

Abstract

Phosphorus-based organocatalysis encompasses several subfields that have undergone rapid growth in recent years. This Outlook gives an overview of its various aspects. In particular, we highlight key advances in three topics: nucleophilic phosphine catalysis, organophosphorus catalysis to bypass phosphine oxide waste, and organophosphorus compound-mediated single electron transfer processes. We briefly summarize five additional topics: chiral phosphoric acid catalysis, phosphine oxide Lewis base catalysis, iminophosphorane super base catalysis, phosphonium salt phase transfer catalysis, and frustrated Lewis pair catalysis. Although it is not catalytic in nature, we also discuss novel discoveries that are emerging in phosphorus(V) ligand coupling. We conclude with some ideas about the future of organophosphorus catalysis.
Short abstract
An Outlook on the rich chemistry of phosphorus, focusing on nucleophilic phosphine catalysis, organophosphorus catalysis to prevent phosphine oxide waste, and phosphorus-mediated radical processes.
1. Introduction
The study and application of organic phosphorus compounds in organic synthesis have a long history that dates back to the Middle Ages.1 Scientific investigations into these types of compounds, especially from a synthetic perspective, have unveiled the rich chemistry of organophosphorus compounds. In this realm, Michaelis and Arbusov, as well as other chemists, were the pioneers who laid the foundations of what we know as phosphorus chemistry today.1 One significant feature of organic phosphorus compounds is that they can be used as ligands, catalysts, or reagents to facilitate various organic reactions.2,3 When asked about the roles of phosphorus compounds in organic synthesis, however, a majority of organic chemists might offer stereotypical answers: ligands in transition-metal-promoted reactions, reagents for Wittig and Mitsunobu reactions, and organocatalysts for reactions that corresponding nitrogen counterparts also facilitate [e.g., the Morita–Baylis–Hillman (MBH) reaction]. In truth, phosphorus-based organocatalysis is a unique, multifaceted field, with many promising research opportunities and applications. For example, chiral phosphoric acid catalysis4 and phosphine oxide Lewis base catalysis5 play important roles in asymmetric organocatalysis. Frustrated Lewis pairs (FLPs), combinations of Lewis acids and Lewis bases, are important species for the activation of small molecules.6 As a green sustainable tool in organic synthesis, phosphonium salt-based phase transfer catalysis is garnering increasing attention.7 Bifunctional phosphonium catalysts have especially great potential in asymmetric synthesis. Chiral iminophosphorane super base catalysts have demonstrated their usefulness in the deprotonation of high-pKa pronucleophiles for enantioselective transformations.8 An abundance of structural and stereochemical diversity among readily available phosphorus compounds remains pivotal for developing novel asymmetric transformations. Notably, nucleophilic phosphine catalysis is being investigated vigorously, with discoveries of novel reaction patterns and asymmetric transformations.3 Although classic transformations that are driven by the formation of phosphine oxides (e.g., Wittig olefination, Mitsunobu, and Appel reactions) are highly popular among synthetic chemists, these reactions have several drawbacks [e.g., the requirement of stoichiometric phosphorus reagents, the use of external oxidants (e.g., azo compounds for Mitsunobu reactions), and the generation of phosphine oxide waste] that limit their use in both academia and industry. Developing catalytic versions of these reactions—in particular to bypass phosphine oxide waste—is highly desirable.3 This area has been studied intensely over the past decade. In addition to those phosphorus-mediated reactions that operate under mechanisms involving electron pair transfer, phosphorus compound-mediated single-electron transfer (SET) processes are a promising, yet undeveloped, area. Because of the rapid development and growing interest in photochemistry and electrochemistry in recent years, studies of organophosphorus compound-mediated SET processes are gaining momentum.9
In the short span of two years since our comprehensive review on organophosphorus catalysis in 2018,3b significant progress has been made in the field. In this Outlook, we highlight the key features of three such topics: nucleophilic phosphine catalysis, organophosphorus catalysis to bypass phosphine oxide waste, and organophosphorus compound-mediated SET processes. We also provide an overview of several other important topics related to phosphorus-based catalysis in the Miscellaneous Catalyses section. We do not go into too much detail regarding these miscellaneous topics but instead draw the reader’s attention to several comprehensive reviews. Moreover, interesting and useful chemistry is emerging in phosphorus-based reactions. For example, phosphorus(V) ligand coupling, although not catalytic in phosphorus, is becoming a method complementary to transition-metal-catalyzed coupling for the functionalization of heterocycles; we discuss these processes briefly. Although this Outlook is not intended to provide a comprehensive review, we are aiming to give an overview of phosphorus-based catalysis, point out the key developments in the past two years, and proffer our perspective about the future of phosphorus-based catalysis, hoping to inspire researchers to consider the rich chemistry of organophosphorus compounds in their own endeavors.
2. Nucleophilic Phosphine Catalysis
Nucleophilic phosphine catalysis typically operates through initial addition of a tertiary phosphine to an electron-deficient multiple bond, generating a reactive zwitterionic species. Various substrates, including alkenes, allenes, alkynes, and MBH alcohol derivatives (MBHADs), can be involved in this type of activation. The generated reactive zwitterionic intermediates can undergo further reactions with a variety of nucleophiles and electrophiles, or a combination of both. Nucleophilic phosphines also play a special role in the activation of C=O, N=N, strained C—C, or strained C—X (e.g., C—O, C—N) bonds. The broad substrate scope, mild reaction conditions, and diverse reaction modes have established nucleophilic phosphine catalysis as a powerful tool in organic synthesis. Since our group’s comprehensive review of phosphine organocatalysis in 2018,3b more than 80 new reports of nucleophilic phosphine catalysis have been published.10 A survey of the progress in nucleophilic phosphine catalysis for the past two years has revealed some interesting features. The main thrusts of the research continue to be the discovery of new patterns of annulation, asymmetric variants of previously reported reactions, and investigations into known reactions using substrates that are much more elaborate than those reported originally. In this Outlook, we classify the reactions of nucleophilic phosphine catalysis based on the nature of their electrophiles with different types of activated multiple bonds—specifically, alkenes, allenes, alkynes, and MBHADs. We further categorize these reactions in terms of the second reaction partner—namely, nucleophiles, electrophiles, and their combinations. In this section, we discuss some recent representative examples of nucleophilic phosphine catalysis to illustrate its unique features and rich chemistry.
2.1. Nucleophilic Phosphine Catalysis of Alkenes
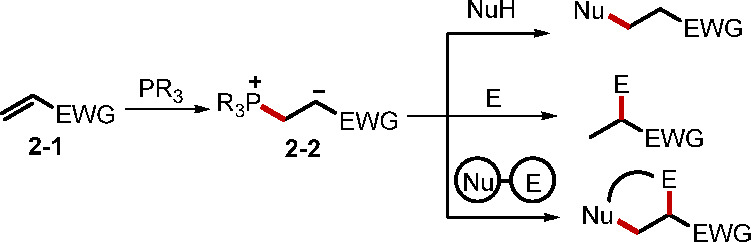
The nucleophilic phosphine catalysis of alkenes involves the addition of phosphine to electron-deficient alkenes 2–1 to form Horner’s zwitterionic adduct 2–2, which has multifarious modes of reaction depending on its reaction partners (Scheme 1). As such, reactions of the zwitterion 2–2 with a pronucleophile (NuH), an electrophile (E), or a hybrid of both can lead to various synthetically useful scaffolds. Recent reports on the reactions of alkenes with pronucleophiles have been limited to the phosphine-catalyzed Michael addition, in which pronucleophiles, such as phosphine oxides and TMSCN, have been introduced to produce β-phosphoryl- and β-cyano-carbonyls, respectively.11 Among the reactions of the zwitterionic adduct with electrophiles, the MBH and Rauhut–Currier (RC) reaction are regularly mentioned as representative classical examples. The broad scope and general applicability of these two reactions for the construction of acyclic and carbo- and heterocyclic compounds have undoubtedly contributed to the significant attention that they have garnered from synthetic chemists. Recent progress in these two reactions has focused mainly on using elaborated alkenes to construct complex polycyclic structures in an enantioselective manner.12,13 In an elegant example, in 2019, Vicario and co-workers reported a catalytic enantioselective transannular MBH reaction to afford various fused bicyclic compounds containing stereogenic tertiary alcohol moieties in excellent yields and enantioselectivities (Scheme 2).13a In their proposed model 2–6 for stereoinduction, hydrogen bonding interactions between the NH functional groups of the bifunctional phosphine catalyst and the electrophilic ketone unit of the substrate were crucial to impart the high enantioselectivity. To further demonstrate its utility, they adopted this new method in the first asymmetric synthesis of the sesquiterpene natural product γ-gurjunene. Specifically, when the racemic ketone 2–7 was subjected to the conditions, kinetic resolution occurred to afford the desired bicyclic adduct 2–8 in 42% yield and 90% ee, paving the way for the total synthesis of γ-gurjunene. In another recent example of reactions between the zwitterionic adduct and electrophiles, Lin and co-workers reported the direct β-acylation of 4-arylidene pyrazolones and 5-arylidene thiazolones with acyl chlorides via phosphine catalysis.13b When an electrophile–nucleophile is reacted with an alkene, annulation usually occurs. For example, Guo and co-workers reported an efficient annulation between γ-sulfonamido-enones and sulfamate-derived cyclic imines to furnish multicyclic imidazolidine derivatives.14
Scheme 1. Nucleophilic Phosphine Catalysis of Alkenes.
Scheme 2. Asymmetric Transannular MBH Reaction.
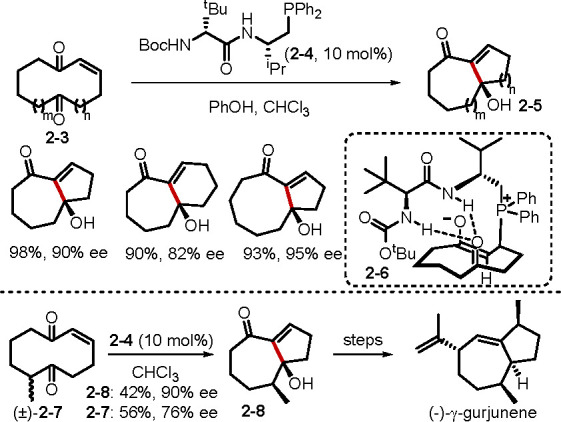
Tertiary phosphines add to electrophiles even in the presence of electron-deficient alkenes. A recent paper by Zhang and co-workers unveiled a phosphine-catalyzed vicinal difunctionalization of α,β-enones with TMSN3 (Scheme 3).15a In this unique transformation, the reaction was initiated by the addition of 1,4-bis(diphenylphosphino)butane (dppb) to TMSN3 to form a phosphazide zwitterion 2–12. Michael addition of this intermediate followed by intramolecular cyclization generated 2–13, whose decomposition formed the β-amino α-diazo carbonyl compounds 2–11. To slow down the decomposition of the phosphazide zwitterionic intermediate 2–12 to the iminophosphorane, the bulky TMS group was intentionally selected. A bifunctional chiral phosphine catalyst 2–14 promoted the asymmetric version of this novel transformation, affording moderate enantioselectivities. In a related study, the same group reported a phosphine-catalyzed [3 + 2] cycloaddition of α-diazoacetates and β-trifluoromethyl enones to access 4-(trifluoromethyl)pyrazolines.15b
Scheme 3. Phosphine-Catalyzed Difunctionalization of α,β-Enones.
2.2. Nucleophilic Phosphine Catalysis of Allenes
Phosphine catalysis of allenes is the most active area of nucleophilic catalysis, due to the diverse modes of reactivity displayed by allenes.3b In particular, highly functionalized allenes that deviate from the generic versions used in classic reactions often result in unforeseen reactivity patterns. Novel reactions continue to be invented, often accompanied by their asymmetric variants mediated by chiral phosphines, reflective of the potpourri of quality chiral phosphines made available in the past decade, specifically for nucleophilic organocatalysis. The phosphine catalysis of allenes reported in the past two years is presented here in three sections, categorized based on the nature of the second starting material: nucleophile, electrophile, or electrophile–nucleophile.
2.2.1. Nucleophilic Phosphine Catalysis of Allenes with Nucleophiles
Reactions of Allenes with Nucleophiles
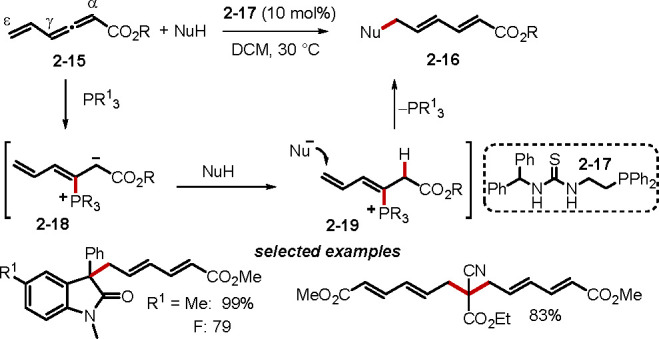
Umpolung addition is the most prevalent mode of reaction between an electron-deficient allene and a nucleophile in the presence of a tertiary phosphine catalyst. Among the umpolung additions of allenes, β′- and γ-umpolung additions are the most commonly observed reaction modes (Scheme 4).3b For allenoates lacking an alkyl substituent at the α-position, γ-umpolung addition may occur; α-alkyl-substituted allenoates undergo β′-umpolung addition. Less common is the Michael addition onto the β-carbon of allenoates, as has been demonstrated with α-fluoro-β-ketoamide nucleophiles.16 Enantioselective γ-umpolung additions of allenes with nitrogen-centered nucleophiles have continued to be studied.17 The Huang group reported a novel phosphine-catalyzed ε-umpolung addition of vinyl allenoates that affords diene carboxylates (Scheme 5).18 Depending on the number of hydrogen atoms on the pronucleophile, either mono or double remote 1,7-addition products can be obtained. Mechanistically, the ε-umpolung addition can be viewed as a vinylogous γ-umpolung addition triggered by the γ-vinyl substituent on the allenoate. Nucleophilic addition of a phosphine catalyst to the γ-vinyl allenoate 2–15 would form the zwitterion 2–18. Subsequent deprotonation of the pronucleophile and nucleophilic addition at the remote ε-carbon atom, followed by proton transfer and β-elimination of the catalyst, would afford the product 2–16. Among the series of phosphines tested, the bifunctional phosphine 2–17 was the best agent. Huang et al. reasoned that the reaction rate was accelerated as a result of additional interactions between the multifunctional phosphine and the substrates.
Scheme 4. β′- and γ-Umpolung Nucleophilic Addition.

Scheme 5. ε-Umpolung Nucleophilic 1,7-Addition of Vinyl Allenoates.
Reactions of Allenes with Dinucleophiles and Trinucleophiles
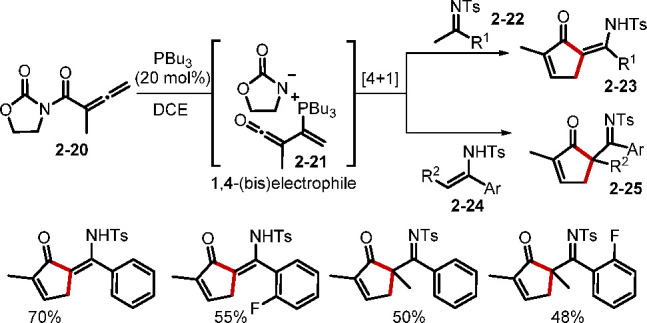
While simple nucleophiles are known to promote umpolung addition of allenes, a distinctive [m + n] mode of annulation can occur when dinucleophiles or trinucleophiles are introduced. Furthermore, when combined with multifunctionalized allenes, these nucleophiles can result in several unique modes of annulation. For example, phosphine-catalyzed [3 + 2] annulations of the dinucleophile 2-sulfonamidomalonate with δ-acetoxy allenoates have allowed access to pyrrolines;19a when vinyl allenoate was reacted with trinucleophiles, complex tetracyclic structures were constructed with the formation of two new rings.19b Along with those novel annulation processes, new patterns of [4 + 1] annulation to form 5-membered carbo- or heterocycles have been discovered.10b,20 In an elegant example, Huang and co-workers reported a novel phosphine-catalyzed [4 + 1] cyclization of allenyl imides with methyl ketimines and enamines (Scheme 6).20a This method provided efficient access to cyclopentenoyl enamines and imines. The α-phosphonium vinyl methyl ketene 2–21 was proposed as the key 1,4-(bis)electrophile intermediate, which reacted with the nucleophiles 2–22 and 2–24 to afford the cyclopentenones. Notably, changing the traditional allenoates to allenyl imides resulted in unforeseen reactivity patterns.
Scheme 6. [4 + 1] Cycloadditions of Allenyl Imides with Methyl Ketimines and Enamines.
2.2.2. Nucleophilic Phosphine Catalysis of Allenes with Electrophiles
Unlike the addition reactions of nucleophiles with allenes, the majority of reactions of allenes with electrophiles forge two new bonds to afford cyclic scaffolds.3b A diverse range of electrophiles (e.g., alkenes, imines, ketones/aldehydes, aziridines, and azomethine imines) can be employed, rendering phosphine-catalyzed reaction of allenes with electrophiles a powerful tool in organic synthesis.
Lu’s Allene–Alkene/Imine [3 + 2] Annulation
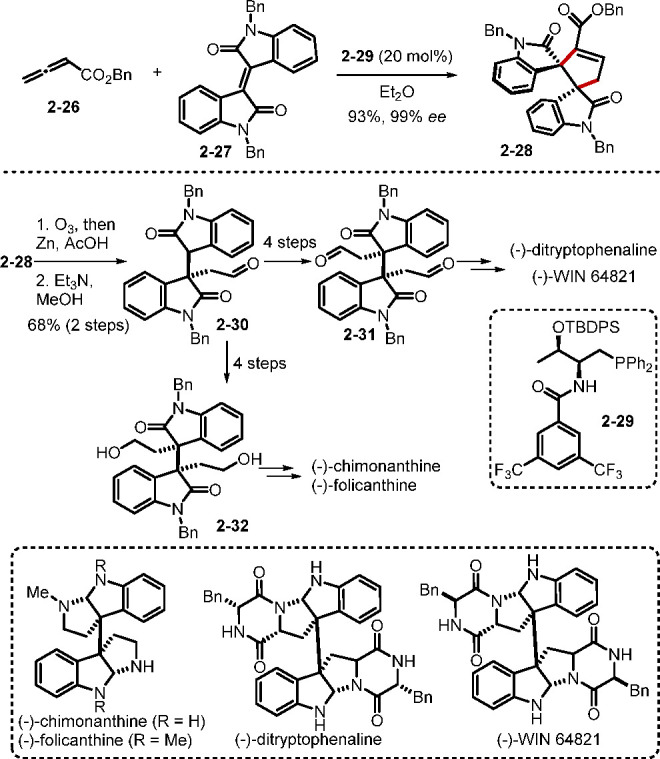
While annulations of allenes with ketones, aziridines, azomethine imines, troponones, and other reaction partners have continued to be explored in this area,21 Lu’s [3 + 2] annulation22 has been the most thoroughly studied and adopted for a variety of synthetic applications.23 Researchers in the field have continued to find new ways to reinvent Lu’s allene–alkene/imine [3 + 2] annulation. Whereas engagement of previously unreported alkenes remains a dominant activity,24−27 significant attention has also been paid to the preparation of enantioenriched cyclopentenes and pyrrolidines.26,27 In 2019, Lu and co-workers reported a phosphine-catalyzed enantioselective [3 + 2] annulation reaction between allenes and isoindigos, resulting in spirooxindoles having two vicinal quaternary stereogenic centers (Scheme 7).26a The threonine-derived bifunctional phosphine 2–29 catalyzed the [3 + 2] annulation to afford dimeric spirocyclic bisindoline motifs in high yields and excellent enantiomeric excesses. Notably, the product 2–28 was transformed into the bisoxindole 2–30, which acted as a common intermediate for the bisoxindoles 2–31 [a known precursor of the natural products (−)-ditryptophenaline and (−)-WIN 64821] and 2–32 [a known precursor of (−)-chimonanthine and (−)-folicanthine].
Scheme 7. [3 + 2] Annulation of Isoindigos with Allenes.
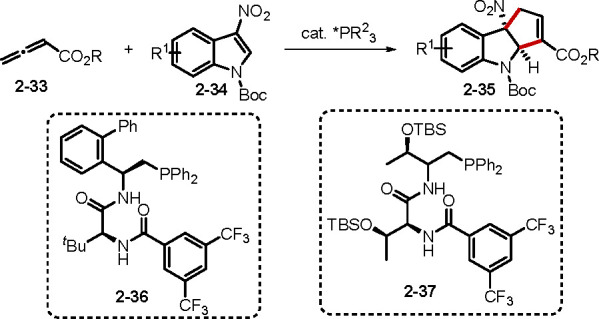
Several examples of allene–nitroolefin [3 + 2] annulations have been reported.24b,25,26 In particular, the engagement of 3-nitroindoles, 3-nitrobenzofurans, and 3-nitrobenzothiophenes resulting in the dearomative functionalization of electron-deficient aromatic rings demonstrates that phosphine organocatalysis is potentially a platform that can be complementary to metal-catalyzed dearomative reactions (Scheme 8).25,26 Two elegant examples have appeared from the Zhang26b and Lu26c groups, who demonstrated the asymmetric dearomative functionalization of electron-deficient allenoates 2–33 with 3-nitroindoles 2–34 catalyzed by the bifunctional phosphines 2–36 and 2–37, respectively. Compared with the preparation of enantioenriched cyclopentenes, examples of enantioselective allene–imine [3 + 2] annulation are rare. To overcome this deficiency, Kwon and co-workers prepared the novel P-stereogenic [2.2.1] bicyclic chiral phosphine CarvoPhos from the natural product carvone27a and applied it to the asymmetric syntheses of a bevy of pyrrolines, including the antiarrhythmogenic small molecule efsevin.
Scheme 8. Dearomative Functionalization through Phosphine Catalysis.
Allene–Electrophile [4 + 2] Annulation
Along with the allene–alkene/imine [3 + 2] cyclization, Kwon’s α-alkylallenoate–imine/alkene [4 + 2] annulation28 is another classic example of phosphine-catalyzed synthesis of cyclic compounds.29 The main development on this reaction since our comprehensive review in 20183b has been in the enantioselective syntheses of tetrahydropyridines and cyclohexenes.30 While Kwon and co-workers reported an enantioselective synthesis of guvacine derivatives through the allene–imine [4 + 2] annulation of imines with α-methylallenoates mediated by [2.2.1] bicyclic exo-(p-anisyl)-HypPhos,30a Lu and co-workers disclosed a chiral bifunctional phosphine-catalyzed asymmetric [4 + 2] reaction to access functionalized tetrahydropyridines.30c 3-Nitroindoles and 3-nitrobenzothiophenes are suitable reaction partners in the α-alkylallenoate–alkene [4 + 2] annulation, as reported by Guo and co-workers for the (S)-SITCP-catalyzed synthesis of dihydrocarbazoles.30d
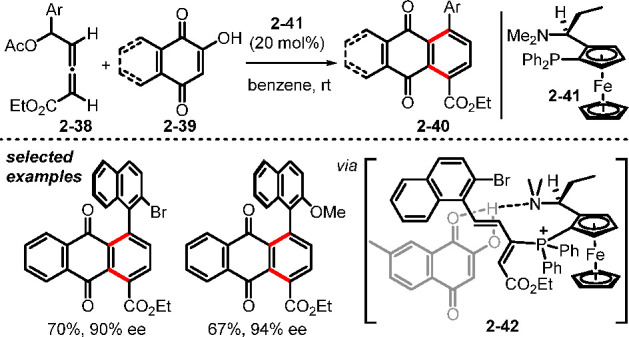
In the context of allene–electrophile [4 + 2] annulation, δ-acetoxy allenoates have been good precursors for the synthesis of benzene rings.31 In a striking example, Tong and co-workers found that the phosphonium diene intermediate formed by addition of the phosphine to δ-acetoxyallenoates could undergo [4 + 2] annulation with 2-hydroxyquinone derivatives to form naphthoquinones 2–40 (Scheme 9).31a The efficient and stereoselective synthesis of axially chiral naphthaquinone atropisomers was facilitated by the cooperative effects of the tertiary phosphine and tertiary amine in the bifunctional chiral phosphine 2–41, as displayed in the intermediate 2–42. In another example, Swamy and co-workers demonstrated that the PPh3-catalyzed reactions between δ-acetoxy allenoates and cyclic N-sulfonyl imines could also generate functionalized benzenes.31c
Scheme 9. Synthesis of Aryl-Naphthaquinone Atropisomers.
Allene–Electrophile Multicycle Annulation
When allenes and/or electrophiles having additional functionalities (e.g., conjugated C=C bonds, a leaving group, or a pronucleophile) are employed, never-before-seen reactions, especially multicycle annulations, may ensue to give complex polycyclic structures. For example, when γ-benzylallenoates were treated with (E)-2-(3-arylallylidene)malononitriles in the presence of a phosphine catalyst, multicycle annulation occurred to give bicyclic[3,3,0]octene derivatives.24a In another example, a C2-symmetric chiral phosphine, NUSIOC-Phos, promoted the reactions between pyrrolidine-2,3-diones and γ-substituted allenoates, resulting in a domino process to form γ-lactams containing five contiguous stereogenic centers.32a Furthermore, Huang and co-workers reported a sequential annulation of δ-sulfonamidoallenoates 2–43 and dienes 2–44 to access highly functionalized hydroisoquinolines 2–45 (Scheme 10).32b The δ-sulfonamidoallenoates acted as novel five-atom units in the sequential annulations with the involvement of an intermediate 2–46 with three nucleophilic sites.
Scheme 10. Domino Annulation of δ-Sulfonamidoallenoates to Access Hydroisoquinoline Derivatives.

δ-Umpolung Addition
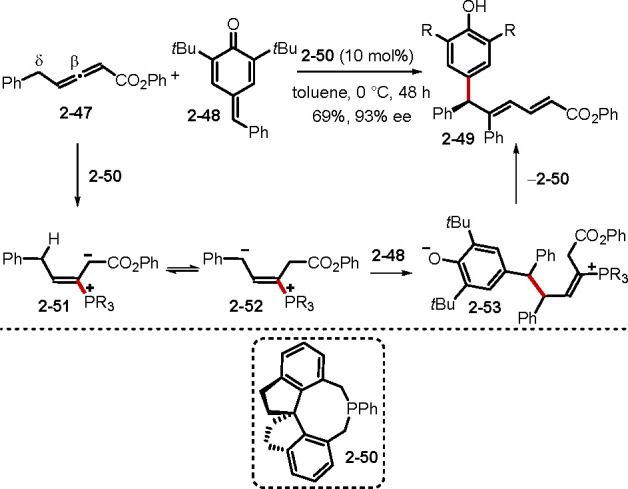
Given that most reactions between allenes and electrophiles have been cyclizations, Xu’s report of a catalytic asymmetric dienylation of para-quinone methides (p-QM) 2–48 with γ-benzylallenoate 2–47 through δ-umpolung addition is noteworthy (Scheme 11).33 Various optically pure diarylmethanes 2–49 were obtained in moderate to good yields through this method. They proposed that the phosphonium dienolate intermediate 2–51 underwent proton transfer to afford the vinylogous ylide 2–52. Subsequent 1,6-conjugate addition to the p-QM 2–48, followed by a series of proton transfers and β-elimination of the catalyst 2–50, afforded the product 2–49.
Scheme 11. δ-Umpolung Addition.
2.2.3. Nucleophilic Phosphine Catalysis of Allenes with Electrophile–Nucleophiles
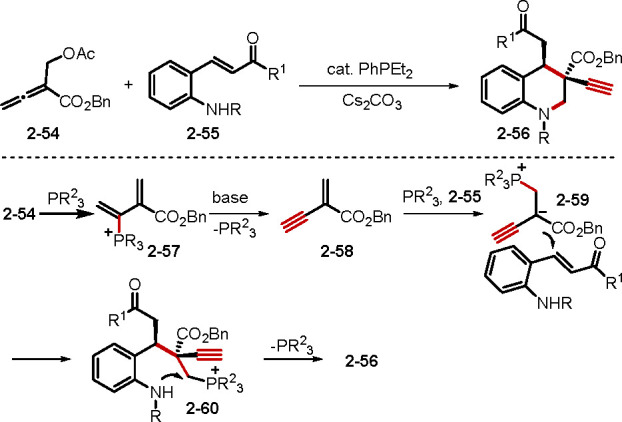
When allenes react with a molecule containing both electrophilic and nucleophilic groups in the presence of catalytic phosphine, annulations typically occur. Representative examples of nucleophile–electrophiles include salicylaldehyde and salicylimine. Recently, Zhou and co-workers introduced an additional pronucleophile, malonate, into the salicylimine structure to give the dinucleophile–electrophile 2-[(2-hydroxybenzylidene)amino] malonate.34a,34b When this dinucleophile–electrophile was treated with benzyl β′-acetoxyallenoate, multicycle annulation occurred to give polycyclic products.34a,34b Mohanan and co-workers found that the annulation of nucleophile–electrophile phenacylmalononitriles and allenoates catalyzed by PBu3 produced functionalized cyclopentenes.34c Another phosphine-catalyzed reaction of nucleophile–electrophile 2-tosylaminochalcones and β′-acetoxyallenoates, reported by the Huang lab, is also notable (Scheme 12).35a The β′-acetoxyallenoate 2–54 was transformed into the enynoate intermediate 2–58 through a substitution/elimination sequence. Nucleophilic addition of the phosphine to 2–58 resulted in the zwitterion 2–59, which underwent 1,4-addition with 2–55 and subsequent intramolecular SN2 displacement to form the tetrahydroquinoline 2–56. The Huang group extended this method to the synthesis of functionalized chromans.35b This report was the first to describe addition across the α- and β′-carbon atoms of α-substituted allenoates.
Scheme 12. Synthesis of 3-Ethynyl-Substituted Tetrahydroquinolines.
2.3. Nucleophilic Phosphine Catalysis of Alkynes
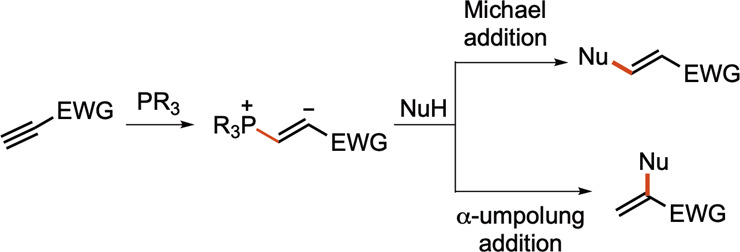
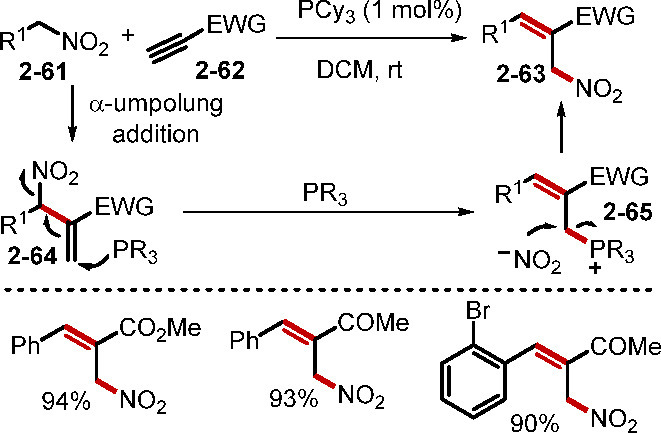
A variety of pronucleophiles, electrophiles, and nucleophile–electrophiles can be involved in the phosphine catalysis of alkynes.3b Although phosphine catalysis with alkynes has received less attention than that with allenes, reports of known reactions with previously unexplored substrates, as well as discoveries of novel reactions, have continued to appear. Among these transformations, Michael addition and α-umpolung addition have been the two major reaction pathways when nucleophiles are present (Scheme 13). In one recent example, Salin and co-workers reported a bishydrophosphorylation of electron-deficient alkynes through nucleophilic phosphine catalysis.36a In another example, Zhang and co-workers demonstrated a chiral bifunctional phosphine-promoted double Michael addition of a tryptamine-derived oxindole dinucleophile onto ynones.36b Contemporarily, the Wang group reported the efficient synthesis of (E)-2-nitromethylcinnamates through phosphine catalysis of alkynes (Scheme 14).37a In this transformation, initial phosphine-catalyzed α-umpolung addition of the alkyne 2–62 with the nitromethane 2–61 formed the 1,2-disubstituted alkene intermediate 2–64. Subsequent phosphine-promoted 1,3-rearrangement of the nitro group generated the more stable trisubstituted alkene 2–63.
Scheme 13. Michael Addition and α-Umpolung Addition in the Phosphine Catalysis of Alkynes.
Scheme 14. Synthesis of (E)-2-Nitromethylcinnamates through Phosphine Catalysis of Alkynes.
Anticarboboration, silaboration, and diboration of alkynoates can be realized using phosphine catalysis.38 A recent example by Santos and co-workers involved PBu3 catalyzing the trans-phosphinoboration of internal alkynes 2–66 (Scheme 15).37b The reaction afforded trans-α-phosphino-β-boryl acrylates 2–67 in moderate to good yields with high regio- and Z-selectivity, providing access to functionalized phosphines that would be challenging to prepare otherwise.
Scheme 15. Trans-Phosphinoboration of Internal Alkynes.
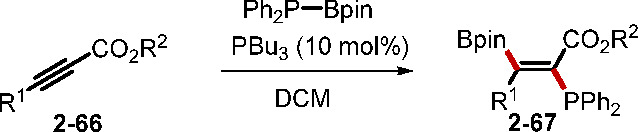
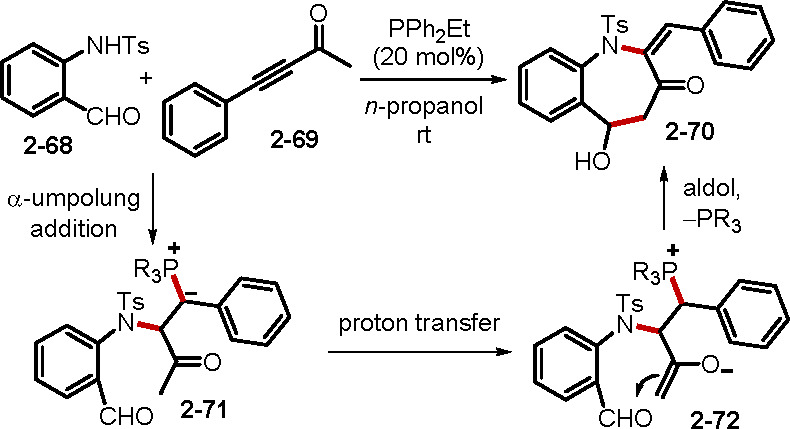
In the presence of nucleophile–electrophiles, alkynes undergo annulations to form a diverse array of carbo- and heterocycles.39 For example, the Kwon group reported the novel phosphine-catalyzed annulation of 2-sulfonamidobenzaldehyes 2–68 and ynones 2–69 (Scheme 16).39b The reaction was initiated through α-umpolung addition of the alkyne 2–68 to form the ylide 2–71. Ensuing proton transfer, followed by an aldol reaction, generated the annulation product benzo[b]azepin-3-one 2–70.
Scheme 16. Synthesis of Benzo[b]azapin-3-ones.
2.4. Nucleophilic Phosphine Catalysis of MBHADs
Together with alkenes, allenes, and alkynes, the MBHADs are also excellent substrates for nucleophilic phosphine catalysis. The MBHADs 2–73 are typically activated by a tertiary phosphine through SN2′ addition to give an active phosphonium intermediate 2–74, which undergoes various reactions when combined with nucleophiles, electrophiles, and nucleophile–electrophiles (Scheme 17). When a nucleophile is present, phosphine catalysis of the MBHAD will involve a unique SN2′–SN2′ process leading to apparent SN2 allylation. As is the case with allenoates, MBHADs can provide three-carbon units in annulation reactions when treated with electrophiles. The known MBHAD–alkene or MBHAD–azo [3 + 2] annulations continue to be reported for asymmetric versions with more elaborate substrates.40 MBHADs undergo the [4 + 1] annulation when mixed with enone/enimine/diene electrophiles in the presence of a phosphine catalyst. Several recent reports have demonstrated the incorporation of various electrophiles, including 2-enoylpyridine (oxide)s, ortho-quinone methides, and α,β-unsaturated imines.41
Scheme 17. Allylic Substitution and Annulations with MBHADs.

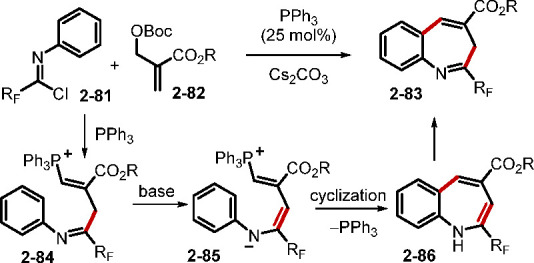
Alongside these developments, several new reactions have been discovered using more elaborate substrates.42 In one such example, the Huang group reported the domino [3 + 3] cyclization of MBH carbonates 2–75 with nucleophile–electrophile para-quinamines 2–76 to access hydroquinolines 2–77 (Scheme 18).42a The reaction was initiated by the SN2′–SN2′ process to afford an allylic amination intermediate 2–78. Subsequent addition with the phosphine, followed by proton transfer, formed the ylide 2–80. Intramolecular Michael addition and elimination of the phosphine generated the hydroquinoline product 2–77. In another example, the same group disclosed a novel phosphine-catalyzed annulation of an MBHAD with perfluoroalkylimidoyl chloride as the electrophile to construct the perfluoroalkylated benzazepines 2–83 (Scheme 19).42b Here, the phosphonium species generated from the MBH carbonate and the phosphine was deprotonated by base; it then added to the fluorinated imidoyl chloride to give the intermediate 2–84. Cyclization of the deprotonated 2–84 and subsequent enamine-to-imine tautomerization afforded the perfluoroalkyl-substituted benzazepines. Notably, the perfluoroalkylimidoyl chlorides provided four atoms of the benzazepine products.
Scheme 18. Synthesis of Hydroquinolines.

Scheme 19. Synthesis of Perfluoroalkylated Benzazepines.
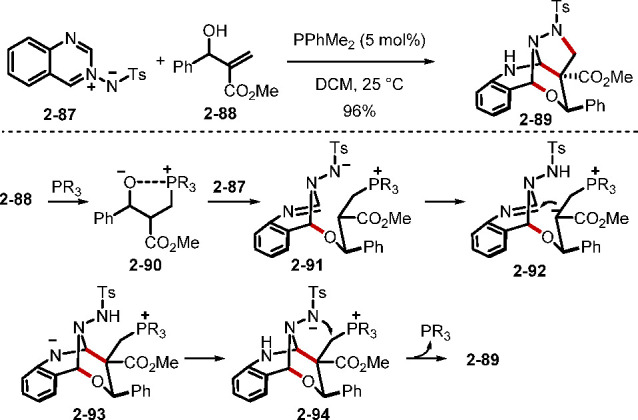
Conventionally, MBH alcohols have been converted to their derivatives, including acetates, carbonates, and halides. A recent report by Guo and co-workers revealed that phosphine catalysis enables the direct reaction of MBH alcohols with azomethine imines to access bicyclic (epoxymethano)-pyrazolo[5,1-b]-quinazolines (Scheme 20).42c They proposed that the MBH alcohol 2–88 was activated through reaction with the phosphine to form the zwitterionic intermediate 2–90. The alkoxide of 2–90 underwent sequential nucleophilic addition to the electron-deficient azomethine imine 2–87, intramolecular Mannich-type reaction, and SN2 displacement of the catalyst to form the bridged tetracycle 2–89.
Scheme 20. Phosphine-Catalyzed Reactions of Unmodified MBH Alcohols.
2.5. Miscellaneous Nucleophilic Phosphine Catalysis
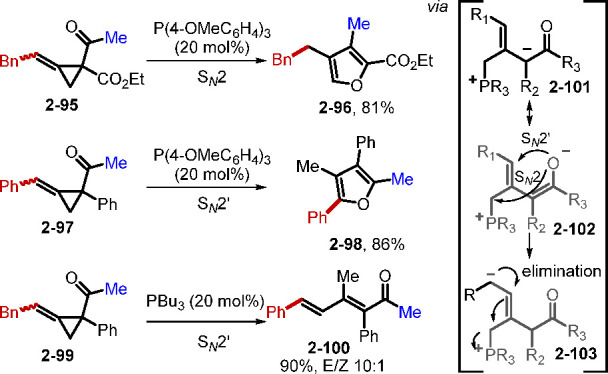
In addition to activated C—C multiple bonds, tertiary phosphine catalysts can also add to C=O and N=N groups and strained-ring electrophiles, resulting in a diverse array of reactions. Several recent examples are noteworthy. For example, the Behrouz and Rad group reported an ultrasound-promoted PPh3-catalyzed three-component condensation of benzil, urea, and aldehyde for the synthesis of 2,4,5-trisubstituted imidazoles.43 Recent progress in phosphine catalysis has unveiled several unique rearrangements of strained cyclopropanes.44 The Xu group demonstrated that phosphine-catalyzed rearrangement of vinylcyclopropylketones can provide access to cycloheptenones.44a Subsequently, the Li and Xu group reported the synthesis of tri- and tetrasubstituted furans, and trisubstituted dienones, through phosphine-catalyzed rearrangements of alkylidenecyclopropylketones (ACPs, Scheme 21).44b The development of this reaction was inspired by a known palladium-catalyzed rearrangement of ACPs.45 In this new study, three types of substrate-controlled rearrangement of alkylidenecyclopropanes occurred to afford three different product types. The results could be explained by considering the allylic phosphonium zwitterions intermediates 2–101, 2–102, and 2–103 that formed upon nucleophilic addition of the phosphine to the ACPs. When R1 was an alkyl group, and R2 was an electron-withdrawing group, an SN2 process gave trisubstituted furan 2–96; when both R1 and R2 were aryl groups, an SN2′ pathway formed tetrasubstituted furan 2–98; when R1 was a primary alkyl group (RCH2) and R2 was an aryl substituent, the intermediate 2–103 underwent elimination and the alkene isomerization to afford the trisubstituted dienone 2–100.
Scheme 21. Phosphine-Catalyzed Rearrangements of Alkylidenecyclopropylketones.
2.6. Summary
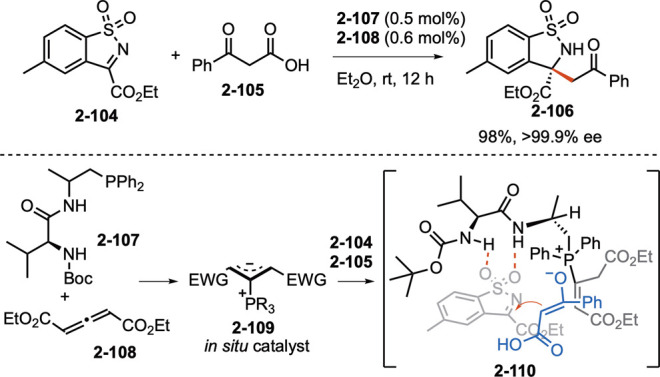
Nucleophilic phosphine catalysis has remained one of the most vigorous areas of research in the field of phosphorus-based organocatalysis. It continues to expand its horizons to the assembly of carbo- and heterocycles through new modes of reaction. Recent progress in this area has shared some common features. Unforeseen reactivity patterns continue to be discovered when highly functionalized substrates, deviating from generic versions, are used in classical reactions (e.g., Schemes 5 and 6). Enantioselective variants of the classical nucleophilic phosphine catalysis processes continue to be reported (e.g., Schemes 7 and 8). Brand new reactions continue to be uncovered, often accompanied by asymmetric variants when using chiral phosphines (e.g., Schemes 9 and 10). The application of nucleophilic phosphine catalysis is a promising approach for the syntheses of functional molecules (e.g., Schemes 2 and 7). In addition, the zwitterionic adducts generated in situ from nucleophilic addition of a phosphine to electron-deficient multiple bonds can be efficient basic catalysts for asymmetric Mannich-type reactions, Strecker reactions, Michael additions, and aldol reactions.46 In a recent example, Wang and co-workers revealed that the zwitterion 2–109 generated from the multifunctional chiral phosphine 2–107 and the allenoate 2–108 was highly efficient in the asymmetric decarboxylative Mannich reactions between the cyclic ketimines 2–104 and the β-keto acids 2–105 (Scheme 22).47 Notably, the reactions could be performed to give high yields and excellent enantioselectivities (from 99.0% to >99.9% ee) at a very low catalyst loading (0.5 mol %). Considering the ready availability of chiral phosphines and electron-deficient alkenes, allenes, and alkynes, this strategy may open a new door for phosphonium-salt catalysis in asymmetric synthesis. Lastly, with its diverse reaction modes, we envision that nucleophilic phosphine catalysis will have many more applications in the syntheses of functional molecules—including biologically active natural products, pharmaceuticals, agrochemicals, and materials.
Scheme 22. Phosphonium Zwitterion-Catalyzed Decarboxylative Mannich-Type Reaction.
3. Phosphine Oxide Catalysis
Whereas the phosphorus reagents toggle between the tertiary phosphine(III) state and quaternary phosphonium(V) species in nucleophilic phosphine catalysis, classical phosphorus(III)-based processes (e.g., Wittig, Mitsunobu, Appel, and Cadogan cyclization reactions) are driven by the formation of strong P(V)=O double bonds. The formation of a phosphine oxide as waste is, however, an issue that has troubled chemists for a long time. Although the issue can be partly solved through immobilization and precipitation of the phosphine oxide,48 catalytic cycling of the phosphorus species would be more atom-economical and environmentally friendly.49 Two strategies, namely, redox-neutral and redox-driven phosphine oxide catalysis, are commonly adopted to recycle phosphorus species in such reactions (Scheme 23).49,50 The redox-neutral catalysis requires in situ activation of the phosphine oxide to form an active P(V) intermediate, which will further react with substrates and regenerate phosphine(V) oxide. In the redox-driven catalysis, P(III) usually reacts with substrate 3–1 to form an active P(V) intermediate, which will further react with the substrate 3–2 to give the product and the phosphine oxide. To turn over the phosphine oxide to P(III), stoichiometric reductant is needed. The harsh conditions required for the activation or reduction of the inert P(V)=O double bond, however, have largely plagued the development of catalytic processes. In the past decade, significant advances have been made in this field.49 Examples include the development of catalytic Wittig-type reactions using arsine or telluride ylides,51 gas evolution (N2, CO2) to assist the redox-neutral catalytic process,49,50,52 and the use of silanes as reductants in the redox-driven catalytic cycle.49,53 Several new advances have emerged since our review in 2018,3b and we highlight them below.
Scheme 23. Redox-Neutral and Redox-Driven Phosphine Oxide Catalysis.

3.1. Redox-Neutral Phosphine Oxide Catalysis
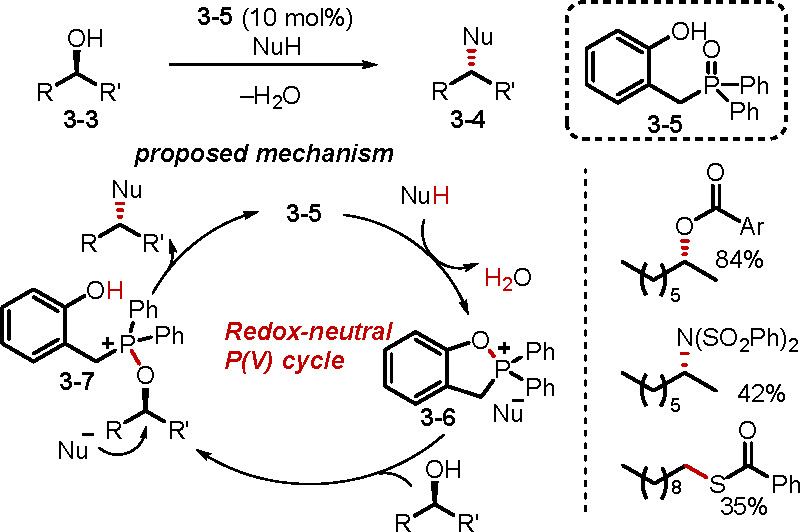
In redox-neutral phosphine oxide catalysis, the phosphine oxide is converted directly into another reactive P(V) species. For example, treatment of a phosphine oxide with an isocyanate or oxalyl chloride produces iminophosphorane52 or chlorophosphonium chloride50 intermediates that have been used in the aza-Wittig or Appel reaction and in the dehydration of oximes to nitriles. The use of this mode of phosphine oxide catalysis, although known since 1962, has lagged behind the thriving activities of redox-derived catalysis.52a Recently, however, in what could be considered one of the most exciting innovations of late, Denton and co-workers developed a redox-neutral organocatalytic Mitsunobu reaction (Scheme 24).54 The Mitsunobu reaction is commonly used for the nucleophilic substitution of primary and secondary alcohols with inversion of their chiral centers in a stereospecific manner. Despite its usefulness, the requirement of stoichiometric amounts of reagents (PPh3, azo oxidant) and the generation of undesired waste have undermined this powerful transformation. Although several attempts have been made to develop a redox-driven organocatalyzed Mitsunobu reaction, a stoichiometric amount of reductant has been required to recycle the phosphine oxide, and a stoichiometric amount of oxidant has been required to regenerate the azo oxidant in cases where a substoichiometric amount of the azo species is used.49f In Denton’s study, a phosphine oxide catalyst 3–5 was employed without any other additives. Notably, the previously required azo oxidant is not necessary here, because the active dihydrobenzooxaphospholium intermediate 3–6 forms with the evolution of benign water as the sole byproduct. Subsequent substitution by the alcohol 3–3 generates the alkoxyphosphonium intermediate 3–7, which then undergoes another substitution with the nucleophile anion to afford the product 3–4, while simultaneously regenerating the catalyst 3–5. By maintaining a constant oxidation state of the phosphorus species, this approach eliminates any need for stoichiometric oxidants/reductants and produces water as the sole byproduct. This process is both scalable and compatible with a variety of pronucleophiles, forming C–O, C–N, and C–S bonds. Furthermore, protocols have been developed to enable one-pot activation of simple alcohols (en route to the synthesis of API and symmetrical ethers) when using p-toluenesulfonic acid or trifluoromethanesulfonic acid as a cocatalyst.
Scheme 24. Catalytic Mitsunobu Reaction.
3.2. Redox-Driven Phosphine Oxide Catalysis
Undoubtedly, O’Brien’s 2009 report on a catalytic Wittig reaction, in which a silane reduces the phosphine oxide byproduct back to phosphine in situ during the reaction, unleashed the imaginations of researchers around the world.53 Consequently, the subsequent decade witnessed numerous accounts of catalytic versions of classical P(V)=O-generating reactions, including Wittig olefination, Appel reaction, Staudinger reduction, Staudinger ligation, aza-Wittig reaction, Mitsunobu reaction, and Cadogan cyclization, with all employing stoichiometric silanes as terminal reductants.3b,49 While studies of redox-driven phosphine oxide catalysis of the aforementioned reactions have continued,55−59 the past two years have been marked by the application of redox-driven phosphine oxide catalysis to lesser-known reactions.
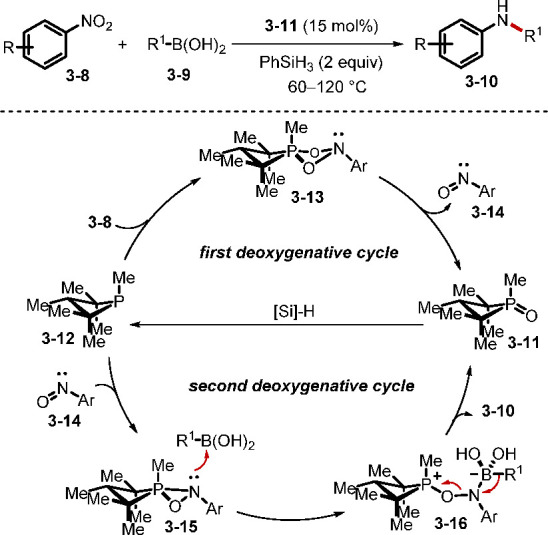
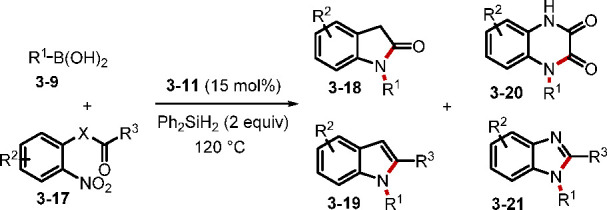
One of the reactions that is mediated by a phosphine, but not widely known, is the cross-coupling between nitrosoarenes and boronic acids for the synthesis of di(hetero)arylamines, reported by Csákÿ.60 In 2018, the Radosevich group disclosed a deoxygenative C–N crosscoupling between nitroarenes and boronic acids (Scheme 25).61a,61b While transition-metal-mediated C–N couplings are used widely, the P(V)/P(III)-catalyzed reaction is a complementary main group redox-catalysis method. In particular, this transformation tolerates various functional groups, including a series of heterocycles and halogen substituents. Radosevich et al. proposed a two-stage deoxygenation sequence for the reductive C–N coupling. Initially, [3 + 1] cheletropic addition of the nitrobenzene 3–8 and the phosphetane 3–12 generated the intermediate 3–13, decomposition of which furnished nitrosoarene and the oxide 3–11. The second deoxygenation began with addition of the phosphetane to the nitrosoarene to form the betaine intermediate 3–16, which underwent 1,2-migration to give the desired product with concomitant generation of the phosphetane oxide 3–11. The Radosevich group recently demonstrated that nitromethane is amenable to the reaction, realizing methylamination of arylboronic acids.61c When the nitroarenes have different ortho substituents, sequential reductive coupling and cyclization could occur efficiently to yield various heterocycles (Scheme 26).62 A tandem coupling/acylation would generate oxindoles 3–18 and quinoxalinediones 3–19, while a tandem coupling/condensation would form indoles 3–20 and benzimidazoles 3–21.
Scheme 25. Reductive C–N Cross-Coupling between Nitroarenes and Boronic Acids.
Scheme 26. Synthesis of Azaheterocycles through Reductive Coupling between Nitroarenes and Boronic Acids.
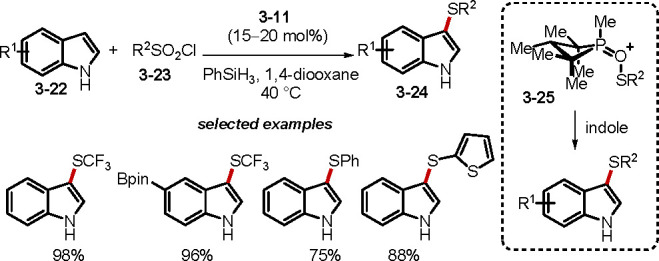
Compared with the oxidation of S(II) species into valuable sulfoxides and sulfones, the deoxygenative transformation of sulfur-containing compounds is less recognized as synthetically useful. The Sharpless group had previously demonstrated that trialkylphosphite could reduce sulfonyl chlorides to corresponding sulfinyl chlorides and, subsequently, to sulfenyl chlorides, and that the sulfinyl chloride could be captured by nucleophiles (e.g., alcohols) to form sulfinate esters.63a The final step in their proposed mechanism for the sequential deoxygenation of sulfonyl chloride was the reaction of the sulfenyl chloride with trialkylphosphite to form trialkoxyl(arylthiol)phosphonium chloride, which underwent a Michaelis–Arbzov reaction to produce the corresponding phosphorothiolate and alkyl chloride.63b Recently, Radosevich and co-workers reported that the phosphetane oxide 3–11 was capable of catalyzing electrophilic sulfenylations through deoxygenation of sulfonyl chlorides with indoles as the nucleophile (Scheme 27).64 The reaction is suitable for a diverse range of indoles and sulfonyl chlorides, providing access to indole derivatives. For this 2-fold deoxygenative sulfenylation, 3–25 was proposed as the key intermediate responsible for the electrophilic substitution. Again, the P(V)/P(III) cycle was driven by phenylsilane-mediated reduction of P(V) oxide to P(III).
Scheme 27. Electrophilic Sulfenylation through Deoxygenation of Sulfonyl Chlorides.
Halophosphonium cations have proven to be effective for deoxygenation in both the Appel reaction and amide formation.65 This behavior inspired the development of phosphetane oxide 3–11-catalyzed didehydration for the preparation of functionalized heterocyclic adducts (Scheme 28).66 The annulation was amenable to both aniline and benzylamine for the construction of six- and seven-membered rings. The key to the success of this sequential annulation is the generation of the halophosphonium cation 3–29 from the in situ-generated phosphetane and the oxidant diethyl bromomalonate (DEBM). Activation of a carboxylic acid with the halophosphonium cation, followed by coupling, resulted in the first dehydration to form an amide intermediate. The amide bond was further activated by the halophosphonium species, with the ensuing SEAr cyclodehydration generating the desired heterocycles.
Scheme 28. Annulation of Amines and Carboxylic Acids through Iterative Dehydration.

Alkene reduction is one of the most fundamental reactions in organic synthesis. While transition metal catalysis dominates the field, organocatalysis approaches may complement the former by providing unique selectivity and functional group compatibility. Indeed, several successful examples of phosphine-mediated reduction of activated double and triple bonds have been reported.67 These reductions have involved addition of the phosphine to the double or triple bond to form the zwitterion 3–32 and a subsequent reaction with water to form the phosphorane 3–33, which collapsed to the product 3–31 and the phosphine oxide (Scheme 29). A stoichiometric quantity of the phosphine was, however, required, producing undesired phosphine oxide waste. Last year, the Werner group reported the first example of a phosphine oxide-catalyzed alkene reduction through silane-mediated P(III)/P(V)=O redox cycling.68 In this transformation, a combination of the strained phosphetane oxide 3–11 and phenylsilane was used to replace the previously employed stoichiometric phosphine. The reduction selectively targeted the activated double bonds without affecting other functional groups susceptible to hydrogenation. Remarkably, water was tolerated by the silane-promoted P(III)/P(V)=O redox cycling.
Scheme 29. Reduction of Activated Alkenes through P(III)/P(V)=O Redox Cycling Catalysis.

3.3. Asymmetric Phosphine Oxide Catalysis
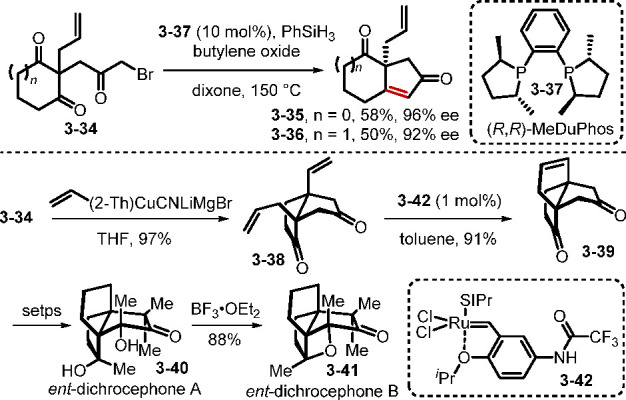
Although chiral phosphines have been recognized as reagents for asymmetric Wittig olefination and Staudinger–aza-Wittig reactions, explorations in this field have been limited by the noneconomical nature of those reactions.69 Today, significant advances in phosphine oxide catalysis have given an impetus for renewed interest in this area, such that enantioselective catalysis processes driven by the P(III)/P(V)=O redox cycling of chiral phosphine oxides are emerging as powerful tools for the synthesis of chiral molecules. In particular, desymmetrization of meso compounds is a valuable strategy for accessing molecules with high optical purity. In 2014, Werner et al. reported the first enantioselective catalytic Wittig reaction through desymmetrization of 2-(3-bromo-2-oxopropyl)-2-methylcyclopentane-1,3-dione with catalytic Me-DuPhos (3–37) and phenylsilane as the terminal reductant.70a They found that Me-DuPhos worked efficiently for asymmetric induction with up to 90% ee, but with less than 10% yield. Three years later, the Christmann group reinvestigated this reaction and found that the desymmetrizing Wittig reaction could provide high enantiomeric excess (92–96% ee) and moderate yields (Scheme 30).70b They found that premature debromination of the triketone, caused by impurities in the starting materials, might have been responsible for the low yield reported initially. Although high temperature (150 °C) was required for the catalytic intramolecular Wittig reaction, good enantioselectivities were obtained when using Me-DuPhos. The optically pure bicyclic enone products were further elaborated into various carbocyclic propellanes through sequential Michael addition with Lipshutz’s cyanocuprates and ring-closing metathesis.70b The propellane 3–39 was applied to the first asymmetric syntheses of ent-dichrocephone A and ent-dichrocephone B.70c
Scheme 30. Synthesis of ent-Dichrocephone A and ent-Dichrocephone B.
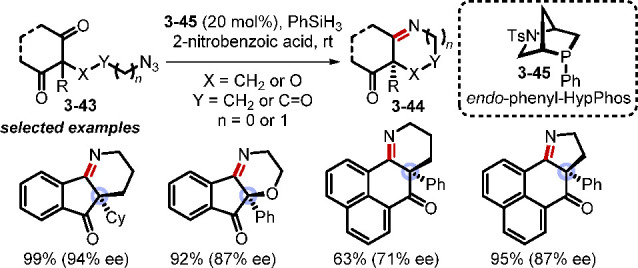
Although good selectivity was obtained in some cases for the asymmetric Wittig olefination at high temperature, mild room-temperature reactions would be more desirable. The inert nature of P(V)=O poses a challenge for its silane-mediated reduction to a trivalent phosphine at ambient temperature. Nevertheless, continued studies in this field have unveiled some factors for lowering the energy barrier of the silane-mediated phosphine oxide reduction, such as the use of the strained small-ring phosphetane oxide 3–11 as the catalyst. Interestingly, strained phosphine oxides can be reduced more readily than unstrained ones. A recent computational study explained why strained phosphetanes and bridged [2.2.1] bicyclic phosphines are privileged structures in redox-driven phosphine oxide catalysis.71 One reason is that one of the C—P—C angles of these phosphines is close to that in the proposed transition state for the intramolecular hydride delivery in the (phosphoniooxy)silcate(IV) adduct formed between the phosphine oxide and the silane. Studies have also demonstrated that both acidic and basic additives can facilitate the silane-mediated phosphine oxide reduction.72 Consolidating these lessons, in 2019, Kwon and co-workers developed a catalytic asymmetric Staudinger–aza-Wittig reaction for the synthesis of chiral cyclic imines 3–44 (Scheme 31).73 In this reaction, the commercially available P-chiral [2.2.1] bicyclic endo-phenyl-HypPhos 3–45 was used to facilitate desymmetrization of the two C=O groups of various 1,3-diketones and to form enantioenriched iminoketones containing a chiral quaternary center. The strained nature of the catalyst, in conjunction with the 2-nitrobenzoic acid additive, enabled room-temperature redox cycling mediated by phenylsilane.
Scheme 31. Catalytic Asymmetric Staudinger–aza-Wittig Reaction.
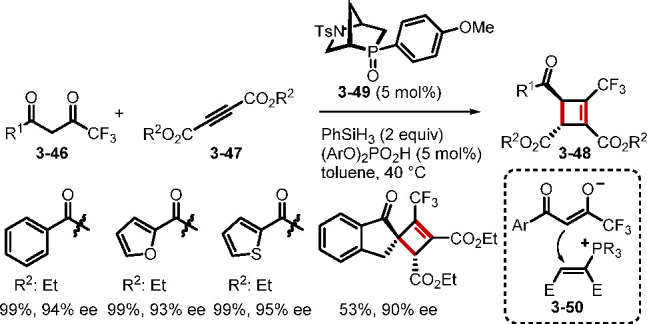
In another recent report, the Voituriez group documented an efficient method to access enantioenriched (trifluoromethyl)cyclobutenes 3–48 through an asymmetric α-umpolung addition–Wittig olefination cascade between the dione 3–46 and the acetylenedicarboxylate 3–47 (Scheme 32).74 The catalyst employed here, exo-anisyl-HypPhos oxide (3–49), is also a strained [2.2.1] bicyclic phosphine oxide, which, along with the phosphoric acid additive, facilitated mild P(III)/P(V)=O redox cycling. This tandem reaction represents another powerful example of phosphine oxide redox cycling as a tool for the synthesis of attractive chiral molecules.
Scheme 32. Enantioselective Synthesis of (Trifluoromethyl)cyclobutenes.
3.4. Summary
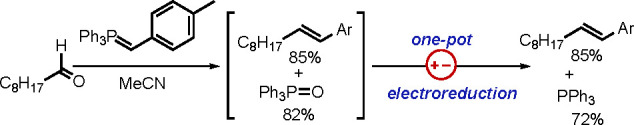
Although significant progress has been made, challenges remain in the area of phosphine oxide catalysis. Most redox-driven phosphine oxide catalysis has been based on silane-mediated in situ reduction. The need for a stoichiometric amount of reductant raises economic and environmental concerns.75 A recent study by Sevov and co-workers revealed that the triphenylphosphine oxide byproduct of the Wittig olefination can be transformed directly into PPh3 through electroreduction (Scheme 33).76 Revitalized adaptation of such unconventional technologies may open new doors for redox-driven phosphine oxide catalysis. Conversely, as demonstrated by Denton and co-workers in their redox-neutral organocatalytic Mitsunobu reaction, designing novel catalysts may also be a promising avenue for green chemistry. Although catalytic versions of those phosphine oxide-driven reactions are appealing, unless their selectivities and yields are comparable with those of the original versions, their utility will be limited. In this regard, there is a great need to develop mild methods for reduction or activation of phosphine oxides. Accordingly, asymmetric phosphine oxide catalysis is another appealing field benefiting from ongoing progress in phosphine oxide catalysis. Although much needs to be done, phosphine oxide catalysis finds itself at the threshold of a bright future.
Scheme 33. One-Pot Wittig Olefination/Electroreduction.
4. Phosphorus-Mediated Radical Processes
Organophosphorus compound-mediated SET processes have received much less attention than their nitrogen counterparts,77 despite trivalent phosphine radical cations and phosphoranyl radicals having been recognized as reaction intermediates as early as the 1950s.78 Progress in this emerging field has established that organophosphorus compounds can act as radical initiators, catalysts, and radical precursors in various SET transformations for the synthesis of important structural motifs.9,79 With the rapid development of photocatalysis in recent years, the use of organophosphorus compounds as radical precursors, especially for the purpose of deoxygenation or desulfurization, is becoming established as a reliable method.9 Several examples of organophosphorus compound-mediated radical processes are discussed below.
4.1. Use of Substoichiometric Phosphine
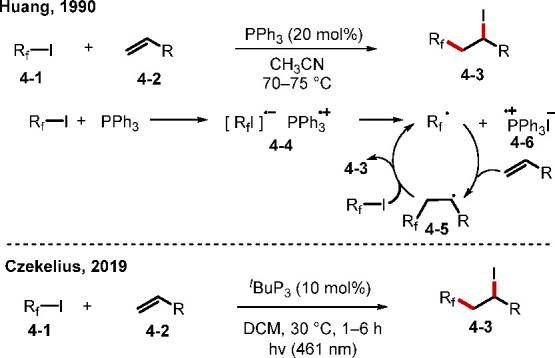
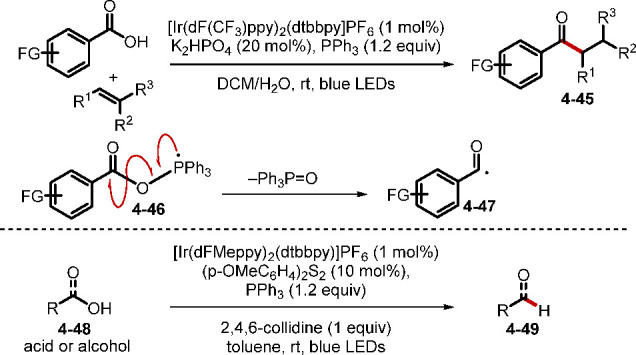
Electron donor–acceptor (EDA) complexes, also known as charge-transfer complexes (CTCs), are typically formed from electron-rich and electron-poor compounds and possess distinct properties differing from those of their parent compounds, enabling some unique transformations.80 In particular, even when the parent compounds cannot absorb visible light, their EDA complexes can, facilitating useful transformations.81 In step with recent attention paid to the study of EDA complexes, especially in the area of photoredox chemistry, the use of phosphorus compounds, which are good electron donors for forming EDA complexes, is gathering momentum. In 1990, the Huang group discovered that a catalytic amount of PPh3 could promote the perfluoroalkylation of simple alkenes with perfluoroalkyl iodides through a radical chain reaction (Scheme 34).82 In this transformation, an EAD complex 4–4, formed from PPh3 and perfluoroiodine 4–1, would decompose to the radical intermediate Rf•. Addition of Rf• to an alkene generates the alkyl radical intermediate 4–5. The reaction of 4–5 with perfluoroiodine forms the product 4–3 and regenerates Rf• for radical chain propagation. While subsequent examinations of this reaction were conducted under more or less the same thermal conditions as those in Huang’s original study,83a−83c Czekelius and co-workers found in 2019 that light also facilitated activation of the EAD complex, allowing the reaction to occur at close to ambient temperature (Scheme 34).83c A systematic study revealed that tBu3P and light from a blue LED (461 nm) were optimal for promoting this radical chain reaction.
Scheme 34. Phosphine-Initiated Perfluoroalkylation of Alkenes.
In the EDA complex-initiated radical processes discussed above, the substrates are involved in the EDA complexes and incorporated into the final products. This strategy requires careful selection of substrates to match the electronic properties of the phosphines, limiting their scope and, consequently, the utility of this strategy. A more general and effective strategy, however, may involve a CTC catalyst that can transfer an electron to a substrate to assist the radical generation and later accept an electron from the late-stage intermediate to complete the catalytic cycle. This strategy was realized by the Fu group in 2019.84 Although most photoredox catalysts involve precious metal complexes or organic dyes, they demonstrated a new type of photocatalytic decarboxylative alkylation using the combination of PPh3 and sodium iodide (Scheme 35). This novel alkylation was realized with several types of alkyl radical precursors (decarboxylative alkylation via redox-active esters, deaminative alkylation via Katritky’s N-alkylpyridinium salts, and trifluoromethylation with Togni’s reagent) and radical acceptors (e.g., silyl enol ethers, activated heterocycles, and 1,1-disubstituted alkenes). In the proposed mechanism, Fu et al. suggested that the CTC 4–10 is formed initially through Coulombic interactions between PPh3, sodium iodide, and the N-(alkylcarbonyl)phthalimide 4–8. SET from iodide to the phthalimide moiety then prompts decomposition and concomitant generation of an alkyl radical, which adds to the radical acceptor. Finally, the transient radical cation complex 4–12 oxidizes the amine radical cation 4–15 to the product 4–16 and turns back to the PPh3–NaI complex 4–10. A chiral phosphoric acid could enable enantioselective Minisci-type reactions with high levels of stereoselectivity. Interestingly, the blue LED light (456 nm) used here is very similar to that (461 nm) used in the perfluoroalkylation of alkenes (Scheme 34), suggesting that related complexes might have formed.
Scheme 35. Photocatalytic Decarboxylative Alkylations Mediated by PPh3 and NaI.

In a related study, the He, Xue, and Chen groups reported a radical C–H arylation of oxazoles with aryl iodides.85 They proposed that the complex D formed from Cs2CO3 and 1,1′-bis(diphenylphosphino)ferrocene (dppf) could serve as a strong electron donor, reducing aryliodine to form the radical anion intermediate 4–20 and further initiating a radical chain reaction (Scheme 36). The radical 4–20 would undergo C–I bond cleavage to form the aryl radical 4–21. Addition of 4–21 to oxazoles, followed by deprotonation, would form the radical anion 4–24. Reduction of the aryl iodide by the radical anion 4–24 would form the product 4–25 and regenerate the radical anion 4–20 for radical chain propagation. The radical anion 4–24 could also reduce D(+1) to D and complete the redox cycle. DFT calculations indicated, however, that dissociating CsCO3– from D(+1) to generate a potential electron acceptor (dppf)+ is thermodynamically challenging. The authors favored the radical chain reaction pathway, while not ruling out the redox cycle pathway.
Scheme 36. dppf/Cs2CO3-Mediated Oxazole C–H Arylation.

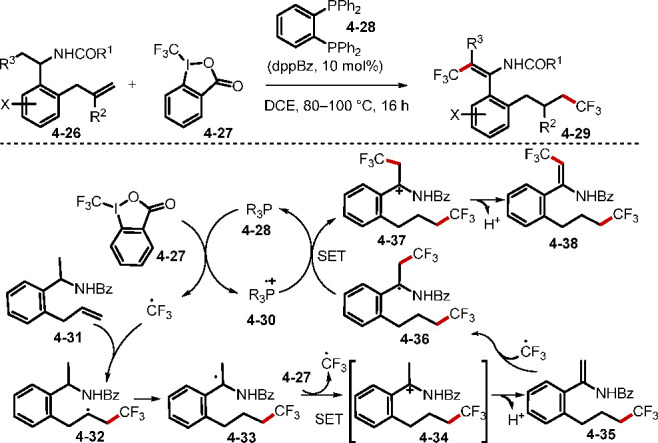
In addition to complexing with other compounds to form catalysts for SET processes, phosphorus compounds themselves can also act as electron shuttles in redox cycles. In 2015, Tan and Liu employed Togni reagent II as the source of trifluoromethyl radicals for the phosphine-catalyzed bistrifluoromethylation of enamides (Scheme 37).86a The reaction involved multiple SET processes, with mechanistic studies indicating that the catalytic cycle was driven by redox cycling of trivalent phosphine and a phosphorus-centered radical cation. Initially, the phosphine 4–28 was oxidized by the Togni reagent II through SET to form the CF3 radical and the phosphorus-centered radical cation 4–30. Addition of the CF3 radical to the alkene, followed by a 1,5-hydride shift, generated the α-amidobenzyl radical 4–33. Subsequent oxidation by the Togni reagent II and deprotonation formed the enamide 4–35. Addition of the CF3 radical to 4–35 afforded the radical intermediate 4–36, which was further oxidized by the phosphorus radical cation and deprotonated to give the product 4–38. The SET oxidation also recycled the phosphine catalyst 4–28. This method was further developed by the authors to allow concomitant trifluoromethylation of the alkene and subsequent remote alcohol or amine α-C–H activation to afford valuable trifluoromethyl-substituted ketones and aldehydes.86b
Scheme 37. Phosphine-Catalyzed Alkene Bistrifluoromethylation.
4.2. Use of Stoichiometric Phosphine
In addition to their roles as radical initiators and catalysts as described above, trivalent phosphorus(III) compounds are increasingly being used as radical precursors for SET transformations.9 Although, at present, stoichiometric reagents (e.g., as ylide or phosphine) are required, the byproducts formed in the deoxygenation or desulfurization process may be recycled in situ, as we have discussed above regarding phosphine oxide catalysis. Mechanistically, fragmentation of phosphoranyl radicals, through either α- or β-scission, is an efficient means of forming radicals for further transformation. The mechanisms of formation of phosphoranyl radicals can be distinguished by whether they involve addition of an external radical to a phosphine or addition of a nucleophile to a phosphorus-centered radical cation, and also by whether light is involved in the formation of the phosphoranyl radicals. Because this emerging field has been summarized in two recent reviews,9 we discuss only selected examples in this Outlook, including the α- and β-scissions of phosphoranyl radicals, light-associated and non-light-associated SET processes, as well as two modes of phosphoranyl radical generation.
Miura and co-workers reported recently the photoredox-driven fragmentation of phosphoranyl radicals through α-scission to generate active radical species.87 They disclosed a practical and efficient synergistic photoredox approach for the preparation of elongated esters from alkenes and stabilized phosphorus ylides under mild conditions (Scheme 38). The phosphoranyl radical intermediate generated through [IrIII*]-mediated reduction of the phosphonium salt 4–42 further fragmented into PPh3 and the radical 4–44 for subsequent addition to alkenes. Notably, the phosphoranyl radical was generated through neither of the methods described above.
Scheme 38. Synthesis of Elongated Alkenes.

The Zhu and Xie group provided an example of photoredox-driven fragmentation of phosphoranyl radicals through β-scission in their efficient approach to ketones through the deoxygenation of aromatic carboxylic acids using synergistic photoredox catalysis in the presence of PPh3 (Scheme 39).88a The reaction was initiated through oxidation of PPh3 to its radical cation, mediated by a photoexcited catalyst. The radical cation coupled with the carboxylate anion to generate the phosphoranyl radical intermediate 4–46, the β-scission of which generated the acyl radical 4–47. The final product was formed through radical addition to an alkene. In addition, corresponding deuterated aldehydes could be formed by the acyl radical intermediate abstracting a deuterium atom from D2O.88b Concurrently, the Doyle group reported the deoxygenation of alcohols and carboxylic acids 4–48 through a similar phosphine-promoted photoredox-catalysis reaction.88c
Scheme 39. Photoredox-Driven Fragmentation of Phosphoranyl Radicals through β-Scission.
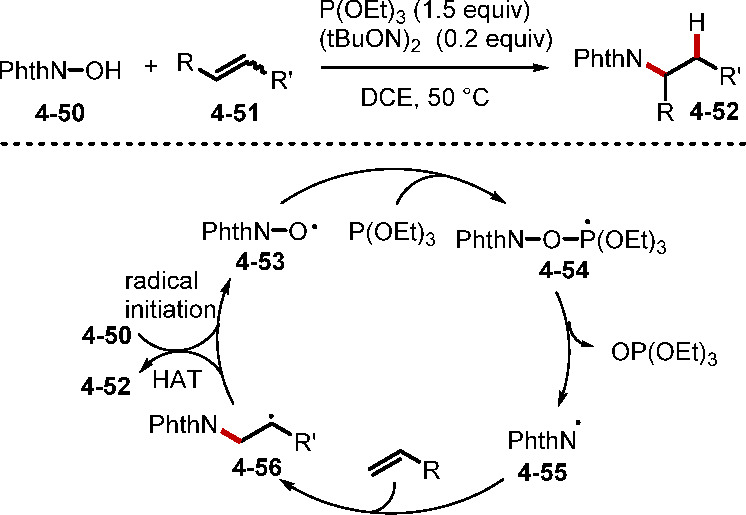
Whereas the examples above involving phosphorus compound-mediated SET were driven by photocatalysis, phosphoranyl radical intermediates can also be formed under thermal conditions. In 2018, the Schmidt group reported an intermolecular anti-Markovnikov hydroamination of alkenes using triethylphosphite and N-hydroxyphthalimide (Scheme 40).89 The reaction was initiated by hydrogen atom abstraction from N-hydroxyphthalimide to produce PhthNO•, which added to triethylphosphite to form the phosphoranyl radical intermediate 4–54. Subsequent β-fragmentation generated the radical PhthN•, which added to the alkene 4–51 to generate the radical 4–56. Abstraction of a hydrogen atom from 4–50 formed the anti-Markovnikov alkene hydroamination product 4–52 and regenerated PhthNO•.
Scheme 40. Anti-Markovnikov Alkene Hydroamination.
4.3. Summary
Organophosphorus compound-promoted SET reactions have reactivity complementary to that of nucleophilic phosphine catalysis. In nucleophilic phosphine catalysis, the substrates are limited to electron-deficient alkenes, whereas inert alkenes (as demonstrated in Schemes 34 and 37–40) and C–H bonds (as demonstrated in Schemes 35–37) can be activated in SET processes. Special attention may be given to the EDA complexes of phosphorus compounds, which are sensitive to light and can either initiate or catalyze SET processes. Exploration of the EDA complexes of phosphorus compound should unveil rich chemistry in this underdeveloped area. A special type of EDA complex, the FLP, can be a powerful tool in the activation of small molecules. Although a two-electron-transfer mechanism is generally recognized,6b recent reports have indicated that SET processes might also play important roles in FLP chemistry.90,91 An interesting study by Stephan and co-workers unveiled that a frustrated radical pair (FRP) [Mes3P•]/[•E(C6F5)3] (E: B/Al) could be formed through a SET process, while the analogous tBu3P pair behaved differently without undergoing such a process.91a This finding, as well as others, provides evidence that the structural features of tertiary phosphines greatly influence the stability of phosphorus-centered radical cations and can even alter the reaction pathway.91 Moreover, a tertiary phosphine itself can function as an electron shuttle in a redox-catalytic cycle. Organophosphorus compound-promoted SET processes, therefore, have huge potential for applications in photochemistry and electrochemistry. The limited number of reports of organophosphorus compound-catalyzed SET reactions may be due to the short half-lives of trivalent phosphine radical cation species.91d Designing phosphines or their complexes with structural frameworks of suitable electronic potential may result in longer half-lives for the open-shell species and benefit this area. The unique role of organophosphorus compounds to generate radical intermediates through SET processes is also appealing.
5. Miscellaneous Catalyses
In addition to nucleophilic phosphine catalysis, catalysis to bypass phosphine oxide waste, and phosphorus-mediated radical processes, organophosphorus compounds are used in multitudes of catalytic processes, including phosphonium salt phase transfer catalysis (PTC), iminophosphorane super base catalysis, asymmetric phosphine oxide Lewis base catalysis, FLP catalysis, and chiral phosphoric acid catalysis. PTC is considered a powerful and green sustainable tool for addressing synthetic challenges.7 In this context, phosphonium-containing organocatalysts, especially bifunctional catalysts, are emerging as powerful phase transfer catalysts for various asymmetric transformations (e.g., alkylation, Michael addition, Darzens reaction, Mannich reaction, and Strecker reaction).7 Deprotonation of pronucleophiles with chiral Brønsted bases (e.g., cinchona alkaloids and derivatives) to induce enantioselective reactions via ion pairs is an established reliable method for realizing various enantiodifferentiating transformations. Although powerful, the application of this method to high-pKa pronucleophiles has been limited. The availability of phosphorus-based super bases [e.g., P-spiro chiral triaminoiminophosphorane, chiral bis(guanidino)iminophosphorane, and bifunctional iminophosphorane], with values of pKBH+ ranging from 22 to 32.9, has opened a new door in this area, realizing various challenging reactions that are difficult to perform using cinchona-based catalysts.8 Another type of phosphorus-related base catalysis is asymmetric phosphine oxide Lewis base catalysis. Generally, a polarized phosphine oxide bond coordinates to chlorosilanes to form pentavalent silicon complexes, thereby enhancing the electrophilicity of the silicon species to activate electrophiles (e.g., carbonyls, epoxides, and imines) for further transformation. In the presence of chiral phosphine oxides (e.g., chiral binaphthyl dioxides and chiral phosphoramides), a variety of asymmetric transformations (e.g., allylation, aldol, hydrophosphonylation, and reduction of unsaturated compounds) can be promoted.5
Alongside base catalyses, phosphorus-based Brønsted acid catalysis has also become established as a reliable and efficient method to form a variety of C–C, C–H, and C–X bonds enantioselectively.4 Since the introduction of chiral phosphoric acid into catalytic asymmetric reactions in 2004,92 the following years have witnessed tremendous progress, resulting in the development of a variety of chiral acids (e.g., BINOL derivatives, TADDOL derivatives, VAPOL derivatives, and spiro phosphoric acids).4 Today, phosphorus-based Brønsted acid catalysis remains an active area of inquiry. Finally, FLPs, formed from combinations of Lewis acids and Lewis bases, have been used in the development of main group element catalysis processes for the activation of small molecules (e.g., H2, CO2, CO, SO2).6 In this context, various phosphines have played important roles in combination with Lewis acids [e.g., B(C6F5)3 and Al(C6F5)3] to form active FLPs.6 Although such studies constitute a large genre of the field of organophosphorus-based catalysis, we cannot cover them in this Outlook, due to limited space; therefore, we direct the reader’s attention to recent reviews on each topic.4−8
Although the scope of organophosphorus-based catalysis is large, most of its topics are “young” in that they have been established as fields of systematic inquiry only within the last 20 years. For example, redox-driven phosphine oxide catalysis began to gain significant interest in 2009 after O’Brien published the catalytic Wittig reaction;53 the phosphorus compound-mediated SET process has been attracting more attention recently as a result of the rapid development of photoredox chemistry;9 FLP chemistry became popular only after Stephan reported, in 2006, that H2 can be activated by an FLP.93 Notably, one of the original functions of chiral phosphoric acid, first reported in 1971, was as a stoichiometric reagent for the resolution of racemic amines.94 The rich chemistry of phosphorus compounds will enable more interesting reactions to be added to the toolbox of organic synthesis. One lesson we can take here is that new chemistry is emerging and that previous stoichiometric reactions may turn into catalytic ones in due course. Although not catalytic in its organophosphorus reagents, phosphorus(V) ligand coupling has been gaining renewed interest recently. We discuss these so-called “contractive coupling” reactions briefly in the next section.
6. Phosphorus(V) Ligand Coupling
Like the other main group elements below the second row of the periodic table (e.g., S, I, Si), phosphorus can adopt hypervalent states featuring an expanded valence-shell, tending to extrude an electron pair to form a more stable octet.95 The collapse of the hypervalent species results in various transformations, one of which is ligand coupling.96 The notion of “ligand coupling” was first introduced by Oae to describe the exclusion of two ligands from hypervalent species to form a stable octet and the coupling product.96,97 It was also termed “contractive coupling” by Newkome earlier in describing a similar process for the synthesis of biheteroaryls.98 While the concept has been applied to various main group elements,97 recent progress has demonstrated that P(V)-mediated ligand coupling can be a powerful method for heterocycle functionalization.99 Many heterocycles, including pyridines, diazines, and benzothiazoles, are important pharmacophores. While transition-metal-mediated functionalization of aromatic rings and electrophilic aromatic substitution are very well established, functionalization of these heterocycles remains a big challenge in terms of site-selectivity and functional group compatibility.100 In this context, phosphorus ligand coupling has found itself very useful in medicinal chemistry. Although a stoichiometric amount of reagent is required at present for phosphorus ligand coupling, the contractive coupling process mimics reductive elimination in transition-metal-catalyzed coupling (Scheme 41). Recently, the main group element-mediated two-electron redox catalysis has demonstrated its potential in coupling reactions.101 The Radosevich group reported a T-shaped P(III) compound as the catalyst for transfer hydrogenation of azobenzenes,101b and Cornella and co-workers unveiled a novel Bi complex that facilitates the fluorination of arylboronic esters through redox catalysis.101c,101d Such progress sheds light on the potential for catalytic phosphorus-mediated ligand coupling. Below, we discuss some recent advances in the area of phosphorus ligand coupling.
Scheme 41. Transition-Metal-Catalyzed Coupling and Phosphorus(V) Ligand Coupling.

The phenomenon of coupling between two ligands has been observed frequently at the phosphorus center.97,98,102,103 For example, phosphine oxides or phosphonium salts bearing two or three 2-pyridyl groups can undergo contractive ligand coupling to afford bipyridines.98,102b,102c The introduction of the heterocycles at the phosphorus center was, however, the most challenging step in those reports. In Newkome’s study, phenyldipyridylphosphine (oxide)s were prepared through reactions between dilithiophenylphosphide and halopyridines or through addition of 2-lithiopyridines to dichlorophenylphosphine.98 The success of ligand coupling requires a reliable method to access the hypervalent pentacoordinated phosphorane. In the case of phosphine oxides, addition of organometallic reagents (e.g., alkoxides, Grignard reagents, and organolithium reagents) can provide ready access to hypervalent pentacoordinated phosphoranes.96−98 For example, Newkome and Hager reported that heating phenyldi(2-pyridyl)phosphine oxides at 100 °C in the presence of EtONa afforded 2,2′-bipyridines in 50–60% yield.98 Phosphonium salts containing two 2-pyridyl groups display similar behavior. Oae and Uchida demonstrated that treatment of benzyltri(2-pyridyl)phosphonium bromide in water or alcohol in the presence or absence of acid could result in ligand coupling.102b−102e They also found that when tri(2-pyridyl)phosphine oxide was treated with nucleophilic organometallic reagents, both the coupling products of the two pyridine ligands and the coupling products of the pyridine ligand and the incoming nucleophile were formed.102b When using benzyl magnesium chloride as the nucleophile, 2-benzylpyridine was formed as the major product.102b Despite these advances, ligand coupling of heterocycles at the phosphorus center has found limited use because no general and mild conditions have been established to access related phosphonium salts bearing heteroaryls.
In the pioneering study reported by the Anders group, pyridine was transformed into a pyridylphosphonium salt in a site-selective and efficient manner through sequential addition of Tf2O, PPh3, and Et3N.104 In one of the limited examples of the application of this finding, they showed that addition of NaN3 as the nucleophile to the pyridylphosphonium salt resulted in coupling of the azido and pyridine groups, followed by a Staudinger reaction to give the iminophosphorane.104a The potential of this methodology in ligand coupling remained unexplored until recently, when McNally and co-workers investigated the site-selective functionalization of pyridines (Scheme 42).99a They found that, in most cases, the original organic base (Et3N) gave good results when forming phosphonium salts, but DBU was superior in some situations. The reaction temperature was also a critical factor of this protocol, with some substrates requiring low temperatures and some others needing higher temperatures. The purification of the phosphonium salts was performed through simple precipitation, without chromatography. The transformation displayed good regioselectivity, with the 4-substituted product being the major product. When the 4-position was blocked, 2-substituted products formed. Various functionalities were tolerated; specifically, pyridines with halogen substituents were introduced smoothly into the phosphonium salt. Addition of sodium alkoxide to the phosphonium salt produced the alkoxylated pyridine. Based on previous reports,96−98 McNally and co-workers proposed the formation of the pentavalent alkoxyphosphorane intermediate 6–5, followed by contractive ligand coupling to construct the C–O bond. They could not, however, rule out the nucleophilic aromatic substitution (SNAr) pathway. The reactions displayed good functional group tolerance and site-selectivity. For example, both nicotine and loratadine were transformed into phosphonium salts and reacted with sodium alkoxide to afford the alkoxylated products in good yields. Furthermore, sodium thiolate, sodium azide, and 2-pyridinelithium were suitable nucleophiles for this phosphorus ligand coupling reaction. Most recently, Vilotijevic and co-workers reported that benzothiazoles can also be functionalized through a phosphorus(V) ligand coupling process.99h Although Anders and co-workers had reported the preparation of benzothiazol-2-yl-triphenylphosphonium triflates previously, they did not fully study the scope and application of these salts.104b
Scheme 42. Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts.

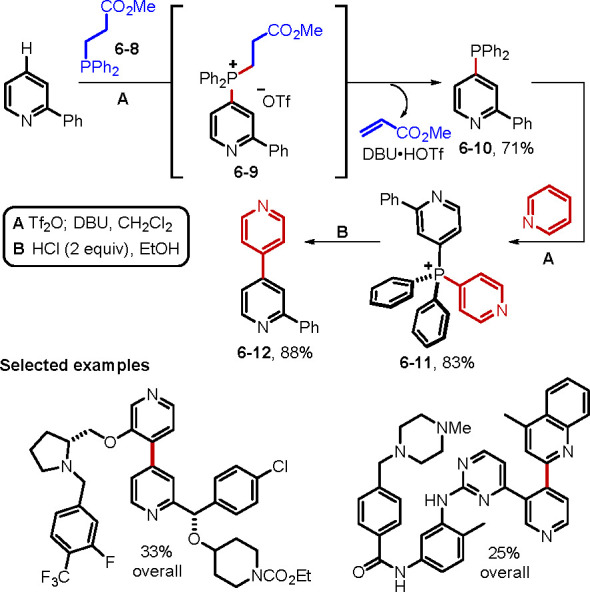
In 2018, McNally and co-workers used phosphonium salts as a platform for the synthesis of heterobiaryls through contractive C–C coupling via P(V) intermediates (Scheme 43).99c Facing the challenge of preparing di(azaaryl)phosphonium salts under mild conditions, they developed a unique route, without using heteroaryl halides or a lithium reagent. The key to the introduction of two heterocycles to the phosphorus center was the use of the fragmentable phosphine 6–8. Thus, the phosphonium triflate 6–9 underwent deprotonation by DBU, accompanied by the loss of methyl acrylate, which then provided the heteroaryl phosphine 6–10, positioning the first coupling partner in place. Upon engagement with a second coupling partner, the dihetarylphosphonium salt 6–11 formed with complete regiochemical control. Finally, an acidic alcohol solution initiated phosphorus ligand coupling to provide the heterobiaryl product 6–12. Numerous examples of the reaction proceeded with synthetically useful overall yields (17–34% over three steps). In addition, the synthesis of complex heterobiaryls is possible, including the use of bioactive molecules as coupling partners. The McNally group has also provided further advances in phosphorus(V) ligand coupling.99d−99g
Scheme 43. Synthesis of Heterobiaryls through P(V) Contractive Coupling.
Although the phenomenon of phosphorus(V) ligand coupling was observed as early as 1948,102a its potential in heterocycle functionalization was unveiled only recently. Contractive phosphorus(V) coupling is a coupling method for heterocycle functionalization that is complementary to transition-metal-mediated reactions in terms of selectivity and functional group tolerance. In transition-metal-catalyzed cross-coupling, redox cycling between the n and n + 2 oxidation states, through oxidative addition and reductive elimination, is crucial for catalysis. The phosphorus contractive coupling resembles reductive elimination, albeit in a noniterative manner. In the three-stage coupling sequence designed by McNally and co-workers, they realized a combination of stoichiometric oxidative addition and reductive elimination. In light of recent progress in the field of main group element redox catalysis, one cannot help but look forward to inventions that will enable P(III)/P(V) redox catalysis for phosphorus ligand coupling.101
7. Conclusion and Outlook
Phosphorus-based organocatalysis is a vast field. This brief Overview of various topics in phosphorus-based catalysis highlights key recent advances in the field, with emphasis on four topics: nucleophilic phosphine catalysis, phosphine oxide catalysis, phosphorus-mediated SET processes, and phosphorus(V) ligand coupling. Below, we provide a summary and outlook for each topic.
-
(1)
In this well-established but still active area, the present focus of nucleophilic phosphine catalysis lies in the development of new patterns of annulations, along with asymmetric variants of known reactions. Unforeseen reactions continue to be discovered when employing highly engineered substrates. The application of nucleophilic catalysis in the synthesis of functional molecules remains promising. Moreover, nucleophilic addition of phosphines to electron-deficient C–C multiple bonds promotes the in situ formation of zwitterionic adducts that can act as efficient base catalysts for various asymmetric transformations. With continued progress, we expect to see the different reactivity patterns of nucleophilic phosphine catalysis merging with transition metal catalysis and photoredox catalysis.
-
(2)
Reactions driven by phosphine oxide formation remain highly popular among synthetic chemists. Catalytic, economical, and environmentally benign versions of these reactions will undoubtedly have a huge impact on synthesis in both academic and industrial settings. Recent advances have unveiled insights into how to improve the in situ activation of strong P(V)=O bonds. Multiple studies have revealed that a strained phosphacycle scaffold can facilitate the in situ reduction of phosphine oxides during redox-driven catalysis. Denton and co-workers found that a carefully designed phosphine oxide catalyst could realize additive-free phosphine oxide catalysis in a redox-neutral setting. Collectively, these findings signify that the design of phosphine (oxide)s with appropriate skeletons will be pivotal in moving the field forward. As demonstrated by Sevov and co-workers, new technologies, including electrochemistry and photoredox chemistry, can also add impetus to the in situ recycling of phosphine oxides. Alongside these advances, asymmetric phosphine oxide catalysis is also appealing for preparing highly valuable chiral molecules. Ultimately, we wish to see catalytic versions of traditional phosphine oxide-generating reactions applied in both academia and industry.
-
(3)
At present, organic phosphorus compound-mediated SET processes remain undeveloped. Although limited in scope, this area is complementary to nucleophilic phosphine catalysis in terms of its mechanisms and functional group tolerance. While nucleophilic phosphine catalysis has been limited to electron-deficient multiple bonds, phosphorus radical catalysis can activate inert alkenes and C–H bonds. The design of phosphines or their complexes with structural frameworks of suitable electronic potential may lead to the open-shell species having longer half-lives, allowing rapid advances in this area. Several recent accounts have revealed that the EDA complexes of phosphines with RfI, NaI, or Cs2CO3 have a good SET ability, especially in the presence of visible light, potentially inspiring more discoveries in this field. Phosphorus-based reagents also facilitate the generation of active radical intermediates through deoxygenation or desulfurization, especially under photoredox catalysis. For the latter, challenges remain in the requirement for stoichiometric phosphorus reagent and the generation of waste. These issues may be addressed through recycling of the phosphine oxide, as we have discussed in Section 3. With recent developments in photochemistry and electrochemistry, we envision phosphorus compound-mediated SET processes to be a promising area.
-
(4)
Phosphorus ligand coupling displays the versatility of phosphorus compounds, and it can serve as a complementary method for transition-metal-mediated coupling for heterocyclic functionalization. Although such reactions remain stoichiometric at present, their reductive elimination steps emulate those of transition-metal-promoted reactions. Together with other main group elements (e.g., S, I, Bi), the realization of catalytic P(V) ligand coupling may be on the horizon.
-
(5)
Finally, several additional topics—phosphonium salt PTCs, iminophosphorane super base catalysis, asymmetric phosphine oxide Lewis base catalysis, FLP catalysis, and chiral phosphoric acid catalysis—are also important aspects of phosphorus-based organocatalysis. With the rich chemistry of organophosphorus compounds, this area continues to progress with the development of novel transformations. Phosphorus-based catalysis is at the threshold of a bright future. With continued invention, we anticipate phosphorus-based catalysis to have many more applications in the chemical and pharmaceutical industries.
Acknowledgments
Financial support by the NIH (R01GM071779) is acknowledged.
Author Present Address
† X.-R.S.: Institute of Organic Chemistry, Jiangxi Science & Technology Normal University, Key Laboratory of Organic Chemistry, Jiangxi Province, Nanchang 330013, China
The authors declare no competing financial interest.
References
- a Kosolapoff G. M.Organophosphorus Compounds; Wiley-VCH: London, 1950. [Google Scholar]; b Arbusow B. A. Michaelis-Arbusow- Und Perkow-Reaktionen. Pure Appl. Chem. 1964, 9, 307–336. 10.1351/pac196409020307. [DOI] [Google Scholar]; c Quin L. D.A Guide to Organophosphorus Chemistry; John Wiley & Sons: New York, 2000. [Google Scholar]
- a Tang W.; Zhang X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3070. 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]; b Pereira M. M.; Calvete M. J. F.; Carrilho R. M. B.; Abreu A. R. Synthesis of Binaphthyl Based Phosphine and Phosphite Ligands. Chem. Soc. Rev. 2013, 42, 6990–7027. 10.1039/c3cs60116a. [DOI] [PubMed] [Google Scholar]; c Fu W.; Tang W. Chiral Monophosphorus Ligands for Asymmetric Catalytic Reactions. ACS Catal. 2016, 6, 4814–4858. 10.1021/acscatal.6b01001. [DOI] [Google Scholar]
- a Ni H.; Chan W.-L.; Lu Y. Phosphine-Catalyzed Asymmetric Organic Reactions. Chem. Rev. 2018, 118, 9344–9411. 10.1021/acs.chemrev.8b00261. [DOI] [PubMed] [Google Scholar]; b Guo H.; Fan Y. C.; Sun Z.; Wu Y.; Kwon O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. 10.1021/acs.chemrev.8b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang Z.; Xu X.; Kwon O. Phosphine catalysis of allenes with electrophiles. Chem. Soc. Rev. 2014, 43, 2927–2940. 10.1039/C4CS00054D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Akiyama T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]; b Parmar D.; Sugiono E.; Raja S.; Rueping M. Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates. Chem. Rev. 2014, 114, 9047–9153. 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]; c Maji R.; Mallojjala S. C.; Wheeler S. E. Chiral Phosphoric Acid Catalysis: From Numbers to Insights. Chem. Soc. Rev. 2018, 47, 1142–1158. 10.1039/C6CS00475J. [DOI] [PubMed] [Google Scholar]; d Xia Z.-L.; Xu-Xu Q.-F.; Zheng C.; You S.-L. Chiral Phosphoric Acid-Catalyzed Asymmetric Dearomatization Reactions. Chem. Soc. Rev. 2020, 49, 286–300. 10.1039/C8CS00436F. [DOI] [PubMed] [Google Scholar]
- a Orito Y.; Nakajima M. Lewis Base Catalyzed Asymmetric Reactions Involving Hypervalent Silicate Intermediates. Synthesis 2006, 2006, 1391–1401. 10.1055/s-2006-926405. [DOI] [Google Scholar]; b Benaglia M.; Rossi S. Chiral Phosphine Oxides in Present-Day Organocatalysis. Org. Biomol. Chem. 2010, 8, 3824–3830. 10.1039/c004681g. [DOI] [PubMed] [Google Scholar]; c Vedejs E.; Denmark S. E.. Lewis Base Catalysis in Organic Synthesis; John Wiley & Sons: Weinheim, 2016. [Google Scholar]; d Lao Z.; Toy P. H.. Use of Phosphine Oxides as Catalysts and Precatalysts. In Nonnitrogenous Organocatalysis; Harned A., Ed.; CRC Press: Boca Raton, 2017; pp 144–163. [Google Scholar]; e Kotani S. Phosphine Oxide-Catalyzed Asymmetric Aldol Reactions and Double Aldol Reactions. Chem. Pharm. Bull. 2019, 67, 519–526. 10.1248/cpb.c19-00015. [DOI] [PubMed] [Google Scholar]; f Ayad T.; Gernet A.; Pirat J.-L.; Virieux D. Enantioselective Reactions Catalyzed by Phosphine Oxides. Tetrahedron 2019, 75, 4385–4418. 10.1016/j.tet.2019.06.042. [DOI] [Google Scholar]
- a Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]; b Stephan D. W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. 10.1021/jacs.5b06794. [DOI] [PubMed] [Google Scholar]; c Stephan D. W. The Broadening Reach of Frustrated Lewis Pair Chemistry. Science 2016, 354, aaf7229 10.1126/science.aaf7229. [DOI] [PubMed] [Google Scholar]; d Bayne J. M.; Stephan D. W. Phosphorus Lewis Acids: Emerging Reactivity and Applications in Catalysis. Chem. Soc. Rev. 2016, 45, 765–774. 10.1039/C5CS00516G. [DOI] [PubMed] [Google Scholar]; e Fontaine F.-G.; Stephan D. W. On the Concept of Frustrated Lewis Pairs. Philos. Trans. R. Soc., A 2017, 375, 20170004. 10.1098/rsta.2017.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lam J.; Szkop K. M.; Mosaferi E.; Stephan D. W. FLP Catalysis: Main Group Hydrogenations of Organic Unsaturated Substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. 10.1039/C8CS00277K. [DOI] [PubMed] [Google Scholar]; g Liu L.; Lukose B.; Jaque P.; Ensing B. Reaction Mechanism of Hydrogen Activation by Frustrated Lewis Pairs. Green Energy Environ. 2019, 4, 20–28. 10.1016/j.gee.2018.06.001. [DOI] [Google Scholar]; h Li N.; Zhang W.-X. Frustrated Lewis Pairs: Discovery and Overviews in Catalysis. Chin. J. Chem. 2020, 38, 1360–1370. 10.1002/cjoc.202000027. [DOI] [Google Scholar]
- a Manabe K. Asymmetric Phase-Transfer Alkylation Catalyzed by a Chiral Quaternary Phosphonium Salt with a Multiple Hydrogen-Bonding Site. Tetrahedron Lett. 1998, 39, 5807–5810. 10.1016/S0040-4039(98)01181-2. [DOI] [Google Scholar]; b He R.; Wang X.; Hashimoto T.; Maruoka K. Binaphthyl-Modified Quaternary Phosphonium Salts as Chiral Phase-Transfer Catalysts: Asymmetric Amination of β-Keto Esters. Angew. Chem., Int. Ed. 2008, 47, 9466–9468. 10.1002/anie.200804140. [DOI] [PubMed] [Google Scholar]; c Liu S.; Kumatabara Y.; Shirakawa S. Chiral Quaternary Phosphonium Salts as Phase-Transfer Catalysts for Environmentally Benign Asymmetric Transformations. Green Chem. 2016, 18, 331–341. 10.1039/C5GC02692J. [DOI] [Google Scholar]; d Golandaj A.; Ahmad A.; Ramjugernath D. Phosphonium Salt in Asymmetric Catalysis: A Journey of a Decade’s Extensive Research Work. Adv. Synth. Catal. 2017, 359, 3676–3706. 10.1002/adsc.201700795. [DOI] [Google Scholar]
- a Uraguchi D.; Ooi T. Development of P-Spiro Chiral Aminophosphonium Salts as a New Class of Versatile Organic Molecular Catalyst. Yuki Gosei Kagaku Kyokaishi 2010, 68, 1185–1194. 10.5059/yukigoseikyokaishi.68.1185. [DOI] [Google Scholar]; b Krawczyk H.; Dziȩgielewski M.; Deredas D.; Albrecht A.; Albrecht Ł. Chiral Iminophosphoranes—An Emerging Class of Superbase Organocatalysts. Chem. - Eur. J. 2015, 21, 10268–10277. 10.1002/chem.201500481. [DOI] [PubMed] [Google Scholar]; c Teng B.; Lim W. C.; Tan C.-H. Recent Advances in Enantioselective Brønsted Base Organocatalytic Reactions. Synlett 2017, 28, 1272–1277. 10.1055/s-0036-1588847. [DOI] [Google Scholar]; d Wang Y.-H.; Cao Z.-Y.; Li Q.-H.; Lin G.-Q.; Zhou J.; Tian P. Activating Pronucleophiles with High pKa Values: Chiral Organo-Superbases. Angew. Chem., Int. Ed. 2020, 59, 8004–8014. 10.1002/anie.201913484. [DOI] [PubMed] [Google Scholar]
- a Rossi-Ashton J. A.; Clarke A. K.; Unsworth W. P.; Taylor R. J. K. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. 10.1021/acscatal.0c01923. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pan D.; Nie G.; Jiang S.; Li T.; Jin Z. Radical Reactions Promoted by Trivalent Tertiary Phosphines. Org. Chem. Front. 2020, 7, 2349–2371. 10.1039/D0QO00473A. [DOI] [Google Scholar]
- a Li E.-Q.; Huang Y. Recent Advances in Phosphine Catalysis Involving γ-Substituted Allenoates. Chem. Commun. 2020, 56, 680–694. 10.1039/C9CC08241G. [DOI] [PubMed] [Google Scholar]; b Zhang H.; Zhou R. Recent Advances in Phosphine-Promoted (4 + 1) Annulation Reactions. Eur. J. Org. Chem. 2020, 2020, 4098–4107. 10.1002/ejoc.202000023. [DOI] [Google Scholar]
- a Salin A. V.; Il’in A. V.; Faskhutdinov R. I.; Galkin V. I.; Islamov D. R.; Kataeva O. N. Tributylphosphine Catalyzed Addition of Diphenylphosphine Oxide to Unsubstituted and Substituted Electron-Deficient Alkenes. Tetrahedron Lett. 2018, 59, 1630–1634. 10.1016/j.tetlet.2018.03.040. [DOI] [Google Scholar]; b Salin A. V.; Il’in A. V. Comparison of Catalytic Activity of Hexamethyltriamino- and Tri-n-Butylphosphines in the Pudovik Reaction. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 355–356. 10.1080/10426507.2018.1542403. [DOI] [Google Scholar]; c Salin A. V.; Islamov D. R. Phosphine-Catalyzed Michael Additions to α-Methylene-γ-Butyrolactones. Org. Biomol. Chem. 2019, 17, 7293–7299. 10.1039/C9OB01401B. [DOI] [PubMed] [Google Scholar]; d Zhou T.; Ji X.; Zhang J.; Liu L. Phosphine-Catalyzed Conjugate Cyanation of β-Trifluoromethyl Enones: Access to α-Trifluoromethyl γ-Carbonyl Nitriles. Org. Chem. Front. 2020, 7, 2644–2648. 10.1039/D0QO00582G. [DOI] [Google Scholar]
- a Li K.; Jin Z.; Chan W.-L.; Lu Y. Enantioselective Construction of Bicyclic Pyran and Hydrindane Scaffolds via Intramolecular Rauhut-Currier Reactions Catalyzed by Thiourea-Phosphines. ACS Catal. 2018, 8, 8810–8815. 10.1021/acscatal.8b02706. [DOI] [Google Scholar]; b Jin Z.; Ni H.; Zhou B.; Zheng W.; Lu Y. Phosphine-Catalyzed Formal Oxa-[4 + 2] Annulation Employing Nitroethylene and Enones: Enantioselective Synthesis of Dihydropyrans. Org. Lett. 2018, 20, 5515–5518. 10.1021/acs.orglett.8b02519. [DOI] [PubMed] [Google Scholar]; c Xiao B.-X.; Jiang B.; Song X.; Du W.; Chen Y.-C. Phosphine-Catalysed Asymmetric Dearomative Formal [4 + 2] Cycloadditions of 3-Benzofuranyl Vinyl Ketones. Chem. Commun. 2019, 55, 3097–3100. 10.1039/C9CC00386J. [DOI] [PubMed] [Google Scholar]
- a Mato R.; Manzano R.; Reyes E.; Carrillo L.; Uria U.; Vicario J. L. Catalytic Enantioselective Transannular Morita-Baylis-Hillman Reaction. J. Am. Chem. Soc. 2019, 141, 9495–9499. 10.1021/jacs.9b03679. [DOI] [PubMed] [Google Scholar]; b Khairnar P. V.; Su Y.-H.; Chen Y.-C.; Edukondalu A.; Chen Y.-R.; Lin W. Organophosphane-Catalyzed Direct β-Acylation of 4-Arylidene Pyrazolones and 5-Arylidene Thiazolones with Acyl Chlorides. Org. Lett. 2020, 22, 6868–6872. 10.1021/acs.orglett.0c02408. [DOI] [PubMed] [Google Scholar]
- Shi W.; Zhou L.; Mao B.; Wang Q.; Wang C.; Zhang C.; Li X.; Xiao Y.; Guo H. Phosphine-Catalyzed [3 + 2] Annulation of β-Sulfonamido-Substituted Enones with Sulfamate-Derived Cyclic Imines. J. Org. Chem. 2019, 84, 679–686. 10.1021/acs.joc.8b02515. [DOI] [PubMed] [Google Scholar]
- a Wang H.; Zhang L.; Tu Y.; Xiang R.; Guo Y.-L.; Zhang J. Phosphine-Catalyzed Difunctionalization of β-Fluoroalkyl α,β-Enones: A Direct Approach to β-Amino α-Diazo Carbonyl Compounds. Angew. Chem., Int. Ed. 2018, 57, 15787–15791. 10.1002/anie.201810253. [DOI] [PubMed] [Google Scholar]; b Li Y.; Wang H.; Su Y.; Li R.; Li C.; Liu L.; Zhang J. Phosphine-Catalyzed [3 + 2] Cycloaddition Reaction of α-Diazoacetates and β-Trifluoromethyl Enones: A Facile Access to Multisubstituted 4-(Trifluoromethyl)Pyrazolines. Org. Lett. 2018, 20, 6444–6448. 10.1021/acs.orglett.8b02757. [DOI] [PubMed] [Google Scholar]
- Vaishanv N. K.; Zaheer M. K.; Kant R.; Mohanan K. Phosphine-Catalyzed β-Selective Conjugate Addition of α-Fluoro-β-Ketoamides to Allenic Esters. Eur. J. Org. Chem. 2019, 2019, 6138–6142. 10.1002/ejoc.201901199. [DOI] [Google Scholar]
- a Wang H.; Guo C. Enantioselective γ-Addition of Pyrazole and Imidazole Heterocycles to Allenoates Catalyzed by Chiral Phosphine. Angew. Chem., Int. Ed. 2019, 58, 2854–2858. 10.1002/anie.201813381. [DOI] [PubMed] [Google Scholar]; b Qiu H.; Chen X.; Zhang J. Design, Synthesis and Application of a New Type of Bifunctional Le-Phos in Highly Enantioselective γ-Addition Reactions of N-Centered Nucleophiles to Allenoates. Chem. Sci. 2019, 10, 10510–10515. 10.1039/C9SC04073K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Huang Y. Phosphine-Catalyzed Remote 1,7-Addition for Synthesis of Diene Carboxylates. ACS Catal. 2020, 10, 3541–3547. 10.1021/acscatal.0c00588. [DOI] [Google Scholar]
- a Wang D.; Liu W.; Hong Y.; Tong X. Phosphine-Catalyzed (3 + 2) Annulation of δ-Acetoxy Allenoates with 2-Sulfonamidomalonate: Synthesis of Highly Substituted 3-Pyrrolines and Mechanistic Insight. Org. Lett. 2018, 20, 5002–5005. 10.1021/acs.orglett.8b02156. [DOI] [PubMed] [Google Scholar]; b Feng J.; Huang Y. Phosphine-Catalyzed (3 + 2)/(2 + 3) Sequential Annulation Involving a Triple Nucleophilic Addition Reaction of γ-Vinyl Allenoates. Chem. Commun. 2019, 55, 14011–14014. 10.1039/C9CC07346A. [DOI] [PubMed] [Google Scholar]
- a Cao Z.-H.; Wang Y.-H.; Kalita S. J.; Schneider U.; Huang Y.-Y. Phosphine-Catalyzed [4 + 1] Cycloadditions of Allenes with Methyl Ketimines, Enamines, and a Primary Amine. Angew. Chem., Int. Ed. 2020, 59, 1884–1890. 10.1002/anie.201912263. [DOI] [PubMed] [Google Scholar]; b Blank B. R.; Andrews I. P.; Kwon O. Phosphine-Catalyzed (4 + 1) Annulation: Rearrangement of Allenylic Carbamates to 3-Pyrrolines through Phosphonium Diene Intermediates. ChemCatChem 2020, 12, 4352–4372. 10.1002/cctc.202000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gao Z.; Wang C.; Zhou L.; Yuan C.; Xiao Y.; Guo H. Phosphine-Catalyzed [8 + 2]-Annulation of Heptafulvenes with Allenoates and Its Asymmetric Variant: Construction of Bicyclo[5.3.0]Decane Scaffold. Org. Lett. 2018, 20, 4302–4305. 10.1021/acs.orglett.8b01734. [DOI] [PubMed] [Google Scholar]; b Liu H.; Lin Y.; Zhao Y.; Xiao M.; Zhou L.; Wang Q.; Zhang C.; Wang D.; Kwon O.; Guo H. Phosphine-Promoted [4 + 3] Annulation of Allenoate with Aziridines for Synthesis of Tetrahydroazepines: Phosphine-Dependent [3 + 3] and [4 + 3] Pathways. RSC Adv. 2019, 9, 1214–1221. 10.1039/C8RA09852B. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qin Z.; Liu R.; Zhou R.; Li R.; He Z. Phosphine-Catalyzed [3 + 2] and [2 + 4] Annulations of γ-Methyl Allenoates with Aryl α-Keto Esters: Stereoselective Syntheses of Functionalized Tetrahydrofurans and 4-Chromanols. Asian J. Org. Chem. 2020, 9, 86–93. 10.1002/ajoc.201900635. [DOI] [Google Scholar]; d Shi W.; Mao B.; Xu J.; Wang Q.; Wang W.; Wu Y.; Li X.; Guo H. Phosphine-Catalyzed Cascade Michael Addition/[4 + 2] Cycloaddition Reaction of Allenoates and 2-Arylidene-1,3-Indanediones. Org. Lett. 2020, 22, 2675–2680. 10.1021/acs.orglett.0c00637. [DOI] [PubMed] [Google Scholar]; e Manzano R.; Romaniega A.; Prieto L.; Díaz E.; Reyes E.; Uria U.; Carrillo L.; Vicario J. L. γ-Substituted Allenic Amides in the Phosphine-Catalyzed Enantioselective Higher Order Cycloaddition with Azaheptafulvenes. Org. Lett. 2020, 22, 4721–4725. 10.1021/acs.orglett.0c01523. [DOI] [PubMed] [Google Scholar]; f Gallardo-Fuentes S.; Ormazábal-Toledo R.; Fernández I. Unraveling the Selectivity Patterns in Phosphine-Catalyzed Annulations of Azomethine Imines and Allenoates. J. Org. Chem. 2020, 85, 9272–9280. 10.1021/acs.joc.0c01272. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Lu X. Phosphine-Catalyzed Cycloaddition of 2,3-Butadienoates or 2-Butynoates With Electron-Deficient Olefins: A Novel [3 + 2] Annulation Approach to Cyclopentenes. J. Org. Chem. 1995, 60, 2906–2908. 10.1021/jo00114a048. [DOI] [Google Scholar]
- Shaikh A. C.; Kwon O. Discussion Addendum for: Phosphine-Catalyzed [3 + 2] Annulation: Synthesis of Ethyl 5-(tert-Butyl)-2-phenyl-1-tosyl-3-pyrroline-3-carboxylate. Org. Synth. 2019, 96, 214–231. 10.15227/orgsyn.096.0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li E.; Jin H.; Huang Y. Phosphine-Catalyzed [3 + 2] Annulation of γ-Substituted Allenoates: Novel Access to Functionalized Cyclopentenes and Bicyclic[3, 3, 0]octene Derivatives. ChemistrySelect 2018, 3, 12007–12010. 10.1002/slct.201802800. [DOI] [Google Scholar]; b Soares M. I. L.; Gomes C. S. B.; Nunes S. C. C.; Pais A. A. C. C.; Melo T. M. V. D. P. e. Phosphane-Catalyzed [3 + 2] Annulation of Allenoates with 3-Nitro-2H-Chromenes: Synthesis of Tetrahydrocyclopenta[c]Chromenes. Eur. J. Org. Chem. 2019, 2019, 5441–5451. 10.1002/ejoc.201900564. [DOI] [Google Scholar]; c Yuan C.; Tan H.; Jiang X.-F.; Liu S.; Jiang L.; Cao Q.-Y.; Xu X.-J.; Deng X.-Y.; Pan G.-N.; Chen J.-Y.; Cui H.-L. Phosphine-Catalyzed [3 + 2] Cycloaddition and Vinylation of Indole-Derived α,α-Dicyanoolefins with γ-Substituted Allenoates. Asian J. Org. Chem. 2019, 8, 1893–1902. 10.1002/ajoc.201900393. [DOI] [Google Scholar]; d Gan Z.; Gong Y.; Chu Y.; Li E.-Q.; Huang Y.; Duan Z. Phosphine-Catalyzed Regiodivergent Annulations of γ-Substituted Allenoates with Conjugated Dienes. Chem. Commun. 2019, 55, 10120–10123. 10.1039/C9CC05207K. [DOI] [PubMed] [Google Scholar]; e Liu R.; Qin Z.; Fan B.; Li R.; Zhou R.; He Z. Phosphine-Catalyzed Chemo- and Diastereoselective [2 + 2 + 2] and [3 + 2] Annulations of γ-Methyl Allenoates with Doubly Activated Olefins: Syntheses of Highly Substituted Cyclohexanes and Cyclopentenes. J. Org. Chem. 2019, 84, 12490–12498. 10.1021/acs.joc.9b01968. [DOI] [PubMed] [Google Scholar]; f Luo W.; Shao B.; Li J.; Song D.; Yi X.; Ling F.; Zhong W. Divergent Synthesis of Spirocyclopentene-Pyrazolones and Pyrano[2,3-c]-Pyrazoles via Lewis Base Controlled Annulation Reactions. Tetrahedron Lett. 2019, 60, 151206. 10.1016/j.tetlet.2019.151206. [DOI] [Google Scholar]
- a Cerveri A.; Faza O. N.; López C. S.; Grilli S.; Monari M.; Bandini M. Phosphine-Catalyzed Stereoselective Dearomatization of 3-NO2-Indoles with Allenoates. J. Org. Chem. 2019, 84, 6347–6355. 10.1021/acs.joc.9b00559. [DOI] [PubMed] [Google Scholar]; b Zhao J.-Q.; Yang L.; You Y.; Wang Z.-H.; Xie K.-X.; Zhang X.-M.; Xu X.-Y.; Yuan W.-C. Phosphine-Catalyzed Dearomative (3 + 2) Annulation of 2-Nitrobenzofurans and Nitrobenzothiophenes with Allenoates. Org. Biomol. Chem. 2019, 17, 5294–5304. 10.1039/C9OB00775J. [DOI] [PubMed] [Google Scholar]; c Liu K.; Wang G.; Cheng S.-J.; Jiang W.-F.; He C.; Ye Z.-S. Phosphine-Catalyzed Dearomative [3 + 2] Annulation of 3-Nitroindoles and Allenoates. Tetrahedron Lett. 2019, 60, 1885–1890. 10.1016/j.tetlet.2019.06.016. [DOI] [Google Scholar]
- a Chan W.-L.; Tang X.; Zhang F.; Quek G.; Mei G.-J.; Lu Y. Phosphine-Catalyzed (3 + 2) Annulation of Isoindigos with Allenes: Enantioselective Formation of Two Vicinal Quaternary Stereogenic Centers. Angew. Chem., Int. Ed. 2019, 58, 6260–6264. 10.1002/anie.201900758. [DOI] [PubMed] [Google Scholar]; b Wang H.; Zhang J.; Tu Y.; Zhang J. Phosphine-Catalyzed Enantioselective Dearomative [3 + 2]-Cycloaddition of 3-Nitroindoles and 2-Nitrobenzofurans. Angew. Chem., Int. Ed. 2019, 58, 5422–5426. 10.1002/anie.201900036. [DOI] [PubMed] [Google Scholar]; c Li K.; Gonçalves T. P.; Huang K.-W.; Lu Y. Dearomatization of 3-Nitroindoles by a Phosphine-Catalyzed Enantioselective [3 + 2] Annulation Reaction. Angew. Chem., Int. Ed. 2019, 58, 5427–5431. 10.1002/anie.201900248. [DOI] [PubMed] [Google Scholar]; d Ni H.; Wong Y. L.; Wu M.; Han Z.; Ding K.; Lu Y. Highly Enantioselective [3 + 2] Annulation of 3-Butynoates with β-Trifluoromethyl Enones Promoted by an Amine-Phosphine Binary Catalytic System. Org. Lett. 2020, 22, 2460–2463. 10.1021/acs.orglett.0c00681. [DOI] [PubMed] [Google Scholar]; e Luo W.; Shao B.; Li J.; Xiao X.; Song D.; Ling F.; Zhong W. Construction of Spirooxindole-Fused Spiropyrazolones Containing Contiguous Three Stereogenic Centres via [3 + 2] Annulation Utilizing a Ferrocene Derived Bifunctional Phosphine Catalyst. Org. Chem. Front. 2020, 7, 1016–1021. 10.1039/D0QO00140F. [DOI] [Google Scholar]
- a Smaligo A. J.; Vardhineedi S.; Kwon O. Carvone-Derived P-Stereogenic Phosphines: Design, Synthesis, and Use in Allene-Imine [3 + 2] Annulation. ACS Catal. 2018, 8, 5188–5192. 10.1021/acscatal.8b01081. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yu Z.; Jin Z.; Duan M.; Bai R.; Lu Y.; Lan Y. Toward a Predictive Understanding of Phosphine-Catalyzed [3 + 2] Annulation of Allenoates with Acrylate or Imine. J. Org. Chem. 2018, 83, 9729–9740. 10.1021/acs.joc.8b01259. [DOI] [PubMed] [Google Scholar]
- Zhu X.-F.; Lan J.; Kwon O. An Expedient Phosphine Catalyzed [4 + 2] Annulation: Synthesis of Highly Functionalized Tetrahydropyridines. J. Am. Chem. Soc. 2003, 125, 4716–4717. 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]
- Shaikh A. C.; Kwon O. Discussion Addendum for: Phosphine-Catalyzed [4 + 2] Annulation: Synthesis of Ethyl 6-Phenyl-1-tosyl-1,2,5,6-tetrahydropyridine-3-carboxylate. Org. Synth. 2019, 96, 110–123. [PMC free article] [PubMed] [Google Scholar]
- a Xu Q.; Dupper N. J.; Smaligo A. J.; Fan Y. C.; Cai L.; Wang Z.; Langenbacher A. D.; Chen J.; Kwon O. Catalytic Enantioselective Synthesis of Guvacine Derivatives through [4 + 2] Annulations of Imines with -Methylallenoates. Org. Lett. 2018, 20, 6089–6093. 10.1021/acs.orglett.8b02489. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Khong S. N.; Xie C.; Wang X.; Tan H.; Kwon O. Chiral Aminophosphines Derived from Hydroxyproline and Their Application in Allene-Imine [4 + 2] Annulation. J. Antibiot. 2019, 72, 389–396. 10.1038/s41429-019-0181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang X.; Lu M.; Su Q.; Zhou M.; Addepalli Y.; Yao W.; Wang Z.; Lu Y. Phosphine-Catalyzed [4 + 2] Cycloadditions of Allenic Ketones: Enantioselective Synthesis of Functionalized Tetrahydropyridines. Chem. - Asian J. 2019, 14, 3409–3413. 10.1002/asia.201901104. [DOI] [PubMed] [Google Scholar]; d Wang H.; Hu Q.; Wang M.; Guo C. Enantioselective [4 + 2] Annulation to the Concise Synthesis of Chiral Dihydrocarbazoles. iScience 2020, 23, 100840. 10.1016/j.isci.2020.100840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen X.; Gao D.; Wang D.; Xu T.; Liu W.; Tian P.; Tong X. Access to Aryl-Naphthaquinone Atropisomers by Phosphine-Catalyzed Atroposelective (4 + 2) Annulations of δ-Acetoxy Allenoates with 2-Hydroxyquinone Derivatives. Angew. Chem., Int. Ed. 2019, 58, 15334–15338. 10.1002/anie.201908923. [DOI] [PubMed] [Google Scholar]; b Xu T.; Wang D.; Liu W.; Tong X. Phosphine-Promoted Divergent Annulations of δ-Acetoxy Allenoates with -Hydroxy---Carbonyl Ester Derivatives: Synthesis of Tetrasubstituted Cyclopentadienes and Benzenes. Org. Lett. 2019, 21, 1944–1947. 10.1021/acs.orglett.8b03808. [DOI] [PubMed] [Google Scholar]; c Arupula S. K.; Qureshi A. A.; Swamy K. C. K. Lewis Base-Switched [3 + 3] and [4 + 2] Annulation Reactions of δ-Acetoxy Allenoates with Cyclic N-Sulfonyl Imines: Divergent Synthesis of Functionalized α-Pyridyl Acetates and Teraryl Scaffolds. J. Org. Chem. 2020, 85, 4130–4144. 10.1021/acs.joc.9b03281. [DOI] [PubMed] [Google Scholar]
- a Wu M.; Han Z.; Li K.; Wu J.; Ding K.; Lu Y. Cyclohexyl-Fused, Spirobiindane-Derived, Phosphine-Catalyzed Synthesis of Tricyclic -Lactams and Kinetic Resolution of L-Substituted Allenoates. J. Am. Chem. Soc. 2019, 141, 16362–16373. 10.1021/jacs.9b07418. [DOI] [PubMed] [Google Scholar]; b Li N.; Jia P.; Huang Y. Facile Access to Highly Functionalized Hydroisoquinoline Derivatives via Phosphine-Catalyzed Sequential [3 + 3]/[3 + 3] Annulation. Chem. Commun. 2019, 55, 10976–10979. 10.1039/C9CC05832J. [DOI] [PubMed] [Google Scholar]
- Wang D.; Song Z.-F.; Wang W.-J.; Xu T. Highly Regio- and Enantioselective Dienylation of p-Quinone Methides Enabled by an Organocatalyzed Isomerization/Addition Cascade of Allenoates. Org. Lett. 2019, 21, 3963–3967. 10.1021/acs.orglett.9b01110. [DOI] [PubMed] [Google Scholar]
- a Dai Z.; Zhu J.; Wang J.; Su W.; Yang F.; Zhou Q. Phosphine-Catalyzed Chemoselective [4 + 3] Cycloaddition of Alminine Esters and β′-Acetoxy Allenoates for Divergent Synthesis of Azepines. Adv. Synth. Catal. 2020, 362, 545–551. 10.1002/adsc.201901132. [DOI] [Google Scholar]; b Dai Z.; Zhu J.; Su W.; Zeng W.; Liu Z.; Chen M.; Zhou Q. Phosphine-Catalyzed Stereoselective Tandem Annulation Reaction for the Synthesis of Chromeno[4,3-b]pyrroles. Org. Lett. 2020, 22, 7008–7012. 10.1021/acs.orglett.0c02558. [DOI] [PubMed] [Google Scholar]; c Vaishanv N. K.; Chandrasekharan S. P.; Zaheer M. K.; Kant R.; Mohanan K. Substrate-Controlled, PBu3-Catalyzed Annulation of Phenacylmalononitriles with Allenoates Enables Tunable Access to Cyclopentenes. Chem. Commun. 2020, 56, 11054–11057. 10.1039/D0CC04688D. [DOI] [PubMed] [Google Scholar]
- a Zhang Q.; Jin H.; Feng J.; Zhu Y.; Jia P.; Wu C.; Huang Y. Sequential Phosphine-Catalyzed [4 + 2] Annulation of 2′-Acetoxy Allenoates: Enantioselective Synthesis of 3-Ethynyl-Substituted Tetrahydroquinolines. Org. Lett. 2019, 21, 1407–1411. 10.1021/acs.orglett.9b00130. [DOI] [PubMed] [Google Scholar]; b Zhu Y.; Wang D.; Huang Y. Phosphine Sequentially Catalyzed Domino 1,6-Addition/Annulation: Access to Functionalized Chromans and Tetrahydroquinolines with an Ethynyl-Substituted All-Carbon Quaternary Center. Org. Lett. 2019, 21, 908–912. 10.1021/acs.orglett.8b03819. [DOI] [PubMed] [Google Scholar]
- a Salin A. V.; Il’in A. V.; Faskhutdinov R. I.; Fayzullin R. R. Phosphine-Catalyzed Bishydrophosphorylation of Electron-Deficient Alkynes. Tetrahedron 2019, 75, 2676–2686. 10.1016/j.tet.2019.03.042. [DOI] [Google Scholar]; b Cong T.; Wang H.; Li X.; Wu H.-H.; Zhang J. Chiral Bifunctional Bisphosphine Enabled Enantioselective Tandem Michael Addition of Tryptamine-Derived Oxindoles to Ynones. Chem. Commun. 2019, 55, 9176–9179. 10.1039/C9CC04357H. [DOI] [PubMed] [Google Scholar]
- a Huang G.; Ren X.; Jiang C.; Wu J.-H.; Gao G.; Wang T. Efficient Synthesis of (E)-2-Nitromethylcinnamates via Phosphine-Catalyzed Tandem α-Addition and 1,3-Rearrangement. Org. Chem. Front. 2019, 6, 2872–2876. 10.1039/C9QO00478E. [DOI] [Google Scholar]; b Fritzemeier R. G.; Nekvinda J.; Vogels C. M.; Rosenblum C. A.; Slebodnick C.; Westcott S. A.; Santos W. L. Organocatalytic Trans Phosphinoboration of Internal Alkynes. Angew. Chem., Int. Ed. 2020, 59, 14358–14362. 10.1002/anie.202006096. [DOI] [PubMed] [Google Scholar]
- a Nagao K.; Ohmiya H.; Sawamura M. Phosphine-Catalyzed Anti-Carboboration of Alkynoates With Alkyl-, Alkenyl-, and Arylboranes. J. Am. Chem. Soc. 2014, 136, 10605–10608. 10.1021/ja506310v. [DOI] [PubMed] [Google Scholar]; b Nagao K.; Ohmiya H.; Sawamura M. Anti-Selective Vicinal Silaboration and Diboration of Alkynoates Through Phosphine Organocatalysis. Org. Lett. 2015, 17, 1304–1307. 10.1021/acs.orglett.5b00305. [DOI] [PubMed] [Google Scholar]
- a Zhang Y.; Sun Y.; Wei Y.; Shi M. Phosphine-Catalyzed Intermolecular Annulations of Fluorinated ortho-Aminophenones with Alkynones: The Switchable [4 + 2] or [4 + 2]/[3 + 2] Cycloaddition. Adv. Synth. Catal. 2019, 361, 2129–2135. 10.1002/adsc.201900082. [DOI] [Google Scholar]; b Zhang K.; Cai L.; Hong S.; Kwon O. Phosphine-Catalyzed α-Umpolung-Aldol Reaction for the Synthesis of Benzo[b]Azapin-3-Ones. Org. Lett. 2019, 21, 5143–5146. 10.1021/acs.orglett.9b01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cui H.-L.; Tang X.; Li M.-F.; Xu X.-J.; Shi Y. Asymmetric [3 + 2] Cycloaddition of Olefins with Morita-Baylis-Hillman Carbonates Catalyzed by BINOL-Based Bifunctional Phosphine. Synlett 2019, 30, 845–850. 10.1055/s-0037-1611752. [DOI] [Google Scholar]; b Zhang J.; Chan W.-L.; Chen L.; Ullah N.; Lu Y. Creation of Bispiro[pyrazolone-3,3′-oxindoles] via a Phosphine-Catalyzed Enantioselective [3 + 2] Annulation of the Morita-Baylis-Hillman Carbonates with Pyrazoloneyldiene Oxindoles. Org. Chem. Front. 2019, 6, 2210–2214. 10.1039/C9QO00471H. [DOI] [Google Scholar]; c Wang C.; Chen Y.; Li J.; Zhou L.; Wang B.; Xiao Y.; Guo H. Phosphine-Catalyzed Asymmetric Cycloaddition Reaction of Diazenes: Enantioselective Synthesis of Chiral Dihydropyrazoles. Org. Lett. 2019, 21, 7519–7523. 10.1021/acs.orglett.9b02800. [DOI] [PubMed] [Google Scholar]
- a Cheng Y.; Fang Z.; Li W.; Li P. Phosphine-Mediated Enantioselective [4 + 1] Annulations between Ortho-Quinone Methides and Morita-Baylis-Hillman Carbonates. Org. Chem. Front. 2018, 5, 2728–2733. 10.1039/C8QO00487K. [DOI] [Google Scholar]; b Wang T.; Zhang P.; Li W.; Li P. Phosphine-Mediated Enantioselective [1 + 4]-Annulation of Morita-Baylis-Hillman Carbonates with 2-Enoylpyridines. RSC Adv. 2018, 8, 41620–41623. 10.1039/C8RA09453E. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qian C.; Zhang P.; Li W.; Li P. Phosphine-Catalyzed Enantioselective [1 + 4] Annulation of Morita-Baylis-Hillman Carbonates with α,β-Unsaturated Imines. Asian J. Org. Chem. 2019, 8 (2), 242–245. 10.1002/ajoc.201800719. [DOI] [Google Scholar]; d Zhang P.; Guo X.; Liu C.; Li W.; Li P. Enantioselective Construction of Pyridine N-Oxides Featuring 2,3-Dihydrofuran Motifs via Phosphine-Catalyzed [4 + 1]-Annulation of 2-Enoylpyridine N-Oxides with Morita-Baylis-Hillman Carbonates. Org. Lett. 2019, 21, 152–155. 10.1021/acs.orglett.8b03612. [DOI] [PubMed] [Google Scholar]
- a Jin H.; Lai J.; Huang Y. Phosphine-Catalyzed Domino [3 + 3] Cyclization of Para-Quinamines with Morita-Baylis-Hillman Carbonates: Access to Hydroquinoline Derivatives. Org. Lett. 2019, 21, 2843–2846. 10.1021/acs.orglett.9b00834. [DOI] [PubMed] [Google Scholar]; b Chen J.; Yin Z.; Huang Y. Phosphine-Catalyzed Divergent [4 + 3] Domino Annulations of CF3-Containing Imines with MBH Carbonates: Construction of Perfluoroalkylated Benzazepines. Org. Lett. 2019, 21, 7060–7064. 10.1021/acs.orglett.9b02626. [DOI] [PubMed] [Google Scholar]; c Zhou L.; Yuan C.; Zeng Y.; Wang Q.; Wang C.; Liu M.; Wang W.; Wu Y.; Zheng B.; Guo H. Direct Activation of Unmodified Morita-Baylis-Hillman Alcohols Through Phosphine Catalysis for Rapid Construction of Three-Dimensional Heterocyclic Compounds. Org. Lett. 2019, 21, 4882–4886. 10.1021/acs.orglett.9b01783. [DOI] [PubMed] [Google Scholar]
- Behrouz S.; Navid Soltani Rad M.; Abdollahzadeh M.; Amin Piltan M. Ultrasound-Promoted Mild, and Efficient Protocol for Three-Component Synthesis of 2,4,5-Trisubstituted Imidazoles Using Urea and PPh3 as the Sources of Nitrogen and Organocatalyst. ChemistrySelect 2020, 5, 7467–7473. 10.1002/slct.202001722. [DOI] [Google Scholar]
- a Wu J.; Tang Y.; Wei W.; Wu Y.; Li Y.; Zhang J.; Zheng Y.; Xu S. Phosphine-Catalyzed Activation of Vinylcyclopropanes: Rearrangement of Vinylcyclopropylketones to Cycloheptenones. Angew. Chem., Int. Ed. 2018, 57, 6284–6288. 10.1002/anie.201800555. [DOI] [PubMed] [Google Scholar]; b He X.; Tang Y.; Wang Y.; Chen J.-B.; Xu S.; Dou J.; Li Y. Phosphine-Catalyzed Activation of Alkylidenecyclopropanes: Rearrangement to Form Polysubstituted Furans and Dienones. Angew. Chem., Int. Ed. 2019, 58, 10698–10702. 10.1002/anie.201903320. [DOI] [PubMed] [Google Scholar]
- Ma S.; Zhang J. Tuning the Regioselectivity in the Palladium(II)-Catalyzed Isomerization of Alkylidene Cyclopropyl Ketones: A Dramatic Salt Effect. Angew. Chem., Int. Ed. 2003, 42, 183–187. 10.1002/anie.200390073. [DOI] [PubMed] [Google Scholar]
- a Wang H.-Y.; Zhang K.; Zheng C.-W.; Chai Z.; Cao D.-D.; Zhang J.-X.; Zhao G. Asymmetric Dual-Reagent Catalysis: Mannich-Type Reactions Catalyzed by Ion Pair. Angew. Chem., Int. Ed. 2015, 54, 1775–1779. 10.1002/anie.201409342. [DOI] [PubMed] [Google Scholar]; b Wang H.-Y.; Zheng C.-W.; Chai Z.; Zhang J.-X.; Zhao G. Asymmetric Cyanation of Imines via Dipeptide-Derived Organophosphine Dual-Reagent Catalysis. Nat. Commun. 2016, 7, 12720. 10.1038/ncomms12720. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Huang B.; Li C.; Wang H.; Wang C.; Liu L.; Zhang J. Phosphine-Catalyzed Diastereo- and Enantioselective Michael Addition of β-Carbonyl Esters to β-Trifluoromethyl and β-Ester Enones: Enhanced Reactivity by Inorganic Base. Org. Lett. 2017, 19, 5102–5105. 10.1021/acs.orglett.7b02365. [DOI] [PubMed] [Google Scholar]; d Yu Q.-W.; Wu L.-P.; Kang T.-C.; Xie J.; Sha F.; Wu X.-Y. Enantioselective Cyanosilylation of α,α-Dialkoxy Ketones by Using Phosphine-Thiourea Dual-Reagent Catalysis. Eur. J. Org. Chem. 2018, 2018, 3992–3996. 10.1002/ejoc.201800459. [DOI] [Google Scholar]; e Du M.; Yu L.; Du T.; Li Z.; Luo Y.; Meng X.; Tian Z.; Zheng C.; Cao W.; Zhao G. N-Protecting Group Tuning of the Enantioselectivity in Strecker Reactions of Trifluoromethyl Ketimines to Synthesize Quaternary α-Trifluoromethyl Amino Nitriles by Ion Pair Catalysis. Chem. Commun. 2020, 56 (10), 1581–1584. 10.1039/C9CC09151C. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Jiang C.; Tan J.-P.; Hu H.-L.; Chen Y.; Ren X.; Zhang H.-S.; Wang T. Highly Enantioselective Construction of Fully Substituted Stereocenters Enabled by In Situ Phosphonium-Containing Organocatalysis. ACS Catal. 2020, 10, 5698–5706. 10.1021/acscatal.0c01079. [DOI] [Google Scholar]
- a Relles H. M.; Schluenz R. W. Chemical Transformations with Regenerable, Polymer-Supported Trisubstituted Phosphine Dichlorides. Efficacious Incorporation of Phosphorus Reagents on Polymer Supports. J. Am. Chem. Soc. 1974, 96, 6469–6475. 10.1021/ja00827a034. [DOI] [Google Scholar]; b Guinó M.; Hii K. K. M. Applications of Phosphine-Functionalised Polymers in Organic Synthesis. Chem. Soc. Rev. 2007, 36, 608–617. 10.1039/B603851B. [DOI] [PubMed] [Google Scholar]; c Batesky D. C.; Goldfogel M. J.; Weix D. J. Removal of Triphenylphosphine Oxide by Precipitation with Zinc Chloride in Polar Solvents. J. Org. Chem. 2017, 82, 9931–9936. 10.1021/acs.joc.7b00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Marsden S. P. In Sustainable Catalysis; Dunn P. J., Hii K. K., Krische M. J., Williams M. T., Eds.; John Wiley & Sons: New York, 2013; pp 339–361. [Google Scholar]; b van Kalkeren H. A.; van Delft F. L.; Rutjes F. P. J. T. Organophosphorus Catalysis to Bypass Phosphine Oxide Waste. ChemSusChem 2013, 6, 1615–1624. 10.1002/cssc.201300368. [DOI] [PubMed] [Google Scholar]; c An J.; Denton R. M.; Lambert T. H.; Nacsa E. D. The Development of Catalytic Nucleophilic Substitution Reactions: Challenges, Progress and Future Directions. Org. Biomol. Chem. 2014, 12, 2993–3003. 10.1039/c4ob00032c. [DOI] [PubMed] [Google Scholar]; d Xu S.; Tang Y. Catalytic Approaches to Stoichiometric Phosphine-Mediated Organic Reactions. Lett. Org. Chem. 2014, 11, 524–533. 10.2174/1570178611666140401223253. [DOI] [Google Scholar]; e Lao Z.; Toy P. H. Catalytic Wittig and Aza-Wittig Reactions. Beilstein J. Org. Chem. 2016, 12, 2577–2587. 10.3762/bjoc.12.253. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Beddoe R. H.; Sneddon H. F.; Denton R. M. The Catalytic Mitsunobu Reaction: A Critical Analysis of the Current State-of-the-Art. Org. Biomol. Chem. 2018, 16, 7774. 10.1039/C8OB01929K. [DOI] [PubMed] [Google Scholar]
- Denton R. M.; An J.; Adeniran B.; Blake A. J.; Lewis W.; Poulton A. M. Catalytic Phosphorus(V)-Mediated Nucleophilic Substitution Reactions: Development of a Catalytic Appel Reaction. J. Org. Chem. 2011, 76, 6749–6767. 10.1021/jo201085r. [DOI] [PubMed] [Google Scholar]
- a Shi L.; Wang W.; Wang Y.; Huang Y. The First Example of a Catalytic Wittig-Type Reaction. Tri-n-Butylarsine-Catalyzed Olefination in the Presence of Triphenyl Phosphite. J. Org. Chem. 1989, 54, 2027–2028. 10.1021/jo00270a001. [DOI] [Google Scholar]; b Huang Y.-Z.; Shi L.-L.; Li S.-W.; Wen X.-Q. An Efficient Olefination Method Catalysed by Dibutyl Telluride. J. Chem. Soc., Perkin Trans. 1 1989, 2397–2399. 10.1039/p19890002397. [DOI] [Google Scholar]
- a Campbell T. W.; Monagle J. J. A New Synthesis of Mono- and Polycarbodiimides. J. Am. Chem. Soc. 1962, 84, 1493–1493. 10.1021/ja00867a033. [DOI] [Google Scholar]; b Campbell T. W.; Monagle J. J.; Foldi V. S. Carbodiimides. I. Conversion of Isocyanates to Carbodiimides with Phospholine Oxide Catalyst. J. Am. Chem. Soc. 1962, 84, 3673–3677. 10.1021/ja00878a015. [DOI] [Google Scholar]; c Marsden S. P.; McGonagle A. E.; McKeever-Abbas B. Catalytic Aza-Wittig Cyclizations for Heteroaromatic Synthesis. Org. Lett. 2008, 10, 2589–2591. 10.1021/ol800921n. [DOI] [PubMed] [Google Scholar]; d Yan Y.-M.; Li H.-Y.; Ren J.; Wang S.; Ding M.-W. One-Pot Selective Synthesis of Multisubstituted Quinoxalin-2(1H)-ones by a Ugi 4CR/Catalytic Aza-Wittig Sequence. Synlett 2018, 29, 1447–1450. 10.1055/s-0037-1609846. [DOI] [Google Scholar]
- a O’Brien C. J.; Tellez J. L.; Nixon Z. S.; Kang L. J.; Carter A. L.; Kunkel S. R.; Przeworski K. C.; Chass G. A. Recycling the Waste: The Development of a Catalytic Wittig Reaction. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]; b O’Brien C. J.; Lavigne F.; Coyle E. E.; Holohan A. J.; Doonan B. J. Breaking the Ring through a Room Temperature Catalytic Wittig Reaction. Chem. - Eur. J. 2013, 19, 5854–5858. 10.1002/chem.201300546. [DOI] [PubMed] [Google Scholar]; c Coyle E. E.; Doonan B. J.; Holohan A. J.; Walsh K. A.; Lavigne F.; Krenske E. H.; O’Brien C. J. Catalytic Wittig Reactions of Semi- and Nonstabilized Ylides Enabled by Ylide Tuning. Angew. Chem., Int. Ed. 2014, 53, 12907–12911. 10.1002/anie.201406103. [DOI] [PubMed] [Google Scholar]
- Beddoe R. H.; Andrews K. G.; Magné V.; Cuthbertson J. D.; Saska J.; Shannon-Little A. L.; Shanahan S. E.; Sneddon H. F.; Denton R. M. Redox-Neutral Organocatalytic Mitsunobu Reactions. Science 2019, 365, 910–914. 10.1126/science.aax3353. [DOI] [PubMed] [Google Scholar]
- a Han X.; Saleh N.; Retailleau P.; Voituriez A. Phosphine-Catalyzed Reaction Between 2-Aminobenzaldehyde and Dialkyl Acetylenedicarboxylayes: Synthesis of 1,2-Dihydroquinoline Derivatives and Toward the Development of a New Olefination Reaction. Org. Lett. 2018, 20, 4584–4588. 10.1021/acs.orglett.8b01870. [DOI] [PubMed] [Google Scholar]; b Longwitz L.; Spannenberg A.; Werner T. Phosphetane Oxides as Redox Cycling Catalysts in the Catalytic Wittig Reaction at Room Temperature. ACS Catal. 2019, 9, 9237–9244. 10.1021/acscatal.9b02456. [DOI] [Google Scholar]; c Grandane A.; Longwitz L.; Roolf C.; Spannenberg A.; Murua Escobar H.; Junghanss C.; Suna E.; Werner T. Intramolecular Base-Free Catalytic Wittig Reaction: Synthesis of Benzoxepinones. J. Org. Chem. 2019, 84, 1320–1329. 10.1021/acs.joc.8b02789. [DOI] [PubMed] [Google Scholar]
- Longwitz L.; Jopp S.; Werner T. Organocatalytic Chlorination of Alcohols by P(III)/P(V) Redox Cycling. J. Org. Chem. 2019, 84, 7863–7870. 10.1021/acs.joc.9b00741. [DOI] [PubMed] [Google Scholar]
- a Lenstra D. C.; Lenting P. E.; Mecinović J. Sustainable Organophosphorus-Catalysed Staudinger Reduction. Green Chem. 2018, 20, 4418–4422. 10.1039/C8GC02136H. [DOI] [Google Scholar]; b Lenstra D. C.; Wolf J. J.; Mecinović J. Catalytic Staudinger Reduction at Room Temperature. J. Org. Chem. 2019, 84, 6536–6545. 10.1021/acs.joc.9b00831. [DOI] [PubMed] [Google Scholar]
- White P. B.; Rijpkema S. J.; Bunschoten R. P.; Mecinović J. Mechanistic Insight into the Catalytic Staudinger Ligation. Org. Lett. 2019, 21, 1011–1014. 10.1021/acs.orglett.8b04035. [DOI] [PubMed] [Google Scholar]
- Nykaza T. V.; Ramirez A.; Harrison T. S.; Luzung M. R.; Radosevich A. T. Biphilic Organophosphorous-Catalyzed Intramolecular Csp2-H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc. 2018, 140, 3103–3113. 10.1021/jacs.7b13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscales S.; Csáky̅ A. G. Synthesis of Di(hetero)arylamines from Nitrosoarenes and Boronic Acids: A General, Mild, and Transition-Metal-Free Coupling. Org. Lett. 2018, 20, 1667–1671. 10.1021/acs.orglett.8b00473. [DOI] [PubMed] [Google Scholar]
- a Nykaza T. V.; Cooper J. C.; Li G.; Mahieu N.; Ramirez A.; Luzung M. R.; Radosevich A. T. Intermolecular Reductive C-N Cross Coupling of Nitroarenes and Boronic Acids by PIII/PV=O Catalysis. J. Am. Chem. Soc. 2018, 140, 15200–15205. 10.1021/jacs.8b10769. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li G.; Nykaza T. V.; Cooper J. C.; Ramirez A.; Luzung M. R.; Radosevich A. T. An Improved PIII/PV=O-Catalyzed Reductive C-N Coupling of Nitroaromatics and Boronic Acids by Mechanistic Differentiation of Rate- and Product-Determining Steps. J. Am. Chem. Soc. 2020, 142, 6786–6799. 10.1021/jacs.0c01666. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Li G.; Qin Z.; Radosevich A. T. P(III)/P(V)-Catalyzed Methylamination of Arylboronic Acids and Esters: Reductive C-N Coupling with Nitromethane as a Methylamine Surrogate. J. Am. Chem. Soc. 2020, 142, 16205–16210. 10.1021/jacs.0c08035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykaza T. V.; Li G.; Yang J.; Luzung M. R.; Radosevich A. T. PIII/PV=O Catalyzed Cascade Synthesis of N-Functionalized Azaheterocycles. Angew. Chem., Int. Ed. 2020, 59, 4505–4510. 10.1002/anie.201914851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Klunder J. M.; Sharpless K. B. Convenient Synthesis of Sulfinate Esters from Sulfonyl Chlorides. J. Org. Chem. 1987, 52, 2598–2602. 10.1021/jo00388a051. [DOI] [Google Scholar]; b Hoffmann F. W.; Moore T. R.; Kagan B. The Reaction Between Triethyl Phosphite and Alkyl and Aryl-Sulfonyl Chlorides. J. Am. Chem. Soc. 1956, 78, 6413–6414. 10.1021/ja01605a033. [DOI] [Google Scholar]
- Ghosh A.; Lecomte M.; Kim-Lee S.-H.; Radosevich A. T. Organophosphorus-Catalyzed Deoxygenation of Sulfonyl Chlorides: Electrophilic (Fluoroalkyl)Sulfenylation by PIII/PV=O Redox Cycling. Angew. Chem., Int. Ed. 2019, 58, 2864–2869. 10.1002/anie.201813919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Kalkeren H. A.; Leenders S. H. A. M.; Hommersom C. R. A.; Rutjes F. P. J. T.; van Delft F. L. In Situ Phosphine Oxide Reduction: A Catalytic Appel Reaction. Chem. - Eur. J. 2011, 17, 11290–11295. 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; b Lenstra D. C.; Rutjes F. P. J. T.; Mecinović J. Triphenylphosphine-Catalysed Amide Bond Formation between Carboxylic Acids and Amines. Chem. Commun. 2014, 50, 5763–5766. 10.1039/c4cc01861c. [DOI] [PubMed] [Google Scholar]
- Lecomte M.; Lipshultz J. M.; Kim-Lee S.-H.; Li G.; Radosevich A. T. Driving Recursive Dehydration by PIII/PV Catalysis: Annulation of Amines and Carboxylic Acids by Sequential C-N and C-C Bond Formation. J. Am. Chem. Soc. 2019, 141, 12507–12512. 10.1021/jacs.9b06277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burnett M. G.; Oswald T.; Walker B. J. Stoichiometric Reduction of Activated Olefins by Co-Ordinated and Free Tertiary Alkyl Phosphines. J. Chem. Soc., Chem. Commun. 1977, 5, 155–156. 10.1039/c39770000155. [DOI] [Google Scholar]; b Cao S.-H.; Zhang X.-C.; Wei Y.; Shi M. Chemoselective Reduction of Isatin-Derived Electron-Deficient Alkenes Using Alkylphosphanes as Reduction Reagents. Eur. J. Org. Chem. 2011, 2011, 2668–2672. 10.1002/ejoc.201100017. [DOI] [Google Scholar]; c Arbuzova S. N.; Glotova T. E.; Dvorko M. Y.; Ushakov I. A.; Gusarova N. K.; Trofimov B. A. Stereoselective Reduction of 1-Acyl-2-phenylacetylenes With Triphenylphosphine in Water: An Efficient Synthesis of E-Chalcones. ARKIVOC 2011, 2011, 183–188. 10.3998/ark.5550190.0012.b16. [DOI] [Google Scholar]
- Longwitz L.; Werner T. Reduction of Activated Alkenes by PIII/PV Redox Cycling Catalysis. Angew. Chem., Int. Ed. 2020, 59, 2760–2763. 10.1002/anie.201912991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The firsts:; a Trost B. M.; Curran D. P. An enantiodirected cyclopentenone annulation. Synthesis of a Useful Building Block for Condensed Cyclopentanoid Natural Products. J. Am. Chem. Soc. 1980, 102, 5699–5700. 10.1021/ja00537a059. [DOI] [Google Scholar]; b Lertpibulpanya D.; Marsden S. P.; Rodriguez-Garcia I.; Kilner C. A. Asymmetric Aza-Wittig Reactions: Enantioselective Synthesis of t-Quaternary Azacycles. Angew. Chem., Int. Ed. 2006, 45, 5000–5002. 10.1002/anie.200601383. [DOI] [PubMed] [Google Scholar]; For reviews:; c Rein T.; Pedersen T. M. Asymmetric Wittig Type Reactions. Synthesis 2002, 2002, 579–594. 10.1055/s-2002-23535. [DOI] [Google Scholar]; d Zhang K.; Lu L.-Q.; Xiao W.-J. Recent Advances in the Catalytic Asymmetric Alkylation of Stabilized Phosphorous Ylides. Chem. Commun. 2019, 55, 8716–8721. 10.1039/C9CC02831E. [DOI] [PubMed] [Google Scholar]
- a Werner T.; Hoffmann M.; Deshmukh S. First Enantioselective Catalytic Wittig Reaction. Eur. J. Org. Chem. 2014, 2014, 6630–6633. 10.1002/ejoc.201402941. [DOI] [Google Scholar]; b Schneider L. M.; Schmiedel V. M.; Pecchioli T.; Lentz D.; Merten C.; Christmann M. Asymmetric Synthesis of Carbocyclic Propellanes. Org. Lett. 2017, 19, 2310–2313. 10.1021/acs.orglett.7b00836. [DOI] [PubMed] [Google Scholar]; c Schmiedel V. M.; Hong Y. J.; Lentz D.; Tantillo D. J.; Christmann M. Synthesis and Structure Revision of Dichrocephones A and B. Angew. Chem., Int. Ed. 2018, 57, 2419–2422. 10.1002/anie.201711766. [DOI] [PubMed] [Google Scholar]
- Kirk A. M.; O’Brien C. J.; Krenske E. H. Why Do Silanes Reduce Electron-Rich Phosphine Oxides Faster than Electron-Poor Phosphine Oxides?. Chem. Commun. 2020, 56, 1227–1230. 10.1039/C9CC08718D. [DOI] [PubMed] [Google Scholar]
- a Hérault D.; Nguyen D. H.; Nuel D.; Buono G. Reduction of Secondary and Tertiary Phosphine Oxides to Phosphines. Chem. Soc. Rev. 2015, 44, 2508–2528. 10.1039/C4CS00311J. [DOI] [PubMed] [Google Scholar]; b Podyacheva E.; Kuchuk E.; Chusov D. Reduction of Phosphine Oxides to Phosphines. Tetrahedron Lett. 2019, 60, 575–582. 10.1016/j.tetlet.2018.12.070. [DOI] [Google Scholar]
- Cai L.; Zhang K.; Chen S.; Lepage R. J.; Houk K. N.; Krenske E. H.; Kwon O. Catalytic Asymmetric Staudinger-Aza-Wittig Reaction for the Synthesis of Heterocyclic Amines. J. Am. Chem. Soc. 2019, 141, 9537–9542. 10.1021/jacs.9b04803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorton C.; Castanheiro T.; Voituriez A. Catalytic and Asymmetric Process via PIII/PV=O Redox Cycling: Access to (Trifluoromethyl)cyclobutenes via a Michael Addition/Wittig Olefination Reaction. J. Am. Chem. Soc. 2019, 141, 10142–10147. 10.1021/jacs.9b02539. [DOI] [PubMed] [Google Scholar]
- van Kalkeren H. A.; Blom A. L.; Rutjes F. P. J. T.; Huijbregts M. A. J. On the Usefulness of Life Cycle Assessment in Early Chemical Methodology Development: The Case of Organophosphorus-Catalyzed Appel and Wittig Reactions. Green Chem. 2013, 15, 1255–1263. 10.1039/c3gc00053b. [DOI] [Google Scholar]
- Manabe S.; Wong C. M.; Sevov C. S. Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine. J. Am. Chem. Soc. 2020, 142, 3024–3031. 10.1021/jacs.9b12112. [DOI] [PubMed] [Google Scholar]
- a Hu J.; Wang J.; Nguyen T. H.; Zheng N. The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. 10.3762/bjoc.9.234. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Beatty J. W.; Stephenson C. R. J. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nakajima K.; Miyake Y.; Nishibayashi Y. Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1946–1956. 10.1021/acs.accounts.6b00251. [DOI] [PubMed] [Google Scholar]; d Jiang H.; Studer A. Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem. 2019, 1, 38–49. 10.31635/ccschem.019.20180026. [DOI] [Google Scholar]
- a Ramirez F.; Dershowitz S. The Structure of Quinone-Donor Adducts. I. The Action of Triphenylphosphine on p-Benzoquinone, 2,5-Dichloro-p-benzoquinone and Chloranil. J. Am. Chem. Soc. 1956, 78, 5614–5622. 10.1021/ja01602a041. [DOI] [Google Scholar]; b Walling C.; Rabinowitz R. The Reaction of Thiyl Radicals with Trialkyl Phosphites. J. Am. Chem. Soc. 1957, 79, 5326–5326. 10.1021/ja01576a077. [DOI] [Google Scholar]
- a Leca D.; Fensterbank L.; Lacôte E.; Malacria M. Recent Advances in the Use of Phosphorus-Centered Radicals in Organic Chemistry. Chem. Soc. Rev. 2005, 34, 858–865. 10.1039/b500511f. [DOI] [PubMed] [Google Scholar]; b Luo K.; Yang W.-C.; Wu L. Photoredox Catalysis in Organophosphorus Chemistry. Asian J. Org. Chem. 2017, 6, 350–367. 10.1002/ajoc.201600512. [DOI] [Google Scholar]
- Kochi J. K. Electron Transfer and Charge Transfer: Twin Themes in Unifying the Mechanisms of Organic and Organometallic Reactions. Angew. Chem., Int. Ed. Engl. 1988, 27, 1227–1266. 10.1002/anie.198812273. [DOI] [Google Scholar]
- a Lima C. G. S.; de M. Lima T.; Duarte M.; Jurberg I. D.; Paixão M. W. Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. 10.1021/acscatal.5b02386. [DOI] [Google Scholar]; b Yuan Y.; Majumder S.; Yang M.; Guo S. Recent Advances in Catalyst-Free Photochemical Reactions via Electron-Donor-Acceptor (EDA) Complex Process. Tetrahedron Lett. 2020, 61, 151506. 10.1016/j.tetlet.2019.151506. [DOI] [Google Scholar]; c Crisenza G. E. M.; Mazzarella D.; Melchiorre P. Synthetic Methods Driven by the Photoactivity of Electron Donor-Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. 10.1021/jacs.0c01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.-Y.; Zhang H.-Z. Reaction of Perfluoroalkyl Iodides with Alkenes Initiated by Organophosphine and Related Compounds. J. Fluorine Chem. 1990, 50, 133–140. 10.1016/S0022-1139(00)82185-5. [DOI] [Google Scholar]
- a Lumbierres M.; Moreno-Mañas M.; Vallribera A. Addition of Perfluorooctyl Iodide to Alkenes. Catalysis by Triphenylphosphane. Tetrahedron 2002, 58, 4061–4065. 10.1016/S0040-4020(02)00254-5. [DOI] [Google Scholar]; b Zhao L.; Huang Y.; Wang Z.; Zhu E.; Mao T.; Jia J.; Gu J.; Li X.-F.; He C.-Y. Organophosphine-Catalyzed Difluoroalkylation of Alkenes. Org. Lett. 2019, 21, 6705–6709. 10.1021/acs.orglett.9b02314. [DOI] [PubMed] [Google Scholar]; c Helmecke L.; Spittler M.; Baumgarten K.; Czekelius C. Metal-Free Activation of C-I Bonds and Perfluoroalkylation of Alkenes with Visible Light Using Phosphine Catalysts. Org. Lett. 2019, 21, 7823–7827. 10.1021/acs.orglett.9b02812. [DOI] [PubMed] [Google Scholar]
- Fu M.-C.; Shang R.; Zhao B.; Wang B.; Fu Y. Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434. 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Li M.; Mou X.-Q.; He G.; Xue X.-S.; Chen G. Radical C-H Arylation of Oxazoles with Aryl Iodides: Dppf as an Electron-Transfer Mediator for Cs2CO3. Org. Lett. 2018, 20, 1684–1687. 10.1021/acs.orglett.8b00530. [DOI] [PubMed] [Google Scholar]
- a Yu P.; Zheng S.-C.; Yang N.-Y.; Tan B.; Liu X.-Y. Phosphine-Catalyzed Remote β-C-H Functionalization of Amines Triggered by Trifluoromethylation of Alkenes: One-Pot Synthesis of Bistrifluoromethylated Enamides and Oxazoles. Angew. Chem., Int. Ed. 2015, 54, 4041–4045. 10.1002/anie.201412310. [DOI] [PubMed] [Google Scholar]; b Li L.; Ye L.; Ni S.-F.; Li Z.-L.; Chen S.; Du Y.-M.; Li X.-H.; Dang L.; Liu X.-Y. Phosphine-Catalyzed Remote α-C-H Bond Activation of Alcohols or Amines Triggered by the Radical Trifluoromethylation of Alkenes: Reaction Development and Mechanistic Insights. Org. Chem. Front. 2017, 4, 2139–2146. 10.1039/C7QO00500H. [DOI] [Google Scholar]
- Miura T.; Funakoshi Y.; Nakahashi J.; Moriyama D.; Murakami M. Synthesis of Elongated Esters from Alkenes. Angew. Chem., Int. Ed. 2018, 57, 15455–15459. 10.1002/anie.201809115. [DOI] [PubMed] [Google Scholar]
- a Zhang M.; Xie J.; Zhu C. A General Deoxygenation Approach for Synthesis of Ketones from Aromatic Carboxylic Acids and Alkenes. Nat. Commun. 2018, 9, 3517. 10.1038/s41467-018-06019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang M.; Yuan X.-A.; Zhu C.; Xie J. Deoxygenative Deuteration of Carboxylic Acids with D2O. Angew. Chem., Int. Ed. 2019, 58, 312–316. 10.1002/anie.201811522. [DOI] [PubMed] [Google Scholar]; c Stache E. E.; Ertel A. B.; Rovis T.; Doyle A. G. Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage-Independent Activation of Strong C-O Bonds. ACS Catal. 2018, 8, 11134–11139. 10.1021/acscatal.8b03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardy S. W.; Schmidt V. A. Intermolecular Radical Mediated Anti-Markovnikov Alkene Hydroamination Using N-Hydroxyphthalimide. J. Am. Chem. Soc. 2018, 140, 12318–12322. 10.1021/jacs.8b06881. [DOI] [PubMed] [Google Scholar]
- Liu L. L.; Stephan D. W. Radicals Derived from Lewis Acid/Base Pairs. Chem. Soc. Rev. 2019, 48, 3454–3463. 10.1039/C8CS00940F. [DOI] [PubMed] [Google Scholar]
- a Liu L.; Cao L. L.; Shao Y.; Ménard G.; Stephan D. W. A Radical Mechanism for Frustrated Lewis Pair Reactivity. Chem. 2017, 3, 259–267. 10.1016/j.chempr.2017.05.022. [DOI] [Google Scholar]; b Merk A.; Großekappenberg H.; Schmidtmann M.; Luecke M.-P.; Lorent C.; Driess M.; Oestreich M.; Klare H. F. T.; Müller T. Single-Electron Transfer Reactions in Frustrated and Conventional Silylium Ion/Phosphane Lewis Pairs. Angew. Chem., Int. Ed. 2018, 57, 15267–15271. 10.1002/anie.201808922. [DOI] [PubMed] [Google Scholar]; c Soltani Y.; Dasgupta A.; Gazis T. A.; Ould D. M. C.; Richards E.; Slater B.; Stefkova K.; Vladimirov V. Y.; Wilkins L. C.; Willcox D.; Melen R. L. Radical Reactivity of Frustrated Lewis Pairs with Diaryl Esters. Cell Rep. Phys. Chem. 2020, 1, 100016. 10.1016/j.xcrp.2020.100016. [DOI] [Google Scholar]; d Holtrop F.; Jupp A. R.; Leest van N. P.; Dominguez M. P.; Williams R. M.; Brouwer A. M.; Bruin de B.; Ehlers A. W.; Slootweg J. C. Photoinduced and Thermal Single-Electron Transfer to Generate Radicals from Frustrated Lewis Pairs. Chem. - Eur. J. 2020, 26, 9005–9011. 10.1002/chem.202001494. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Holtrop F.; Jupp A. R.; Kooij B. J.; Leest van N. P.; Bruin de B.; Slootweg J. C. Single-Electron Transfer in Frustrated Lewis Pair Chemistry. Angew. Chem., Int. Ed. 2020, 59, 22210–22216. 10.1002/anie.202009717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T.; Itoh J.; Yokota K.; Fuchibe K. Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- Jacques J.; Fouquey C.; Viterbo R. Enantiomeric Cyclic Binaphthyl Phosphoric Acids as Resolving Agents. Tetrahedron Lett. 1971, 12, 4617–4620. 10.1016/S0040-4039(01)97544-6. [DOI] [Google Scholar]
- Hoffmann R.; Howell J. M.; Muetterties E. L. Molecular Orbital Theory of Pentacoordinate Phosphorus. J. Am. Chem. Soc. 1972, 94, 3047–3058. 10.1021/ja00764a028. [DOI] [Google Scholar]
- Oae S.; Uchida Y. Ligand-Coupling Reactions of Hypervalent Species. Acc. Chem. Res. 1991, 24, 202–208. 10.1021/ar00007a003. [DOI] [Google Scholar]
- Finer J.-P.Ligand Coupling Reactions with Heteroaromatic Compounds. Tetrahedron Organic Chemistry Series; Pergamon Press: Oxford, 2009; Vol. 18, p 95. [Google Scholar]
- Newkome G. R.; Hager D. C. A New Contractive Coupling Procedure. Convenient Phosphorus Expulsion Reaction. J. Am. Chem. Soc. 1978, 100, 5567–5568. 10.1021/ja00485a053. [DOI] [Google Scholar]
- a Hilton M. C.; Dolewski R. D.; McNally A. Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc. 2016, 138, 13806–13809. 10.1021/jacs.6b08662. [DOI] [PubMed] [Google Scholar]; b Anderson R. G.; Jett B. M.; McNally A. Selective Formation of Heteroaryl Thioethers via a Phosphonium Ion Coupling Reaction. Tetrahedron 2018, 74, 3129–3136. 10.1016/j.tet.2017.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hilton M. C.; Zhang X.; Boyle B. T.; Alegre-Requena J. V.; Paton R. S.; McNally A. Heterobiaryl Synthesis by Contractive C-C Coupling via P(V) Intermediates. Science 2018, 362, 799–804. 10.1126/science.aas8961. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Anderson R. G.; Jett B. M.; McNally A. A Unified Approach to Couple Aromatic Heteronucleophiles to Azines and Pharmaceuticals. Angew. Chem., Int. Ed. 2018, 57, 12514–12518. 10.1002/anie.201807322. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Patel C.; Mohnike M.; Hilton M. C.; McNally A. A Strategy to Aminate Pyridines, Diazines, and Pharmaceuticals via Heterocyclic Phosphonium Salts. Org. Lett. 2018, 20, 2607–2610. 10.1021/acs.orglett.8b00813. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Boyle B. T.; Hilton M. C.; McNally A. Nonsymmetrical Bis-Azine Biaryls from Chloroazines: A Strategy Using Phosphorus Ligand-Coupling. J. Am. Chem. Soc. 2019, 141, 15441–15449. 10.1021/jacs.9b08504. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Levy J. N.; Alegre-Requena J. V.; Liu R.; Paton R. S.; McNally A. Selective Halogenation of Pyridines Using Designed Phosphine Reagents. J. Am. Chem. Soc. 2020, 142, 11295–11305. 10.1021/jacs.0c04674. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Zi Y.; Schömberg F.; Wagner K.; Vilotijevic I. C-H Functionalization of Benzothiazoles via Thiazol-2-yl-Phosphonium Intermediates. Org. Lett. 2020, 22, 3407–3411. 10.1021/acs.orglett.0c00882. [DOI] [PubMed] [Google Scholar]
- Dolewski R. D.; Hilton M. C.; McNally A. 4-Selective Pyridine Functionalization Reactions via Heterocyclic Phosphonium Salts. Synlett 2018, 29, 8–14. 10.1055/s-0036-1591850. [DOI] [Google Scholar]
- a Dohi T.; Kita Y. Hypervalent Iodine Reagents as a New Entrance to Organocatalysts. Chem. Commun. 2009, 16, 2073–2085. 10.1039/b821747e. [DOI] [PubMed] [Google Scholar]; b Dunn N. L.; Ha M.; Radosevich A. T. Main Group Redox Catalysis: Reversible PIII/PV Redox Cycling at a Phosphorus Platform. J. Am. Chem. Soc. 2012, 134, 11330–11333. 10.1021/ja302963p. [DOI] [PubMed] [Google Scholar]; c Planas O.; Wang F.; Leutzsch M.; Cornella J. Fluorination of Arylboronic Esters Enabled by Bismuth Redox Catalysis. Science 2020, 367, 313–317. 10.1126/science.aaz2258. [DOI] [PubMed] [Google Scholar]; d Janssen-Müller D.; Oestreich M. Transition-Metal-Like Catalysis with a Main-Group Element: Bismuth-Catalyzed C-F Coupling of Aryl Boronic Esters. Angew. Chem., Int. Ed. 2020, 59, 8328–8330. 10.1002/anie.201914729. [DOI] [PubMed] [Google Scholar]
- a Mann F. G.; Watson J. Conditions of Salt Formation in Polyamines and Kindred Compounds. Salt Formation in the Tertiary 2-Pyridylamine, Phosphines and Arsines. J. Org. Chem. 1948, 13, 502–531. 10.1021/jo01162a007. [DOI] [Google Scholar]; b Uchida Y.; Onoue K.; Tada N.; Nagao F.; Oae S. Ligand Coupling Reaction on the Phosphorus Atom. Tetrahedron Lett. 1989, 30, 567–570. 10.1016/S0040-4039(00)95256-0. [DOI] [Google Scholar]; c Uchida Y.; Kozawa H. Formation of 2,2′-Bipyridyl by Ligand Coupling on the Phosphorus Atom. Tetrahedron Lett. 1989, 30, 6365–6368. 10.1016/S0040-4039(01)93895-X. [DOI] [Google Scholar]; d Uchida Y.; Onoue K.; Tada N.; Nagao F.; Kozawa H.; Oae S. Reactions of 2-Pyridyl Substituted Phosphine Oxides and Phosphonium Salts with Organometallic Reagents and in Aqueous Media. Heteroat. Chem. 1990, 1, 295–306. 10.1002/hc.1990.1.4.295. [DOI] [Google Scholar]; e Uchida Y.; Echikawa N.; Oae S. Reactions of Heteroaryllithium Compounds with Phosphorus Trichloride, Phosphorus Oxychloride, and Thionyl Chloride. Formation of Heterocyclic Biaryls. Heteroat. Chem. 1994, 5, 409–413. 10.1002/hc.520050414. [DOI] [Google Scholar]
- a Wittig G.; Maercker A. Zur Reaktionsweise Aromatischer Spirophosphorane. Chem. Ber. 1964, 97, 747–768. 10.1002/cber.19640970319. [DOI] [Google Scholar]; b Seyferth D.; Fogel J.; Heeren J. K. Studies in Phosphinemethylene Chemistry. XV. The Reaction of Tetraarylphosphonium Bromides with Vinylic Organolithium Reagents1. J. Am. Chem. Soc. 1966, 88, 2207–2212. 10.1021/ja00962a024. [DOI] [Google Scholar]
- a Anders E.; Markus F. Neue Methode Zur Regiospezifischen Substitution Einiger Reaktionsträcer N-Heteroaromatischer Ringsysteme. Tetrahedron Lett. 1987, 28, 2675–2676. 10.1016/S0040-4039(00)96178-1. [DOI] [Google Scholar]; b Anders E.; Markus F. Chemie Der Triphenyl-(Oder Tri-n-Butyl-)Pyridylphosphoniumsalze, 1 Neue Methode Zur Regioselektiven Einführung von Phosphoniumgruppen in N-Heteroaromatische Ringsysteme. Chem. Ber. 1989, 122, 113–118. 10.1002/cber.19891220118. [DOI] [Google Scholar]; c Haase M.; Goerls H.; Anders E. Synthesis of PO(OR)2- and PR3+-Disubstituted Pyridines via N-(Trifluoromethylsulfonyl)Pyridinium Triflates. Synthesis 1998, 1998, 195–200. 10.1055/s-1998-2012. [DOI] [Google Scholar]