

Abstract
Genetic mutations or regulatory failure underlies cellular malfunction in many diseases including colorectal cancer (CRC) and Inflammatory bowel diseases (IBD). However, mutational defects alone fail to explain the complexity of such disorders. Epigenetic regulation – control of gene action through chemical and structural changes of chromatin – provides a platform to integrate multiple extracellular inputs and prepares the cellular genome for appropriate gene expression responses. Coregulation by Polycomb Repressive Complex 2 (PRC2) mediated trimethylation of Lysine 27 on Histone 3 (H3K27me3) and DNA methylation has emerged as one of the most influential epigenetic control in CRC and many other diseases, but molecular details remain inadequate. Here we review the molecular interplay of these epigenetic features in relation to gastrointestinal development, homeostasis, and disease biology. We discuss other epigenetic mechanisms pertinent to the balance of H3K27me3 and DNA methylation and their action in gastrointestinal cancers. We also review the current molecular understanding of chromatin control in the pathogenesis of IBD.
Keywords: Colorectal cancer, Inflammatory bowel disease, DNA methylation, PRC2, H3K27me3, epigenetic regulation, intestinal stem cells
INTRODUCTION
Gastrointestinal stem cells produce billions of epithelial cells of discrete types each day. In intestine and colon, these cells continuously make precise cell fate decisions through hundreds of well-orchestrated gene expression changes to maintain tissue homeostasis and in response to injuries. While doing so, the cells receive various extracellular stimuli from the chemical and microbial milieu of the lumen, and from the underlying mesenchyme. Cell response to the external environment is significantly influenced by intrinsic cellular variables including the genome and the epigenome. While the genome is largely stable, epigenome allows for dynamic control of gene expression and cellular identity. Accordingly, varying levels of genetic as well as epigenetic changes contribute to gastrointestinal diseases such as colorectal cancer (CRC) and Inflammatory bowel disease (IBD).
Methylation of DNA and histone modifications such as methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation are incorporated by various enzymes as post-translational modifications (PTMs). Depending on their co-occupancy and placement along the genome, these modifications influence the surrounding chromatin structure, access to transcription factors (TFs) and enzymes, and gene expression. Both DNA methylation and PRC2-mediated Histone 3 Lysine 27 trimethylation (H3K27me3) are important epigenetic modifications associated with cell-type-specific gene repression during tissue development and homeostasis. Additionally, their functional interaction with each other and many other chromatin features has been identified as a major contributing factor to CRC initiation and progression1. Here we focus on how their combined action influences intestinal function and diseases.
GENE CONTROL MECHANISMS OF PRC2 AND DNA METHYLATION
Recent studies have provided unprecedented detail regarding PRC2 complex composition and its effect on chromatin modification and function. PRC2 is a multimeric protein complex that consists of core subunits EZH1/2, EED, SUZ12, and RBBP4/7. The catalytic domain of PRC2 is encoded by SET-domain proteins, EZH1 or EZH22–5. However, the enzymatic activity of PRC2 is also dependent on EED, SUZ12 and histone-binding proteins, making PRC2 an obligate quadromeric complex. PRC2 can mono-, di-, or tri-methylate lysine 27 of histone H3 and is the only identified H3K27 methyltransferase. PRC2-mediated H3K27me3 deposition at gene promoters and gene bodies forms repressive chromatin state. Thus, appropriate distribution of H3K27me3 across the genome is essential for cell-type-specific gene expression and formation of cell identity during differentiation in embryonic and adult stem cells6, 7. There are two recognized subtypes of PRC2, PRC2.1 and PRC2.2; they differ in composition of their accessory proteins3, 8. PRC2.1 is associated with PCL1/PHF1, PCL2/MTF2, or PCL3/PHF19 and EPOP or PALI1. On the other hand, PRC2.2 contains JARID2 and AEBP2 as accessory proteins. PRC2.1 and PRC2.2 synergy is critical for proper disposition of H3K27me3 and inhibition of their associated proteins cause redistribution of H3K27me33. Although, majority of the PRC2.1 and PRC2.2 target sites overlap, they direct H3K27me3 deposition in an independent manner. PRC2.2 chromatin localization is dependent on PRC1 activity and mediated through binding of JARID2-to H2AK119Ub3, 9. The structure and the mechanism of PRC2 is reviewed extensively elsewhere10.
DNA methylation represents one of the most studied and well-understood heritable epigenetic changes of the DNA. De-novo DNA methylases DNAMT3A and DNMT3B modify more than 28 million CpG bases across the human genome through covalent addition of a methyl group, and DNMT1 maintains the modification by faithfully copying these patterns over cell divisions. DNA methylation can be actively removed through oxidation by TET1, −2, and −3 enzymes (ten-eleven translocations)11. Traditionally, studies have focused on understanding the effects of DNA methylation patterns at genes and their promoters: unmethylated promoters of active genes, methylation across gene bodies, hypermethylation at promoters leading to gene repression in cancers. A significant addition to this regulatory view of DNA methylation was the observation that c/s-regulatory regions or enhancers are hypomethylated12. This local demethylation is induced by binding of DNA methylation insensitive TFs, which further enables binding of other methylation sensitive TFs. Thus, tissue-specific enhancer formation is linked with DNA methylation changes dependant on co-regulatory TF action.
Studies examining PRC2 targeting and H3K27me3 deposition in intestinal development, homeostasis, and intestinal stem cell differentiation reveal how PRC2 and DNA methylation cogovern tissue fucntion13, 14. Additionally, studies in embryonic stem (ES) and other cells have revealed novel compositions of the PRC2 complex and significant mechanistic details regarding its function10. Below we discuss the relevant studies and their implications for future exploration of epigenetic control in diseases including CRC and IBD.
PRC2 and DNA methylation in gastrointestinal regulation
The roles of DNA and H3K27 methylation have been tested in detail in the intestine. Loss of DNA methylation through disruption of methyltransferase Dnmt1 activity in mouse intestinal epithelium causes crypt expansion due to delayed differentiation, but intestinal function remains unperturbed15. Ezh2, the enzymatic subunit of PRC2, is expressed at the highest level in the crypt of the intestine where all the cycling cells, including the intestinal stem cells, reside16. Genomic deletion of Eed causes complete loss of PRC2 action and leads to cell cycle arrest in crypts, which can be attributed to the loss of H3K27me3 based repression at tumor suppressor Cdkn2a14>, 16–18. Surprisingly, very few genes involved in intestinal stem cell differentiation and homeostasis are controlled by PRC2. Instead, genes expressed during the intestinal development are silenced by H3K27me3 in adult epithelium and get reactivated in absence of PRC2 action13, 16. This reactivation is highly influenced by functional interactions of H3K27me3 with other histone modifications and DNA methylation. Activating histone modification H3K4me3 is found at many gene promoters decorated with H3K27me3 resulting in formation of bivalent domains. The amount of H3K4me3 mark at bivalent promoters of developmental genes can predict the extent of gene activation upon PRC2 loss14, 16. Moreover, decommissioned enhancers of these developmental genes maintain DNA hypomethylation in adult cells, and get activated in PRC2 null cells13. These novel observations in the adult and developmental intestine reveal principles of PRC2 function in adult mammalian cells. However, many fundamental questions, including how mammalian PRC2 is recruited to its genomic target sites, are still a point of contention in the field of epigenomics.
In Drosophila, the polycomb complex is recruited to specific genomic regions called the polycomb responsive elements, but no such regions are identified in mammalian genomes19. Sequence specificity is likely not a major factor in recruitment of PRC2 to the mammalian genome since no recurrent motif has been associated with PRC2 binding. This further emphasises the degree of complexity regulating PRC2 recruitment and activity; the underlying molecular mechanism likely involve DNA methylation, nucleosomal localization, as well as the ability of H3K27me3 to co-localize with both permissive and repressive histone modifications like H3K4me3 and H3K9me3, respectively20–22. Additionally, H3K27me3 can also act to restrict spreading of epigenetic modifications such as the activating mark H3K36me223, or form ‘bivalent’ enhancers hosting both H3K27me3 and H3K4me1 at intergenic regions14, 16, 20, 24. Bivalent states likely poise expression of developmental genes in pluripotent and multipotent cells. Abnormal PRC2 activity can disrupt the balance of activation and deactivation of these unique chromatin states affecting stem cell differentiation and proliferation, and resulting in adverse outcomes including tumor formation. It is not surprising that modulation of PRC2 and H3K27me3, through mutations or indirect chromatin deregulation, is a recurrent observation in many cancer types including CRC.
Coregulation by PRC2 and DNA methylation – Molecular lessons from development and beyond
More than half of all mammalian genes contain high density CpG regions (termed CpG islands - CGIs) around their promoters. H3K27me3-mediated repression is a recurrent event at the hypomethylated CGI-promoters (Figure 1A), which highlights the tug of war between DNA methylation and H3K27me3 for transcriptional repression21, 25. The dilemma of the competition between these two repressive epigenetic modifications for genomic real estate is an active field of research21, 26–28. The mutual antagonism of H3K27me3 and DNA methylation at CGI-promoters suggests that PRC2 targeted CGI-promoters require protection from DNA methylation by chromosomal proteins, as in the case of FBXL10 in mouse embryos28, or other active histone modifications, as in the case of H3K4me3 at bivalent promoters21, 29 (Figure 1B). Perhaps, the dual influence of H3K27me3 and DNA methylation at CGI-promoters tune stem cell plasticity where H3K27me3 and DNA methylation advocate plastic and rigid cellular identity, respectively. After all, H3K27me3 plays a critical role in maintaining bivalency at promoters, a major contributor to cell plasticity required for differentiation, in both embryonic20 and adult stem cells16. Moreover, expression of many of the developmental regulatory genes including SOX2, NODAL, EOMES, are regulated by H3K27me3 in ESCs, mesenchymal stem cells, and neurospheres30. Even in naïve pluripotent cells, H3K27me3 and DNA methylation regulate each other’s distribution and dictate higher order chromatin structure26, 27, 31.
Figure 1. Epigenetic interplay involving H3K27 methylation and its consequence on gene expression.

A) PRC2 complex deposits H3K27me3 at hypomethylated gene promoters leading to a repressed state. Such PRC2 based chromatin and gene control is prevalent in facultative heterochromatin located near the center of the nucleus. H3K9me3 decorated constitutive heterochromatin is found along the nuclear periphery. B) Co-occupancy of activating H3K4me3 and repressive H3K27me3 at hypomethylated promoters keeps genes form ‘bivalent’ domains. Genes with such dual histone marking are in poised state with reduced or no gene expression. C) Mutual exclusivity of activation associated H3K36me3 and H3K27me3 restricts spreading of H3K27me3 around expressed genes. D) PRC1-mediated H2a119Ub is involved in recruitment of PRC2 at many gene promoters, which leads subsequent gene suppression.
DNA methylation also co-occurs with epigenetic layers that restrict spreading of repressive H3K27me3, such as H3K36 methylation23, 32, 33 (Figure 1C). DNA methylation and H3K36me2/3, modifications associated with active gene bodies and non-coding regions of euchromatin are positively correlated: H3K36me2/3 mediates recruitment of de novo DNA methyltransferase DNMT3A through direct binding in ESCs as well as in head and neck squamous cell carcinoma32, 33, where changes in H3K36 methylation patterns induce flux in DNA methylation. Sequential ChIP-bisulfite assays show increased H3K27me3 occupancy in the absence of DNMTs at CpG dense regions hypermethylated in ESCs31. However, expected H3K27me3 and DNA methylation antagonism is not obvious when the relationship between H3K27me3 and DNA methylation is examined globally, as reported across 111 human reference epigenome profiles19. From a genome wide vantage point, considering CGIs and non-CGI regions of the genome, H3K27me3 enrichment is positively correlated with DNA methylation in most cells; an observation that is concurred multiple times in human ESCs21, 31. This contradictory relationship suggests that the association between DNA methylation and H3K27me3 is highly dependent on CpG density and genomic features (e.g. CGIs and promoters)21, 24, 29, 31, where H3K27me3 and DNA methylation are antagonistic at CpG dense regions (i.e. CGIs) and co-occur at orphan CpGs or CpG poor regions. These observations put forward the importance of other epigenetic layers and complexes which act to selectively recruit or block either polycomb complexes or DNMTs. Moreover, they call to attention that disruption in either DNA methylation or H3K27me3 distort the landscape of the other.
Together, these findings emphasize that understanding the mechanistic interdependence of various chromatin layers, particularly as manifested in developmental and homeostatic processes, has significant implications for precisely investigating the failure of this machinery in CRC. The majority of CRCs show a well defined mutational progression in genes such as the adenomatous polyposis coli (APC), Kirsten rat sarcoma viral oncogene (KRAS), and tumor protein p53 (TP53)34. While the effect of DNA methylation changes on CRC is well studied, the role of PRC2 in CRC may be profoundly linked to DNA methylation rather than mutation-induced changes to its function, as observed in other cancers. We start by highlighting a few such cases that are pertinent to understandings the role of PRC2 in CRC, which is detailed in the following section.
PRC2 AND CANCER
Deregulation and/or mutation of various PRC2 components has been observed in numerous diseases, including cancers of blood, prostate, brain, and gastrointestinal tissues35–40. However, to date, no common pattern of disruption for the PRC2 subunits in cancer has emerged. This is indicative of the delicate and context-specific H3K27me3 regulation by PRC2 action that allows for its dual activity – both as a tumor suppresser and as an oncogene. Broadly, PRC2 perturbances contributing to cancer initiation and progression can be stratified into 3 groups: (1) activating and deactivating mutations of PRC2 subunits, (2) modulation of PRC2 activity by up-or down-regulation of PRC2 subunit genes, and (3) mutations in histone H3 protein (oncohistone) that alter H3K27 methylation patterns. Both activating and deactivating mutations of the catalytic subunit EZH2 are distinctly present in various cancer types. For example, in germinal cell-derived large B-cell lymphoma, a neomorphic mutation is detected in the C-terminal catalytic SET domain of EZH2. This mutation most likely causes an increase in H3K27me3 during germinal B-cell formation leading to altered expression of proliferation-checkpoint and terminal-differentiation genes41; accordingly, EZH2 inhibition prevents proliferation of the tumor35, 40, 42. In contrast, inactivating mutations of EZH2, SUZ12 and EED are observed in many cancers, including myelodyplastic patient genomes, that lead to a decrease in global H3K27me3 levels and increase in expression of genes such as HOXA9, suppressed by the polycomb action39, 43–48. Furthermore, abnormal expression of PRC2 components in the absence of mutations may lead to abnormal levels of H3K27me3 resulting in disrupted cell proliferation and differentiation in many cancers including lung and liver’49–54. Thus, increased expression of EZH2 is linked to promotion of proliferation, invasion, and metastasis through suppression of differentiation linked Gata3 in triple negative breast cancer55–61. In contrast, inhibition of EZH2 in cases where it is overexpressed rescues the proliferative phenotype and impedes malignant cell growth, as seen in breast and small-cell lung cancers55, 58, 59. In the case of small-cell lung cancer, PRC2-mediated suppression of MHC-1 antigen processing pathway (APP) genes allows the cancer cells to evade antitumor immunity, which is a hallmark feature in these cancers62. Inhibition of EZH2 respectively removes repressive H3K27me3 at promoters of MHC-1 APP genes and promotes anti-tumor activity. Finally, latest discoveries have identified a mutation in substrate of PRC2, a methionine substitution of lysine 27 on amino terminal of histone 3 (H3K27M), which inhibits EZH2 by binding to the pocket of the SET domain where lysine residue is normally housed61. H3K27M leads to rewiring of the H3K27me3 landscape in cells causing tumor development, particularly gliomas63–67. Surprisingly, despite local inhibition of H3K27me3 due to H3K27M, several loci, including tumor-suppressor genes, show increased H3K27me3 enrichment in pediatric gliomas. EZH2 has already been shown to be a potential therapeutic target in these instances66. The variety of causes for change in PRC2 activity and its wide-ranging effects across cancers have powerful implications for our understanding of PRC2 action in CRC.
PRC2 in initiation, progression, and metastasis of CRC
Only ~2.5% of CRC harbor EZH2 mutations; however, studies using patient tissue specimens, CRC cell lines, as well as ex vivo organoid tissue culture indicate involvement of PRC2 in tumor initiation and progression68–70. Proliferating Lgr5+ stem cells in the gastrointestinal crypts are the source of all functional cells in the epithelium and their transformation into cancer stem cells (CSCs) is essential for tumor initiation and metastasis71. Wnt/β-catenin signaling is necessary for stem cell proliferation and over 80% of CRCs carry non-synonymous mutations in APC, a component of Wnt-pathway-antagonist cytoplasmic protein complex, resulting in constitutively active Wnt/β–catenin pathway72, 73. EZH2 promotes colorectal stem-like cells by activating the Wnt/β–catenin pathway74 as EZH2 expression negatively correlates with p21Cpi1, an antagonist of Wnt pathway. Moreover, knock down of EZH2 restores p21Cpi1 expression and suppresses β–catenin in vitro74, 75. Recent evidence suggests that PRC2 aids self-renewal of tumor initiating cells by inhibiting differentiation through H3K27me3 deposition at genes such as Indian Hedgehog (IHH) (Figure 2A), a canonical marker of colonocyte differentiation in normal gastrointestinal developmental, that is also marked with activating modifications such as the H3K4me370.
Figure 2. Interdependent changes in histone modifications, DNA methylation, chromatin compaction, 3D genomic structure, and their impact on gene expression in CRC.
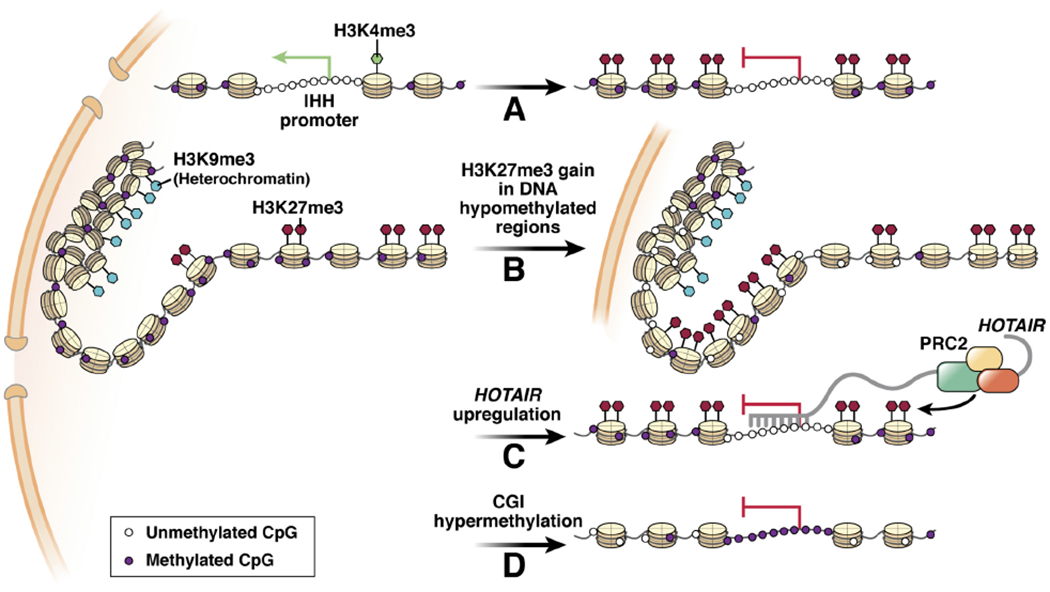
A) Abnormal H3K27me3 spreading during differentiation causes repression of associated genes. B) Large scale loss of DNA methylation allows spreading of H3K27me3 that contributes to rearrangement of compacted chromatin. C) Abnormally high expression of noncoding RNAs such as the HOTAIR is involved in aberrant PRC2 recruitment and target gene suppression. D) Gain of DNA methylation at promoter associated CGIs is a common feature in CRC that leads to abnormal expression profile in cancer cells.
EZH2-mediated repression of cell-cell contact protein E-cadherin is correlated with increased cell mobility and thought to contribute to metastasis of CRC74, 76. Furthermore, Immunohistochemical studies using CRC tissues revealed that increased cellular EZH2 expression positively correlates with Ki67, a marker of cell proliferation, during progression of adenoma to carcinoma75. However, how EZH2 overexpression affects H3K27me3 distribution across the genome and its contribution to this transformation remains unknown. Interestingly, normal primary colon cells adjacent to the cancerous cells express significantly lower levels of EZH2 (specifically the less differentiated ones)77. In fact, cells located at the center of the tumor possess higher expression of EZH2 and levels of H3K27me3 compared to those found at the tumor invasion front78. Furthermore, CRC cells metastasized to liver show significant reduction in H3K27me2, a suppressive histone modification covering inter- and intragenic regions, in comparison to the primary tumors79.
Recent studies highlight epigenetic changes in not just the elementary features of DNA methylation and histone modification, but suggest widespread rearrangements in nuclear architecture in gastrointestinal tumors80, 81. The affected features include chromatin looping that allows enhancer promoter interactions influencing gene activity, topological domains that isolate set of genes and their cis-regulatory control regions, as well as the nuclear compartmentalization that isolates inactive heterochromatin along nuclear periphery while maintaining active euchromatin towards center of the nucelus81 (Figure 2B). Notably, one study observed a mega base reorganization of H3K27me3 enriched regions associated with broad DNA hypomethylation, a hallmark of CRC, located at interface between canonical euchromatin and heterochromatin compartments in CRC cell nuclei81. What causes such broad H3K27me3 gains and the large-scale topological changes in CRC remains to be determined. In contrast to the traditional view of epigenetic changes aiding cancer progression, transcriptional changes associated with the higher order changes in nuclear architecture seem to be tumor suppressive, highlighting the interplay of various chromatin layers in controlling the transcriptional output in a context dependant manner.
Crosstalk of PRC2 and epigenome in CRC
PRC2 activity is known to be dependent on its interactions with other epigenetic enzyme complexes and modifications. PRC1, a related polycomb repressive complex that catalyzes H2AK119Ub, is critical for PRC2 recruitment at target genes and appropriate H3K27me3 deposition in ESCs and several adult cell types including gastrointestinal epithelium82, 83 (Figure 1D). PRC1 sustains Wnt/β–catenin signaling by repressing many non-lineage-specific transcription factors that may adversely affect the signaling through β–catenin/Tcf transcriptional control82. Interestingly, Bmi1, a component of PRC1 is critical for progression and maintenance of CRC; loss of Bmi1 relieves repression of the target tumor suppressor gene Arf84. BMI1 also promotes invasion and proliferation of CRC by inducing epithelial to mesenchymal transition (EMT) and through downregulation of E-cadherin based cell-cell adhesion, both in vitro and in vivo85, 86. In addition to these direct links to CRC, PRC1 is known to control various aspects of chromatin structure along with PRC2. For example, PRC1 occupancy correlates with compacted chromatin domains and their appropriate rearrangements are crucial for proper ESC differentiation87. Taken together, the cross-talk between PRC1 and PRC2 complexes, their influence on various stem cell functions, and link to the chromosomal topology in gastrointestinal tumors80 further increase the plausibility of H3K27me3 and related chromosomal 3D structure modulation influencing CRC progression.
Another important epigenetic crosstalk involving PRC2 that is relevant to gastrointestinal homeostasis and cancer is its interaction with long non-coding RNA (lncRNA) HOTAIR, which silences its target genes by recruiting PRC288, 89 (Figure 2C). In CRC patient samples, the expression of HOTAIR and PRC2 components such as SUZ12, EZH2, and the resulting H3K27me3 marks are highly correlated90. Moreover, HOTAIR overexpression is associated with induction of EMT and an increased capacity for metastasis and invasion in primary colon tumors and in cultured cells, respectively90–93. This further implicates PRC2 action and changes in H3K27me3 distribution in various stages of CRC. Remodelling of H3K27me3 landscape can also be a consequence of perturbance in other epigenetic modifications like H3K36me3 as in the case of undifferentiated sarcomas harbouring H3K36M (methionine substitution of lysine)23. Methylation of H3K36 restricts H3K27me3 spreading across the genome; therefore, change in H3K36 methylation may cause alteration in global level and genomic occupancy of H3K27me3.
Collectively, these observations make a compelling argument that PRC2 and H3K27me3 are clinically relevant in CRC biology. Even if mutated in a small percentage of CRC cases, PRC2 action has profound effect on various layers of chromatin structure, thus directly or indirectly controlling gene expression, cellular transformation, and cancer progression. In this context, one of the most important and influential coregulatory epigenetic mechanisms in CRC is DNA methylation.
DNA METHYLATION AND CANCER
As early as the discovery that DNA methylation patterns may influence gene expression, it was predicted that changes in these patterns may be involved in tumorigenesis94. Indeed, over the last several decades, DNA methylation has been studied in context of all cancer types1, 95–98. Mutations in the methylase enzymes or in substrate CpGs may change global DNA methylation patterns, as reviewed in detail previously1. DNA methylation change may also be linked to oncogenic mutations, loss of tumor suppressor action, and flux in other epigenetic features. However, the mechanisms through which these changes take place and influence gene expression are still understudied; here we focus on these in the context of CRC.
DNA Hypomethylation and hypermethylation in CRC
Early studies identified large scale DNA hypomethylation in many cancers including CRC99–102. Latest advances in whole genome bisulfite sequencing (WGBS) allow analysis of DNA methylation at the single CpG level. Such studies have identified large hypomethylated regions in CRC with average CpG methylation of 40%-60% as compared to the average 80% across the genome103, 104. Many of these hypomethylated regions are larger than 100 kb, which covers almost half of the genome. It has been known for some time now that such hypomethylation occurs relatively early in CRC progression34. Recent revelations suggest that alterations in DNA methylation may be related to fat content in the diet105. However, the exact causes and consequences of this broad change remain understudied. The reduced methylation in CRC and other cancers is thought to cause genomic instability and aid aneuploidy106, 107. Indeed, mouse embryonic stem cells devoid of DNA methylase Dnmt1 activity show elevated rate of mutations due to reduced global DNA methylation and the resulting mitotic recombination and chromosomal loss108, 109. Although DNMT1 gene mutations are found in CRC, these events are very rare and many do not explain the prevalence of DNA hypomethylation in CRC110. Hypomethylation may also lead to increased transcription of transposable elements, repeat regions, and proto-oncogenes in CRC106, 111, 112.
The DNA is largely unmethylated at CGIs, which are prevalent at gene promoters, representing relatively accessible chromatin that is favourable for gene expression when needed. Up to 10% promoter linked CGIs are hypermethylated in many cancers including CRC (Figure 2D), making it one of the hallmarks of cancer that also defines a subclass called CpG island methylator phenotype (CIMP)113–115 . While most genes display DNA methylation across the gene body without consequences for the gene activity, the promoter hypermethylation can drive gene repression and can impact tumorigenesis by shutting down tumor suppressor and other genes linked to cell fate choices and signaling116–118. In CRC, deactivating hypermethylation events have been identified in known tumor suppressors such as P16 (Cell cycle)119, and CDX2 (critical transcription factor linked to differentiation)120–122, as well as AXIN2 and ASCL2 (regulators and effectors of Wnt singling pathway)123–125. Moreover, DNA methylation indirectly contributes to proliferation and cell migration by regulating expression of miRNA that modulates β-catenin signaling pathway126. DNA mismatch repair is an essential part of cellular proliferation as it ensures accurate copying of genome over multiple rounds of cell divisions. Mutation in hMLH1, a gene involved in mismatch repair process, causes microsatellite instability (MSI) in colorectal cancers, which leads to almost 100 fold increase in mutation rate as compared to the normal cells127–130. Remarkably, in absence of hMLH1 mutations, hypermethylation of CpG island 5’ of hMLH1 can cause gene repression that leads to MSI131–133. This highlights the impact of epigenetic silencing events as an alternative to genetic mutations and exemplifies the widespread effects of promoter hypermethylation. DNA methylation changes in CRC may also derail gene control networks as evident through loss of imprinting (LOI) at the IGF2 gene134. DNA hypermethylation at a nearby insulator gene H19 causes loss of its action which triggers biallelic expression of IGF2 and overproduction of growth inducing IGF2 protein. DNA methylation driven silencing blocks differentiation in APC mutant patient derived CRC organoids by supressing differentiation associated genes such as Ndrg4 and Stox2135. Intriguingly, the differentiation capacity can be restored by active TET induced hypomethylation. DNA hypermethylation and hypomethylation events during early CRC evolution are thought to trigger LOI during early cancer progression 34. In addition, Recent evidence suggests that cancer associated microbiota can trigger oncogenic DNA methylation signatures in vivo136. Beginning with these initial changes in tumorigenesis, DNA methylation evolves through coordinated action with other epigenetic features and genetic lesions, molecular dynamics of which remain unexplored.
Interplay of DNA methylation and epigenome in CRC
Multiple cell intrinsic and extrinsic factors dictate how DNA methylation patterns change in CRC and impact gene expression and cellular transformation. Two of the most important considerations are involvement of complementary epigenetic features and specification of boundaries within which these features interact to influence surrounding chromatin and gene expression. For example, in normal stroma, most gene promoters contain CpGs at high density (CpG islands) that lack DNA methylation. This allows for more accessible or open chromatin that has features such as nucleosome depletion, presence of histone variant H2A.Z in flanking nucleosomes, and activity associated histone modifications such as the H3K4 methylation, and H3K9 and H3K14 acetylation116. Recent studies report that DNMTs interact with and are recruited by lncRNAs to suppress genes via promoter hypermethylation and consequently promote metastatic CRC137. These observations along with latest reports of de-novo methylation at H3K36me3 marked promoter CGIs are testimony to the complex relationship of DNA methylation with the chromatin landscape138.
Bordering gene promoters upstream and downstream, CpGs are usually distributed at a lower density and are almost fully methylated. This significant switch in DNA methylation is also accompanied by removal of the active histone modifications and addition of repressive marks such as H3K9me2, which indicates coordinated recruitment and activity of specific chromatin modifiers across genomic regions separated by clear boundaries1, 116. Recent single cell multiomics analysis of primary CRC tumors revealed that the hypomethylation expands to regions encompassing H3K9me3 and long interspersed nuclear element 1 (LINE-1); interestingly, the DNA hypo-methylation state of LINE-1 shows linear correlation with cancer stage111, 139, 140. Both, the interplay of chromatin features and the boundaries that separate chromatin domains, are perturbed in CRC1. Hypomethylated regions (as discussed above) can be identified as mega base-pair length “blocks” stretching across many genes, and several genes within these blocks display promoter associated CpG islands hypermethylation103, 139, 141. Interestingly, the hypomethylation blocks also show broad changes in histone modifications and other chromatin features. Understanding this interplay of epigenetic features can provide insights into how the CRC epigenome is established and evolves, and its implications for gene expression and cellular transformation.
Early clues for coregulation of CRC chromatin by histone modifications and DNA methylation came from the observation that the regions of focal hypermethylation in CRC are pre-marked with H3K27me3 in normal tissue142. Interestingly, these H3K27me3 patterns are established during early development and consist of activating H3K4me3 mark along side the repressive H3K27me3 modifications forming a bivalent state in ES cells143. Moreover, these hypermethylated CpG islands and the surrounding mega-base hypomethylated domains are associated with nuclear lamina linked heterochromatin in CRC104, suggesting a concordant impact of DNA and histone methylation flux on the chromatin structure in cancer cells. Most direct proof for this chromatin coregulation comes from recent studies revealing changes in chromatin 3D architecture through analysis of genome topology (Hi-C assays) in CRC81. Using transmission electron microscopy for analysis of heterochromatin distribution and DNA fluorescence in situ hybridization to determine positioning of specific genomic regions, these studies report reduction in nuclear peripheral heterochromatin (canonically H3K9me3 marked), and growth of H3K27me3 marked DNA hypomethylated regions (Figure 2B). In addition to providing insights into how histone and DNA methylation are involved in the large-scale rearrangements, these studies also call into question the canonical views of PRC2 targeting being restricted to unmethylated regions and the causal relationship between DNA and H3K27 methylation. Collectively, molecular dependencies involving DNA and H3K27 methylation are critical for gastrointestinal gene regulation; most significant ones are listed in Table 1. Given the high interest in understanding these dependencies, many mouse models have been generated in which genes coding for their enzyme complexes have been mutated; Table 2 summarizes such mouse models and the related phenotypes in context of the gastrointestinal tissue.
Table 1.
Epigenetic interplay involving DNA and H3K27 methylation relevant to gene regulation and colorectal cancer
| Epigenetic interaction | Molecular event | Biological relevance or consequence | Reference |
|---|---|---|---|
| PRC2-PRC1 | H3K27me3 and H2AK119Ub co-dependently recruit respective complexes | Recurrent deregulation in CRC | 55,56 |
| Loss of Bmi1, a component of PRC1, relieves repression of the target genes | Promotion of invasion and epithelial to mesenchymal transition | 58,59 | |
| PRC2-ncRNA | HOTAIR guides PRC2 to target genes destined for suppression | Increased invasion and metastasis in CRC | 64–67 |
| PRC2-H3K4me3 | H3K27me3 and H3K4me3 co-occupancy poise expression of developmental genes | Abnormal differentiation and proliferation | 44 |
| PRC2-DNA methylation | Mega base H3K27me3 enriched regions associated with broad DNA hypomethylation | Compromised higher order chromatin structure in CRC | 54 |
| Hypomethylated developmental enhancers maintained by PRC2 | Ectopic reactivation of enhancers in adult tissue upon PRC2 loss | 115 | |
| DNA methylation-H3K9me3 | Cooccurrence of H3K9me3 and DNA methylation at heterochromatin region | Reduction in nuclear peripheral heterochromatin accompanied by loss of DNA methylation in CRC | 54 |
Table 2.
Consequence of deletion of PRC and DNMT genes on normal intestinal development
| Mouse Model | Complex | Modification | Phenotypic Anomaly | References |
|---|---|---|---|---|
| Ezh2 KO | PRC2 | H3K27me1/2/3 | No Significant difference in weight and intestinal morphology compare to WT | 14, 17 |
| Eed KO | PRC2 | H3K27me1/2/3 | Significant weight loss and severely degraded crypt | 13, 18 |
| Ring1a/b KO | PRC1 | H2AK119UB | Significant weight loss and severely degraded crypt | 149 |
| Bmi1 KO | PRC1 | H2AK119UB | Significantly shorter small intestine and reduced cycling crypt cell | 150 |
| Dnmt1 KO | Dnmt1 | Methylated Cytosine | Smaller in size with shorter GI track, villus atrophy in neo-natal, and degeneration of GI smooth muscle | 15, 150–152 |
| Dnmt3b and Dnmt1 KO | Dnmt1 and Dnmt3 | Methylated Cytosine | Significant reduction in weight. Increase apoptosis and degradation of villi and crypt | 153 |
Levels of H3K27me3 and DNA methylation can be utilized for prognosis and to guide treatment of CRC144, 145. Increased levels of H3K27me3 are correlated with improved drug sensitivity in CRC patients146. However, results regarding prognostic capability of EZH2 are contrary at best. This suggests that even though there has been a comprehensive effort to classify colorectal cancers147, there is a need to better understand CRC based on epigenetic modifications. This may also indicate potential cancer-specific role of EZH2 independent of H3K27me3. All these considerations will have important implications for clinical application of PRC2 inhibitors148.
EPIGENTICS OF INFLAMMATORY BOWEL DISEASES
IBDs and relevant cell types
Inflammatory bowel diseases represent heterogeneous forms of pathologies characterized by chronic inflammation in the lining of the gut, with Crohn’s disease (CD) and ulcerative colitis (UC) being the most well studied subtypes. Although a few inherited gene mutations such as IL23R, IL-10R, and NOD2 are considered causal154, 155, increased risk of the disease is associated with complex regulation through genetic variations (Single nucleotide polymorphisms - SNPs) and environmental factors. Molecular mechanisms underlying these complex diseases, particularly the involvement of epigenetic changes, are poorly appreciated, owing to inadequately representative models and incomplete understanding of relevant cell types156. Although the causes of IBD are indeterminate, the pathogenicity of the disease is recognized to be driven by immune cell responses that fight off harmful pathogens invading the epithelium and aid in healing of the epithelial lining. In the case of IBD, the immune response that usually clears invading bacteria attacks the body’s normal and healthy microbiome.
Immune cells in the lamina propria communicate with the intestinal and the colonic epithelial cells and mount an immune response by secreting cytokines and signals that recruit additional immune cells to the site, resulting in inflammation. The immune infiltration mechanistically contributes to the breakdown of the barrier tissue which worsens symptoms of IBD. On the other hand, epithelial stem cells are equipped with receptors to sense the epithelial barrier breach and secrete cytokines like IL-33 to actively recruit immune cells to prevent spread of bacteria and to repair the damage157.
Much of the molecular understanding of IBD has come from studying T cells. T cells arise in the bone marrow and undergo somatic rearrangements to produce a unique antigen reactive receptor in thymus. These trained T cells attack and eradicate specific targets such as virally infected or cancerous cells (CD8+) and invading pathogens (CD4+), while some cells work to quiet immune response (CD4+ T regulatory Cells – Tregs). T helper cells (CD4+) remain naive when leaving the thymus and differentiate into subsets including Th1, Th2, and Th17 upon activation in gut, where they drive the chronic inflammation and pathogenicity. The essential role of T cells in IBD pathogenesis is apparent from the observations in patients with depleted CD4 cells, such as the HIV+ve individuals, who have milder IBD symptoms158, and activated T cells, once they become pathogenic, are sufficient to sustain the disease159.
In spite of the increased understanding of the cell types, cell-cell interactions, and signaling events involved in IBD, molecular mechanisms controlling the epithelial cell response, timing of the disease, and maintenance of inflammation remain severely understudied. In absence of clear genetic drivers, hundreds of susceptibility loci predicted by GWAS studies may be attractive leads, however, most of these SNP loci are in non-coding regions, suggesting that gene control failure is involved in IBD. Interestingly, recent examples offer hints of how cellular epigenetic states and chromatin dynamics influence many of the cells involved in IBD during homeostasis and disease. We review the molecular aspects of such controls highlighting the chromatin features involved in inflammatory cell responses in IBD.
Epigenetic changes in IBD – Role of DNA methylation and histone modifications
DNA methylation changes have been an attractive avenue for mapping regulatory epigenetic changes in IBDs. An epigenome-wide association study (EWAS) using intestinal biopsies from monozygotic twin pairs with discordant UC status reported 61 differentially methylated loci associated with aberrantly expressed transcripts160. Many of the novel candidates, such as CFI, SPINK4, and THY1, are involved in immune regulation, highlighting the use of this approach160. Furthermore, DNMT1 and DNMT3b are highly expressed in colonic epithelial cells with active inflammation compared to non-inflamed colons from UC patients, which also suggests increase in corresponding demethylase activity with increased inflammatory status. Surprisingly, intestinal epithelial cells (IECs) from patients with active or inactive UC display global DNA hypomethylation compared to cells from healthy individuals161. These changes are accelerated with the IBD linked gain of IEC proliferation, which may further predispose these individuals to develop colorectal neoplasms161. DNA methylation in IECs from pediatric patients show disease subtype-specific patterns for Crohn’s disease and ulcerative colitis that are linked to their unique transcriptional profiles162.
A recent study revealed that reduced levels of SETDB1, a histone methyltransferase responsible for H3K9me3, in IBD patients promotes IBD pathogenesis163. Given the role of H3K9me3 in chromatin compaction and compartmentalization, loss of SETDB1 causes genome instability and reactivation of repressed endogenous retroviruses that leads to necroptosis. Collectively, these studies highlight that IBD linked epigenetic changes are disease and stage-specific and may outlast the inflammation, However, molecular and clinical implications of these sustained effects remain to be elucidated.
Epigenetic control of T cell activity and inflammation in IBD
T cells are critical in directing intestinal stem cells (ISC) during homeostasis as well as in IBD. In mice, Tregs produce immune suppressive cytokines like IL-10 to promote ISC self-renewal, while multiple types of T-helper cells produce proinflammatory cytokines such as IFNγ (Th1), IL-13 (Th2), and IL-17 (Th17) to promote rapid differentiation and repair of breaches during IBD164. To establish these regulatory circuits, naïve T cell differentiation, activation, and cytokine production are highly coordinated and tightly controlled by transcription factors (TFs) and precise chromatin modulation. Transcription factors T-bet and Gata3 induce Th1 and Th2 cell fates, respectively165, 166. T-bet inhibits Th2 cytokines and directs chromatin remodeling at individual IFNγ alleles; accordingly, loss of T-bet in mice causes increased IL-4 production in an induced colitis model where symptoms could be ameliorated with anti-IL-4 antibody therapy165. On the other hand, GATA3 consolidates Th2 development by shutting down the Th1 developmental program through IL-12 gene repression. Additionally, it upregulates its own expression along with Th2 cytokines IL-4 and IL-13 through direct binding and remodeling of chromatin at the target loci166–168. While TFs enforce the lineage choices through chromatin modulation, additional epigenetic changes stabilize the fate choices over cell divisions. For example, in naive T cells, inactive cytokine genes are located in accessible chromatin regions, where as in differentiated Th subsets those genes are in highly silenced centromeric heterochromatin169, thus securing the differentiated T cell states and lineages. Additionally, activating histone acetylation on cytokine genes IFNγ (in Th1) and IL-4 (in Th2) allows action of lineage-specific TFs and reinforces cell fate decisions170. Epigenetic changes thus control the plasticity and stability of Th phenotypes and are foundational to immune responses.
T cells can have deleterious effect on intestinal epithelium as their infiltration around intestinal crypts can cause ISC damage through IFNγ during allogenic bone marrow transplant and graft-versus-host disease171. Moreover, CD is associated with Th1 differentiation, while UC is linked to Th2 fate, further highlighting the importance of precise epigenetic control of T cell fate and cytokine production. IFNγ is the most potent pro-inflammatory cytokine, and the gene encoding IFNγ is one of many cytokines identified as IBD risk loci. Recent studies mapped the chromatin activation in stimulated human CD4+ T cells in the presence of cytokines by profiling the open chromatin regions with ATACseq (Assay for Transposase-Accessible Chromatin using sequencing) and ChIPseq for activating histone modification H3K27ac172; interestingly, the magnitude of T cell activation induced chromatin changes is fine-tuned by particular cytokines. Importantly, SNP variant loci associated with IBD are predominantly accessible and H3K27ac marked in naïve T cells induced with Th1 cytokines like IFNγ or in Th1 cells, but not in macrophages or B cells. These findings illustrate the potential for functional understanding of GWAS identified IBD linked loci through epigenetic investigation.
PRC2-mediated gene repression in T cells and IBD
Recent studies show that T cell activation is accompanied by increased PRC2 activity and chemical inhibition of PRC2 reduces allergy inflammation. In the intestine, Tregs produce anti-inflammatory cytokines like IL-10 and TGF- β that quiet the immune response and allow the epithelium to recover from inflammation173. Ezh2 works as an essential cofactor with Treg-specific TF Foxp3 for targeted deposition of repressive H3K27me3 at genes part of an immune suppression program; reduced FOXP3-EZH2 interaction is associated with early onset of IBD174 (Figure 3A). In addition to this role in inflammation control, appropriate distribution of H3K27me3 is also essential for proper T cell differentiation. Loss of Kdm6b, a stress-inducible H3K27 demethylase, inhibits naïve T cell differentiation into Th1 cells and promotes differentiation into Th2 and Th17 subtypes in the small intestine and colon175 (Figure 3B). Additionally, in Th17 cells, inhibition of Kdm6b leads to suppression of Proinflammatory cytokine production including IL-17176.
Figure 3. Epigenetic changes functionally linked to IBD through control of immune cell differentiation, activation, and inflammatory response.

A) Treg cell-specific transcription factor FoxP3 directs Ezh2 and the PRC2 complex to specific genomic loci causing H3K27me3 deposition and repression of surrounding target genes. This epigenetic gene control is essential for immune suppression during inflammation. B) T cell differentiation and lineage determination is linked to cell-type-specific activation of H3K27me3 repressed genes in naïve T cell. Targeted action of H3K27 demethylase Kdm6b enables formation of appropriate gene expression patterns and production of Th1 or Th2 and Th17 cell types.
Epigenetic control of macrophages
Macrophages are part of the innate immune system and use toll-like receptors (TLR) to detect and bind microbial lipopolysaccarides (LPS), which activates them leading to production of antimicrobial peptides and activation of inflammation promoting genes. Various conditions including prolonged stimulation with LPS can cause macrophages to become unresponsive or ‘LPS tolerant’ 177, a state in which they preferentially produce antimicrobial peptides. A deeper look into the epigenetic regulation of promoters found that newly activated macrophages contain activating H3K4me3 and H4 acetylation modifications on both proinflammatory and antimicrobial genes. However, tolerant macrophages lose the H4 acetylation at the promoters associated with the class of harmful pro-inflammatory genes while maintaining both H3K4me and H3 acetylation at antimicrobial peptide genes. This unique use of selective epigenetic control allows these cells to be primed for quicker response upon secondary encounters with LPS178.
Control of epithelial and immune cells by microbiome
Commensal bacteria greatly influence the gut epithelium and supporting niche cells during homeosis, inflamed conditions in IBD, and in CRC. These effects are widely attributed to microbial metabolite production and microbial dysbiosis, and are reviewed elsewhere179–181. Interestingly, for the epithelial and immune cells discussed here, the microbial impact involves modulation of cell differentiation and direct action on cellular epigenome. Dendritic cells in the colon can induce naive CD4+ T helper cells to polarize into inflammation driving TH1 and TH17 cell types. Recent studies reveal that bacteria consume host-derived chemicals such as the bile acids and generate biologically active molecules including 3β-hydroxydeoxycholic acid (isoDCA), which reduces immunostimulatory capacity of the dendritic cells182. Moreover, this isoDCA action influences peripheral Tregs (pTregs) development as it directs these cells into a RORγt+ pTreg fate, further reducing the inflammation. Recent studies testing transplantation of fecal matter from CRC patients and normal individuals into mice colon reveal that the CRC-specific microbiota causes hypermethylation of several genes, such as WIF1, PENK, and NPY, linked to CRC pathogenesis136. Expression of many pro-inflammatory mediators like TNF-α and NOS2, and anti-inflammatory mediator IL10 are tightly controlled by deposition of activating histone modification H3K9ac and its erasure by histone deacetylase (HDAC) in many tissues including the gastrointestinal track183–185. In mouse models of microbial dysbiosis and related colitis, HDAC inhibition caused induction of anti-inflammatory Tregs along with suppression of proinflammatory Th1 and Th17 cells184, 185. These reports highlight the molecular and cellular impact of microbiome modulation and the underlying involvement of epigenetic changes.
IBDs are complex diseases in which regulatory failure may occur in various mesenchymal and/or epithelial cell types. With the role of epigenetic controls in individual cell development and function under evaluation, two of the bigger priorities for future research are: A) to understand how epithelial and mesenchymal cells together allow formation and maintenance of a chronic inflammation, and B) to illustrate the epigenetic control at the known IBD risk loci.
CONCLUDING REMARKS:
Epigenetic regulation involving DNA and histone methylations are critical for control of gene expression changes in gastrointestinal development and homeostasis. These chromatin features operate in context of other epigenetic layers to establish cellular epigenome. Deregulation of epigenomic structure and function is involved in many diseases including CRC and IBDs. CRC, with a well-established chronology of mutational events, and IBDs, with few known causal mutations and many complex variables, both show strong involvement of epigenetic modulation in development and progression of the disease. However, many mechanistic details of epigenetic dependencies in these diseases remain unexplored, further limiting their clinical use for prognostic or drug development purposes. Recent studies have revealed unprecedented details of how coregulatory alliance among various epigenetic layers is established during development and how they influence the homeostatic cell behavior. This knowledge presents unique opportunities to understand how their mis-regulation contributes to the disease.
Acknowledgments
Funding:
This work was supported by the National Institutes of Health grant K01DK113067 to Unmesh Jadhav and by grants CA72851, CA184792, CA187956, and CA227602 from the National Cancer Institute to Ajay Goel.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1.Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montgomery ND, Yee D, Chen A, et al. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol 2005;15:942–7. [DOI] [PubMed] [Google Scholar]
- 3.Healy E, Mucha M, Glancy E, et al. PRC2.1 and PRC2.2 Synergize to Coordinate H3K27 Trimethylation. Mol Cell 2019;76:437–452.e6. [DOI] [PubMed] [Google Scholar]
- 4.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 2004;15:57–67. [DOI] [PubMed] [Google Scholar]
- 5.Kasinath V, Faini M, Poepsel S, et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Science 2018;359:940–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011;469:343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, Hu B, Hou Y, et al. Silencing of developmental genes by H3K27me3 and DNA methylation reflects the discrepant plasticity of embryonic and extraembryonic lineages. Cell Res 2018;28:593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holoch D, Margueron R. Mechanisms Regulating PRC2 Recruitment and Enzymatic Activity. Trends Biochem Sci 2017;42:531–542. [DOI] [PubMed] [Google Scholar]
- 9.Cooper S, Grijzenhout A, Underwood E, et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun 2016;7:13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laugesen A, Højfeldt JW, Helin K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol Cell 2019;74:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev 2011;25:2436–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domcke S, Bardet AF, Adrian Ginno P, et al. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015;528:575–9. [DOI] [PubMed] [Google Scholar]
- 13.Jadhav U, Cavazza A, Banerjee KK, et al. Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA. Mol Cell 2019;74:542–554 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jadhav U, Manieri E, Nalapareddy K, et al. Replicational Dilution of H3K27me3 in Mammalian Cells and the Role of Poised Promoters. Mol Cell 2020;78:141–151.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheaffer KL, Kim R, Aoki R, et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev 2014;28:652–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jadhav U, Nalapareddy K, Saxena M, et al. Acquired Tissue-Specific Promoter Bivalency Is a Basis for PRC2 Necessity in Adult Cells. Cell 2016;165:1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppens MA, Bounova G, Gargiulo G, et al. Deletion of Polycomb Repressive Complex 2 From Mouse Intestine Causes Loss of Stem Cells. Gastroenterology 2016;151:684–697.e12. [DOI] [PubMed] [Google Scholar]
- 18.Chiacchiera F, Rossi A, Jammula S, et al. PRC2 preserves intestinal progenitors and restricts secretory lineage commitment. EMBO J 2016;35:2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassis JA, Brown JL. Polycomb group response elements in Drosophila and vertebrates. Adv Genet 2013;81:83–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125:315–26. [DOI] [PubMed] [Google Scholar]
- 21.Lorzadeh A, Bilenky M, Hammond C, et al. Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility. Cell Rep 2016;17:2112–2124. [DOI] [PubMed] [Google Scholar]
- 22.Pellacani D, Bilenky M, Kannan N, et al. Analysis of Normal Human Mammary Epigenomes Reveals Cell-Specific Active Enhancer States and Associated Transcription Factor Networks. Cell Rep 2016;17:2060–2074. [DOI] [PubMed] [Google Scholar]
- 23.Lu C, Jain SU, Hoelper D, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016;352:844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kundaje A, Meuleman W, Ernst J, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jermann P, Hoerner L, Burger L, et al. Short sequences can efficiently recruit histone H3 lysine 27 trimethylation in the absence of enhancer activity and DNA methylation. Proc Natl Acad Sci U S A 2014;111:E3415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLaughlin K, Flyamer IM, Thomson JP, et al. DNA Methylation Directs Polycomb-Dependent 3D Genome Re-organization in Naive Pluripotency. Cell Rep 2019;29:1974–1985.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Inoue A, Jiang L, Lu F, et al. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017;547:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulard M, Edwards JR, Bestor TH. FBXL10 protects Polycomb-bound genes from hypermethylation. Nat Genet 2015;47:479–85. [DOI] [PubMed] [Google Scholar]
- 29.Neri F, Krepelova A, Incarnato D, et al. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013;155:121–34. [DOI] [PubMed] [Google Scholar]
- 30.Xie W, Schultz MD, Lister R, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 2013;153:1134–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinkman AB, Gu H, Bartels SJ, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 2012;22:1128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinberg DN, Papillon-Cavanagh S, Chen H, et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 2019;573:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baubec T, Colombo DF, Wirbelauer C, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015;520:243–7. [DOI] [PubMed] [Google Scholar]
- 34.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 35.McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012;492:108–12. [DOI] [PubMed] [Google Scholar]
- 36.Jain P, Di Croce L. Mutations and deletions of PRC2 in prostate cancer. Bioessays 2016;38:446–54. [DOI] [PubMed] [Google Scholar]
- 37.Cooney E, Bi W, Schlesinger AE, et al. Novel EED mutation in patient with Weaver syndrome. Am J Med Genet A 2017;173:541–545. [DOI] [PubMed] [Google Scholar]
- 38.Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 2014;46:1170–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueda T, Sanada M, Matsui H, et al. EED mutants impair polycomb repressive complex 2 in myelodysplastic syndrome and related neoplasms. Leukemia 2012;26:2557–60. [DOI] [PubMed] [Google Scholar]
- 40.Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010;42:181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Béguelin W, Rivas MA, Calvo Fernández MT, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun 2017;8:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCabe MT, Graves AP, Ganji G, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012;109:2989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan SN, Jankowska AM, Mahfouz R, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013;27:1301–9. [DOI] [PubMed] [Google Scholar]
- 44.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 2010;42:722–6. [DOI] [PubMed] [Google Scholar]
- 45.Brecqueville M, Cervera N, Adélaïde J, et al. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J 2011;1:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puda A, Milosevic JD, Berg T, et al. Frequent deletions of JARID2 in leukemic transformation of chronic myeloid malignancies. Am J Hematol 2012;87:245–50. [DOI] [PubMed] [Google Scholar]
- 47.Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 2010;42:665–7. [DOI] [PubMed] [Google Scholar]
- 48.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nature Genetics 2014;46:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Béguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013;23:677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wasenang W, Puapairoj A, Settasatian C, et al. Overexpression of polycomb repressive complex 2 key components EZH2/SUZ12/EED as an unfavorable prognostic marker in cholangiocarcinoma. Pathol Res Pract 2019;215:152451. [DOI] [PubMed] [Google Scholar]
- 51.Sato T, Kaneda A, Tsuji S, et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep 2013;3:1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue C, Wang K, Jiang X, et al. The Down-Regulation of SUZ12 Accelerates the Migration and Invasion of Liver Cancer Cells via Activating ERK1/2 Pathway. J Cancer 2019;10:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu PP, Xu YJ, Dai SK, et al. Polycomb Protein EED Regulates Neuronal Differentiation through Targeting SOX11 in Hippocampal Dentate Gyrus. Stem Cell Reports 2019;13:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herrera-Merchan A, Arranz L, Ligos JM, et al. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun 2012;3:623. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Zhu D, Zhang Z, et al. EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. Oncogene 2020;39:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emran AA, Chatterjee A, Rodger EJ, et al. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends Immunol 2019;40:328–344. [DOI] [PubMed] [Google Scholar]
- 57.Hirukawa A, Smith HW, Zuo D, et al. Targeting EZH2 reactivates a breast cancer subtype-specific anti-metastatic transcriptional program. Nat Commun 2018;9:2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yomtoubian S, Lee SB, Verma A, et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep 2020;30:755–770.e6. [DOI] [PubMed] [Google Scholar]
- 59.Yi X, Guo J, Sun S, et al. EZH2-mediated epigenetic silencing of TIMP2 promotes ovarian cancer migration and invasion. Sci Rep 2017;7:3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chien YC, Liu LC, Ye HY, et al. EZH2 promotes migration and invasion of triple-negative breast cancer cells via regulating TIMP2-MMP-2/−9 pathway. Am J Cancer Res 2018;8:422–434. [PMC free article] [PubMed] [Google Scholar]
- 61.Justin N, Zhang Y, Tarricone C, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 2016;7:11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burr ML, Sparbier CE, Chan KL, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019;36:385–401.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harutyunyan AS, Krug B, Chen H, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun 2019;10:1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen KY, Bush K, Klein RH, et al. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun Biol 2020;3:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jain SU, Rashoff AQ, Krabbenhoft SD, et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohammad F, Weissmann S, Leblanc B, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 2017;23:483–492. [DOI] [PubMed] [Google Scholar]
- 67.Pathania M, De Jay N, Maestro N, et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017;32:684–700 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu YL, Gao X, Jiang Y, et al. Expression and clinicopathological significance of EED, SUZ12 and EZH2 mRNA in colorectal cancer. J Cancer Res Clin Oncol 2015;141:661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang CG, Ye YJ, Yuan J, et al. EZH2 and STAT6 expression profiles are correlated with colorectal cancer stage and prognosis. World J Gastroenterol 2010;16:2421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lima-Fernandes E, Murison A, da Silva Medina T, et al. Targeting bivalency de-represses Indian Hedgehog and inhibits self-renewal of colorectal cancer-initiating cells. Nat Commun 2019;10:1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017;543:676–680. [DOI] [PubMed] [Google Scholar]
- 72.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781–810. [DOI] [PubMed] [Google Scholar]
- 73.Muzny D, Bainbridge M, Chang K, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen JF, Luo X, Xiang LS, et al. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/β-catenin signaling. Oncotarget 2016;7:41540–41558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohuchi M, Sakamoto Y, Tokunaga R, et al. Increased EZH2 expression during the adenoma-carcinoma sequence in colorectal cancer. Oncol Lett 2018;16:5275–5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee CC, Chen WS, Chen CC, et al. TCF12 protein functions as transcriptional repressor of E-cadherin, and its overexpression is correlated with metastasis of colorectal cancer. J Biol Chem 2012;287:2798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Z, Yang P, Li W, et al. Expression of EZH2 is associated with poor outcome in colorectal cancer. Oncol Lett 2018;15:2953–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Böhm J, Muenzner JK, Caliskan A, et al. Loss of enhancer of zeste homologue 2 (EZH2) at tumor invasion front is correlated with higher aggressiveness in colorectal cancer cells. J Cancer Res Clin Oncol 2019;145:2227–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tamagawa H, Oshima T, Numata M, et al. Global histone modification of H3K27 correlates with the outcomes in patients with metachronous liver metastasis of colorectal cancer. Eur J Surg Oncol 2013;39:655–61. [DOI] [PubMed] [Google Scholar]
- 80.Flavahan WA, Drier Y, Johnstone SE, et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019;575:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnstone SE, Reyes A, Qi Y, et al. Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer. Cell 2020;182:1474–1489.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chiacchiera F, Rossi A, Jammula S, et al. Polycomb Complex PRC1 Preserves Intestinal Stem Cell Identity by Sustaining Wnt/β-Catenin Transcriptional Activity. Cell Stem Cell 2016;18:91–103. [DOI] [PubMed] [Google Scholar]
- 83.Tamburri S, Lavarone E, Fernández-Pérez D, et al. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol Cell 2020;77:840–856.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maynard MA, Ferretti R, Hilgendorf KI, et al. Bmi1 is required for tumorigenesis in a mouse model of intestinal cancer. Oncogene 2014;33:3742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yanai H, Atsumi N, Tanaka T, et al. Intestinal cancer stem cells marked by Bmi1 or Lgr5 expression contribute to tumor propagation via clonal expansion. Sci Rep 2017;7:41838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang Z, Bu X, Chen H, et al. Bmi-1 promotes the invasion and migration of colon cancer stem cells through the downregulation of E-cadherin. Int J Mol Med 2016;38:1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kundu S, Ji F, Sunwoo H, et al. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell 2018;71:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010;329:689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010;464:1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kogo R, Shimamura T, Mimori K, et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res 2011. ;71:6320–6. [DOI] [PubMed] [Google Scholar]
- 91.Wu ZH, Wang XL, Tang HM, et al. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol Rep 2014;32:395–402. [DOI] [PubMed] [Google Scholar]
- 92.Luo ZF, Zhao D, Li XQ, et al. Clinical significance of HOTAIR expression in colon cancer. World J Gastroenterol 2016;22:5254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y, Chen X, Liu J, et al. Long non-coding RNA HOTAIR knockdown enhances radiosensitivity through regulating microRNA-93/ATG12 axis in colorectal cancer. Cell Death Dis 2020;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Srinivasan PR, Borek E. Enzymatic Alteration of Nucleic Acid Structure. Science 1964;145:548–53. [DOI] [PubMed] [Google Scholar]
- 95.Kerachian MA, Javadmanesh A, Azghandi M, et al. Crosstalk between DNA methylation and gene expression in colorectal cancer, a potential plasma biomarker for tracing this tumor. Sci Rep 2020;10:2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao SG, Chen WS, Li H, et al. The DNA methylation landscape of advanced prostate cancer. Nat Genet 2020;52:778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luo H, Zhao Q, Wei W, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med 2020;12. [DOI] [PubMed] [Google Scholar]
- 98.Raut JR, Guan Z, Schrotz-King P, et al. Fecal DNA methylation markers for detecting stages of colorectal cancer and its precursors: a systematic review. Clin Epigenetics 2020;12:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–92. [DOI] [PubMed] [Google Scholar]
- 100.Bedford MT, van Helden PD. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res 1987;47:5274–6. [PubMed] [Google Scholar]
- 101.Lin CH, Hsieh SY, Sheen IS, et al. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res 2001;61:4238–43. [PubMed] [Google Scholar]
- 102.Kim YI, Giuliano A, Hatch KD, et al. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994;74:893–9. [DOI] [PubMed] [Google Scholar]
- 103.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011. ;43:768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berman BP, Weisenberger DJ, Aman JF, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 2011. ;44:40–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li R, Grimm SA, Mav D, et al. Transcriptome and DNA Methylome Analysis in a Mouse Model of Diet-Induced Obesity Predicts Increased Risk of Colorectal Cancer. Cell Rep 2018;22:624–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ehrlich M, Lacey M. DNA hypomethylation and hemimethylation in cancer. Adv Exp Med Biol 2013;754:31–56. [DOI] [PubMed] [Google Scholar]
- 107.Zhang W, Klinkebiel D, Barger CJ, et al. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen RZ, Pettersson U, Beard C, et al. DNA hypomethylation leads to elevated mutation rates. Nature 1998;395:89–93. [DOI] [PubMed] [Google Scholar]
- 109.Gaudet F, Hodgson JG, Eden A, et al. Induction of tumors in mice by genomic hypomethylation. Science 2003;300:489–92. [DOI] [PubMed] [Google Scholar]
- 110.Kanai Y, Ushijima S, Nakanishi Y, et al. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett 2003;192:75–82. [DOI] [PubMed] [Google Scholar]
- 111.Hur K, Cejas P, Feliu J, et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014;63:635–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mima K, Nowak JA, Qian ZR, et al. Tumor LINE-1 methylation level and colorectal cancer location in relation to patient survival. Oncotarget 2016;7:55098–55109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Rijnsoever M, Grieu F, Elsaleh H, et al. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut 2002;51:797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tapial S, Olmedillas-Lopez S, Rueda D, et al. Cimp-Positive Status is More Representative in Multiple Colorectal Cancers than in Unique Primary Colorectal Cancers. Sci Rep 2019;9:10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell 2013;153:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tse JWT, Jenkins LJ, Chionh F, et al. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer 2017;3:698–712. [DOI] [PubMed] [Google Scholar]
- 118.Guo X, Song C, Fang L, et al. FLRT2 functions as Tumor Suppressor gene inactivated by promoter methylation in Colorectal Cancer. J Cancer 2020;11:7329–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shima K, Nosho K, Baba Y, et al. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: Cohort study and literature review. Int J Cancer 2011;128:1080–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baba Y, Nosho K, Shima K, et al. Relationship of CDX2 loss with molecular features and prognosis in colorectal cancer. Clin Cancer Res 2009;15:4665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hryniuk A, Grainger S, Savory JG, et al. Cdx1 and Cdx2 function as tumor suppressors. J Biol Chem 2014;289:33343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Graule J, Uth K, Fischer E, et al. CDX2 in colorectal cancer is an independent prognostic factor and regulated by promoter methylation and histone deacetylation in tumors of the serrated pathway. Clin Epigenetics 2018;10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kandimalla R, Linnekamp JF, van Hooff S, et al. Methylation of WNT target genes AXIN2 and DKK1 as robust biomarkers for recurrence prediction in stage II colon cancer. Oncogenesis 2017;6:e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.de Sousa EMF, Colak S, Buikhuisen J, et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011;9:476–85. [DOI] [PubMed] [Google Scholar]
- 125.Kleeman SO, Koelzer VH, Jones HJ, et al. Exploiting differential Wnt target gene expression to generate a molecular biomarker for colorectal cancer stratification. Gut 2020;69:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu X, Chen X, Zeng K, et al. DNA-methylation-mediated silencing of miR-486-5p promotes colorectal cancer proliferation and migration through activation of PLAGL2/IGF2/beta-catenin signal pathways. Cell Death Dis 2018;9:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–38. [DOI] [PubMed] [Google Scholar]
- 128.Liu B, Nicolaides NC, Markowitz S, et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 1995;9:48–55. [DOI] [PubMed] [Google Scholar]
- 129.Sheaffer KL, Elliott EN, Kaestner KH. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev Res (Phila) 2016;9:534–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hu G, Qin L, Zhang X, et al. Epigenetic Silencing of the MLH1 Promoter in Relation to the Development of Gastric Cancer and its use as a Biomarker for Patients with Microsatellite Instability: a Systematic Analysis. Cell Physiol Biochem 2018;45:148–162. [DOI] [PubMed] [Google Scholar]
- 131.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A 1998;95:6870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 1998;95:8698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shan T, Uyar DS, Wang LS, et al. SOX11 hypermethylation as a tumor biomarker in endometrial cancer. Biochimie 2019;162:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaneda A, Feinberg AP. Loss of imprinting of IGF2: a common epigenetic modifier of intestinal tumor risk. Cancer Res 2005;65:11236–40. [DOI] [PubMed] [Google Scholar]
- 135.Tran TQ, Hanse EA, Habowski AN, et al. alpha-Ketoglutarate attenuates Wnt signaling and drives differentiation in colorectal cancer. Nat Cancer 2020;1:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sobhani I, Bergsten E, Couffin S, et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc Natl Acad Sci U S A 2019;116:24285–24295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang C, Wang L, Jin C, et al. Long non-coding RNA Lnc-LALC facilitates colorectal cancer liver metastasis via epigenetically silencing LZTS1. Cell Death Dis 2021;12:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Masalmeh RHA, Taglini F, Rubio-Ramon C, et al. De novo DNA methyltransferase activity in colorectal cancer is directed towards H3K36me3 marked CpG islands. Nat Commun 2021;12:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bian S, Hou Y, Zhou X, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018;362:1060–1063. [DOI] [PubMed] [Google Scholar]
- 140.Antelo M, Balaguer F, Shia J, et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS One 2012;7:e45357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hon GC, Hawkins RD, Caballero OL, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res 2012;22:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007;39:232–6. [DOI] [PubMed] [Google Scholar]
- 143.Ohm JE, McGarvey KM, Yu X, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007;39:237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Carvalho S, Freitas M, Antunes L, et al. Prognostic value of histone marks H3K27me3 and H3K9me3 and modifying enzymes EZH2, SETDB1 and LSD-1 in colorectal cancer. J Cancer Res Clin Oncol 2018;144:2127–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lam K, Pan K, Linnekamp JF, et al. DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim Biophys Acta 2016;1866:106–20. [DOI] [PubMed] [Google Scholar]
- 146.Wang Q, Chen X, Jiang Y, et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J Mol Cell Biol 2020;12:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fessler E, Medema JP. Colorectal Cancer Subtypes: Developmental Origin and Microenvironmental Regulation. Trends Cancer 2016;2:505–518. [DOI] [PubMed] [Google Scholar]
- 148.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016;22:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chiacchiera F, Rossi A, Jammula S, et al. Polycomb Complex PRC1 Preserves Intestinal Stem Cell Identity by Sustaining Wnt/beta-Catenin Transcriptional Activity. Cell Stem Cell 2016;18:91–103. [DOI] [PubMed] [Google Scholar]
- 150.Lopez-Arribillaga E, Rodilla V, Pellegrinet L, et al. Bmi1 regulates murine intestinal stem cell proliferation and self-renewal downstream of Notch. Development 2015;142:41–50. [DOI] [PubMed] [Google Scholar]
- 151.Jorgensen BG, Berent RM, Ha SE, et al. DNA methylation, through DNMT1, has an essential role in the development of gastrointestinal smooth muscle cells and disease. Cell Death Dis 2018;9:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu DH, Gadkari M, Zhou Q, et al. Postnatal epigenetic regulation of intestinal stem cells requires DNA methylation and is guided by the microbiome. Genome Biol 2015; 16:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Elliott EN, Sheaffer KL, Kaestner KH. The ‘de novo’ DNA methyltransferase Dnmt3b compensates the Dnmt1-deficient intestinal epithelium. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rivas MA, Beaudoin M, Gardet A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 2011;43:1066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhu L, Shi T, Zhong C, et al. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterology Res 2017;10:65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kiesler P, Fuss IJ, Strober W. Experimental Models of Inflammatory Bowel Diseases. Cellular and Molecular Gastroenterology and Hepatology 2015;1:154–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Monticelli LA, Osborne LC, Noti M, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 2015;112:10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Viazis N, Vlachogiannakos J, Georgiou O, et al. Course of inflammatory bowel disease in patients infected with human immunodeficiency virus. Inflammatory Bowel Diseases 2009;16:507–511. [DOI] [PubMed] [Google Scholar]
- 159.Shin B, Kress RL, Kramer PA, et al. Effector CD4 T cells with progenitor potential mediate chronic intestinal inflammation. J Exp Med 2018;215:1803–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hasler R, Feng Z, Backdahl L, et al. A functional methylome map of ulcerative colitis. Genome Research 2012;22:2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Glória L, Cravo M, Pinto A, et al. DNA hypomethylation and proliferative activity are increased in the rectal mucosa of patients with long-standing ulcerative colitis. Cancer 1996;78:2300–2306. [DOI] [PubMed] [Google Scholar]
- 162.Howell KJ, Kraiczy J, Nayak KM, et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells From Pediatric Patients With Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate With Outcome. Gastroenterology 2018;154:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang R, Li H, Wu J, et al. Gut stem cell necroptosis by genome instability triggers bowel inflammation. Nature 2020;580:386–390. [DOI] [PubMed] [Google Scholar]
- 164.Biton M, Haber AL, Rogel N, et al. T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 2018;175:1307–1320.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Neurath MF, Weigmann B, Finotto S, et al. The Transcription Factor T-bet Regulates Mucosal T Cell Activation in Experimental Colitis and Crohn’s Disease. Journal of Experimental Medicine 2002;195:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhou M, Ouyang W. The Function Role of GATA-3 in Th1 and Th2 Differentiation. Immunologic Research 2003;28:25–38. [DOI] [PubMed] [Google Scholar]
- 167.Agarwal S, Avni O, Rao A. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity 2000;12:643–52. [DOI] [PubMed] [Google Scholar]
- 168.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity 1998;9:765–75. [DOI] [PubMed] [Google Scholar]
- 169.Grogan JL, Mohrs M, Harmon B, et al. Early Transcription and Silencing of Cytokine Genes Underlie Polarization of T Helper Cell Subsets. Immunity 2001;14:205–215. [DOI] [PubMed] [Google Scholar]
- 170.Avni O, Lee D, Macian F, et al. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nature Immunology 2002;3:643–651. [DOI] [PubMed] [Google Scholar]
- 171.Takashima S, Martin ML, Jansen SA, et al. T cell-derived interferon-gamma programs stem cell death in immune-mediated intestinal damage. Sci Immunol 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Soskic B, Cano-Gamez E, Smyth DJ, et al. Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nature Genetics 2019;51:1486–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Huber S, Gagliani N, Esplugues E, et al. Th17 Cells Express Interleukin-10 Receptor and Are Controlled by Foxp3− and Foxp3+ Regulatory CD4+ T Cells in an Interleukin-10-Dependent Manner. Immunity 2011;34:554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Arvey A, Van Der Veeken J, Samstein RM, et al. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nature Immunology 2014;15:580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Li Q, Zou J, Wang M, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nature Communications 2014;5:5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Cribbs AP, Terlecki-Zaniewicz S, Philpott M, et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proceedings of the National Academy of Sciences 2020;117:6056–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.West MA, Heagy W. Endotoxin tolerance: a review. Crit Care Med 2002;30:S64–73. [PubMed] [Google Scholar]
- 178.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007;447:972–978. [DOI] [PubMed] [Google Scholar]
- 179.Allen J, Sears CL. Impact of the gut microbiome on the genome and epigenome of colon epithelial cells: contributions to colorectal cancer development. Genome Med 2019;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 2014;12:661–72. [DOI] [PubMed] [Google Scholar]
- 181.Chen J, Pitmon E, Wang K. Microbiome, inflammation and colorectal cancer. Semin Immunol 2017;32:43–53. [DOI] [PubMed] [Google Scholar]
- 182.Campbell C, McKenney PT, Konstantinovsky D, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020;581:475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Patnala R, Arumugam TV, Gupta N, et al. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol Neurobiol 2017;54:6391–6411. [DOI] [PubMed] [Google Scholar]
- 184.Alrafas HR, Busbee PB, Chitrala KN, et al. Alterations in the Gut Microbiome and Suppression of Histone Deacetylases by Resveratrol Are Associated with Attenuation of Colonic Inflammation and Protection Against Colorectal Cancer. J Clin Med 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Alrafas HR, Busbee PB, Nagarkatti M, et al. Resveratrol modulates the gut microbiota to prevent murine colitis development through induction of Tregs and suppression of Th17 cells. J Leukoc Biol 2019;106:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]