

Abstract
Protein tyrosine phosphorylation is a key regulatory mechanism in eukaryotic cell physiology. Aberrant expression or function of protein tyrosine kinases and protein tyrosine phosphatases can lead to serious human diseases, including cancer, diabetes, as well as cardiovascular, infectious, autoimmune, and neuropsychiatric disorders. Here, we give an overview of the protein tyrosine phosphatase superfamily with its over 100 members in humans. We review their structure, function, and implications in human diseases, and discuss their potential as novel drug targets, as well as current challenges and possible solutions to developing therapeutics based on these enzymes.
Keywords: Tyrosine phosphorylation, Phosphatases, PTP, Structure, Function, Substrate recognition, Specificity, Human disease, PTP1B, SHP2, Inhibitors
1. Introduction
Protein phosphorylation is a fundamental regulatory mechanism for numerous important aspects of cell physiology throughout all kingdoms of life [1–6]. Protein phosphorylation is a rapidly reversible posttranslational modification, controlled by the opposing activities of protein kinases and protein phosphatases. A change in phosphorylation state can be the result of a change in the activity or access of either enzyme. In eukaryotes, about one third of the entire proteome is phosphorylated, predominantly on serine, threonine, and tyrosine [7]. While phosphorylation on serine/threonine is most abundant (~98 %), tyrosine phosphorylation [8], which accounts for less than 2 % of the total phosphoproteome, is a key regulatory mechanism in numerous important aspects of eukaryotic cell physiology [9–17]. The importance of tyrosine phosphorylation in normal cell physiology is highlighted by the fact that many human diseases are the result of aberrant protein tyrosine kinase (PTK) [18] or protein tyrosine phosphatase (PTP) function [9, 13, 19]. Historically, research had long been focused on the role of PTKs in signaling, as it was generally believed that PTPs functioned merely as indiscriminate “housekeeping” enzymes with broad specificities. Now, we know that PTPs are highly specific and tightly regulated, both in space and time [9, 13]. Here, we review the PTP family, describe their classification into subfamilies, discuss their common structural features and shared catalytic mechanism, analyze their substrate recognition and specificity, and finally review their implication in human disease and highlight their potential as novel drug targets and challenges that need to be overcome in order to develop therapeutics based on these pivotal enzymes.
2. The Human PTP Superfamily
PTPs constitute the largest family of phosphatase genes and are defined by their active-site signature motif C(X)5R. The catalytic mechanism of PTPs (as discussed in more detail below) is based on a nucleophilic cysteine residue that is part of this motif. The complement of human PTP genes can be divided into three distinct families based on the sequence of their PTP domains: class I PTPs (100 genes), class II PTPs (1 gene), and class III PTPs (3 genes) (Table 1) [9]. The class I PTPs constitute the vast majority of human PTP genes and can be divided into the classical, phosphotyrosine (pTyr)- specific PTP, and the VH1-like or dual specificity protein phosphatase (DSP, DUSP) subfamilies. The pTyr-specific PTPs (37 genes) are further divided into the transmembrane receptor-like PTPs (RPTPs) and the intracellular nonreceptor-like PTPs (NRPTPs) [9, 13, 20]. The DUSP subfamily (63 genes) is the most diverse group in terms of substrate specificity. This group includes the phosphothreonine (pThr)/pTyr-specific mitogen- activated protein (MAP) kinase phosphatases (MKPs), the pThr/pTyr- or mRNA-specific atypical DSPs, the phosphoserine (pSer)-specific slingshots, the pTyr-specific phosphatases of regenerating liver (PRLs), the pSer/pThr-specific CDC14s, the PTENs, which are specific for the 3’-phosphate of the inositol ring in phosphatidylinositol-(3,4,5)-triphosphate, the myotubularins, which dephosphorylate phosphatidylinositol3-phosphate and phosphatidylinositol-(3,5)-bisphosphate, as well as the inositol 4- phosphatases, which catalyze the hydrolysis of the 4’-phosphate of phosphatidylinositol-(3,4)-bisphosphate, inositol-(1,3,4)-trisphosphate, and inositol(3,4)-bisphosphate [9, 21, 22]. All class I PTPs appear to have evolved from a common ancestor, based on their similar structural folds [20]. The class II PTP family is represented by only a single gene, which encodes a pTyr-specific low molecular weight PTP (LMPTP) [23] that is structurally related to bacterial arsenate reductases and appears to be more ancient than class I PTPs. Representatives of this family are found in all major phyla, including plants, numerous prokaryotes, and archaea [9]. The class III PTP family comprises the pThr/pTyr-specific CDC25s, which appear to have evolved from bacterial rhodanese- like enzymes [20].
Table 1.
The human PTPome
| Gene | Protein, synonymes | UniProt | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transmembrane receptor-like PTPs (RPTPs) | PTPRA | RPTPα | P18433 | X | X | ||||||||||||||||||||||||||||||||||
| PTPRB | RPTPβ | B2RU80 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRC | CD45, LCA | P08575 | X | X | X | ||||||||||||||||||||||||||||||||||
| PTPRD | RPTPδ | P23468 | X | X | X | X | |||||||||||||||||||||||||||||||||
| PTPRE | RPTPε | P23469 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRF | LAR | P10586 | X | X | X | X | |||||||||||||||||||||||||||||||||
| PTPRG | RPTPγ | P23470 | X | X | X | X | |||||||||||||||||||||||||||||||||
| PTPRH | SAP1 | Q9HD43 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRJ | DEP1, CD148, RPTPη | Q12913 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRK | RPTPκ | Q15262 | X | X | X | X | X | X | |||||||||||||||||||||||||||||||
| PTPRM | RPTPμ | P28827 | X | X | X | X | X | X | |||||||||||||||||||||||||||||||
| PTPRN | IA-2, Islet cell antigen 512 | Q16849 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRN2 | PTPRP, RPTPπ, IA-2β, phogrin | Q92932 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRO | GLEPP1, PTP-U2, PTPROτ isoforms A/B/C | Q16827 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRQ | PTPS31 | Q9UMZ3 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRR | PTP-SL, PCPTP, PTPBR7, PC12-PTP1 | Q15256 | X | X | |||||||||||||||||||||||||||||||||||
| PTPRS | RPTPσ | Q13332 | X | X | X | X | |||||||||||||||||||||||||||||||||
| PTPRT | RPTPρ | Q99M80 | X | X | X | X | X | X | |||||||||||||||||||||||||||||||
| PTPRU | PTPJ/PTP-U1/PTPRomicron isoforms 1/2/3 | Q92729 | X | X | X | X | X | X | |||||||||||||||||||||||||||||||
| PTPRZ | RPTPζ | P23471 | X | X | X | X | |||||||||||||||||||||||||||||||||
| Intracellular nonreceptor-like PTPs (NRPTPs) | PTPN1 | PTP1B | P18031 | X | |||||||||||||||||||||||||||||||||||
| PTPN2 | TCPTP, MPTP, PTP-S | P17706 | X | ||||||||||||||||||||||||||||||||||||
| PTPN3 | PTPH1 | P26045 | X | X | X | ||||||||||||||||||||||||||||||||||
| PTPN4 | PTP-MEG1, TEP | P29074 | X | X | X | ||||||||||||||||||||||||||||||||||
| PTPN5a | STEP | P54829 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN6 | SHP1, PTP1C, SH-PTP1, HCP | P29350 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN7 | HePTP, LCPTP | P35236 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN9 | PTP-MEG2 | P43378 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN11 | SHP2, SH-PTP2, Syp, PTP1D, PTP2C, SH-PTP3 | Q06124 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN12 | PTP-PEST, PTP-P19, PTPG1 | Q05209 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN13 | PTP-BAS, FAP-1, PTP1E, RIP, PTPL1, PTP-BL | Q12923 | X | X | X | ||||||||||||||||||||||||||||||||||
| PTPN14 | PTP36, PEZ, PTPD2 | Q15678 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN18 | PTP-HSCF, PTP20, BDP | Q99952 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN20 | TypPTP | Q4JDL3 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN21 | PTPD1, PTP2E, PTP-RL10 | Q16825 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN22 | LYP | Q9Y2R2 | X | X | |||||||||||||||||||||||||||||||||||
| PTPN23 | HD-PTP, HDPTP, PTP-TD14, KIAA1471 | Q9H3S7 | X | X | X | X | |||||||||||||||||||||||||||||||||
| Map kinase phosphatases (MKPs) | DUSP1 | MKP-1, 3CH134, PTPN10, erp, CL100/HVH1 | P28562 | X | X | ||||||||||||||||||||||||||||||||||
| DUSP2 | PAC-1 | Q05923 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP4 | MKP-2, hVH2/TYP1 | Q13115 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP5 | hVH3/B23 | Q16690 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP6 | MKP-3, PYST1 | Q16828 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP7 | MKP-X, PYST2, B59 | Q16829 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP8 | hVH5, M3/6, HB5 | Q13202 | X | X | X | ||||||||||||||||||||||||||||||||||
| DUSP9 | MKP-4, Pyst3 | Q99956 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP10 | MKP-5 | Q9Y6W6 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP16 | MKP-7, MKP-M | Q9BY84 | X | X | X | ||||||||||||||||||||||||||||||||||
| DUSP24 | MK-STYX, MKSTYX, DUSP24, STYXL1 | Q9Y6J8 | X | X | |||||||||||||||||||||||||||||||||||
| Atypical dual-specificity phosphatases | DUSP3 | VHR, T-DSP11 | P51452 | X | |||||||||||||||||||||||||||||||||||
| DUSP11 | PIR1 | O75319 | X | ||||||||||||||||||||||||||||||||||||
| DUSP12 | HYVH1, GKAP, LMW-DSP4 | Q9UNI6 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP13Ab | BEDP | Q6B8I1 | X | ||||||||||||||||||||||||||||||||||||
| DUSP13Bb | TMDP, TS-DSP6 | Q9UII6 | X | ||||||||||||||||||||||||||||||||||||
| DUSP14 | MKP6, MKP-L | O95147 | X | ||||||||||||||||||||||||||||||||||||
| DUSP15 | VHY, Q9H1R2 | Q9H1R2 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP18 | DUSP20, LMW-DSP20 | Q8NEJ0 | X | ||||||||||||||||||||||||||||||||||||
| DUSP19 | DUSP17, SKRP1, LDP-2, TS-DSP1 | Q8WTR2 | X | ||||||||||||||||||||||||||||||||||||
| DUSP21 | LMW-DSP21, BJ-HCC-26 tumor antigen | Q9H596 | X | ||||||||||||||||||||||||||||||||||||
| DUSP22 | VHX, MKPX, JSP1, LMW-DSP2, TS-DSP2, JKAP | Q9NRW4 | X | X | |||||||||||||||||||||||||||||||||||
| DUSP23 | VHZ, DUSP25, FLJ20442, LMW-DSP3 | Q9BVJ7 | X | ||||||||||||||||||||||||||||||||||||
| DUSP26 | MKP-8, DUSP24, LDP4, NATA1, SKRP3 | Q9BV47 | X | ||||||||||||||||||||||||||||||||||||
| PTPMT1 | MOSP, PLIP | Q8WUK0 | X | ||||||||||||||||||||||||||||||||||||
| DUSP27c | FMDSP, DUPD1, ‘similar to cyclophilin’, | Q5VZP5 | X | ||||||||||||||||||||||||||||||||||||
| DUSP28 | VHP, DUSP26 | Q4G0W2 | X | ||||||||||||||||||||||||||||||||||||
| EPM2A | Laforin | O95278 | X | X | |||||||||||||||||||||||||||||||||||
| RNGTT | mRNA capping enzyme | O60942 | X | X | |||||||||||||||||||||||||||||||||||
| STYX | STYX | Q8WUJ0 | X | ||||||||||||||||||||||||||||||||||||
| Slingshots | SSH1 | SSH1, slingshot 1 | Q8WYL5 | X | X | ||||||||||||||||||||||||||||||||||
| SSH2 | SSH2, slingshot 2 | Q76I76 | X | X | |||||||||||||||||||||||||||||||||||
| SSH3 | SSH3, slingshot 3 | Q8TE77 | X | X | |||||||||||||||||||||||||||||||||||
| PRLs | PTP4A1 | PRL-1 | Q93096 | X | X | ||||||||||||||||||||||||||||||||||
| PTP4A2 | PRL-2, OV-1 | Q12974 | X | X | |||||||||||||||||||||||||||||||||||
| PTP4A3 | PRL-3 | O75365 | X | X | |||||||||||||||||||||||||||||||||||
| CDC14s | CDC14A | CDC14A | Q9UNH5 | X | X | ||||||||||||||||||||||||||||||||||
| CDC14B | CDC14B | O60729 | X | X | |||||||||||||||||||||||||||||||||||
| CDKN3 | KAP | Q16667 | X | ||||||||||||||||||||||||||||||||||||
| PTPDC | PTPDC, PTP9Q22 | A2A3K4 | X | ||||||||||||||||||||||||||||||||||||
| PTENs | PTEN | PTEN, MMAC1, TEP1 | P60484 | X | X | X | |||||||||||||||||||||||||||||||||
| TPIP | TPIPα, TPTE and PTEN homologous | Q6XPS3 | X | X | X | ||||||||||||||||||||||||||||||||||
| TPTE | PTEN-like, PTEN2 | P56180 | X | X | X | ||||||||||||||||||||||||||||||||||
| TNS1 | Tensin-1, TNS | Q9HBL0 | X | X | X | ||||||||||||||||||||||||||||||||||
| TENC1 | C1-TEN, TENC1, KIAA1075, TNS2 | Q63HR2 | X | X | X | X | |||||||||||||||||||||||||||||||||
| TNS3 | Tensin 3 | Q68CZ2 | X | X | X | ||||||||||||||||||||||||||||||||||
| Myotubularins | MTM1 | Myotubularin | Q13496 | X | X | X | X | ||||||||||||||||||||||||||||||||
| MTMR1 | MTMR1 | Q13613 | X | X | X | X | |||||||||||||||||||||||||||||||||
| MTMR2 | MTMR2 | Q13614 | X | X | X | X | |||||||||||||||||||||||||||||||||
| MTMR3 | MTMR3, FYVE-DSP1 | Q13615 | X | X | X | X | |||||||||||||||||||||||||||||||||
| MTMR4 | MTMR4, FYVE-DSP2 | Q9NYA4 | X | X | X | X | |||||||||||||||||||||||||||||||||
| MTMR5 | MTMR5, SBF1 | O95248 | X | X | X | X | X | ||||||||||||||||||||||||||||||||
| MTMR6 | MTMR6 | Q9Y217 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR7 | MTMR7 | Q9Y216 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR8 | MTMR8 | Q96EF0 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR9 | MTMR9, LIP-STYX | Q96QG7 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR10 | MTMR10 | Q9NXD2 | X | X | |||||||||||||||||||||||||||||||||||
| MTMR11 | MTMP11, CRA α/β | A4FU01 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR12 | MTMR12, 3-PAP | Q9C0I1 | X | X | X | ||||||||||||||||||||||||||||||||||
| MTMR13 | MTMR13, SBF2, CMT4B2 | Q86WG5 | X | X | X | X | X | ||||||||||||||||||||||||||||||||
| MTMR14d | hJumpy, FLJ22075, hEDTP | Q8NCE2 | X | ||||||||||||||||||||||||||||||||||||
| Inositol 4-phosphatases | INPP4A | Inositol 4-phosphatase type I | Q96PE3 | X | |||||||||||||||||||||||||||||||||||
| INPP4B | Inositol 4-phosphatase type II | O15327 | X | ||||||||||||||||||||||||||||||||||||
| Class II PTP | ACP1 | LMPTP, low Mr PTP, LMWPTP, BHPTP | P24666 | X | |||||||||||||||||||||||||||||||||||
| Class III PTPs | CDC25A | CDC25A | P30304 | X | |||||||||||||||||||||||||||||||||||
| CDC25B | CDC25B | P30305 | X | ||||||||||||||||||||||||||||||||||||
| CDC25C | CDC25C | P30307 | X | ||||||||||||||||||||||||||||||||||||
Current data indicate that there are 104 PTP genes in humans, including 100 class I, one class II, and three class III PTP genes
The earlier predicted human OST-PTP (Andersen et al., 2004, FASEB J, 18, 8–30) is not included here. OST-PTP is an active PTP in mouse and rat, but the human OST-PTP cDNA sequence exhibits numerous disablements, indicating that it does not code for a PTP but is rather a pseudogene (Cousin et al., 2004, Biochem Biophys Res Commun, 321, 259–65)
The earlier predicted myotubularin-related phosphatase MTMR15 (Alonso et al., 2004, Cell, 117, 699–711) is not included here. The protein (KIAA1018/FAN1) was identified as a DNA repair nuclease (MacKay et al., 2010, Cell, 142, 65–76)
STEP exists as two major alternatively spliced isoforms, STEP61 and STEP46. STEP61 includes additional 172 amino acids in its amino-terminal region, which contains two transmembrane domains and two proline-rich motifs
The human DUSP13 gene contains two DSP genes: DUSP13A (BEDP; 3 exons), two noncoding exons, followed by DUSP13B (TMDP; 3 exons). In the mouse, the two genes are designated as separate genes
Formerly DUPD1, but does not contain an N-terminal cyclophilin domain, as originally annotated. The mouse ortholog also lacks cyclophilin domain. Thus, the name DUPD1 is a misnomer
hJumpy lacks any of the typical domains characteristic of the myotubularin family, but it does contain the myotubularin-specif WDR motif within the P-loop and does have phosphatidylinositol-3-phosphate and phosphatidylinositol-(3,5)-bisphosphate phosphatase activity (Tosch et al., 2006, Hum Mol Genet, 15, 3098–106)
Domains and motifs are ordered by frequency of occurrence:
1 PTP domain, 2 Pseudo-PTP domain, 3 Fibronectin type III-like domain, 4 Coiled-coil motif, 5 Pleckstrin homology-glucosyltransferase, Rab-like GTPase activator and myotubularin (PH-GRAM) domain, 6 CDC25-homolgy (CH2) domain, 7 Immunoglobulin-like domain, 8 Proline-rich motif, 9 FERM domain, 10 Phosphotyrosine binding domain, 11 Src-homology-2 (SH2) domain, 12 Cadherin-like juxtamembrane sequence, 13 Meprin/A5/μ domain (MAM), 14 PDZ-binding sequence, 15 C2 domain, 16 CAAX sequence/farnesylation site, 17 Kinase-interaction motif (KIM), 18 PDZ domain, 19 Slingshot domain, 20 Carbonic anhydrase-like domain, 21. DENN domain, 22 DSP-like domain, 23 FYVE domain, 24 Heavily glycosylated, 25 Potential myristoylation site, 26 Putative transmembrane segment, 27 RDGS-adhesion recognition motif, 28 BRO1-homolgy domain, 29 C1 domain, 30 Carbohydrate-binding domain, 31 Cysteine-rich zinc-binding domain, 32 Guanylyltransferase domain, 33 Histidine domain, 34 Pleckstrin homology (PH) domain, 35 SEC14/cellular retinaldehyde-binding protein-like domain
3. The Mechanism of PTP Catalysis
The mechanism of PTP catalysis was elucidated by site-directed mutagenesis and kinetic analyses [24–26], in conjunction with structural information [27–29, 44, 58]. All PTPs share a common catalytic mechanism based on a nucleophilic cysteine with a low pKa that forms a thiophosphate intermediate during catalysis (Fig. 1). The transition state of the reaction is stabilized by an invariant arginine, and hydrolysis is assisted by a catalytic acid/base aspartate. The ordered uni–bi two-step reaction is facilitated by an optimal arrangement of these residues, which are located in a number of highly conserved loops that form the active site. In the first step of the reaction, the catalytic cysteine of the phosphate-binding loop (P-loop) initiates the breaking of the phosphorus–oxygen bond via a nucleophilic attack on the phosphorous atom, while the catalytic aspartate of the WPD-loop acts as a general acid, donating a proton to the OH-leaving group. This step results in the formation of a phosphocysteine intermediate and the release of the dephosphorylated substrate. In the second, rate-limiting step, the thiophosphate intermediate is hydrolyzed, again with the assistance of the catalytic aspartate in the WPD-loop, which now acts as a general base to abstract a proton from a water molecule, thus facilitating hydrolysis of the scissile phosphorous-sulfur bond and producing free phosphate. In classical PTPs, the water molecule used for hydrolysis is positioned by a conserved glutamine located in the Q-loop. The guanidinium group of an invariant arginine of the P-loop coordinates the phosphate group during substrate binding and, more importantly, it stabilizes the transition state of the first step of the enzymatic reaction. While mutational studies have shown that PTPs lacking the aspartate or arginine still have residual phosphatase activity, PTPs with mutations in the catalytic cysteine are completely inactive [30]. Interestingly, despite a highly conserved active site, different PTPs, even within one subfamily, show different catalytic efficiencies toward the general PTP substrate p-nitrophenylphosphate (pNPP). For example, the Yersinia PTP YopH [31] is approximately tenfold more catalytically efficient than human PTP1B [64], which is in turn approximately tenfold more catalytically efficient than human hematopoietic PTP (HePTP) [32]. Thus, even small changes in the active site microenvironment can significantly affect the efficiency of PTP catalysis. One known determinant of the catalytic rate is the flexibility of the WPD-loop, which cycles between an open conformation in the apo structure, and a closed, active conformation when substrate is bound in the active site [33]. Point mutations or specific ligands that decrease the flexibility of this loop result in substantially lower PTP catalytic activity [34, 35].
Fig. 1.
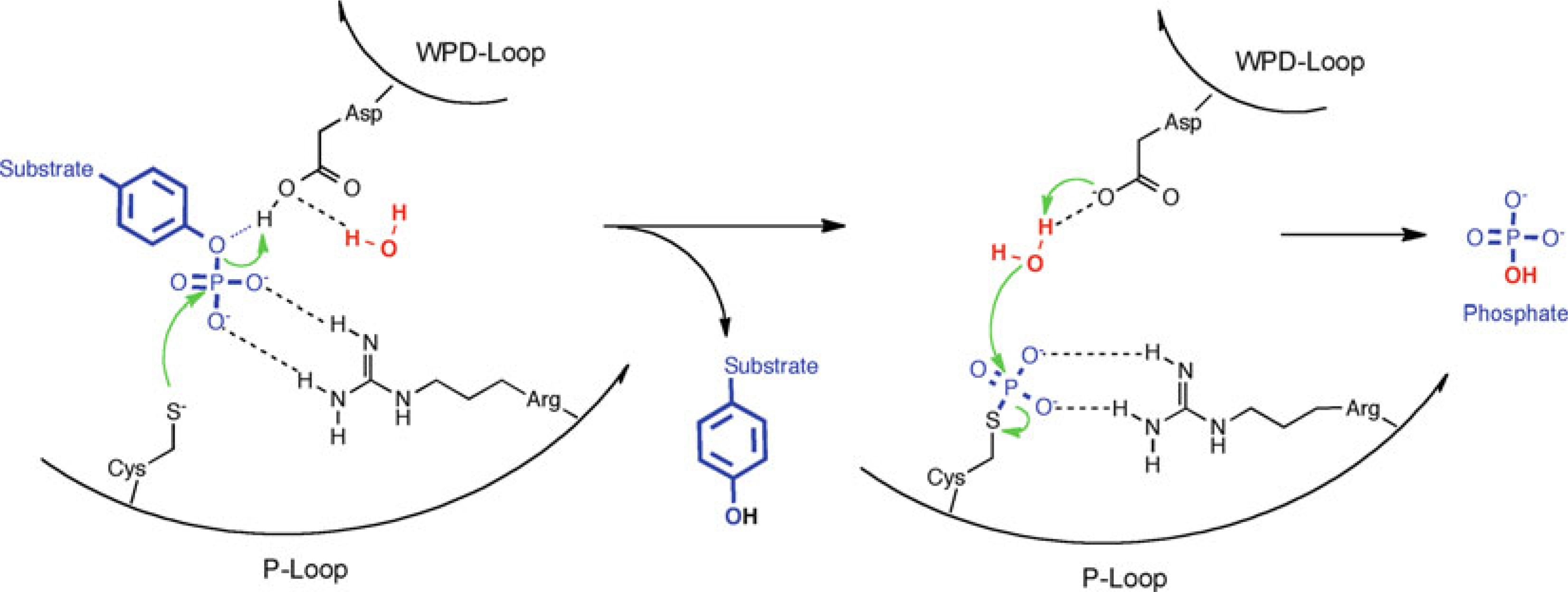
Common PTP catalytic mechanism
4. Modular Domain Arrangement of Human PTPs
Alongside the ~280 residue PTP catalytic domain, most human PTPs also contain additional non-catalytic domains or motifs (Table 1), which provide a variety of different functions. For example, MKPs contain CDC25-homology (CH2) domains that mediate binding to their MAP kinase substrates. Similarly, HePTP (PTPN7), STEP (PTPN5), and PTP-SL (PTPRR) contain a 16-residue kinase interaction motif (KIM) that is essential for their ability to bind to MAP kinase substrates. In contrast, binding of a proline-rich motif in LYP (PTPN22) to the Src-homology 3 (SH3) domain in C-src kinase (CSK) dictates subcellular localization and restrains LYP substrate access [36]. Other examples of PTPs with domains/motifs that dictate subcellular localization include PTPH1 (PTPN3), PTP-MEG1 (PTPN4), and PTP-BAS (PTPN13), which contain PDZ domains for targeting to the cytoskeleton, as well as PAC-1 (DUSP2), MKP-1 (DUSP1), MKP-2 (DUSP4), MKP-3 (DUSP6), and MKP-7 (DUSP16), which contain signals for nuclear localization. Interestingly, some non-catalytic domains/motifs play multiple roles in PTP regulation. For example, SHP1 (PTPN6) and SHP2 (PTPN11) each contain two tandem Src-homology 2 (SH2) domains for targeting to specific tyrosine-phosphorylated proteins upon cell stimulation [37]. However, in unstimulated cells, one of the two SH2 domains folds upon the PTP domain and auto-inhibits SHP1/2 activity by blocking access to the active site [38, 39]. RPTPs have extracellular domains such as immunoglobulin, fibronectin, MAM, and carbonic anhydrase domains that are utilized to interact with the extracellular milieu in a receptor-like fashion. Many RPTPs also contain a second PTP domain that lacks phosphatase activity due to a single or multiple mutations of catalytically essential amino acids. These C-terminal pseudo-PTP domains presumably function as regulatory [40] or substrate-binding [41, 42] domains. Intriguingly, the multidomain structure of PTPs is in stark contrast to the architecture of protein serine/threonine phosphatases (PSPs). The PSP superfamily constitutes only ten apoenzymes, consisting of a small catalytic subunit that binds regulatory or targeting subunits encoded by separate genes [43]. Together, these subunits form a large number of distinct holoenzymes with different biological functions. Presumably, this combinatorial principle can generate more diversity and flexibility among the PSPs, but may lack some of the specificity and tight regulation possible with single-chain multidomain PTPs [9]. In this context it is noteworthy that several members of the atypical DSP subgroup of PTPs consist of only a very small catalytic core structure and do not contain any other domains or motifs [9]. It is possible that these enzymes participate in multisubunit complexes, similarly to PSPs, but no examples have yet been found. To summarize, most PTPs have a modular arrangement of individual extracatalytic domains and motifs that provide substrate specificity, dictate subcellular localization, furnish receptor-like functions, and even alter phosphatase activity.
5. Structure of the PTP Domain
Significant progress has been made in the past two decades toward understanding PTP specificity on a molecular level. Numerous atomic resolution structures of human PTP catalytic domains are now available, with representatives from every human PTP family and subfamily (Fig. 2). PTP1B was the first PTP whose structure was determined to high resolution [44]. With >100 structures solved to date, PTP1B often serves as a reference for PTP structures, the overall fold of which is extremely well conserved, consisting of a central twisted β-sheet surrounded by an arrangement of α-helices (Fig. 3). The active site architecture is formed by several loop regions: (1) the phosphate-binding loop (P-loop), which contains the PTP signature motif with the catalytic cysteine and the invariant arginine; (2) the WPD-loop, which contains the catalytic acid/base aspartate; (3) the Q-loop, which contains a conserved glutamine that coordinates the water molecule necessary for hydrolysis; (4) the pTyr-recognition loop (pTyr-loop), which contains a conserved motif that provides specificity for pTyr in classical PTPs; and (5) the E-loop, which contains multiple conserved residues that appear to coordinate the dynamics of the WPD-loop. Interestingly, PTP function has different structural requirements among the various subfamilies. In fact, the P-loop is the only active site feature that is present in all PTPs. For instance, the WPD-loop with the catalytic aspartate, although highly conserved, is not found in the myotubularins and CDC25s, and apparently is not necessary for PTP activity in these enzymes. The pTyr-recognition loop is only present in the classical, pTyr-specific PTPs, which use this loop to form a deep catalytic cleft in order to discriminate between pTyr and the shorter pSer/pThr residues. In contrast, DSPs, which do not possess an equivalent loop, have a much shallower active site, allowing pSer/pThr to fully penetrate the catalytic center.
Fig. 2.
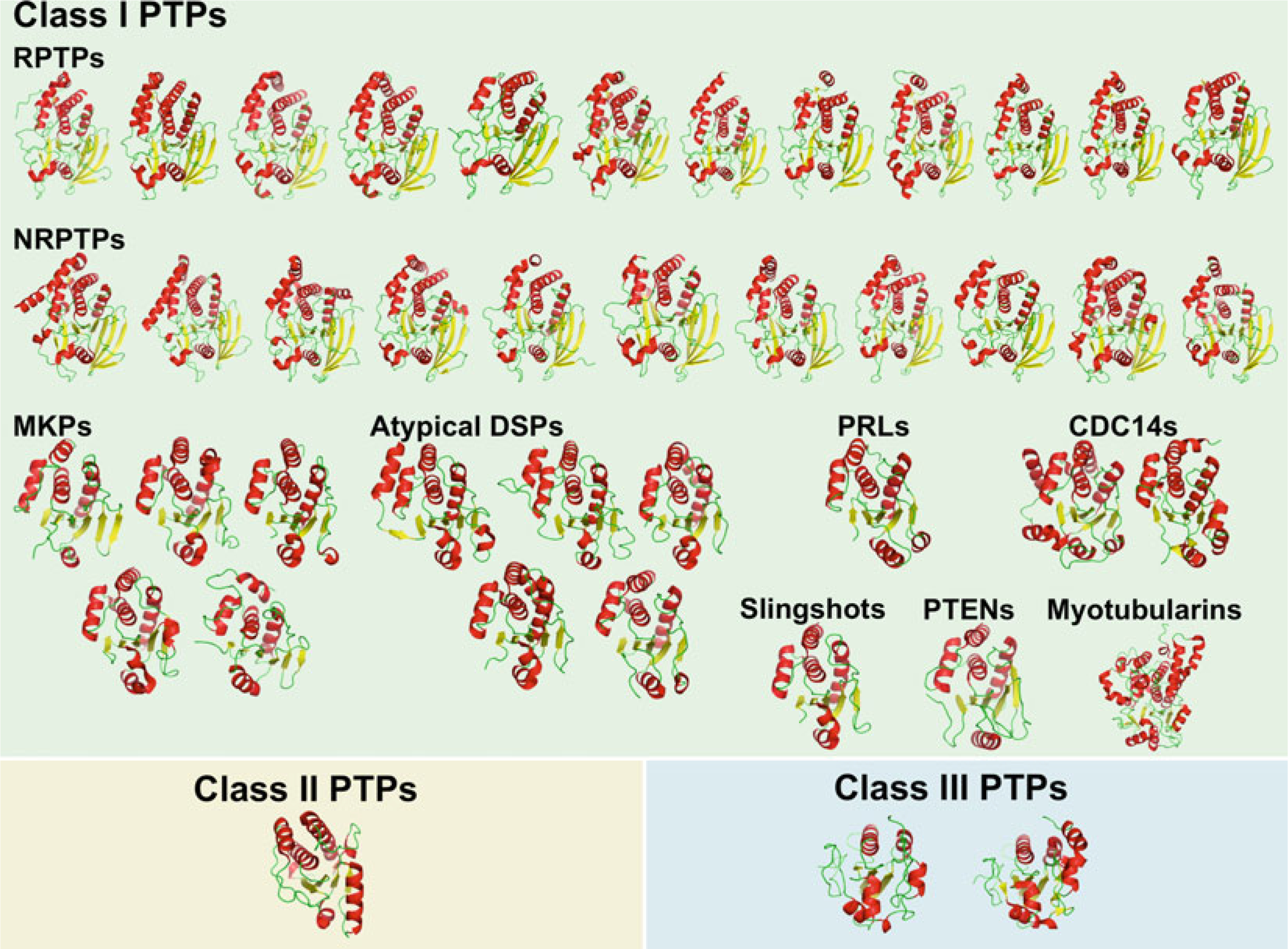
Structures of human PTP catalytic domains. Cartoon representation of human PTP catalytic domain structures, divided by family/subfamily, and colored by secondary structure (α-helices in red, β-strands in yellow, loops in green)
Fig. 3.
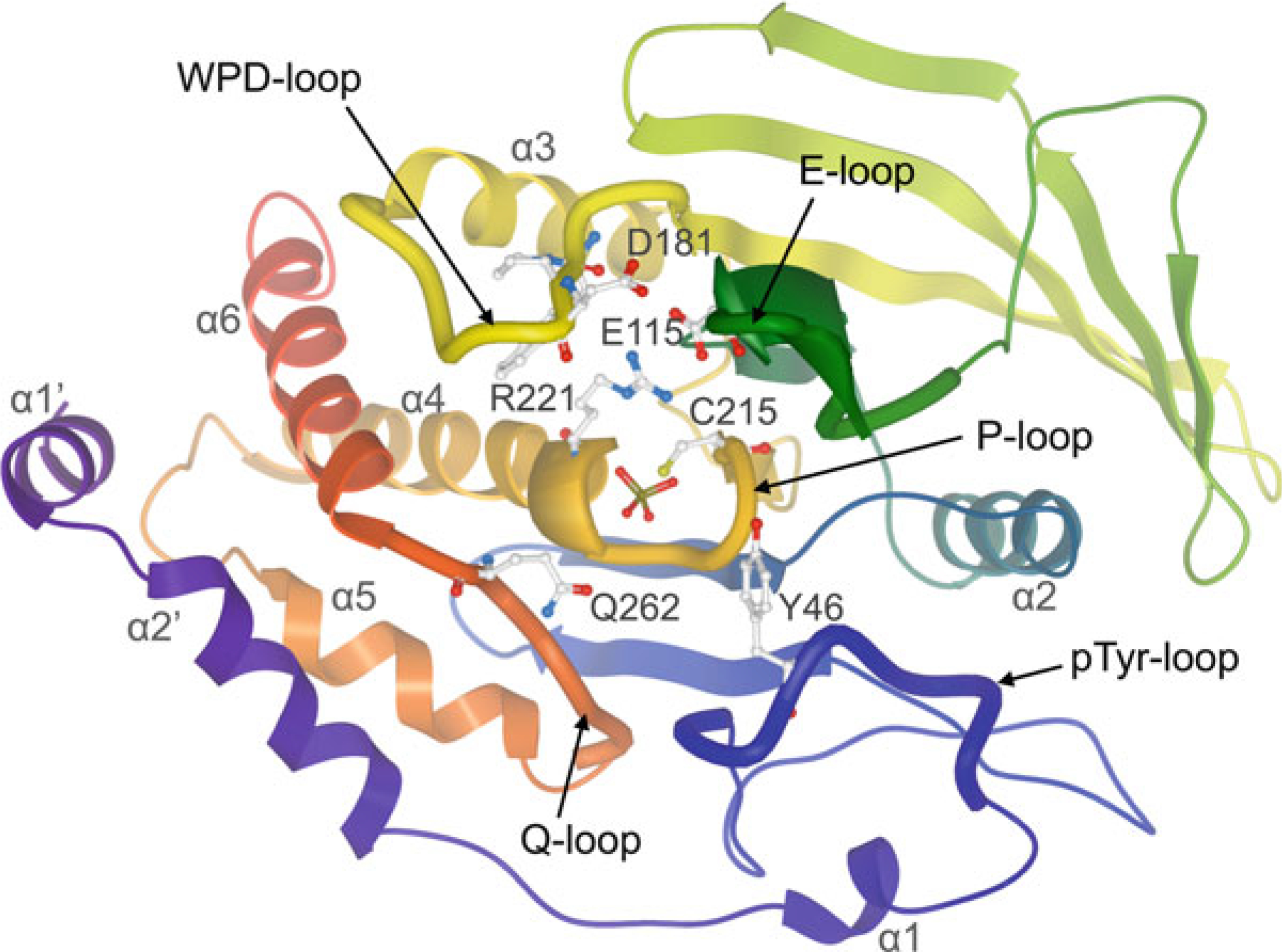
Ribbon representation of the classical class I PTP catalytic domain, colored by NtoC, with tungstate (shown in ball-and-stick representation) bound into the active site (PTP1B; PDB ID: 2HNQ). Conserved residues important for catalysis are highlighted in ball-and-stick representation: catalytic cysteine (C215) and invariant arginine (R221) of the P-loop; WPD-loop residues, including the catalytic acid/base aspartate (D181); conserved glutamine (Q262) of the Q-loop; tyrosine (Y46) of the pTyr-recognition loop (pTyr-loop); conserved glutamate (E115) of the E-loop
5.1. The P-Loop
The base of the active site of all PTPs is formed by the P-loop, which contains the conserved PTP signature motif C(X)5R. The backbone nitrogen atoms of P-loop residues project into the catalytic pocket, creating a positively charged microenvironment that is further enhanced by the guanidinium group of the conserved P-loop arginine. The positively charged environment of the catalytic pocket has two functions: (1) to provide exceptional affinity for tetrahedral oxyanions such as the phosphate group, and (2) to lower the pKa of the catalytic cysteine by stabilizing the thiolate anion (Cys-S− ). A deprotonated thiol group of the catalytic cysteine is crucial for its role as nucleophile in the catalytic mechanism (see Fig. 1). While cysteine residues within proteins usually have pKa values of ~8.5, which would not allow for effective catalysis due to the low nucleophilicity under physiological conditions, the unique environment of the active site confers an unusually low pKa (between 4.5 and 5.5), permitting the enzymatic reaction to be efficient at physiological pH (or even at the pH optimum of 5–6) [45]. On the other hand, due to its low pKa, the catalytic cysteine is also highly susceptible to oxidation [46], nitrosylation [47], and sulfhydration [48], resulting in reversible or irreversible modifications that abrogate its nucleophilic function and thereby inactivate enzyme activity.
5.2. The WPD-Loop
The WPD-loop is present in almost all PTPs, apart from a few exceptions, which include the myotubularins and the CDC25s. This loop is located approximately 30–40 residues upstream of the PTP signature motif in the primary structures of both pTyr-specific PTPs and DSPs. The single class II PTP LMPTP is the exception, with the P-loop located near the N-terminus, and the WPD-loop ~120 amino acids downstream from there. The WPD-loop is so named because in classical PTPs it contains the highly conserved tryptophan–proline–aspartate (WPD) motif. Interestingly, both the tryptophan and proline residues (100 and 97 % conserved in the pTyr-specific PTPs) are not present in the corresponding loops of the DSPs and LMPTP. This is somewhat surprising, as in classical PTPs the tryptophan has been identified as an essential hinge residue important for loop flexibility [49, 50]. On the other hand, the aspartate residue, which functions as the general acid/base during catalysis, is conserved in all PTP subfamilies. The WPD-loop acts as a flexible gate to the active site and has been observed in the “closed” (active) conformation, as well as a variety of “open” (inactive) conformations (Fig. 4) [33]. In the absence of substrate, the WPD-loop likely fluctuates between the open and closed conformations [51]. However, substrate binding can only occur when the loop is in the open state. Upon substrate binding, the WPD-loop closes about the active site so that the catalytic aspartate is positioned to participate in catalysis. Compromised mobility of the WPD-loop results in substantially lower catalytic activity, as shown by studies with mutated tryptophan hinge residue [49, 50], or with allosteric inhibitors that decrease WPD-loop flexibility [34, 35]. PTPs with a different amino acid at the position of the catalytic aspartate, including the RPTPs IA-2 (PTPRN; D→A) [52], PTPS31 (PTPRQ; D→E) [53], and PTPλ (PTPRU; D→E) [54], and the NRPTP HD-PTP (PTPN23; D→E) [55], are often inactive or have a very low activity. Exceptions are PTPD1 (PTPN21; D→E), which has been shown to effectively dephosphorylate Src at Tyr527 [56], and PTPS31 (PTPRQ; D→E), which can dephosphorylate phosphoinositite [53]. A different amino acid at the position of the conserved proline in the WPD motif is only found in IA-2β (PTPRN2, P→Y), an RPTP that has no PTP activity [57].
Fig. 4.
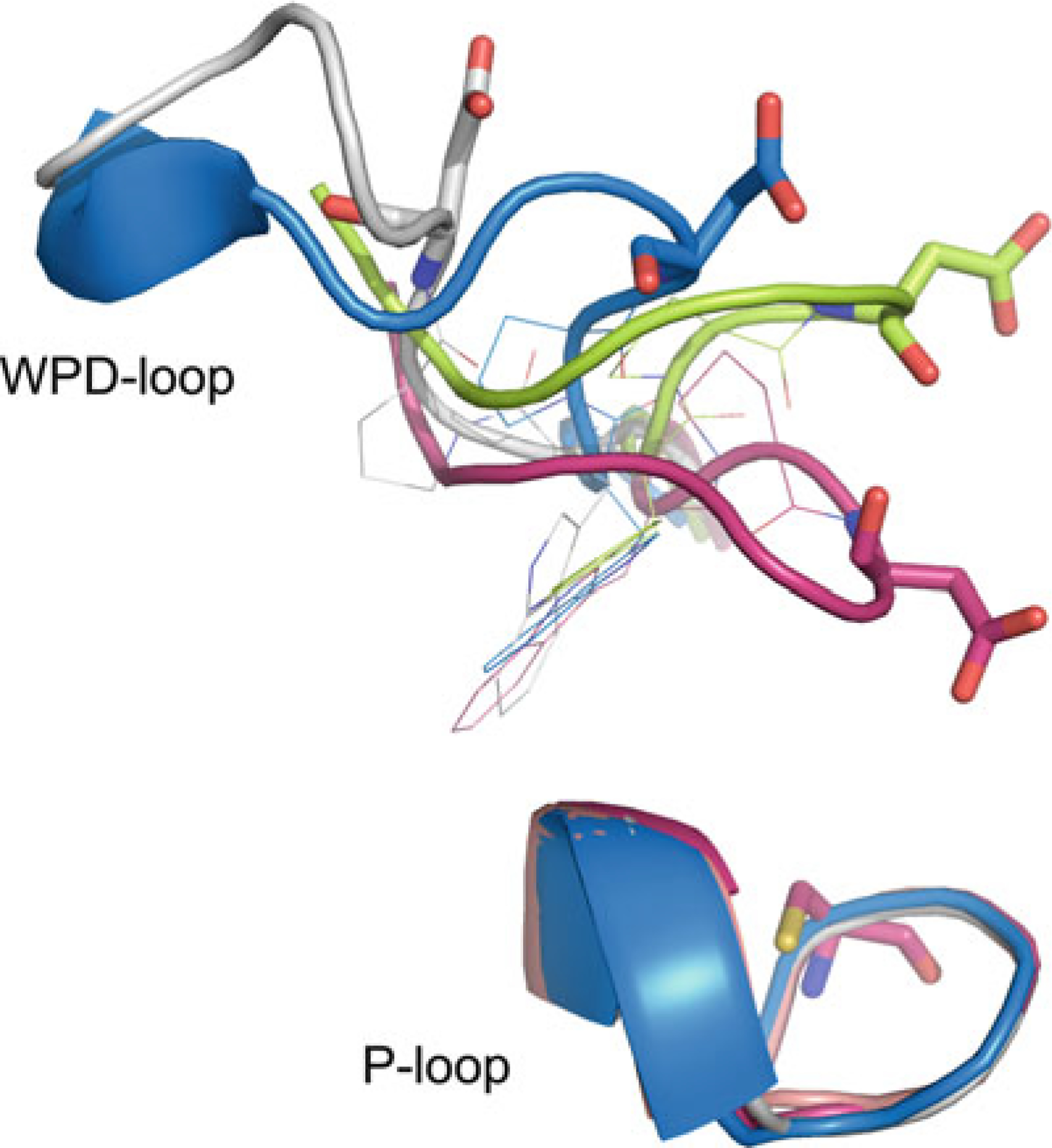
WPD-loop conformations. PTP1B closed state (magenta; PDB ID: 1SUG), PTP1B open state (lime; PDB ID: 2HNP), STEP atypical open state (blue, PDB ID: 2BV5), and LYP atypical open state (white; PDB ID: 2P6X). Catalytic cysteine and catalytic aspartate residues shown in stick representation, conserved tryptophan and proline shown in line representation
5.3. The pTyr-Recognition Loop (pTyr-Loop)
The pTyr-loop, which sometimes also is referred to as the substrate-binding loop and is present in all classical PTPs, defines the depth of the catalytic pocket, thereby creating selectivity for pTyr over the shorter pSer/pThr (Fig. 5). The loop contains the pTyr-recognition motif KNRY, with the 84 % conserved tyrosine positioned such that its side chain acts a causeway to the catalytic pocket, defining its considerable depth of ~9 Å, and ensuring that only pTyr can reach the bottom of the pocket. In addition to defining the depth of the phosphate-binding pocket, the conserved tyrosine also forms aromatic π−π interactions with the substrate pTyr residue, thereby facilitating substrate binding to the active site [58]. The guanidinium group of the 100 % conserved arginine of the KNRY motif interacts with nearby backbone oxygen atoms, thereby stabilizing the loop conformation. In addition, the surface-exposed arginine side chain may also act as substrate binding site. There are six PTPs in which the tyrosine of the KNRY motif is replaced by another amino acid [20]. Interestingly, five of these six phosphatases, IA-2 (PTPRN; Y->H), IA-2β (PTPRN2, Y->S), PTPS31 (PTPRQ; Y->F), PTPλ (PTPRU; Y->Q), and PTPD1 (PTPN21; Y->F), also have altered WPD motifs. Thus, the function of these PTPs may have changed during the course of evolution in a concerted fashion, i.e., through mutations in both the WPD- and pTyr-loops. Two residues C-terminal of the conserved tyrosine of the pTyr-loop KNRY motif is an aspartate or asparagine residue (84 % conserved). This residue forms a bipartite hydrogen bond interaction with the backbone amide nitrogen atoms of the substrate pTyr and adjacent residue, providing additional stabilization to the substrate pTyr in the active site (Fig. 5).
Fig. 5.
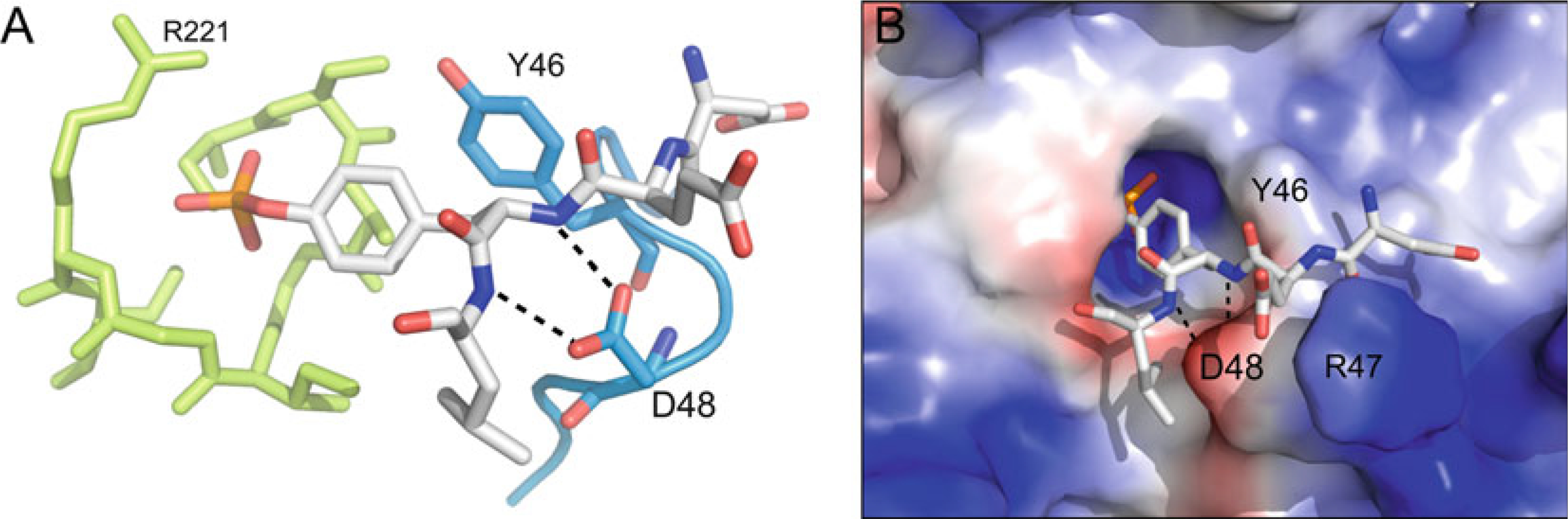
The pTyr-loop. (a) pTyr-loop (blue) relative to the P-loop (lime), with bound pTyr-peptide (white) (PTP1B; PDB ID: 1PTT). The conserved tyrosine (Y46 in PTP1B) defines the depth of the catalytic pocket and facilitates pTyr binding through aromatic π−π interactions. The conserved aspartate or asparagine (D48 in PTP1B) stabilizes substrate binding through bipartite hydrogen bonding interaction with backbone nitrogen atoms of the substrate peptide. (b) Same complex as in (a) but with PTP1B rendered in surface representation (blue, most positive; red, most negative). (In addition to the interactions listed in (a), R47 of the PTP1B pTyr-loop is labeled, the side chain of which interacts with the glutamate in the −1 position of the substrate peptide, highlighting the fact that PTP1B favors substrates with acidic residues at this position)
5.4. The E-Loop
In classical PTPs, the E-loop contains a 100 % conserved glutamate residue and an 89 % conserved lysine residue. In >85 % of known classical PTP structures, the E-loop forms a tight β-hairpin, with the conserved glutamate forming a bipartite hydrogen bonding interaction with the side chain of the invariant P-loop arginine, presumably stabilizing the guanidinium group in a position that favors phosphate binding to the P-loop (Fig. 6a). Although the E-loop has also been observed in multiple unique conformations [59–61] or to be completely disordered [32, 62], the glutamate-arginine interaction is found in nearly all of the reported classical PTP structures. The conserved lysine often is observed to hydrogen bond with the catalytic aspartate of the WPD-loop when this loop is in the closed conformation, an interaction that likely stabilizes the WPD-loop in its active, substrate-bound conformation (Fig. 6b). Indeed, mutating this residue to alanine results in reduced catalytic efficiency in HePTP [63]. Interestingly, there appears to be no equivalent function of the E-loop outside of the classical PTP family. Although all PTP subfamilies have a somewhat similarly positioned loop, the conserved glutamate and lysine residues are only present in the pTyr-specific PTPs. No other conserved residues are found at the corresponding positions in DSPs.
Fig. 6.
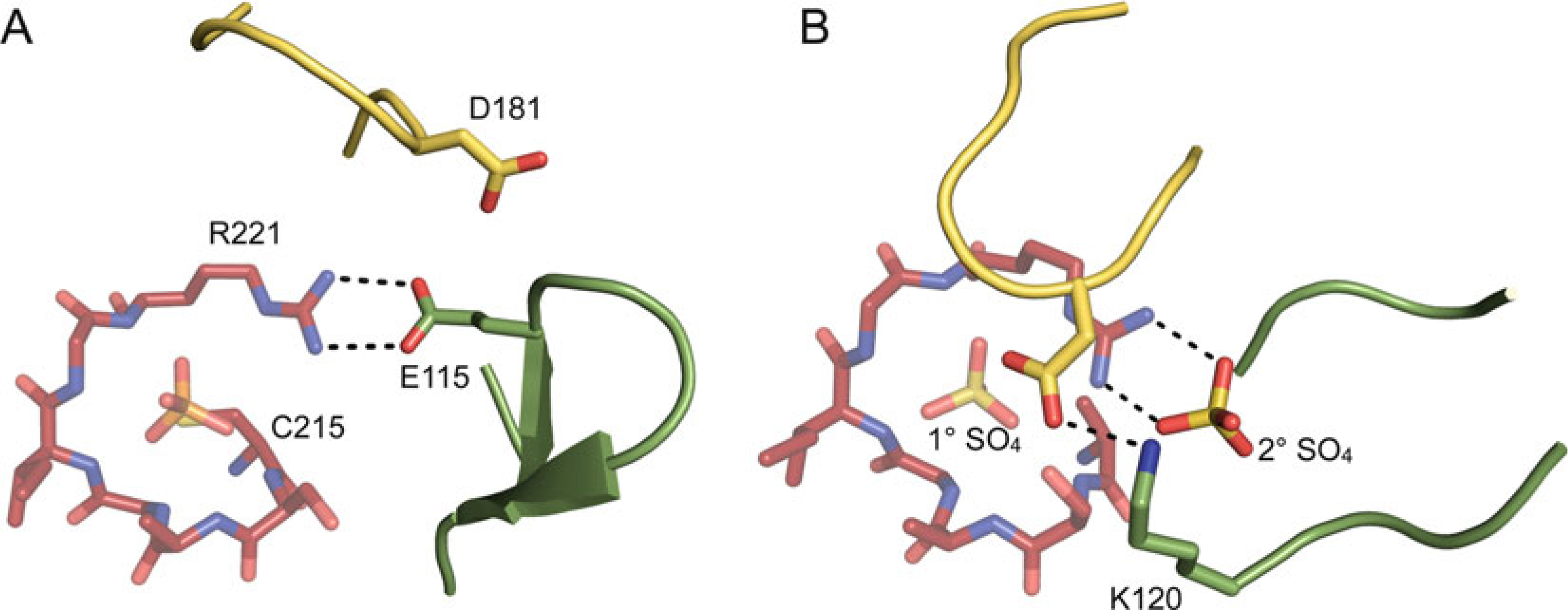
The E-loop. (a) The E-loop (green) relative to the P-loop (red) and WPD-loop (yellow) (PTP1B; PDB ID: 2HNQ). In this structure, the WPD-loop is in the open conformation and the E-loop forms a tight β-hairpin, with the E-loop glutamate (E115, shown in stick representation) neutralizing the charge of the conserved P-loop arginine (R221). (b) The E-loop (green) relative to the P-loop (red) and WPD-loop (yellow) (PTP1B; PDB ID: 2B4S). In this structure, the WPD-loop is in the closed conformation and the E-loop does not form a β-hairpin and is partially disordered. The E-loop lysine (K120) and catalytic aspartate of the WPD-loop (both shown in stick representation) form a hydrogen bond, and a sulfate molecule is bound at the active site (1° SO4) and at a secondary binding site (2° SO4), which neutralizes the charge of the P-loop arginine
5.5. The Q-Loop
The Q-loop is present in all classical PTPs and is so named because it contains a 97 % conserved glutamine residue. (A similarly positioned loop is also present in DSPs, but does not contain a glutamine or equivalently conserved residue.) Kinetic studies [64–66] in conjunction with structural analyses [27–29, 44, 58] revealed that the glutamine side chain, which reaches toward the catalytic center, positions the water molecule that is used for the phosphoester hydrolysis. Interestingly, LMPTP (which does not contain an equivalent loop) or DSPs (which do not have the glutamine) catalyze phosphoryl transfer also to alcohols in addition to water [65]. In contrast, in the classical PTPs, the Q-loop is responsible for maintaining strict hydrolytic activity. Mutations of the glutamine to residues that cannot hydrogen bond with the nucleophilic water result in less restrictive enzymes and confer phosphotransferase activity [65]. Thus, the Q-loop prevents classical PTPs from acting as kinase-like phosphotransferases, which otherwise may phosphorylate indiscriminately. In addition to this function, it was proposed that the water molecule positioned by the Q-loop glutamine might also be important for closure of the WPD-loop [33].
5.6. Structural Comparison Between Class I, Class II, and Class III PTPs
Based on their conserved P-loop region, including the signature motif C(X)5R, all PTPs have evolved from a common ancestor gene. While the more ancient class II PTPs are structurally related to bacterial arsenate reductases, the class III PTPs likely stem from bacterial rhodaneses. In fact, both arsenate reductases and rhodaneses also contain the same C(X)5R P-loop structural element, suggesting that these three enzyme classes may have evolved from an ancestral oxyanion-binding protein. The structure of the class II PTP domain shares key similarities with that of the classical class I PTP domain. Like that of the class I PTPs, the class II PTP domain consists of a central twisted β-sheet surrounded by an arrangement of α-helices, with the base of the active site formed by residues of the P-loop. Also like the class I PTPs, the class II PTP domain contains a WPD-loop with an aspartate residue that functions as a catalytic acid/base. Additionally, although not directly corresponding to the pTyr-loop in classical class I PTPs, the class II PTPs contain a similarly positioned loop that defines the depth of the PTP active site [67]. Only a single member, LMPTP (ACP1), represents the class II PTP family in humans [23]. The primary transcript of LMPTP has been shown to be alternatively spliced, resulting in excision of either exon 3 (LMPTP isoform A) or exon 4 (LMPTP isoform B) [68], or both exons 3 and 4 (LMPTP isoform C) [69]. The latter splice variant encodes an approximately 15 kDa protein with no detectable in vitro phosphatase activity, whereas LMPTP splice variants lacking either exon 3 or exon 4 alone encode proteins with pTyr-specific phosphatase activity [70]. Exons 3 and 4 encode amino acids 39–75, which includes the loop region that corresponds to the pTyr-loop in the classical class I PTPs, as well as the substrate-binding region that is responsible for the differing substrate specificities of the two isoenzymes [68].
The major difference between the class I and class II PTPs is the sequential order of the typical PTP structural elements. Overall, the positioning of α-helices, β-sheets, and active site loops is reminiscent of class I PTPs. However, crystal structures of LMPTP cannot be superimposed onto those of classical PTPs because of the quite different sequential alignment of these structural elements. For instance, the P-loop containing the PTP signature sequence is located at the extreme N-terminus of LMPTP. In addition, the molecular weight of catalytically active LMPTP is only approximately 18 kDa; in contrast, the classical class I PTP catalytic domains exceed 30 kDa in size. Nevertheless, LMPTP-A and LMPTP-B are both highly active pTyr-specific phosphatases with important and nonredundant functions in human cell physiology [23].
The class III PTP family is represented in humans by three members, CDC25A, CDC25B, and CDC25C, each of which have different splice variants [71]. The CDC25 proteins are typified by their N-terminal and C-terminal regions. The N-terminal region is highly divergent in sequence and contains multiple sites for posttranslational modifications, which regulate expression levels [72, 73], protein–protein interactions [74–76], and/or catalytic activity [77, 78]. CDC25 homology with other PTPs is limited entirely to the C-terminal region, which, unsurprisingly, contains the PTP signature sequence. Crystal structures have been determined of the PTP catalytic domain from CDC25A [79] and CDC25B [80]. The class III PTP catalytic domain fold is most similar to that of the rhodanese sulfur transport protein from mitochondria and some bacteria [81]. Similar to the class II PTPs, the class III PTP catalytic domain is considerably smaller than that of the classical class I PTPs, which is attributed to a narrower central β-sheet surrounded by fewer, shorter α-helices. Importantly, and in contrast to the classical class I PTP catalytic domain, the class III PTP catalytic domain does not contain equivalents of the pTyr-, WPD-, or Q-loops. As such, the comparatively shallow CDC25 active site permits access of both pTyr- and pThr-containing substrates to the catalytic cysteine.
6. PTP Substrate Recognition and Specificity
Despite the highly conserved structure of the PTP catalytic domain, PTPs have distinct substrate preferences from one another. For example, PTP1B and TCPTP preferentially dephosphorylate receptor tyrosine kinases and related adaptor molecules, whereas the KIM-family PTPs HePTP, STEP, and PTP-SL dephosphorylate specific MAP kinases. While the presence of distinct non-catalytic domains/motifs facilitate specific localization or binding to substrate proteins, PTP substrate specificity is also dictated by differences within the PTP catalytic domain itself.
6.1. Active Site Substrate Specificity Determinants
The most effective active site determinant is the pTyr-loop in the classical PTPs, which dictates strict specificity for pTyr over pSer/pThr in this PTP subfamily (see Subheading 5.3). In addition to excluding substrates other than pTyr from reaching the catalytic residues at the bottom of the catalytic pocket, the pTyr-loop also controls specificity among the classical PTPs. In 84 % of classical PTPs, the pTyr-loop contains an aspartate or asparagine residue at the KNRY+2-position. The side chain of this residue forms a bipartite hydrogen bond interaction with backbone residues of the substrate peptide, and thereby facilitates substrate binding at the active site (Fig. 5) [82]. However, in a small subset of PTPs, the aspartate/asparagine is replaced with an alanine, proline, or threonine residue, which are incapable of forming the equivalent interaction, and thereby restrain substrate binding at the active site in these PTPs. For example, the KIM-family PTPs HePTP, STEP, and PTP-SL are unique in that they have a threonine at the corresponding position. Studies with HePTP have shown that the threonine (Thr106 in HePTP) serves as a negative determinant, which reduces phosphatase activity towards pTyr-containing substrates other than MAP kinases [83]. Mutation of Thr106 in HePTP to aspartate or asparagine results in a 10- to 70-fold increase in catalytic efficiency towards several pTyr-peptides, including a peptide corresponding to the activation loop of its direct substrate ERK2. This is in agreement with earlier studies that indicated the importance of the common aspartate/asparagine at this position for the catalytic efficiency in PTP1B [82]. In contrast, dephosphorylation of full-length ERK2 by HePTP is not affected by the Thr106 mutations. In fact, catalytic efficiency of wild-type HePTP and aspartate/asparagine mutants is >300-times greater towards full- length ERK2 than towards the ERK2 peptide, owing to the presence of the ERK2-binding KIM in the N-terminal region of HePTP [83]. Thus, the threonine in the pTyr-loop in HePTP (and probably also in STEP and PTP-SL) acts as a negative determinant that restrains binding of pTyr-containing proteins and, in combination with a non-catalytic motif (the KIM and also the kinase specificity sequence (KIS) [84, 85]), provides specificity for the MAP kinases.
Altered substrate specificity and increased phosphatase activity upon substrate binding has also been reported for the MAP kinase phosphatase-3 (MKP-3), a DSP that dephosphorylates the activation loop pThr and pTyr residues specifically in ERK1/2. When tested against the corresponding ERK2 peptide (DHTGFLpTEpYVATR), MKP-3 catalytic efficiency was very low (kcat/Km = 5.0 M−1 s−1), and only pTyr was dephosphorylated [86]. In contrast, MKP3-catalyzed hydrolysis of the intact ERK2 protein was 106-fold more efficient (kcat/Km = 3.8 × 106 M−1 s−1), and both pTyr and pThr were dephosphorylated [87]. Structural and biochemical studies suggested that binding of ERK to MKP-3 causes a significant change of the active site loop conformations, resulting in much higher catalytic activity and also a change in substrate phospho-amino acid recognition (reviewed in ref. 30). In light of these results, it is not surprising that numerous published specificity studies utilizing combinatorial peptide libraries fail to identify true substrate sequence specificity among the PTPs.
Finally, sequence motifs within the P-loop also appear to govern substrate specificity. For instance, all 15 myotubularins share a Trp-Asp-Arg (WDR) sequence as part of the PTP signature motif (C(X)3WDR). This sequence seems to confer specificity for their substrates phosphatidylinositol-3-phosphate and phosphatidylinositol-(3,5)-bisphosphate. Interestingly, because the myotubularins do not contain a WPD-loop, the aspartate residue of the WDR motif may function as the general acid/base during catalysis; mutating the corresponding Asp422 in MTMR2 to alanine renders the enzyme inactive [88].
6.2. Gateway Residues, Secondary Substrate- Binding Pocket, and Second-Site Loop
Based on a second-site binding pocket first identified in PTP1B [89], the combination of structural information, sequence alignment, and Cα-regiovariation score analysis led to the identification of non-conserved but similarly located putative binding pockets in other PTPs [20]. In PTP1B, this “second site” is important for substrate recognition [90], accommodating a second pTyr residue at the substrate +1 position. Commonly, the second site is defined by residues of helix α2′ and the loop connecting helix α2′ and helix α1 (Fig. 7). Residues of this loop, which is also termed the “second-site loop” [33], are not conserved among the PTPs and, together with loop length and conformation, determine the diverse shape and nature of the PTP second site. In PTP1B, Arg24 and Arg254 are responsible for the positively charged nature of the second-site pocket, facilitating strong binding interactions with the second pTyr residue [89]. Access to the second site is controlled by a so-called gateway residue (Gly259 in PTP1B), which is located in the Q-loop and is a key determinant in substrate recognition and catalysis [91]. The small size of Gly259 in PTP1B allows unhindered access to the second site, whereas in CD45, for instance, a leucine residue (Leu869) blocks the narrow cleft and impedes access. Based on the nature of and access to the second site, Barr and colleagues have proposed to group PTPs into five different categories [33], a classification that should be very useful for the future development of specific PTP inhibitors.
Fig. 7.
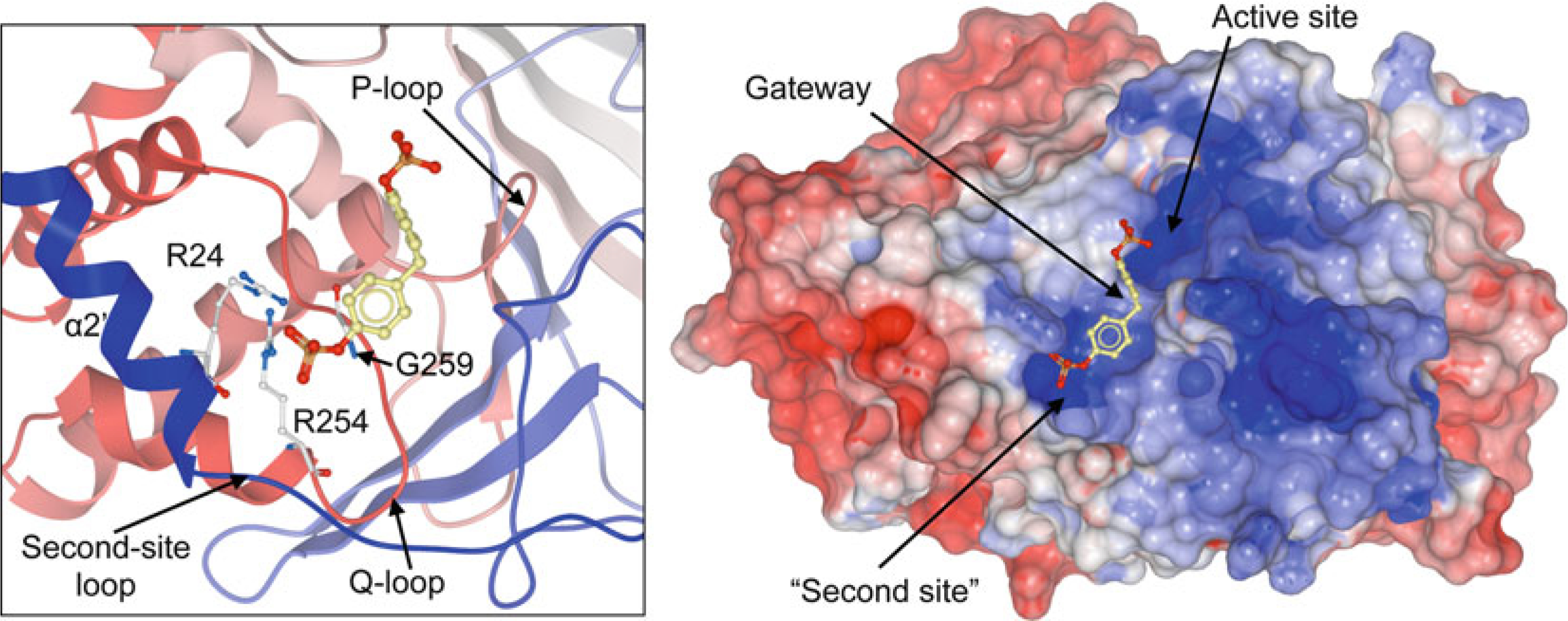
Binding of a small-molecule phosphate (methylenebis(4,1-phenylene) bis(dihydrogen phosphate); shown in stick representation) to the “second site” in PTP1B (PDB ID: 1AAX; shown as ribbon diagram (left panel) and surface representation (right panel; blue, most positive; red, most negative). Second-site residues Arg24 and Arg 254, as well as gateway residue Gly259 are shown in stick representation (left panel)
6.3. Tools to Determine PTP Substrate Recognition by X-ray Crystallography
In order to study the complex of a PTP with its phosphorylated substrate using X-ray crystallography, the PTP must be inactivated so that a stable PTP:substrate complex is formed. Inactivation of PTPs is achieved by mutating the residues directly involved in catalysis, including the catalytic cysteine of the P-loop, the catalytic aspartate of the WPD-loop, and the conserved glutamine of the Q-loop. Collectively, these mutants are called substrate-trapping mutants (STMs) [92]. In some cases, effective STMs have been formed by mutating the catalytic cysteine, either to serine [58, 93, 94] or alanine [90, 95]. In other cases, effective STMs have been formed by mutating the catalytic aspartate alone [92, 96] or in combination with the conserved glutamine [64]. STMs have been used not only to crystallize PTP:substrate complexes [58, 61, 90, 93, 94], but also to identify novel PTP targets in intact cells [92, 97, 98]. Thus, STMs represent invaluable tools for investigating PTP substrate recognition both in vitro and in vivo.
Apo PTP crystals can often be readily obtained by including tetrahedral oxyanion, such as phosphate, sulfate or tungstate, in the crystallization drop. This almost always results in binding of the oxyanion at the PTP active site (i.e., P-loop). For this reason, in order to obtain crystals of PTP:substrate complexes, crystallization precipitants containing tetrahedral oxyanions should be avoided, as they will compete with substrates for binding the PTP active site. Still, apo PTP crystals containing oxyanion at the active site can actually be depleted of active site-bound oxyanion [63, 99, 100], after which alternative ligands can populate the active site. In one example, crystals of the classical PTP HePTP were obtained in the presence of sulfate and contained sulfate bound at the active site with the WPD-loop in the closed conformation [63]. Transfer of these crystals from the original crystallization drop into drops containing decreasing concentrations of sulfate, as well as increasing concentrations of the oxyanion tartrate, resulted in gradual depletion of active site-bound sulfate (PDB ID: 3O4T), opening of the WPD-loop, and subsequent repopulation of the active site by tartrate (PDB ID: 3O4U). Importantly, in this HePTP crystal form, the WPD-loop faced a solvent channel, which was distal to crystallographic symmetry mates, a critical feature for opening of the WPD-loop without concomitant disintegration of the crystal. It is expected that, for other PTPs, the WPD-loop must also be distal from symmetry mates in order to allow for movement of the WPD-loop and depletion/repopulation of the active site using this methodology.
7. PTPs in Human Disease
Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation of signaling molecules is known to be crucial for the development of many human diseases, ranging from cancer to cardiovascular, immunological, infectious, neurological, and metabolic diseases (reviewed in refs. 13, 19, 21, 101–104). In cancer, this is best exemplified by the common loss of PTEN in many malignancies including breast and prostate cancer [105]. Similarly, SHP1 (PTPN6) is frequently lost in myelodysplastic syndrome (MDS) [106] and lymphomas [107]. However, PTPs not only act as tumor suppressors but also as “positive” components of signaling pathways and cell functions, illustrated by a growing number of examples of overexpressed or hyperactive PTPs in cancer cells [103]. For instance, SHP2 (PTPN11) is a well-known oncogene with various gain-of-function mutants occurring in several forms of leukemia [108, 109]; overexpression of PRL3 (PTP4A3) and MKP1 (DUSP1) has been found in metastatic colon cancer [110] and prostate cancer [111], respectively; CDC25, which is a rate-limiting enzyme for cyclin-dependent kinase (CDK)-dependent transition from G1 to S phase and G2 to M phase during cell cycle, is overexpressed in a number of cancers [112]; or PTP1B (PTPN1), first discovered as a crucial negative regulator of insulin and leptin signaling [113], was recently found to be a positive regulator of ErbB2 (HER2/neu) induced signals that trigger breast tumorigenesis and metastasis [114–116]. The lymphoid tyrosine phosphatase LYP (PTPN22), which is best known for its causative role in numerous autoimmune diseases including type 1 diabetes (T1D) [117], rheumatoid arthritis (RA) [118], systemic lupus erythematosus (SLE) [119], and others (reviewed in refs. 17, 102, 120), was recently found overexpressed in chronic lymphocytic leukemia (CLL) [121], a common malignancy of autoreactive B cells. Interestingly, autoimmunity is related to a single nucleotide polymorphism (SNP) in LYP, whereas inhibition of antigen-induced apoptosis and activation of anti- apoptotic pathways in CLL occurs independently of the SNP. HePTP (PTPN7), one of the three KIM-family PTPs and a critical negative regulator of the MAP kinases ERK1/2 and p38 in hematopoietic cells [122], is upregulated in acute myeloid leukemia (AML) and in T cell acute lymphoblastic leukemia (T-ALL) [123, 124]. Additionally, the HePTP gene is often duplicated in bone marrow cells from patients with MDS [125, 126]. Another KIM-family PTP, the brain-specific STEP (PTPN5), which modulates key signaling molecules involved in synaptic plasticity and neuronal function, has been implicated in a number of neuropsychiatric disorders, including Alzheimer’s disease, schizophrenia, fragile X syndrome, epileptogenesis, alcohol-induced memory loss, Huntington’s disease, drug abuse, stroke/ischemia, and inflammatory pain (reviewed in ref. 104). Genetic polymorphisms of the ubiquitously expressed LMPTP (ACP1) is linked to several common diseases, including allergy, asthma, obesity, myocardial hypertrophy, and Alzheimer’s disease [23, 127]. Loss of the DSP laforin (EPM2A) causes Lafora’s epilepsy [128], whereas loss of myotubularin (MTM1) leads to an X-linked muscle dystrophy [129]. Mutations in myotubularin-related proteins 2 and 13 (MTMR2 and MTMR13) are associated with the inherited nerve myelination disease Charcot-Marie-Tooth syndrome type 4B [130, 131]. Loss of the hematopoietic-specific transmembrane PTP CD45 (PTPRC), which regulates Src kinases required for T and B cell activation, has been linked to severe combined immunodeficiency disease [132]. These are just a few examples of the rapidly growing number of human diseases associated with aberrant PTP function. Not surprisingly, this has begun to elicit growing interest in novel therapeutics that target specific PTPs. In the following we highlight three of the currently most interesting/validated PTP drug targets.
7.1. PTP1B (PTPN1)
In 1999 Elchebly and colleagues reported increased insulin sensitivity and obesity resistance in mice lacking the PTP1B gene [133]. It was this paper that truly ignited the quest for PTP inhibitors. PTPN1−/− mice showed increased phosphorylation of the insulin receptor and were resistant to weight gain and insulin insensitivity when fed a high-fat diet. The data suggested that inhibition of PTP1B would alleviate insulin resistance in type 2 diabetes and would improve the effects of insulin on both glucose balance and fatty acid metabolism. Laboratories in both industry and academia heavily invested in the development of PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. As a result, a large number of PTP1B inhibitors have been developed during the last decade (reviewed in refs. 134–137). However, only few compounds progressed into clinical trials, and so far none have advanced beyond phase II. The reasons for failure and common challenges in PTP inhibitor design are discussed in Subheading 8.
More recently, PTP1B was found as an activator of ErbB2 (HER2/neu)-induced signals that trigger breast tumorigenesis and metastasis [114–116]. Genetic or pharmacological deletion of PTP1B activity in transgenic mice containing an activating mutation in ErbB2 (NDL2 mice, ref. 138) resulted in significant mammary tumor latency and resistance to lung metastasis [114, 115], providing proof-of-concept and validating PTP1B as a novel drug target in breast cancer. The PTP1B gene is located at the chromosomal region 20q13 that is frequently amplified in breast and ovarian cancer and is associated with poor prognosis [139, 140]. PTP1B overexpression was found in 72 % of human breast tumors, correlating with ErbB2 overexpression [141], as well as in ErbB2- transformed human breast epithelial cells and tumors derived from such cells [142]. The underlying molecular mechanism of PTP1B function in tumorigenesis is not fully understood and is somewhat controversial. Studies conducted with breast cancer cell lines suggest that PTP1B function is required for c-Src activation by dephosphorylation of the inhibitory Tyr527, thereby positively affecting ErbB2 signaling [143, 144]. However, in animal studies, no change of Src phosphorylation and activity was observed with overactive ErbB2 receptors [114, 115]. Consistent with these findings, an earlier study demonstrated that Src kinase function is not necessary for ErbB2-mediated tumorigenesis [145]. Instead, loss of PTP1B function led to reduced Ras/MAP kinase pathway and PI3 kinase/AKT pathway activation [114, 115]. Indeed, PTP1B is required for the activation of the small GTPases Ras and Rac, which are typically associated with increased cell proliferation and motility [146, 147]. To bypass Src activation, an alternative mechanism was suggested, in which p62Dok, an inhibitor of the Ras/MAPK pathway, is deactivated by PTP1B, and loss of PTP1B function leads to hyperphosphorylation of p62Dok with subsequent inactivation of Ras and its downstream effectors [147, 148].
ErbB2 is overexpressed in 20–30 % of early-stage breast cancers and is associated with poor prognosis [149, 150]. The current therapeutic approach in ErbB2 positive tumors is a combination of chemotherapy and a monoclonal antibody that selectively binds ErbB2 (trastuzumab, ref. 151). However, the development of trastuzumab resistances in the majority of patients has limited the use of the drug [152]. Nonetheless, the therapeutic efficacy of trastuzumab is strong evidence for the critical role of ErbB2 in human breast cancer and clearly justifies more targeted approaches downstream of the ErbB2 receptor, including PTP1B.
7.2. SHP2 (PTPN11)
Src homology 2 domain-containing protein tyrosine phosphatase 2 (SHP2) is a widely expressed tyrosine-specific PTP that appears to have a net positive role in cell activation in response to growth factors, cytokines, and hormones, regulating cell survival, growth, and differentiation [12, 153]. In addition, SHP2 modulates cell adhesion-induced signal transduction and plays a role in cell migration and motility [154]. Germ-line mutations in PTPN11 were first observed in Noonan syndrome (NS) [155], an autosomal dominant disorder that is associated with craniofacial abnormalities, cardiac defects, short stature, and learning disabilities [156–158]. NS affects 1 in 1,000–2,500 live births [155, 159], 40–50 % of which carry mutations in SHP2. Similar germ-line mutations cause two related genetic disorders, Noonan-like disorder with multiple giant cell lesion syndrome and LEOPARD syndrome [160]. Somatic mutations in PTPN11 occur in ~35 % of cases of juvenile myelomonocytic leukemia (JMML) [161, 162], a rare but aggressive myeloid neoplasm of childhood, clinically characterized by overproduction of monocytic cells that can infiltrate organs including spleen, liver, gastrointestinal tract, and lung. Somatic mutations in PTPN11 have also been found in solid tumors [162] and other types of leukemia (reviewed in refs. 108, 153, 163, 164).
Under basal conditions, SHP2 activity is inhibited via intramolecular interactions between the N-terminal SH2 domain (N-SH2) and the PTP domain (Fig. 8) [39, 165]. Activation of SHP2 in response to extracellular stimuli involves binding of pTyr residues via the tandem SH2 domains, resulting in a conformational change that allows substrate access to the catalytic site. Most of the PTPN11 mutations in NS and JMML (as well as other leukemias) affect amino acid residues at the interface between the N-SH2 and PTP domains and directly interfere with the autoinhibitory state of SHP2. The resulting gain-of-function (GOF) effect of the mutant SHP2 proteins promotes sustained activation of ERK MAP kinases in transfected cells and in animal models [166–171]. Hyperactivation of ERK most likely is the underlying cause of the developmental dysfunctions seen in NS patients. Indeed, mutations in other components of the Ras/ERK pathway, such as KRAS, RAF, and SOS1, have been identified in NS patients lacking PTPN11 mutations [172–175]. Similarly, 75–85 % of JMML cases directly result from GOF-mutations of components of the Ras/ERK signaling cascade (NRAS, KRAS, NF1, SHP2). Interestingly, PTPN11 mutations in JMML appear to be mutually exclusive of NRAS/KRAS2 or other genetic lesions [176]. Moreover, while PTPN11 mutations observed at disease presentation were undetectable at remission or in control individuals, the same mutations could be detected at initial diagnosis and relapse [176]. These data suggest that PTPN11 mutations represent events that directly contribute to leukemogenesis.
Fig. 8.
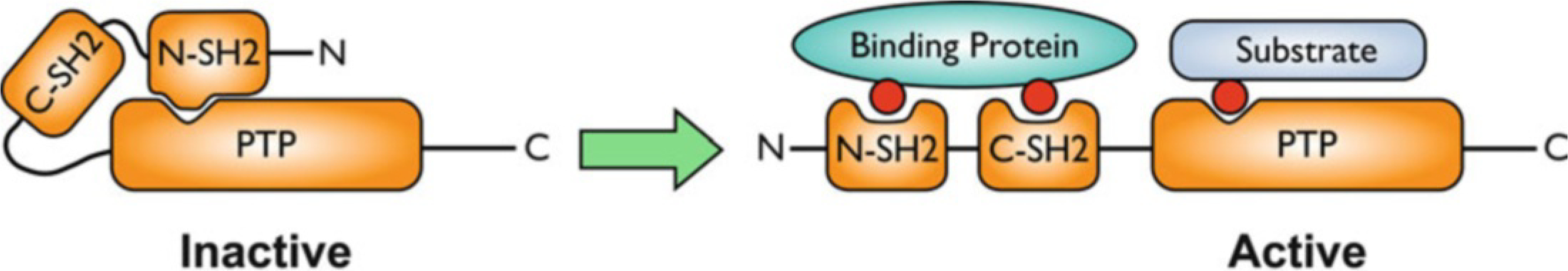
Activation of SHP2 by binding of specific pTyr-containing proteins
In hematopoietic cells, SHP2 appears to be involved in various signaling pathways (Fig. 9), with the positive regulatory role generally ascribed to activation of the ERK pathway upstream of Ras [177, 178]. This function could be mediated by inhibiting the recruitment of Ras-GAP or the DOK proteins, or more indirectly by inhibiting CSK recruitment to CBP/PAG and thus promoting Src family kinase activity [179]. One of the primary groups of cytosolic adapters which recruit SHP2 are the GAB proteins, which amplify PI3K signaling and likely place SHP2 in the proximity of appropriate targets [179]. Additional identified interaction partners include growth factor/cytokine receptors, SIRPα/SHPS-1, PZR, GRB2, FRS, IRS-1, p85, STAT5/3/1, and Sprouty proteins [180]. However, none of the putative substrates identified to date can fully account for the various signaling effects of SHP2 or its oncogenic potential.
Fig. 9.
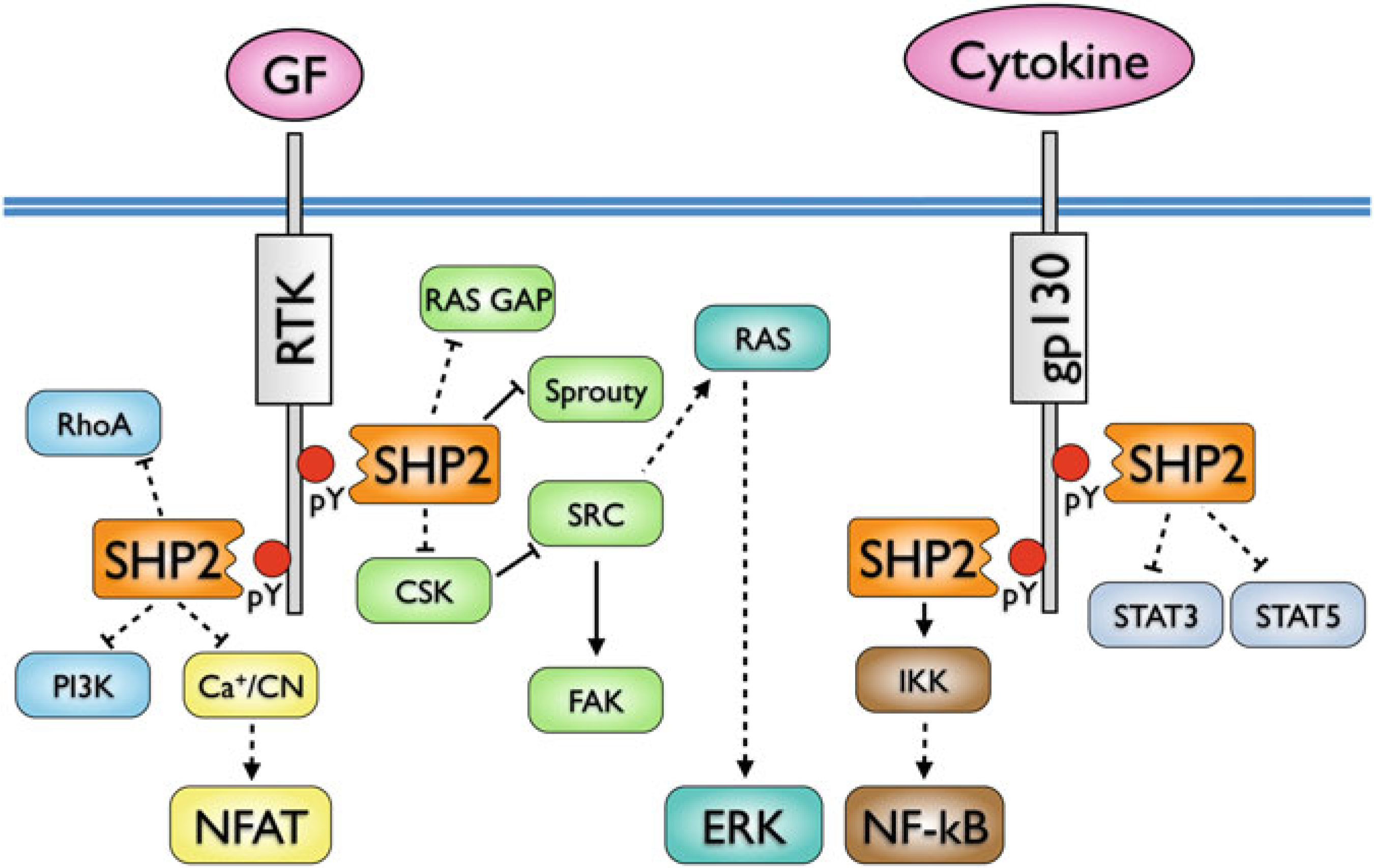
SHP2 interactions and signaling pathways in hematopoietic cells. Direct interactions are indicated by solid lines, indirect interactions by dashed lines (adapted from ref. 158)
JMML is unresponsive to radiation or existing chemotherapy. The current standard of care relies on allogeneic hematopoietic stem cell transplant, resulting in ~50 % survival rate [181]. However, relapse is the most frequent cause of treatment failure. Several therapeutic strategies targeting the Ras/RAF/ERK signaling pathway have been investigated, including inhibition of farnesyltransferase [181], inhibition of RAF (Sorafenib is currently in early phase testing), and inhibition of MEK [182]. In particular the disappointing results from the MEK inhibition trials suggest that aberrant signaling through ERK is not sufficient to drive leukemogenesis, at least not in JMML. Inhibiting constitutively active SHP2 seems to be more intriguing, given its causal role in disease development and the complexity of interaction networks. Several inhibitors of SHP2 have been reported [183–188]. However, most of these compounds suffer from either poor selectivity for SHP2, lack of efficacy in cell-based assays, or both. Future efforts need to address these shortcomings in order to generate specific SHP2 inhibitors with efficacy in vivo.
7.3. STEP (PTPN5)
Striatal-enriched protein tyrosine phosphatase (STEP) is a brain-specific PTP that exists as two major alternatively spliced isoforms, STEP61 and STEP46, which appear to have distinct substrate specificities and functions (reviewed in ref. 131, 189). Membrane-associated STEP61 is targeted to the postsynaptic density, extrasynaptic sites, and the endoplasmic reticulum; STEP46 is primarily found in the cytoplasm. STEP function is regulated by several mechanisms, including phosphorylation, cleavage, dimerization, and ubiquitination. STEP recently gained attention when Lombroso and colleagues reported that genetic deletion of STEP attenuates the cognitive and cellular deficits observed in 6-months old 3 × Tg-AD mice, a triple transgenic Alzheimer’s disease (AD) model [190]. Previous studies indicated that STEP levels are elevated in the frontal cortex of AD patients and in three transgenic AD mouse models [191, 192]. STEP was shown to dephosphorylate glutamate receptor subunits, resulting in the internalization of NMDA and AMPA glutamate receptors that control synaptic plasticity and memory function [193, 194]. The classical hypothesis of AD proposes that accumulation of β-amyloid (Aβ) in the brain is responsible for disease-related pathology. Synaptic function is thought to be disrupted through Aβ-induced internalization of NMDA and AMPA receptors [195, 196]. The current data suggest an advanced model in which Aβ-induced internalization of glutamate receptors is mediated through STEP (Fig. 10). More precisely, STEP activity is increased due to (1) Aβ-mediated activation of calcineurin, resulting in dephosphorylation and activation of STEP [191], and (2) Aβ-inhibition of the proteasome, resulting in decreased degradation of STEP [197]. As a result, STEP activity is increased in AD, leading to loss of NMDA and AMPA receptors from synaptosomal membranes. Collectively, these studies suggest that inhibition of STEP activity may be beneficial in AD treatment, and validate STEP as a novel drug target in AD (and perhaps other neuropsychiatric disorders, reviewed in ref. 131).
Fig. 10.
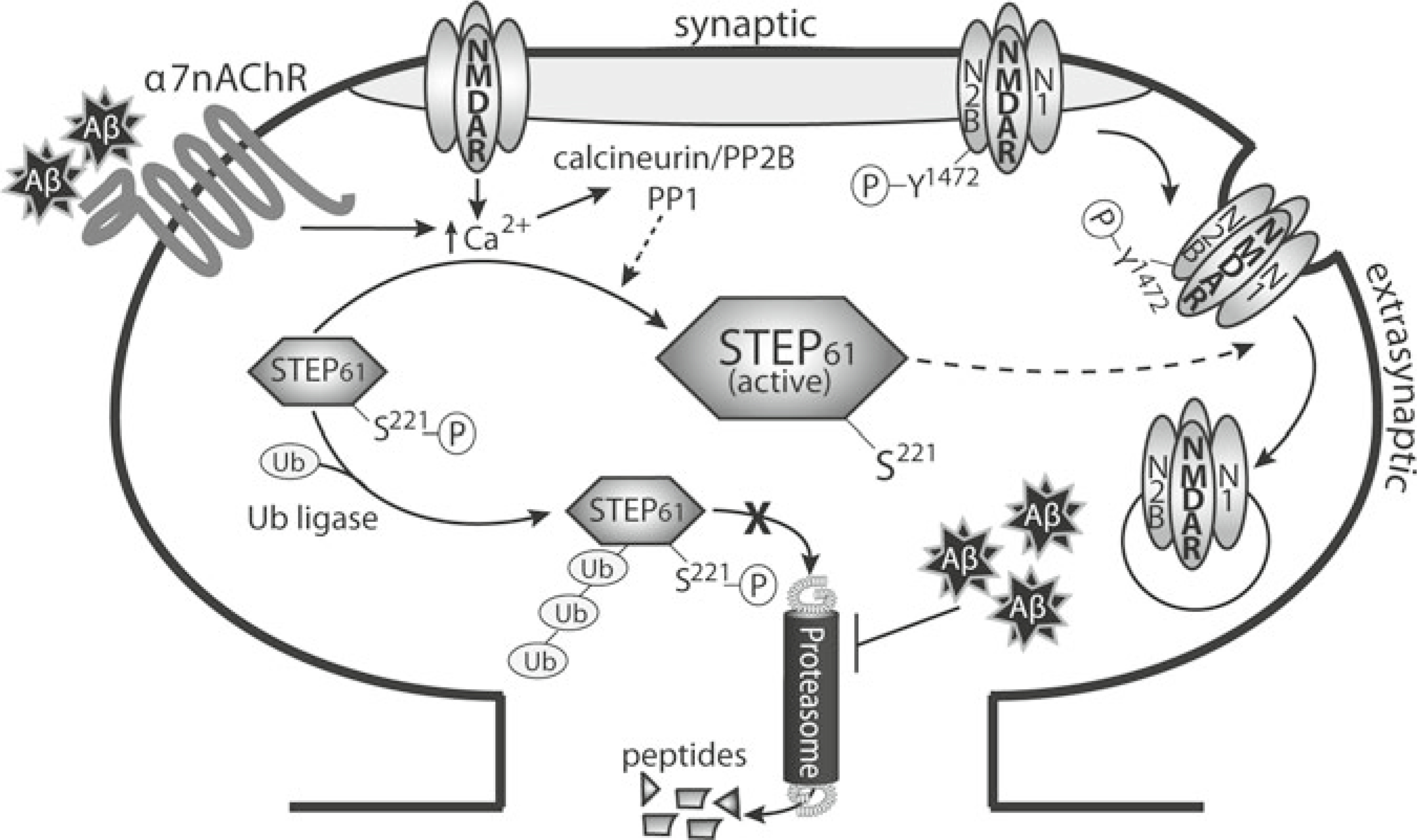
Model of glutamate receptor internalization through Aβ-mediated activation of STEP. Aβ activates the α7 nicotinic receptor, leading to Ca2+ influx and activation of calcineurin [197]. Calcineurin subsequently dephosphorylates and activates STEP. Concomitantly, Aβ also elevates STEP protein levels through inhibition of the ubiquitin proteasome system [203]. STEP dephosphorylates a regulatory tyrosine residue in both the NR2B and GluR2 glutamate receptor subunits, leading to internalization of the receptors [199, 200]. As a result, synaptic function is disrupted (Figure from ref. 131, with permission)
8. PTP Inhibitor Development
PTPs have been considered as therapeutic targets for more than a decade, and many promising compounds have been published (reviewed in refs. 19, 136, 198–201). However, past efforts to develop drugs targeting a specific PTP have been plagued by issues related to bioavailability and selectivity. This is due to the fact that the majority of PTP inhibitors carry a pTyr-mimicking group that provides most of the binding energy through interaction with the highly conserved active site residues. In early efforts, high-affinity peptide substrates were converted into competitive PTP inhibitors by changing the pTyr moiety to a nonhydrolyzable pTyr mimetic such as a (fluoro)phosphonate group [202]. However, despite the incredible potency of some of these compounds in vitro, little to no cell-based activity could be achieved, owing to the multi-charged nature of the phosphonates and their poor drug-likeness according to Lipinski’s rules [203], resulting in the lack of cell membrane permeability of these molecules. Subsequently, small molecules containing pTyr mimetics with only one negative charge (e.g., a carboxylic acid or tetrazole group) have been developed; these compounds are often not as potent as the phosphonates, but usually have better drug-like properties. Prodrug strategies to deliver PTP inhibitors more easily into cells have also been utilized [204, 205]. However, a significant challenge yet to be mastered is posed by the highly conserved catalytic core structure among the members of the PTP family. This makes it difficult to generate inhibitors with selectivity for a particular target, and to generate drugs without serious side effects. Although evidence in the form of many solved 3D structures has now made it clear that there are indeed differences in surface topology and charge distribution in the terrain that surrounds the catalytic pocket [33], examples of truly specific inhibitors are still missing. The fact that the majority of PTP inhibitors owe most of their binding energy to their interactions within the highly conserved phosphate-binding pocket (catalytic pocket) illustrates the challenge in designing potent and at the same time selective inhibitors. Taking advantage of unique amino acid residues and surface features peripheral of the catalytic pocket has resulted in inhibitors with somewhat increased selectivity; however, the trade off is usually expanded size and molecular weight of the inhibitor, making the compound less likely to enter cells or to be absorbed in the gut. Clearly, new approaches are needed to overcome this significant hurdle in the development of novel PTP-based treatment strategies in human disease.
8.1. Open State Binding
Recent co-crystal structures solved by Zhong-Yin Zhang’s group and structure-based computational approaches by our laboratory have shown that small-molecule inhibitors can bind a PTP with its WPD-loop in the inactive, open conformation (see Subheading 5.2) and can stabilize the loop in this position [187, 206–208]. Although crystal structures with the WPD-loop in open conformation have long been available, it was previously thought that ligand binding to the P-loop always causes the WPD-loop to close, restricting the ligand binding mode to resemble the one of the natural substrate. As shown by recent crystal structures of LYP:inhibitor [206], PTP1B:inhibitor [207], and SHP2:inhibitor [187] complexes, this is not necessarily the case. These findings suggest that it is indeed possible to generate molecules that bind and inhibit PTPs in a way that locks the WPD-loop in its open, inactive state. Such a binding mode has profound implications on the design of inhibitors. Surface properties of the active site are much more diverse among PTPs with the WPD-loop in the open state conformation, suggesting that compounds could be generated that specifically bind the open state of the target PTP of choice. Moreover, the active site pocket in the open state not only is less conserved, but also significantly larger, allowing the design of inhibitors that bind entirely in the pocket. A comparison of PTP1B:inhibitor complexes in closed and open conformations is shown in Fig. 11. In the closed-state binding mode, only the head group of the inhibitor binds into the catalytic pocket (Fig. 11a, b). In the open-state binding mode, the entire inhibitor occupies the very large depression between P-loop and WPD- loop in PTP1B (Fig. 11c, d). A comparison of open state structures of different PTPs reveals significant differences in shape and electrostatic potential of the large pocket (Fig. 11f–j). Why “open state binding” has only been observed in a few cases so far is not entirely clear. It may be more difficult for a small molecule to stabilize a rather flexible loop in the open state. Or co- crystallization may be more successful with compounds that bind the closed state, which in turn may favor closed state binders in the selection of lead compounds. Regardless, given the existence of many crystal structures in the open conformation, structure-based methods such as virtual ligand screening may be employed in the search for hits that specifically bind the open state [208].
Fig. 11.
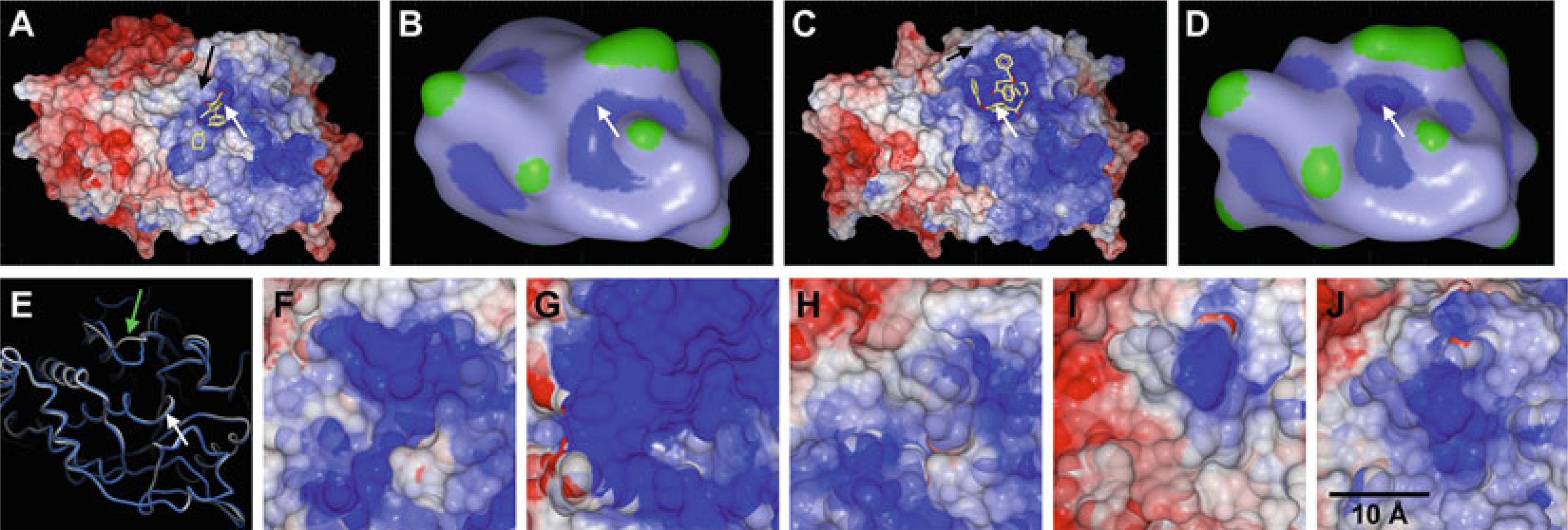
(a–d) Comparison of ligand binding to PTP1B in closed state (a/b; PDB ID: 2QBS) and open state (c/d; PDB ID: 3EAX). The protein surface in (a) and (c) is colored by electrostatic potential as calculated and rendered in ICM (blue, most positive; red, most negative; the colors were capped at ±5 kcal/electron units). Macroshape representations as rendered in ICM (blue, deepest depression; green, largest protrusion) illustrate the differences between the active site pockets in closed (b) and open conformation (d). White arrows indicate the position of the P-loop, black arrows indicate the WPD-loop in closed state (a) and open state (c). (e) Comparison of PTP1B with WPD-loop in closed (blue; PDB ID 2QBS) and open (grey; PDB ID 3EAX) conformation. The white arrow indicates the P-loop, the green arrow indicates the WPD-loop. (f–j) Comparison of open state conformation in PTP1B (f; PDB ID 3EB1), TCPTP (g; PDB ID 1L8K), LYP (h; PDB ID 2P6X), LAR (i; PDB ID 1LAR), and RPTPγ (j; PDB ID 2H4V). The protein surface is colored by electrostatic potential as calculated and rendered in ICM (blue, most positive; red, most negative; the colors were capped at ±5 kcal/electron units)
8.2. Allosteric Inhibition
Another solution to the inherent selectivity problem could be to target allosteric sites at the PTP protein surface. So far allosteric inhibitors have only been reported for PTP1B [34]. In their paper, Wiesmann and colleagues determined the crystal structure of the complex between PTP1B and 3-(3,5-dibromo-4-hydroxybenzoyl)-2-ethyl- N -(4-sulfamoylphenyl) benzofuran-6-sulfonamide (“compound 2,” Fig. 12a). They found that compound 2 binds to a site located ~20 Å away from the catalytic site at the “back” of the phosphatase. They showed that compound 2 acts as an allosteric inhibitor (IC50 = 25 μM) by blocking the mobility of the WPD-loop and thereby preventing the formation of the active, closed form of the enzyme. Interestingly, the residues that form the allosteric site in PTP1B are not conserved, and the surface properties of this site differ even between closely related PTPs (Fig. 12b). In agreement with this notion, compound 2, which also enhanced insulin signaling in cells, exhibited good selectivity for PTP1B. In a follow-up study, Kamerlin and colleagues used molecular dynamics simulations to study WPD-loop mobility in the presence of 3-(3,5-dibromo-4-hydroxybenzoyl)-2-ethyl- N,N-dimethylbenzofuran −6- sulfonamide (“compound 1” in ref. 34, IC50 = 350 μM) [35]. Their data suggest that the reduced flexibility of the WPD-loop may be the result of the allosteric inhibitor making the α3-helix more rigid and blocking a potential contraction of the helix that is necessary for WPD-loop flexibility. Additionally, their data shows that the reduced flexibility in the WPD-loop is accompanied by a reduced flexibility in the S-loop, which is the loop that follows the α3-helix and precedes a β-sheet that connects to the P-loop. Thus, allosteric inhibition of PTP1B, and perhaps of other PTPs, may not be restricted to binding of compounds to the site identified by Wiesmann and colleagues, but may also be mediated through binding of compounds to additional surface areas that involve the α3-helix and the S-loop. In fact, Zhang and Bishop used scanning-insertional mutagenesis to engineer mutants of TCPTP in the search for potential allosteric sites [209]. Specifically, they employed a small molecule fluorescein arsenical hairpin binder (FlAsH) and a FlAsH-binding peptide (TetraCys: CCPGCC) inserted at various loop regions in the catalytic domain (Fig. 12c). They then tested the phosphatase activity in the presence or absence of FlAsH. They found that TCPTP catalytic efficiency was decreased by more than threefold in the presence of FlAsH when TetraCys was inserted at the Ala79 position. Interestingly, Ala79 is located near the S-loop in the TCPTP 3D structure, suggesting that the allosteric effect of FlAsH-binding could be due to reduced flexibility of the WPD-loop via stabilization of the S-loop/α3-helix. Collectively, these studies provide proof of principle for an allosteric approach in PTP inhibitor development and justify future studies that specifically search for ligands that bind corresponding sites in PTP1B, TCPTP, or other PTPs.
Fig. 12.
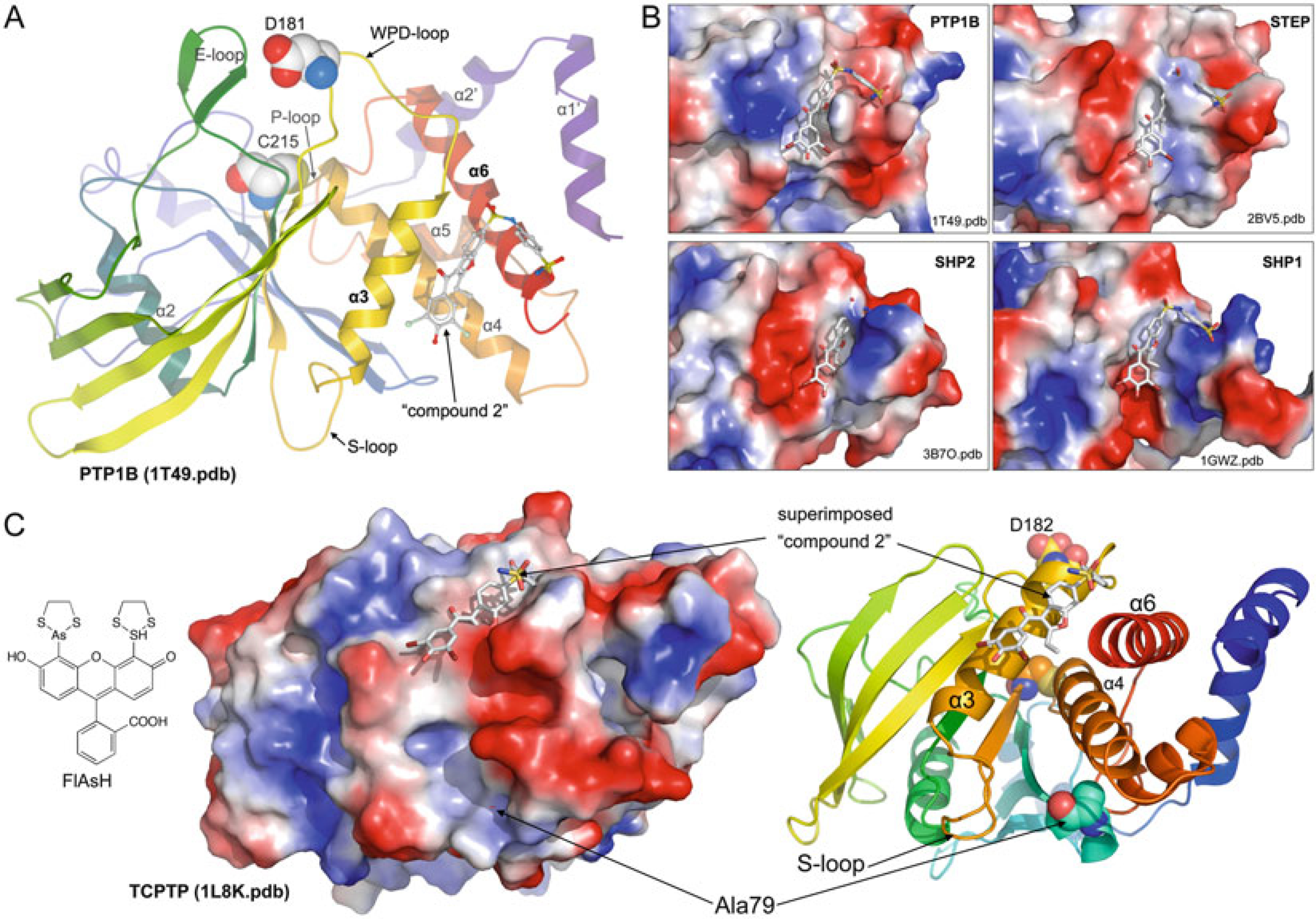
Allosteric inhibition of PTPs. (a) Crystal structure of PTP1B complexed with an allosteric inhibitor (compound 2, shown in ball-and-stick representation); catalytic Cys215 and Asp181 are shown as spheres (PDB ID: 1T49, ref. 34). (b) Comparison of the allosteric site in PTP1B (as in (a)) with corresponding sites in STEP (PDB ID: 2BV5), SHP2 (PDB ID: 3B7O), and SHP1 (PDB ID: 1GWZ). Structures are superimposed; proteins are shown in surface representation (blue, most positive; red, most negative); compound 2 is shown as reference in all structures. (c) Scanning-insertional mutagenesis using FlAsH and a FlAsH-binding peptide (TetraCys: CCPGCC) inserted at position Ala79 identifies a potential allosteric site in TCPTP. To illustrate the location of the identified allosteric site, the crystal structure of TCPTP (PDB ID: 1L8K) is shown as surface and ribbon representation; Ala79, Cys216, and D182 are shown as spheres. The PTP allosteric inhibitor (compound 2) is superimposed for orientation
8.3. Possible Alternative Approaches
To date, numerous screening and medicinal chemistry campaigns have failed to produce highly specific and efficacious PTP inhibitors with drug-like properties. However, PTP specificity in vivo is indisputable, raising the question whether the commonly used assay systems [210] are capable of identifying suitable compounds that can be developed into PTP-based small-molecule therapeutics. Given that binding of intact protein substrates appears to determine PTP substrate specificity and even changes the nature of the PTP active site (see Subheading 6.1), it seems reasonable to develop assay systems that employ intact protein substrates and PTP proteins that, in addition to the minimal catalytic domain, also include all relevant substrate binding domains/motifs. A limiting factor for such an approach will be the availability of suitable recombinant proteins in quantities necessary for high-throughput screening (HTS). While recombinant PTP catalytic domains can be easily expressed and isolated from bacteria in high yields and purity, larger protein entities that include additional domains/motifs are not as easily accessible or may not be attainable at all. A second limiting factor will be the implementation of a reliable PTP assay system. Km values of intact substrate proteins are usually in the nanomolar range (e.g., 610 nM for ERK2/pTpY with HePTP). Using the protein substrates at Km concentration in the screening assay will produce free phosphate at levels below the detection limit of the commonly used malachite green-based reagents [210]. Thus, enzyme-coupled or pTyr antibody-based assays with much greater sensitivity will need to be adopted and implemented for HTS. Besides active site inhibitors with possibly greater selectivity for the PTP of interest, an assay that uses intact protein substrates with suitable PTPs is expected to also identify compounds that interfere with substrate binding distal from the active site. Because the distal substrate binding sites, such as the “second site” (see Subheading 6.2), appear not nearly as conserved as the active site-proximal substrate interface, such inhibitors are expected to be more selective for the PTP of interest.
A major drawback in PTP activity-based HTS assays is that the nucleophilic cysteine (i.e., thiolate) is extremely susceptible to inactivation through alkylation [211–213], oxidation [46], nitrosylation [47], and sulfhydration [48]. For instance, trace amounts of Cu2+ (widely used as a catalyst in synthetic chemistry) can effectively abrogate PTP activity via oxidation that is only partially impeded/reversed by the use of reducing agents such as dithiothreitol (DTT) [214]. In fact, from our own experience in utilizing libraries of commercial compounds for HTS in the search for PTP inhibitors, we find that the majority of hit compounds, when repurchased as powders from commercial sources, contain impurities that are responsible for the observed PTP inhibition. Thus, significantly improved quality of compound libraries would greatly enhance the quality of the data from PTP activity-based HTS assays. Alternatively, assays could be applied that do not rely on measuring PTP activity, but instead measure ligand binding to proteins. One such technology suitable for HTS is differential scanning fluorimetry (DSF; aka Thermofluor or fluorescence thermal shift assay). DSF is a rapid and inexpensive screening method to identify small-molecule ligands that bind and stabilize globular proteins [215]. Applicable to 384- and even 1536-well formats, DSF uses relatively little protein and provides a fluorescence read-out measurement of protein melting temperatures, which correlate with ligand binding. Confirmed binders can subsequently be repurchased, repurified, and tested in PTP activity assays. By blocking the active site during the DSF measurement (e.g., with orthovanadate), it should also be possible to search specifically for ligands that do not bind to the active site and possibly interfere with protein substrate binding or act as allosteric inhibitors.
Another advantage of using an assay system for HTS that does not rely on measuring PTP activity is the possibility to utilize PTPs in an inactive, e.g., oxidized state, and specifically screen for molecules that bind the protein in this state. Under physiological conditions, optimal tyrosine phosphorylation responses are also controlled by reactive oxygen species (ROS), which transiently inactivate PTPs by oxidizing the active site cysteine [216, 217]. ROS can oxidize reactive thiol groups to sulfenic acid (−SOH, oxidation state +1) [218], a transient modification that is reversed by thiol-containing reductants such as glutathione [219]. However, under stronger oxidizing conditions, sulfenic acid can be further oxidized to sulfinic acid (−SO2H, oxidation state +2) and sulfonic acid (− SO3H, oxidation state +4). Both of these oxidation states cannot be reversed with reducing agents. Given that reversibility is essential for normal cell signaling, different mechanisms are in place that prevent further oxidation of the active site cysteine in PTPs. In many PTPs, an additional cysteine residue is located in close proximity to the active site cysteine. For a number of these PTPs, including CDC25, PTEN, LMPTP, MKP-3, DUSP12, SHP1, SHP2, and LYP, it was shown that disulfide bond formation between the two cysteine residues prevents irreversible oxidation (reviewed in ref. 220). Active site cysteine residues in PTPs that do not contain an additional cysteine near the active site can be protected by formation of a cyclic sulphenyl-amide, in which the cysteine sulfur atom is covalently linked to the backbone nitrogen atom of an adjacent residue. This was first observed in PTP1B [221, 222], and later also in the membrane distal pseudo-PTP domain (D2) in RPTPα [223]. Interestingly, sulphenyl-amide formation is accompanied by a major rearrangement of the active site P- and pTyr-loops (Fig. 13). In fact, Tonks and colleagues recently reported the generation of antibodies that specifically stabilize PTP1B in the oxidized, sulphenyl-amide state and thereby inhibit PTP1B function [224]. Expression of these conformation- sensing antibodies enhanced insulin-induced tyrosine phosphorylation of the insulin receptor and its substrate IRS-1 and increased insulin-induced phosphorylation of PKB/AKT. Importantly, the antibodies were specific for PTP1B and did not recognize oxidized TCPTP, a closely related PTP. These data suggest that stabilization of the oxidized, inactive form of PTP1B may (1) be beneficial in the treatment of type 2 diabetes, and (2) be attained very specifically. Judging from the crystal structure of the sulphenyl-amide form of PTP1B, the surface properties of the active site not only appear dramatically different from the reduced form, but also seem to be amenable to high affinity small-molecule interaction (Fig. 13). Importantly, due to the conformational change of the P-loop, the highly charged active site pocket in the reduced, active state (Fig. 13b) does not exist as such in the oxidized state (Fig. 13c). As a consequence of these major changes, active site PTP inhibitors are not expected to bind to the oxidized state and vice versa. Furthermore, the conformational change of the pTyr-loop, which leads to a major rearrangement of the conserved tyrosine residue (Tyr46 in PTP1B), opens up a cleft that could be exploited for small-molecule binding (Fig. 13c). Collectively, these data provide rationale for developing a novel screening strategy, which seeks to identify compounds that specifically bind PTP1B in the oxidized state. Non-activity-based HTS assays such as DSF could be employed for this purpose. Such assays will also benefit from a mutant form of PTP1B (PTP1B-CASA), in which the catalytic cysteine and adjacent serine residues are mutated to alanine. Due to the loss of two critical hydrogen bonding interactions, PTP1B-CASA adopts a stable P-loop conformation that is identical to PTP1B in the oxidized, sulphenyl-amide state [224]. Targeting the oxidized state would circumvent the common difficulties related to the highly charged PTP active site inhibitors, because high-affinity binding would no longer depend on pTyr- mimicking groups, which usually account for low membrane permeability and nonselective inhibition of PTPs. If such efforts yield potent and efficacious compounds, and if future studies show that under physiological conditions sulphenyl-amide formation is a general feature of the PTP family, PTP inhibitor development could be up for a major paradigm shift.
Fig. 13.
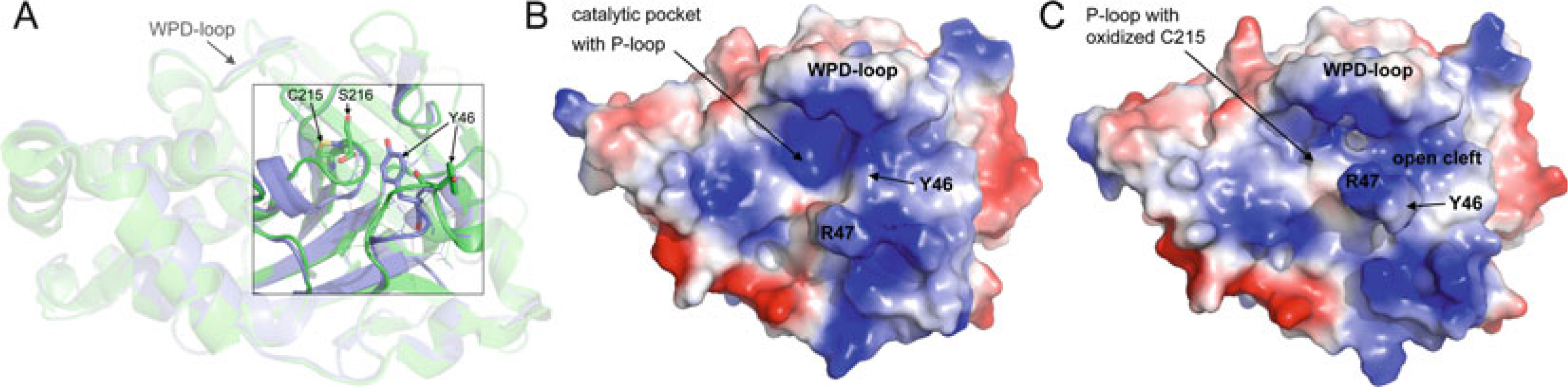
Comparison of reduced and oxidized states of PTP1B. (a) Ribbon diagram of PTP1B in reduced state (blue; PDB ID: 2HNP) and PTP1B in oxidized, sulphenyl-amide state (green, PDB ID: 1OEM). P-loop Cys215 and Ser216 residues, which form the sulphenyl-amide five-membered ring, and pTyr-loop Tyr46 residues are shown in stick representation. (b/c) Surface representation (blue, most positive; red, most negative) of PTP1B in the reduced state (b) and oxidized state (c)
Acknowledgments
This work was supported by NIH grants R03MH095532, R03MH084230, and R21CA132121 (to L.T.). We thank Robert Parker for his help in preparing Table 1.
References
- 1.Hunter T (2000) Signaling—2000 and beyond. Cell 100:113–127 [DOI] [PubMed] [Google Scholar]
- 2.Pawson T, Scott JD (2005) Protein phosphorylation in signaling—50 years and counting. Trends Biochem Sci 30:286–290 [DOI] [PubMed] [Google Scholar]
- 3.Luan S (2003) Protein phosphatases in plants. Annu Rev Plant Biol 54:63–92 [DOI] [PubMed] [Google Scholar]
- 4.Deutscher J, Saier MHJ (2005) Ser/Thr/Tyr protein phosphorylation in bacteria—for long time neglected, now well established. J Mol Microbiol Biotechnol 9:125–131 [DOI] [PubMed] [Google Scholar]
- 5.Feher Z, Szirak K (1999) Signal transduction in fungi—the role of protein phosphorylation. Acta Microbiol Immunol Hung 46:269–271 [DOI] [PubMed] [Google Scholar]
- 6.Moorhead GB, De Wever V, Templeton G et al. (2009) Evolution of protein phosphatases in plants and animals. Biochem J 417:401–409 [DOI] [PubMed] [Google Scholar]
- 7.Olsen JV, Blagoev B, Gnad F et al. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635–648 [DOI] [PubMed] [Google Scholar]
- 8.Hunter T, Sefton BM (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A 77:1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter T (1998) The role of tyrosine phosphorylation in cell growth and disease. Harvey Lect 94:81–119 [PubMed] [Google Scholar]
- 10.Larsen M, Tremblay ML, Yamada KM (2003) Phosphatases in cell-matrix adhesion and migration. Nat Rev Mol Cell Biol 4:700–711 [DOI] [PubMed] [Google Scholar]
- 11.Alonso A, Sasin J, Bottini N et al. (2004) Protein tyrosine phosphatases in the human genome. Cell 117:699–711 [DOI] [PubMed] [Google Scholar]
- 12.Mustelin T, Vang T, Bottini N (2005) Protein tyrosine phosphatases and the immune response. Nat Rev Immunol 5:43–57 [DOI] [PubMed] [Google Scholar]
- 13.Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7:833–846 [DOI] [PubMed] [Google Scholar]
- 14.Halle M, Tremblay ML, Meng TC (2007) Protein tyrosine phosphatases: emerging regulators of apoptosis. Cell Cycle 6:2773–2781 [DOI] [PubMed] [Google Scholar]
- 15.Pao LI, Badour K, Siminovitch KA et al. (2007) Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol 25:473–523 [DOI] [PubMed] [Google Scholar]
- 16.Hunter T (2009) Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 21:140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhee I, Veillette A (2012) Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol 13:439–447 [DOI] [PubMed] [Google Scholar]
- 18.Cohen P (2002) Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 1:309–315 [DOI] [PubMed] [Google Scholar]
- 19.Tautz L, Pellecchia M, Mustelin T (2006) Targeting the PTPome in human disease. Expert Opin Ther Targets 10:157–177 [DOI] [PubMed] [Google Scholar]
- 20.Andersen JN, Mortensen OH, Peters GH et al. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol 21:7117–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pulido R, Hooft van Huijsduijnen R (2008) Protein tyrosine phosphatases: dual-specificity phosphatases in health and disease. FEBS J 275:848–866 [DOI] [PubMed] [Google Scholar]
- 22.Patterson KI, Brummer T, O’Brien PM et al. (2009) Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J 418:475–489 [DOI] [PubMed] [Google Scholar]
- 23.Bottini N, Bottini E, Gloria-Bottini F et al. (2002) Low-molecular-weight protein tyrosine phosphatase and human disease: in search of biochemical mechanisms. Arch Immunol Ther Exp (Warsz) 50:95–104 [PubMed] [Google Scholar]
- 24.Zhang ZY, Wang Y, Dixon JE (1994) Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc Natl Acad Sci U S A 91:1624–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang ZY (1995) Kinetic and mechanistic characterization of a mammalian protein-tyrosine phosphatase, PTP1. J Biol Chem 270:11199–11204 [DOI] [PubMed] [Google Scholar]
- 26.Denu JM, Stuckey JA, Saper MA et al. (1996) Form and function in protein dephosphorylation. Cell 87:361–364 [DOI] [PubMed] [Google Scholar]
- 27.Denu JM, Lohse DL, Vijayalakshmi J et al. (1996) Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis. Proc Natl Acad Sci U S A 93:2493–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuckey JA, Schubert HL, Fauman EB et al. (1994) Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature 370:571–575 [DOI] [PubMed] [Google Scholar]
- 29.Hengge AC, Denu JM, Dixon JE (1996) Transition-state structures for the native dual-specific phosphatase VHR and D92N and S131A mutants. Contributions to the driving force for catalysis. Biochemistry 35: 7084–7092 [DOI] [PubMed] [Google Scholar]
- 30.Zhang ZY (2002) Protein tyrosine phosphatases, structure and function, substrate specificity, and inhibitor development. Annu Rev Pharmacol Toxicol 42:209–234 [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZY, Maclean D, McNamara DJ et al. (1994) Protein tyrosine phosphatase substrate specificity: size and phosphotyrosine positioning requirements in peptide substrates. Biochemistry 33:2285–2290 [DOI] [PubMed] [Google Scholar]
- 32.Mustelin T, Tautz L, Page R (2005) Structure of the hematopoietic tyrosine phosphatase (HePTP) catalytic domain: structure of a KIM phosphatase with phosphate bound at the active site. J Mol Biol 354:150–163 [DOI] [PubMed] [Google Scholar]
- 33.Barr AJ, Ugochukwu E, Lee WH et al. (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136:352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiesmann C, Barr KJ, Kung J et al. (2004) Allosteric inhibition of protein tyrosine phosphatase 1B. Nat Struct Mol Biol 11:730–737 [DOI] [PubMed] [Google Scholar]
- 35.Kamerlin SC, Rucker R, Boresch S (2007) A molecular dynamics study of WPD-loop flexibility in PTP1B. Biochem Biophys Res Commun 356:1011–1016 [DOI] [PubMed] [Google Scholar]
- 36.Vang T, Liu WH, Delacroix L et al. (2012) LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol 8:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorenz U (2009) SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228:342–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pei D, Lorenz U, Klingmuller U et al. (1994) Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: a new function for Src homology 2 domains. Biochemistry 33: 15483–15493 [DOI] [PubMed] [Google Scholar]
- 39.Hof P, Pluskey S, Dhe-Paganon S et al. (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92:441–450 [DOI] [PubMed] [Google Scholar]
- 40.Johnson P, Ostergaard HL, Wasden C et al. (1992) Mutational analysis of CD45. A leukocyte- specific protein tyrosine phosphatase. J Biol Chem 267:8035–8041 [PubMed] [Google Scholar]
- 41.Streuli M, Krueger NX, Thai T et al. (1990) Distinct functional roles of the two intracellular phosphatase like domains of the receptor-linked protein tyrosine phosphatases LCA and LAR. EMBO J 9:2399–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kashio N, Matsumoto W, Parker S et al. (1998) The second domain of the CD45 protein tyrosine phosphatase is critical for interleukin- 2 secretion and substrate recruitment of TCR-zeta in vivo. J Biol Chem 273: 33856–33863 [DOI] [PubMed] [Google Scholar]
- 43.Shi Y (2009) Serine/threonine phosphatases: mechanism through structure. Cell 139: 468–484 [DOI] [PubMed] [Google Scholar]
- 44.Barford D, Flint AJ, Tonks NK (1994) Crystal structure of human protein tyrosine phosphatase 1B. Science 263:1397–1404 [PubMed] [Google Scholar]
- 45.Zhang ZY, Dixon JE (1993) Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry 32: 9340–9345 [DOI] [PubMed] [Google Scholar]
- 46.Tonks NK (2005) Redox redux: revisiting PTPs and the control of cell signaling. Cell 121:667–670 [DOI] [PubMed] [Google Scholar]
- 47.Chen YY, Chu HM, Pan KT et al. (2008) Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J Biol Chem 283:35265–35272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishnan N, Fu C, Pappin DJ et al. (2011) H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal 4:ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keng YF, Wu L, Zhang ZY (1999) Probing the function of the conserved tryptophan in the flexible loop of the Yersinia protein- tyrosine phosphatase. Eur J Biochem 259:809–814 [DOI] [PubMed] [Google Scholar]
- 50.Hoff RH, Hengge AC, Wu L et al. (2000) Effects on general acid catalysis from mutations of the invariant tryptophan and arginine residues in the protein tyrosine phosphatase from Yersinia. Biochemistry 39:46–54 [DOI] [PubMed] [Google Scholar]
- 51.Kurkcuoglu Z, Bakan A, Kocaman D et al. (2012) Coupling between catalytic loop motions and enzyme global dynamics. PLoS Comput Biol 8:e1002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui L, Yu WP, DeAizpurua HJ et al. (1996) Cloning and characterization of islet cell antigen- related protein-tyrosine phosphatase (PTP), a novel receptor-like PTP and autoantigen in insulin-dependent diabetes. J Biol Chem 271:24817–24823 [PubMed] [Google Scholar]
- 53.Seifert RA, Coats SA, Oganesian A et al. (2003) PTPRQ is a novel phosphatidylinositol phosphatase that can be expressed as a cytoplasmic protein or as a subcellularly localized receptor-like protein. Exp Cell Res 287: 374–386 [DOI] [PubMed] [Google Scholar]
- 54.Cheng J, Wu K, Armanini M et al. (1997) A novel protein-tyrosine phosphatase related to the homotypically adhering kappa and mu receptors. J Biol Chem 272:7264–7277 [DOI] [PubMed] [Google Scholar]
- 55.Gingras MC, Zhang YL, Kharitidi D et al. (2009) HD-PTP is a catalytically inactive tyrosine phosphatase due to a conserved divergence in its phosphatase domain. PLoS One 4:e5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cardone L, Carlucci A, Affaitati A et al. (2004) Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol 24:4613–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rabin DU, Pleasic SM, Shapiro JA et al. (1994) Islet cell antigen 512 is a diabetes-specific islet autoantigen related to protein tyrosine phosphatases. J Immunol 152: 3183–3188 [PubMed] [Google Scholar]
- 58.Jia Z, Barford D, Flint AJ et al. (1995) Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268:1754–1758 [DOI] [PubMed] [Google Scholar]
- 59.Eswaran J, von Kries JP, Marsden B et al. (2006) Crystal structures and inhibitor identification for PTPN5, PTPRR and PTPN7: a family of human MAPK-specific protein tyrosine phosphatases. Biochem J 395:483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iversen LF, Moller KB, Pedersen AK et al. (2002) Structure determination of T cell protein- tyrosine phosphatase. J Biol Chem 277:19982–19990 [DOI] [PubMed] [Google Scholar]
- 61.Yang J, Cheng Z, Niu T et al. (2000) Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1. J Biol Chem 275:4066–4071 [DOI] [PubMed] [Google Scholar]
- 62.Asante-Appiah E, Patel S, Desponts C et al. (2006) Conformation-assisted inhibition of protein-tyrosine phosphatase-1B elicits inhibitor selectivity over T-cell protein-tyrosine phosphatase. J Biol Chem 281:8010–8015 [DOI] [PubMed] [Google Scholar]
- 63.Critton DA, Tautz L, Page R (2011) Visualizing active-site dynamics in single crystals of HePTP: opening of the WPD loop involves coordinated movement of the E loop. J Mol Biol 405:619–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie L, Zhang YL, Zhang ZY (2002) Design and characterization of an improved protein tyrosine phosphatase substrate-trapping mutant. Biochemistry 41:4032–4039 [DOI] [PubMed] [Google Scholar]
- 65.Zhao Y, Wu L, Noh SJ et al. (1998) Altering the nucleophile specificity of a protein-tyrosine phosphatase-catalyzed reaction. Probing the function of the invariant glutamine residues. J Biol Chem 273:5484–5492 [DOI] [PubMed] [Google Scholar]
- 66.Pedersen AK, Guo XL, Moller KB et al. (2004) Residue 182 influences the second step of protein-tyrosine phosphatase- mediated catalysis. Biochem J 378:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zabell AP, Schroff ADJ, Bain BE et al. (2006) Crystal structure of the human B-form low molecular weight phosphotyrosyl phosphatase at 1.6-A resolution. J Biol Chem 281: 6520–6527 [DOI] [PubMed] [Google Scholar]
- 68.Bryson GL, Massa H, Trask BJ et al. (1995) Gene structure, sequence, and chromosomal localization of the human red cell-type low-molecular- weight acid phosphotyrosyl phosphatase gene, ACP1. Genomics 30:133–140 [DOI] [PubMed] [Google Scholar]
- 69.Tailor P, Gilman J, Williams S et al. (1999) A novel isoform of the low molecular weight phosphotyrosine phosphatase, LMPTP-C, arising from alternative mRNA splicing. Eur J Biochem 262:277–282 [DOI] [PubMed] [Google Scholar]
- 70.Ramponi G, Manao G, Camici G et al. (1989) The 18 kDa cytosolic acid phosphatase from bovine live has phosphotyrosine phosphatase activity on the autophosphorylated epidermal growth factor receptor. FEBS Lett 250: 469–473 [DOI] [PubMed] [Google Scholar]
- 71.Rudolph J (2007) Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry 46:3595–3604 [DOI] [PubMed] [Google Scholar]
- 72.Mailand N, Falck J, Lukas C et al. (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288: 1425–1429 [DOI] [PubMed] [Google Scholar]
- 73.Donzelli M, Squatrito M, Ganoth D et al. (2002) Dual mode of degradation of Cdc25 A phosphatase. EMBO J 21:4875–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conklin DS, Galaktionov K, Beach D (1995) 14–3-3 proteins associate with cdc25 phosphatases. Proc Natl Acad Sci U S A 92:7892–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Forrest A, Gabrielli B (2001) Cdc25B activity is regulated by 14–3-3. Oncogene 20: 4393–4401 [DOI] [PubMed] [Google Scholar]
- 76.Giles N, Forrest A, Gabrielli B (2003) 14–3-3 acts as an intramolecular bridge to regulate cdc25B localization and activity. J Biol Chem 278:28580–28587 [DOI] [PubMed] [Google Scholar]
- 77.Hoffmann I, Clarke PR, Marcote MJ et al. (1993) Phosphorylation and activation of human cdc25-C by cdc2–cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J 12:53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoffmann I, Draetta G, Karsenti E (1994) Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J 13:4302–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fauman EB, Cogswell JP, Lovejoy B et al. (1998) Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 93:617–625 [DOI] [PubMed] [Google Scholar]
- 80.Reynolds RA, Yem AW, Wolfe CL et al. (1999) Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J Mol Biol 293:559–568 [DOI] [PubMed] [Google Scholar]
- 81.Hofmann K, Bucher P, Kajava AV (1998) A model of Cdc25 phosphatase catalytic domain and Cdk-interaction surface based on the presence of a rhodanese homology domain. J Mol Biol 282:195–208 [DOI] [PubMed] [Google Scholar]
- 82.Sarmiento M, Zhao Y, Gordon SJ et al. (1998) Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J Biol Chem 273:26368–26374 [DOI] [PubMed] [Google Scholar]
- 83.Huang Z, Zhou B, Zhang ZY (2004) Molecular determinants of substrate recognition in hematopoietic protein-tyrosine phosphatase. J Biol Chem 279:52150–52159 [DOI] [PubMed] [Google Scholar]
- 84.Francis DM, Rozycki B, Tortajada A et al. (2011) Resting and Active States of the ERK2:HePTP Complex. J Am Chem Soc 133:17138–17141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Francis DM, Rozycki B, Koveal D et al. (2011) structural basis of p38a regulation by hematopoietic tyrosine phosphatase. Nat Chem Biol 7:916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wiland AM, Denu JM, Mourey RJ et al. (1996) Purification and kinetic characterization of the mitogen-activated protein kinase phosphatase rVH6. J Biol Chem 271: 33486–33492 [DOI] [PubMed] [Google Scholar]
- 87.Zhao Y, Zhang ZY (2001) The mechanism of dephosphorylation of extracellular signal-regulated kinase 2 by mitogen-activated protein kinase phosphatase 3. J Biol Chem 276:32382–32391 [DOI] [PubMed] [Google Scholar]
- 88.Begley MJ, Taylor GS, Kim SA et al. (2003) Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Mol Cell 12:1391–1402 [DOI] [PubMed] [Google Scholar]
- 89.Puius YA, Zhao Y, Sullivan M et al. (1997) Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proc Natl Acad Sci U S A 94:13420–13425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salmeen A, Andersen JN, Myers MP et al. (2000) Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell 6:1401–1412 [DOI] [PubMed] [Google Scholar]
- 91.Peters GH, Iversen LF, Branner S et al. (2000) Residue 259 is a key determinant of substrate specificity of protein-tyrosine phosphatases 1B and alpha. J Biol Chem 275: 18201–18209 [DOI] [PubMed] [Google Scholar]
- 92.Flint AJ, Tiganis T, Barford D et al. (1997) Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A 94:1680–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sarmiento M, Puius YA, Vetter SW et al. (2000) Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition. Biochemistry 39:8171–8179 [DOI] [PubMed] [Google Scholar]
- 94.Song H, Hanlon N, Brown NR et al. (2001) Phosphoprotein-protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol Cell 7:615–626 [DOI] [PubMed] [Google Scholar]
- 95.Wang S, Tabernero L, Zhang M et al. (2000) Crystal structures of a low-molecular weight protein tyrosine phosphatase from Saccharomyces cerevisiae and its complex with the substrate p-nitrophenyl phosphate. Biochemistry 39:1903–1914 [DOI] [PubMed] [Google Scholar]
- 96.Garton AJ, Flint AJ, Tonks NK (1996) Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol 16:6408–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blanchetot C, Chagnon M, Dube N et al. (2005) Substrate-trapping techniques in the identification of cellular PTP targets. Methods 35:44–53 [DOI] [PubMed] [Google Scholar]
- 98.Agazie YM, Hayman MJ (2003) Development of an efficient “substrate-trapping” mutant of Src homology phosphotyrosine phosphatase 2 and identification of the epidermal growth factor receptor, Gab1, and three other proteins as target substrates. J Biol Chem 278:13952–13958 [DOI] [PubMed] [Google Scholar]
- 99.Sohn J, Parks JM, Buhrman G et al. (2005) Experimental validation of the docking orientation of Cdc25 with its Cdk2-CycA protein substrate. Biochemistry 44:16563–16573 [DOI] [PubMed] [Google Scholar]
- 100.Buhrman G, Parker B, Sohn J et al. (2005) Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry 44:5307–5316 [DOI] [PubMed] [Google Scholar]
- 101.Ostman A, Hellberg C, Bohmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6:307–320 [DOI] [PubMed] [Google Scholar]
- 102.Vang T, Miletic AV, Arimura Y et al. (2008) Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol 26:29–55 [DOI] [PubMed] [Google Scholar]
- 103.Julien SG, Dube N, Hardy S et al. (2011) Inside the human cancer tyrosine phosphatome. Nat Rev Cancer 11:35–49 [DOI] [PubMed] [Google Scholar]
- 104.Goebel-Goody SM, Baum M, Paspalas CD et al. (2012) Therapeutic implications for striatal- enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev 64:65–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keniry M, Parsons R (2008) The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 27: 5477–5485 [DOI] [PubMed] [Google Scholar]
- 106.Mena-Duran AV, Togo SH, Bazhenova L et al. (2005) SHP1 expression in bone marrow biopsies of myelodysplastic syndrome patients: a new prognostic factor. Br J Haematol 129: 791–794 [DOI] [PubMed] [Google Scholar]
- 107.Zhang Q, Wang HY, Marzec M et al. (2005) STAT3- and DNA methyltransferase 1- mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A 102:6948–6953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohi MG, Neel BG (2007) The role of Shp2 (PTPN11) in cancer. Curr Opin Genet Dev 17:23–30 [DOI] [PubMed] [Google Scholar]
- 109.Xu R (2007) Shp2, a novel oncogenic tyrosine phosphatase and potential therapeutic target for human leukemia. Cell Res 17:295–297 [DOI] [PubMed] [Google Scholar]
- 110.Saha S, Bardelli A, Buckhaults P et al. (2001) A phosphatase associated with metastasis of colorectal cancer. Science 294:1343–1346 [DOI] [PubMed] [Google Scholar]
- 111.Loda M, Capodieci P, Mishra R et al. (1996) Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol 149:1553–1564 [PMC free article] [PubMed] [Google Scholar]
- 112.Jiang ZX, Zhang ZY (2008) Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev 27:263–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bourdeau A, Dube N, Tremblay ML (2005) Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Curr Opin Cell Biol 17:203–209 [DOI] [PubMed] [Google Scholar]
- 114.Julien SG, Dube N, Read M et al. (2007) Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet 39:338–346 [DOI] [PubMed] [Google Scholar]
- 115.Bentires-Alj M, Neel BG (2007) Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res 67:2420–2424 [DOI] [PubMed] [Google Scholar]
- 116.Tonks NK, Muthuswamy SK (2007) A brake becomes an accelerator: PTP1B—a new therapeutic target for breast cancer. Cancer Cell 11:214–216 [DOI] [PubMed] [Google Scholar]
- 117.Bottini N, Musumeci L, Alonso A et al. (2004) A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36:337–338 [DOI] [PubMed] [Google Scholar]
- 118.Begovich AB, Carlton VE, Honigberg LA et al. (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75:330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kyogoku C, Langefeld CD, Ortmann WA et al. (2004) Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75:504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stanford SM, Mustelin TM, Bottini N (2010) Lymphoid tyrosine phosphatase and autoimmunity: human genetics rediscovers tyrosine phosphatases. Semin Immunopathol 32: 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Negro R, Gobessi S, Longo PG et al. (2012) Overexpression of the autoimmunity- associated phosphatase PTPN22 promotes survival of antigen-stimulated CLL cells by selectively activating AKT. Blood 119:6278–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Saxena M, Williams S, Brockdorff J et al. (1999) Inhibition of T cell signaling by mitogen- activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP). J Biol Chem 274:11693–11700 [DOI] [PubMed] [Google Scholar]
- 123.Zanke B, Squire J, Griesser H et al. (1994) A hematopoietic protein tyrosine phosphatase (HePTP) gene that is amplified and overexpressed in myeloid malignancies maps to chromosome 1q32.1. Leukemia 8:236–244 [PubMed] [Google Scholar]
- 124.Sergienko E, Xu J, Liu WH et al. (2012) Inhibition of hematopoietic protein tyrosine phosphatase augments and prolongs ERK1/2 and p38 activation. ACS Chem Biol 7: 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fonatsch C, Haase D, Freund M et al. (1991) Partial trisomy 1q. A nonrandom primary chromosomal abnormality in myelodysplastic syndromes? Cancer Genet Cytogenet 56: 243–253 [DOI] [PubMed] [Google Scholar]
- 126.Mamaev N, Mamaeva SE, Pavlova VA et al. (1988) Combined trisomy 1q and monosomy 17p due to translocation t(1;17) in a patient with myelodysplastic syndrome. Cancer Genet Cytogenet 35:21–25 [DOI] [PubMed] [Google Scholar]
- 127.Souza AC, Azoubel S, Queiroz KC et al. (2009) From immune response to cancer: a spot on the low molecular weight protein tyrosine phosphatase. Cell Mol Life Sci 66:1140–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Minassian BA, Lee JR, Herbrick JA et al. (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 20: 171–174 [DOI] [PubMed] [Google Scholar]
- 129.Laporte J, Hu LJ, Kretz C et al. (1996) A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet 13: 175–182 [DOI] [PubMed] [Google Scholar]
- 130.Bolino A, Muglia M, Conforti FL et al. (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet 25:17–19 [DOI] [PubMed] [Google Scholar]
- 131.Azzedine H, Bolino A, Taieb T et al. (2003) Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet 72:1141–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kung C, Pingel JT, Heikinheimo M et al. (2000) Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nat Med 6: 343–345 [DOI] [PubMed] [Google Scholar]
- 133.Elchebly M, Payette P, Michaliszyn E et al. (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283: 1544–1548 [DOI] [PubMed] [Google Scholar]
- 134.Zhang S, Zhang ZY (2007) PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov Today 12: 373–381 [DOI] [PubMed] [Google Scholar]
- 135.Lee S, Wang Q (2007) Recent development of small molecular specific inhibitor of protein tyrosine phosphatase 1B. Med Res Rev 27:553–573 [DOI] [PubMed] [Google Scholar]
- 136.Barr AJ (2010) Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Med Chem 2:1563–1576 [DOI] [PubMed] [Google Scholar]
- 137.Popov D (2011) Novel protein tyrosine phosphatase 1B inhibitors: interaction requirements for improved intracellular efficacy in type 2 diabetes mellitus and obesity control. Biochem Biophys Res Commun 410: 377–381 [DOI] [PubMed] [Google Scholar]
- 138.Siegel PM, Ryan ED, Cardiff RD et al. (1999) Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J 18:2149–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tanner MM, Grenman S, Koul A et al. (2000) Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 6:1833–1839 [PubMed] [Google Scholar]
- 140.Ginestier C, Cervera N, Finetti P et al. (2006) Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res 12:4533–4544 [DOI] [PubMed] [Google Scholar]
- 141.Wiener JR, Kerns BJ, Harvey EL et al. (1994) Overexpression of the protein tyrosine phosphatase PTP1B in human breast cancer: association with p185c-erbB-2 protein expression. J Natl Cancer Inst 86:372–378 [DOI] [PubMed] [Google Scholar]
- 142.Zhai YF, Beittenmiller H, Wang B et al. (1993) Increased expression of specific protein tyrosine phosphatases in human breast epithelial cells neoplastically transformed by the neu oncogene. Cancer Res 53:2272–2278 [PubMed] [Google Scholar]
- 143.Bjorge JD, Pang A, Fujita DJ (2000) Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem 275:41439–41446 [DOI] [PubMed] [Google Scholar]
- 144.Arias-Romero LE, Saha S, Villamar-Cruz O et al. (2009) Activation of Src by protein tyrosine phosphatase 1B Is required for ErbB2 transformation of human breast epithelial cells. Cancer Res 69:4582–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kaminski R, Zagozdzon R, Fu Y et al. (2006) Role of SRC kinases in Neu-induced tumorigenesis: challenging the paradigm using Csk homologous kinase transgenic mice. Cancer Res 66:5757–5762 [DOI] [PubMed] [Google Scholar]
- 146.Dadke S, Chernoff J (2003) Protein-tyrosine phosphatase 1B mediates the effects of insulin on the actin cytoskeleton in immortalized fibroblasts. J Biol Chem 278:40607–40611 [DOI] [PubMed] [Google Scholar]
- 147.Dube N, Cheng A, Tremblay ML (2004) The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci U S A 101: 1834–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kashige N, Carpino N, Kobayashi R (2000) Tyrosine phosphorylation of p62dok by p210bcr-abl inhibits RasGAP activity. Proc Natl Acad Sci U S A 97:2093–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Slamon DJ, Clark GM, Wong SG et al. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182 [DOI] [PubMed] [Google Scholar]
- 150.Bange J, Zwick E, Ullrich A (2001) Molecular targets for breast cancer therapy and prevention. Nat Med 7:548–552 [DOI] [PubMed] [Google Scholar]
- 151.Carter P, Presta L, Gorman CM et al. (1992) Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 89:4285–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nahta R, Esteva FJ (2007) Trastuzumab: triumphs and tribulations. Oncogene 26: 3637–3643 [DOI] [PubMed] [Google Scholar]
- 153.Grossmann KS, Rosario M, Birchmeier C et al. (2010) The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res 106:53–89 [DOI] [PubMed] [Google Scholar]
- 154.Liu X, Qu CK (2011) Protein tyrosine phosphatase SHP-2 (PTPN11) in hematopoiesis and leukemogenesis. J Signal Transduct 2011:195–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tartaglia M, Mehler EL, Goldberg R et al. (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465–468 [DOI] [PubMed] [Google Scholar]
- 156.Noonan JA (1968) Hypertelorism with turner phenotype. a new syndrome with associated congenital heart disease. Am J Dis Child 116: 373–380 [DOI] [PubMed] [Google Scholar]
- 157.Allanson JE (1987) Noonan syndrome. J Med Genet 24:9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marino B, Digilio MC, Toscano A et al. (1999) Congenital heart diseases in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr 135:703–706 [DOI] [PubMed] [Google Scholar]
- 159.Opitz JM (1985) The Noonan syndrome. Am J Med Genet 21:515–518 [DOI] [PubMed] [Google Scholar]
- 160.Tartaglia M, Gelb BD (2005) Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet 6:45–68 [DOI] [PubMed] [Google Scholar]
- 161.Tartaglia M, Niemeyer CM, Fragale A et al. (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148–150 [DOI] [PubMed] [Google Scholar]
- 162.Bentires-Alj M, Paez JG, David FS et al. (2004) Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 64: 8816–8820 [DOI] [PubMed] [Google Scholar]
- 163.Chan RJ, Feng GS (2007) PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 109:862–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chan G, Kalaitzidis D, Neel BG (2008) The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 27:179–192 [DOI] [PubMed] [Google Scholar]
- 165.Barford D, Neel BG (1998) Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 6:249–254 [DOI] [PubMed] [Google Scholar]
- 166.Araki T, Mohi MG, Ismat FA et al. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10:849–857 [DOI] [PubMed] [Google Scholar]
- 167.Chan RJ, Leedy MB, Munugalavadla V et al. (2005) Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood 105:3737–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fragale A, Tartaglia M, Wu J et al. (2004) Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat 23:267–277 [DOI] [PubMed] [Google Scholar]
- 169.Krenz M, Gulick J, Osinska HE et al. (2008) Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc Natl Acad Sci U S A 105:18930–18935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mohi MG, Williams IR, Dearolf CR et al. (2005) Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell 7:179–191 [DOI] [PubMed] [Google Scholar]
- 171.Yu WM, Daino H, Chen J et al. (2006) Effects of a leukemia-associated gain-of-function mutation of SHP-2 phosphatase on interleukin- 3 signaling. J Biol Chem 281:5426–5434 [DOI] [PubMed] [Google Scholar]
- 172.Carta C, Pantaleoni F, Bocchinfuso G et al. (2006) Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 79:129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schubbert S, Zenker M, Rowe SL et al. (2006) Germline KRAS mutations cause Noonan syndrome. Nat Genet 38:331–336 [DOI] [PubMed] [Google Scholar]
- 174.Roberts AE, Araki T, Swanson KD et al. (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 39:70–74 [DOI] [PubMed] [Google Scholar]
- 175.Tartaglia M, Pennacchio LA, Zhao C et al. (2007) Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 39:75–79 [DOI] [PubMed] [Google Scholar]
- 176.Tartaglia M, Martinelli S, Cazzaniga G et al. (2004) Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 104:307–313 [DOI] [PubMed] [Google Scholar]
- 177.Shi ZQ, Yu DH, Park M et al. (2000) Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol Cell Biol 20:1526–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nguyen TV, Ke Y, Zhang EE et al. (2006) Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre- TCR and TCR signals. J Immunol 177:5990–5996 [DOI] [PubMed] [Google Scholar]
- 179.Matozaki T, Murata Y, Saito Y et al. (2009) Protein tyrosine phosphatase SHP-2: a proto-oncogene product that promotes Ras activation. Cancer Sci 100:1786–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Neel BG, Gu H, Pao L (2003) The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293 [DOI] [PubMed] [Google Scholar]
- 181.Loh ML (2011) Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br J Haematol 152:677–687 [DOI] [PubMed] [Google Scholar]
- 182.Le DT, Kong N, Zhu Y et al. (2004) Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 103:4243–4250 [DOI] [PubMed] [Google Scholar]
- 183.Chen L, Sung SS, Yip ML et al. (2006) Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol 70:562–570 [DOI] [PubMed] [Google Scholar]
- 184.Noren-Muller A, Reis-Correa IJ, Prinz H et al. (2006) Discovery of protein phosphatase inhibitor classes by biology-oriented synthesis. Proc Natl Acad Sci U S A 103: 10606–10611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hellmuth K, Grosskopf S, Lum CT et al. (2008) Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci U S A 105:7275–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lawrence HR, Pireddu R, Chen L et al. (2008) Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J Med Chem 51:4948–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang X, He Y, Liu S et al. (2010) Salicylic acid based small molecule inhibitor for the oncogenic Src homology-2 domain containing protein tyrosine phosphatase-2 (SHP2). J Med Chem 53:2482–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu S, Yu Z, Yu X et al. (2011) SHP2 is a target of the immunosuppressant tautomycetin. Chem Biol 18:101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Baum ML, Kurup P, Xu J et al. (2010) A STEP forward in neural function and degeneration. Commun Integr Biol 3:419–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang Y, Kurup P, Xu J et al. (2010) Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107: 19014–19019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Snyder EM, Nong Y, Almeida CG et al. (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058 [DOI] [PubMed] [Google Scholar]
- 192.Chin J, Palop JJ, Puolivali J et al. (2005) Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci 25: 9694–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang Y, Venkitaramani DV, Gladding CM et al. (2008) The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci 28:10561–10566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Y, Kurup P, Xu J et al. (2011) Reduced levels of the tyrosine phosphatase STEP block beta amyloid-mediated GluA1/GluA2 receptor internalization. J Neurochem 119: 664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185 [DOI] [PubMed] [Google Scholar]
- 196.Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6:487–498 [DOI] [PubMed] [Google Scholar]
- 197.Kurup P, Zhang Y, Xu J et al. (2010) Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci 30:5948–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bialy L, Waldmann H (2005) Inhibitors of protein tyrosine phosphatases: next-generation drugs? Angew Chem Int Ed Engl 44:3814–3839 [DOI] [PubMed] [Google Scholar]
- 199.Vintonyak VV, Antonchick AP, Rauh D et al. (2009) The therapeutic potential of phosphatase inhibitors. Curr Opin Chem Biol 13: 272–283 [DOI] [PubMed] [Google Scholar]
- 200.Sobhia ME, Paul S, Shinde R et al. (2012) Protein tyrosine phosphatase inhibitors: a patent review (2002–2011). Expert Opin Ther Pat 22:125–153 [DOI] [PubMed] [Google Scholar]
- 201.He R, Zeng LF, He Y et al. (2012) Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J 280: 731–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Burke TRJ, Kole HK, Roller PP (1994) Potent inhibition of insulin receptor dephosphorylation by a hexamer peptide containing the phosphotyrosyl mimetic F2Pmp. Biochem Biophys Res Commun 204:129–134 [DOI] [PubMed] [Google Scholar]
- 203.Lipinski CA, Lombardo F, Dominy BW et al. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26 [DOI] [PubMed] [Google Scholar]
- 204.Andersen HS, Olsen OH, Iversen LF et al. (2002) Discovery and SAR of a novel selective and orally bioavailable nonpeptide classical competitive inhibitor class of protein-tyrosine phosphatase 1B. J Med Chem 45:4443–4459 [DOI] [PubMed] [Google Scholar]
- 205.Erbe DV, Klaman LD, Wilson DP et al. (2009) Prodrug delivery of novel PTP1B inhibitors to enhance insulin signalling. Diabetes Obes Metab 11:579–588 [DOI] [PubMed] [Google Scholar]
- 206.Yu X, Sun JP, He Y et al. (2007) Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci U S A 104:19767–19772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Liu S, Zeng LF, Wu L et al. (2008) Targeting inactive enzyme conformation: aryl diketoacid derivatives as a new class of PTP1B inhibitors. J Am Chem Soc 130:17075–17084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wu S, Bottini M, Rickert RC et al. (2009) In silico screening for PTPN22 inhibitors: active hits from an inactive phosphatase conformation. ChemMedChem 4:440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zhang XY, Bishop AC (2007) Site-specific incorporation of allosteric-inhibition sites in a protein tyrosine phosphatase. J Am Chem Soc 129:3812–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tautz L, Mustelin T (2007) Strategies for developing protein tyrosine phosphatase inhibitors. Methods 42:250–260 [DOI] [PubMed] [Google Scholar]
- 211.Pot DA, Dixon JE (1992) Active site labeling of a receptor-like protein tyrosine phosphatase. J Biol Chem 267:140–143 [PubMed] [Google Scholar]
- 212.Zhang ZY, Davis JP, Van Etten RL (1992) Covalent modification and active site-directed inactivation of a low molecular weight phosphotyrosyl protein phosphatase. Biochemistry 31:1701–1711 [DOI] [PubMed] [Google Scholar]
- 213.Liu S, Zhou B, Yang H et al. (2008) Aryl vinyl sulfonates and sulfones as active site-directed and mechanism-based probes for protein tyrosine phosphatases. J Am Chem Soc 130: 8251–8260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kim JH, Cho H, Ryu SE et al. (2000) Effects of metal ions on the activity of protein tyrosine phosphatase VHR: highly potent and reversible oxidative inactivation by Cu2+ ion. Arch Biochem Biophys 382:72–80 [DOI] [PubMed] [Google Scholar]
- 215.Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2:2212–2221 [DOI] [PubMed] [Google Scholar]
- 216.Rhee SG, Chang TS, Bae YS et al. (2003) Cellular regulation by hydrogen peroxide. J Am Soc Nephrol 14:S211–S215 [DOI] [PubMed] [Google Scholar]
- 217.Miki H, Funato Y (2012) Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem 151:255–261 [DOI] [PubMed] [Google Scholar]
- 218.Roos G, Messens J (2011) Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med 51: 314–326 [DOI] [PubMed] [Google Scholar]
- 219.Poole LB, Karplus PA, Claiborne A (2004) Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol 44:325–347 [DOI] [PubMed] [Google Scholar]
- 220.Ostman A, Frijhoff J, Sandin A et al. (2011) Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem 150: 345–356 [DOI] [PubMed] [Google Scholar]
- 221.Salmeen A, Andersen JN, Myers MP et al. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423:769–773 [DOI] [PubMed] [Google Scholar]
- 222.van Montfort RL, Congreve M, Tisi D et al. (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423:773–777 [DOI] [PubMed] [Google Scholar]
- 223.Yang J, Groen A, Lemeer S et al. (2007) Reversible oxidation of the membrane distal domain of receptor PTPalpha is mediated by a cyclic sulfenamide. Biochemistry 46: 709–719 [DOI] [PubMed] [Google Scholar]
- 224.Haque A, Andersen JN, Salmeen A et al. (2011) Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 147:185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]