

Abstract
Neurodegenerative diseases are characterized by the accumulation of misfolded proteins. This protein aggregation suggests that abnormal proteostasis contributes to aging-related neurodegeneration. A better fundamental understanding of proteins that regulate proteostasis may provide insight into the pathophysiology of neurodegenerative disease and may perhaps reveal novel therapeutic opportunities. The 26S proteasome is the key effector of the ubiquitin-proteasome system responsible for degrading polyubiquitinated proteins. However, additional factors, such as valosin-containing protein (VCP/p97/Cdc48) and C9orf72, play a role in regulation and trafficking of substrates through the normal proteostasis systems of a cell. Nonhuman AAA+ ATPases, such as the disaggregase Hsp104, also provide insights into the biochemical processes that regulate protein aggregation. X-ray crystallography and cryo-electron microscopy (cryo-EM) structures not bound to substrate have provided meaningful information about the 26S proteasome, VCP, and Hsp104. However, recent cryo-EM structures bound to substrate have provided new information about the function and mechanism of these proteostasis factors. Cryo-EM and cryo-electron tomography data combined with biochemical data have also increased the understanding of C9orf72 and its role in maintaining proteostasis. These structural insights provide a foundation for understanding proteostasis mechanisms with near-atomic resolution upon which insights can be gleaned regarding the pathophysiology of neurodegenerative diseases.
Keywords: Alzheimer disease, Amyotrophic lateral sclerosis, ClpX, Fibrils, Frontotemporal degeneration, Multisystem proteinopathy, Vacuolar tauopathy
INTRODUCTION
Protein aggregates are a hallmark feature of many neurodegenerative diseases, including Alzheimer disease, Lewy body diseases, amyotrophic lateral sclerosis (ALS), and frontotemporal degeneration (FTD). While protein aggregation is central to these aging-related diseases, the field has looked beyond the aggregates themselves for insights into disease pathogenesis and potential therapeutic targets. The proteostasis process has been a logical area of inquiry, as aggregation indicates a dysfunction in the proper regulation of the proteome. Aggregates may either cause proteostatic dysfunction or result from proteostatic dysfunction, and their specific role likely depends on the disease process in question.
At the heart of human proteostasis is the 26S proteasome (1). The 26S proteasome is the final step of the ubiquitin proteasome system (UPS), degrading proteins that have been marked as misfolded or aggregated, as is the case in many neurodegenerative diseases (2). However, the 26S proteasome represents only the final arbiter of UPS-mediated degradation. In normal cellular functioning, before marked proteins can be degraded, they need to be removed from the organelles, membranes, complexes, or aggregates in which they are found. Proteins like valosin-containing protein (VCP) play the role of separating or disaggregating proteins so that they can be reused or degraded in healthy cells (3–6). Complementing the UPS system are autophagic processes which degrade intracellular components, including protein aggregates. Regulation of autophagy likely plays an important role in neurodegenerative disease through factors such as C9orf72. C9orf72’s complete function remains nebulous, but has been linked with autophagy defects, UPS, and a variety of neurodegenerative diseases (7, 8). When dysfunction occurs within these processes that regulate and turn over the proteome, protein aggregation can occur. Aggregation can lead to cellular toxicity and death as is seen in neurodegenerative diseases, such as ALS and FTD (Table ).
TABLE.
Summary of Proteostasis Factors Valosin-Containing Protein (VCP), 26S Proteasome, Hsp104, and C9orf72 Role in Proteostasis, Their Substrates, and Mutation-Associated Diseases (PDB: 6OA9, 6MSD, 5VJH, 6LT0; EMDB: 7479)
| Proteostasis Factor | Role in Proteostasis | Associated Substrates | Mutation-Associated Diseases |
|---|---|---|---|
|
Valosin-containing protein
|
|
Tau, transactive response DNA-binding protein 43 kDa (TDP-43) | Vacuolar tauopathy, multisystem proteinopathy |
|
26S proteasome
|
|
Amyloid precursor protein, Tau, α-synuclein, polyQ aggregates, poly-GA aggregates | N/A |
|
Hsp104
|
|
Seminal amyloid, TDP-43, α-synuclein, FUS, stress granules | N/A (yeast protein) |
|
C9orf72
|
|
Rab8a, Rab11a, Arf1, Arf5, Arf6, 26S proteasome (binding partner with 65 other neurodegenerative associated proteins) | Amyotrophic lateral sclerosis, frontotemporal degeneration |
Naturally, the 26S proteasome, VCP, C9orf72, and similar factors have emerged as candidates for investigation into the mechanism and treatment of neurodegenerative diseases due to their key role in maintaining functional proteostasis. However, nonhuman proteins, such as Hsp104, also present interesting therapeutic opportunities. Hsp104 is not found in humans naturally, but it has been found to disaggregate neurodegenerative disease-relevant aggregates more effectively than human homologs (9–12) (Table ). If the effective disaggregation ability of Hsp104 can be harnessed in humans, it may be an enticing potential therapy for neurodegenerative disease.
TECHNOLOGICAL ADVANCES IN CRYO-ELECTRON MICROSCOPY
Structural insight into the proteins of the protein regulatory pathway is important to understanding neurodegenerative disease mechanisms and exploiting these factors for therapeutic potential. The advent and improvement of cryo-electron microscopy (cryo-EM) techniques has been central to the boom of published structures relevant to proteostasis. X-ray crystallography was previously the preferred method for obtaining near-atomic and atomic resolution structures of proteins. However, crystallography requires the growth of crystals, which can be unpredictable and may not represent the native state of a protein in solution, as was the case for crystallized proteostasis factors (13–15). Unlike crystallography, cryo-EM allows for the study of protein complexes in isolation without crystal packings and can capture multiple conformations of those complexes (16). The concept of cryo-EM has been around since the 1970s (17); however, until recently the technology has been insufficient to attain near-atomic or atomic resolution reconstructions. Since 2013, advancements in cryo-EM technology have allowed for near-atomic and even atomic resolution (∼1.2 Å) (18) reconstructions. New microscopes allow for highly coherent 300 kV illumination, automatic sample insertion that can store and handle several grids at one time, automatic liquid nitrogen refilling system, and increased imaging stability from an extensive lens system. Direct electron detectors are more sensitive and substantially faster than traditional cameras, capable of taking many frames per second as opposed to requiring long exposure times for a single image. The stack of frames can be analyzed to correct for specimen movement upon electron exposure to improve image resolution. Single-particle cryo-EM is useful for obtaining atomic structure of in vitro proteins and complexes. Single-particle cryo-EM relies on many copies of identical complexes that can be imaged from many different orientations on a grid. The different orientations are used to generate a 3D structure of the complex. Cryo-electron tomography (cryo-ET) takes a different approach by rotating samples to obtain 3D information about each subject. Cryo-ET is well suited for in situ structural determination that is not possible with other structural determination techniques. Cryo-ET obtains more information about each individual target than single-particle cryo-EM at the sacrifice of number of targets imaged, generally resulting in lower-resolution reconstructions than single-particle cryo-EM.
Here, we review recent cryo-EM and cryo-ET structures of several proteostasis factors relevant to neurodegenerative disease: VCP, 26S proteasome, Hsp104, and C9orf72 (Table).
VCP/p97/Cdc48
Valosin-containing protein (p97/VCP in mammals, Cdc48 in yeast) plays an essential role in cellular processes and is necessary for cell viability, which has led to its establishment as a target for cancer therapy (19, 20). The main function of VCP is removing individual polyubiquitinated polypeptides from organelle membranes, protein complexes, chromatin, and ribosomes (3–5), although VCP can process some substrates in the absence of ubiquitin (21, 22).
There is growing evidence that VCP may contribute to the clearance of protein aggregates (4, 6). Mutations in VCP can cause neurodegenerative proteinopathies, including vacuolar tauopathy and multisystem proteinopathy (MSP; characterized by variable phenotypes including inclusion body myositis, Paget disease of bone, ALS, and frontotemporal dementia [IBMPFD]) (6, 23–25). A variety of cellular pathway defects have been reported with these neurodegenerative mutations, including defects in endosomal trafficking, autophagy, mammalian target of rapamycin regulation, and mitochondrial homeostasis, and this is likely an incomplete list of cellular processes that are affected by VCP activity (26–29).
VCP also extracts polyubiquitinated misfolded proteins from the endoplasmic reticulum (ER) and transfers them to the 26S proteasome in a process called ER-associated protein degradation (ERAD) (30, 31). Unlike the 26S proteasome which requires a flexible polypeptide to initiate its proteolytic processing, VCP does not require its substrates to be flexible or to be a partially unfolded polypeptide (32–34). As a result, VCP can directly unfold or segregate substrates that cannot be directly degraded by the 26S proteasome due to their lack of a flexible region. This ability to bypass the need for a flexible region furthers the hypothesis that VCP may prepare well-folded proteins for processing by the proteasome (31, 35). While 26S proteasome recognizes and initiates translocation using flexible regions in the polypeptide substrate as a signal, it remains not fully clear how VCP is able to recognize a broad range of substrates in the absence of a flexible region. Polyubiquitin chains appear to be one signal that VCP uses to recognize some substrates (36).
VCP is classified as part of the AAA+ family of ATPases. It has an N-terminal domain (NTD) and 2 ATPase domains named D1 and D2 (13, 37). Similar to other AAA+ ATPases, VCP is a homohexamer with a central pore (Fig. 1A, B). The NTD on the “cis” side of the ring formation can be in an “up” conformation (above the D1 ring) or “down” conformation (coplanar with the D1 ring) (Fig. 1D). These 2 conformations play a role in cofactor binding, discussed further below. D2 resides beneath D1 on the “trans” side of the ring formation (Fig. 1A, B, D) (13, 37). Most missense mutations associated with MSP lie at the NTD/D1 interface (Fig. 1C) (38).
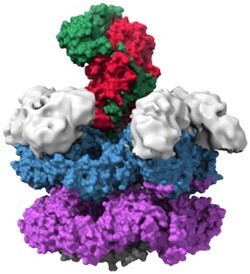
FIGURE 1.

Structures of valosin-containing protein (VCP) processing substrate, its mutations, and different conformational arrangements sampled by VCP during its function. (A) VCP bound to Npl4 (red) and ubiquitinated substrate (green) with N-domains (light gray) modeled in. N-terminal domains (NTD) (light gray) sit on top of the D1 ATPase ring (blue) that sits on top of the D2 ATPase ring (purple). VCP has a C-terminal tail (dark gray) on the trans side of the ring (Atomic structure: PDB 6OA9, N-domains: EMDB 7479). (B) Cut-through of VCP bound to ubiquitinated substrate (green). The substrate is bound to Npl4 (red) on the cis side of VCP and travels through the pore, contacting the D1 ATPase ring (blue) and the D2 ATPase ring (purple) (PDB: 6OA9). (C) Disease-related VCP mutations shown on a VCP monomer. The multisystem proteinopathy (MSP) mutations (orange) are associated with a variety of clinical phenotypes. The MSP mutations generally are near the interface of the NTD (light gray) and the D1 ATPase ring (blue) and may change VCP to have a preference for the “up” conformation. The newly discovered D395G mutation (pink) is associated with vacuolar tauopathy (PDB: 5C19). (D) On the left is the “down” conformation of VCP that has the NTD (light gray) coplanar with the D1 ATPase ring (blue). The “down” conformation can bind some VCP cofactors such as VIMP. The right is the “up” conformation of VCP that has the NTD (light gray) above the D1 ATPase ring (blue). The “up” conformation is required for binding of the Ufd1/Npl4 (UN) cofactors (PDB: 5FTN, 5FTM). (E) The VCP D1 and D2 pore loop staircase processing substrate with hypothesized nucleotide bound states of the D2 pore (D1 nucleotide bound states and how they correlate to D2 nucleotide bound states are still being investigated). Each side chain color represents a monomer that moves down a position in the staircase and eventually returns to the top of the staircase. ATP is bound to all monomers except those at the bottom of the staircase and in process of moving to the top of the staircase. W562 and Y561 are the key aromatic residues in pore loop 2 that bind to substrate in the staircase. (PDB: 6OAB).
Cofactors of VCP determine its substrate specificity and cellular localization. All known cofactors of VCP bind the NTD or the C-terminal tail (39). The Ufd1/Npl4 (UN) cofactor complex allows VCP to recognize and process Lys48-linked polyubiquitin chains, but can also recognize more complex polyubiquitin chains (Fig. 1A, B) (40, 41). However, VCP without cofactors can still interact with polyubiquitin and in some situations can unfold substrate without ubiquitin (21, 42). UN is needed for VCP’s role in ERAD and is the only cofactor of VCP essential for cell viability in yeast (40, 43–46). Npl4 has an N-terminal ubx-like domain (UBXL), 2 Zn2+-finger domains (zf-Npl4), and an Mpr1/Pad1 N-terminal domain (MPN) (36, 46). Notably, mammalian Npl4, but not yeast Npl4 has an additional C-terminal Zn2+-finger domain that binds ubiquitin (47, 48). Ufd1 has an Ufd1 truncation 3 (UT3) ubiquitin-binding domain, flexible UT6 domain that has an Npl4 binding site, and 2 short suppressor of high-copy PP1-containing motifs that bind to VCP. UN has also been shown to bind ubiquitin without VCP (49), which may indicate UN binding to ubiquitin precedes UN binding to VCP in vivo. In general, when VCP is complexed with the UN cofactor it is able to recruit substrates by binding to polyubiquitin chains, requiring at least 5 Lys48-linked ubiquitins for efficient binding and subsequent substrate processing (50, 51).
In vitro experiments suggest that D2 uses ATP hydrolysis to move a polypeptide substrate through the central pore, unfolding substrate in the process (51). However, until recently, there was doubt that VCP could move a polypeptide through its pore due to the narrowness of the pore as seen by X-ray crystallography (13). Cryo-EM has completely changed our understanding of how VCP works. Within the central pore, D2 contains conserved pore loops with aromatic residues that are used for polypeptide translocation by VCP and similar ATPases (52). D1, however, lacks aromatic residues in its pore loops, and in vitro evidence suggests ATP needs only to bind, and does not need be hydrolyzed, to allow substrate processing (41, 51). To date, no evidence has demonstrated how VCP is able to pull the polypeptide substrate through D1 to reach the strong aromatic residues of D2. D1 ATP hydrolysis is thought to be involved with substrate release, potentially in conjunction with another cofactor, DUB, that can trim the polyubiquitin chain, but the details of this process remain to be elucidated (51).
In 2019, Twomey et al and Cooney et al published complementary structures of VCP that further elucidate how VCP processes and recognizes substrate. Twomey used a known in vitro substrate, while the Cooney structures show an unidentifiable substrate from yeast (14, 15).
The Twomey study used Cdc48(E588Q) (a yeast VCP mutant that can bind but not unfold substrate) and UN with a polyubiquitinated substrate that contains an Eos fluorescent protein which reports whether or not a protein is still folded. Unfortunately, in the Twomey structure, UT6, the flexible Ufd1 domain with an Npl4 binding site, was unable to be modeled and there was no density seen for UT3, the Ufd1 ubiquitin-binding domain. Without substrate, the β-strand finger of the MPN domain of Npl4 is above the pore, but with substrate bound the β-strand finger is no longer above the pore leaving it open for substrate processing (14, 36). Two folded ubiquitins were modeled at the top of the Npl4 tower with an unfolded ubiquitin extending through an Npl4 groove and then through D1 and D2 (Fig. 1A, B). Amino acids within the Npl4 groove are conserved between species, and mutations of these amino acids decreases the ability to unfold polypeptides without decreasing the ability of ubiquitin to bind to Npl4 or Ufd1 (14). Hydrogen-deuterium exchange mass spectrometry corroborates that UN binds 1 unfolded and 3–4 folded ubiquitins, which explains the observation that a minimum chain length of 5 ubiquitins is necessary for efficient binding (51). Notably, a structure published with human VCP (p97) did not show ubiquitin unfolding, which may suggest important differences in substrate engagement and recognition between Cdc48 and wild-type (WT) p97 (48). However, more structures of Cdc48 and p97 will need to be solved with substrate engaged in order to show whether there are in fact differences between yeast and humans and whether these differences are physiologically significant.
The structure of VCP’s ATP-bound state has been controversial, similar to those of other AAA ATPases. In their structure using ATP, Twomey et al found that D1 is bound to ATP in all monomers. Ubiquitin can be threaded through both D1 and D2 rings without ATP hydrolysis. The unfolded ubiquitin only interacts with 2 of 6 monomers in the D1 ring with both interactions being at methionine residue 288 in the D1 pore. D2, alternatively, connects to the unfolded ubiquitin at 4 of the 6 monomers, which form a staircase positioned to pull the substrate through the pore to the trans-side of VCP. The 3 highest positioned monomers are ATP bound and the lowest monomer bound to ubiquitin is ADP bound. D2 is positioned further downward compared to D1, which was also seen previously without substrate when D1 and D2 were bound with ATPγS (a nonhydrolyzable ATP analog that approximates just before ATP hydrolysis) (14, 37).
Twomey et al also published a structure with ADP-BeFx (an analog that approximates just after ATP hydrolysis). In this structure, they found 5 monomers in contact with the polypeptide to make a staircase. Four of 5 monomers are bound to ADP-BeFx and 1 monomer is likely ADP bound (ADP is left over from purification and not able to be removed from solution). They hypothesize that the ADP-BeFx structure likely represents one step further in the mechanism than what is seen in the structure with ATP. The D2 ring of VCP contacts the polypeptide backbone of the substrate with tryptophan at residue position 561 and tyrosine at position 562 in the D2 pore loop. These aromatic residues “pinch” every other peptide bond indicating that the hydrolysis of each ATP molecule by D2 moves the peptide 2 amino acids further through the pore (Fig. 1E).
Cooney et al show that the amino acid side chains of substrate intermingle between pore loop 1 residues, which may add to the grip strength of VCP by increasing pore loop contact with substrate. However, VCP primarily binds to the substrate polypeptide backbone with aromatic residues in its pore loops. Binding to the polypeptide backbone allows for universality of VCP binding, eliminating the need for specific amino acid sequences to function.
When a VCP monomer bound to substrate is at the lowest position on the staircase, ATP is hydrolyzed and the monomer prepares to move back to the top of the staircase to bind the backbone of another amino acid (Fig. 1E) (14). A similar mechanism has been seen with different single- and double-ringed ATPases (33, 34, 53–55). VCP, however, is distinct because it has 2 aromatic rings that pinch the substrate instead of 1 (Fig. 1E). The additional aromatic ring that pinches the polypeptide backbone may provide VCP extra strength to unfold folded substrates, unlike the 26S proteasome that needs a flexible polypeptide region. In a similar ATPase to VCP, ClpX, it has been shown that more aromatic rings increase grip of the protein (56). Of note, addition of aromatic rings in the D1 domain of VCP causes lethality in yeast (57). Lethality with additional aromatic rings in the D1 domain could indicate that too much grip in the D1 domain may be detrimental, potentially due to problems in substrate release specifically. Future experiments will need to be done to determine whether a change in the number of aromatic residues in D2 affects survival. The structure containing ADP-BeFx also has a D1 staircase not seen in the ATP structure or any previous structure (13, 36, 37). The additional D1 staircase may be an artifact or transient state due to ADP-BeFx fixing the D1/D2 interaction such that they cannot act independently.
VCP is the only complex known to unfold ubiquitin. For ubiquitin to reach D2, it must be unfolded at least partially, thus its unfolding is likely not solely due to the extra aromatic residues in the D2 pore loop. Atomic force microscopy has shown that pulling K48 in a ubiquitin chain can unfold ubiquitin (58), and Npl4 may use its groove to lock K48 so that VCP can unfold ubiquitin (14). Unfolding ubiquitin exemplifies the power that VCP is able to generate and may play a role in how the 26S proteasome could recognize substrates after VCP processing. The 26S proteasome generally also requires ubiquitin, so if ubiquitin is not cleaved and able to refold on the trans side of VCP, it may be used by the 26S proteasome for recognition (discussed further below).
Cooney et al used co-immunoprecipitation to purify Cdc48 with Shp1 (a substrate-recruiting cofactor) directly from Saccharomyces cerevisiae using ADP-BeFx. Shp1 is not found in humans, but human analogs of Shp1 (p37, p47, and UBXN2a) dissociate the inactive PP1-SDS22-I3 complex (21). The NTD of VCP was shown to be in an “up” conformation (Fig. 1D) similar to that shown in the structure published by Twomey et al, consistent with the fact that this conformation is necessary for substrate processing. Five of the monomers have helical symmetry and grip substrate, similar to the ADP-BeFx structure from Twomey et al. The sixth monomer does not have helical symmetry with the others and has 2 distinct conformations that show the transition from substrate release to binding another amino acid at the top of the staircase. They hypothesize that the interactions between monomers are stabilized by ATP binding in D2. Therefore, hydrolysis in the fourth monomer allows the fifth monomer to transition to the asymmetric position of the sixth monomer. The sixth monomer is no longer bound to ADP and is moving higher to grab another segment of substrate. When the sixth monomer binds ATP, it becomes monomer 1 (Fig. 1E) (15).
Twomey et al hypothesize that the cofactor DUB is necessary to cut ubiquitin and allow for substrate release. The Otu1 domain from DUB can bind to VCP and cleave the ubiquitin chain near the D1 domains, triggering the NTD to move into the “down” conformation and release UN. Otu1 can release substrate with up to 10 ubiquitin molecules (51). If the released ubiquitin can be refolded on the trans side of VCP, the 26S proteasome may be able to immediately recognize and continue processing the same substrate as the 26S proteasome only needs 4 ubiquitins for recognition (59). However, as VCP unfolds its substrate, the 26S proteasome may begin processing the same substrate before VCP has released it, as there is evidence that the 26S proteasome and VCP can cooperate in the absence of DUB (31). In one in situ study of neurodegenerative aggregates, VCP was not found near the 26S proteasome, so further investigations will need to be done to elucidate the interaction of VCP and the 26S proteasome in physiologic systems (60).
As previously mentioned, VCP mutations have been linked to neurodegenerative disease. Recently, Blythe et al used cryo-EM to elucidate how these mutations alter VCP’s structure and function. It had been shown that, in the absence of substrate, MSP mutants of VCP increase ATPase activity in D2 relative to WT VCP (Fig. 1C) (6, 61–64). However, there was debate about whether this increased ATPase activity was productive, detrimental, or inconsequential for substrate processing (65, 66). First, it was shown that the A232E mutation has increased substrate processing, then later it was shown that all MSP mutants process substrate and ATP faster than WT (41, 67). Patients with MSP mutations are generally heterozygous for the mutations and thus likely have hexamers that contain mixed mutant and WT monomers (23). In vitro experiments have shown that heterohexamers of A232E VCP mixed with WT VCP have intermediate substrate processing ability between that of homohexamer A232E VCP and WT VCP (67).
A near-atomic resolution structure of A232E VCP shows VCP bound to the UN cofactor (67). The A232E mutant structure resembles the overall conformation of previously reported WT VCP structures without substrate (36, 37). The A232E VCP structure has its NTD in the “up” conformation. This conformation was first reported in a crystal structure of VCP with D2 removed and ATPγS bound (68). Since then, it has been shown to be the preferred conformation of full-length WT VCP when ATP analogs are bound (14, 15, 36, 37). The “up” conformation is thought to represent the active form of VCP (Fig. 1D). This is supported by the fact that a structure of VCP bound to an allosteric inhibitor demonstrated that the inhibitor worked by preventing VCP from forming the “up” conformation (37). It has also been shown that the “down” conformation would cause a steric clash when VCP interacts with its binding partner VIMP, suggesting that this conformation may be incompatible with activity in at least some scenarios (Fig. 1D) (69).
MSP mutants favor the “up” conformation regardless of their nucleotide bound state, unlike WT VCP (63, 69–73). UN binds A232E VCP 12 times stronger than it binds WT VCP with ADP bound and 65 times stronger with ATP bound. The difference notably only applies to the “on”-rate, with no change in the “off”-rate; therefore, the increased binding affinity is likely due to a preference of MSP mutants for the “up” conformation. Similar results were shown with R155H and T262A VCP mutants (Fig. 1C) (69).
Using 2D class averages from single-particle cryo-EM, Blythe et al classified A232E and WT VCP into groups of “up,” “down,” or “mobile” NTD’s. When ATP is bound, they found that WT may be up, down, or mobile, but A232E VCP is only found in the up or mobile states. With ADP bound, WT was never up, but A232E can still be up with a large number of particles being in the mobile state. Using 2D class averages, Blythe et al were able to show that MSP mutants stabilize the “up” conformation or destabilize the “down” conformation (67). UN binds to VCP in the “up” conformation, thus the MSP mutants have a faster “on”-rate of UN because they are more likely to be in the “up” conformation. The preference for the “up” conformation by the MSP mutants even when bound to ADP supports data that show MSP mutants did not decrease their affinity for cofactor VIMP when bound to ADP as compared to ATPγS (69).
The affinity for the “up” conformation seems to indicate that the MSP mutations of VCP are in fact a gain of function (GOF) mutation, in opposition to the proposed “dominant negative” theory that proposes MSP mutations lead to a loss of function in VCP even with one WT VCP allele (65, 66). How the GOF seen in MSP mutations relates to human MSP disease is less clear. Blythe et al 2019 suggest that MSP mutants could outpace the 26S proteasome, thus leading to a build-up of product from VCP that is not further degraded by the 26S proteasome. Their theory is supported by the fact that aggregates are ubiquitinated and thus may have already been processed by VCP (74). However, recent cryo-EM structures and studies of neurodegenerative protein aggregates have not definitively concluded whether ubiquitination precedes or postcedes aggregation in all cases (75–77).
Blythe et al also calculate that MSP mutant VCP binds most of the cellular UN, leaving no free UN within cells. Mitophagy may need free UN to initiate substrate binding (78). Substrate may also need to bind UN before it can be processed by VCP in a cellular environment, though this is not supported by in vitro data (41, 51, 67). No matter the exact mechanism, MSP mutations may lead to less free VCP and/or UN and cause a loss of function in other VCP or UN pathways. For example, UBXD1 has a decreased affinity for MSP mutant VCP, as UBXD1 prefers the “down” conformation for binding (28, 71, 72). Analysis of patients with MSP mutations has suggested that the lysosomal degradation pathway mediated by UBXD1 may fail in the context of the MSP mutations (27, 28, 79, 80). A dysfunction in the lysosomal degradation pathway supports that the MSP mutations may cause failure of other UN or VCP pathways. Additionally, the cellular localization of VCP is affected by its cofactors, thus if VCP is bound solely or predominantly to UN, it may not be able to fulfill its function in other parts of the cell (43, 49, 81–84).
A novel VCP mutation, D395G, was recently reported to present with tau pathology (Fig. 1C) (6). Interestingly, the D395G mutation shows lower ATPase activity and less ability to process substrate than WT or MSP mutant VCP (6). The pathology seen in patients with the D395G mutation is distinct from the pathology seen with MSP mutations. This mutation has not been characterized as fully as the MSP mutations, so there remains much to learn. However, these findings may suggest therapeutic potential or mechanistic relevance for VCP in more common neurodegenerative diseases such as Alzheimer disease.
Though cryo-EM structures have indicated how ubiquitinated substrates are able to be recognized and processed by VCP and how mutants may affect VCP structure and function, we have only recently gained insight into how VCP may recognize and process substrates that are not ubiquitinated (22). VCP has been shown to be able to recognize and unfold substrates lacking ubiquitin, which may be related to the fact that this activity is dependent on p37, suggesting that nonubiquitinated substrate processing may be cofactor dependent (21). Therefore, this phenomenon may be limited to specific substrates VCP has evolved to recognize independent of ubiquitin. Future structures may shed light on the ubiquitin-independent process and demonstrate whether or not VCP interacts with different substrates in different manners. As of yet, no structure of VCP with an identifiable natural VCP substrate has been published. This is a notable gap in our understanding of VCP as physiologic substrates may have different mechanisms of action with VCP than those substrates studied in vitro.
Specifically, understanding how VCP recognizes and processes highly stable neurodegenerative aggregates may be crucial in understanding proteostatic dysfunction in neurodegenerative diseases. Mutations of VCP seem to be causative in some aggregation-related neurodegenerative diseases. A full understanding of how these mutations affect VCP in vivo and the proteostasis pathway in general will likely lead to a more complete understanding of neurodegenerative disease than we currently have.
26S PROTEASOME
The UPS is a central component of a cell’s proteostasis network, with roles in regulation of the cell cycle, apoptosis, immune responses, inflammation, and response to proteotoxic stress (85–87). The UPS is responsible for most of the regulated protein degradation in eukaryotic cells (88). UPS is responsible for marking, removing, and degrading misfolded or aggregated proteins, including those associated with neurodegenerative diseases (2). UPS is at the heart of the proteostasis network, and so a build-up of defective protein aggregates suggests an issue with the proteasome. UPS has been specifically implicated in the breakdown of amyloid precursor protein, tau, α-synuclein, and polyQ aggregates, which are implicated in a variety of neurodegenerative diseases (89–94). It is also thought that aggregates may overwhelm or even inhibit the UPS, compounding proteostasis issues within in a cell (60, 91, 95, 96).
Many structures of 26S proteasome (referred to as proteasome here) have been published since the advent of cryo-EM (33, 34, 60, 97–114). The proteasome protein complex is particularly well-suited to cryo-EM due to its large number of subunits and its conformational heterogeneity. Similar to VCP, structures published in the past few years have significantly changed the way that the proteasome complex is viewed (33, 34, 97). Previously, there were only low-resolution (>9.0 Å) structures of the proteasome degrading substrate or structures of the proteasome with various ATP analogs without substrate. These structures allowed for vague hypotheses on the conformational changes and steps of substrate degradation by the proteasome. New, near-atomic resolution structures of the proteasome have allowed for a clear understanding of the steps in the proteasomal degradation process (33, 34). Additionally, new structures of the interface of the AAA+ ATPase ring and core particle (CP) gate have altered the understanding of how subunits regulate its opening (97).
The proteasome is composed of the 20S CP that is responsible for proteolysis and 1 or 2 19S regulatory particles (RPs) that cap the CP on one or both ends (Fig. 2A) (115, 116). The RP controls access to the CP to prevent unregulated proteolysis within a cell. The CP is made of a stack of 4 heptameric rings: α1–7-β1–7-β1–7-α1–7. The N-terminal extensions of the α rings control entry to the CP and the β rings form the catalytic chamber for proteolysis (Fig. 2A, D) (117, 118). Proteins are targeted to the UPS by ubiquitin, which is covalently bound at lysines. Ubiquitin is then recognized and eventually removed by the RP (Fig. 2B, C) (119). Unlike VCP, the proteasome requires a flexible region of approximately 20–30 amino acids to initiate processing by the RP (120–122). As discussed above, VCP may act in conjunction with the proteasome. VCP can unfold substrates to allow the proteasome to initiate processing with substrates that it could not otherwise process (5, 31, 51).
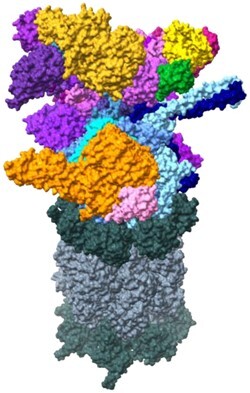
FIGURE 2.

The 26S proteasome structure with substrate processing and steps of substrate processing in the AAA+ ATPase ring. Core particle (CP) gate regulation by AAA+ ATPase C-terminal tails is also shown. (A) The 26S proteasome with one regulatory particle (RP) on top of the CP. The RP is composed of a lid (different shades of pink/purple) and a base (different shades of orange/yellow) that contains an AAA+ ATPase (blue). Rpn11 is specifically noted as the subunit that first interacts with ubiquitin in substrate recognition by the RP. The CP below the RP has a set of α-rings (dark green) on either side of the pair of stacked β-rings (slate gray) that make up the catalytic chamber (PDB: 6MSD). (B) The 26S proteasome with ubiquitinated substrate bound to the regulatory particle. Rpn11 (purple) binds ubiquitin (green) and holds it so substrate (red) can be fed through the AAA+ ATPase ring (blue) of the RP. After going through the RP, substrate passes through the CP gate created by the α-ring of the CP (dark green), before entering the catalytic chamber made of the β-rings of the CP (slate gray) (PDB: 6EF3). (C) The 3 modes of nucleotide processing in AAA+ ATPase (blue) of regulatory particle. Mode 1 corresponds to ubiquitin binding and engagement of substrate. Mode 2 is the initiation of translocation and release of ubiquitin. Mode 3 is the sequential ATP hydrolysis that powers substrate through the central pore (PDB: 6MSG, 6MSD, 6MSE, 6MSH, 6MSJ, 6MSK). (D) A model of C-terminal tail regulation of core particle gate. The N-terminal extensions of the α-ring subunits (dark green) keep the CP gate closed until Rpt subunits dock their C-terminal tails between monomers. Rpt3, Rpt2, and Rpt5 are necessary, but not sufficient for CP gate opening. Rpt1 and Rpt6 C-terminal tails docking between α-ring subunits are what allows for the CP gate to open (PDB: 6MSE, 6MSH).
The RP is further categorized into a base and a lid complex. The lid has 9 RP non-ATPase (Rpn) subunits: Rpn 3, 5–9, 11, 12, and 15/Sem1 (86). Rpn11 is a deubiquitinase located above the central pore that removes ubiquitin from the protein substrate after recognition (Fig. 2A, B) (102, 103, 123–125). Rpn11 is essential for cell viability and proper proteolytic function of the proteasome (126–128). Rpn10 bridges the lid and the base of the RP (129, 130). The base of the RP contains 3 ubiquitin receptors (Rpn 1, 2, 13) and 6 distinct AAA+ ATPases that form a ring (Rpt1–6) (131). The AAA+ ATPase ring acts as the motor to unfold substrates, similar to VCP, and feed them into the CP (Fig. 2A–C) (132). The Rpt subunits act to regulate CP gate opening as will be discussed further below (Fig. 2D) (133, 134).
Similar to VCP, the proteasome changes conformation when bound to nucleotides and/or substrates. In the absence of substrate, the proteasome likely adopts the “s1” configuration in vivo, referring to the proteasome conformation that was determined without substrate and without nucleotide present (101, 103, 107). In contrast, a low-resolution structure of the proteasome processing substrate was the first to show that the proteasome undergoes significant conformational change upon substrate processing (111). Here, we will focus on substrate bound structures of the proteasome.
Substrate binding and initiation is regulated through ubiquitin and the RP. Ubiquitin initially binds Rpt4-Rpt5 coiled coil (CC) domain near Rpn11 and then is transferred to Rpn11 (Fig. 2A, B) (33). Ubiquitin interfaces with Rpn11 in a hydrophobic binding pocket (33, 135). The Rpt5 N-loop, in conjunction with the insert-1 (Ins1) region of Rpn11, guides ubiquitin toward the catalytic zinc-binding site within Rpn11, moving the isopeptide bond to a proper alignment for deubiquitination. The Rpt5 N-loop is disordered in other steps of the cycle and the isopeptide bond is not visualized after this step, indicating that ubiquitin is cleaved in this step (33). The Ins1 region is maintained in an inactive state but switches to an active state with ubiquitin binding, likely serving to ensure that only substrates that are committed to going through the proteasome are deubiquitinated (135). The substrate maintains contact with Rpn11 in a hydrophobic binding groove even after ubiquitin is gone.
To initiate translocation of substrate, Rpt6 rotates out of the plane of the ATPase ring to open up the central ATPase pore to allow substrate to enter, since prior to this rotation the pore is too narrow for translocation to occur (33). Dong et al hypothesize that ATP hydrolysis and release cause an “iris-like” movement in the ATPase ring which allows for substrate entry. Additionally, their series of structures show coordinated ATP hydrolysis in the subunit directly across the ring, which may increase the conformational flexibility of the ATPase ring (Fig. 2C). Rpt1-Rpt2 help initiate translocation by vertically rotating to the substrate at the top of the pore loop staircase as ATP binding at Rpt6 triggers pore-1 loop to bind to substrate at the top of the staircase. The binding of pore-1 loop to substrate moves substrate one peptide couplet forward through the pore. ATP binding at Rpt1 and Rpt2 initiates their engagement with substrate and repeats the cycle of movement of dipeptide through the pore (Fig. 2C) (33). During the steps of ADP release and ATP binding to Rpt1 and Rpt3, ubiquitin is seen to dissociate from Rpn11. When ubiquitin dissociates, the ATPase ring repositions above the CP, resetting the proteasome for subsequent protein degradation (33). Notably, unlike Dong et al, de la Peña et al see ubiquitin bound to Rpn11 in all the translational states of the proteasome, which may be due to their method of stalling substrate in the proteasome by inhibiting Rpn11 (34). Both sets of structures, though, do support that deubiquitination occurs during translocation when a substrate is committed to going through the proteasome, not before translocation (Fig. 2C). Notably, there is evidence of ubiquitin-independent processing by the proteasome, so the mechanism of substrate recognition and initiation of translocation seen by Dong et al may not be the only possible mechanism (136, 137).
During substrate translocation, the substrate spirals through the central pore of the ATPase ring contacting the pore-1 loop aromatic residues. The staircase formed by the pore loops stays consistent in location, but the subunit at each position changes throughout the process as substrate rotates down through the pore (33, 34). The pore-1 loop aromatic residues use hydrophobic interactions to contact a dipeptide region, reminiscent of the mechanism seen in VCP (14, 15, 33, 34). Distinct from VCP, however, the pore-1 loops interact with the amino acid side-chains but not peptide backbone of the substrate (14, 15, 33, 34). These distinct substrate interactions between VCP and the proteasome could decrease the amount of force the proteasome can generate, affecting what substrates it is able to process compared to VCP.
As substrate moves through the ATPase pore, the ADP bound states rotate counterclockwise sequentially through the 6 Rpt subunits (Fig. 2C) (33, 34). The major conformational change, however, does not occur with ATP hydrolysis and phosphate release, but phosphate release loosens interactions between subunits that prepares them for a major conformational change observed with ADP-ATP exchange (33, 34). Three adjacent substrates coordinate nucleotide processing to continually translocate substrate, 2 peptides at a time, similar to VCP. One subunit hydrolyzes ATP, releasing a phosphate and destabilizing its inter-subunit interactions, preparing itself to release substrate and move to the top of the staircase. A second subunit releases ADP and rotates to the top of the staircase. A third subunit binds ATP and engages substrate at the top of the staircase (Fig. 2C) (33, 34). Vertical rotation of the subunit exchanging ADP for ATP simultaneously causes the other subunits to move down as a rigid body to translocate substrate (33, 34). A similar rigid body movement is seen with ClpX, the hexameric protease of Escherichia coli (138, 139). Each Rpt subunit of the proteasome takes a turn rotating to the top of the staircase and then gradually moves in a conveyor belt style to the bottom of the staircase.
The mechanism of substrate processing by the proteasome is complex, but it can be simplified into 3 discrete and sequential modes of substrate processing, as described by Dong et al: Mode 1, Mode 2, and Mode 3. Beginning with substrate binding, Mode 1 describes 2 oppositely positioned ATPase subunits coordinate ATP hydrolysis to initiate substrate binding. This pattern is similar to that seen previously in the nucleotide-binding pattern of a substrate-free proteasome structure and ClpX (99, 140). Next, in Mode 2, ATP hydrolysis in adjacent subunits initiates substrate translocation and CP gate opening (discussed further below). Finally, in Mode 3, ATP is hydrolyzed sequentially by subunits with a cyclical fashion (Fig. 2C).
De la Peña et al note that one of their structures has an “off-pathway” nucleotide bound state. The “off-pathway” structure shows both Rpt5 and Rpt1 bound to ADP and not bound to substrate. They hypothesize that this structure could be due to Rpt5 prematurely releasing a substrate. The Rpt5 pore-1 loop has a methionine rather than a lysine, which may cause a weaker interaction with substrate that could be prone to erroneous release, especially in the case of stalled substrate that is seen in the de la Peña structures (34, 136). De la Peña et al also note the “off-pathway” structure may represent a “failed nucleotide exchange” by Rpt1 at the top of the staircase. Considering the structures from Dong et al that were published after those of de la Peña et al, it is possible that the “off-pathway” state is in fact capturing Mode 2, initiation of substrate translocation, rather than an “off-pathway” state of Mode 3, continued substrate translocation. Adjacent subunits hydrolyze ATP simultaneously in initialization of translocation (Fig. 2C). Though the substrate is already translocated in “off-pathway” structure, the stalling of the proteasome by an Rpn11 inhibitor could cause an erroneous hydrolysis of ATP by adjacent subunits. This may happen because ubiquitin was not cleaved; therefore, the proteasome still seems to be in an initialization of translocation mode, Mode 2, rather than a pure translocation mode, Mode 3.
De la Peña et al also hypothesize that during Mode 3 there are 12 possible conformations of the proteasome, in which one subunit is not bound to substrate and is bound to either ADP or ATP. They note that even though they only report 4 conformations of the helical staircase in Mode 3, previous structures without substrate have captured all of the other conformations, except those in which Rpt6 is not bound to substrate (97, 99, 100, 109).
Beneath the RP, CP gate regulation has been an area of intense focus. It has been shown that the C-terminal tails of the Rpt subunits facilitate CP gate opening by docking between α subunits (97, 98, 134). Three subunits (Rpt2, Rpt3, Rpt5) contain an HbYX motif that was previously proposed to mediate CP opening as determined by studies of the archaeal proteasome and biochemical studies with HbYX peptides (133, 134). However, structures of the proteasome showed Rpt2, Rpt3, and Rpt5 C-terminal tail docking in the α ring of the CP without an open CP gate (Fig. 2D) (34, 99, 100, 109, 111). Structures of the proteasome with open CP gates show that Rpt1 and Rpt6 are also docked in the α ring (Fig. 2D) (34, 97, 99). Rpt1 and Rpt6 do not have the HbYX motif, but they do have a conserved region in the same area that may function similarly to the HbYX motif. The structural finding that open CP gates occur with Rpt1 and Rpt6 tails docked in the α ring, with support from biochemical studies of Rpt1 and Rpt6 C-terminal tail mutations, suggests that Rpt1 and Rpt6 are actually the C-terminal tails that regulate CP gate opening (97). Likely Rpt2, Rpt3, and Rpt5 need to have their C-terminal tails docked for the CP gate to open, but Rpt1 and Rpt6 are the C-terminal tails that regulate whether or not the CP gate is open (Fig. 2D). Knowledge of which C-terminal tails regulate CP opening may help with specifically targeting the proteasome in disease modifying therapies.
Though there are many similarities between VCP and the proteasome, one difference that may affect function and substrate choice is that the proteasome requires a flexible region for initiation. Many proteins within a cell contain flexible regions of more than 30 amino acids (141). However, proteins that do not have long flexible regions still require a degradation pathway. Biochemical evidence suggests that VCP can unfold proteins, so they can be degraded by the proteasome (31). Elucidating how these 2 proteins work together will be critical to understanding the mechanism and regulation of the degradation pathway for proteins lacking a flexible region or that are too difficult for the proteasome to degrade, such as neurodegenerative aggregates.
VCP and the proteasome are likely intertwined in their neurodegeneration-related dysfunction. However, further research is necessary to understand this dysfunctional relationship. Mechanistic understanding of the VCP-proteasome interaction could be harnessed to develop new therapeutics for neurodegenerative disease or understand how neurodegenerative disease initiates and progresses.
Hsp104
Hsp104 is an AAA+ ATPase and part of the Hsp100 class of chaperone proteins. It functions as a disaggregase and is found in yeast as well as in all nonmetazoan eukaryotes, eubacteria, and some archaebacteria (142, 143). Metazoa use alternatives to Hsp104 to achieve similar functions. Disaggregases such as Hsp104 or similarly engineered proteins may present therapeutic potential in the prevention or treatment of diseases of protein aggregation that currently have no effective therapies (144–152).
Variants of Hsp104 have been shown to disaggregate seminal amyloid fibrils that promote HIV infection, in addition to aggregates of TDP-43, α-synuclein, and FUS that are highly correlated with neurodegenerative disease (9–12). Not only were these Hsp104 mutants able to dissolve fibrils, but they were also able to rescue dopaminergic degeneration from α-synuclein in Caenorhabditis elegans and reduce FUS toxicity in mammalian cells (FUS is an RNA-binding protein whose aggregates are associated with ALS) (11, 153). In yeast, Hsp104 enables proteasomal degradation of proteins and extracts insoluble proteins of ERAD, similar to the function of VCP, and also imports cytosolic proteins into the mitochondria for degradation (154–157). Hsp104 also plays a role in the phase properties of membrane-less organelles, or stress granules that have been implicated in the pathophysiology of neurodegenerative diseases such as ALS, FTD, and AD (158–160). Hsp104 has been shown to remove stress granules after stress is removed (158, 159, 161).
Hsp104 is composed of an NTD, ATPase domain 1 (D1), middle domain (MD), D2, and C-terminal domain (CTD) (Fig. 3A) (143). The NTD has been implicated in substrate engagement and the CTD assists with hexamerization (162–164). Similar to VCP and the proteasome, D1 and D2 have flexible pore loops that bind to substrate and give Hsp104 its functionality (143, 165). The D1 and D2 domains allow Hsp104 to remove proteins from a wide variety of aggregations or complexes, including preamyloid oligomers, phase-transitioned gels, disordered aggregates, amyloids, and yeast prions (149, 158, 159, 166–177). Yeast prions are neither infectious proteins nor disease related, but rather drive different phenotypes in yeast and are named prions based on their prion-like behavior on a biophysical level.
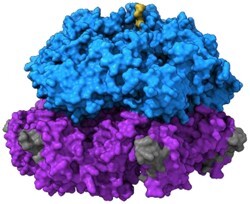
FIGURE 3.

The Hsp104 structure, different modes of translocation, and sequential processing of substrate using its pore loop staircase. (A) Upper: the surface view of Hsp104 ATPase domain 1 ring (D1, blue) on top of D2 ATPase ring (purple) bound to substrate (gold) (PDB: 5VJH). Lower: the domain architecture plot of Hsp104 including domains not in the structure shown above (N-terminal domain [NTD]: red, D1: blue, MD: green, D2: purple, CTD: dark gray). (B) Models of partial and complete modes of substrate translocation by Hsp104. Partial translocation does not pull substrate (gold) all the way through the pore, but releases it separated from other parts of the complex. Complete translocation unfolds substrate pulling it all the way through the Hsp104 pore, dissociating each monomer. (C) The possible order of events for Hsp104 staircase in substrate processing that shows a monomer binding to substrate (gold) at the top of the pore loop staircase prior to a monomer releasing substrate at the bottom of the pore loop staircase. The side chain colors correspond to each monomer and its movement through the staircase. Y257 is the key residue in pore loop 1 and Y662 is the key residue in pore loop 2 (PDB: 5JVH, 5VYA).
Distinct from the proteasome and what has been shown so far with VCP, Hsp104 can have partial or complete translocation of substrate across its pore (Fig. 3B). Complete or partial translocation of substrate by Hsp104 is thought to be correlated with aggregate stability (165, 167, 178–182). Less stable aggregates are thought to have a noncooperative mechanism of disaggregation by Hsp104 subunits, while more stable aggregates, like amyloids, have a cooperative mechanism of disaggregation by Hsp104 subunits (167, 174). In the cooperative mechanism, Hsp104 subunits work together to unfold aggregates. Hsp104 can also initiate disaggregation at an internal segment of protein aggregates (Fig. 3B) (180, 182). Neither VCP nor the proteasome is known to initiate substrate processing at an internal segment, thus Hsp104 may possess a unique ability that expands its disaggregation potential. It remains possible that VCP, and less likely the proteasome (due its lid subunits, Fig. 2A), may be able to initiate processing in a similar manner, but this has never been observed.
The disaggregase activity of Hsp104 is enhanced by the Hsp70 chaperone system that includes Hsp70 itself in addition to Hsp40 and Hsp110 (167, 170, 173, 183–186). Hsp70 binds to the MD of Hsp104 and is thought to bind polypeptides in order to deliver partially unfolded stands of polypeptides to Hsp104 (Fig. 3A) (187–190). Though Hsp70 stimulates the disaggregase activity of Hsp104, unlike VCP and the AAA+ ATPases of the proteasome which require cofactors or additional subunits to engage substrate, Hsp104 has an intrinsic ability to engage and process specific aggregates (11, 166, 167, 172, 191–193). Hsp70, however, may still be important for targeting certain types of aggregates, and thus should be considered in proposals to use Hsp104 as a therapy for diseases of protein aggregation (181, 186, 190, 194).
Structures of Hsp104 by cryo-EM at high resolution have been generally difficult to determine. However, Gates et al were able to capture Hsp104 processing casein, a model substrate, at about 4.0 Å resolution to elucidate one mechanism Hsp104 uses to process substrate (Fig. 3A). Yokom et al published a relatively high-resolution (5.6 Å) structure of Hsp104 bound to AMP-PNP; however, this structure likely represents an inactive state for Hsp104 when compared to the Hsp104 structure bound to casein reported in Gates et al. Gates et al found 2 structures of relatively equivalent proportions in their samples that they deemed “closed” and “extended” states for Hsp104. The structures resemble those seen for VCP and the proteasome, indicating that at least one mode of processing by Hsp104 is similar to that of VCP and the proteasome.
The “closed” structure shows 5 subunits contacting casein within the pore, while the sixth subunit breaks the helical structure at an intermediate elevation compared to the other 5 subunits (Fig. 3C). The structured pore loops show that aromatic residues Y257 and Y662 are important for substrate contact and thus translocation of substrate through the central pore. Similar to VCP and the proteasome, every other amino acid is bound by adjacent subunits so that each power stroke of the hexamer moves the 2 substrate amino acids through the pore.
The “extended” conformation shows an intermediate step in the processing pathway that has not been seen in proteasome or VCP structures, but may exist in their respective pathways, depending on the order of the top monomer binding substrate and the bottom monomer releasing substrate. The “extended” conformation shows all 6 subunits bound to substrate (Fig. 3C). The “extended” conformation is likely the state immediately before or after the “closed” conformation, in which the subunit at the top of the staircase has just contacted substrate and the subunit at the bottom of the staircase has yet to release substrate. However, the “extended” conformation may also represent a distinct step from the proteasome pathway. Structures of the proteasome have shown 2 subunits unbound from substrate, raising the question of which step comes first: the release of substrate or the engagement of substrate at the top of the helical staircase (34). Additionally, either the proteasome structure with 2 unbound subunits or the Hsp104 structure with 6 bound subunits may represent a nonphysiologic state due to the addition of ATPγS or a deubiquitination inhibitor (34, 165). No matter which theory is true, the “extended” state is a state that has been seen in Hsp104 by cryo-EM, but not with either the proteasome or VCP.
The nucleotide state of the “extended” structure is reminiscent of that seen with VCP, where 5 subunits are bound to ATP and are either bound to substrate or are primed to bind substrate, with the sixth subunit bound to ADP, preparing to disengage from substrate (14, 165). The nucleotide state of the “closed” structure may provide insight into the order in which the 2 ATPase rings hydrolyze ATP to prepare for disengagement. The “closed” structure shows 4 D1 subunits bound to ATP and 3 D2 subunits bound to ATP, indicating that D2 may hydrolyze ATP before D1 to release substrate at the bottom of the staircase (165).
Hsp104 likely needs both complete and partial translocations to be an effective disaggregase (Fig. 3B) (167, 195). Gates et al propose that switching between the inactive or “open” state seen bound to ADP or AMP-PNP and the “closed” state they present may allow for the partial translocation or “pulling” on substrates (165, 196). It is additionally thought that maybe Hsp104 can adapt to different substrates (167). As previously noted, Hsp104 has a unique ability to initiate disaggregation at an internal segment (Fig. 3B) (180, 182). Though no structure to date has shown this explicitly, the seam seen between the top subunit and its counterclockwise neighbor may allow for substrate insertion from the side of Hsp104 rather than through the top of the pore (165, 196, 197). As has been seen in structures of VCP, the proteasome, and Hsp104, when ATP is hydrolyzed there is increased ring flexibility to allow for large movements of a subunit to the top of the ring (14, 15, 33, 34, 165). The increased flexibility may allow for the ring to open enough for substrate to enter from the side, conveying an expanded array of substrates that can be processed. If Hsp104 indeed has a greater ability to process aggregates and help cells cope with stress, it may be of great benefit to treating or preventing neurodegenerative diseases associated with aggregates.
C9orf72
A hexanucleotide GGGGCC repeat expansion (sometimes referred to as G4C2) in the 5′ noncoding region of the gene C9orf72 accounts for most familial cases and some sporadic cases of ALS/FTD (198–200). The effects of the C9orf72 hexanucleotide repeats are still actively studied but have generally been put into 2 boxes: loss of function and GOF. Loss-of-function effects include decreased C9orf72 transcript and protein (198, 199, 201, 202). C9orf72 is essential for monocyte function and normal axonal actin function in motor neurons, with restoration of WT C9orf72 rescuing model neurons (202–204). GOF effects include the generation of dipeptide repeat proteins and also perhaps RNA G-quadruplexes that disrupt neural function and lead to neural death (60, 205–215). Although the GGGGCC repeat is found in a noncoding region, there are sense and antisense transcripts translated in all reading frames which produce 5 different DPR’s (GA, GR, GP, PR, PA), with inclusions mostly containing poly-GA (215–219). Poly-GA has been shown to be toxic both in primary neuron cultures and in mice (220–225). With poly-GA toxicity, aggregates sequester UPS components and cells show an impairment in the UPS (221, 223–225). Parts of the C9orf72 complex itself have also been shown to bind 65 proteins involved in neurodegenerative disease, including the 26S proteasome and other UPS components (8). C9orf72 is unique because it is involved in both regulating proteostasis and aggregation, bridging 2 domains of neurodegenerative disease pathophysiology.
C9orf72, SMCR8, and WDR41 form a complex (CSW) that helps to regulate autophagy through interactions with ULK1 and other proteins (Fig. 4A) (7, 8, 226–228). Knocking out C9orf72 causes a defect in autophagy, indicating that C9orf72 is a positive regulator of this process. In contrast, an SMCR8 knockout causes increased ULK1 expression, suggesting that it is a negative regulator (226, 229, 230). WDR41 has been localized to the ER and is thought to target the CSW complex to lysosomes via PQLC2 interaction (231–234). C9orf72 and SMCR8 have DENN domains and are both predicted to be part of the DENN family of proteins that are best known for their role as guanine nucleotide exchange factors (GEFs) for Rab GTPases (Fig. 4A) (235–240). Rab proteins are GTPases generally involved in membrane trafficking and are key in a diversity functions depending on their location. The C9orf72/SMCR8 complex has been shown to regulate Rab-positive endosomes, which regulate protein trafficking, and Rab8a/Rab39b in membrane transport that may be involved in axonal growth and ciliogenesis in neurons (226, 241–244). Though the complete function of C9orf72 remains elusive, 2 independently solved structures of the CSW complex using cryo-EM led to similar conclusions about the function of C9orf72 and the CSW complex (Fig. 4A) (245, 246).
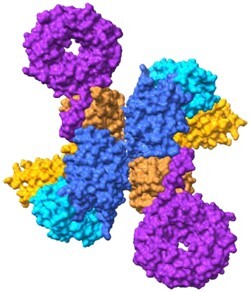
FIGURE 4.
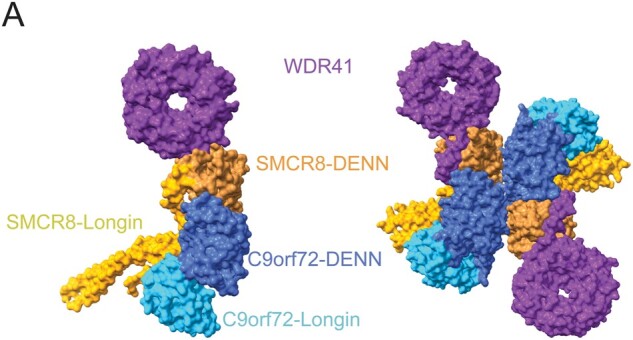
Structure of C9orf72 in complex with SMCR8, WDR41. (A) Monomeric and dimeric structures of C9orf72 (blue), SMCR8 (orange), WDR41 (purple) complex with key domains labeled. The DENN domains are predicted to be guanine nucleotide exchange factors for Rab GTPases. The DENN domains are the interface of C9orf72 and SMCR8 (PDB: 6LT0, 6V4U).
The 2 recently solved structures of the CSW complex found very similar structures, except that one was a dimer of trimers and the other was a solitary trimer (Fig. 4A) (245, 246). This difference may be due to in vitro artifacts, but further studies are required to determine the physiologic relevance and function of the dimerization. Each trimer showed that C9orf72 uses its DENN domain to bind to the DENN domain of SMCR8. WDR41 binds the DENN domain of SMCR8 but does not physically contact C9orf72 (Fig. 4A). C9orf72 alone cannot bind WDR41 (245). SMCR8 was shown to cluster with PQLC2 in cells under starved conditions, but only if it was able to bind to WDR41, indicating that this interaction between SMCR8 and WDR41 is necessary to target the CSW complex to lysosomes (246).
The dimer of trimers solved by Tang et al shows that the dimer has an almost 2-fold rotational symmetry and the dimer interface is located at the C-terminus of C9orf72 and the DENN of SMCR8 (Fig. 4A). A ΔCTR mutant (deletion of the C-terminal region of C9orf72) was able to make the singular trimeric CSW complex, with the same affinity of C9orf72/SMCR8 to WDR41. However, this mutant prevented dimerization (245). The C-terminal region of C9orf72 (amino acids 461–481) is highly conserved between species, thus there may be conserved functionality of this region (245). If the dimerization of the trimeric CSW complex is physiologic, the C-terminal region of C9orf72 likely has a key role in mediating the complex’s structure.
Predicted as a likely a GEF due to the DENN domains, structural comparison to known functional GTPase activating proteins (GAPs) showed that maybe the CSW complex had a different function than what was predicted due to similar motifs (245–249). Further characterization showed that CSW is not actually a GEF as predicted by the DENN domains in C9orf72 and SMCR8, but is in fact a GAP (245, 246). CSW is a GAP for Rab8a/11a and Arf1/5/6. Rab8a and Rab11a play a role in ciliogenesis and axonal growth in neurons (243, 244). Arf’s are found on golgi, endosomes, plasma membranes, and cytoskeleton and within the cytosol (250). CSW, however, did not act as a GAP for lysosomal Arf-like proteins Ar18a and Ar18b (246).
C9orf72 alone was not sufficient to stimulate Rab hydrolysis of GTP. However, the CSW complex was able to affect Rab8a and Rab11a with or without WDR41 (245). Additionally, the CSW complex was able to stimulate Rab8a and Rab11a as either a single trimer or a dimer of trimers, leaving the functional relevance of a dimer of the CSW complex still unresolved (245).
Though complex, subtle, and largely still a mystery, determining the correct function of the CSW complex is important in understanding the mechanism of disease, how to make effective models, and how to effectively treat disease. The recent structures of the CSW complex show the power of cryo-EM to provide insight into protein function. Already, the published structures of the CSW complex have shed light on how C9orf72 and the CSW complex may affect autophagy and the proteostasis pathway indirectly. Whatever the role of C9orf72 in neurodegeneration, the functional data indicate that it likely does not act alone. SMCR8 has emerged as a key factor in C9orf72-mediated activity. With much focus on C9orf72 as a causative agent in ALS/FTD, SMCR8 and other proteins that interact with C9orf72 may be of great interest in resolving the mechanism of disease development and progression.
In addition to in vitro characterization of the CSW complex, in situ structural analysis of cells similar to those seen in C9orf72 patients has provided insight on the mechanism of disease. Using GFP-tagged poly-GA in rat neurons, Guo et al created aggregates of similar size and intensity to those seen in C9orf72 patient tissues (60). Cryo-ET was used to image the neurons and visualize the aggregates in situ. Unlike single-particle cryo-EM, cryo-ET allows for imaging inside of cells in an even more physiologic environment than in vitro experiments. Densities near the poly-GA aggregates were identified as the 26S proteasome. The proteasome was found at about 30-fold higher concentration in the region of aggregates compared to the body of the neuron and to cellular processes of control neurons that were previously imaged by cryo-ET (60, 251). Proteasome expression did not increase, thus the increase in concentration was due to sequestration of the UPS in the region of the aggregates. Guo et al deemed many proteasome structures to be “stalled” on the aggregates (60). This insight provided by in situ analysis may explain the dysfunction seen in the UPS with DPRs. If the UPS is being sequestered and even stalled by aggregates without a compensatory increase in expression, then the UPS cannot effectively perform normal proteostasis in other parts of the cell. Without proper UPS function, cells are at risk for aggregation and general imbalance of their proteome, which may lead to neurodegenerative diseases such as ALS and FTD.
C9orf72 has an interesting role in the pathophysiology of ALS and FTD as a promoter of normal proteostasis in the CSW complex and in the inhibition of normal proteostasis due to pathologic aggregates that impair or sequester the UPS. Unlike the other proteins reviewed here (VCP, proteasome, Hsp104), our understanding of C9orf72 protein function in disease pathophysiology is relatively less understood, and indeed it is yet uncertain whether C9orf72 mutations cause disease through GOF mechanisms that are distinct from the normal function of C9orf72 protein.
CONCLUSION
Our structural understanding of proteostasis factors has greatly expanded within the past 4 years thanks to the effective implementation of new cryo-EM technologies. This enhanced structural understanding of VCP, the 26S proteasome, Hsp104, and C9orf72 has also provided insight into their function and their broad roles in cellular functions (Table). Knowledge of the structure and function of these factors moves the field of neurodegeneration one step closer to understanding how the proteostasis pathway fails and why it fails, an understanding that is likely necessary to effectively combat neurodegenerative disease. Understanding how these protein pathways function may allow for better disease models. Current disease models in animals or cells may not take into account the proteostasis pathways to most effectively model true human-relevant neurodegenerative disease.
With our aging demographic and the increasing burden of neurodegenerative diseases, effective therapies are lacking. Current therapies do not target pathophysiologic mechanisms. Thus far, direct targeting of protein aggregates has not proven therapeutically effective. As has been discussed here, other factors besides aggregates may play a driving or primary role in disease course. Targeting these proteostasis factors may prove fruitful in terms of understanding disease mechanisms and developing novel therapeutics.
This study was supported by NIH (R01NS095792, R56AG063344, P01AG066597, P01AG010124, P01AG062418, U54NS115322, and U19AG062418 to E.B.L.) and a David and Lucile Packard Fellowship for Science and Engineering (2019-69645 to Y.-W.C.).
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Bard JAM, Goodall EA, Greene ER, et al. Structure and function of the 26S proteasome. Annu Rev Biochem 2018;87:697–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciechanover A, Brundin P.. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003;40:427–46 [DOI] [PubMed] [Google Scholar]
- 3. Xia D, Tang WK, Ye Y.. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016;583:64–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van den Boom J, Meyer H.. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 2018;69:182–94 [DOI] [PubMed] [Google Scholar]
- 5. Ye Y, Tang WK, Zhang T, et al. A mighty “protein extractor” of the cell: Structure and function of the p97/CDC48 ATPase. Front Mol Biosci 2017;4: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Darwich NF, Phan JM, Kim B, et al. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science 2020;370:eaay8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cali CP, Patino M, Tai YK, et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol 2019;138:795–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodier JL, Soares AO, Pereira GC, et al. C9orf72-associated SMCR8 protein binds in the ubiquitin pathway and with proteins linked with neurological disease. Acta Neuropathol Commun 2020;8:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castellano LM, Bart SM, Holmes VM, et al. Repurposing Hsp104 to antagonize seminal amyloid and counter HIV infection. Chem Biol 2015;22:1074–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackrel ME, Shorter J.. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis Model Mech 2014;7:1175–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackrel ME, DeSantis ME, Martinez BA, et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014;156:170–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo L, Kim HJ, Wang H, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 2018;173:677–92.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davies JM, Brunger AT, Weis WI.. Improved structures of full-length p97, an AAA ATPase: Implications for mechanisms of nucleotide-dependent conformational change. Structure 2008;16:715–26 [DOI] [PubMed] [Google Scholar]
- 14. Twomey EC, Ji Z, Wales TE, et al. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019;365:eaax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cooney I, Han H, Stewart MG, et al. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 2019;365:502–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benjin X, Ling L.. Developments, applications, and prospects of cryo‐electron microscopy. Protein Sci Publ Protein Soc 2020;29:872–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taylor KA, Glaeser RM.. Electron diffraction of frozen, hydrated protein crystals. Science 1974;186:1036–7 [DOI] [PubMed] [Google Scholar]
- 18. Nakane T, Kotecha A, Sente A, et al. Single-particle cryo-EM at atomic resolution. Nature 2020;587:152–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peters J, Walsh M, Franke W.. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J 1990;9:1757–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson DJ, Le Moigne R, Djakovic S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015;28:653–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weith M, Seiler J, van den Boom J, et al. Ubiquitin-independent disassembly by a p97 AAA-ATPase complex drives PP1 holoenzyme formation. Mol Cell 2018;72:766–77.e6 [DOI] [PubMed] [Google Scholar]
- 22. Kracht M, van den Boom J, Seiler J, et al. Protein phosphatase-1 complex disassembly by p97 is initiated through multivalent recognition of catalytic and regulatory subunits by the p97 SEP-domain adapters. J Mol Biol 2020;432:6061–74 [DOI] [PubMed] [Google Scholar]
- 23. Watts GDJ, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–81 [DOI] [PubMed] [Google Scholar]
- 24. Kimonis VE, Fulchiero E, Vesa J, et al. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim Biophys Acta 2008;1782:744–8 [DOI] [PubMed] [Google Scholar]
- 25. Johnson JO, Mandrioli J, Benatar M, et al. ; ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ching JK, Elizabeth SV, Ju J-S, et al. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet 2013;22:1167–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ju J-S, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 2009;187:875–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ritz D, Vuk M, Kirchner P, et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 2011;13:1116–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang T, Mishra P, Hay BA, et al. Valosin-containing protein (VCP/p97) inhibitors relieve mitofusin-dependent mitochondrial defects due to VCP disease mutants. eLife 2017;6:e17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu X, Rapoport TA.. Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 2018;53:22–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olszewski MM, Williams C, Dong KC, et al. The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun Biol 2019;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prakash S, Tian L, Ratliff KS, et al. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol 2004;11:830–7 [DOI] [PubMed] [Google Scholar]
- 33. Dong Y, Zhang S, Wu Z, et al. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019;565:49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de la Peña AH, Goodall EA, Gates SN, et al. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 2018;362:eaav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beskow A, Grimberg KB, Bott LC, et al. A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J Mol Biol 2009;394:732–46 [DOI] [PubMed] [Google Scholar]
- 36. Bodnar NO, Kim KH, Ji Z, et al. Structure of the cdc48 atpase with its ubiquitin-binding cofactor ufd1–npl4. Nat Struct Mol Biol 2018;25:616–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Banerjee S, Bartesaghi A, Merk A, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016;351:871–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Saracino D, Clot F, Camuzat A, et al. ; French research network on FTD/FTD-ALS. Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol Aging 2018;72:187.e11–e14. [DOI] [PubMed] [Google Scholar]
- 39. Hänzelmann P, Schindelin H.. The interplay of cofactor interactions and post-translational modifications in the regulation of the AAA+ ATPase p97. Front Mol Biosci 2017;4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruderer RM, Brasseur C, Meyer HH.. The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1-Npl4 and p47, through a common bipartite binding mechanism. J Biol Chem 2004;279:49609–16 [DOI] [PubMed] [Google Scholar]
- 41. Blythe EE, Olson KC, Chau V, et al. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc Natl Acad Sci U S A 2017;114:E4380–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dai RM, Li C.. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin–proteasome degradation. Nat Cell Biol 2001;3:740–4 [DOI] [PubMed] [Google Scholar]
- 43. Ye Y, Meyer HH, Rapoport TA.. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001;414:652–6 [DOI] [PubMed] [Google Scholar]
- 44. Meyer HH, Shorter JG, Seemann J, et al. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 2000;19:2181–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pye VE, Beuron F, Keetch CA, et al. Structural insights into the p97-Ufd1-Npl4 complex. Proc Natl Acad Sci U S A 2007;104:467–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Isaacson RL, Pye VE, Simpson P, et al. Detailed structural insights into the p97-Npl4-Ufd1 interface. J Biol Chem 2007;282:21361–9 [DOI] [PubMed] [Google Scholar]
- 47. Alam SL, Sun J, Payne M, et al. Ubiquitin interactions of NZF zinc fingers. EMBO J 2004;23:1411–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pan M, Zheng Q, Yu Y, et al. Seesaw conformations of Npl4 in the human p97 complex and the inhibitory mechanism of a disulfiram derivative. Nat Commun 2021;12:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ye Y, Meyer HH, Rapoport TA.. Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 2003;162:71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein A, Ruggiano A, Carvalho P, et al. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014;158:1375–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bodnar NO, Rapoport TA.. Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 2017;169:722–35.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. White SR, Lauring B.. AAA+ ATPases: Achieving diversity of function with conserved machinery. Traffic 2007;8:1657–67 [DOI] [PubMed] [Google Scholar]
- 53. Puchades C, Rampello AJ, Shin M, et al. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 2017;358:eaao0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Han H, Monroe N, Sundquist WI, et al. The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 2017;6:e31324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lo Y-H, Sobhany M, Hsu AL, et al. Cryo-EM structure of the essential ribosome assembly AAA-ATPase Rix7. Nat Commun 2019;10:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodriguez-Aliaga P, Ramirez L, Kim F, et al. Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat Struct Mol Biol 2016;23:974–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esaki M, Islam MT, Tani N, et al. Deviation of the typical AAA substrate-threading pore prevents fatal protein degradation in yeast Cdc48. Sci Rep 2017;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carrion-Vazquez M, Li H, Lu H, et al. The mechanical stability of ubiquitin is linkage dependent. Nat Struct Biol 2003;10:738–43 [DOI] [PubMed] [Google Scholar]
- 59. Thrower JS, Hoffman L, Rechsteiner M, et al. Recognition of the polyubiquitin proteolytic signal. EMBO J 2000;19:94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guo Q, Lehmer C, Martínez-Sánchez A, et al. In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell 2018;172:696–705.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chou T-F, Bulfer SL, Weihl CC, et al. Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J Mol Biol 2014;426:2886–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Halawani D, LeBlanc AC, Rouiller I, et al. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol Cell Biol 2009;29:4484–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niwa H, Ewens CA, Tsang C, et al. The role of the N-domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J Biol Chem 2012;287:8561–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang X, Gui L, Zhang X, et al. Altered cofactor regulation with disease-associated p97/VCP mutations. Proc Natl Acad Sci U S A 2015;112:E1705–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ju J-S, Weihl CC.. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum Mol Genet 2010;19:R38–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tang WK, Xia D.. Mutations in the human AAA+ chaperone p97 and related diseases. Front Mol Biosci 2016;3:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Blythe EE, Gates SN, Deshaies RJ, et al. Multisystem proteinopathy mutations in VCP/p97 article multisystem proteinopathy mutations. Structure 2019;27:1820–9.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tang WK, Li D, Li C, et al. A novel ATP-dependent conformation in p97 N–D1 fragment revealed by crystal structures of disease-related mutants. EMBO J 2010;29:2217–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tang WK, Zhang T, Ye Y, et al. Structural basis for nucleotide-modulated p97 association with the ER membrane. Cell Discov 2017;3:17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huang R, Ripstein ZA, Rubinstein JL, et al. Cooperative subunit dynamics modulate p97 function. Proc Natl Acad Sci U S A 2019;116:158–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schuetz AK, Kay LE.. A dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 2016;5:e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schütz AK, Rennella E, Kay LE.. Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proc Natl Acad Sci U S A 2017;114:E6822–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tang WK, Xia D.. Altered intersubunit communication is the molecular basis for functional defects of pathogenic p97 mutants. J Biol Chem 2013;288:36624–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated diseases: Inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 2011;45:522–31 [DOI] [PubMed] [Google Scholar]
- 75. Arakhamia T, Lee CE, Carlomagno Y, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 2020;180:633–44.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ye Y, Klenerman D, Finley D.. N-terminal ubiquitination of amyloidogenic proteins triggers removal of their oligomers by the proteasome holoenzyme. J Mol Biol 2020;432:585–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schweighauser M, Shi Y, Tarutani A, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020;585:464–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kimura Y, Fukushi J, Hori S, et al. Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 2013;18:1131–43 [DOI] [PubMed] [Google Scholar]
- 79. Kirchner P, Bug M, Meyer H.. Ubiquitination of the N-terminal region of caveolin-1 regulates endosomal sorting by the VCP/p97 AAA-ATPase. J Biol Chem 2013;288:7363–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Papadopoulos C, Kirchner P, Bug M, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 2017;36:135–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Richly H, Rape M, Braun S, et al. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 2005;120:73–84 [DOI] [PubMed] [Google Scholar]
- 82. Braun S, Matuschewski K, Rape M, et al. Role of the ubiquitin-selective CDC48UFD1/NPL4 chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J 2002;21:615–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Song EJ, Yim S-H, Kim E, et al. Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol Cell Biol 2005;25:2511–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kondo H, Rabouille C, Newman R, et al. p47 is a cofactor for p97-mediated membrane fusion. Nature 1997;388:75–8 [DOI] [PubMed] [Google Scholar]
- 85. Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, et al. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res 2016;26:869–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Finley D, Chen X, Walters KJ.. Gates, channels, and switches: Elements of the proteasome machine. Trends Biochem Sci 2016;41:77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Moon SL, Morisaki T, Stasevich TJ, et al. Coupling of translation quality control and mRNA targeting to stress granules. J Cell Biol 2020;219:e202004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tomko RJ, Hochstrasser M.. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem 2013;82:415–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kaneko M, Koike H, Saito R, et al. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci 2010;30:3924–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee MJ, Lee JH, Rubinsztein DC.. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013;105:49–59 [DOI] [PubMed] [Google Scholar]
- 91. Keck S, Nitsch R, Grune T, et al. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem 2003;85:115–22 [DOI] [PubMed] [Google Scholar]
- 92. Bedford L, Hay D, Devoy A, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci 2008;28:8189–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Paine SML, Anderson G, Bedford K, et al. Pale body-like inclusion formation and neurodegeneration following depletion of 26S proteasomes in mouse brain neurones are independent of α-synuclein. PLoS One 2013;8:e54711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wahl C, Kautzmann S, Krebiehl G, et al. A comprehensive genetic study of the proteasomal subunit S6 ATPase in German Parkinson’s disease patients. J Neural Transm (Vienna) 2008;115:1141–8 [DOI] [PubMed] [Google Scholar]
- 95. Tseng BP, Green KN, Chan JL, et al. Aβ inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 2008;29:1607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mayer RJ, Lowe J, Lennox G, et al. Intermediate filaments and ubiquitin: A new thread in the understanding of chronic neurodegenerative diseases. Prog Clin Biol Res 1989;317:809–18 [PubMed] [Google Scholar]
- 97. Eisele MR, Reed RG, Rudack T, et al. Expanded coverage of the 26S proteasome conformational landscape reveals mechanisms of peptidase gating. Cell Rep 2018;24:1301–15.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen S, Wu J, Lu Y, et al. Structural basis for dynamic regulation of the human 26S proteasome. Proc Natl Acad Sci U S A 2016;113:12991–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhu Y, Wang WL, Yu D, et al. Structural mechanism for nucleotide-driven remodeling of the AAA-ATPase unfoldase in the activated human 26S proteasome. Nat Commun 2018;9:1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wehmer M, Rudack T, Beck F, et al. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc Natl Acad Sci U S A 2017;114:1305–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schweitzer A, Aufderheide A, Rudack T, et al. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci U S A 2016;113:7816–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lasker K, Förster F, Bohn S, et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci U S A 2012;109:1380–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lander GC, Estrin E, Matyskiela ME, et al. Complete subunit architecture of the proteasome regulatory particle. Nature 2012;482:186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. da Fonseca PCA, He J, Morris EP.. Molecular model of the human 26S proteasome. Mol Cell 2012;46:54–66 [DOI] [PubMed] [Google Scholar]
- 105. Huang X, Luan B, Wu J, et al. An atomic structure of the human 26S proteasome. Nat Struct Mol Biol 2016;23:778–85 [DOI] [PubMed] [Google Scholar]
- 106. Luan B, Huang X, Wu J, et al. Structure of an endogenous yeast 26S proteasome reveals two major conformational states. Proc Natl Acad Sci U S A 2016;113:2642–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Beck F, Unverdorben P, Bohn S, et al. Near-atomic resolution structural model of the yeast 26S proteasome. Proc Natl Acad Sci U S A 2012;109:14870–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Śledź P, Unverdorben P, Beck F, et al. Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc Natl Acad Sci U S A 2013;110:7264–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ding Z, Fu Z, Xu C, et al. High-resolution cryo-EM structure of the proteasome in complex with ADP-AlFx. Cell Res 2017;27:373–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Unverdorben P, Beck F, Śledź P, et al. Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc Natl Acad Sci U S A 2014;111:5544–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Matyskiela ME, Lander GC, Martin A.. Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol 2013;20:781–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Dambacher CM, Worden EJ, Herzik MA, et al. Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. eLife 2016;5:e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. da Fonseca PCA, Morris EP.. Cryo-EM reveals the conformation of a substrate analogue in the human 20S proteasome core. Nat Commun 2015;6:7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen X, Dorris Z, Shi D, et al. Cryo-EM reveals unanchored M1-ubiquitin chain binding at hRpn11 of the 26S proteasome. Structure 2020;28:1206–17.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Groll M, Ditzel L, Löwe J, et al. Structure of 20S proteasome from yeast at 2.4Å resolution. Nature 1997;386:463–71 [DOI] [PubMed] [Google Scholar]
- 116. Lu Y, Wu J, Dong Y, et al. Conformational landscape of the p28-bound human proteasome regulatory particle. Mol Cell 2017;67:322–33.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Baumeister W, Walz J, Zühl F, et al. The proteasome: Paradigm of a self-compartmentalizing protease. Cell 1998;92:367–80 [DOI] [PubMed] [Google Scholar]
- 118. Groll M, Bajorek M, Köhler A, et al. A gated channel into the proteasome core particle. Nat Struct Biol 2000;7:1062–7 [DOI] [PubMed] [Google Scholar]
- 119. Komander D, Rape M.. The ubiquitin code. Annu Rev Biochem 2012;81:203–29 [DOI] [PubMed] [Google Scholar]
- 120. Lu Y, Lee B, King RW, et al. Substrate degradation by the proteasome: A single-molecule kinetic analysis. Science 2015;348:1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fishbain S, Prakash S, Herrig A, et al. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat Commun 2011;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Inobe T, Fishbain S, Prakash S, et al. Defining the geometry of the two-component proteasome degron. Nat Chem Biol 2011;7:161–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yao T, Cohen RE.. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002;419:403–7 [DOI] [PubMed] [Google Scholar]
- 124. Verma R, Aravind L, Oania R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002;298:611–5 [DOI] [PubMed] [Google Scholar]
- 125. Glickman MH, Rubin DM, Coux O, et al. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 1998;94:615–23 [DOI] [PubMed] [Google Scholar]
- 126. Maytal-Kivity V, Reis N, Hofmann K, et al. MPN+, a putative catalytic motif found in a subset of MPN domain proteins from eukaryotes and prokaryotes, is critical for Rpn11 function. BMC Biochem 2002;3:28– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Guterman A, Glickman MH.. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem 2004;279:1729–38 [DOI] [PubMed] [Google Scholar]
- 128. Rinaldi T, Pick E, Gambadoro A, et al. Participation of the proteasomal lid subunit Rpn11 in mitochondrial morphology and function is mapped to a distinct C-terminal domain. Biochem J 2004;381:275–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. van Nocker S, Sadis S, Rubin DM, et al. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol Cell Biol 1996;16:6020–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Deveraux Q, Ustrell V, Pickart C, et al. A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem 1994;269:7059–61 [PubMed] [Google Scholar]
- 131. Glickman MH, Rubin DM, Fried VA, et al. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol Cell Biol 1998;18:3149–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tomko RJ, Funakoshi M, Schneider K, et al. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: Implications for proteasome structure and assembly. Mol Cell 2010;38:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rabl J, Smith DM, Yu Y, et al. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell 2008;30:360–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Smith DM, Chang S-C, Park S, et al. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s α ring opens the gate for substrate entry. Mol Cell 2007;27:731–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Worden EJ, Dong KC, Martin A.. An AAA motor-driven mechanical switch in Rpn11 controls deubiquitination at the 26S proteasome. Mol Cell 2017;67:799–811.e8 [DOI] [PubMed] [Google Scholar]
- 136. Zhang F, Hu M, Tian G, et al. Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell 2009;34:473–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Erales J, Coffino P.. Ubiquitin-independent proteasomal degradation. Biochim Biophys Acta 2014;1843:216–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Iosefson O, Nager AR, Baker TA, et al. Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat Chem Biol 2015;11:201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Iosefson O, Olivares AO, Baker TA, et al. Dissection of axial-pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Rep 2015;12:1032–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Glynn SE, Martin A, Nager AR, et al. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 2009;139:744–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. van der Lee R, Lang B, Kruse K, et al. Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep 2014;8:1832–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Erives AJ, Fassler JS.. Metabolic and chaperone gene loss marks the origin of animals: Evidence for Hsp104 and Hsp78 chaperones sharing mitochondrial enzymes as clients. PLoS One 2015;10:e0117192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sweeny EA, Shorter J.. Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J Mol Biol 2016;428:1870–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Carmichael J, Chatellier J, Woolfson A, et al. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc Natl Acad Sci U S A 2000;97:9701–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Satyal SH, Schmidt E, Kitagawa K, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A 2000;97:5750–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bao YP, Cook LJ, O'Donovan D, et al. Mammalian, yeast, bacterial, and chemical chaperones reduce aggregate formation and death in a cell model of oculopharyngeal muscular dystrophy. J Biol Chem 2002;277:12263–9 [DOI] [PubMed] [Google Scholar]
- 147. Vacher C, Garcia-Oroz L, Rubinsztein DC.. Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum Mol Genet 2005;14:3425–33 [DOI] [PubMed] [Google Scholar]
- 148. Perrin V, Régulier E, Abbas-Terki T, et al. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington’s disease. Mol Ther 2007;15:903–11 [DOI] [PubMed] [Google Scholar]
- 149. Lo Bianco C, Shorter J, Régulier E, et al. Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J Clin Invest 2008;118:3087–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Cushman-Nick M, Bonini NM, Shorter J.. Hsp104 suppresses polyglutamine-induced degeneration post onset in a Drosophila MJD/SCA3 model. PLoS Genet 2013;9:e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shorter J. Engineering therapeutic protein disaggregases. Mol Biol Cell 2016;27:1556–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Shorter J. Designer protein disaggregases to counter neurodegenerative disease. Curr Opin Genet Dev 2017;44:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Yasuda K, Clatterbuck-Soper SF, Jackrel ME, et al. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol 2017;216:1015–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lee DH, Goldberg AL.. Hsp104 is essential for the selective degradation in yeast of polyglutamine expanded ataxin-1 but not most misfolded proteins generally. Biochem Biophys Res Commun 2010;391:1056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Taxis C, Hitt R, Park S-H, et al. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem 2003;278:35903–13 [DOI] [PubMed] [Google Scholar]
- 156. Preston GM, Guerriero CJ, Metzger MB, et al. Substrate insolubility dictates Hsp104-dependent endoplasmic-reticulum-associated degradation. Mol Cell 2018;70:242–53.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ruan L, Zhou C, Jin E, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017;543:443–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wallace EWJ, Kear-Scott JL, Pilipenko EV, et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 2015;162:1286–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kroschwald S, Maharana S, Mateju D, et al. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015;4:e06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Wolozin B, Ivanov P.. Stress granules and neurodegeneration. Nat Rev Neurosci 2019;20:649–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Cherkasov V, Hofmann S, Druffel-Augustin S, et al. Coordination of translational control and protein homeostasis during severe heat stress. Curr Biol 2013;23:2452–62 [DOI] [PubMed] [Google Scholar]
- 162. Rosenzweig R, Farber P, Velyvis A, et al. ClpB N-terminal domain plays a regulatory role in protein disaggregation. Proc Natl Acad Sci U S A 2015;112:E6872–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Doyle SM, Hoskins JR, Wickner S.. DnaK chaperone-dependent disaggregation by caseinolytic peptidase B (ClpB) mutants reveals functional overlap in the N-terminal domain and nucleotide-binding domain-1 pore tyrosine. J Biol Chem 2012;287:28470–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mackay RG, Helsen CW, Tkach JM, et al. The C-terminal extension of Saccharomyces cerevisiae Hsp104 plays a role in oligomer assembly. Biochemistry 2008;47:1918–27 [DOI] [PubMed] [Google Scholar]
- 165. Gates SN, Yokom AL, Lin J, et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017;357:273–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Shorter J, Lindquist S.. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004;304:1793–7 [DOI] [PubMed] [Google Scholar]
- 167. DeSantis ME, Leung EH, Sweeny EA, et al. Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012;151:778–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kroschwald S, Munder MC, Maharana S, et al. Different material states of Pub1 condensates define distinct modes of stress adaptation and recovery. Cell Rep 2018;23:3327–39 [DOI] [PubMed] [Google Scholar]
- 169. Parsell DA, Kowal AS, Singer MA, et al. Protein disaggregation mediated by heat-shock protein Hspl04. Nature 1994;372:475–8 [DOI] [PubMed] [Google Scholar]
- 170. Glover JR, Lindquist S.. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 1998;94:73–824 [DOI] [PubMed] [Google Scholar]
- 171. Liu Y-H, Han Y-L, Song J, et al. Heat shock protein 104 inhibited the fibrillization of prion peptide 106–126 and disassembled prion peptide 106–126 fibrils in vitro. Int J Biochem Cell Biol 2011;43:768–74 [DOI] [PubMed] [Google Scholar]
- 172. Shorter J, Lindquist S.. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell 2006;23:425–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Shorter J, Lindquist S.. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J 2008;27:2712–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. DeSantis ME, Shorter J.. Hsp104 drives “protein-only” positive selection of Sup35 prion strains encoding strong [PSI+]. Chem Biol 2012;19:1400–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Klaips CL, Hochstrasser ML, Langlois CR, et al. Spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. eLife 2014;3:e04288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Park Y-N, Zhao X, Yim Y-I, et al. Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot Cell 2014;13:635–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Zhao X, Rodriguez R, Silberman RE, et al. Heat shock protein 104 (Hsp104)-mediated curing of [PSI+] yeast prions depends on both [PSI+] conformation and the properties of the Hsp104 homologs. J Biol Chem 2017;292:8630–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Lum R, Tkach JM, Vierling E, et al. Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 2004;279:29139–46 [DOI] [PubMed] [Google Scholar]
- 179. Lum R, Niggemann M, Glover JR.. Peptide and protein binding in the axial channel of Hsp104 insights into the mechanism of protein unfolding. J Biol Chem 2008;283:30139–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Haslberger T, Zdanowicz A, Brand I, et al. Protein disaggregation by the AAA+ chaperone ClpB involves partial threading of looped polypeptide segments. Nat Struct Mol Biol 2008;15:641–50 [DOI] [PubMed] [Google Scholar]
- 181. Tessarz P, Mogk A, Bukau B.. Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol Microbiol 2008;68:87–97 [DOI] [PubMed] [Google Scholar]
- 182. Sweeny EA, Jackrel ME, Gupta K, et al. The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol Cell 2015;57:836–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Cashikar AG, Duennwald M, Lindquist SL.. A chaperone pathway in protein disaggregation Hsp26 alters the nature of protein aggregates to facilitate reactivation by Hsp104. J Biol Chem 2005;280:23869–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Haslbeck M, Miess A, Stromer T, et al. Disassembling protein aggregates in the yeast cytosol the cooperation of Hsp26 with SSA1 and HSP104. J Biol Chem 2005;280:23861–8 [DOI] [PubMed] [Google Scholar]
- 185. Shorter J. The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 2011;6:e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kaimal JM, Kandasamy G, Gasser F, et al. Coordinated Hsp110 and Hsp104 activities power protein disaggregation in Saccharomyces cerevisiae. Mol Cell Biol 2017;37:e00027-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Haslberger T, Weibezahn J, Zahn R, et al. M domains couple the ClpB threading motor with the DnaK chaperone activity. Mol Cell 2007;25:247–60 [DOI] [PubMed] [Google Scholar]
- 188. Oguchi Y, Kummer E, Seyffer F, et al. A tightly regulated molecular toggle controls AAA+ disaggregase. Nat Struct Mol Biol 2012;19:1338–46 [DOI] [PubMed] [Google Scholar]
- 189. DeSantis ME, Sweeny EA, Snead D, et al. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J Biol Chem 2014;289:848–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Mogk A, Kummer E, Bukau B.. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci 2015;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Doyle SM, Shorter J, Zolkiewski M, et al. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat Struct Mol Biol 2007;14:114–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Zhang T, Kedzierska-Mieszkowska S, Liu H, et al. Aggregate-reactivation activity of the molecular chaperone ClpB from Ehrlichia chaffeensis. PLoS One 2013;8:e6245423667479 [Google Scholar]
- 193. Krajewska J, Modrak-Wójcik A, Arent ZJ, et al. Characterization of the molecular chaperone ClpB from the pathogenic spirochaete Leptospira interrogans. PLoS One 2017;12:e0181118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Weibezahn J, Tessarz P, Schlieker C, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 2004;119:653–65 [DOI] [PubMed] [Google Scholar]
- 195. Li T, Weaver CL, Lin J, et al. Escherichia coli ClpB is a non-processive polypeptide translocase. Biochem J 2015;470:39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Yokom AL, Gates SN, Jackrel ME, et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat Struct Mol Biol 2016;23:830–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Shorter J, Southworth DR.. Spiraling in control: Structures and mechanisms of the Hsp104 disaggregase. Cold Spring Harb Perspect Biol 2019;11:a034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 2011;72:245–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Renton AE, Majounie E, Waite A, et al. ; ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Majounie E, Renton AE, Mok K, et al. ; ITALSGEN Consortium. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol 2012;11:323–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Haeusler AR, Donnelly CJ, Periz G, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014;507:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 2018;24:313–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. O'Rourke JG, Bogdanik L, Yáñez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016;351:1324–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Sivadasan R, Hornburg D, Drepper C, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci 2016;19:1610–8 [DOI] [PubMed] [Google Scholar]
- 205. Wen X, Tan W, Westergard T, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014;84:1213–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kwon I, Xiang S, Kato M, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014;345:1139–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Conlon EG, Lu L, Sharma A, et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. eLife 2016;5:e17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Zhang Y-J, Guo L, Gonzales PK, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science 2019;363:eaav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Lin Y, Mori E, Kato M, et al. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 2016;167:789–802.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Lee K-H, Zhang P, Kim HJ, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 2016;167:774–88.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015;525:56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Freibaum BD, Lu Y, Lopez-Gonzalez R, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015;525:129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Chew J, Gendron TF, Prudencio M, et al. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015;348:1151–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Mizielinska S, Grönke S, Niccoli T, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014;345:1192–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Mori K, Weng S-M, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013;339:1335–8 [DOI] [PubMed] [Google Scholar]
- 216. Ash PEA, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013;77:639–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Gendron TF, Bieniek KF, Zhang Y-J, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol 2013;126:829–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Mori K, Arzberger T, Grässer FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 2013;126:881–9 [DOI] [PubMed] [Google Scholar]
- 219. Zu T, Liu Y, Bañez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 2013;110:E4968–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Jovičić A, Mertens J, Boeynaems S, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 2015;18:1226–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. May S, Hornburg D, Schludi MH, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 2014;128:485–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Schludi MH, Becker L, Garrett L, et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol 2017;134:241–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yamakawa M, Ito D, Honda T, et al. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet 2015;24:1630–45 [DOI] [PubMed] [Google Scholar]
- 224. Zhang Y-J, Jansen-West K, Xu Y-F, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 2014;128:505–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Zhang Y-J, Gendron TF, Grima JC, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 2016;19:668–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2016;2:e1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Amick J, Roczniak-Ferguson A, Ferguson SM.. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell 2016;27:3040–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun 2016;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Zhang Y, Burberry A, Wang J-Y, et al. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev 2018;32:929–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Jung J, Nayak A, Schaeffer V, et al. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. eLife 2017;6:e23063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Xu C, Min J.. Structure and function of WD40 domain proteins. Protein Cell 2011;2:202–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Amick J, Tharkeshwar AK, Amaya C, et al. WDR41 supports lysosomal response to changes in amino acid availability. Mol Biol Cell 2018;29:2213–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Ge L, Melville D, Zhang M, et al. The ER–Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013;2:e00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Amick J, Tharkeshwar AK, Talaia G, et al. PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J Cell Biol 2020;219:e201906076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Zhang D, Iyer LM, He F, et al. Discovery of novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet 2012;3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013;29:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Chaineau M, Ioannou MS, McPherson PS.. Rab35: GEFs, GAPs and effectors. Traffic 2013;14:1109–17 [DOI] [PubMed] [Google Scholar]
- 238. Barr F, Lambright DG.. Rab GEFs and GAPs. Curr Opin Cell Biol 2010;22:461–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Wu X, Bradley MJ, Cai Y, et al. Insights regarding guanine nucleotide exchange from the structure of a DENN-domain protein complexed with its Rab GTPase substrate. Proc Natl Acad Sci U S A 2011;108:18672–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Marat AL, Dokainish H, McPherson PS.. DENN domain proteins: Regulators of Rab GTPases. J Biol Chem 2011;286:13791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Sellier C, Campanari M-L, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J 2016;35:1276–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 2014;23:3579–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Knödler A, Feng S, Zhang J, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A 2010;107:6346–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Furusawa K, Asada A, Urrutia P, et al. Cdk5 regulation of the GRAB-mediated Rab8-Rab11 cascade in axon outgrowth. J Neurosci 2017;37:790–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Tang D, Sheng J, Xu L, et al. Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc Natl Acad Sci U S A 2020;117:9876–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Su M-Y, Fromm SA, Zoncu R, et al. Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 2020;585:251–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Shen K, Huang RK, Brignole EJ, et al. Architecture of the human GATOR1 and GATOR1–Rag GTPases complexes. Nature 2018;556:64–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Lawrence RE, Fromm SA, Fu Y, et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019;366:971–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Shen K, Rogala KB, Chou H-T, et al. Cryo-EM structure of the human FLCN-FNIP2-Rag-ragulator complex. Cell 2019;179:1319–29.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Sztul E, Chen P-W, Casanova JE, et al. ARF GTPases and their GEFs and GAPs: Concepts and challenges. Mol Biol Cell 2019;30:1249–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Asano S, Fukuda Y, Beck F, et al. A molecular census of 26S proteasomes in intact neurons. Science 2015;347:439–42 [DOI] [PubMed] [Google Scholar]