

Abstract
To enable large-scale analyses of transcription regulation in model species, we developed DeepArk, a set of deep learning models of the cis-regulatory activities for four widely studied species: Caenorhabditis elegans, Danio rerio, Drosophila melanogaster, and Mus musculus. DeepArk accurately predicts the presence of thousands of different context-specific regulatory features, including chromatin states, histone marks, and transcription factors. In vivo studies show that DeepArk can predict the regulatory impact of any genomic variant (including rare or not previously observed) and enables the regulatory annotation of understudied model species.
Deciphering the regulatory function of the noncoding genome remains a grand challenge of modern biology. Model species have long been at the forefront of biological discovery and biomedical innovation, but our knowledge of the cis-regulatory logic remains incomplete (Manolio et al. 2017). Many important questions remain: How should we mutate a fly enhancer to change its activity in a tissue-specific manner? Which regulatory variants for mouse disease genes are functional? How can we predictively edit the genome to efficiently guide experimentation? Answering these questions requires interpreting specific effects of any genomic variant, including changes to chromatin states, histone modifications, and binding of transcription factors (TFs). Addressing this challenge across the entire spectrum of genomic variation requires generalizing from the experimental studies (e.g., ChIP-seq data) to learn the regulatory code and thus enable the prediction of effects for any genomic variant. These effects must be predicted in specific contexts, including developmental stage, cell and tissue type, and drug treatments.
Existing approaches for model organisms fall short of this goal. A common approach is to scan for highly conserved binding sites with position weight matrices. However, such motifs have limited contextual information and fail to consider the multiple interacting factors that frequently delineate histone marks or chromatin accessibility (Zhou and Troyanskaya 2015; Wagih et al. 2018). In contrast, newer sequence-based deep learning models have been successfully used in human genomics to learn this context-specific cis-regulatory code from large-scale sequencing data without the use of hand-engineered features. In particular, the many successive convolutional layers used in these models allow them to learn relatively complex motifs and, we presume, interactions between them (LeCun et al. 2015; Avsec et al. 2021). This flexibility, combined with an efficiency that allows these models to be applied at a truly whole-genome scale, and a growing ecosystem of open-source software and supporting resources (e.g., the Kipoi model archive) (Avsec et al. 2019) have made deep learning a potent and useful tool for genomics and computational life sciences in general (Ching et al. 2018). For instance, in the context of human biology, sequence-based deep learning models have been successfully used to estimate gene expression from DNA sequences (Kelley et al. 2018; Zhou et al. 2018), predict the splicing and processing of pre-mRNA (Jaganathan et al. 2019; Park et al. 2021), and even improve human variant pathogenicity prioritization by integrating information from both human and non-human primate sources (Sundaram et al. 2018). These models have also been proven to be powerful for the complex task of predicting regulatory activity from human genomic sequences (Zhou and Troyanskaya 2015; Kelley et al. 2016). Nevertheless, the use of such models to predict regulatory activity in model organisms remains largely undemonstrated aside from one application in mice (Kelley 2020).
In what follows, we introduce “DeepArk,” a set of convolutional neural networks (CNNs) that model the cis-regulatory functions of four model organisms: Caenorhabditis elegans, Danio rerio, Drosophila melanogaster, and Mus musculus. As we show, DeepArk is broadly useful for genomics research in both model organisms and related species and, furthermore, enables potent computational and experimental analyses, such as predictive genome editing and the interpretation of the regulatory effects of genomic variants.
Results
We developed a set of sequence-based deep CNNs, which we collectively named “DeepArk,” modeling the cis-regulatory activities of four of the most widely studied model organisms: C. elegans, D. rerio, D. melanogaster, and M. musculus (Fig. 1A). To the best of our knowledge, DeepArk is the first such resource for these model organisms. DeepArk provides an in silico ChIP-seq capability: Given a genomic sequence as input, DeepArk's CNNs predict the activity of a total of 6562 regulatory features, including histone marks, different TFs, RNA polymerases, and chromatin accessibility (Supplemental Table S1). DeepArk leverages a wide sequence context of 4095 bp to provide accurate predictions for broad regulatory features with complex regulatory origins (e.g., chromatin accessibility); wide sequence context has been established as important for prediction accuracy (Zhou and Troyanskaya 2015; Kelley et al. 2016, 2018). DeepArk's multitask approach to modeling also allows it to make such predictions efficiently. Many predictions are made in specific contexts—larval or adult stages, specific tissues or cell types—and under particular treatments (e.g., lipopolysaccharide stimulation). In total, DeepArk provides predictions for 554 individual TFs and 62 distinct histone marks, as well as RNA polymerase profiles across 61 different cell types and chromatin accessibility across 95 cell types. For most of the organisms and regulatory features considered, DeepArk is the first method capable of predicting regulatory activity from genomic sequence and the regulatory effects of genomic variants (Lee et al. 2015; Kelley 2020).
Figure 1.

Overview of DeepArk models and their predictive accuracy. (A) The DeepArk architecture (Supplemental Fig. S1) uses convolutional layers to scan an input sequence for regulatory motifs and uses maximum pooling layers to perform dimensionality reduction. By using many successive layers, DeepArk is able to extract complex motifs while presumably leveraging interactions between motifs (LeCun et al. 2015; Avsec et al. 2021) and can use a wide sequence context of 4095 bp. Key applications enabled by DeepArk include prioritizing observed genomic variants by their putative regulatory effects (top right), exposing the predictive sequence features for regulatory events through in silico saturated mutagenesis (middle right), and predicting the regulatory effects of novel variants for prospective experiments (bottom right). (B) Performance on test chromosomes from each organism, as quantified by the area under the curve (AUC) of the receiver operating characteristic (ROC) curve (Supplemental Table S1). Only regulatory features with at least 50 positive test examples are included. For each box plot, the center line marks the median, and the top and bottom edges of the box mark the first and third quartiles, respectively. The top and bottom whiskers extend to 1.5× the interquartile range (IQR), with data points outside of this range considered outliers and plotted individually. (C) DeepArk's performance on the test chromosomes from each organism, here quantified by the AUC for the precision-recall curve (PRC) (Supplemental Table S1). Only regulatory features with at least 50 positive test examples are shown. For each box plot, the center line marks the median, and the top and bottom edges of the box mark the first and third quartiles, respectively. The top and bottom whiskers extend to 1.5× the IQR. Data points outside of this range are considered outliers and plotted individually. (D) Performance on the test chromosomes from each organism in terms of the log2 fold-change in the AUC for the PRC relative to the feature-specific baselines (Supplemental Table S1). Only regulatory features with at least 50 positive test examples are shown. For each box plot, the center line marks the median, and the top and bottom edges of the box mark the first and third quartiles, respectively. The top and bottom whiskers extend to 1.5× the IQR. Data points outside of this range are considered as outliers and plotted individually. DeepArk's performance never falls below the baseline.
We trained each DeepArk model on publicly-available genome-wide measurements of regulatory activity (i.e., ChIP-seq of TFs and histone marks, DNase-seq, and ATAC-seq) from its respective species (Supplemental Table S1) and tested its performance on chromosomes that were not used during training (Methods) (Supplemental Table S2). Training data sets were carefully filtered to retain only data meeting various quality thresholds (Methods). We found, consistent with the faithful inference of cis-regulatory logic, that each DeepArk model accurately predicted the regulatory activity of the test sequences (Fig. 1B–D; Supplemental Table S1). Performance appeared strong across each category of regulatory features, and there were a few interesting, albeit expected, trends. For instance, DeepArk's performance for TFs appeared to have the highest variance of any class of regulatory features (Fig. 1B–D). In terms of species-specific trends, DeepArk's performance for D. rerio seemed especially strong and narrowly distributed (Fig. 1B–D). This may be because its genome is quite large (∼1.67 Gbp) and because the training data for D. rerio were all produced by a single consortium (Tan et al. 2016). TFs also seem to have the highest intra-species variance in performance, which may be owing to the wide variety of regulatory features (e.g., pioneer factors vs. chromatin remodelers) that this category encompasses. Nevertheless, performance for all regulatory feature categories in all species was higher than baseline, which indicates the relevance of DeepArk's predictions.
At its core, DeepArk provides a mapping from DNA sequences to their predicted regulatory activity. By comparing DeepArk's predictions for separate sequences, we can identify how sequence differences may lead to regulatory differences. As a consequence, we can predict whether a variant might increase or decrease regulatory activity for any of DeepArk's 6562 regulatory features. This ability to predict the cis-regulatory effects of genomic variants is an important step forward for model species genomics, as there is a paucity of such methods available.
To assess DeepArk's ability to guide the interpretation of regulatory variants, we compared its predictions for the regulatory effects of variants in an enhancer of ALDOB with their actual effects as measured by a massively parallel in vivo reporter assay (MPRA) in murine livers from Patwardhan et al. (2012). By barcoding each variant and quantifying enhancer activity with RNA-sequencing, the Patwardhan et al. (2012) MPRA tested the expression-modulating effects of all possible single-nucleotide polymorphisms (SNPs) in the ALDOB enhancer. DeepArk's mouse model's variant effect predictions were significantly correlated with the expression effects of the SNPs measured in the ALDOB enhancer MPRA (Pearson's r = 0.714, P = 3.58 × 10−122 and Spearman's ρ = 0.587, P = 2.91 × 10−73, n = 777) (Fig. 2A,B), further showing that DeepArk's predictions reflect in vivo observations.
Figure 2.

DeepArk's variant effect predictions are well correlated with variant expression effects measured in a massively parallel in vivo reporter assay (MPRA) of enhancer activity. (A) The plot shows DeepArk's predictions for a liver-specific DNase-seq experiment (accession no. SRX3201109) for all possible variants (blue circles) in the ALDOB enhancer (hg19: Chr 9: 104,195,570–104,195,828) and the expression effects measured by the massively parallel reporter assay from Patwardhan et al. (2012), which are significantly correlated (Pearson's r = 0.714, P = 3.58 × 10−122, n = 777 and Spearman's ρ = 0.587, P = 2.91 × 10−73, n = 777). Note that the high degree of correlation with this DeepArk feature and the reported expression change is representative of the high correlation witnessed for liver-specific DNase-seq predictions in general, as shown in the next panel in comparison to other features. (B) The blue line in the plot shows the empirical complementary cumulative distribution function of the Pearson's correlation between the reported expression change in the MPRA of the ALDOB enhancer performed by Patwardhan et al. (2012) and DeepArk's variant effect predictions for each regulatory feature for M. musculus. The red circles correspond to liver-specific DNase-seq experiments from mice under control conditions (accession numbers SRX188645, SRX681492, SRX681493, SRX681494, SRX681495, SRX681496, SRX681497, SRX681498, SRX681499, SRX191053, and SRX3201109). The correlations of these liver-specific DNase-seq features are especially strong (average Pearson's correlation of 0.7046, n = 11 features), which is appropriate because the MPRA in question also measures the effects on expression levels in murine livers.
DeepArk can also be deployed to investigate regulatory loci at the genome- or chromosome-wide scale. For example, a researcher interested in identifying loci guiding the spreading of the dosage compensation complex (DCC) of C. elegans, a complex that both binds and spreads along the inactivated X Chromosome (Csankovszki et al. 2004), could use DeepArk to investigate the DCC computationally and identify sites involved in the DCC's initial recruitment. First, the researcher identifies a region as a highly-probable site of DCC binding by scanning all of Chromosome X for binding of several protein components of the DCC (e.g., DPY-27) in vivo (Supplemental Table S3; Supplemental Fig. S2). Then, the researcher conducts in silico saturated mutagenesis of the putative DCC-bound region for DCC members and visualizes the results for SDC-3 binding (NCBI Sequence Read Archive [SRA: https://www.ncbi.nlm.nih.gov/sra] accession no. SRX2228883), which reveals a single highly constrained sequence (TCGCGCAGGGAA) that is necessary for DCC binding in vivo (Supplemental Fig. S3). This site appears to be a near-perfect match to the consensus sequence for the “recruitment elements on X” or “rex” motif, a critical sequence for DCC binding (McDonel et al. 2006; Jans et al. 2009). Repeating this analysis at two additional high-probability DCC binding sites reveals similar trends and gives the researcher further confidence in their findings (Supplemental Figs. S4, S5). This illustrates how DeepArk may be used to interpret the binding patterns of even relatively complicated protein complexes.
As another application, DeepArk can directly assist in studying regulatory genomics. We used the DeepArk model for D. melanogaster to investigate the regulatory effects of mutations in the mesodermal enhancer of the T48 gene (Lim et al. 2017), whose timely expression regulates gastrulation in flies (Supplemental Fig. S7; Supplemental Tables S4, S5; Kölsch et al. 2007; Lim et al. 2017) and relies on the binding of zelda (Zld), a pioneer factor (Dufourt et al. 2018; Yamada et al. 2019). DeepArk predicted that the original suboptimal Zld binding site would have the lowest probability for Zld binding, whereas the variants CTT > CTA and CTT > GTA would have moderate probabilities and the CTT > CTG variant would have the largest positive effect on Zld binding. To test the predictive capabilities of DeepArk, we examined the in vivo expression of these three variants in live embryos. Experimental quantification of the total transcriptional output of the T48 enhancer variants clearly shows that DeepArk's predictions were accurate and that, as expected, an increase in Zld's binding probability correlates with an increase in gene activation in vivo (Fig. 3A–C). This finding is further supported by the correlation (average Pearson's correlation of 0.7217, n = 6 features) between the enhancer's observed in vivo expression and DeepArk's predictions for the binding of twist (Twi), a TF known to drive Zld-mediated mesodermal enhancers (Lim et al. 2017), during nuclear cycle 14 (Supplemental Table S4). Altogether, these findings experimentally confirm DeepArk's predictions and show its utility in designing genome editing experiments.
Figure 3.
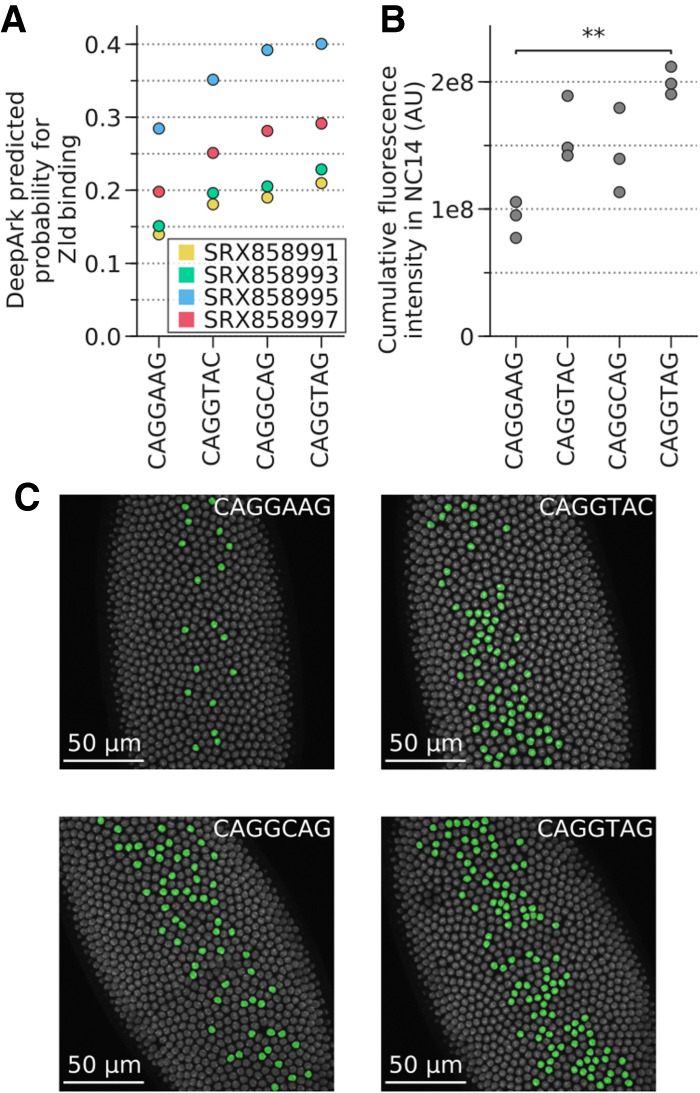
DeepArk's predicted effects for the different T48 mesodermal enhancer variants correlate with in vivo results. (A) Plot of all DeepArk predictions for Zld binding during nuclear cycle 14 for each of the four enhancer alleles. Each point represents a DeepArk prediction for a specific Zld ChIP-seq experiment and a specific allele. The CAGGTAG allele has the highest predicted probability of binding, whereas the reference allele CAGGAAG has the lowest. (B) The total transcriptional output for each of the four alleles, as quantified with in vivo MS2-GFP tagging during nuclear cycle 14. Each point in the graph represents the total transcription output (Methods) of all nuclei in a single embryo. Note that CAGGAAG and CAGGTAG have the lowest and highest transcriptional outputs, respectively, which is consistent with DeepArk's predictions. Bonferroni-corrected two-sided t-test with unequal variances: (**) P = 4.139 × 10−3; all others, P > 5 × 10−2. (C) False-color nuclei with active transcription in Drosophila embryos during minute 20 of nuclear cycle 14 illustrate the distinct levels of transcriptional activation induced by each allele (Supplemental Fig. S6). Three replicates were imaged for each allele. (AU) Arbitrary units, which represent the intensity of the pixels that correspond to the fluorescence of the MS2-GFP-tagged foci relative to the background GFP signal (Methods). (NC14) Nuclear cycle 14.
DeepArk may also be particularly useful for researchers of understudied model organisms without available regulatory data. Presently, pairwise alignments of regulatory regions allow for the detection of constrained noncoding sequences in the absence of regulatory assays, but they are only a proxy for regulatory function and their interpretation can be confounded. For instance, some regulatory elements are enriched for recent evolution (Moon et al. 2019), whereas other highly conserved noncoding regions have no known function (Ahituv et al. 2007). By directly predicting regulatory activity from sequence, DeepArk directly tackles this challenge. To that end, we used the DeepArk model for the model organism D. rerio to predict chromatin accessibility and H3K4me3 marks during development in the genome of Oryzias latipes, or medaka, a fish that diverged from D. rerio an estimated 314–332 myr ago (Kasahara et al. 2007). Even after filtering conserved loci, we find that DeepArk accurately predicts ChIP-seq and ATAC-seq peaks for developing O. latipes (average ROC AUC of 0.927) using only their genomic sequence as model input (Supplemental Table S6; Supplemental Fig. S8). Thus, DeepArk may also be used to help annotate the genomes of understudied organisms when whole-genome assays of regulatory features do not already exist (Supplemental Figs. S8, S9).
Discussion
In summary, we described DeepArk: a deep learning model for regulatory genomics in model organisms. Through computational evaluations and experimental verification, we demonstrated DeepArk's utility for a number of diverse tasks, including investigating the regulatory landscape of model species and related organisms, predictive genome editing, and variant effect prediction. DeepArk can also be used to uncover sequences critical to regulatory activity through in silico saturated mutagenesis. We have shown several examples illustrating the application of DeepArk, a resource that we have made publicly available through a dynamic, interactive interface (https://DeepArk.princeton.edu).
We developed DeepArk in a transparent and open-source manner, so it can be readily extended and repurposed for other tasks via transfer learning. Furthermore, DeepArk could be used as a scoring function in a sequence optimization and design pipeline in synthetic biology (Cuperus et al. 2017), as inputs for models of more complicated regulatory events such as enhancer–promoter looping (Fudenberg et al. 2020) or gene expression (Zhou et al. 2018), and in high-resolution trait–loci association mapping within animal models as it becomes widespread (Parker et al. 2016).
DeepArk can predict thousands of regulatory features and provides more expansive coverage of regulatory features than any previous sequence-based deep learning models for regulatory genomics. However, there is still much about regulation that is unknown. Thus, despite DeepArk's inclusion of thousands of different regulatory features, there are certainly regulatory features that DeepArk has not yet modeled because of the lack of data. For instance, a user may be interested in a TF's binding in a rare cell type that has not been modeled by DeepArk. The user may consider DeepArk's predictions for their TF—or another feature known to correlate with the TF—in a related cell type. However, in the absence of a well-characterized causal relationship, features correlated in one context may not be correlated in other contexts. For example, a passenger motif associated with a tissue-specific pioneer factor may be critical to a TF's binding in one tissue, but that passenger motif may be irrelevant to said TF's binding in other tissues. Complex and tissue-specific relationships such as this can make analyses difficult because they require analyzing more than the canonical motif for the TF, but they can also generate novel hypotheses to be tested with mechanistic follow-up experiments. Along those lines, cross-tissue predictions that systematically incorporate information about causal relationships (e.g., known regulatory networks, physical TF interactions) may also be a fruitful avenue for future research (Nair et al. 2019). In the meantime, to mitigate this issue of missing regulatory features, we plan to update and expand DeepArk as further training data become available. Based on DeepArk's strong performance for the data from the DANIO-CODE consortium (Tan et al. 2016), we expect future releases of DeepArk to benefit from continued improvements in quality control and data standards for experiments in general. Thus, DeepArk's relevance and utility will only grow as time goes on.
Future work on DeepArk and other sequence-based models should draw on new data modalities as well. We suspect that the proliferation of single-cell sequencing will be especially useful to these endeavors. Besides providing regulatory insights in a range of cell types, the resolution enabled by these protocols may lead to increased tissue-specific and cell type–specific chromatin accessibility data for C. elegans (Durham et al. 2021) and D. melanogaster (Cusanovich et al. 2018). At present, these two organisms have relatively little tissue-specific chromatin accessibility data from bulk sequencing compared with D. rerio or M. musculus. Increasingly high-resolution trait–loci association mapping within model organisms (Parker et al. 2016) and improvements in MPRA methods may also provide additional means of validating or interpreting DeepArk's variant effect predictions in the future as well.
Although DeepArk performs well (Fig. 1B–D; Supplemental Table S1), there is still room for further improvements. We suspect that novel CNN architectures will prove central to such endeavors. For instance, a wide sequence context is known to be important for accurate prediction of regulatory activities (Zhou and Troyanskaya 2015; Kelley et al. 2016, 2018), and we did find that a model with a 4095-bp input sequence length outperformed one with a 2047-bp input sequence length in all four organisms (Methods). However, it is possible that other sequence lengths may do better still and that each organism we considered may benefit from distinct sequence lengths owing to their regulatory differences. Thus, we expect that future deep learning–based models of regulatory activity are also likely to benefit from continued improvements in hardware (Jouppi et al. 2018) and network architectures (Tan and Le 2019), as well as more efficient algorithms for neural architecture search (Tan and Le 2019; Zhang et al. 2021a,b).
In total, DeepArk is a model capable of enhancing regulatory genomics research, and we expect that it will be used to quickly and efficiently generate in silico hypotheses, which researchers can follow up and validate with more mechanistic in vivo studies.
Methods
DeepArk model architecture
DeepArk is a collection of four deep CNNs, each modeling the activity of different regulatory features in a separate model organism. In total, DeepArk is capable of making predictions for 6562 context-specific regulatory features. In what follows, we detail the design and structure of the DeepArk model.
Each DeepArk model takes a 4095-bp genomic sequence as input and predicts the probability that the centermost base of this sequence is covered by a peak for each regulatory feature of interest. This input sequence is encoded as a 4095 × 4 one-hot matrix with columns corresponding to each base in the sequence and with rows corresponding to adenine, cytosine, guanine, and thymine, respectively. The output of each DeepArk model is a vector of length N, where N is the number of features for that model's given organism (Supplemental Table S1). DeepArk is a multitask model, which means it jointly learns the sequence-specific activities of multiple regulatory features simultaneously, rather than modeling each regulatory feature separately. The DeepArk architecture was fixed across organisms (Supplemental Fig. S1), but we learned distinct model parameters and hyperparameters for each organism (Supplemental Table S7).
We chose a sequence length of 4095 bp, as prior works modeling human regulatory features from sequence inputs have repeatedly shown that longer sequence lengths improve predictive accuracy for deep sequence–based models of regulatory features (Zhou and Troyanskaya 2015; Kelley et al. 2018; Zhou et al. 2018). We also trained models using 2047-bp input sequence lengths. Consistent with prior literature, the 4095-bp sequence length achieved lower validation loss than the 2047-bp models in each organism. Other motivating factors for choosing 4095 bp in particular included the fact that it is odd (i.e., so that the center base of the reverse complement is also the center base of the forward strand) and close to a power of two (i.e., for GPU memory alignment).
The DeepArk architecture (Supplemental Fig. S1) consists of a deep CNN, wherein the network's output is the functional composition of many linear and nonlinear transformations, called “layers.” The specific parameters of these transformations are selected during training to optimize the objective function. We consider five types of transformations in our network: convolutional layers, maximum pooling layers, batch normalization layers, and the rectified linear unit (ReLU) and sigmoid activation functions. The basic unit of our model is a multilayer “convolutional unit,” which contains, in order, a batch normalization layer, a ReLU layer, and a convolution layer. We further organize our model into five multilayered convolutional blocks. We used maximum pooling at the start of each convolutional block, as we found that spatial invariance and reduced training time allowed us to improve our model. The output of the final convolutional block is fed into a length-one convolution with output channels equal to the number output features of the model, as well as fed into the sigmoid activation function.
We designed the DeepArk architecture to regularize it and avoid overfitting. First, we averaged predictions made for the forward and reverse complement of sequences. Second, we leveraged spatial dropout (Tompson et al. 2014), which randomly zeros out channels in the input to a convolutional layer. Typically, dropout (Srivastava et al. 2014) randomly sets input values to zero with probability P, which has the effect of forcing the model to overcome perturbations in internal values (i.e., without altering the sequence input) to make correct predictions. However, highly correlated sequence positions in CNN inputs may diminish the effectiveness of dropout and slow training. Conversely, spatial dropout mitigates this by zeroing out entire channels in the convolutional layer's input.
Training examples
Training examples are two-tuples of a 4095-bp genomic sequence and a label vector. For each example, a given feature's entry in the label vector is positive if the center base of the 4095-bp sequence is overlapped by a peak from the feature's corresponding ChIP-seq, DNase-seq, or ATAC-seq experiment. With the exception of ENCODE blacklisted regions (Amemiya et al. 2019), all positions in the genome were considered valid examples.
Nonintersecting training, validation, and testing sets were generated by whole-chromosome holdout (Supplemental Table S2). We chose one validation and one test chromosome for each organism. Chromosomes were selected for validation and testing that were representatively sized and were near the median chromosome length for their respective organism. Validation data were generated by randomly drawing 64,000 examples from a given species’ set of validation chromosomes. Training and validation examples are drawn uniformly and with replacement. Each species’ test set consisted of 1 million examples drawn uniformly and without replacement from the held-out test chromosomes for said species. Only features with at least 50 positive examples in the held-out test data were considered when calculating performance metrics.
Training DeepArk
We used stochastic gradient descent with momentum and mini-batches of 128 examples to select network weights that optimized the model objective function during training. Specifically, our objective function was the sum of the binary cross-entropy (BCE) loss and an L2 regularization term,
where yi is the vector of target labels for example i, is DeepArk's prediction for example i, λ is the weight decay hyperparameter, W is the weight matrix, and N is the mini-batch size. Model validation performance was evaluated every 5000 training steps with a validation set drawn randomly from a set of held-out validation chromosomes (see “Training examples” section in Methods). When minimum validation loss failed to decrease for five consecutive epochs, we decrease the learning rate by 20% of its current value. We terminated training when validation loss stopped decreasing for a sustained period of time. Hyperparameters for SGD and model training (Supplemental Table S7) were selected based on each model's performance on its respective validation set. DeepArk was implemented and trained with Selene (Chen et al. 2019).
Training data preparation
Labels for training, validation, and testing data were constructed using publicly-available ChIP-seq, DNase-seq, and ATAC-seq for C. elegans, D. rerio, D. melanogaster, and M. musculus. For C. elegans, D. melanogaster, and M. musculus, we used peak intervals from ChIP-Atlas (Oki et al. 2018), a large compendium of uniformly processed high-throughput sequencing experiments sourced from the NCBI Sequence Read Archive (SRA), the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home), and the DNA Data Bank of Japan (DDBJ; https://www.ddbj.nig.ac.jp). Specifically, we used peaks called with a maximum Q-value cutoff of 1 × 10−5. ChIP-Atlas intervals for C. elegans, D. melanogaster, and M. musculus data were downloaded from the following URLs:
http://dbarchive.biosciencedbc.jp/kyushu-u/ce10/allPeaks_light/allPeaks_light.ce10.05.bed.gz,
http://dbarchive.biosciencedbc.jp/kyushu-u/dm3/allPeaks_light/allPeaks_light.dm3.05.bed.gz, and
http://dbarchive.biosciencedbc.jp/kyushu-u/mm9/allPeaks_light/allPeaks_light.mm9.05.bed.gz.
To keep our methodology consistent, we called our own peaks for D. rerio. Specifically, we downloaded aligned BAMs for ChIP-seq and ATAC-seq experiments from the DANIO-CODE website (Supplemental Table S8; Tan et al. 2016). Peaks were called for these BAMs using MACS2 (Gaspar 2018) with a maximum Q-value cutoff of 1 × 10−5 and an effective genome size of 8.1 × 108 bp. This approximate effective genome size was calculated by counting the number of unambiguous bases in the Genome Reference Consortium Zebrafish Build 11 (GRCz11), without including repeats (Howe et al. 2013). The repeat-masked genome was downloaded from the UCSC Genome Browser annotation database (Haeussler et al. 2019).
To ensure that we only considered high-quality experiments, we removed those with too few peaks, an insufficient number of mapped reads, or average read length <32 bp (Supplemental Table S9). We also removed experiments that did not list a specific antibody target. We manually curated sample metadata regarding strains, cell lines, genetic modifications, and sample treatment. Because there exists a wide range of mouse cell lines and strains with extensive genetic and phenotypic diversity among them, we removed mouse experiments that did not reference a specific strain or cell line.
Finally, we removed experiments in which there was duplication or redundancy between SRA, ENA, and DDBJ. We considered experiments to be duplicates if they were from the same species, differed by fewer than 100 peaks, and had the same number of unmapped, mapped, and duplicate-free reads. We manually inspected the FASTQ files to ensure true duplication. In cases in which both accessions had the same metadata, we discarded one of the duplicate accessions at random. If the duplicates did not have the same antibody or biological source (e.g., cell type) listed, we discarded all of them.
Analysis of massively parallel reporter assay
To evaluate DeepArk's accuracy, we used it to predict the regulatory effects of all possible variants in the ALDOB enhancer (Patwardhan et al. 2012). We downloaded variant effects for the MPRA of the ALDOB enhancer from MaveDB (Esposito et al. 2019; https://www.mavedb.org/scoreset/urn:mavedb:00000006-a-1/). The predicted functional effect of each variant was calculated with in silico saturated mutagenesis. Specifically, the change in chromatin accessibility at the center of the 259-bp ALDOB enhancer (hg19: Chr 9: 104,195,570–104,195,828) was predicted for all possible variants within the 4095-bp window, and those reported in the MPRA were retained for analysis.
Predicting the binding of the DCC in C. elegans
To identify a high-confidence binding site for the DCC, we scanned the entire X Chromosome and made predictions every 200 bp with DeepArk. We took the mean probability across all features corresponding to DCC components (Supplemental Table S3) as a proxy of DCC binding probability. The top three sites with the maximum mean probabilities across DCC components were then analyzed with in silico saturated mutagenesis (Supplemental Tables S10–S12). Finally, we annotated locations within the in silico saturated mutagenesis input sequences that appeared to be canonical DCC recruitment sites, generally known as “recruitment elements on X” or rex sites, by scanning the sequences with FIMO (Grant et al. 2011). When running FIMO, we used FIMO's default parameters and sourced the rex motif's position weight matrix from Jans et al. (2009). The practice of subtracting the mean probability across alleles at a given position in the in silico saturated mutagenesis visualizations was based on the methods of Shrikumar et al. (2019). Joint visualization of the outputs from FIMO and the in silico saturated mutagenesis predictions enabled comparison between the two.
Cloning of T48 enhancer MS2 reporter alleles
To clone the four different T48 enhancer MS2 reporters, the T48 enhancer was first cut with NotI from the T48 > MS2 > yellow plasmid (Lim et al. 2017) and subcloned into a pGEM-T Easy vector. Site-directed mutagenesis was then performed by amplifying the pGEM-T Easy T48 enhancer vector (Supplemental Table S13). The different PCR reactions were digested with Dpn and transformed into Escherichia coli in order to obtain clones of the four different T48 alleles. These plasmids were then individually subcloned into the pbphi-evePr-MS2-yellow vector (Fukaya et al. 2016) using NotI.
Live imaging of T48 enhancer alleles
To visualize live transcription of the T48 MS2 reporters, female fly virgins carrying the MCP-GFP and His2Av-mRFP fusion proteins (Lim et al. 2018) were crossed to males carrying the MS2 reporter genes inserted on a landing site of the third chromosome (strain 9450, Bloomington stock center). The resulting embryos were dechorionated and mounted between a semipermeable membrane and a coverslip with Halocarbon oil 27 (Sigma-Aldrich). Embryos were imaged from the beginning of nuclear cycle 14 up to the onset of gastrulation using a Zeiss LSM 880 confocal microscope and a Plan-Apochromat 40×/1.3 NA oil-immersion objective. For each time point, a stack of 21 images separated by 0.5 μm with a final time resolution of 14 sec was acquired at 16 bit. Two laser lines at 488 nm and 561 nm were used to excite the green and red fluorophores, respectively. The same imaging conditions were used across the three replicates and the four different reporter lines.
Image analysis and transcription quantification
To quantify the fluorescent signal resulting from the embryo's live transcription, the 21 images corresponding to each time point were converted into maximum projections. The subsequent analysis was processed by segmenting the nuclei using the His2Av-mRFP channel and by tracking the segmented individual nuclei during nuclear cycle 14. To record the MS2-GFP fluorescent signal corresponding to the transcription foci, an average of the signal for the three brightest pixels within each nucleus was determined after subtracting the background GFP signal. Total transcriptional output was calculated by adding the transcription foci signal for each active nucleus for the first 200 time frames of each embryo after the onset of nuclear cycle 14 (for a more detailed description of the image analysis methods, see Fukaya et al. 2016).
Interspecies regulatory prediction
To illustrate DeepArk's ability to make robust predictions in novel species (i.e., not C. elegans, D. rerio, D. melanogaster, or M. musculus), we used the DeepArk model for D. rerio to predict regulatory activity of sequences from the genome of O. latipes, which diverged from D. rerio between 314 and 332 myr ago (Kasahara et al. 2007). Specifically, we used extant ATAC-seq and H3K4me1 ChIP-seq data from O. latipes.
To generate testing examples for O. latipes, we randomly drew 1 million locations from the O. latipes reference genome without replacement. We ignored regions that contained an excess (i.e., more than 50) of ambiguous bases. To ensure the model was truly generalizing to the O. latipes genome, we removed test sequences in the O. latipes genome that were conserved between D. rerio and O. latipes. To identify conserved bases, we used a multiple whole-genome alignment of eight vertebrates, including O. latipes, to the D. rerio reference genome. We downloaded this alignment from the UCSC Genome Browser website (https://hgdownload.soe.ucsc.edu/goldenPath/danRer7/multiz8way/multiz8way.maf.gz). To enable comparisons between the two fish, morphological stages of O. latipes development were matched to their corresponding stages in D. rerio (Tena et al. 2014; Marlétaz et al. 2018).
Labels for the testing examples were assigned using existing ATAC-seq and ChIP-seq data from O. latipes. We downloaded unprocessed FASTQ files from SRA using the SRA toolkit (Supplemental Table S6). We filtered and clipped reads using TrimGalore. We used BWA-MEM (Li 2013) to align reads to the O. latipes reference genome (Kasahara et al. 2007). Following alignment, we used SAMtools (Li et al. 2009) to index and sort the BAM files and used the “MarkDuplicates” command from Picard Tools to identify and remove duplicate reads in each BAM file. Finally, we used MACS2 (Gaspar 2018) to call peaks with a Q-value cutoff of 1 × 10−5 and an effective genome size of 8.18 × 108 bp.
Lastly, RNA-seq data for D. rerio (accession no. SRX3353221) and O. latipes (accession no. SRX3353227) were used to visualize changes in expression and compare changes in histone modifications at promoters (Supplemental Fig. S9). We downloaded the unprocessed FASTQ files for these data from SRA using the SRA toolkit. Using HISAT2 (Kim et al. 2019), the processed reads for O. latipes were aligned to its reference genome (Kasahara et al. 2007), and the reads for D. rerio were aligned to GRCz11 (Howe et al. 2013). Coverage was quantified as counts per million mapped reads (CPM) using the “bamCoverage” command from deepTools (Ramírez et al. 2016).
Software availability
The code to run DeepArk locally is available as Supplemental Code and has also been uploaded to GitHub (https://github.com/FunctionLab/DeepArk). DeepArk is also freely accessible through our user-friendly web server (https://DeepArk.princeton.edu).
Data access
Raw videos from imaging are available on Zenodo (https://doi.org/10.5281/zenodo.3759736). DeepArk predictions for DCC component binding along the C. elegans X Chromosome are also available on Zenodo (https://doi.org/10.5281/zenodo.4663161, https://doi.org/10.5281/zenodo.3759699). DeepArk variant effect predictions of M. musculus regulatory features for the ALDOB MPRA experiment from Patwardhan et al. (2012) are available on Zenodo as well (https://doi.org/10.5281/zenodo.4060298). The training data for DeepArk are also available on Zenodo (https://doi.org/10.5281/zenodo.4647691).
Supplementary Material
Acknowledgments
We acknowledge all members of the Troyanskaya and Levine laboratories for their helpful discussions, as well as the SCC center at the Flatiron Institute. We thank Beryl M. Jones, Siena Dumas Ang, and Rachel Kaletsky for their feedback regarding the DeepArk web server. We also thank Teka H. Nicholas for proofreading the manuscript. We acknowledge that this work was performed using the high-performance computing resources at the Simons Foundation and the TIGRESS computer center at Princeton University. E.M.C. was supported by National Institutes of Health (NIH) grant T32 HG003284 and the National Science Foundation Graduate Research Fellowship Program (NSF-GRFP). This work was supported by NIH grant R01 GM071966 (O.G.T.) and R35 GM118147 (M.S.L.).
Author contributions: E.M.C., C.L.T., and O.G.T. conceived the idea. E.M.C. performed all computational analyses and modeling. E.M.C. designed and implemented the DeepArk model architecture. E.M.C., C.L.T., J.R., O.G.T., and M.S.L. jointly conceptualized the in vivo validation study. J.R. designed and executed all imaging experiments and quantified the transcription output from videos. Y.Y. designed and prepared the T48 reporters. E.M.C., A.T., and A.K.W. designed, implemented, and deployed the DeepArk web server, with feedback regarding website design from all other authors. E.M.C., C.L.T., J.R., M.S.L., and O.G.T. wrote the manuscript with feedback from all other authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.266171.120.
Freely available online through the Genome Research Open Access option.
Competing interest statement
The authors declare no competing interests.
References
- Ahituv N, Zhu Y, Visel A, Holt A, Afzal V, Pennacchio LA, Rubin EM. 2007. Deletion of ultraconserved elements yields viable mice. PLoS Biol 5: e234. 10.1371/journal.pbio.0050234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, Boyle AP. 2019. The ENCODE blacklist: identification of problematic regions of the genome. Sci Rep 9: 9354. 10.1038/s41598-019-45839-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsec Ž, Kreuzhuber R, Israeli J, Xu N, Cheng J, Shrikumar A, Banerjee A, Kim DS, Beier T, Urban L, et al. 2019. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat Biotechnol 37: 592–600. 10.1038/s41587-019-0140-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsec Ž, Weilert M, Shrikumar A, Kreuger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, et al. 2021. Base-resolution models of transcription factor binding reveal soft motif syntax. Nat Genet 53: 354–366. 10.1038/s41588-021-00782-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KM, Cofer EM, Zhou J, Troyanskaya OG. 2019. Selene: a PyTorch-based deep learning library for sequence data. Nat Methods 16: 315–318. 10.1038/s41592-019-0360-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching T, Himmelstein DS, Beaulieu-Jones BK, Kalinin AA, Do BT, Way GP, Ferrero E, Agapow P, Zietz M, Hoffman MM, et al. 2018. Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface 15: 20170387. 10.1098/rsif.2017.0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, McDonel P, Meyer BJ. 2004. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science 303: 1182–1185. 10.1126/science.1092938 [DOI] [PubMed] [Google Scholar]
- Cuperus JT, Groves B, Kuchina A, Rosenberg AB, Jojic N, Fields S, Seelig G. 2017. Deep learning of the regulatory grammar of yeast 5′ untranslated regions from 500,000 random sequences. Genome Res 27: 2015–2024. 10.1101/gr.224964.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Reddington JP, Garfield DA, Daza RM, Aghamirzaie D, Marco-Ferreres R, Pliner HA, Christiansen L, Qiu X, Steemers FJ, et al. 2018. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555: 538–542. 10.1038/nature25981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufourt J, Trullo A, Hunter J, Fernandez C, Lazaro J, Dejean M, Morales L, Nait-Amer S, Schulz KN, Harrison MM, et al. 2018. Temporal control of gene expression by the pioneer factor Zelda through transient interactions in hubs. Nat Commun 9: 5194. 10.1038/s41467-018-07613-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham TJ, Daza RM, Gevirtzman L, Cusanovich DA, Bolonduro O, Noble WS, Shendure J, Waterston RH. 2021. Comprehensive characterization of tissue-specific chromatin accessibility in L2 Caenorhabditis elegans nematodes. Genome Res 10.1101/gr.271791.120 [DOI] [PMC free article] [PubMed]
- Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, Fowler DM, Rubin AF. 2019. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol 20: 223. 10.1186/s13059-019-1845-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudenberg G, Kelley DR, Pollard KS. 2020. Predicting 3D genome folding from DNA sequence with Akita. Nat Methods 17: 1111–1117. 10.1038/s41592-020-0958-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, Levine MS. 2016. Enhancer control of transcriptional bursting. Cell 166: 358–368. 10.1016/j.cell.2016.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar JM. 2018. Improved peak-calling with MACS2. bioRxiv 10.1101/496521 [DOI]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, et al. 2019. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res 47: D853–D858. 10.1093/nar/gky1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496: 498–503. 10.1038/nature12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al. 2019. Predicting splicing from primary sequence with deep learning. Cell 176: 535–548.e24. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Jans J, Gladden JM, Ralston EJ, Pickle CS, Michel AH, Pferdehirt RR, Eisen MB, Meyer BJ. 2009. A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev 23: 602–618. 10.1101/gad.1751109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouppi NP, Young C, Patil N, Patterson D. 2018. A domain-specific architecture for deep neural networks. Commun ACM 61: 50–59. 10.1145/3154484 [DOI] [Google Scholar]
- Kasahara M, Naruse K, Sasaki S, Nakatani Y, Qu W, Ahsan B, Yamada T, Nagayasu Y, Doi K, Kasai Y, et al. 2007. The medaka draft genome and insights into vertebrate genome evolution. Nature 447: 714–719. 10.1038/nature05846 [DOI] [PubMed] [Google Scholar]
- Kelley DR. 2020. Cross-species regulatory sequence activity prediction. PLoS Comput Biol 16: e1008050. 10.1371/journal.pcbi.1008050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Snoek J, Rinn JL. 2016. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 26: 990–999. 10.1101/gr.200535.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Reshef YA, Bileschi M, Belanger D, McLean CY, Snoek J. 2018. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res 28: 739–750. 10.1101/gr.227819.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kölsch V, Seher T, Fernandez-Ballester GJ, Serrano L, Leptin M. 2007. Control of Drosophila gastrulation by apical localization of adherens junctions and RhoGEF2. Science 315: 384–386. 10.1126/science.1134833 [DOI] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, Hinton G. 2015. Deep learning. Nature 521: 436–444. 10.1038/nature14539 [DOI] [PubMed] [Google Scholar]
- Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA. 2015. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet 47: 955–961. 10.1038/ng.3331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio.GN].
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Levine M, Yamazaki Y. 2017. Transcriptional pre-patterning of Drosophila gastrulation. Curr Biol 27: 286–290. 10.1016/j.cub.2016.11.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim B, Heist T, Levine M, Fukaya T. 2018. Visualization of transvection in living Drosophila embryos. Mol Cell 70: 287–296.e6. 10.1016/j.molcel.2018.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Fowler DM, Starita LM, Haendel MA, MacArthur DG, Biesecker LG, Worthey E, Chisholm RL, Green ED, Jacob HJ, et al. 2017. Bedside back to bench: building bridges between basic and clinical genomic research. Cell 169: 6–12. 10.1016/j.cell.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlétaz F, Firbas PN, Maeso I, Tena JJ, Bogdanovic O, Perry M, Wyatt CDR, de la Calle-Mustienes E, Bertrand S, Burguera D, et al. 2018. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 564: 64–70. 10.1038/s41586-018-0734-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonel P, Jans J, Peterson BK, Meyer BJ. 2006. Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature 444: 614–618. 10.1038/nature05338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JM, Capra JA, Abbot P, Rokas A. 2019. Signatures of recent positive selection in enhancers across 41 human tissues. G3 9: 2761–2774. 10.1534/g3.119.400186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S, Kim DS, Perricone J, Kundaje A. 2019. Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. Bioinformatics 35: i108–i116. 10.1093/bioinformatics/btz352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, Kawaji H, Nakaki R, Sese J, Meno C. 2018. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep 19: e46255. 10.15252/embr.201846255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Zhou J, Wong AK, Chen KM, Theesfeld CL, Darnell RB, Troyanskaya OG. 2021. Genome-wide landscape of RNA-binding protein target site dysregulation reveals a major impact on psychiatric disorder risk. Nat Genet 53: 166–173. 10.1038/s41588-020-00761-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Gopalakrishnan S, Carbonetto P, Gonzales NM, Leung E, Park YJ, Aryee E, Davis J, Blizard DA, Ackert-Bicknell CL, et al. 2016. Genome-wide association study of behavioral, physiological and gene expression traits in outbred CFW mice. Nat Genet 48: 919–926. 10.1038/ng.3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee S-I, Cooper GM, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–270. 10.1038/nbt.2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrikumar A, Tian K, Avsec Ž, Shcherbina A, Banerjee A, Sharmin M, Nair S, Kundaje A. 2019. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) version 0.5.6.5. arXiv:1811.00416 [cs.LG].
- Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R. 2014. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15: 1929–1958. [Google Scholar]
- Sundaram L, Gao H, Padigepati SR, McRae JF, Li Y, Kosmicki JA, Fritzilas N, Hakenberg J, Dutta A, Shon J, et al. 2018. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50: 1161–1170. 10.1038/s41588-018-0167-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Le Q. 2019. EfficientNet: rethinking model scaling for convolutional neural networks. Proc Mach Learn Res 97: 6105–6114. [Google Scholar]
- Tan H, Onichtchouk D, Winata C. 2016. DANIO-CODE: toward an encyclopedia of DNA elements in zebrafish. Zebrafish 13: 54–60. 10.1089/zeb.2015.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tena JJ, González-Aguilera C, Fernández-Miñán A, Vázquez-Marín J, Parra-Acero H, Cross JW, Rigby PWJ, Carvajal JJ, Wittbrodt J, Gómez-Skarmeta JL, et al. 2014. Comparative epigenomics in distantly related teleost species identifies conserved cis-regulatory nodes active during the vertebrate phylotypic period. Genome Res 24: 1075–1085. 10.1101/gr.163915.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompson J, Goroshin R, Jain A, LeCun Y, Bregler C. 2014. Efficient object localization using convolutional networks. arXiv:1411.4280 [cs.CV].
- Wagih O, Merico D, Delong A, Frey BJ. 2018. Allele-specific transcription factor binding as a benchmark for assessing variant impact predictors. bioRxiv 10.1101/253427 [DOI]
- Yamada S, Whitney PH, Huang S-K, Eck EC, Garcia HG, Rushlow CA. 2019. The Drosophila pioneer factor Zelda modulates the nuclear microenvironment of a Dorsal target enhancer to potentiate transcriptional output. Curr Biol 29: 1387–1393.e5. 10.1016/j.cub.2019.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Park CY, Theesfeld CL, Troyanskaya OG. 2021a. An automated framework for efficiently designing deep convolutional neural networks in genomics. Nat Mach Intell 10.1038/s42256-021-00316-z [DOI]
- Zhang Z, Cofer EM, Troyanskaya OG. 2021b. AMBIENT: accelerated convolutional neural network architecture search for regulatory genomics. bioRxiv 10.1101/2021.02.25.432960 [DOI]
- Zhou J, Troyanskaya OG. 2015. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods 12: 931–934. 10.1038/nmeth.3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG. 2018. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet 50: 1171–1179. 10.1038/s41588-018-0160-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.