

Abstract
Purpose:
Multiple myeloma is a malignancy of plasma cells. Extensive genetic and transcriptional characterization of myeloma has identified subtypes with prognostic and therapeutic implications. In contrast, relatively little is known about the myeloma epigenome.
Experimental Design:
CD138+CD38+ myeloma cells were isolated from fresh bone marrow aspirate or the same aspirate after freezing for one to six months. Gene expression and chromatin accessibility were compared between fresh and frozen samples by RNA-seq and ATAC-seq. Chromatin accessible regions were used to identify regulatory RNA expression in over 700 samples from newly diagnosed patients in the MMRF CoMMpass trial (NCT01454297).
Results:
Gene expression and chromatin accessibility of cryopreserved myeloma recapitulated that of freshly isolated samples. ATAC-seq performed on a series of biobanked specimens identified thousands of chromatin accessible regions with hundreds being highly coordinated with gene expression. Over 4,700 of these chromatin accessible regions were transcribed in newly diagnosed myelomas from the CoMMpass trial. Regulatory element activity alone recapitulated myeloma gene expression subtypes, and in particular myeloma subtypes with IGH translocations were defined by transcription of distal regulatory elements. Moreover, enhancer activity predicted oncogene expression implicating gene regulatory mechanisms in aggressive myeloma.
Conclusions:
These data demonstrate the feasibility of using biobanked specimens for retrospective studies of the myeloma epigenome and illustrate the unique enhancer landscapes of myeloma subtypes that are coupled to gene expression and disease progression.
Introduction
Multiple myeloma is a malignancy of differentiated B cells, known as plasma cells, that secrete high levels of immunoglobulin. Tremendous progress in the treatment and management of myeloma has been made over the past 25 years resulting in a doubling of life expectancy with a 5-year survival rate now well above 50% (1,2). These improved outcomes are largely due to the introduction of novel therapeutic agents and regimens that target plasma cell biology rather than myeloma-specific mutations (3,4). Nonetheless, over 32,000 people are diagnosed with myeloma in the U.S. every year – and despite improved outcomes and extended remissions – the majority will develop disease that is refractory to treatment and thus incurable (2).
The genetic basis of multiple myeloma has been extensively studied leading to a molecular classification with prognostic implications (5–7). Approximately half of myelomas have translocations that juxtapose strong immunoglobulin heavy chain (IGH) enhancers proximal to one of the three cyclin D genes (CCND1–3), the histone methyltransferase NSD2 (also known as MMSET, WHSC1), or one of the transcription factors and proto-oncogenes MAF, MAFB, or MAFA (8–10). Myelomas without IGH translocations most often have trisomies of odd-numbered chromosomes referred to as hyperdiploidy (11). Almost all myelomas also have secondary genetic events such as copy number alterations commonly including del(13p) (12) and amp(1q) (13,14), structural rearrangements of MYC (15–18), translocations of either immunoglobulin light chain Kappa (IGK) or Lambda (IGL) (19), inactivation of TP53 (20), mutations in common and disease-specific oncogenes (21), and complex structural rearrangements (22). These genetic alterations partially underlie distinct gene expression subtypes that reflect the biology and sometimes the disease course of myeloma (23,24). However, genetic alterations fail to fully explain the high-risk proliferation gene expression subtype and miss a substantial number of myeloma patients that experience poor outcomes.
Epigenetic dysregulation including aberrant DNA methylation and altered histone modifications has been implicated in myeloma for many years (25–28). The most well studied example stems from t(4;14) translocations that result in IGH enhancer-driven NSD2 overexpression and excessive histone 3 lysine 36 dimethylation (9,29–31). While t(4;14) translocations are a marker of high-risk disease (5,6), the prognostic implications of other epigenetic alterations are less well understood.
One challenge that has hindered epigenetic studies of primary myeloma is the use of assays that require large amounts of specimen. For example, ChIP-seq is often performed using millions of cells. The assay for transpose accessible chromatin-sequencing (ATAC-seq) overcomes this limitation by identifying regions of chromatin accessibility using the Tn5 transposase on small numbers of cells (32). Still, intermittent acquisition of samples makes it difficult to plan and conduct large epigenetic studies. Thus, an approach that allows for the use of biobanked samples would facilitate retrospective studies of the myeloma epigenome. To this end, we recently showed that cryopreserved human peripheral B cells could be used to faithfully characterize the accessible chromatin profile from limiting cell numbers (33). We therefore sought to determine whether such a technique is amenable to multiple myeloma cells from cryopreserved bone marrow aspirates.
Here, we compared RNA-seq and ATAC-seq from CD138+CD38+ myeloma specimens obtained from both fresh and biobanked bone marrow aspirates. Myeloma cells isolated from cryopreserved aspirates recapitulated both the chromatin accessibility and mRNA characteristics of those obtained from fresh samples. We identified hundreds of cis-regulatory elements where accessibility was predictive of gene expression and confirmed these in an independent cohort. Chromatin accessible regions were combined with H3K27ac ChIP-seq data to interrogate transcription of intergenic regulatory elements using RNA-seq data from 768 newly diagnosed patients from the Multiple Myeloma Research Foundation (MMRF) CoMMpass trial. These data identified over 4,700 active regulatory elements that reflected myeloma subtype and identified a program of transcribed enhancers in patients with poor outcome.
Materials and Methods
Myeloma sample isolation
Written patient informed consent was obtained and collection of multiple myeloma bone marrow aspirates followed approved protocols from the Emory University Institutional Review Board and comply with the Belmont Report and the U.S. Common Rule. Processing bone marrow aspirates has previously been described (34). Specifically, bone marrow aspirates were enrichment for mono-nuclear cells by Ficoll gradient centrifugation by first diluting aspirates to 25 mL in PBS and carefully pipetting 10 mL of Ficoll lymphocyte separation medium (Corning 25–072-Cl) under the aspirate prior to centrifugation for 30 minutes at 400 g with no deceleration resistance. The mono-nuclear cell layer (“buffy coat”) was collected by graduated transfer pipette (Fisherbrand 13–711-9AM), washed in PBS and re-suspended in RPMI-1640 (Corning #14–030-CV) with 10% FBS (GeminiBio 97068-085), 1% Pen-strep (Corning 30-002-CI), 1% L-glutamine (Corning 25005CI), and 1% HEPES (Corning 25–060-CI).
After processing, bone marrow aspirate cells were either cryopreserved or analyzed within 6 hours. Cryopreservation used 1–5 million cells per mL in 10% DMSO followed by immediately cooling to −80°C buffered by isopropyl alcohol (Thermo Scientific 51000001) and transfer to −140°C liquid nitrogen freezers within 1–4 days. Both fresh and cryopreserved samples were stained with anti-CD138-FITC (Becton Dickinson 552723), anti-CD38-BV450 (Becton Dickinson 561378), anti-CD45-APC-Cy7 Becton Dickinson 348795), and propidium iodide (PI; Sigma-Aldrich P4170) prior to analysis and isolation on a FACS Aria II (Becton Dickinson) (Supplementary Fig. S1).
mRNA-seq
mRNA-seq analysis was performed similarly to previously described (35). 50,000 cells were FACS isolated directly into 600 μL of RLT lysis buffer with 1% β-ME (Sigma-Aldrich M6250) prior to vortexing 1 min at max speed and freezing in a dry ice ethanol mixture. RNA was isolated (Qiagen RNeasy Mini 74104) and quality assessed using an Agilent BioAnalyzer. Stranded mRNA-seq libraries were made using the mRNA HyperPrep kit (Kapa 08098115702) following the manufacturer’s protocol and using custom short TruSeq compatible sequencing adapters (IDT) (Supplementary Table S1) (36). Barcodes were added to each library using long PCR primers (Supplementary Table S1) during 8 cycles of PCR amplification. Samples were sequenced on a HiSeq 4000 (Illumina).
mRNA-seq processing
FASTQ sequencing files were quality and adapter trimmed using Trim Galore! (v0.6.4; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and CutAdapt (v2.5; https://github.com/marcelm/cutadapt/) prior to mapping the GRCh37 genome accounting for the Ensembl GRCh37.74 transcription database using the STAR aligner (v2.5.3a) (37). Putative PCR duplicate reads were marked in BAM files using Samtools (v1.7) (38). Gene read counts were calculated in R (v3.6.2) (39) by reading in BAM files using the function readGAlignmentPairs and counting the number of reads that overlap any exonic region of an Ensemble (v74) gene using the function summarizeOverlaps of the GenomicAlignments package (v1.2) (40). Gene expression data was normalized for sequencing depth using reads per million (RPM) or fragments per kilobase per million reads (FPKM) where read depth was the total number of reads on autosomal chromosomes only (Supplementary Data S6).
mRNA-seq analysis
Genes that did not have ≥1 FPKM in expression in at least one sample were removed from downstream analyses as not expressed. Principle component analysis used the R function prcomp of the stats package and using log2(FPKM+1) gene expression values from autosomes only. Hierarchical clustering used the hclust function also of the stats package as well as a Euclidean distance dissimilarity metric of the log2(FPKM+1) expression values again from only the autosomes. Plots of genomic regions used rtracklayer (v1.44.4) (41) to read in summarized files and custom R code to plot sequencing depth.
ATAC-seq
ATAC-seq was performed similarly to previously described (33,42). Here, 20,000 CD138+CD38+PI- viable myeloma cells were sorted into PBS and kept on ice. Cells were isolated by centrifugation at 500 × g for 10 minutes at 4°C and the supernatant was carefully removed by pipet. Cells were resuspended in 50 μL of nuclei-lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3mM MgCl2, 0.1% IGEPAL) prior to centrifugation at 500 × g for 30 minutes at 4°C and careful removal of nuclei-lysis buffer by pipet. Nuclei were resuspended in 25 μL of tagmentation mix consisting of 10.5 μL H2O, 12.5 μL TD Buffer (Illumina) and 2 μL Tn5 transposase (Illumina) and incubated at 37 °C for 1 hour. After tagmentation, DNA was isolated by digesting with 20 mg Proteinase K for 1 hour in 25 μL of tagmentation clean-up buffer (326 mM NaCl, 109 mM EDTA, 0.63% SDS). High molecular weight DNA was excluded by 0.6 × volume negative selection with SPRI beads (Kapa Biosystems) followed by a 1.2 × SPRI bead positive selection, which was repeated twice. ATAC libraries were amplified for 12 PCR cycles using Nextera adapters (Illumina) prior to sequencing on an Illumina HiSeq 4000.
ATAC-seq processing
ATAC-seq FASTQ files were quality and adapter trimmed using Trim Galore! (v0.6.4; https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and CutAdapt (v2.5; https://github.com/marcelm/cutadapt/) prior to mapping the GRCh37 genome using bowtie2 (v2.2.6) for alignment (43). Aligned SAM files were converted to BAM files and putative PCR duplicates were marked with Samtools (v1.7) (38). Regions of chromatin accessibility were identified in each sample using MACS2 (v2.1.0.20151222) (44). The union of all accessible regions was determined and reads mapping into these regions were determined for each sample using the summarizeOverlaps of the GenomicAlignments package (v1.2) in R (v3.6.2). Regions that overlapped ENCODE blacklisted regions were removed (45). As a quality control metric the number of reads in peaks was determined and used to calculate a normalized accessibility score as reads per peak million (RPPM) as previously described (42) according to the following formula:
ATAC-seq data is listed Supplementary Data S7.
ATAC-seq analysis
The chromatin accessibility signal was log2(RPPM+1) transformed and used for PCA analysis using the prcomp function in R, hierarchical clustering using the hclust function also in R, as well as Pearson correlation comparisons. Chromatin stretch enhancer analysis was performed as previously described (46). Briefly, regions of chromatin accessibility that did not overlap promoters (>2,500 bp from the TSS) but were within 12,500 bp of each other were stitched together. The RPPM normalized read count of these stretch regions was determined for all samples and they were ranked by the average RPPM signal with regions past the inversion point being considered ‘super enhancers’. Gene ontology analysis of genes most proximal to these regions was performed using the GOstats (v2.50.0) R packages to determine enriched biological processes (47).
ATAC-seq correlation with mRNA-seq
The edgeR package (v3.26.8) (48) was used to correlate regions of chromatin accessibility with the 5% most variably expressed genes between samples. Here, a ‘digital gene expression’ list (DGEList) using read counts for all ATAC-seq autosomal regions of chromatin accessibility was used to estimate dispersion in edgeR. Next, for each gene the DGEList object was subsetted for regions within 100 kb of the transcribed region of the gene and ATAC-seq reads were correlated with the log2(FPKM+1) gene expression using the glmQLFit and glmQLFTest functions. Data were compiled for all genes tested and P-values were corrected for multiple hypothesis testing using a Benjamini-Hochberg FDR correction.
Replication of myeloma ATAC-seq and mRNA-seq and meta-analysis of ChIP-seq data
mRNA-seq, ATAC-seq, and H3K27ac ChIP-seq data from Jin et al. (49) was downloaded from the European nucleotide archive for project PRJEB25605 (https://www.ebi.ac.uk/ena/data/search?query=PRJEB25605). mRNA-seq and ATAC-seq data were processed as described above in a harmonized fashion as the data we generated. ChIP-seq data were processed as described for ATAC-seq data above except normalization used RPM instead of RPPM.
Analysis of CoMMpass RNA-seq and regulatory element transcription
Access to CoMMpass data was granted through the dbGaP data access committee and data was downloaded from project (phs000748.v7.p4). Processing of CoMMpass was as previously described (19) and used the same reference genome to harmonize the data.
Regulatory element regions were defined as the union of overlapping regions with H3K27ac enrichment in any primary myeloma sample from Jin et al. (49) and regions showing chromatin accessibility for any primary myeloma sample analyzed here. Regulatory element regions were trimmed so that they did not overlap any region within 500 bp upstream of a TSS to 5 kb downstream of the TTS. Regulatory element transcription was determined by counting the number of CoMMpass RNA-seq reads using the countOverlaps function of the GenomicRanges (v1.36.1) package in R. To discriminate signal from noise, regulatory element transcription for each region in each sample was compared to the transcription detected from that same sized region shuffled 1,000 times across the genome following the same rules (i.e. cannot overlap regions 500 bp upstream of TSS to 5 kbp downstream of TTS). Detection of regulatory element transcription was defined as regions that had more reads in the actual regulatory region as compared to the shuffled regions (P ≤0.01).
Gene set enrichment analysis of regulatory element transcription used GSEA (v4.03) and the hallmarks MSigDb gene set (v7.0) (50). Here, the regulatory elements were annotated to the closest gene and the GSEA Preranked gene set option was used based on the –log10(P-value) × sign(odds ratio) where P-value and odds ratio were determined by Fisher’s exact test of enhancer expression based on the number of samples in which transcription of the regulatory element was detected.
Correlation of regulatory element transcription and proximal mRNA expression used edgeR (v3.26.8) as described in the “ATAC-seq correlation with mRNA-seq” methods section above. Gene expression subtypes of CoMMpass data were determined by consensus clustering as previously described (19) using the ConsensusClusterPlus package (v1.48.0) (51). t-SNE analysis of CoMMpass mRNA and enhancer transcription used the Rtsne package (v0.15) (52) based on the log2(FPKM+1) expression values.
Gene expression prognostic of outcome was determined using a cox proportional hazard regression implemented using the R function ‘coxph’ of the survival package (v3.1–8). Here, the log2(FPKM+1) expression values for each gene were regressed on censored survival data from CoMMpass interim analysis 15. P-values from the cox proportional hazards Wald test were corrected for multiple hypothesis correction using a Benjamini-Hochberg FDR.
Data Access
RNA-seq and ATAC-seq raw data as well as summarized data have been deposited in the Gene Expression Omnibus under access GSE167969.
Results
mRNA content and chromatin accessibility are preserved in viable frozen myeloma specimens
To determine the feasibility of performing transcriptional and epigenetic studies on biobanked myeloma specimens, we compared CD138+CD38+ myeloma cells obtained from fresh bone marrow aspirates to those obtained from cryopreserved bone marrow cells from the same aspirate (see methods). Both fresh and cryopreserved cells were isolated by fluorescence activated cell sorting (FACS) to obtain viable CD138+CD38+ cells (Supplementary Fig. S1A). Cryopreserved bone marrow aspirates had reduced overall viability relative to fresh isolates (Supplementary Fig. S1B), but CD138+CD38+ myeloma cells were obtained in both cases (Supplementary Fig. S1C).
RNA-seq was performed on FACS-isolated myeloma cells from both fresh and frozen bone marrow aspirates for three patients. Principal component analysis (PCA) and hierarchical clustering of RNA expression indicated a strong similarity between fresh and frozen samples from the same patient as compared to samples from other patients (Fig. 1A, Supplementary Fig. S2A). In parallel, we also isolated 20,000 CD138+CD38+ myeloma cells for ATAC-seq. Regions of chromatin accessibility for each sample were determined and normalized reads in all accessible regions were calculated (RPPM: reads per peak million). Similar to the RNA expression, both PCA and hierarchical clustering of chromatin accessibility data indicated that fresh and frozen samples from each patient were very similar to each other but distinct from specimens from other patients (Fig. 1B, Supplementary Fig. S2B). Pairwise comparison of both gene-specific expression and chromatin accessibility levels for all six samples indicated that fresh and frozen samples from the same patient consistently had a higher Pearson correlation coefficient (R) as opposed to inter-patient comparisons (Supplementary Fig. S2C–D). RNA-seq at the immunoglobulin light chains IGK and IGL reflected the clonal nature of myeloma where both fresh and frozen specimens from the same patient expressed variable, joining (J), and constant (C) regions from either IGK (1563) or IGL (1557 and 1562), but not both (Supplementary Fig. S2E, top and middle) – a phenomenon known as light chain restriction. Additionally, both fresh and frozen samples for each patient showed consistent expression of a unique immunoglobulin heavy chain (IGH) variable, diversity (D), joining (J), and constant (C) chain (Supplementary Fig. S2E).
Figure 1.
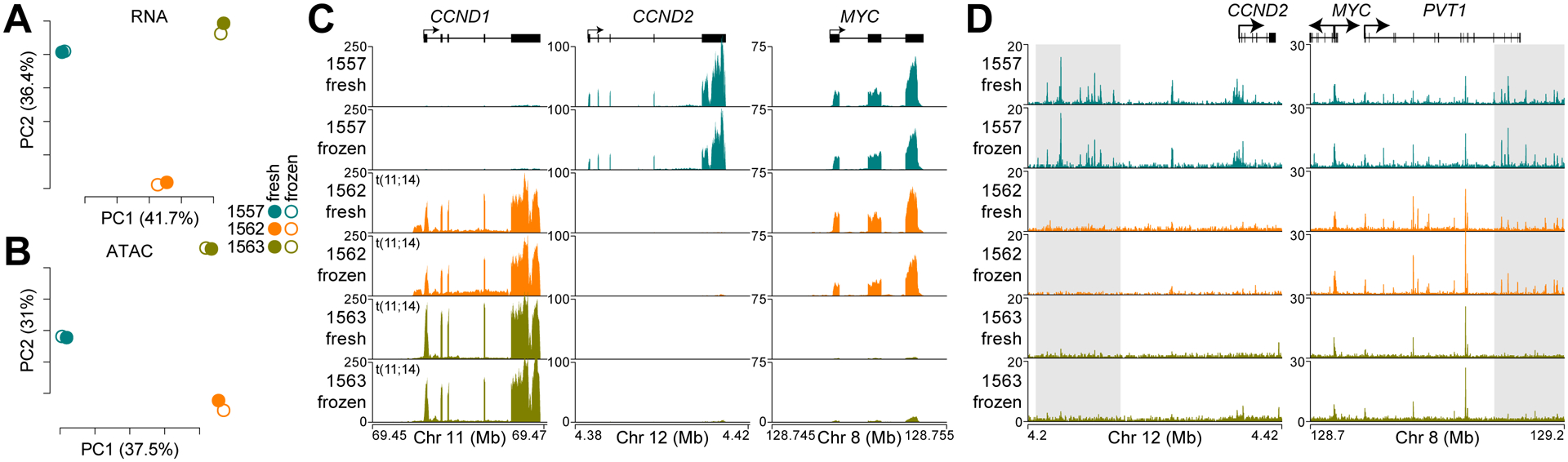
The mRNA content and chromatin accessibility of myeloma is maintained in cryopreserved specimens. A-B, Principle component analysis of mRNA expression (A) and chromatin accessibility (B). Principle components (PC) 1 and 2 are shown with the percent of variation explained by each component denoted in parentheses. C, mRNA-seq reads for CCND1, CCND2, and MYC genes. Note that specimens 1562 and 1563 were FISH positive for t(11;14). D, ATAC-seq data for fresh and frozen replicates at CCND2 (left) and MYC (right). Gray shading denotes regions of differences between the samples. The scale is either reads per million (RPM; C), log2(RPM+1) (A), reads per peak million (RPPM; D) or log2(RPPM+1) (B).
Inspection of the myeloma oncogene CCND1 showed that two of the patients expressed very high levels and this was consistent in both fresh and frozen specimens (Fig. 1C, left). Fluorescence in-situ hybridization data indicated that both patients had a t(11;14) translocation that juxtaposes the strong IGH enhancer(s) to the CCND1 locus (8). The specimens from patient 1557 did not express high levels of CCND1, but did express high levels of CCND2 – which is consistent with previous reports that myeloma aberrantly express one of the Cyclin D genes (Fig. 1D, middle) (53). Additionally, inspection of the MYC oncogene showed that it was highly expressed in two of the patients and this was consistent between fresh and frozen specimens (Fig. 1D). We queried chromatin accessibility near CCND2 and MYC to gain insight into the regulation of these oncogenes. This showed that the ATAC-seq signal was similar between fresh and frozen samples for all patients, but distinct from samples from other patients as well as identifying regions of chromatin accessibility that corresponded with CCND2 and MYC expression (Fig. 1D, see shaded regions). Overall, these data indicate that the mRNA content and chromatin accessibility state are preserved in viably frozen cells.
cis-regulatory elements predict gene expression in myeloma.
To better understand the distinct cis-regulatory elements coordinating gene expression in multiple myeloma we performed paired RNA-seq and ATAC-seq on eight cryopreserved myeloma specimens. This identified 91,632 autosomal regions of chromatin accessibility that were present in at least one sample (Fig. 2A). The largest regions of chromatin accessibility were cataloged by calculating the number of ATAC-seq reads in stretches of accessible regions that were within 12.5 kb of each other – similar to the “super-enhancer” analyses performed on H3K27ac or MED1 ChIP-seq data (46). Cumulatively across all the samples, the regions with the largest stretches of accessible chromatin were found at several myeloma lineage-defining genes including IRF4, CD38, SLAMF7, IGH, IGL, and MYC (Fig. 2B). Annotation of genes proximal to these large regions of chromatin accessibility indicated functions in RNA biosynthesis, nucleotide metabolism, transcription, and protein modification (Fig. 2C) – processes required to support the copious amounts of immunoglobulin synthesis and growth that are hallmarks of multiple myeloma. Inspection of IRF4, CD38, and SLAMF7 loci showed that samples shared the same regions of chromatin accessibility (Fig. 2D).
Figure 2.
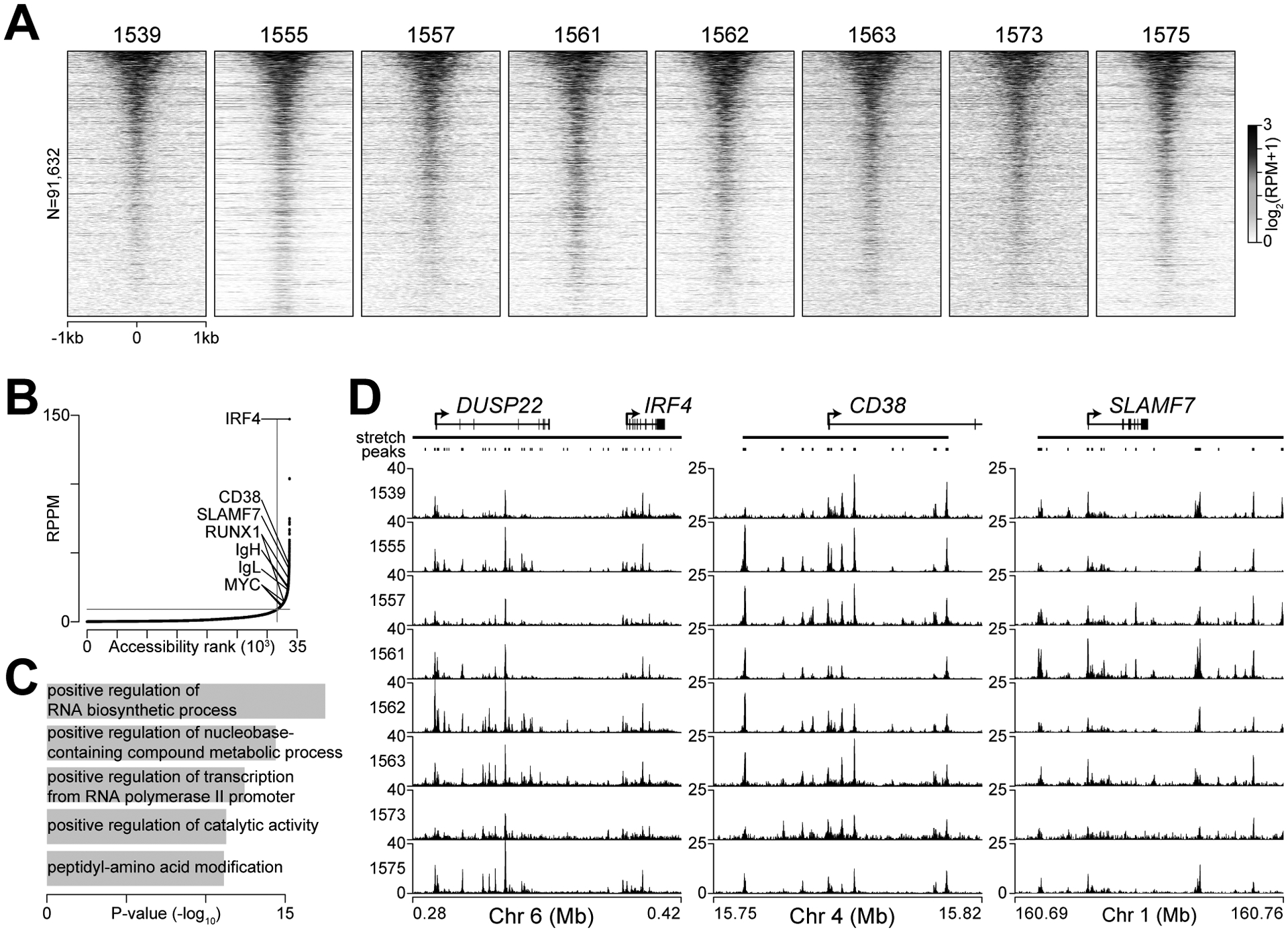
Chromatin accessibility clusters at highly expressed genes in myeloma. A, Heatmap of ATAC-seq data at 91,632 chromatin accessible autosomal regions in eight CD138+CD38+ myeloma specimens from bone marrow aspirates. Accessible regions were sorted from most (top) to least (bottom) accessible. B, ATAC-seq signal (reads per peak million; RPPM) in stretch regions of chromatin accessibility C, Top 5 gene ontology biological processes for genes proximal to stretch regions of chromatin accessibility. D, Genome plot of stretch regions of chromatin accessibility at IRF4, CD38, and SLAMF7 shown for all eight samples.
To identify gene regulatory elements, we correlated chromatin accessibility to expression for regions within 100 kb of a gene. Here, we focused on the most variably expressed genes in our cohort (top 5%), which also showed significantly higher variation in expression across samples in larger studies (Supplementary Fig. S3A–B). For example, there were four regions of chromatin accessibility within 100 kb of TLR1 (Fig. 3A), but only the most distal region showed a significant correspondence between chromatin accessibility and TLR1 expression (Fig. 3A–B, see red arrow). Cumulatively, 16,822 comparisons identified 833 regions where chromatin accessibility significantly correlated with expression of 376 genes (FDR ≤0.05; Supplementary Data S1) including several genes that have been implicated in the pathology of myeloma such as CCND2, FRZB, GSTT1, MS4A1 (encodes CD20), NCAM1, PAX5, RRAS2, and WNT5A (Supplementary Fig. S4). These results were confirmed in an independent data set by repeating these analyses using RNA-seq and ATAC-seq data from 18 myeloma samples from Jin et al. (49). This resulted in 1,190 regions that correlated with proximal gene expression, including 207 of those found in our cohort (Supplementary Fig. S3C). The composition of the different study populations and modest sample sizes may account for some of the differences observed, but it is important to note that there was a significant overlap of regions where chromatin accessibility predicted gene expression between the two studies (P=8.4×10−62, Fisher’s exact test).
Figure 3.
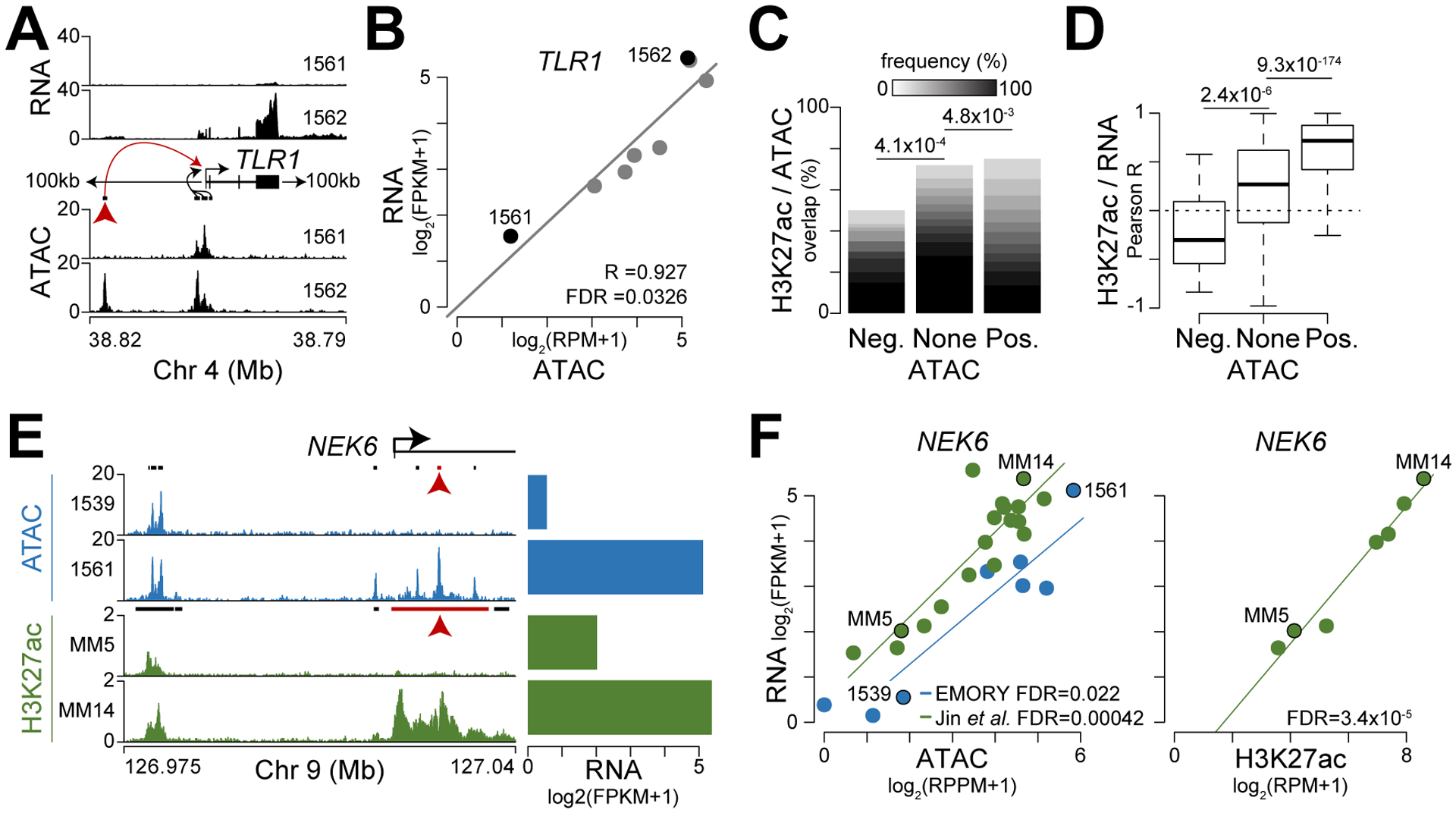
Chromatin accessibility and H3K27ac correspond with gene expression. A, Schematic of analysis where RNA expression (top) was correlated with regions of chromatin accessibility (ATAC; bottom) within 100 kb of the top 5% of variably expressed genes (see Supplementary Figure S3). B, Correlation of ATAC and RNA shown for the region identified by the red arrow in part A. C, Frequency of H3K27ac overlap and accessible regions that are negatively (Neg.) or positively (Pos.) associated with gene expression. The frequency of patients with overlapping H3K27ac enriched regions are shown in a gray scale and P-values for significant differences are shown on top (Fisher’s exact test). D, Correlation (Pearson R) of H3K27ac level and proximal gene expression for regions that overlap chromatin accessibility which is negatively or positively associated with gene expression. P-values for significant differences in correlation distribution are shown on top (Mann Whitney U-test). E, cis-regulatory elements identified upstream of the promoter and in the first intron (see red arrow) of NEK6 using both chromatin accessibility (top; blue) and H3K27ac from Jin et al. (bottom; green). The scale is RPPM (ATAC) or RPM (ChIP-seq) and RNA expression is shown (right). F, Scatterplot of chromatin accessibility and gene expression (left) or H3K27ac enrichment and gene expression (right) for the loci shown in part E (see red arrow) with samples from Emory (blue) and Jin et al. (green) denoted by color.
To understand what discriminates regions of chromatin accessibility that correspond with gene expression from those that do not, we assessed the genomic overlap with the activating modification histone 3 lysine 27 acetylation (H3K27ac) using ChIP-seq data from primary myeloma samples (49). Regions where chromatin accessibility negatively correlated with gene expression were depleted of H3K27ac and conversely those regions where accessibility positively corresponded with gene expression were enriched for H3K27ac as compared to regions not indicative of gene expression (Fig. 3C). Furthermore, H3K27ac enrichment negatively correlated with expression at regions where accessibility also negatively corresponded with expression, and H3K27ac positively correlated with expression at regions where chromatin accessibility did as well (Fig. 3D). For example, distinct regions of chromatin accessibility and H3K27ac were present both upstream of the promoter and in the first intron of the mitosis related kinase NEK6 (Fig. 3E, left). However, in the two ATAC and ChIP-seq samples shown only the intronic enhancer was strongly correlated with expression (Fig. 3E, see red arrow), and this was generally true across all the samples analyzed (Fig. 3F). Cumulatively, these data identify a set of cis-regulatory elements implicated in myeloma gene regulation.
Transcription of regulatory elements reflects myeloma gene expression subtype
We sought to expand this analysis by interrogating samples from the CoMMpass trial (NCT01454297), a longitudinal study of over 1,000 myeloma patients. Since epigenetic data have yet to be included in CoMMpass, we used RNA-seq data available for 768 samples to interrogate transcription of regulatory elements, a phenomenon linked to active enhancers (54,55). Here, the overlapping regions of chromatin accessibility and H3K27ac (49) defined above were assessed for transcription using CoMMpass RNA-seq. To avoid contamination from exonic mRNAs or intronic pre-mRNAs, intragenic regions were removed as well as 500 bp upstream of TSSs and 5 kb downstream of transcription termination sites. This identified 13,452 putative intergenic regulatory elements to query for transcription (Supplementary Data S2). To distinguish signal from noise we compared the RNA-seq reads at each regulatory element for each sample to the same size region shuffled 1,000 times across the genome (see methods). Thus, detection of transcription could be determined for each regulatory element in each sample by comparing the actual number of reads with those obtained from the permuted regions. Plotting the frequency of detected RNA for each regulatory element showed an asymmetrical distribution (Fig. 4A) with some that were consistently transcribed as well as those that were only transcribed in a subset of samples. In total, transcription was detected at 4,729 regulatory elements in 5% or more of samples. Gene set enrichment analysis of the genes closest in proximity to the most commonly transcribed regulatory elements indicated the top enriched gene set was protein secretion (Fig. 4B, Supplementary Data S3), which is consistent with the primary function of plasma cells from which myelomas are derived. As an example, there are three regions between PIK3C2B and MDM4 that contain overlapping H3K27ac and chromatin accessibility as well as evidence of transcription in both our cohort (generated using stranded RNA-seq) and that from CoMMpass (Fig. 4C, left – see shaded areas). Notably, paired-end RNA-seq reads from the middle enhancer region were all contained within that transcribed element indicating that it is not spliced with MDM4 or some other gene, but rather is a distinct transcriptional unit. The stranded RNA-seq data also indicated that the two promoter proximal transcribed regions near PIK3C2B and MDM4 are on the antisense strand respective to each gene, and are thus not part of the canonical gene, but likely represent bi-directional transcription. Conversely, inspection of the FOS locus (Fig. 4C, right) shows a regulatory region immediately upstream of the FOS TSS that is transcribed on the same strand as FOS. Importantly, paired-end RNA-seq reads did not span this promoter proximal cis-regulatory region with those coming from FOS, again indicating that this is a distinct transcriptional product.
Figure 4.
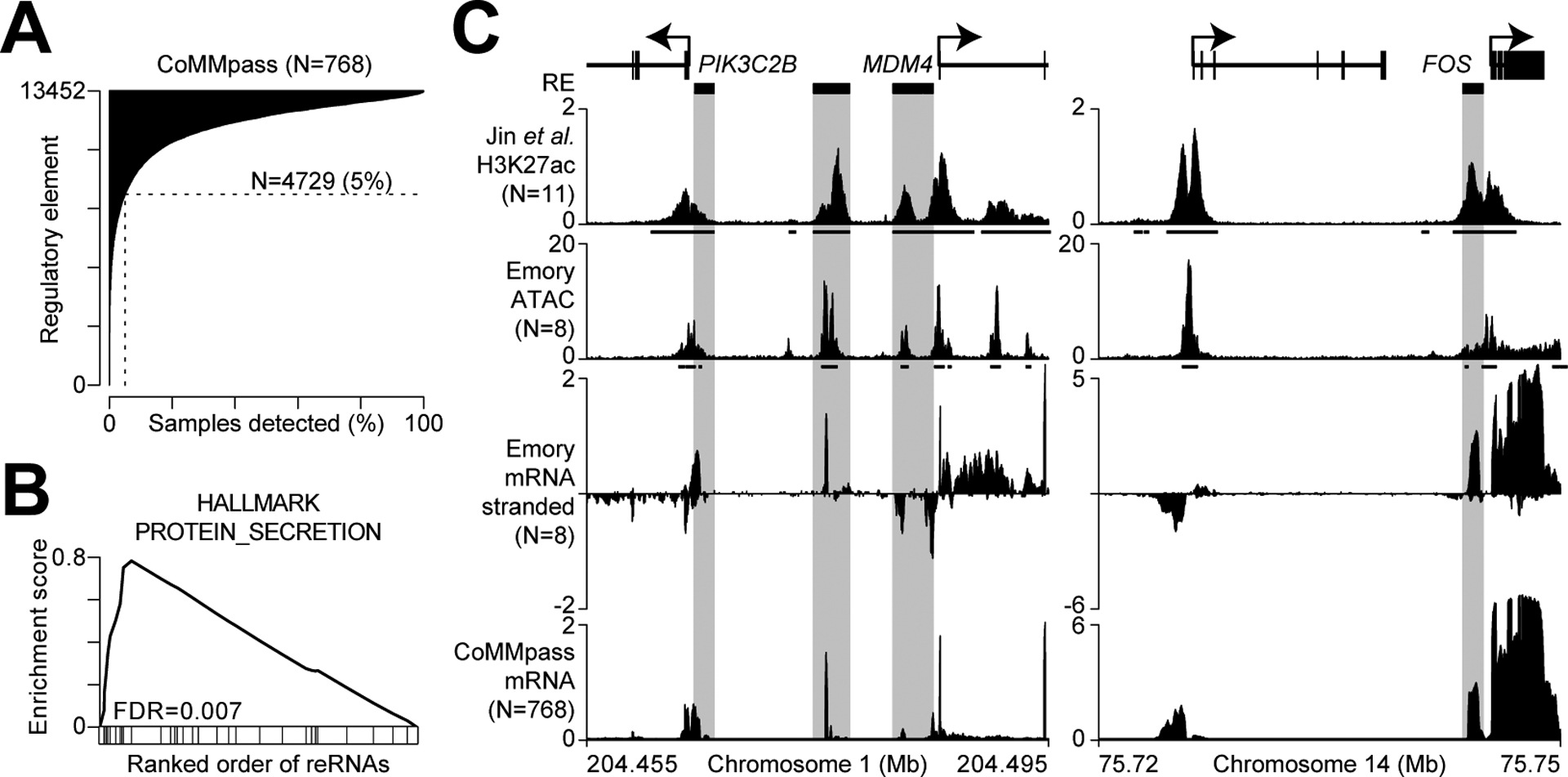
Regulatory element transcription reflects the myeloma gene expression program. A, Frequency of detected transcription at 13,452 regulatory elements (RE) in 768 newly diagnosed myeloma patients from the CoMMpass study. Detection for each sample and regulatory element is determined based on the actual signal compared to 1,000 permutated enhancers (P ≤0.01; see methods). B, Gene set enrichment analysis of the top pathway enriched at genes proximal to transcribed regulatory elements. C, Genome plot of the PIK3CB and MDM4 locus (left) and the FOS locus (right) with regulatory elements (grayed boxes) defined as overlapping regions with both H3K27ac ChIP-seq (from Jin et al.) and chromatin accessibility (ATAC) that are 500 bp from the TSS and 5 kb from TTS. Both stranded mRNA-seq data (Emory) and unstranded mRNA-seq data (CoMMpass) are shown. The scale for ChIP-seq is reads per million (RPM), ATAC-seq is reads per peak million (RPPM), and mRNA-seq is log2(RPM+1), all tracks represent a composite of all samples analyzed.
Distal regulatory elements define specific myeloma subtypes
The above examples at PIK3C2B, MDM4, and FOS suggest that these regulatory regions are composed of both promoter proximal and distal elements, with the latter more likely representing enhancer elements. To understand how these distinct active regulatory elements were coupled to gene expression, we used previously defined gene expression subtypes that reflect key oncogenic drivers and genomic alterations in myeloma (19). T-distributed stochastic neighbor embedding (t-SNE) analysis was performed for gene expression data as well as for transcription at both promoter proximal (≤2500 bp of TSS) and distal regulatory elements with each sample colored by gene expression subtype (Fig. 5A). As expected, t-SNE analysis of gene expression data grouped expression subtypes together. Additionally, both sets of regulatory elements tended to group myeloma subtypes together, however the promoter proximal elements separated the MMSET (MS), CCND (CD), and MAF (MF) subsets into multiple clusters (Fig. 5A, middle – see red, gold, and turquoise colors, respectively), whereas the distal regulatory elements clustered these subsets with other samples from the same subset (Fig. 5A, right). Indeed, quantifying the t-SNE distances between all samples within a given group showed that the average distance between MMSET, CCND, and MAF samples was significantly less when using distal regulatory elements as opposed to promoter proximal elements (Fig. 5B). To a lesser extent this is also observed in the Proliferation (PR) and low-bone disease (LB) groups. Subsequently, we determined uniquely transcribed regulatory elements for each myeloma subtype and focused on several hundred elements that distinguished the MMSET and MAF subtypes from others (Supplementary Data S3). Integrating these data with expression of genes within 1 MB identified distal regulatory elements highly predictive of genes uniquely regulated in the MMSET and MAF subtypes. For instance, a regulatory element ~154 kb upstream of CCND2 was uniquely transcribed in the MAF subtype and this was highly correlated with MAF expression (Fig. 5C–D). These data identify active distal regulatory elements that are coordinately regulated with the unique gene expression programs of myeloma subtypes.
Figure 5.
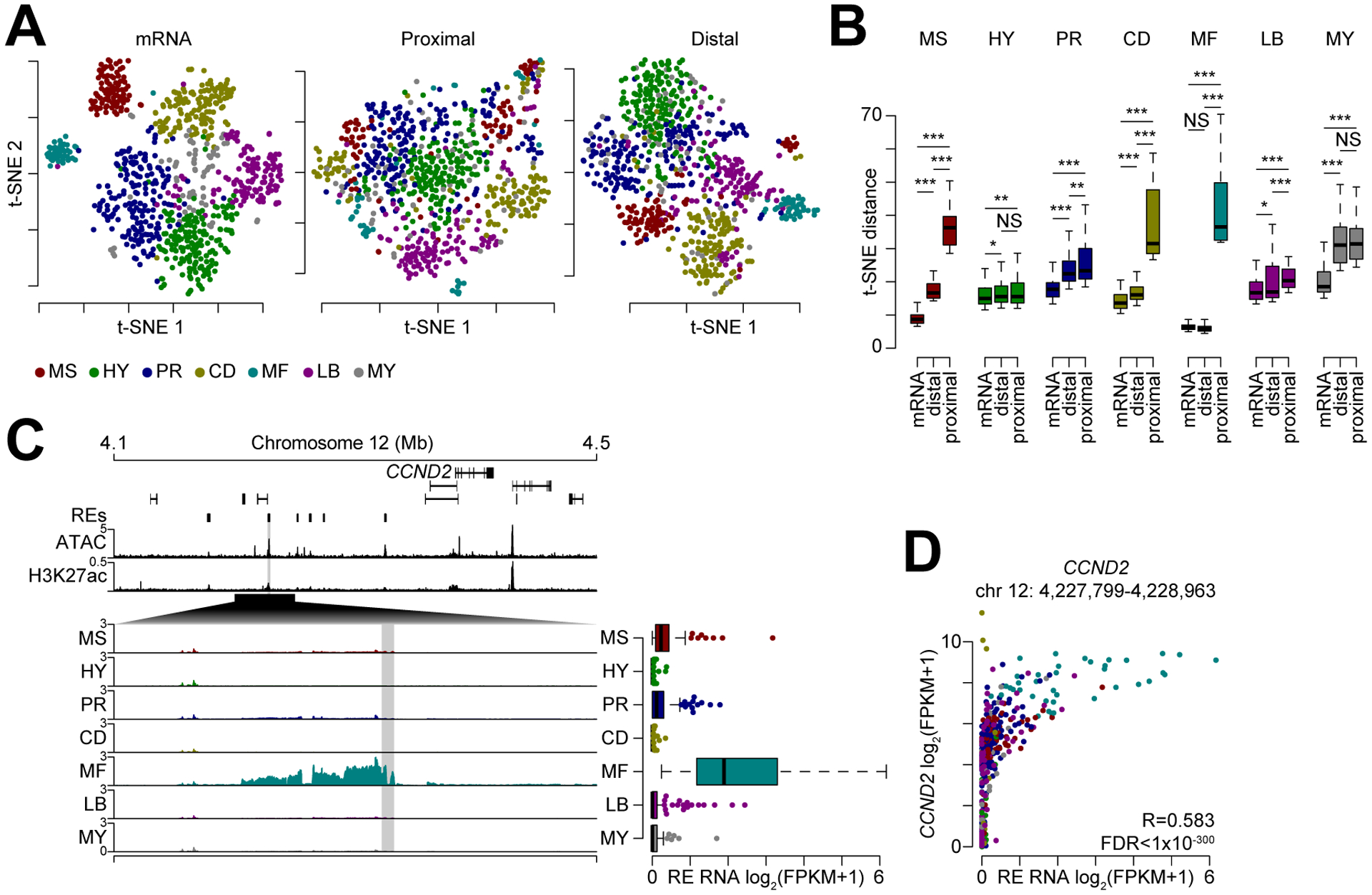
Distal regulatory elements define myeloma subtype gene expression. A, t-SNE analysis of gene expression data (left), transcription at promoter proximal (middle) and distal (right) regulatory elements. Samples are colored by myeloma gene expression subtype (key bottom). B, t-SNE distance between samples within a given subtype (denoted by color) for mRNA as well as promoter proximal and distal regulatory elements. ***P<0.001, **P<0.01, *P<0.05, NS: not significant; determined by a paired Mann-Whitney U test. C, Genome plot of regions distinctly regulated between myeloma subtypes near the CCND2 locus. Regulatory elements (REs) are denoted on top with cumulative ATAC (RPPM) and H3K27ac (RPM) signal shown and mean transcription is shown (log2(RPM+1)) for each myeloma subtype. Transcription for the shaded region is shown (right). D, Scatterplot of CCND2 expression and regulatory element transcription.
Distal regulatory element activity predicts myeloma gene expression and outcome
Given that transcription at regulatory elements predicted gene expression, we wondered if regulatory element activity could be used to identify putative mechanisms of molecular pathogenesis and poor outcome. First, we identified gene expression that was prognostic of overall survival (OS) in CoMMpass patients using a Cox proportional hazards model. Gene expression prognostic of OS are represented in a volcano plot of the OS hazard ratio (HR) for each gene compared to the significance of association with outcome (Fig. 6A). For example, reduced expression of CD27 and SLAMF7 or increased expression of RUNX2 or SUZ12 correspond with poor outcome. Next, distal regulatory RNA levels within 1 MB of genes prognostic of outcome were correlated with expression identifying 42% of regulatory elements that positively corresponded with gene expression (Fig. 6B, see green; Supplementary Data S4). Plotting the frequency of transcribed distal regulatory elements relative to genes prognostic of outcome indicated that approximately 1.5 active regulatory elements per MB per gene could be detected (Fig. 6C). In total, 1,308 active distal regulatory elements were identified within 1 MB of genes prognostic of OS and 609 of these were positively correlated with gene expression (Fig. 6C, green; Supplementary Data S5). We asked if these regions were enriched or depleted for transcription factor binding consensus motifs, which identified five transcription factor families enriched in these regions including Nuclear Respiratory Factor (NRF), Zinc Finger (ZF), Nuclear Receptor (NR), Helix-Loop-Helix (HLH), and Activating Protein 2 (AP2) families, whereas only Interferon Response Factor (IRF) binding motifs were depleted (Fig. 6D–E; Supplementary Data S6). Additionally, gene expression of the genes that encode NRF1, SP1, and HIF-1β (encoded by ARNT1) were also associated with worse outcome as shown by a hazard ratio greater than one (Fig. 6F), further indicating these factors may be driving a gene expression program of aggressive myeloma. Indeed, NRF1 has previously been linked to proteasome inhibitor resistance (56,57), and similarly both SP1 and HIF-1β have also been linked to myeloma pathogenesis (58,59). An example of one of these distal regulatory elements is shown for RUNX2 (Fig. 6G). Here, genes are shown on top with RUNX2 in red and the correlation of regulatory element transcription to gene expression (RE/GX) is denoted by the height of the curves connecting each regulatory element to the RUNX2 promoter; regulatory elements, ATAC-seq, and H3K27ac ChIP-seq data are shown below. The specific regulatory element demarcated by the red correlation curve and the region harboring the regulatory element is enlarged to show RNA levels stratified by quartile. This particular regulatory element contained both SP1 and HIF-1b binding motifs (Fig. 6G, bottom). Regulatory element expression quartiles are quantitated on a box-and-whisker plot and the corresponding expression of RUNX2 is shown for each quartile (Fig. 6H), indicating a strong concurrence in regulation as denoted by a positive Pearson correlation (R=0.35) which was highly significant (FDR = 9.4×10−28). Cumulatively, these data identify putative distal regulatory elements and transcription factors whose activity are highly coordinated with the gene expression program of aggressive myeloma.
Figure 6.
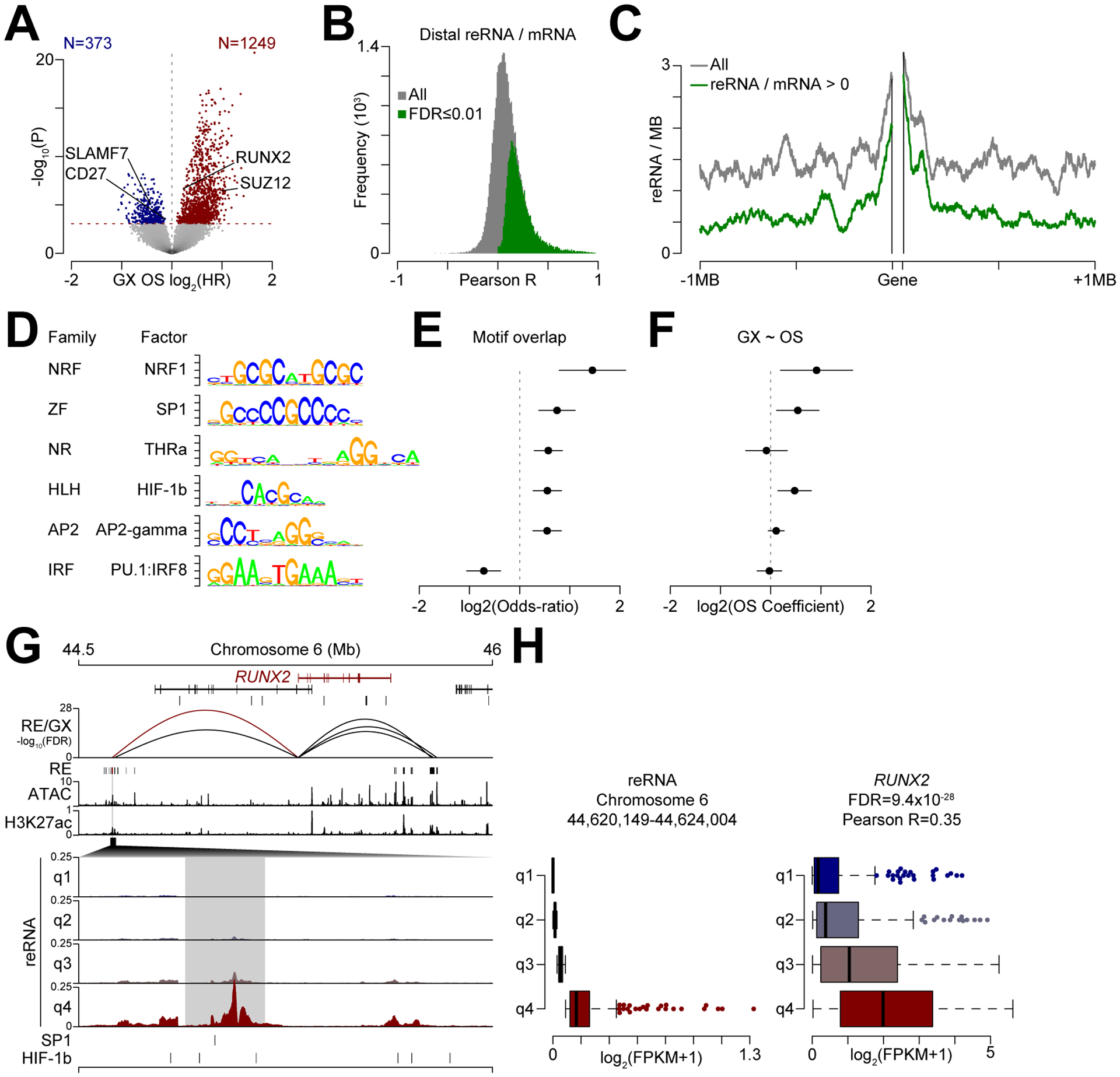
Distal regulatory element activity predicts expression of genes prognostic of myeloma outcome. A, Volcano plot of gene expression (GX) associated with overall survival (OS). The y-axis represents the hazard ratio (HR) of OS given gene expression with genes significantly associated with poor outcome denoted in blue (less expression) or red (more expression). The significance line (FDR ≤0.01) is shown (dashed red line). B, Correlation (Pearson R) between transcribed distal regulatory elements and gene expression. C, Frequency of regulatory elements relative to the average gene. All expressed distal regulatory elements are denoted in gray and those that are positively associated with gene expression are shown in green (FDR ≤0.01). D, Top transcription factor consensus binding motifs enriched in distal regulatory elements that predict gene expression associated with poor outcome. Only the top factor for each family is shown (see Supplementary Data S6 for full results). NRF: Nuclear Respiratory Factor; ZF: Zinc Finger; NR: Nuclear Receptor; HLH: Helix-Loop-Helix; AP2: Activating Protein 2; IRF: Interferon Regulatory Factor. E, Overlap odds ratio of transcription factor binding motifs in regulatory elements positively associated with gene expression indicative of poor outcome relative to all transcribed regulatory elements (95% confidence intervals are shown). F, Overall survival (OS) hazard ratio of expression of the transcription factors (95% confidence intervals are shown). G, Genome plot of RUNX2 (denoted in red). The correlation of regulatory element transcription with gene expression (GX) is shown below the genes with the height of each line denoting the significance of correlation. Regulatory elements, and cumulative ATAC and H3K27ac signal are shown below. A specific distal regulatory region is enlarged as denoted by a black polygon with regulatory element transcription stratified by quartile of expression. H, Regulatory element transcription quartiles (left) and corresponding gene transcription for RUNX2. The Pearson correlation (R) and significance (FDR) of reRNA and RUNX2 expression is shown.
Discussion
Here, we show that cryopreserved bone marrow aspirates can be used to isolate multiple myeloma cells that faithfully recapitulate the mRNA content and chromatin accessibility of those isolated from fresh samples. This approach was applied to a series of cryopreserved samples that were interrogated to identify regions of chromatin accessibility that were predictive of gene expression – results that were corroborated using a published study in an independent cohort of myeloma specimens. Intergenic chromatin accessible regions were used to query transcription in over 700 myelomas from newly diagnosed patients to identify active regulatory elements. Analysis of these active regulatory elements provided interesting insights into gene regulation in this disease. First while regulatory RNAs were able to predict gene expression-defined myeloma subtypes, they were in some cases not as good as the mRNA expression. This might be expected as the subtypes were defined by the mRNA expression. However, closer examination reveals that distal active regulatory elements better predicted mRNA expression than promoter elements that likely represent bidirectional transcription. This suggests that it is the activity of these distal elements that is driving the gene expression-based subtypes. Thus, while myeloma has been appreciated to be a “enhanceropathy” for some time now (8,60), our data suggest that this relationship goes well beyond enhancers that are physically juxtaposed to oncogenes during initiating translocation events, and include the impact of many distal regulatory elements. Nevertheless, such initiating events appear to influence enhancer usage. While the activity of distal regulatory elements was much better at identifying gene expression-defined myeloma subtypes, this relationship was most evident amongst myelomas that have a primary IGH translocation (CCND, MMSET or MAF). In contrast, there was little difference in the ability of distal vs. proximal regulatory element activity in distinguishing myelomas of subtypes that were not exclusively associated with an IGH translocation, such as the hyperdiploid (HY), low bone disease (LB), and proliferation (PR). It is possible that this reflects a distinct gene regulatory program between hyperdiploid and non-hyperdiploid myeloma. Regardless, understanding how oncogenic changes are controlling these distal regulatory elements may provide insights into new pathways to target myeloma based on the transcriptional mechanisms propagating a malignant phenotype, as well as explain the molecular basis of therapeutic response to current backbone therapies that work in part by targeting transcription factor activity (e.g. IMiDs, steroids and indirectly, proteasome inhibitors).
Interrogating current RNA-seq data sets for regulatory RNAs has emerged as a powerful tool in deciphering gene regulatory mechanisms. Enhancer transcription from TCGA has provided unique insight into a range of solid and some hematological tumors revealing both ubiquitously expressed and cancer specific enhancer transcription (61,62). While multiple myeloma was not included in the TCGA project, use of data from the MMRF CoMMpass study has allowed us to provide the largest study of enhancer transcription in myeloma to date. Median depth of mRNA-sequencing for CoMMpass specimens was over 230 million mapped reads per sample, which is significantly higher than most TCGA data, and provides sufficient power to detect regulatory element transcription. Our discovery of 4,729 transcribed regulatory elements in 5% or more of myeloma samples is highly congruent with TCGA analysis that found on average 4,591 eRNAs expressed in 10% or more of samples per cancer type (61). Others have found ~20% of all detectable reRNAs are ubiquitous across cancer types whereas the remaining 80% are either partially shared or unique to specific cancer types (62). We anticipate that myeloma is similar in this regard. For instance transcribed enhancers predictive of MDM4 expression have previously been reported, albeit the regulatory element reported here is distinct from that previously described (62). Further insights into the unique molecular architecture of myeloma – and therefore potential therapeutic targets – will likely be provided by understanding the regulatory elements that are unique to myeloma versus those that are common across other cancer and/or cell types.
Despite the powerful insights gleaned, interrogating regulatory element transcription from RNA-seq is not without caveats. For instance, most RNA-seq data sets, including those used here, primarily only capture poly-adenylated RNAs, and only a portion of regulatory RNAs such as enhancer RNAs (eRNAs) are poly-adenylated (55,63). Additionally, it is not currently feasible to interrogate intragenic regions thereby missing more than half of all regulatory elements. Furthermore, transcription through the annotated transcription termination site often occurs and contaminates 3’ regulatory elements with RNA that is a byproduct of transcription rather than a signal of enhancer function. For these reasons it will be important to conduct large-scale genetic, epigenetic, and transcriptional studies to better interrogate the regulatory mechanisms and identify molecular susceptibilities of myeloma. In this regard, the high-level of RNA and chromatin accessibility correspondence between fresh and biobanked samples should provide confidence that such studies can be conducted using viably frozen bone marrow aspirates. While it is important to note that these samples were only frozen for 4 months – and longer term studies are needed – this approach potentially has the advantage of isolating immune or stromal cells from the same bone marrow aspirate, thus allowing for analysis of both tumor, stromal and/or immune compartments.
Large scale genetic, epigenetic, and transcriptional studies have the potential to change our understanding of myeloma pathogenesis and ultimately allow us to alter the disease course. Ensuring an adequate sample size will be of paramount importance in dissecting the distinct molecular pathology of the many myeloma subtypes. Such analyses should shed light on effective modes of oncogene regulation. For instance, some Hyperdiploid cases of myeloma express high levels of CCND1 despite not having t(11;14) IGH translocations (64), and while we were not able to identify candidate cis-elements regulating CCND1 expression outside of the promoter in these myelomas this may be due to such an element residing in a intron or an enhancer that does not efficiently transcribe eRNA. In the case of CCND2, we were able to identify an element highly coordinated with CCND2 expression in the MAF subtype, but CCND2 is also aberrantly expressed in the MMSET and Proliferation subtypes, which showed much more modest transcription at the same enhancer. Thus, it still remains to be shown if the MMSET and Proliferation subtypes regulate CCND2 expression through the same regulatory elements (albeit with less transcription) or if there are other elements not discernable by this analysis that contribute to CCND2 dysregulation in these subtypes. Further studies of myeloma epigenetic and molecular programming may provide insight into why these types of myeloma exhibit aberrant expression of such oncogenes. Likewise, epigenetic studies may provide insight into the cell differentiation state from which a myeloma originated. For instance, B cells undergo rapid epigenetic remodeling as they become activated and differentiate into germinal center B cells and plasma cells (35,42,65); thus epigenetic analysis of myeloma may provide insights into disease etiology. Indeed, the CD2 myeloma subtype derives its name for Cyclin D and CD20, the latter being a B cell marker normally not retained on plasma cells or other myeloma subtypes (23). Thus, epigenetic analysis of this subtype may reveal remnants of the B cell molecular program. Such epigenetic studies will identify important cis-regulatory regions for myeloma pathology and help prioritize transcription factors and signaling pathways for a myeloma subtype precision medicine approach.
Supplementary Material
Acknowledgements
This study was supported in part by the Emory Integrated Genomics Core of Winship Cancer Institute of Emory University and NIH/NCI award number P30 CA138292. This work was supported by an Answer Fund award from the MMRF and 3R01 CA192844–04W1 to LHB. CCH is supported by NIH/NCI award 5R01CA201382–03. BGB is supported by Developmental Funds from the Winship Cancer Institute of Emory University, post-doctoral fellowship PF-17–109-1-TBG from the American Cancer Society, and a Research Fellow Award from the Multiple Myeloma Research Foundation (MMRF). We thank Dr. Leif Bergsagel for constructive conversations. We thank the patients, researchers and clinicians involved in the MMRF CoMMpass trial and the entire Multiple Myeloma Research Consortium.
Conflicts of Interest
CCH has received research grants from Oncolytics Biotech, research and personal grants from Janssen, BMS, Sanofi. Nektar, and Karyopharm, and personal grants from Imbrium and Oncopeptides over the last 24 months all outside the submitted work. AKN declares advisory roles and compensation from Amgen, Takeda, Janssen, BMS, GSK, Adaptive, Karyopharm and Oncopeptides. SL is a consultant for Takeda, Celgene, Amgen, Novartis, and BMS. LHB declares consultancy for Abbvie and Genentech, and honoraria and research funding from AstraZeneca.
References
- 1.Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia. Nature Publishing Group; 2014;28:1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin [Internet]. 2020;70:7–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31912902 [DOI] [PubMed] [Google Scholar]
- 3.Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood [Internet]. 2014. [cited 2015 Oct 13];124:1873–9. Available from: http://www.bloodjournal.org/content/124/12/1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of origin and genetic alterations in the pathogenesis of multiple myeloma. Front Immunol [Internet]. 2019;10. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2019.01121/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonial S, Boise LH, Kaufman J. How I treat high-risk myeloma. Blood [Internet]. 2015. [cited 2015 Oct 12];126:1536–43. Available from: http://www.bloodjournal.org/content/126/13/1536 [DOI] [PubMed] [Google Scholar]
- 6.Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J Clin Oncol. 2015;33:2863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia [Internet]. Nature Publishing Group; 2009. [cited 2018 Dec 12];23:2210–21. Available from: http://www.nature.com/articles/leu2009174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergsagel PL, Chesi M, Nardini E, Brents LA, Kirby SL, Kuehl WM. Promiscuous translocations into immunoglobulin heavy chain switch regions in multiple myeloma. Proc Natl Acad Sci U S A [Internet]. 1996;93:13931–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8943038%5Cnhttp://www.pnas.org/content/93/24/13931.full.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chesi M, Nardini E, Lim RSC, Smith KD, Michael Kuehl W, Bergsagel PL. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood [Internet]. 1998. [cited 2017 Mar 9];92:3025–34. Available from: http://www.bloodjournal.org.proxy.library.emory.edu/content/92/9/3025 [PubMed] [Google Scholar]
- 10.Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood [Internet]. 1998;91:4457–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9616139 [PubMed] [Google Scholar]
- 11.Fonseca R, Debes-Marun CS, Picken EB, Dewald GW, Bryant SC, Winkler JM, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood [Internet]. 2003. [cited 2017 Jan 28];102:2562–7. Available from: http://www.bloodjournal.org.proxy.library.emory.edu/content/102/7/2562 [DOI] [PubMed] [Google Scholar]
- 12.Avet-Loiseau H, Andree-Ashley LE, Moore D, Mellerin MP, Feusner J, Bataille R, et al. Molecular cytogenetic abnormalities in multiple myeloma and plasma cell leukemia measured using comparative genomic hybridization. Genes Chromosom Cancer. 1997;19:124–33. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt T, Barwick B, Joseph N, Heffner LT, Hofmeister C, Bernal L, et al. Gain of Chromosome 1q is Associated with Early Progression in Multiple Myeloma Patients Treated with Lenalidomide, Bortezomib, and Dexamethasone. Clin Lymphoma Myeloma Leuk [Internet]. Springer US; 2019;19:e76–7. Available from: 10.1038/s41408-019-0254-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fonseca R, Van Wier SA, Chng WJ, Ketterling R, Lacy MQ, Dispenzieri A, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20:2034–40. [DOI] [PubMed] [Google Scholar]
- 15.Affer M, Chesi M, Chen WD, Keats JJ, Demchenko YN, Tamizhmani K, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia [Internet]. 2014. [cited 2017 Mar 9];28:1725–35. Available from: http://www.nature.com/leu/journal/v28/n8/full/leu201470a.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A [Internet]. 2000. [cited 2018 Feb 18];97:228–33. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=26645&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demchenko Y, Roschke A, Chen WD, Asmann Y, Bergsagel PL, Kuehl WM. Frequent occurrence of large duplications at reciprocal genomic rearrangement breakpoints in multiple myeloma and other tumors. Nucleic Acids Res [Internet]. Oxford University Press; 2016. [cited 2019 Jan 2];44:8189–98. Available from: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, et al. MYC dysregulation in the progression of multiple myeloma. Leukemia [Internet]. Springer US; 2020;34:322–6. Available from: 10.1038/s41375-019-0543-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun [Internet]. Nature Publishing Group; 2019. [cited 2019 Apr 23];10:1911. Available from: http://www.nature.com/articles/s41467-019-09555-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia [Internet]. Nature Publishing Group; 2019. [cited 2019 Mar 4];33:159–70. Available from: http://www.nature.com/articles/s41375-018-0196-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker BA, Mavrommatis K, Wardell CP, Cody Ashby T, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun [Internet]. Springer US; 2019;10:1–12. Available from: 10.1038/s41467-019-11680-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, et al. The molecular classification of multiple myeloma. Blood [Internet]. 2006. [cited 2016 May 27];108:2020–8. Available from: http://www.bloodjournal.org/content/108/6/2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broyl A, Hose D, Lokhorst H, De Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood [Internet]. American Society of Hematology; 2010. [cited 2019 Mar 4];116:2543–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20574050 [DOI] [PubMed] [Google Scholar]
- 25.Ng MHL, Chung YF, Lo KW, Wickham NWR, Lee JCK, Huang DP. Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood [Internet]. 1997. [cited 2016 Jul 4];89:2500–6. Available from: http://www.bloodjournal.org/content/89/7/2500 [PubMed] [Google Scholar]
- 26.Kaiser MF, Johnson DC, Wu P, Walker BA, Brioli A, Mirabella F, et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood [Internet]. 2013. [cited 2016 Aug 6];122:219–26. Available from: http://www.bloodjournal.org/content/122/2/219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agirre X, Castellano G, Pascual M, Heath S, Kulis M, Segura V, et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res [Internet]. 2015. [cited 2015 Feb 18];25:478–87. Available from: http://genome.cshlp.org/content/early/2015/02/02/gr.180240.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood [Internet]. 2011. [cited 2016 Jul 4];117:553–62. Available from: http://www.bloodjournal.org/content/117/2/553 [DOI] [PubMed] [Google Scholar]
- 29.Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, et al. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet [Internet]. 2014. [cited 2015 Oct 21];10:e1004566. Available from: 10.1371/journal.pgen.1004566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, et al. NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol Cell [Internet]. Elsevier Inc.; 2011;44:609–20. Available from: 10.1016/j.molcel.2011.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood [Internet]. 2008. [cited 2016 Aug 10];111:3145–54. Available from: http://www.bloodjournal.org/content/111/6/3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods [Internet]. 2013. [cited 2014 Apr 14];10:1213–8. Available from: http://www.nature.com/nmeth/journal/v10/n12/full/nmeth.2688.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scharer CD, Blalock EL, Barwick BG, Haines RR, Wei C, Sanz I, et al. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naïve SLE B cells. Sci Rep [Internet]. 2016. [cited 2016 Jul 1];6:27030. Available from: http://www.nature.com/articles/srep27030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matulis SM, Gupta VA, Neri P, Bahlis NJ, Maciag P, Leverson JD, et al. Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia [Internet]. Springer US; 2019;33:1291–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30679802%0Ahttp://www.nature.com/articles/s41375-018-0374-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barwick BG, Scharer CD, Bally APR, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol [Internet]. 2016. [cited 2016 Aug 9];17:1216–25. Available from: http://www.nature.com/doifinder/10.1038/ni.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowman SK, Simon MD, Deaton AM, Tolstorukov M, Borowsky ML, Kingston RE. Multiplexed Illumina sequencing libraries from picogram quantities of DNA. BMC Genomics [Internet]. 2013. [cited 2014 May 20];14:466. Available from: http://www.biomedcentral.com/1471-2164/14/466/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics [Internet]. 2013. [cited 2016 Oct 26];29:15–21. Available from: http://bioinformatics.oxfordjournals.org/content/29/1/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics [Internet]. 2009. [cited 2016 Dec 29];25:2078–9. Available from: https://academic.oup.com/bioinformatics/article/25/16/2078/204688/The-Sequence-Alignment-Map-format-and-SAMtools [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol [Internet]. 2004. [cited 2013 Feb 2];5:R80. Available from: http://genomebiology.com/2004/5/10/R80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, et al. Software for Computing and Annotating Genomic Ranges. PLoS Comput Biol [Internet]. 2013. [cited 2015 Jun 2];9:e1003118. Available from: http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence M, Gentleman R, Carey V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics [Internet]. 2009. [cited 2013 Dec 4];25:1841–2. Available from: http://bioinformatics.oxfordjournals.org/content/25/14/1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barwick BG, Scharer CD, Martinez RJ, Price MJ, Wein AN, Haines RR, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun [Internet]. Nature Publishing Group; 2018. [cited 2018 May 15];9:1900. Available from: http://www.nature.com/articles/s41467-018-04234-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods [Internet]. 2012. [cited 2015 Apr 17];9:357–9. Available from: http://www.nature.com.proxy.library.emory.edu/nmeth/journal/v9/n4/full/nmeth.1923.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol [Internet]. 2008. [cited 2012 Sep 25];9:R137. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18798982%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2592715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature [Internet]. 2012. [cited 2012 Sep 9];489:57–74. Available from: http://www.nature.com/doifinder/10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell [Internet]. 2013. [cited 2014 Jan 13];153:307–19. Available from: http://www.sciencedirect.com/science/article/pii/S0092867413003929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics [Internet]. 2007. [cited 2014 Aug 29];23:257–8. Available from: http://bioinformatics.oxfordjournals.org/content/23/2/257 [DOI] [PubMed] [Google Scholar]
- 48.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics [Internet]. 2009. [cited 2016 Oct 25];26:139–40. Available from: http://bioinformatics.oxfordjournals.org.proxy.library.emory.edu/content/26/1/139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin Y, Chen K, De Paepe A, Hellqvist E, Krstic AD, Metang L, et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood [Internet]. American Society of Hematology; 2018. [cited 2018 Jun 20];131:2138–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29519805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A [Internet]. 2005. [cited 2014 Aug 29];102:15545–50. Available from: http://www.pnas.org/content/102/43/15545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkerson MD, Hayes DN. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics [Internet]. 2010. [cited 2017 Dec 13];26:1572–3. Available from: http://www.bioconductor.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.García-Alonso CR, Pérez-Naranjo LM, Fernández-Caballero JC. Multiobjective evolutionary algorithms to identify highly autocorrelated areas: The case of spatial distribution in financially compromised farms. Ann Oper Res. 2014;219:187–202. [Google Scholar]
- 53.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol [Internet]. 2005. [cited 2016 Aug 14];23:6333–8. Available from: http://jco.ascopubs.org.proxy.library.emory.edu/content/23/26/6333.full.pdf [DOI] [PubMed] [Google Scholar]
- 54.Tuan D, Kong S, Hu K. Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci U S A. 1992;89:11219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lam MTY, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci [Internet]. Elsevier Ltd; 2014;39:170–82. Available from: 10.1016/j.tibs.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu Y, Barwick BG, Shanmugam M, Hofmeister CC, Kaufman J, Nooka A, et al. Downregulation of PA28α induces proteasome remodeling and results in resistance to proteasome inhibitors in multiple myeloma. Blood Cancer J [Internet]. Springer US; 2020;10:125. Available from: 10.1038/s41408-020-00393-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription Factor Nrf1 Mediates the Proteasome Recovery Pathway after Proteasome Inhibition in Mammalian Cells. Mol Cell [Internet]. Elsevier; 2010. [cited 2021 Feb 18];38:17–28. Available from: http://www.cell.com/article/S1097276510002406/fulltext [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulciniti M, Amin S, Nanjappa P, Rodig S, Prabhala R, Li C, et al. Significant biological role of Sp1 transactivation in multiple myeloma. Clin Cancer Res. 2011;17:6500–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu C, Yang T, Liu Y, Lu Y, Yang Y, Liu X, et al. ARNT/HIF-1β links high-risk 1q21 gain and microenvironmental hypoxia to drug resistance and poor prognosis in multiple myeloma. Cancer Med. 2018;7:3899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet [Internet]. 1997. [cited 2016 Aug 10];16:260–4. Available from: http://www.nature.com.proxy.library.emory.edu/ng/journal/v16/n3/abs/ng0797-260.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Caesar-Johnson SJ, et al. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell [Internet]. Elsevier; 2018. [cited 2018 Dec 14];173:386–399.e12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29625054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Z, Lee JH, Ruan H, Ye Y, Krakowiak J, Hu Q, et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat Commun [Internet]. Springer US; 2019;10:1–12. Available from: 10.1038/s41467-019-12543-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Natoli G, Andrau J-C. Noncoding Transcription at Enhancers: General Principles and Functional Models. Annu Rev Genet. 2012;46:1–19. [DOI] [PubMed] [Google Scholar]
- 64.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J. Cyclin D dysregulation: An early and unifying pathogenic event in multiple myeloma. Blood [Internet]. 2005. [cited 2016 Aug 10];106:296–303. Available from: http://www.bloodjournal.org.proxy.library.emory.edu/content/106/1/296?sso-checked=true [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kulis M, Merkel A, Heath S, Queirós AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet [Internet]. 2015. [cited 2015 Jun 14];47:746–56. Available from: http://www.nature.com.proxy.library.emory.edu/ng/journal/vaop/ncurrent/full/ng.3291.html [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.