lett. 2014, 24, 917–922. [DOI] [PubMed] [Google Scholar]
- (146).Rai MA; Pannek S; Fichtenbaum CJ Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerging Drugs 2018, 23, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Feng M; Wang D; Grobler JA; Hazuda DJ; Miller MD; Lai MT In vitro resistance selection with doravirine (MK1439), a novel nonnucleoside reverse transcriptase inhibitor with distinct mutation development pathways. Antimicrob. Agents Chemother. 2015, 59, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Kawamoto A; Kodama E; Sarafianos SG; Sakagami Y; Kohgo S; Kitano K; Ashida N; Iwai Y; Hayakawa H; Nakata H; et al. 2’-deoxy-4’-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [DOI] [PubMed] [Google Scholar]
- (149).Michailidis E; Marchand B; Kodama EN; Singh K; Matsuoka M; Kirby KA; Ryan EM; Sawani AM; Nagy E; Ashida N; et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4’-Ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Michailidis E; Huber AD; Ryan EM; Ong YT; Leslie MD; Matzek KB; Singh K; Marchand B; Hagedorn AN; Kirby KA; et al. 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol. Chem. 2014, 289, 24533–24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Murphey-Corb M; Rajakumar P; Michael H; Nyaundi J; Didier PJ; Reeve AB; Mitsuya H; Sarafianos SG; Parniak MA Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4’-ethynyl-2-fluoro-2’deoxyadenosine in vitro and in vivo. Antimicrob. Agents Chemother. 2012, 56, 4707–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Zhang W; Parniak MA; Sarafianos SG; Empey PE; Rohan LC In vitro transport characteristics of EFdA, a novel nucleoside reverse transcriptase inhibitor using Caco-2 and MDCKII cell monolayers. Eur. J. Pharmacol. 2014, 732, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Stoddart CA; Galkina SA; Joshi P; Kosikova G; Moreno ME; Rivera JM; Sloan B; Reeve AB; Sarafianos SG; Murphey-Corb M; et al. Oral administration of the nucleoside EFdA (4’-ethynyl-2-fluoro-2’-deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob. Agents Chemother. 2015, 59, 4190–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Kirby KA; Singh K; Michailidis E; Marchand B; Kodama EN; Ashida N; Mitsuya H; Parniak MA; Sarafianos SG The sugar ring conformation of 4′-ethynyl-2-fluoro-2′-deoxyadenosine and its recognition by the polymerase active site of HIV reverse transcriptase. Cell. Mol. Biol. 2011, 57, 40–46. [PMC free article] [PubMed] [Google Scholar]
- (155).Kirby KA; Michailidis E; Fetterly TL; Steinbach MA; Singh K; Marchand B; Leslie MD; Hagedorn AN; Kodama EN; Marquez VE; et al. Effects of substitutions at the 4’ and 2 positions on the bioactivity of 4’-ethynyl-2-fluoro-2’-deoxyadenosine. Antimicrob. Agents Chemother. 2013, 57, 6254–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Salie ZL; Kirby KA; Michailidis E; Marchand B; Singh K; Rohan LC; Kodama EN; Mitsuya H; Parniak MA; Sarafianos SG Structural basis of HIV inhibition by translocation-defective RT inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9274–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Markowitz M; Sarafianos SG 4’-Ethynyl-2-fluoro-2’deoxyadenosine, MK-8591: a novel HIV-1 reverse transcriptase translocation inhibitor. Curr. Opin. HIV AIDS 2018, 13, 294–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Grobler JA; Fillgrove K; Hazuda D; Huang Q; Lai MT; Matthews RP; Rudd DJ; Vargo R MK-8591 potency and PK provide high inhibitory quotients at low doses QD and QW. In Conference on Retroviruses and Opportunistic Infections (CROI), Seattle, WA, 2019; 481, http://www.croiconference.org/sessions/mk-8591potency-and-pk-provide-high-inhibitory-quotients-low-doses-qd-andqw (accessed 2020-07-13). [Google Scholar]
- (159).Namasivayam V; Vanangamudi M; Kramer VG; Kurup S; Zhan P; Liu X; Kongsted J; Byrareddy SN The journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J. Med. Chem. 2019, 62, 4851–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Al-Salama ZT Elsulfavirine: first global approval. Drugs 2017, 77, 1811–1816. [DOI] [PubMed] [Google Scholar]
- (161).Winters MA; Shafer RW; Jellinger RA; Mamtora G; Gingeras T; Merigan TC Human immunodeficiency virus type 1 reverse transcriptase genotype and drug susceptibility changes in infected individuals receiving dideoxyinosine monotherapy for 1 to 2 years. Antimicrob. Agents Chemother. 1997, 41, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Harrigan PR; Stone C; Griffin P; Najera I; Bloor S; Kemp S; Tisdale M; Larder B Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J. Infect. Dis. 2000, 181, 912–920. [DOI] [PubMed] [Google Scholar]
- (163).Margot NA; Lu B; Cheng A; Miller MD Resistance development over 144 weeks in treatment-naive patients receiving tenofovir disoproxil fumarate or stavudine with lamivudine and efavirenz in Study 903. HIV Med. 2006, 7, 442–450. [DOI] [PubMed] [Google Scholar]
- (164).Das K; Bandwar RP; White KL; Feng JY; Sarafianos SG; Tuske S; Tu X; Clark AD Jr.; Boyer PL; Hou X; et al. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and tenofovir resistance. J. Biol. Chem. 2009, 284, 35092–35100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Das K; Arnold E HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013, 3, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Stone C; Ait-Khaled M; Craig C; Griffin P; Tisdale M Human immunodeficiency virus type 1 reverse transcriptase mutation selection during in vitro exposure to tenofovir alone or combined with abacavir or lamivudine. Antimicrob. Agents Chemother. 2004, 48, 1413–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).St Clair MH; Martin JL; Tudor-Williams G; Bach MC; Vavro CL; King DM; Kellam P; Kemp SD; Larder BA Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse transcriptase. Science 1991, 253, 1557–1559. [DOI] [PubMed] [Google Scholar]
- (168).Martin JL; Wilson JE; Haynes RL; Furman PA Mechanism of resistance of human immunodeficiency virus type 1 to 2’,3′-dideoxyinosine. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6135–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Sarafianos SG; Das K; Clark AD Jr.; Ding J; Boyer PL; Hughes SH; Arnold E Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 10027–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Gao HQ; Boyer PL; Sarafianos SG; Arnold E; Hughes SH The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 2000, 300, 403–418. [DOI] [PubMed] [Google Scholar]
- (171).Das K; Xiong X; Yang H; Westland CE; Gibbs CS; Sarafianos SG; Arnold E Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Shirasaka T; Kavlick MF; Ueno T; Gao WY; Kojima E; Alcaide ML; Chokekijchai S; Roy BM; Arnold E; Yarchoan R; et al. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2398–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Kavlick MF; Wyvill K; Yarchoan R; Mitsuya H Emergence of multi-dideoxynucleoside-resistant human immunodeficiency virus type 1 variants, viral sequence variation, and disease progression in patients receiving antiretroviral chemotherapy. J. Infect. Dis. 1998, 177, 1506–1513. [DOI] [PubMed] [Google Scholar]
- (174).Larder BA; Kemp SD Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 1989, 246, 1155–1158. [DOI] [PubMed] [Google Scholar]
- (175).Arion D; Kaushik N; McCormick S; Borkow G; Parniak MA Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 1998, 37, 15908–15917. [DOI] [PubMed] [Google Scholar]
- (176).Meyer PR; Matsuura SE; So AG; Scott WA Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13471–13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Tu X; Das K; Han Q; Bauman JD; Clark AD Jr.; Hou X; Frenkel YV; Gaffney BL; Jones RA; Boyer PL; et al. Structural basis of HIV-1 resistance to AZT by excision. Nat. Struct. Mol. Biol. 2010, 17, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Scott WA Structures of reverse transcriptase pre- and postexcision complexes shed new light on HIV-1 AZT resistance. Viruses 2011, 3, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Boyer PL; Sarafianos SG; Arnold E; Hughes SH Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 2001, 75, 4832–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Boucher CA; O’Sullivan E; Mulder JW; Ramautarsing C; Kellam P; Darby G; Lange JM; Goudsmit J; Larder BA Ordered appearance of zidovudine resistance mutations during treatment of 18 human immunodeficiency virus-positive subjects. J. Infect. Dis. 1992, 165, 105–110. [DOI] [PubMed] [Google Scholar]
- (181).Jeeninga RE; Keulen W; Boucher C; Sanders RW; Berkhout B Evolution of AZT resistance in HIV-1: the 41–70 intermediate that is not observed in vivo has a replication defect. Virology 2001, 283, 294–305. [DOI] [PubMed] [Google Scholar]
- (182).Arion D; Parniak MA HIV resistance to zidovudine: the role of pyrophosphorolysis. Drug Resist. Updates 1999, 2, 91–95. [DOI] [PubMed] [Google Scholar]
- (183).Yahi N; Tamalet C; Tourres C; Tivoli N; Ariasi F; Volot F; Gastaut JA; Gallais H; Moreau J; Fantini J Mutation patterns of the reverse transcriptase and protease genes in human immunodeficiency virus type 1-infected patients undergoing combination therapy: survey of 787 sequences. J. Clin. Microbiol. 1999, 37, 4099–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Garcia-Lerma JG; MacInnes H; Bennett D; Weinstock H; Heneine W Transmitted human immunodeficiency virus type 1 carrying the D67N or K219Q/E mutation evolves rapidly to zidovudine resistance in vitro and shows a high replicative fitness in the presence of zidovudine. J. Virol. 2004, 78, 7545–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Harrigan PR; Kinghorn I; Bloor S; Kemp SD; Najera I; Kohli A; Larder BA Significance of amino acid variation at human immunodeficiency virus type 1 reverse transcriptase residue 210 for zidovudine susceptibility. J. Virol. 1996, 70, 5930–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Hooker DJ; Tachedjian G; Solomon AE; Gurusinghe AD; Land S; Birch C; Anderson JL; Roy BM; Arnold E; Deacon NJ An in vivo mutation from leucine to tryptophan at position 210 in human immunodeficiency virus type 1 reverse transcriptase contributes to high-level resistance to 3′-azido-3′deoxythymidine. J. Virol. 1996, 70, 8010–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Imamichi T; Berg SC; Imamichi H; Lopez JC; Metcalf JA; Falloon J; Lane HC Relative replication fitness of a high-level 3′-azido-3′-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr–>Gly) at codon 69. J. Virol. 2000, 74, 10958–10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Boyer PL; Imamichi T; Sarafianos SG; Arnold E; Hughes SH Effects of the Delta67 complex of mutations in human immunodeficiency virus type 1 reverse transcriptase on nucleoside analog excision. J. Virol. 2004, 78, 9987–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Mas A; Parera M; Briones C; Soriano V; Martinez MA; Domingo E; Menendez-Arias L Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 2000, 19, 5752–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Boyer PL; Sarafianos SG; Arnold E; Hughes SH Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves ATP-mediated excision. J. Virol. 2002, 76, 9143–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Meyer PR; Lennerstrand J; Matsuura SE; Larder BA; Scott WA Effects of dipeptide insertions between codons 69 and 70 of human immunodeficiency virus type 1 reverse transcriptase on primer unblocking, deoxynucleoside triphosphate inhibition, and DNA chain elongation. J. Virol. 2003, 77, 3871–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).White KL; Chen JM; Margot NA; Wrin T; Petropoulos CJ; Naeger LK; Swaminathan S; Miller MD Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 2004, 48, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Lowe DM; Aitken A; Bradley C; Darby GK; Larder BA; Powell KL; Purifoy DJ; Tisdale M; Stammers DK HIV-1 reverse transcriptase: crystallization and analysis of domain structure by limited proteolysis. Biochemistry 1988, 27, 8884–8889. [DOI] [PubMed] [Google Scholar]
- (194).Ren J; Esnouf R; Hopkins A; Ross C; Jones Y; Stammers D; Stuart D The structure of HIV-1 reverse transcriptase complexed with 9-chloro-TIBO: lessons for inhibitor design. Structure 1995, 3, 915–926. [DOI] [PubMed] [Google Scholar]
- (195).Das K; Ding J; Hsiou Y; Clark AD Jr.; Moereels H; Koymans L; Andries K; Pauwels R; Janssen PA; Boyer PL; et al. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [DOI] [PubMed] [Google Scholar]
- (196).Ren J; Nichols CE; Chamberlain PP; Weaver KL; Short SA; Stammers DK Crystal structures of HIV-1 reverse transcriptases mutated at codons 100, 106 and 108 and mechanisms of resistance to non-nucleoside inhibitors. J. Mol. Biol. 2004, 336, 569–578. [DOI] [PubMed] [Google Scholar]
- (197).Rhee SY; Liu T; Ravela J; Gonzales MJ; Shafer RW Distribution of human immunodeficiency virus type 1 protease and reverse transcriptase mutation patterns in 4,183 persons undergoing genotypic resistance testing. Antimicrob. Agents Chemother. 2004, 48, 3122–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Vingerhoets J; Tambuyzer L; Azijn H; Hoogstoel A; Nijs S; Peeters M; de Bethune MP; De Smedt G; Woodfall B; Picchio G Resistance profile of etravirine: combined analysis of baseline genotypic and phenotypic data from the randomized, controlled Phase III clinical studies. AIDS 2010, 24, 503–514. [DOI] [PubMed] [Google Scholar]
- (199).Tambuyzer L; Azijn H; Rimsky LT; Vingerhoets J; Lecocq P; Kraus G; Picchio G; de Bethune MP Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antivir. Ther. 2009, 14, 103–109. [PubMed] [Google Scholar]
- (200).Tambuyzer L; Vingerhoets J; Azijn H; Daems B; Nijs S; de Bethune M-P; Picchio G Characterization of genotypic and phenotypic changes in HIV-1-infected patients with virologic failure on an etravirine-containing regimen in the DUET-1 and DUET-2 clinical studies. AIDS Res. Hum. Retroviruses 2010, 26, 1197–1205. [DOI] [PubMed] [Google Scholar]
- (201).Rimsky L; Vingerhoets J; Van Eygen V; Eron J; Clotet B; Hoogstoel A; Boven K; Picchio G Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. JAIDS, J. Acquired Immune Defic. Syndr. 2012, 59, 39–46. [DOI] [PubMed] [Google Scholar]
- (202).Bacheler LT; Anton ED; Kudish P; Baker D; Bunville J; Krakowski K; Bolling L; Aujay M; Wang XV; Ellis D; et al. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob. Agents Chemother. 2000, 44, 2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Zhang Z; Xu W; Koh YH; Shim JH; Girardet JL; Yeh LT; Hamatake RK; Hong Z A novel nonnucleoside analogue that inhibits human immunodeficiency virus type 1 isolates resistant to current nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2007, 51, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Reuman EC; Rhee SY; Holmes SP; Shafer RW Constrained patterns of covariation and clustering of HIV-1 nonnucleoside reverse transcriptase inhibitor resistance mutations. J. Antimicrob. Chemother. 2010, 65, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Johnson BC; Pauly GT; Rai G; Patel D; Bauman JD; Baker HL; Das K; Schneider JP; Maloney DJ; Arnold E; et al. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology 2012, 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Melikian GL; Rhee SY; Varghese V; Porter D; White K; Taylor J; Towner W; Troia P; Burack J; Dejesus E; et al. Non-nucleoside reverse transcriptase inhibitor (NNRTI) cross-resistance: implications for preclinical evaluation of novel NNRTIs and clinical genotypic resistance testing. J. Antimicrob. Chemother. 2014, 69, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Theys K; Camacho RJ; Gomes P; Vandamme AM; Rhee SY Predicted residual activity of rilpivirine in HIV-1 infected patients failing therapy including NNRTIs efavirenz or nevirapine. Clin. Microbiol. Infect. 2015, 21, 607. [DOI] [PubMed] [Google Scholar]
- (208).Balzarini J; Pelemans H; Esnouf R; De Clercq E A novel mutation (F227L) arises in the reverse transcriptase of human immunodeficiency virus type 1 on dose-escalating treatment of HIV type 1-infected cell cultures with the nonnucleoside reverse transcriptase inhibitor thiocarboxanilide UC-781. AIDS Res. Hum. AIDS Res. Hum. Retroviruses 1998, 14, 255–260. [DOI] [PubMed] [Google Scholar]
- (209).Sarafianos SG; Das K; Hughes SH; Arnold E Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV1 reverse transcriptases. Curr. Opin. Struct. Biol. 2004, 14, 716–730. [DOI] [PubMed] [Google Scholar]
- (210).Feng M; Sachs NA; Xu M; Grobler J; Blair W; Hazuda DJ; Miller MD; Lai MT Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob. Agents Chemother. 2016, 60, 2241–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Ren J; Nichols CE; Chamberlain PP; Weaver KL; Short SA; Chan JH; Kleim JP; Stammers DK Relationship of potency and resilience to drug resistant mutations for GW420867X revealed by crystal structures of inhibitor complexes for wild-type, Leu100Ile, Lys101Glu, and Tyr188Cys mutant HIV-1 reverse transcriptases. J. Med. Chem. 2007, 50, 2301–2309. [DOI] [PubMed] [Google Scholar]
- (212).Eshleman SH; Jones D; Galovich J; Paxinos EE; Petropoulos CJ; Jackson JB; Parkin N Phenotypic drug resistance patterns in subtype A HIV-1 clones with nonnucleoside reverse transcriptase resistance mutations. AIDS Res. Hum. Retroviruses 2006, 22, 289–293. [DOI] [PubMed] [Google Scholar]
- (213).Parkin NT; Gupta S; Chappey C; Petropoulos CJ The K101P and K103R/V179D mutations in human immunodeficiency virus type 1 reverse transcriptase confer resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2006, 50, 351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Balamane M; Varghese V; Melikian GL; Fessel WJ; Katzenstein DA; Shafer RW Panel of prototypical recombinant infectious molecular clones resistant to nevirapine, efavirenz, etravirine, and rilpivirine. Antimicrob. Agents Chemother. 2012, 56, 4522–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Kulkarni R; Babaoglu K; Lansdon EB; Rimsky L; Van Eygen V; Picchio G; Svarovskaia E; Miller MD; White KL The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. JAIDS, J. Acquired Immune Defic. Syndr. 2012, 59, 47–54. [DOI] [PubMed] [Google Scholar]
- (216).Xu HT; Colby-Germinario SP; Asahchop EL; Oliveira M; McCallum M; Schader SM; Han Y; Quan Y; Sarafianos SG; Wainberg MA Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob. Agents Chemother. 2013, 57, 3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Singh K; Marchand B; Rai DK; Sharma B; Michailidis E; Ryan EM; Matzek KB; Leslie MD; Hagedorn AN; Li Z; et al. Biochemical mechanism of HIV-1 resistance to rilpivirine. J. Biol. Chem. 2012, 287, 38110–38123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Gupta S; Fransen S; Paxinos EE; Stawiski E; Huang W; Petropoulos CJ Combinations of mutations in the connection domain of human immunodeficiency virus type 1 reverse transcriptase: assessing the impact on nucleoside and nonnucleoside reverse transcriptase inhibitor resistance. Antimicrob. Antimicrob. Agents Chemother. 2010, 54, 1973–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Azijn H; Tirry I; Vingerhoets J; de Bethune MP; Kraus G; Boven K; Jochmans D; Van Craenenbroeck E; Picchio G; Rimsky LT TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob. Agents Chemother. 2010, 54, 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Lai MT; Feng M; Falgueyret JP; Tawa P; Witmer M; DiStefano D; Li Y; Burch J; Sachs N; Lu M; et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 1652–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Basson AE; Rhee SY; Parry CM; El-Khatib Z; Charalambous S; De Oliveira T; Pillay D; Hoffmann C; Katzenstein D; Shafer RW; et al. Impact of drug resistance-associated amino acid changes in HIV-1 subtype C on susceptibility to newer nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2015, 59, 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Chellappan S; Kiran Kumar Reddy GS; Ali A; Nalam MN; Anjum SG; Cao H; Kairys V; Fernandes MX; Altman MD; Tidor B; et al. Design of mutation-resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem. Biol. Drug Des. 2007, 69, 298–313. [DOI] [PubMed] [Google Scholar]
- (223).Nalam MN; Ali A; Altman MD; Reddy GS; Chellappan S; Kairys V; Ozen A; Cao H; Gilson MK; Tidor B; et al. Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J. Virol. 2010, 84, 5368–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Ozen A; Sherman W; Schiffer CA Improving the resistance profile of Hepatitis C NS3/4A inhibitors: dynamic substrate envelope guided design. J. Chem. Theory Comput. 2013, 9, 5693–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Tuske S; Sarafianos SG; Clark AD Jr.; Ding J; Naeger LK; White KL; Miller MD; Gibbs CS; Boyer PL; Clark P; et al. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004, 11, 469–474. [DOI] [PubMed] [Google Scholar]
- (226).White KL; Margot NA; Wrin T; Petropoulos CJ; Miller MD; Naeger LK Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob. Agents Chemother. 2002, 46, 3437–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Deval J; White KL; Miller MD; Parkin NT; Courcambeck J; Halfon P; Selmi B; Boretto J; Canard B Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem. 2004, 279, 509–516. [DOI] [PubMed] [Google Scholar]
- (228).Das K; Martinez SE; Arnold E Structural insights into HIV reverse transcriptase mutations Q151M and Q151M complex that confer multinucleoside drug resistance. Antimicrob. Agents Chemother. 2017, 61, 00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Hung M; Tokarsky EJ; Lagpacan L; Zhang L; Suo Z; Lansdon EB Elucidating molecular interactions of L-nucleotides with HIV-1 reverse transcriptase and mechanism of M184V-caused drug resistance. Commun. Biol. 2019, 2, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Marchand B; Tchesnokov EP; Gotte M The pyrophosphate analogue foscarnet traps the pre-translocational state of HIV-1 reverse transcriptase in a Brownian ratchet model of polymerase translocation. J. Biol. Chem. 2007, 282, 3337–3346. [DOI] [PubMed] [Google Scholar]
- (231).Christ F; Voet A; Marchand A; Nicolet S; Desimmie BA; Marchand D; Bardiot D; Van der Veken NJ; Van Remoortel B; Strelkov SV; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [DOI] [PubMed] [Google Scholar]
- (232).Zhao Q; Ma L; Jiang S; Lu H; Liu S; He Y; Strick N; Neamati N; Debnath AK Identification of N-phenyl-N’-(2,2,6,6tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 2005, 339, 213–225. [DOI] [PubMed] [Google Scholar]
- (233).Madani N; Schon A; Princiotto AM; Lalonde JM; Courter JR; Soeta T; Ng D; Wang L; Brower ET; Xiang SH; et al. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 2008, 16, 1689–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Hayashi H; Takamune N; Nirasawa T; Aoki M; Morishita Y; Das D; Koh Y; Ghosh AK; Misumi S; Mitsuya H Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by darunavir. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12234–12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Koh Y; Matsumi S; Das D; Amano M; Davis DA; Li J; Leschenko S; Baldridge A; Shioda T; Yarchoan R; et al. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 2007, 282, 28709–28720. [DOI] [PubMed] [Google Scholar]
- (236).Jurado KA; Wang H; Slaughter A; Feng L; Kessl JJ; Koh Y; Wang W; Ballandras-Colas A; Patel PA; Fuchs JR; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8690–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Deng N; Hoyte A; Mansour YE; Mohamed MS; Fuchs JR; Engelman AN; Kvaratskhelia M; Levy R Allosteric HIV-1 integrase inhibitors promote aberrant protein multimerization by directly mediating inter-subunit interactions: Structural and thermodynamic modeling studies. Protein Sci. 2016, 25, 1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Tachedjian G; Aronson HE; de los Santos M; Seehra J; McCoy JM; Goff SP Role of residues in the tryptophan repeat motif for HIV-1 reverse transcriptase dimerization. J. Mol. Biol. 2003, 326, 381–396. [DOI] [PubMed] [Google Scholar]
- (239).Mulky A; Sarafianos SG; Jia Y; Arnold E; Kappes JC Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are required for subunit interaction using infectious virions. J. Mol. Biol. 2005, 349, 673–684. [DOI] [PubMed] [Google Scholar]
- (240).Tintori C; Corona A; Esposito F; Brai A; Grandi N; Ceresola ER; Clementi M; Canducci F; Tramontano E; Botta M Inhibition of HIV-1 reverse transcriptase dimerization by small molecules. ChemBioChem 2016, 17, 683–688. [DOI] [PubMed] [Google Scholar]
- (241).Grohmann D; Corradi V; Elbasyouny M; Baude A; Horenkamp F; Laufer SD; Manetti F; Botta M; Restle T Small molecule inhibitors targeting HIV-1 reverse transcriptase dimerization. ChemBioChem 2008, 9, 916–922. [DOI] [PubMed] [Google Scholar]
- (242).Tachedjian G; Orlova M; Sarafianos SG; Arnold E; Goff SP Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 7188–7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Figueiredo A; Moore KL; Mak J; Sluis-Cremer N; de Bethune MP; Tachedjian G Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006, 2, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Ballandras-Colas A; Maskell DP; Serrao E; Locke J; Swuec P; Jonsson SR; Kotecha A; Cook NJ; Pye VE; Taylor IA; et al. A supramolecular assembly mediates lentiviral DNA integration. Science 2017, 355, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Passos DO; Li M; Jozwik IK; Zhao XZ; SantosMartins D; Yang R; Smith SJ; Jeon Y; Forli S; Hughes SH; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Shaw-Reid CA; Munshi V; Graham P; Wolfe A; Witmer M; Danzeisen R; Olsen DB; Carroll SS; Embrey M; Wai JS; et al. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 2003, 278, 2777–2780. [DOI] [PubMed] [Google Scholar]
- (247).Corona A; Di Leva FS; Thierry S; Pescatori L; Cuzzucoli Crucitti G; Subra F; Delelis O; Esposito F; Rigogliuso G; Costi R; et al. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by Diketo acid derivatives. Antimicrob. Agents Chemother. 2014, 58, 6101–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Vernekar SK; Liu Z; Nagy E; Miller L; Kirby KA; Wilson DJ; Kankanala J; Sarafianos SG; Parniak MA; Wang Z Design, synthesis, biochemical, and antiviral evaluations of C6 benzyl and C6 biarylmethyl substituted 2-hydroxylisoquinoline-1,3diones: dual inhibition against HIV reverse transcriptase-associated RNase H and polymerase with antiviral activities. J. Med. Chem. 2015, 58, 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Xi Z; Wang Z; Sarafianos SG; Myshakina NS; Ishima R Determinants of active-site inhibitor interaction with HIV-1 RNase H. ACS Infect. Dis. 2019, 5, 1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Wang L; Tang J; Huber AD; Casey MC; Kirby KA; Wilson DJ; Kankanala J; Parniak MA; Sarafianos SG; Wang Z 6-Biphenylmethyl-3-hydroxypyrimidine-2,4-diones potently and selectively inhibited HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem. 2018, 156, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Kankanala J; Kirby KA; Huber AD; Casey MC; Wilson DJ; Sarafianos SG; Wang Z Design, synthesis and biological evaluations of N-Hydroxy thienopyrimidine-2,4-diones as inhibitors of HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem. 2017, 141, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Kirby KA; Myshakina NA; Christen MT; Chen YL; Schmidt HA; Huber AD; Xi Z; Kim S; Rao RK; Kramer ST; et al. A 2-hydroxyisoquinoline-1,3-dione active-site RNase H inhibitor binds in multiple modes to HIV-1 reverse transcriptase. Antimicrob. Agents Chemother. 2017, 61, 01351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Vernekar SKV; Tang J; Wu B; Huber AD; Casey MC; Myshakina N; Wilson DJ; Kankanala J; Kirby KA; Parniak MA; et al. Double-Winged 3-Hydroxypyrimidine-2,4-diones: potent and selective inhibition against HIV-1 RNase H with significant antiviral activity. J. Med. Chem. 2017, 60, 5045–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Esposito F; Tramontano E Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: single and dual active site inhibitors. Antivir. Chem. Chemother. 2014, 23, 129–144. [DOI] [PubMed] [Google Scholar]
- (255).Tramontano E; Corona A; Menendez-Arias L Ribonuclease H, an unexploited target for antiviral intervention against HIV and hepatitis B virus. Antiviral Res. 2019, 171, 104613. [DOI] [PubMed] [Google Scholar]
- (256).Kankanala J; Kirby KA; Liu F; Miller L; Nagy E; Wilson DJ; Parniak MA; Sarafianos SG; Wang Z Design, synthesis, and biological evaluations of hydroxypyridonecarboxylic acids as inhibitors of HIV reverse transcriptase associated RNase H. J. Med. Chem. 2016, 59, 5051–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Kuroda DG; Bauman JD; Challa JR; Patel D; Troxler T; Das K; Arnold E; Hochstrasser RM Snapshot of the equilibrium dynamics of a drug bound to HIV-1 reverse transcriptase. Nat. Chem. 2013, 5, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Bohacek RS; McMartin C; Guida WC The art and practice of structure-based drug design: a molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [DOI] [PubMed] [Google Scholar]
- (259).Lonsdale R; Ward RA Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [DOI] [PubMed] [Google Scholar]
- (260).Singh J; Petter RC; Baillie TA; Whitty A The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- (261).Baillie TA Targeted covalent inhibitors for drug design. Angew. Chem., Int. Ed. 2016, 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- (262).Barf T; Kaptein A Irreversible protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [DOI] [PubMed] [Google Scholar]
- (263).Shannon DA; Weerapana E Covalent protein modification: the current landscape of residue-specific electrophiles. Curr. Opin. Chem. Biol. 2015, 24, 18–26. [DOI] [PubMed] [Google Scholar]
- (264).Jackson PA; Widen JC; Harki DA; Brummond KM Covalent modifiers: A chemical perspective on the reactivity of alpha,beta-unsaturated carbonyls with thiols via hetero-michael addition reactions. J. Med. Chem. 2017, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Pettinger J; Jones K; Cheeseman MD Lysine-targeting covalent inhibitors. Angew. Chem., Int. Ed. 2017, 56, 15200–15209. [DOI] [PubMed] [Google Scholar]
- (266).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Forli S; Huey R; Pique ME; Sanner MF; Goodsell DS; Olson AJ Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Sliwoski G; Kothiwale S; Meiler J; Lowe EW Jr Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Chan AH; Lee WG; Spasov KA; Cisneros JA; Kudalkar SN; Petrova ZO; Buckingham AB; Anderson KS; Jorgensen WL Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9725–9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Lee WG; Chan AH; Spasov KA; Anderson KS; Jorgensen WL Design, conformation, and crystallography of 2naphthyl phenyl ethers as potent anti-HIV agents. ACS Med. Chem. Lett. 2016, 7, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Bauman JD; Patel D; Arnold E Fragment screening and HIV therapeutics. Top. Curr. Chem. 2011, 317, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Patel D; Bauman JD; Arnold E Advantages of crystallographic fragment screening: functional and mechanistic insights from a powerful platform for efficient drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Bauman JD; Patel D; Dharia C; Fromer MW; Ahmed S; Frenkel Y; Vijayan RS; Eck JT; Ho WC; Das K; et al. Detecting allosteric sites of HIV-1 reverse transcriptase by X-ray crystallographic fragment screening. J. Med. Chem. 2013, 56, 2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).La J; Latham CF; Tinetti RN; Johnson A; Tyssen D; Huber KD; Sluis-Cremer N; Simpson JS; Headey SJ; Chalmers DK; et al. Identification of mechanistically distinct inhibitors of HIV-1 reverse transcriptase through fragment screening. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6979–6984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Ly JK; Margot NA; MacArthur HL; Hung M; Miller MD; White KL The balance between NRTI discrimination and excision drives the susceptibility of HIV-1 RT mutants K65R, M184V and K65R+M184V. Antivir. Chem. Chemother. 2007, 18, 307–316. [DOI] [PubMed] [Google Scholar]
- (276).Wolf K; Walter H; Beerenwinkel N; Keulen W; Kaiser R; Hoffmann D; Lengauer T; Selbig J; Vandamme AM; Korn K; et al. Tenofovir resistance and resensitization. Antimicrob. Agents Chemother. 2003, 47, 3478–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Whitcomb JM; Parkin NT; Chappey C; Hellmann NS; Petropoulos CJ Broad nucleoside reverse-transcriptase inhibitor cross-resistance in human immunodeficiency virus type 1 clinical isolates. J. Infect. Dis. 2003, 188, 992–1000. [DOI] [PubMed] [Google Scholar]
- (278).Borroto-Esoda K; Parkin N; Miller MD A comparison of the phenotypic susceptibility profiles of emtricitabine and lamivudine. Antivir. Chem. Chemother. 2007, 18, 297–300. [DOI] [PubMed] [Google Scholar]
- (279).Gregson J; Rhee SY; Datir R; Pillay D; Perno CF; Derache A; Shafer RS; Gupta RK Human immunodeficiency virus-1 viral load is elevated in individuals with reverse-transcriptase mutation M184V/I during virological failure of first-line antiretroviral therapy and is associated with compensatory mutation L74I. J. Infect. Dis. 2020, 222, 1108–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Kulkarni R; Feng JY; Miller MD; White KL Dead-end complexes contribute to the synergistic inhibition of HIV-1 RT by the combination of rilpivirine, emtricitabine, and tenofovir. Antiviral Res. 2014, 101, 131–135. [DOI] [PubMed] [Google Scholar]
- (281).Hachiya A; Reeve AB; Marchand B; Michailidis E; Ong YT; Kirby KA; Leslie MD; Oka S; Kodama EN; Rohan LC; et al. Evaluation of combinations of 4’-Ethynyl-2-Fluoro-2’Deoxyadenosine with clinically used antiretroviral drugs. Antimicrob. Agents Chemother. 2013, 57, 4554–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Smith SJ; Pauly GT; Akram A; Melody K; Ambrose Z; Schneider JP; Hughes SH Rilpivirine and doravirine have complementary efficacies against NNRTI-resistant HIV-1 mutants. JAIDS, J. Acquired Immune Defic. Syndr. 2016, 72, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Sarafianos SG; Hughes SH; Arnold E Designing anti-AIDS drugs targeting the major mechanism of HIV-1 RT resistance to nucleoside analog drugs. Int. J. Biochem. Cell Biol. 2004, 36, 1706–1715. [DOI] [PubMed] [Google Scholar]
- (284).Han Q; Sarafianos SG; Arnold E; Parniak MA; Gaffney BL; Jones RA Synthesis of AZTpSpCX2ppSA and AZTpSpCX2ppSAZT: hydrolysis-resistant potential inhibitors of the AZT excision reaction of HIV-1 RT. Org. Lett. 2007, 9, 5243–5246. [DOI] [PubMed] [Google Scholar]
- (285).Takamatsu Y; Tanaka Y; Kohgo S; Murakami S; Singh K; Das D; Venzon DJ; Amano M; Higashi-Kuwata N; Aoki M; et al. 4’-modified nucleoside analogs: potent inhibitors active against entecavir-resistant hepatitis B virus. Hepatology 2015, 62, 1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Hashimoto M; Taguchi K; Ishiguro T; Kohgo S; Imoto S; Yamasaki K; Mitsuya H; Otagiri M Pharmacokinetics studies of 4’-cyano-2’-deoxyguanosine, a potent inhibitor of the hepatitis B virus, in rats. J. Pharm. Pharmacol. 2018, 70, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Agostini ML; Andres EL; Sims AC; Graham RL; Sheahan TP; Lu X; Smith EC; Case JB; Feng JY; Jordan R; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, 00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Higashi-Kuwata N; Hayashi S; Das D; Kohgo S; Murakami S; Hattori SI; Imoto S; Venzon DJ; Singh K; Sarafianos SG; et al. CMCdG, a novel nucleoside analog with favorable safety features, exerts potent activity against wild-type and entecavir-resistant hepatitis B virus. Antimicrob. Agents Chemother. 2019, 63, 02143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Eastman RT; Roth JS; Brimacombe KR; Simeonov A; Shen M; Patnaik S; Hall MD Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. ACS Cent. Sci. 2020, 6, 672–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Gordon CJ; Tchesnokov EP; Woolner E; Perry JK; Feng JY; Porter DP; Gotte M Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Yasutake Y; Hattori SI; Tamura N; Matsuda K; Kohgo S; Maeda K; Mitsuya H Active-site deformation in the structure of HIV-1 RT with HBV-associated septuple amino acid substitutions rationalizes the differential susceptibility of HIV-1 and HBV against 4’-modified nucleoside RT inhibitors. Biochem. Biophys. Res. Commun. 2019, 509, 943–948. [DOI] [PubMed] [Google Scholar]
- (292).Klumpp K; Mirzadegan T Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr. Pharm. Des. 2006, 12, 1909–1922. [DOI] [PubMed] [Google Scholar]
- (293).Ilina T; Labarge K; Sarafianos SG; Ishima R; Parniak MA Inhibitors of HIV-1 reverse transcriptase-associated Ribonuclease H activity. Biology (Basel, Switz.) 2012, 1, 521–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Cao L; Song W; De Clercq E; Zhan P; Liu X Recent progress in the research of small molecule HIV-1 RNase H inhibitors. Curr. Med. Chem. 2014, 21, 1956–1967. [DOI] [PubMed] [Google Scholar]
- (295).Wang X; Gao P; Menendez-Arias L; Liu X; Zhan P Update on recent developments in small molecular HIV-1 RNase H inhibitors (2013–2016): opportunities and challenges. Curr. Med. Chem. 2018, 25, 1682–1702. [DOI] [PubMed] [Google Scholar]
- (296).Wang L; Sarafianos SG; Wang Z Cutting into the substrate dominance: pharmacophore and structure-based approaches toward inhibiting Human Immunodeficiency Virus reverse transcriptase-associated Ribonuclease H. Acc. Chem. Res. 2020, 53, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Barrett SE; Teller RS; Forster SP; Li L; Mackey MA; Skomski D; Yang Z; Fillgrove KL; Doto GJ; Wood SL; et al. Extended-duration MK-8591-eluting implant as a candidate for HIV treatment and prevention. Antimicrob. Agents Chemother. 2018, 62, 01058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Markowitz M; Gettie A; St Bernard L; Andrews CD; Mohri H; Horowitz A; Grasperge BF; Blanchard JL; Niu T; Sun L; et al. Once-weekly oral dosing of MK-8591 protects male rhesus macaques from intrarectal challenge with SHIV109CP3. J. Infect. Dis. 2020, 221, 1398–1406. [DOI] [PubMed] [Google Scholar]
- (299).Schurmann D; Rudd DJ; Zhang S; De Lepeleire I; Robberechts M; Friedman E; Keicher C; Huser A; Hofmann J; Grobler JA; et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: an open-label, phase 1b, consecutive-panel trial. Lancet HIV 2020, 7, e164–e172. [DOI] [PubMed] [Google Scholar]
- (300).Gunawardana M; Remedios-Chan M; Miller CS; Fanter R; Yang F; Marzinke MA; Hendrix CW; Beliveau M; Moss JA; Smith TJ; et al. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob. Agents Chemother. 2015, 59, 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (301).Bekker L-G; Li SS; Tolly B; Marzinke MA; Mgodi N; Justman JE; Swaminathan S; Adeyeye A; Farrior JH; Sista N Hptn 076:Tmc278 ls safe, tolarable, and acceptable for HIV preexposure prophylaxis. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, WA, 2017. [Google Scholar]
- (302).Janssen Products; Latina, Italy, 2011. [Google Scholar]
- (303).Hawkins T; Veikley W; St Claire RL III; Guyer B; Clark N; Kearney BP Intracellular pharmacokinetics of tenofovir diphosphate, carbovir triphosphate, and lamivudine triphosphate in patients receiving triple-nucleoside regimens. JAIDS, J. Acquired Immune Defic. Syndr. 2005, 39, 406–411. [DOI] [PubMed] [Google Scholar]
- (304).Stevens RC; Blum MR; Rousseau FS; Kearney BP Intracellular pharmacology of emtricitabine and tenofovir. Clin. Infect. Dis. 2004, 39, 877–878 author reply 878–879. [DOI] [PubMed] [Google Scholar]
- (305).Ruiz FX; Hoang A; Das K; Arnold E Structural basis of HIV-1 inhibition by nucleotide-competing reverse transcriptase inhibitor INDOPY-1. J. Med. Chem. 2019, 62, 9996–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Jochmans D; Deval J; Kesteleyn B; Van Marck H; Bettens E; De Baere I; Dehertogh P; Ivens T; Van Ginderen M; Van Schoubroeck B; et al. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J. Virol. 2006, 80, 12283–12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Ehteshami M; Scarth BJ; Tchesnokov EP; Dash C; Le Grice SF; Hallenberger S; Jochmans D; Gotte M Mutations M184V and Y115F in HIV-1 reverse transcriptase discriminate against ″nucleotide-competing reverse transcriptase inhibitors″. J. Biol. Chem. 2008, 283, 29904–29911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Xu HT; Colby-Germinario SP; Quashie PK; Bethell R; Wainberg MA Subtype-specific analysis of the K65R substitution in HIV-1 that confers hypersusceptibility to a novel nucleotide-competing reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 2015, 59, 3189–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]