

SUMMARY
Development and function of conventional dendritic cell (cDC) subsets, cDC1 and cDC2, depend on transcription factor (TF)s IRF8 and IRF4 respectively. Since IRF8 and IRF4 can each interact with TF BATF3 at AP1-IRF composite elements (AICEs) and with TF PU.1 at Ets-IRF composite elements (EICEs), it is unclear how these factors exert divergent actions. Here, we determined the basis for distinct effects of IRF8 and IRF4 in cDC development. Genes expressed commonly by cDC1 and cDC2 used EICE-dependent enhancers that were redundantly activated by low amounts of either IRF4 or IRF8. By contrast, cDC1-specific genes relied on AICE-dependent enhancers which required high IRF concentrations, but were activated by either IRF4 or IRF8. IRF8 was specifically required only by a minority of cDC1-specific genes, such as Xcr1, which could distinguish between IRF8 and IRF4 DNA-binding domains. Thus, these results explain how BATF3-dependent Irf8 autoactivation underlies emergence of the cDC1-specific transcriptional program.
Keywords: Dendritic cell identity, transcriptional program, interferon regulatory factor (IRF) 4, IRF8, cis-regulatory element, Ets-IRF composite element (EICE), AP-1-IRF composite element (AICE)
eTOC blurb
Development and function of conventional dendritic cell (cDC) subsets, cDC1 and cDC2, depends on IRF8 and IRF4. Kim et al. demonstrate that the basis for their distinct effects results from the higher level of IRF expressed in cDC1which is required to selectively engage AICEs that determine cDC1 identity.
Graphical Abstract
INTRODUCTION
The two main branches of conventional dendritic cells (cDCs), cDC1 and cDC2, exert distinct in vivo immune functions, but the transcriptional basis for their difference remains unclear (Miller et al., 2012; Durai and Murphy, 2016). cDC1 and cDC2 differ in their dependence on transcription factors for their development. Development of cDC1 requires the Batf3 and Irf8 transcription factors, while that of cDC2 does not (Hildner et al., 2008; Schiavoni et al., 2002; Ginhoux et al., 2009). Studies of transcription factor mutants in which development of cDC1 and cDC2 are genetically perturbed show that they exert distinct immune functions (Durai and Murphy, 2016). For example, experiments in Batf3−/− mice show that cDC1 are required for in vivo cross-presentation and for responses to many viruses and tumors (Hildner et al., 2008; Durai et al., 2019), but that cDC2 are sufficient for defense against helminths or certain bacteria, such Citrobacter rodentium (Tussiwand et al., 2015; Satpathy et al., 2013). cDC1 and cDC2 express clearly distinct transcriptional programs (Miller et al., 2012). For example, cDC1 express several key signature genes, such as Xcr1, Snx22, and Tlr3, not expressed by cDC2 or other immune lineages (Crozat et al., 2011; Brahler et al., 2017). However, there is currently no explanation for transcriptional differences between cDC1 and cDC2.
The transcription factors ETV6, PU.1, BATF3, IRF4, and IRF8 are important for cDC development. In cDC1, loss of Etv6 reduced but does not abolish cDC1 development, reduces a subset of genes, and causes partial de-repression of others, potentially by interacting with IRF8 (Lau et al., 2018; Kuwata et al., 2002; Humblin et al., 2017). The conditional deletion of Sfpi1 in early hematopoietic progenitors leads to impaired myeloid cell development, including the loss of the common DC progenitor (CDP) in the bone marrow (BM) and all cDCs in the peripheral tissues (Carotta et al., 2010). Recently, PU.1 has been shown to directly bind to and control some of cDC-specific genes with repression of pDC genes at the same time (Chopin et al., 2019), suggesting that PU.1 may be important for controlling the lineage decision between cDCs and pDCs and for maintaining cDC identity at steady state. PU.1 binds to the Flt3 promoter and is necessary for Flt3 expression (Carotta et al., 2010), potentially contributing to the loss of cDCs upon PU.1 deletion. However, PU.1 is broadly expressed in myeloid and lymphoid lineages (Chen et al., 1995; Klemsz et al., 1990), and is also required for development of macrophages, neutrophils, B cells, and T cells (McKercher et al., 1996). Batf3−/− and Irf8−/− mice lack cDC1 development (Hildner et al., 2008; Schiavoni et al., 2002). Irf4−/− mice develop cDC2, but these exhibit abnormal cDC2 function (Bajana et al., 2012; Vander et al., 2014; Persson et al., 2013; Schlitzer et al., 2013). Dual germline deletion of Irf4 and Irf8 deletes both CD4+ and CD8+ cDCs, currently identified as cDC2 and cDC1 respectively, in the spleen (Tamura et al., 2005), although it is possible that only these splenic cDC markers, but not the entire lineage, are affected. In fact, this is the case for Ciita−/− mice, which has been reported to lack all cDCs, but instead only lose expression of MHC class II (MHCII) used to identify the cDC lineage (Anderson, III et al., 2017).
IRF4 and IRF8 are closely related IRF family proteins and consist of DNA-binding domain (DBD), linker domain, and IRF-associated domain (IAD) (Taniguchi et al., 2001). IRF4 and IRF8 can bind to DNA using several types of motifs; as homodimers or heterodimers with other IRF members, such as IRF1, to interferon-stimulated response element (ISRE) motifs, characterized by the sequence GAAANNGAAA (Driggers et al., 1990), as heterodimers with the Ets family members, such as PU.1, to Ets-IRF composite elements (EICEs), characterized by the sequence GGAANNGAAA (Eisenbeis et al., 1995), or in a complex with a JUN/BATF heterodimer binding to an AP-1-IRF composite elements (AICEs), which arise in two forms, an AICE1 (TTTCNNNNTGASTCA) or AICE2 (GAAATGASTCA) (Ciofani et al., 2012; Glasmacher et al., 2012; Li et al., 2012). Among these different binding motifs, ISRE has been suggested to be of relatively lower affinity for IRFs compared to EICE or AICE (Ochiai et al., 2013; Krishnamoorthy et al., 2017). However, the relative affinities of AICEs to EICEs have not been established, nor have differences been reported between the cis-acting element DNA sequences recognized by IRF4 and IRF8.
There is currently no explanation for how the specific cDC1 program is achieved. IRF8 is more highly expressed in cDC1 than cDC2, due to the autoactivation of the Irf8 +32 kb enhancer, which binds a JUN-BATF3-IRF8 complex (Durai et al., 2019). Some studies have suggested that the identity of the IRF determines the fate choice between cDC1 vs. cDC2 (Tamura et al., 2005), although amounts of IRF have also been considered as a potential factor (Collin and Bigley, 2018). However, neither model has experimentally tested, nor is there currently a mechanism to explain how the identity or amount of IRF8 functions in the cDC1 transcriptional signature. Here, we show that the cDC1-specific transcriptional program relies on the selective engagement of AICE-dependent enhancers that require the high amount of IRF8 that is achieved only by BATF3-dependent Irf8 autoactivation, but that are not activated by the amounts of IRF8 or IRF4 expressed in cDC2.
RESULTS
Irf8 and Irf4 are redundant in promoting cDC2 development
In this study, we wanted to determine if IRF8 and IRF4, which have similar protein sequences but different concentrations in cDC1 and cDC2, are functionally interchangeable in cDCs. We first compared the contributions of Irf4 and Irf8 to cDC development in the periphery (Figure S1). Consistent with previous reports (Kanno et al., 2005; Tamura et al., 2005; Bajana et al., 2012), Irf4−/− Irf8−/− mice completely lack CD11c+ MHCII+ cells in the spleen, lung and small intestine and do not simply lack the CD8 and CD4 cDC surface markers which have been used previously for their identification (Figures S1A-S1D). Irf8−/− mice selectively lacked cDC1 in both spleen and peripheral tissues, as expected, but retained cDC2 (Figures S1A-S1D). Irf4−/− mice retained both cDC1 and cDC2 in the spleen, and retained the CD24− CD172+ cDC2 subset but not the CD24+ CD172a+ subset in the lung (Bajana et al., 2016). In the small intestine, they retained the CD11b+ CD103− subset but not the CD103+ CD11b+ subset of cDC2 (Persson et al., 2013; Schlitzer et al., 2013). These results show that Irf8 is sufficient to support cDC2 development but that Irf4 is necessary for the differentiation of the full complement of cDC2 subsets described to date.
Irf4 and Irf8 are redundant for formation of the cDC2 progenitor and Zbtb46 expression
To determine the developmental stage for Irf4 and Irf8 actions, we examined cDC BM progenitors in WT, Irf4−/−, Irf8−/−, and Irf4−/−Irf8−/− mice (Figures 1A, 1B, and S1E). We found that Irf4 was not required for the development of any progenitor population except for a slight decrease in the number of cDC2 progenitors (CD135+ SiglecH− CD117lo CD115+) (Figure 1B) and no decrease in Ly6C+ SiglecH− cDC2 primed pre-DCs (Schlitzer et al., 2015) (Figure S1E). By contrast, loss of Irf8 alone decreased development of CD135+ cells, SiglecH+ pDC progenitors, CDPs, and cDC2 progenitors (Grajales-Reyes et al., 2015; Sichien et al., 2016) but did not impair the development of macrophage and DC progenitor (MDP)s (CD135+ SiglecH− CD117hi CD115+) (Figures 1A, 1B, and S1E). However, loss of both Irf4 and Irf8 led to nearly complete loss of CDPs and cDC2 progenitors by the absolute cell number, but not of MDPs (Figures 1A and 1B). Irf8−/− and Irf4−/−Irf8−/− BM both contained Ly6C− SiglecH− populations, previously described as pre-cDC1 progenitors, but neither contained Ly6C+ SiglecH+ pre-DCs (Figure S1E) (Schlitzer et al., 2015). These results indicate that endogenous Irf8 acts earlier in DC progenitor development compared to Irf4, and that Irf4 and Irf8 are redundant for the formation of the cDC2 progenitor.
Figure 1. Irf8 and Irf4 are redundant in promoting cDC2 development and Zbtb46 expression.

(A and B) Flow cytometric analysis of BM progenitors isolated from WT, Irf4−/−, Irf8−/−, and Irf4−/−Irf8−/− mice. Pre-gate: lineage− CD127− cells. Scatter plots in (B) show numbers of CD135+ cells (top left), SiglecH+ pDC progenitors (top right), CD117hi CD115+ MDPs (middle left) and CD117int CD115+ CDPs (middle right) CD117lo CD115+ pre-cDC2s (bottom left) of the indicated genotypes. Results shown are analysis of ~4 million whole BM cells (bar = average number of cells, n = 3~7). (C) Flow cytometric analysis for Zbtb46GFP-expressing cells in the spleen (top, pre-gate: F4/80− cells) and BM (bottom, pre-gate: lineage− cells) of the indicated genotypes. (D) Scatter plots showing percentages of Zbtb46GFP-expressing cells in the spleen (left) and in BM (right) of the indicated genotypes (bar = average %, n = 3). Numbers in the two-color histograms indicate percentages of the gated cells. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 (Student’s t-test). See also Figure S1.
To test for cDC progenitors that may not have expressed the markers included in our analysis, we examined expression of the cDC-specific gene Zbtb46 in Irf4−/−Irf8−/− mice using Zbtb46GFP reporter mice (Satpathy et al., 2012). Previously we reported that the individual loss of Irf8 or Irf4 does not eliminate all Zbtb46 expression (Grajales-Reyes et al., 2015; Briseno et al., 2016). However, we found that combined loss of Irf4 and Irf8 together completely eliminated Zbtb46GFP expression in both BM and spleen (Figures 1C and 1D). In addition, retroviral expression of either Irf4 or Irf8 into CD117hi BM progenitors from Zbtb46GFP/+Irf4−/−Irf8−/− mice restored expression of Zbtb46GFP in B220− cells, which included cDCs (Figure S1F). These results show that the combined lack of Irf4 and Irf8 completely eliminates all cDC progenitors and that these factors redundantly regulate Zbtb46 expression.
cDC1 development requires a high amount of IRF protein
We next compared the capacities of Irf4 and Irf8 to promote cDC development by retrovirally expressing each into Irf4−/−Irf8−/− BM progenitors (Figures 2 and 3). Irf4 and Irf8 each increased the percentages of cDC1 from WT BM progenitors, and equally restored cDCs (B220− CD11c+ MHCII+) and cDC2s (CD24− CD172a+ or CD24+ CD172a+) from Irf4−/−Irf8−/− BM (Figures 2A, 2B left, S2A and S2C). However, although Irf8 was slightly more efficient than Irf4, neither Irf4 nor Irf8 restored cDC1 (CD24+ CD172a−) development to wild type percentages (Figures 2B, left panels and S2B). We previously reported that retroviral Irf8 expression in Irf8−/− BM progenitors did not achieve the same high amount of IRF8 protein, as is expressed in WT cDC1 (Grajales-Reyes et al., 2015). This may account for the inefficient restoration of cDC1 in Irf4−/−Irf8−/− BM progenitors by our retroviral IRF constructs.
Figure 2. cDC1 development and function can be induced by high IRF4 expression.

(A-C) Flow cytometric analysis showing cDCs differentiated from WT, Irf4−/−, Irf8−/−, or Irf4−/− Irf8−/− CD117hi BM progenitors retrovirally expressing either Irf4 or Irf8 without or with Batf3 co-expression. (A) CD11c+ MHCII+ cDCs (pre-gate: Bst2− B220− cells) and (B) CD24+ CD172a− cDC1 and CD172a+ cDC2 (pre-gate: Bst2− B220− CD11c+ MHCII+ cells). WT cDCs expressing empty-RVs are shown as controls. (C) Expression of IRF4 (top) or IRF8 (bottom) in cDCs of the indicated genotypes. WT DC1 and DC2 show endogenous amounts of IRF8 and IRF4, respectively. Geometric MFI of the cells (parenthesis: MFI for the gated cells) is indicated in the single-color histograms. Data shown is one of two similar experiments. (D) Cross-presentation ability of Irf4+/− Irf8−/− cDCs restored by low or high Irf4 or Irf8 achieved without or with Batf3 co-expression. HKLM-OVA: heat-killed Listeria monocytogenes expressing ovalbumin used as antigens. (E) The percentages of proliferated OT-I T cells under the indicated conditions, L4 (LOW IRF4), H4 (HIGH IRF4), L8 (LOW IRF8), and H8 (HIGH IRF8) are shown as a bar graph (average % ± SD, n = 4). (F-I) Microarray analysis of sort-purified WT cDC1, WT cDC2, Irf4−/− Irf8−/− cDCs restored by low or high amounts of either Irf4 or Irf8. Numbers of differentially expressed genes (> 3 fold, > 9 fold) are indicated on the left top of each scatter plot. (J) Flow cytometric analysis showing CD103 expression on Irf4−/− Irf8−/− cDCs restored by low or high Irf4 or Irf8 achieved without or with Batf3 co-expression. (K) A bar graph showing the percentages of CD103+ cells of the restored Irf4−/− Irf8−/− cDCs (average % ± SD, n = 4). * P < 0.05, ** P < 0.01 **** P < 0.0001 (Student’s t-test). Numbers in the two-color histograms indicate percentages of the gated cells. See also Figure S2.
Figure 3. A minor set of cDC1 genes relies on IRF8-specific DNA binding domain.

(A-D) Microarray analysis of Irf4−/− Irf8−/− cDCs restored by retroviral expression of low or high amounts of either Irf4 or Irf8 achieved without or with Batf3 co-expression. (A) Scatter plots comparing gene expression of sort-purified Irf4−/− Irf8−/− cDCs restored at high IRF4 condition to low IRF4 condition (top) and high IRF8 condition to low IRF8 condition (bottom). Numbers of differentially expressed genes (> 3 fold, > 9 fold) are indicated at the bottom of each scatter plot. (B-D) Heatmaps showing increased genes (> 3 fold) (B) at high IRF4 condition compared to low IRF4 condition, (C) at high IRF8 condition compared to low IRF8 condition, and (D) differentially expressed genes (> 3 fold) between high IRF8 and high IRF4 conditions. Highlighted genes in red and blue indicate cDC1- and cDC2-specific genes, respectively. (E) Flow cytometric analysis showing expression of XCR1 and CD172a on Irf4−/− Irf8−/− cDCs restored at the indicated conditions. (F) Flow cytometric analysis showing expression of XCR1 and CD172a on Irf4+/− Irf8−/− cDCs restored by high amounts of Irf8 (IRF888), Irf4 (IRF444), or chimeras for Irf4-Irf8 (IRF844 and IRF488) achieved with Batf3 co-expression. Structures of IRF4, IRF8, and the chimeric proteins are depicted on the top. DBD, LK, and IAD denote the DNA-binding domain, linker region, and IRF-associated domain. Numbers in the two-color histograms indicate the percentages of gated cells. Data shown is one of three similar experiments. See also Figures S3 and S4.
We noticed that retroviral expression of Batf3 increased the MFI of the GFP retroviral marker (Figure S2D), potentially by acting on the viral long terminal repeat (Hess et al., 1989; Shih et al., 1992), so we wondered if co-expression of Batf3 would increase retroviral IRF expression. Consistently, Batf3 co-expression with Irf4 and Irf8 increased the amount of IRF4 and IRF8 protein derived from retrovirus in Irf4−/− or Irf8−/− BM progenitors respectively (Figure 2C). In addition, we observed that expression of endogenous BATF3 was higher in cDC1 than in cDC2 (Figure S2E), so we reasoned that as well as increasing the amounts of IRF4 and IRF8, Batf3 co-expression would have the advantage of normalizing the BATF3 amounts during cDC development. Therefore, we expressed Batf3 along with Irf4 or Irf8 in Irf4−/−Irf8−/− BM cultures (Figures 2B and S2B). As a control, retroviral Batf3 had no impact on cDC1 and cDC2 development from WT BM and did not restore cDC development in Irf4−/−Irf8−/− BM cultures (Figures 2B, top right panel, and S2C). However, in the context of exogenously expressed Batf3, both Irf4 and Irf8 expressed in Irf4−/− Irf8−/− BM progenitors restored cDC1 development to wild type amounts (Figures 2B, middle and bottom right panels and S2B).
As a second method to control the amount of retroviral Irf4 and Irf8 expression, we used an integrating doxycycline-inducible retroviral expression system (Figure S2F). We first identified a concentration of doxycycline that could induce retroviral IRF8 to the same amount of IRF8 as in wild type cDC1 (Figure S2G). Both Irf4 and Irf8 were capable of restoring cDC1 development in Irf4−/− Irf8−/− BM in a doxycycline dose-dependent manner (Figure S2H). Together these data support the hypothesis that a high amount of IRF protein is required for cDC1 development and that either Irf4 or Irf8 can work.
IRF4 and IRF8 are functionally interchangeable for cross-presentation
Antigen cross-presentation is a hallmark function of cDC1s (Hildner et al., 2008; Durai et al., 2019), so we asked whether both Irf4 and Irf8 could support antigen cross-presentation (Figures 2D and 2E). cDC1, but not cDC2, derived from Flt3L-cultured WT BM, cross-presented HKLM-OVA to OT-I T cells (Figures 2D, top, and 2E). As a negative control, cells generated by Flt3L culture of Irf4+/− Irf8−/− BM, with only one allele of Irf4, were not able to cross-present. cDCs derived from Irf4+/− Irf8−/− BM progenitors transduced with high amounts of either Irf4 or Irf8 restored cross-presentation to approximately 50% of WT cDC1 activity, but cDCs containing low amounts of Irf4 or Irf8 had cross-presenting capacity similar to that of cDC2s (Figures 2D, bottom, and 2E). This result is consistent with the finding that monocyte-derived DCs (MoDC), which express high amounts of IRF4, can cross-present cell-associated antigen (Briseno et al., 2016; Theisen et al., 2019). Thus, high amounts of either IRF4 or IRF8 restored both the development and the cross-presentation function of cDC1.
IRF4 and IRF8 have equivalent cDC transcriptional impact
Since high amounts of both IRF4 and IRF8 could restore development and function of cDC1, we next compared the transcriptional impact of differing amounts of Irf4 and Irf8. For this, we analyzed gene expression of cDCs differentiated from Irf4−/−Irf8−/− BM progenitors retrovirally expressing either Irf4 or Irf8 using microarrays (Figures 2F–2I, and S3A–S3D). As a baseline difference, there are 518 genes (splenic cDC) and 489 genes (in vitro cultured cDC) more than 3-fold differentially expressed between WT cDC1 and cDC2 (Figure 2F). 428 genes are more than 3-fold differentially expressed between cDCs restored by high Irf8 compared to low Irf4 (Figure 2G), which was comparable to the difference between WT cDC1 and cDC2. Similar expression of several cDC1-specific genes, such as Tlr3 and Snx22, were obtained using either high Irf8 or high Irf4 (Figure S3A). cDCs restored by low amounts of Irf4 compared to low amounts of Irf8 were transcriptionally similar, with only 126 genes differentially expressed greater than 3-fold (Figure 2H). Likewise, cDCs restored by high amounts of Irf4 compared to high amounts of Irf8 were transcriptionally similar with only 157 genes differentially expressed more than 3-fold (Figure 2I).
Ex vivo derived WT cDC1 or WT cDC2s showed large transcriptional changes compared to Flt3L-derived WT cDC1 or WT cDC2s (Figure S3D). We expected that the restored cDCs should more closely resemble Flt3L-derived cDCs since they were derived in Flt3L cultures. We compared restored cDCs to both ex vivo and Flt3L-derived cDC1s and cDC2s by gene expression and Spearman’s rank correlation coefficient analysis (Figures S3B, S3C, Tables S1 and S2). These analyses confirmed that there was a high correlation between cDC1-specific genes and genes restored by high Irf4 or by high Irf8, as well as a high correlation between cDC2-specific genes and genes expressed in cDC2 restored by low Irf4, but less with low Irf8 (Figures S3B and S3C).
This suggests that Irf4 and Irf8 induced very similar transcriptional programs when expressed at similar amounts and that approximately 70% of the transcriptional differences between cDC1 and cDC2 are a result of the amount, but not the identity, of the IRF protein being expressed. Consistent with this hypothesis, Itgae encoding CD103, a cDC1 specific gene, was restored by high but not low Irf4 expression, as well as by Irf8 expression (Figures 2J and 2K).
A minor set of cDC1 genes relies on the IRF8 DNA-binding domain
While approximately 70% of the cDC1 signature was similarly induced by either high Irf4 or Irf8, some genes showed a preference for either Irf4 or Irf8 (Figures 2 and 3). 168 genes were differentially expressed more than 3-fold by high Irf4 expression compared to low Irf4 expression, including many cDC1-specific genes, such as Snx22, Tlr3, and Itgae (Figures 3A, 3B, and Table S1). 181 genes were differentially expressed more than 3-fold by high Irf8 compared to low Irf8 (Figures 3A and 3C and Table S1). However, the gene set induced by high Irf8 did not completely overlap with that induced by high Irf4 (Figure 3D). Gene pathway analysis using the Metascape platform (Tripathi et al., 2015) predicted that these patterns of specificity may be functionally relevant, with Irf4-specific gene targets being largely associated with regulation of cytokine production (Adam8, Ccr2, Il17ra, and Mmp12), T cell activation (Cd3g, Bcl11a, and Mafb), and regulation of defense response (Cd37, Ccr2, and Fcgr2b), while Irf8-specific targets are associated with regulation of defense response (Bst1, Cd36, Cd1d1, and Tlr12), cell adhesion (Cd81, Ctla4, and Btla), and positive regulation of cytosolic calcium ion concentration (Xcr1, Cxcr3, and Ccr9) (Figure S3E).
Both Tlr11 and Tlr12 are specifically expressed in cDC1 and are required for Toxoplasma gondii recognition (Koblansky et al., 2013; Raetz et al., 2013) (Figure S4A). Tlr12 was specifically controlled by Irf8 while Tlr11 was induced by high amounts of either Irf4 or Irf8. In agreement, cDC production of IL-12 p40 by soluble T. gondii lysate antigen (STAg) specifically relied on Irf8, consistent with the requirement for Tlr12 for sufficient response to T. gondii antigens (Figure S4C). In contrast, both Irf4 and Irf8, at low amounts, increased Tlr4 expression in cDCs and allowed for production of IL-12 p40 upon LPS and IFNγ stimulation to a similar extent (Figures S4B and S4D). This indicates that the specificity of Irf4 or Irf8 for IL-12 production from cDCs relies on their specific control of Tlr genes.
Expression of XCR1, a chemokine receptor selectively expressed by cDC1, in Irf4−/− Irf8−/− BM progenitors was weakly induced by low Irf8 and strongly induced by high Irf8, but was not induced at all by either low or high Irf4 (Figures S2G, 3D, 3E, S3A, and S3F). To understand the basis for this discrimination, we generated a series of chimeric IRF proteins composed of combinations of IRF4 and IRF8 DBD, linker regions, and IAD (Figures 3F and S4E). We expressed these chimeric IRF proteins at low and high amounts in Irf4+/− Irf8−/− BM progenitors to determine their ability to induce CD11c+ MHCII+ B220− cDCs and markers of cDC subsets. All chimeric proteins efficiently restored cDC development based on CD11c and MHCII expression (Figure S4E). However, XCR1 expression was restored only by chimeric proteins containing the IRF8 DBD, but not by chimeric proteins containing the IRF4 DBD (Figure 3F). These data imply that cis-acting elements controlling expression of the specific genes can discriminate between IRF4 and IRF8.
An AICE-dependent gene program is specific to cDC1
To determine the basis by which the IRF amounts impact cDC gene expression, we used ChIP-seq to identify the DNA motifs bound by IRF8 and IRF4 in cDCs (Figure 4). IRF4 and IRF8 are known to bind DNA at three kinds of motifs, ISRE (Bovolenta et al., 1994; Ochiai et al., 2013), EICE (Eisenbeis et al., 1995; Scott et al., 1994) or AICE (Glasmacher et al., 2012; Tussiwand et al., 2012). We first identified gene clusters that were selectively expressed by dendritic cells (cDCs or plasmacytoid DCs (pDCs)) but not macrophages (Figures 4A and 4B, Table S2). Of 853 genes expressed in cDCs, pDCs, or macrophages, but not expressed in other immune lineages, 462 genes were expressed in cDCs or pDCs (cluster I) and 391 were macrophage-specific (cluster II) (Figure 4A and Table S2). cDCs and pDCs are developmentally related and share some expression programs, including reliance on Irf8 which is highly expressed in both (Sichien et al., 2016), thus they express many genes in common with cDCs. To define cDC-specific genes more precisely from the 462 DC genes shared with pDCs, cluster I was further divided into 5 clusters comprising genes expressed selectively by cDC1 (I-a), both cDC1 and cDC2 (I-b), cDC2 alone (I-c), cDC1 and pDCs (I-d), or pDCs alone (I-e) (Figure 4B and Table S2).
Figure 4. DNA motifs bound by IRF4 and IRF8 within cDC-specific genes.

(A) Heatmap for 853 selected genes showing specific expression in DCs (cDC and pDC) or macrophages in the indicated tissues. Sp: spleen, LN: lymph node, Bl: blood, RP-MAC: red pulp macrophages, PC-MAC: peritoneal cavity macrophages, and AvMAC: alveolar macrophages. (B) Hierarchical clustering of 462 DC-specific genes in the cluster I of (A). Gene expression was shown after normalization by Z-score. (C-E) De novo DNA motif analysis using IRF8 ChIP-seq peaks in cDC1 and IRF4 ChIP-seq peaks in cDC2 merged with the cDC-specific genes (gene body ± 50 kb). (C) A Venn diagram showing numbers of shared or specific peaks for IRF8 and/or IRF4 associated with 127 common genes for cDC1 and cDC2 in cluster I-b (top). De novo motif analysis using (1) 79 shared peaks by IRF8 and IRF4, (2) 158 specific peaks for IRF8, or (3) 30 specific peaks for IRF4 (bottom). (D) De novo DNA motif analysis using IRF4 peaks associated with 22 cDC2 genes (cluster I-c). (E) De novo DNA motif analysis using IRF8 peaks associated with 89 cDC1-specific genes (cluster I-a). # Tg seq. and % Tg/Bg denote the number of total target sequences and percentage of target sequences/percentage of background sequences, respectively.
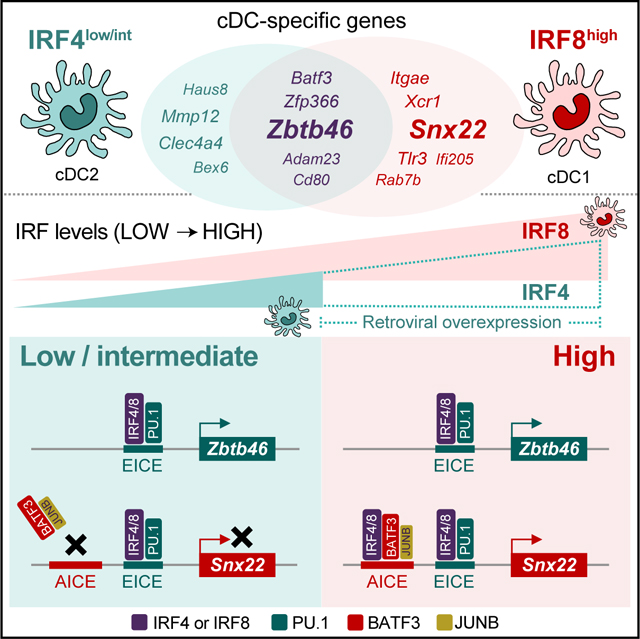
We determined binding motifs associated with genes in the cDC-specific clusters (I-a, I-b, and I-c) using IRF8 and IRF4 ChIP-seq data (Figure 4). While EICE motifs were enriched in both IRF8 and IRF4 binding peaks, AICE motifs were only enriched in peaks that were specific to IRF8. Specifically, there were 267 peaks binding either IRF4 or IRF8 that were associated with the 127 common genes for cDC1 and cDC2 in cluster I-b (Figure 4C). Of these, the 79 peaks that bound both IRF8 and IRF4 were enriched for EICE (PU.1-IRF) and ETS (PU.1or SpiB) motifs and the 30 peaks that bound only IRF4 were enriched for EICE motifs. However, peaks that bound only IRF8 (158 peaks) were enriched for AICE (AP-1-IRF) along with ETS motifs. Similarly, the 25 IRF4 binding peaks associated with the 22 cDC2-specific genes in cluster I-c were enriched only for EICE motifs (Figure 4D). Notably, the 230 IRF8 binding peaks associated with the 89 cDC1-specific genes in cluster I-a, were enriched for AICE along with EICE and ETS motifs (Figure 4E). Thus, these results imply that the common cDC transcriptional program relies on EICE-dependent enhancers that function at low IRF amounts, while the cDC1-specific program relies on high IRF amounts and engagement of AICE-dependent enhancers.
cDC2 gene expression is BATF-independent
To test whether AICE activation requires high concentrations of IRFs, we used ChIP-seq to compare BATF3 binding to AICEs in cDC1, which express a high amount of IRF8, and cDC2, which express a low amount of IRF4 and IRF8 (Figures 5A–5D). First, we identified 18,658 peaks for BATF3 in cDC1, which strongly overlapped with the 14,717 IRF8 peaks, consistent with AICE use (Figure 5A, left). By contrast, there were only 981 BATF3 peaks present in cDC2, compared to 8,448 IRF4 peaks (Figure 5A, right). In agreement, the tag counts for BATF3 were substantially lower in cDC2 compared to cDC1 (Figures 5B and 5C). The small number of peaks binding both IRF4 and BATF3 in cDC2 were enriched for AICE (AP-1-IRF) motifs (Figure S5A). An AICE similar motif was also enriched in peaks from genes preferentially induced by high IRF4 compared to low IRF4 (Figure S5B), and in peaks associated with genes induced by high IRF8 compared to high IRF4 (Figure S5C). Consistently, we found that both IRF4 and IRF8 could bind to an AICE probe along with BATF3 and JUNB in a dose-dependent manner (Figure S5D). Similarly, both IRF4 and IRF8 could bind to an EICE probe along with PU.1 in a dose-dependent manner (Figure S5D). Moreover, the deletion of an AICE-containing enhancer in Irf8 resulted in insufficient amounts of IRF8 to allow for cDC1 development (Figure S5E) (Durai et al., 2019).
Figure 5. cDC2 gene expression is BATF-independent.

(A) Venn diagrams showing the numbers of specific or overlapping ChIP-seq peaks for BATF3 and IRF8 in cDC1 (left) and for BATF3 and IRF4 in cDC2 (right). (B) Violin plots showing the tag counts (log2) for total BATF3 peaks in cDC1 or cDC2 (yellow), for BATF3 peaks merged with IRF8 peaks in cDC1 (orange), or BATF3 peaks merged with IRF4 peaks in cDC2 (green). (C) Tag counts for BATF3 peaks merged with 238 genes (cluster I-a, I-b, and I-c in Figure 4B) showing specific expression in cDC1 and cDC2 (gene body ± 50 kb). The numbers of BATF3 peaks used for tag count analysis were shown in parenthesis below each violin plot. **** P < 0.0001 (Student’s t-test). (D) UCSC genome browser images showing IRF8, IRF4, BATF3, and PU.1 bindings within cDC1- and cDC2-common genes (Zbtb46 and Zfp366) or cDC1-specific genes (Snx22 and Xcr1). Peaks containing potential EICE and AICE were indicated by blue and red arrowheads on the top, respectively. (E) Microarray analysis of splenic cDC2 isolated from WT or Batf−/− Batf3−/− mice. Splenic cDC1 was used as a control to show differentially expressed genes between cDC2 and cDC1. Scatter plots shown are comparisons of gene expression between resting cDC2 from WT and Batf−/− Batf3−/− mice (left), between activated cDC2 from WT and Batf−/− Batf3−/− mice (middle), or between the resting cDC2 and cDC1 from WT mice (right). The number of differentially expressed genes (> 2 fold) between the compared cells is indicated on the top left of each scatter plot. (F) Heatmap showing the expression of 238 cDC genes by the resting and activated cDC2 from WT or Batf−/− Batf3−/− mice. See also Figure S5.
Zbtb46 and Zfp366 are expressed by all cDCs (Satpathy et al., 2012; Chopin et al., 2019), whereas Snx22 and Xcr1 are expressed only by cDC1 (Brahler et al., 2017; Miller et al., 2012). In cDC1s, there were overlapping peaks for BATF3 and IRF8 in genomic regions for these four genes, but no BATF3 binding in cDC2s (Figure 5D). However, there were PU.1 peaks that overlapped with IRF4 and IRF8 binding in both cDC1 and cDC2 in genomic regions near these genes. This was consistent with EICE-based enhancers for cDC genes, with additional AICE-based enhancers for cDC1-specific genes and minimal AICE use in cDC2s.
The minimal use of AICEs in cDC2 predicts that BATF proteins do not control gene expression in cDC2. Batf3 is expressed in both cDC1 and cDC2, but is required only for cDC1 development (Grajales-Reyes et al., 2015). But it has never been fully established that the cDC2 developing in Batf3−/− mice are normal, particularly since we discovered that endogenous Batf expression partially compensates for Batf3 in some settings (Tussiwand et al., 2012). Therefore, we compared the transcriptome of resting and activated cDC2 isolated from WT and Batf−/− Batf3−/− mice (Figure 5E). There were only 115 genes that differed by more than 2-fold between WT and Batf−/− Batf3−/− cDC2 (Figure 5E left, Table S3), compared to the 1210 genes that differ more than 2 fold between WT cDC1 and cDC2 (Figure 5E, right). Only 43 genes differed more than 2-fold between activated WT and Batf−/− Batf3−/− cDC2 (Figure 5E, middle). Importantly, of 238 genes that are expressed specifically in cDCs (Table S2), there was no clear difference in expression between WT and Batf−/− Batf3−/− cDC2 either at homeostasis or upon treatment with IFNγ and LPS (Figure 5F, Table S3). Thus, in agreement with ChIP-seq analysis, cDC2 gene expression appears unresponsive to BATFs and largely does not rely on AICE-containing enhancers.
Zbtb46 expression relies on an EICE-dependent enhancer redundantly controlled by IRF4 or IRF8
The above results suggest that common cDC genes are controlled by EICE-dependent enhancers while cDC1-specific genes additionally use AICE-dependent enhancers. We wished to test these predictions for specific genes in the context of authentic cDC development. For this we examined the enhancer dependence of two genes, Zbtb46 which was expressed by both cDC1 and cDC2, and Snx22 expressed only by cDC1 (Figures 6 and 7). We first identified the genomic regions with bindings of either IRF4 or IRF8 in the Zbtb46 locus. Three peaks were located at +23 kb, +32 kb and +48 kb from the Zbtb46 transcription start site (TSS) (Figure 6A). We tested their individual enhancer activity using an integrating retroviral reporter (Grajales-Reyes et al., 2015) (Figures 6B and S6A). The +23 kb region was active in cDC1, cDC2 and MoDCs, was poorly active in pDCs, and was inactive in neutrophils, macrophages, and splenic B cells (Figures 6B, and S6B), reflecting the expression of endogenous Zbtb46 (Satpathy et al., 2012; Briseno et al., 2016). The +32 kb region was weakly active in cDC1, and less active in cDC2 and pDC (Figure 6B). The +48 kb element was nearly inactive in all cells tested (Figures 6B and S6B).
Figure 6. Zbtb46 expression relies on an EICE-dependent enhancer redundantly controlled by IRF4 or IRF8.

(A) ATAC-seq and ChIP-seq tracks display open chromatin areas and bindings of IRF8, BATF3, IRF4, H3K27ac, H3K4me1, or PU.1 around Zbtb46 locus. Boxed areas at +23 kb, +32 kb, or +48 kb from Zbtb46 TSS indicate regions assessed for enhancer activity. (B) Flow cytometric analysis showing GFP-reporter activities in cDC1, cDC2, and pDCs transduced with empty retrovirus (black) or retroviruses expressing each enhancer element (red). A bar graph below shows averages of MFI fold changes (MFI of enhancer element/MFI of empty) ± SD (n = 4). (C) FIMO analysis depicting p-values of the four predicted EICEs (blue boxes a, b, c, and d) in mouse Zbtb46 chr2: 181,436,313–181,436,629 (+23 kb from Zbtb46 TSS). (D) Flow cytometric analysis showing GFP-reporter activities in cDC1 and cDC2 expressing Zbtb46 +23 kb enhancer or mutants with internal deletion of the indicated EICE(s). Data shown is one of three similar experiments. (E) Flow cytometric analysis showing Zbtb46GFP expression in cDC1, cDC2, or pDC differentiated from Zbtb46GFP/+ Rosa26Cas9-GFP/+ CD117hi BM progenitors expressing scramble RNA or sgRNA(s) targeting Zbtb46 +23 kb EICE_b as depicted above the single-color histograms and in 6C. Targeting sites for EICE_b by sgRNAs are indicated with black arrowheads. Numbers in the histograms indicate the percentage of Zbtb46GFP-negative (Zbtb46GFP−) cells. The average percentages of Zbtb46GFP− cells ± SD (n = 3) are shown as a bar graph below. ** P < 0.01, **** P < 0.0001 (Student’s t-test). See also Figures S6 and S7.
Figure 7. Snx22 expression relies on IRF8 control of both EICE- and AICE-dependent enhancers.

(A) ATAC-seq and ChIP-seq tracks display open chromatin areas and bindings of IRF8, BATF3, IRF4, H3K27ac, H3K4me1, or PU.1 around Snx22 locus. Boxed areas at –12 kb and –3 kb from Snx22 TSS indicate regions assessed for enhancer activity. (B) Flow cytometric analysis showing GFP-reporter activities in cDC1, cDC2, pDCs, or BM-derived macrophages (MAC) transduced with empty retrovirus (black) or retroviruses expressing each enhancer element. A bar graph below shows averages of MFI fold changes (MFI of enhancer element/MFI of empty) ± SD (n = 4). (C) FIMO analysis depicting p-values of two predicted AICEs (red boxes, a and b) in mouse Snx22 chr9: 66,082,049–66,082,328 (–12 kb from Snx22 TSS) (top), or two predicted EICEs (blue boxes, c and d) in Snx22 chr9: 66,072,841–66,073,121 (–3 kb from Snx22 TSS) (bottom). (D) Flow cytometric analysis showing GFP-reporter activities in cDC1 expressing Snx22 –12 kb, Snx22 –3 kb, or their mutants with internal deletion of the AICEs (red boxes a and/or b in 5C) or EICEs (blue boxes c and/or d in 5C). Data shown is one of three similar experiments. (E) Flow cytometric analysis showing Snx22GFP expression in cDC1 (green and light green) and cDC2 (grey) differentiated from Snx22GFP/+ Rosa26Cas9-GFP/+ CD117hi BM progenitors expressing scramble RNA or sgRNA(s) targeting Snx22 –12 kb AICE_b or Snx22 –3 kb EICE_c and d as depicted above the single-color histograms and in 7C. Targeting sites for AICE_b (by gRNA1 or gRNA2) or EICE_c and d (by gRNA3 and gRNA4) are indicated with black arrowheads. Numbers in the histograms indicate the percentage of Snx22GFP-negative (Snx22GFP−) cells. The average percentages of Snx22GFP− cells ± SD (n = 3) are shown in the bar graphs on the right. ** P < 0.01, *** P < 0.001, **** P < 0.0001 (Student’s t-test). See also Figure S6.
The +23 kb Zbtb46 enhancer was contained within 320 bp region harboring 4 potential EICEs, which we labeled as sites a-d (Figure 6C). A 10 bp internal deletion of site b reduced enhancer activity 3-fold in cDC1 and in cDC2 (Figure 6D). Additional deletion of sites a, c, or d further reduced enhancer activity, and deletion of all four sites completely eliminated enhancer activity, suggesting that Zbtb46 expression relies on several EICEs in this enhancer region. To test this, we used CRISPR/Cas9 genome editing to target this enhancer in Zbtb46GFP/+ Rosa26Cas9-GFP/+ mice (Platt et al., 2014). For this analysis, the loss of Zbtb46GFP expression would indicate the importance of this enhancer. Importantly, the signal from the Cas9-GFP did not interfere with this assay, because its GFP MFI was only 2.5 × 102, much lower than Zbtb46GFP MFI, which is 3 × 103 (Figures S6C and S6E). Single guide (sg) RNAs targeted to the EICEs in the +23 kb enhancer caused a substantial reduction in Zbtb46GFP, both in cDC1 and cDC2, compared to little effect of the scramble sgRNA control (Figure 6E). Further, Zbtb46GFP expression was completely BATF-independent, and thus, AICE-independent, since there was no reduction in Zbtb46GFP expression by cDC2 isolated from Batf−/−Batf3−/− Zbtb46GPF/+ mice compared with cDC2 from WT Zbtb46GPF/+ mice (Figure S7). These results show that Zbtb46GFP expression relies on an EICE-dependent enhancer, but not on AICE-dependent enhancers.
Snx22 expression relies on IRF8 control of both EICE- and AICE-dependent enhancers
Since regulation of Zbtb46, a gene expressed by both cDC1 and cDC2, relies only on an EICE-dependent enhancer, we next asked whether regulation of Snx22, a cDC1-specific gene, would have the same or different properties. We identified two peaks that bound IRF4 or IRF8 located at –12 kb and –3 kb from the Snx22 TSS (Figure 7A), and tested their individual enhancer activity as above (Figure 7B). Both regions had significant activity that was higher in cDC1 compared with cDC2 or pDCs, with no activity in macrophages (Figure 7B), reflecting the expression of endogenous Snx22. The –12 kb enhancer contained two potential AICEs (sites a and b), and the –3 kb enhancer contained two potential EICEs (sites c and d) (Figure 7C). Internal deletion of site b in the –12 kb enhancer reduced its activity of by 50%, with no additional reduction upon deletion of site a (Figure 7D). Internal deletion of either site c or d in the –3 kb enhancer partially reduced activity, with deletion of both sites causing a complete loss of enhancer activity (Figure 7D).
We next targeted these sites using CRISPR/Cas9 genome editing in Snx22GFP/+ Rosa26Cas9-GFP/+ mice (Figure 7E). As before, there was no interference by Cas9-GFP, since its MFI of 2.5 × 102 is lower than MFI of Snx22GFP, which is 1 × 104 (Figures S6D and S6F). We next analyzed the role of the AICE in the –12 kb enhancer and the two EICEs in the –3 kb for Snx22 gene expression (Figure 7E). sgRNAs targeted to the site b AICE at –12 kb enhancer caused a substantial reduction in Snx22GFP expression in cDC1, and sgRNAs targeted to the EICEs in the –3 kb enhancer also caused a substantial loss of Snx22GFP expression. Thus, Snx22 expression may rely on the AICE site b in the –12 kb enhancer and the EICE site c and d in the –3 kb enhancer. Together, these data suggest that expression of cDC1-specific genes, exemplified by Snx22, requires activation of both EICE- and AICE-dependent enhancers, while expression of common genes for cDC1and cDC2, exemplified by Zbtb46, relies on EICE-dependent enhancers.
DISCUSSION
In this study, we have provided a molecular basis for distinct transcriptional signatures of cDC1 and cDC2. Differential expression of genes by cDC1 and cDC2 may explain many of their distinct functions, such as the selective expression of Tlr3and Tlr12 by cDC1 in promoting this subset’s sensitivity to infections by viruses or T. gondii (Miller et al., 2012; Mashayekhi et al., 2011). However, the underlying basis for this differential gene expression has not been fully explained (Durai and Murphy, 2016). IRF8 and IRF4 are each expressed in both cDC1 and in cDC2 and are known to contribute to their development and function (Tamura et al., 2005), although the mechanism underlying their distinct actions was not known. In fact, discrepant claims have argued that IRF4 and IRF8 exert either redundant or private actions (Yamamoto et al., 2011; Ochiai et al., 2013). Further, both the identity and the amount of expression can influence the transcriptional impact of IRF factors (Collin and Bigley, 2018), but which of these causes the cDC1 and cDC2 functional differences is unknown. BATF3 is also expressed in both cDC1 and cDC2 (Miller et al., 2012), but is required only for cDC1 development (Hildner et al., 2008), because of its role in Irf8 autoactivation (Grajales-Reyes et al., 2015; Durai et al., 2019). Explanations for the cDC1-specific requirement for BATF3 assume that IRF8 is the cDC1-determining factor, but do not distinguish whether it is IRF8 itself, or alternately only its high amount of expression, that determines cDC1 identity.
Here we have shown that the distinct cDC1 and cDC2 gene signatures resulted primarily because of high amounts of IRF8, and not its specific identity relative to IRF4, and that this accounted for approximately 75% of the cDC1 signature. The high IRF8 amount was required for activation of an AICE-dependent enhancer program, which could also be activated by high amounts of IRF4. AICEs were identified by ChIP-seq analysis only in cDC1, and not in cDC2, and they were located near genes selectively expressed by cDC1. However, similar genes were induced when amounts of IRF4 expression were increased beyond their normal low amounts. In addition to the transcriptional signature, one important function of cDC1 was antigen cross-presentation, which we showed could be achieved with high amounts of IRF4 as well as IRF8. This finding may explain how antigen cross-presentation is achieved in MoDCs, which express high amounts of IRF4, but not IRF8. Also, since antigen cross-presentation by MoDCs appears to be independent of BATF (Briseno et al., 2016), there may be additional cis-acting elements besides AICEs that require high amounts of IRF4 and IRF8. IRF2 deficiency is reported to impact cDC2 homeostasis, and is reversed by additional deficiency in type I interferon signaling, although the underlying mechanism of these phenomenon remains obscure (Ichikawa et al., 2004; Honda et al., 2004). Also, IRF4 and IRF8 are highly conserved between human and mouse, and appear to regulate development and function of human cDCs (Collin and Bigley, 2018; Lee et al., 2017; Schlitzer et al., 2013). Mutations in IRF8 in humans are associated with defects in cDC1 (Hambleton et al., 2011).
There is precedent for concentration-dependent IRF function, although the precise basis for this phenomenon was not determined. For example, IRF4 can exert divergent effects on the fate choice between GC B cells or plasma cells, based on the amounts of IRF4 expression during an immune response (Ochiai et al., 2013). An ISRE-dependent program involved in plasma cells was engaged only at high IRF4 amounts, suggesting a model in which ISREs have lower affinity for IRF4 binding compared EICEs. Graded amounts of IRF4 has also been shown to influence expansion versus effector function in CD8+ T cells, although the mechanism for this have not been determined (Nayar et al., 2014; Krishnamoorthy et al., 2017). One potential mechanism is that variable flanking regions surrounding AICEs can influence their affinity for IRF4-BATF complexes (Iwata et al., 2017). Thus, AICEs with different affinities could allow for differences in gene expression, depending on the amount of IRF4.
Here, we also have shown that genes that are expressed commonly between cDC1 and cDC2, such as Zbtb46 and Itgax, rely on an EICE-dependent program that largely treats IRF8 and IRF4 interchangeably. Previously, IRF8 and IRF4 have been reported to exert redundancy in other immune lineages (Yamamoto et al., 2011). For example, IRF4 or IRF8 function redundantly in supporting the transition from pre-B to B cells during development (Lu et al., 2003). This redundancy is based on the ability of either factor to induce transcription of the germline κ light chain locus in pre-B cells (Ma et al., 2006). IRF4 and IRF8 are also redundant in supporting development of CD11c+ MHCII+ cDCs (Tamura et al., 2005). While the basis for this redundancy has not been determined, evidence for specific actions has been reported (Tamura et al., 2005). Similarly, a study of macrophage development observed both redundant and specific actions of IRF4 and IRF8 mediated through EICEs (Yamamoto et al., 2011).
Finally, we have found that a minority of cDC-specific genes are activated by high amounts of IRF8, but do not respond to high amounts of IRF4. For example, Xcr1 is a cDC1-specific chemokine receptor that is induced selectively by high IRF8, but not IRF4. Among the nine IRF family members, IRF4 and IRF8 are most closely related to each other. They differ substantially in the sequences within their IADs, which mediates protein-protein interactions (Veals et al., 1993; Meraro et al., 1999). In this region, mouse IRF4 and mouse IRF8 are 39% identical. By contrast, the amino terminal region containing the DBD are much more highly conserved, being 79% identical up to residues 130 and 115 of IRF4 and IRF8, respectively. Despite this great similarity, our analysis of chimeric IRF4-IRF8 molecules mapped the specificity for XCR1 expression to the IRF8 DBD. We have not yet determined whether this discrimination relies on an ISRE (Driggers et al., 1990), EICE (Eisenbeis et al., 1995) or AICE (Glasmacher et al., 2012) cis-acting element. Nonetheless, our data suggests that variations in one or more of these DNA binding motifs can mediate the minor fraction of IRF8-specific cDC1 gene signature, but is a topic for future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth M. Murphy (kmurphy@wustl.edu)
Material Availability
All animal strains used in this study are available from The Jackson Laboratory or Taconic Biosciences. No new animal strains were generated for this study.
Data and Code Availability
Microarrays are available on the Gene Expression Omnibus (GEO) database with the accession no. GSE140451 and GSE140452. Following data sets were downloaded and reanalyzed: microarrays for splenic cDC (GSE110789) (Durai et al., 2018), ChIP-seq data sets for IRF4, IRF8, BATF3, H3K4me1, H3K27ac (GSE66899) (Grajales-Reyes et al., 2015), for PU.1 (GSE57563) (Lin et al., 2015), Immunological Genome Project (ImmGen) data sets for ATAC-seq (GSE100738), and microarrays (GSE15907 and GSE37448) (Heng et al., 2008).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Wild type C57BL/6 and 129 SvEv/Tac mice were obtained from The Jackson Laboratory, Taconic Biosciences, or bred in our facility. Zbtb46GFP/GFP mice (B6.129S6(C)-Zbtb46tm1.1Kmm/J, JAX: 027618) (Satpathy et al., 2012), Snx22 GFP/GFP mice (B6.129S6-Snx22tm1.1Kmm/J, JAX: 031837) (Brahler et al., 2017), Irf8 +32 kb−/− mice (C57BL/6J-Irf8em1Kmm/J, JAX: 032744) (Durai et al., 2019), OT-I mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J, JAX: 003831), and Rosa26Cas9-GFP/Cas9-GFP mice (B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J, JAX: 026179) (Platt et al., 2014) were all maintained on the C57BL/6 background. Batf−/− mice (129S-Batftm1.1Kmm/J, JAX: 013757) (Tussiwand et al., 2012) and Batf3−/− mice (129S-Batf3tm1.1Kmm/J, JAX: 013596) (Tussiwand et al., 2012) were on a mixed 129SvEv/C57BL/6 background. Irf8−/− mice were generated by crossing Irf8 exon2f/f mice (B6(Cg)-Irf8tm1.1Hm/J, JAX: 014175) with CMV-Cre mice (B6.C-Tg(CMV-cre)1Cgn/J, JAX: 006054) (Grajales-Reyes et al., 2015). Irf4−/− mice were generated by crossing Irf4f/f mice (B6.129S1-Irf4tm1Rdf/J, JAX: 009380) first to CMV-Cre mice and then to CMV-Flp1 mice (B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/JRainJ, JAX: 009086) (Briseno et al., 2016). All mice were maintained in a specific pathogen-free animal facility following institutional guidelines and with protocols approved by the Animal Studies Committee at Washington University in St. Louis. Most of the experiments were performed with mice 8–12 weeks of age using sex-matched littermates.
Cell isolation and culture
Culture of BM-derived cDC, macrophage, neutrophil, MoDC, and splenic B cell
BM cells were isolated as previously described (Grajales-Reyes et al., 2015). Lineage-committed cells were depleted using anti-CD3, anti-CD19, anti-Ly6G, anti-TER-119, and anti-CD105 antibodies, and MagniSort™ streptavidin negative selection beads. To differentiate BM-derived cDC, lineage− whole BM cells or lineage− CD117hi BM progenitor cells were suspended in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS, 1% Penicillin Streptomycin solution, 1% Sodium Pyruvate, 1% MEM non-essential amino acid, 1% L-glutamine solution, and 55 μM β-mercaptoethanol (complete IMDM) and were cultured with 5% Flt3L conditioned medium for 7 to 8 days. For generating BM-derived macrophages, lineage− whole BM cells were cultured in the complete IMDM supplemented with 25 ng/ml murine recombinant macrophage-colony stimulating factor (M-CSF) for 4 days. Neutrophils were differentiated from granulocyte-macrophage progenitors (GMPs). Lineage− CD117+ Sca-1− CD16/32+ GMPs (Akashi et al., 2000) were sorted and cultured in complete IMDM supplemented with 10 ng/ml murine recombinant granulocyte macrophage-colony stimulating factor (GM-CSF), 10 ng/ml murine recombinant stem cell factor (SCF), and 10 ng/ml murine recombinant interleukin (IL)-3 for 5 days. Monocyte-derived DCs (MoDCs) were differentiated from common monocyte progenitors (cMoPs). Lineage− CD117+ CD115+ Flt3− Ly6C+ CD11b− cMoPs (Hettinger et al., 2013) were sorted and cultured in complete IMDM supplemented with 20 ng/ml murine recombinant GM-CSF and 20 ng/ml murine recombinant IL-4 for 5 days. To isolate and culture splenic B cells, spleens were homogenized and passed through 70 μm cell strainers (Fisher). After lysing red blood cells, the whole splenocytes were cultured with 1 μg/ml E. coli LPS for 3 days.
Isolation of DCs from the spleen, lung, and small intestine
Splenocytes were isolated as previously (Grajales-Reyes et al., 2015). Briefly, excised spleens were incubated in complete IMDM supplemented with 250 μg/ml collagenase B and 30 U/ml DNaseI at 37˚C for 30 min, and red blood cells were lysed with ammonium-chloride-potassium buffer. CD11c+ cells were enriched using CD11c MicroBeads (Miltenyi Biotec). Bst2− B220− CD11c+ MHCII+ CD24+ CD172a− cDC1 and Bst2− B220− CD11c+ MHCII+ CD172a+ cDC2 were sorted from the enriched CD11c+ cell fraction. For microarray analysis, some of the sorted cDC2 were primed with 100 ng/ml interferon (IFN)-γ for 1 h and stimulated with Escherichia coli LPS (055:B5) for another 3 h. RNA was purified from the stimulated or unstimulated cDCs using a NucleoSpin RNA XS kit and then subjected to microarray analysis. Cell suspensions from the lung or lamina propria were prepared as described previously (Satpathy et al., 2013) with minor modifications. Briefly, excised lungs were suspended in Hank’s Balanced Salt Solution (HBSS) containing 15 mM HEPES, 2mM EDTA, 2% FBS, 250 μg/ml collagenase D, and 30 U/ml DNaseI, and were incubated at 37˚C for 1 h with stirring. Cell suspensions from the small intestines were prepared after removing Peyer’s patches and remaining fat tissues. After removing fecal contents, the small intestines were cut into ~0.5 cm pieces and incubated in HBSS containing 15 mM HEPES and 2mM EDTA at 37˚C for 40 min and washed with PBS twice. The recovered lamina propria tissues were minced and incubated in HBSS containing 15 mM HEPES, 2mM EDTA, 250 μg/ml collagenase B, 250 μg/ml collagenase D, and 30 U/ml DNaseI at 37˚C for 30 min with stirring. The cells were suspended in 40% Percoll and overlaid on 70% Percoll, centrifuged at 729 ×g for 10 min without brake, and pelleted. The cells were washed with MACS buffer, stained, and analyzed by flow cytometry.
Construction of retroviral vectors (RVs)
Plasmid T2a-Thy1.1 was constructed using overlapping PCR products containing T2a and Thy1.1 cDNA and cloned into the Murine Stem Cell Virus (MSCV) RV backbone. PCR products containing Irf4 and Irf8 cDNA with FLAG sequences at N-terminus were cloned in frame with T2a-Thy1.1 (MSCV-Irf4-T2a-Thy1.1 and MSCV-Irf8-T2a-Thy1.1). Chimera constructs for Irf4 and Irf8 were generated by overlapping PCR products and cloned into T2a-Thy1.1 RV. Doxycycline-inducible retroviral expression constructs for Irf4 and Irf8 were constructed as follows: The TetO-CMVmin-pA cassette from pTetO-CMVmin-pA-zeocin (Lindsley et al., 2008) was inserted in the reverse orientation into MSCV T2a-Thy1.1. A PCR fragment containing rtTA-advanced from MA2640, a gift from Mikhail Alexeyev (Addgene plasmid # 25434; http://n2t.net/addgene:25434; RRID: Addgene_25434) (Alexeyev et al., 2010), was cloned in frame with T2a-Thy1.1. PCR fragments containing Irf4 and Irf8 with FLAG sequences at N-terminus were inserted between the CMVmin and pA sequences. Construction of the MSCV-Batf3-IRES-GFP, and retroviral reporter vector (Thy1.1 pA GFP CMVp_min PmeI MSCV) for assessing transcriptional activities of enhancer elements for Zbtb46 or Snx22 are described in our previous studies (Tussiwand et al., 2012; Grajales-Reyes et al., 2015; Durai et al., 2019). Sequences of oligonucleotides used for cloning the enhancer elements (300–350 bp) and the mutants are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV510 or V450 rat anti-mouse CD45R/B220 (clone: RA3–6B2) | BD Biosciences | Cat#: 563103 Cat#: 560473 |
| PE mouse anti-rat CD90/mouse CD90.1 (clone: OX-7) | BD Biosciences | Cat#: 554898 |
| V500 rat anti-mouse I-A/I-E (clone: M5/114.15.2) | BD Biosciences | Cat#: 562366 |
| V450 rat anti-mouse CD45 (clone: 30-F11) | BD Biosciences | Cat#: 560501 |
| APC or BUV395 or biotin hamster anti-mouse CD11c (clone: HL3) | BD Biosciences | Cat#: 561119 Cat#: 744180 Cat#: 553800 |
| FITC, APC-Cy™ 7, Alexa Fluor® 700 or biotin rat anti-mouse Ly6C (clone: AL-21) | BD Biosciences | Cat#: 553104 Cat#: 560596 Cat#: 561237 Cat#: 557359 |
| PE-Cy™ 7 rat anti-mouse CD8a (clone: 53–6.7) | BD Biosciences | Cat#: 552877 |
| PE-Cy™ 7 or BUV 395 rat anti-mouse CD117 (clone: 2B8) | BD Biosciences | Cat#: 558163 Cat#: 564011 |
| PE-CF594 rat anti mouse-Flt3 (clone: A2F10.1) | BD Biosciences | Cat#: 562537 |
| BV510 rat anti-mouse CD16/32 (clone: 2.4G2) | BD Biosciences | Cat#: 740111 |
| Biotin rat anti-mouse CD19 (clone: 1D3) | BD Biosciences | Cat#: 553784 |
| Purified rat anti-mouse CD16/CD32 (clone: 2.4G2) | BD Biosciences | Cat#: 553142 |
| Brilliant Violet 510™ anti-mouse CD8a (clone: 53–6.7) | BioLegend | Cat#: 100752 |
| PerCP/Cyanine5.5 or Alexa Fluor® 700 anti-rat CD90/mouse CD90.1 (clone: OX-7) | BioLegend | Cat#: 202516 Cat#: 202527 |
| PE/Cyanine7 anti-mouse CD24 (clone: M1/69) | BioLegend | Cat#: 101822 |
| PE anti-mouse XCR1 (clone: ZET) | BioLegend | Cat#: 148204 |
| PE anti-mouse CD103 (clone: 2E7) | BioLegend | Cat#: 121406 |
| APC anti-mouse CD172a (clone: P84) | BioLegend | Cat#: 144014 |
| PE/Dazzle™ 594 anti-mouse/human CD45R/B220 (clone: RA3–6B2) | BioLegend | Cat#: 103258 |
| Brilliant Violet 605™ anti-mouse Ep-CAM (clone: G8.8) | BioLegend | Cat#: 118227 |
| Alexa Fluor® 488 or Brilliant Violet 711™ anti-mouse CD115 (clone: AFS98) | BioLegend | Cat#: 135512 Cat#: 135515 |
| Brilliant Violet 605™ anti-mouse CD64 (clone: X54–5/7.1) | BioLegend | Cat#: 139323 |
| Alexa Fluor® 647 anti-mouse CD11c (clone: N418) | BioLegend | Cat#: 117312 |
| Biotin anti-mouse CD3ε (clone: 145–2C11) | BioLegend | Cat#: 100304 |
| FITC or Biotin anti-mouse Ly6G (clone: 1A8) | BioLegend | Cat#: 127606 Cat#: 127604 |
| Biotin anti-mouse TER-119 (clone: TER-119) | BioLegend | Cat#: 116204 |
| Alexa Fluor® 488 anti-GFP (clone: 1GFP63) | BioLegend | Cat#: 338007 |
| APC or eFluor® 450 anti-mouse CD317 (clone: eBio927) | eBioscience | Cat#: 17–3172-82 Cat#: 48–3172-82 |
| PE or APC-eFluor® 780 anti-mouse CD11c (clone: N418) | eBioscience | Cat#: 12–0114-83 Cat#: 47–0114-80 |
| V450 anti-mouse I-A/I-E (clone: M5/114.15.2) | eBioscience | Cat#: 48–5321-82 |
| PerCP-eFluor™ 710 anti-mouse CD172a (clone: P84) | eBioscience | Cat#: 46–1721-82 |
| APC-eFluor® 780 anti-mouse F4/80 (clone: BM8) | eBioscience | Cat#: 47–4801-82 |
| PerCP-Cyanine5.5 anti-mouse CD11b (clone: M1/70) | eBioscience | Cat#: 45–0112-82 |
| eFluor® 450 anti-mouse NK1.1 (clone: PK136) | eBioscience | Cat#: 48–5941-82 |
| APC-eFluor® 780 anti-mouse CD4 (clone: RM4–5) | eBioscience | Cat#: 47–0042-82 |
| PE-Cyanine7 anti-mouse Sca-1 (clone: D7) | eBioscience | Cat#: 25–5981-82 |
| PE-Cyanine7 anti-mouse CD45.1 (clone: A20) | eBioscience | Cat#: 25–0453-82 |
| eFluor450® anti-mouse CD45 (clone: 30-F11) | eBioscience | Cat#: 48–0451-82 |
| PE anti-mouse Vα2 TCR (clone: B20.1) | eBioscience | Cat#: 12–5812-82 |
| PerCP-eFluor™ 710 anti-mouse SiglecH (clone: eBio440c) | eBioscience | Cat#: 46–0333-82 |
| APC-eFluor® 780 anti-mouse CD44 (clone: IM7) | eBioscience | Cat#: 47–0441-82 |
| PE-anti-human/mouse IRF4 (clone: 3E4) | eBioscience | Cat#: 12–9858-82 |
| PerCP-eFluor™ 710-anti human/mouse-IRF8 (clone: V3GYWCH) | eBioscience | Cat#: 46–9852-82 |
| Biotin-anti-mouse CD105 (clone: MJ7/18) | eBioscience | Cat#: 13–1051-82 |
| PE-IL-12/IL-23 p40 monoclonal antibody (clone: C17.8) | eBioscience | Cat#: 12–7123-82 |
| PE anti-mouse CD115 (clone: AFS98) | TONBO biosciences | Cat#: 50–1152 |
| PE anti-mouse Ly6G (clone: 1A8) | TONBO biosciences | Cat#: 50–1276 |
| Rabbit IgG PE-conjugated antibody | R&D systems | Cat#: F0110 |
| Rabbit anti-BATF3 | Tussiwand 2012 | N/A |
| Bacterial and Virus Strains | ||
| Subcloning Efficiency™ DH5α™ competent cells | Invitrogen™ | Cat#: 18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TransIT-LTI | MIRUS Bio | Cat#: MIR 2300 |
| Brefeldin A | Sigma-Aldrich | Cat#: B6542 |
| 20% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | Cat#: 15713-S |
| Saponin | Sigma-Aldrich | Cat#: S7900 |
| Lipopolysaccharide from Escherichia coli (055:B5) | Sigma-Aldrich | Cat#: L2880 |
| Polyinosinic–polycytidylic acid sodium salt_Poly (I:C) | Sigma-Aldrich | Cat#: P1530 |
| Poly(deoxyinosinic-deoxycytidylic) acid sodium salt | Sigma-Aldrich | Cat#: P4929 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#: D9891 |
| EasyTides® dCTP | PerkinElmer | Cat#: BLU513Z500UC |
| Bovine Serum Albumin Endotoxin-Free, BSA | Akron Biotech | Cat#: AK8917 |
| Iscove’s Modified Dulbecco’s Medium (IMDM) | Gibco | Cat#: 12440–046 |
| Opti-MEM Reduced Serum Medium | Gibco | Cat#: 31985–070 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | Cat#: H9268 |
| Fetal Bovine Serum (Characterized) | HyClone | Cat#: SH30071.03 |
| Sodium pyruvate | Corning | Cat#: 25–000-CI |
| L-Glutamine | Gibco | Cat#: 25030–164 |
| Pen Strep (Penicillin Streptomycin) | Gibco | Cat#: 15140–122 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat#: M3148 |
| MEM Non-essential Amino Acid Solution (100X) | Sigma-Aldrich | Cat#: M7145 |
| Dnase I | Sigma-Aldrich | Cat#: D4527 |
| Collagenase B | Sigma-Aldrich | Cat#: COLLB-RO |
| Collagenase D | Sigma-Aldrich | Cat#: COLLD-RO |
| Aprotinin bovine recombinant | Sigma-Aldrich | Cat#: A6103 |
| Leupeptin hydrochloride microbial, ≥90% (HPLC) | Sigma-Aldrich | Cat#: L9783 |
| HEPES | Sigma-Aldrich | Cat#: H3375 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | Cat#: E9884 |
| Hanks’ Balanced Salt Solution (HBSS) 10X | Gibco | Cat#: 14185–052 |
| Percoll ® | Sigma-Aldrich | Cat#: P4937 |
| Normal rat serum | STEM CELL Technologies | Cat#: 13551 |
| CFSE [5-(and 6)-carboxyfluorescein diaetate succinimidyl ester] | eBiosciences | Cat#: 65–0850-84 |
| Recombinant murine IFN-gamma | PeproTech | Cat#: 315–05 |
| Recombinant murine GM-CSF | PeproTech | Cat#: 315–03 |
| Recombinant murine IL-4 | PeproTech | Cat#: 214–14 |
| Recombinant murine IL-3 | PeproTech | Cat#: 213–13 |
| Recombinant murine M-CSF | PeproTech | Cat#: 315–02 |
| Recombinant murine SCF | PeproTech | Cat#: 250–03 |
| Soluble Toxoplasma gondii antigen (STAg) | Tussiwand et al., 2012 | N/A |
| Resources and Reagents | ||
| MagniSort™ streptavidin negative selection beads | Invitrogen™ | Cat#: MSNB-6002 |
| CD11c MicroBeads UltraPure (mouse) | Miltenyi Biotec | Cat#: 130–108-338 |
| Qdot™ 605 Streptavidin conjugate | Invitrogen™ | Cat#: Q10101MP |
| Brilliant Violet 711™-Streptavidin | BioLegend | Cat#: 405241 |
| PE-Cy™7 Streptavidin | BD Biosciences | Cat#: 557598 |
| Critical Commercial Assays | ||
| NucleoSpin RNA XS Kit | Machery-Nagel | Cat#: 740902.50 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat#: 00–5523-00 |
| WT-Pico Kit | Applied Biosystems™ | Cat#: 902622 |
| Deposited Data | ||
| Microarray | This study |
GSE140451 GSE140452 |
| Microarray | Durai et al., 2017 | GSE110789 |
| Microarray | ImmGen |
GSE15079 GSE37448 |
| ATAC-seq | ImmGen | GSE100738 |
| ChIP-seq (IRF4, IRF8, BATF3, H3K4me1, H3K27ac) | Grajales-Reyes et al., 2015 | GSE66899 |
| ChIP-seq (PU.1) | Allhoff et al., 2014 | GSE57563 |
| Experimental Models: Cell Lines | ||
| Platinum-E retroviral packaging cell line | Morita et al., 2000 | N/A |
| HEK293FT cell line | Invitrogen™ | Cat#: R70007 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | JAX: 000664 |
| Mouse: 129S6/SvEv Tac | Taconic Biosciences | 129SVE-(F or M) |
| Mouse: B6.129S6(C)-Zbtb46tm1.1Kmm/J (Zbtb46GFP/GFP) | Jackson Laboratory | JAX: 027618 |
| Mouse: B6.129S6-Snx22tm1.1Kmm/J (Snx22GFP/GFP) | Jackson Laboratory | JAX: 031837 |
| Mouse: B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Rosa26Cas9-GFP /Cas9-GFP) | Jackson Laboratory | JAX: 026179 |
| Mouse: B6(Cg)-Irf8tm1.1Hm/J | Jackson Laboratory | JAX: 014175 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | Jackson Laboratory | JAX: 006054 |
| Mouse: B6.129S1-Irf4tm1Rdf/J | Jackson Laboratory | JAX: 009380 |
| Mouse: B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/JRainJ | Jackson Laboratory | JAX: 009086 |
| Mouse: C57BL/6J-Irf8em1Kmm/J (Irf8 +32 kb–/–) | Jackson Laboratory | JAX: 032744 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) | Jackson Laboratory | JAX: 003831 |
| Mouse: 129S-Batftm1.1Kmm/J | Jackson Laboratory | JAX: 013757 |
| Mouse: 129S-Batf3tm1.1Kmm/J | Jackson Laboratory | JAX: 013596 |
| Oligonucleotides | ||
| Sequences of primers used for cloning Zbtb46 enhancer elements and mutants (5’ > 3’) | ||
| +23 kb (FW): CATGAAGCTTACAAACCCCTGACTGAAAAG | Sigma-Aldrich | N/A |
| +23 kb (RV): TCGAGGATCCAGAACTTTGCTTCCACTC | Sigma-Aldrich | N/A |
| +32 kb (FW): CATGAAGCTT ACAAGACCCAAGTTTGCTG | Sigma-Aldrich | N/A |
| +32 kb (RV): TCGAGGATCCTCAAATGGAAGAAATCACCT | Sigma-Aldrich | N/A |
| +48 kb (FW): CATGATCCATCCTTTGGTTTAAGCACCTC | Sigma-Aldrich | N/A |
| +48 kb (RV): TCGAGGATCCTAAATGTAGCATTCAACTC | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_a (FW): CATAAGATAGTGAGCAACCAAAGT | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_a (RV): ACTTTGGTTGCTCACTATCTTATG | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_b (FW): GCCAGGCCGGTGCAGAGAAGGGTAGCACCGAGAG | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_b (RV): TCTCGGTGCTACCCTTCTCTGCACCGGCCTGGC | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_c (FW): GTAGCACCGAGAAGGGCAAGTGGT | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_c (RV): ACCACTTGCCCTTCTCGGTGCTAC | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_d (FW): AGTGGTGTATGGTGGGCAGGCATC | Sigma-Aldrich | N/A |
| +23 kb ΔEICE_d (RV): GATGCCTGCCCACCATACACCACT | Sigma-Aldrich | N/A |
| Sequences of primers used for cloning Snx22 enhancer elements and mutants (5’ > 3’) | ||
| –12 kb (FW): CATGAAGCTTAGCCAGCAGCTTATCTTCCA | Sigma-Aldrich | N/A |
| –12 kb (RV): TCGAGGATCCAGTGGCTATTGGGGACACAC | Sigma-Aldrich | N/A |
| –3 kb (FW): ATGAAGCTTTGTGAGAGAGAAGGAAACAGAGG | Sigma-Aldrich | N/A |
| –3 kb (RV): TCGAGGATCCGGCAGAGGACATCCTTACCA | Sigma-Aldrich | N/A |
| –12 kb ΔAICE_a (FW): ACCCACAGAAAAACCCAGGACCTTTCCCTA | Sigma-Aldrich | N/A |
| –12 kb ΔAICE_a (FW): TAGGGAAAGGTCCTGGGTTTTTCTGTGGGT | Sigma-Aldrich | N/A |
| –12 kb ΔAICE_b (FW): GAGATCCGACAACTGTTCCCCCTGTCAGCT | Sigma-Aldrich | N/A |
| –12 kb ΔAICE_b (FW): AGCTGACAGGGGGAACAGTTGTCGGATCTC | Sigma-Aldrich | N/A |
| –3 kb ΔEICE_c (FW): ACATCCCAAAGACCCCAGCACTTC | Sigma-Aldrich | N/A |
| –3 kb ΔEICE_c (FW): GAAGTGCTGGGGTCTTTGGGATGT | Sigma-Aldrich | N/A |
| –3 kb ΔEICE_d (FW): TGGTAGGGGTCAAGAGACCACACC | Sigma-Aldrich | N/A |
| –3 kb ΔAICE_d (FW): GGTGTGGTCTCTTGACCCCTACCA | Sigma-Aldrich | N/A |
| Sequences of sgRNA for targeting Zbtb46 enhancer (+23 kb EICE_b) (5’ > 3’) | ||
| sgRNA1 (FW): CACCGGGTGCAGAGGGAACTGAGAA | Sigma-Aldrich | N/A |
| sgRNA1 (RW): AAACTTCTCAGTTCCCTCTGCACCC | Sigma-Aldrich | N/A |
| sgRNA2 (FW): CACCGAGAAGCCAGGCCGGTGCAGA | Sigma-Aldrich | N/A |
| sgRNA2 (RW): AAACTCTGCACCGGCCTGGCTTCTC | Sigma-Aldrich | N/A |
| Sequences of sgRNA for targeting Snx22 enhancers (–12 kb AICE) (5’ > 3’) | ||
| sgRNA1 (FW): CACCGAGTCATTTGGAAACAGTTGT | Sigma-Aldrich | N/A |
| sgRNA1 (RW): AAACACAACTGTTTCCAAATGACTC | Sigma-Aldrich | N/A |
| sgRNA2 (FW): CACCGGACAGGGGGAAATAGTCATT | Sigma-Aldrich | N/A |
| sgRNA2 (RW): AAACAATGACTATTTCCCCCTGTCC | Sigma-Aldrich | N/A |
| Sequences of sgRNA for targeting Snx22 enhancers (–3 kb EICE) (5’ > 3’) | ||
| sgRNA3 (FW): CACCGCCAGCACTTCTCCTTGGTAG | Sigma-Aldrich | N/A |
| sgRNA3 (RW): AAACCTACCAAGGAGAAGTGCTGGC | Sigma-Aldrich | N/A |
| sgRNA4 (FW): CACCGAAGGGAAATGACCCCTACCA | Sigma-Aldrich | N/A |
| sgRNA4 (RW): AAACTGGTAGGGGTCATTTCCCTTC | Sigma-Aldrich | N/A |
| Sequences of EMSA probes (5’ > 3’) | ||
| Snx22 –12 kb AICE (FW): CAGGGGGAAATAGTCATTTGGAAACAGTTGT | Sigma-Aldrich | N/A |
| Snx22 –12 kb AICE (RV): CCGACAACTGTTTCCAAATGACTATTTCC | Sigma-Aldrich | N/A |
| Snx22 –3 kb EICE (FW): GTGCTGGGGTTTCATTTCCTCTTTGGGAT | Sigma-Aldrich | N/A |
| Snx22 –12 kb EICE (RV): GATCCCAAAGAGGAAATGAAACCCCAG | Sigma-Aldrich | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v10 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| FACSDiva | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| ArrayStar 15 | DNASTAR | https://www.dnastar.com/ |
| Metascape | Tripathi et al., 2015 | https://metascape.org/gp/index.html#/main/step1 |
| Morpheus | Broad Institute |
https://software.broadinstitute.org/morpheus/ |
Platinum-E retroviral packaging cell line (Plat-E) and HEK293FT cell line
Plat-E cells (Morita et al., 2000) and HEK293FT cells (Invitrogen™) were cultured in complete IMDM. When reaching 70–80% confluence, the cells were split by trypsinization.
Bacterial strains
Subcloning Efficiency™ DH5α™ competent cells were purchased from Invitrogen.
METHOD DETAILS
Flow cytometry
Cells were suspended in PBS supplemented with 0.5% BSA and 2 mM EDTA (MACS buffer) and kept on ice during the whole staining process. Cells were incubated with anti-mouse CD16/32 antibodies (2.4G2, BD Biosciences) or normal rat serum (STEMCELL Technologies) for 5 min to block Fc receptors and then stained with fluorochrome-conjugated or biotinylated antibodies. All the antibodies and streptavidin conjugates with fluorochrome used for this study are listed in the key resources table. Cells were washed with MACS buffer once and analyzed on a FACSCanto II or FACSAria Fusion flow cytometer (BD Biosciences). All the flow cytometric data were analyzed using FlowJo V10 software (TreeStar).
For analysis of intracellular expression of IL-12 p40 in cDCs, cells were stimulated with E. coli LPS or soluble Toxoplasma gondii antigens (STAg) for 6 h in the presence of Brefeldin A (250 ng/ml). The cells were stained for surface markers on ice, fixed with PBS containing 2% paraformaldehyde, and then permeabilized with MACS buffer containing 0.5% saponin at room temperature. The cells were then stained with anti-mouse IL-12 p40 antibodies for 30 min at room temperature, washed, and analyzed by flow cytometry. For analysis of intracellular expression of IRF8, IRF4, and BATF3 in cDCs, cells were stained for surface markers on ice and then fixed/permeabilized using transcription factor staining buffer set (eBioscience) at room temperature. The cells were then stained for 1 h at room temperature with anti-human/mouse IRF8, anti-human/mouse IRF4, or rabbit anti-BATF3 antibodies (prepared in-house) (Tussiwand et al., 2012) in the presence of 2% normal rat serum. For BATF3 detection, the cells were stained with PE-conjugated anti-rabbit IgG for another 1 h at room temperature. The cells were washed and analyzed by flow cytometry.
Transfection and transduction
Plat-E cells were plated in 6- or 12-well cell culture plates at a density of ~0.4 × 106 cells/ml and incubated overnight. Retroviral plasmid DNAs mixed with TransIT-LT1 (Mirus Bio) in Opti-MEM™ reduced serum medium were transfected into the Plat-E cells and incubated overnight. The culture media was changed, and the supernatant containing retroviruses was collected 24 h later. For retroviral transduction, lineage− BM progenitors (CD117hi cells, cMoPs, or GMPs) and splenic B cells were used. BM progenitors were sort-purified and cultured in complete IMDM supplemented with appropriate cytokines overnight. After removing the culture media, the cells were transduced with the supernatant containing retroviruses in the presence of 2 μg/ml polybrene by spinoculation at 729 ×g for 1 h at room temperature. The culture media was changed 24 h later, and the cells were further cultured. Details for the cell culture are described in the cell isolation and culture section.
CRISPR-Cas9 deletion of enhancer elements
Oligonucleotides containing guide RNA sequences with BbsI compatible overhangs were annealed and ligated into single guide RNA (sgRNA) vector Thy1.1-hU6-gRNA-BbsI stuffer-MSCV (Theisen et al., 2018). Oligonucleotide sequences for generating sgRNA are listed in the key resources table. Plasmids containing either scramble RNA or guide RNA were transfected into Plat-E packaging cells and retroviruses were collected 48 h later as described in the ‘Transfection and transduction’ section. Lineage− CD117hi BM progenitors sort-purified from either Zbtb46GFP/+ Rosa26Cas9-GFP/ Cas9-GFP mice or Snx22GFP/+ Rosa26Cas9-GFP/+ mice were transduced with the retroviruses and then cultured with 5% Flt3L conditioned medium for 7 days.
Cross-presentation assay
In vitro cross-presentation assay was performed as described previously (Kretzer et al., 2016; Theisen et al., 2018). Briefly, 10,000 sort-purified cDCs were co-cultured with 25,000 CFSE-labeled OT-I T cells in the absence or presence of heat-killed Listeria monocytogenes expressing ovalbumin (HKLM-OVA) (2 × 108 CFU/ml) (a gift from H. Shen, University of Pennsylvania, Philadelphia, PA) for 3 days and then CFSE dilution was analyzed by flow cytometry. HKLM-OVA was prepared as described previously (Kretzer et al., 2016). cDCs used for this assay were generated as follows. CD117hi BM progenitors from WT or Irf4+/− Irf8−/− mice were sort-purified and transduced with Irf4- or Irf8-expressing retroviruses with or without Batf3-expressing retrovirus, and cultured with 5% Flt3L for 7 days. CD11c+ MHCII+ cDCs were sort-purified from the transduced cell populations and co-cultured with OT-I T cells in the absence or presence of HKLM-OVA. cDC1 differentiated from untransduced WT BM progenitors was used as a positive control, and cDC2 differentiated from untransduced WT or Irf4+/− Irf8−/− BM progenitors were used as negative controls for OT-I T cell proliferation.
Electromobility shift assay (EMSA)
EMSA was performed as previously described (Tussiwand et al., 2012; Iwata et al., 2017). Oligonucloetide sequences for EMSA probes are available in the key resources table. Forward and reverse oligonucleotides were annealed and labeled with 32P-dCTP using Klenow polymerase. HEK293FT cells were transiently transfected with MSCV-Sfpi1-IRES-GFP (PU.1), MSCV-Batf3-IRES-GFP/MSCV-Junb-IRES-GFP (BATF3/JUNB), MSCV-Irf4-T2a-Thy1.1 (IRF4) or MSCV-Irf8-T2a-Thy1.1 (IRF8) usingTransIT-LT1 and incubated for 24 h and then the culture media was changed. After another 24 h, the cells were lysed with buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2 and 10 mM KCl) containing 0.2% NP40 and protease inhibitors (aprotinin and leupeptin) and centrifuged to obtain nuclei. The pelleted nuclei were resuspended in buffer C (20mM HEPES-KOH, pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA and 25% glycerol) and centrifuged to obtain nuclear extracts. EMSA was performed using combinations of the nuclear extracts, 0.5 μg poly dI-dC, and 32P-labeled probes in 20 μl binding reactions for 30 min on ice. Reactions were separated on 7%T 3.3%C polyacrylamide mini-gels in 0.4× TBE for 35min at 250 V at 4 °C and analyzed by autoradiography.
Microarray analysis
RNA was isolated by NucleoSpin RNA XS (Macherey-Nagel) and amplified with WT Pico System (Affymetrix) and hybridized to GeneChip Mouse Gene 1.0 ST microarrays (Affymetrix) for 18 h at 45°C in a GeneChip Hybridization Oven 640. The data was analyzed with the Affymetrix GeneChip Command Console. Microarray expression data was processed using Command Console (Affymetrix, Inc) and the raw (.CEL) files generated were analyzed using Expression Console software with Affymetrix default RMA Gene analysis settings (Affymetrix, Inc). Probe summarization (Robust Multichip Analysis, RMA), quality control analysis, and probe annotation were performed according to recommended guidelines (Expression Console Software, Affymetrix, Inc.). Data were normalized by robust multi-array average summarization and underwent quantile normalization with ArrayStar software (DNASTAR). Gene pathway analysis was performed using Metascape platform (Tripathi et al., 2015) with gene ontology (GO) biological process, GO cellular component, and GO molecular function database. Unsupervised hierarchical clustering of differentially expressed genes, heatmap, and Spearman’s rank correlation coefficient analysis were computed with Morpheus software from Broad Institute.
Computational analysis for ChIP-seq and ATAC-Seq
ChIP-seq and ATAC-seq data sets were aligned and mapped on the mouse reference genome (GRCm38/mm10) using Bowtie software (version 1.1.1) with the following command: --sam --best -p 4 -m 1 mm10 --chunkmbs 1000. Making tag directories, peak calling, and de novo motif analysis were performed by HOMER (version 4.9) software. Tag directories for ChIP-seq and ATAC-seq were generated by ‘makeTagDirectory’ and duplicated reads in each data set were discarded using the argument -tbp 1. Peak calling for transcription factor ChIP-seq and ATAC-seq was performed by ‘findPeaks’ with following options: -style factor -size 100 -poisson 1e-10. For histone mark ChIP-seq, -style histone -size 500 -poisson 1e-10 were used as arguments. Browser Extensible Data (BED) files with genomic locations for each specific gene set were generated using ‘table browser’ tool in the UCSC genome browser web site. The transcriptional regions (gene body) were extended by 50 kb upstream and downstream using ‘slopBed’ in BedTools2 software. ChIP-seq or ATAC-seq peaks merged with genomic regions for specific gene sets were extracted by ‘intersectBed’. De novo DNA motifs were analyzed by ‘findMotifsGenome.pl’ with following arguments: -mask -bits -size 100. Bedgraph files for visualizing as UCSC genome browser tracks were created by ‘makeUCSCfile’ with following arguments: -fsize 5e7 -res 1. Venn diagrams displaying the number of ChIP-seq peaks and violin plots showing tag counts (log2) for ChIP-seq peaks were generated using ‘venneuler’ and ‘vioplot’ packages of R, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with GraphPad Prism 7.0 (GraphPad Software).
Supplementary Material
Table S1 Genes preferentially induced by retroviral reconstitution of Irf4 or Irf8 with or without co-expression of BATF3, related to Figure 3
Table S2 Genes specific for DC and macrophage, related to Figure 4
Table S3 Genes differentially expressed in splenic cDC2 from WT and Batf−/− Batf3−/− mice, related to Figure 5
Highlights.
cDC identity relies on cis-regulatory elements redundantly controlled by IRF4 and IRF8.
cDC1 is distinguished from cDC2 by the use of AICE-dependent gene program.
AICE-dependent gene program requires high levels of either IRF4 or IRF8.
Specific cis-regulatory elements distinguish between IRF4 and IRF8 DNA-binding domains.
ACKNOWLEDGEMENTS
We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by the National Cancer Institute’s Cancer Center Support grant no. P30 CA91842 to the Siteman Cancer Center and by the Institute of Clinical and Translational Sciences/Clinical and Translational Science Award grant no. UL1TR000448 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of the NCRR or NIH. This work benefitted from data assembled by the ImmGen consortium (Heng et al., 2008). This work was supported by the Howard Hughes Medical Institute (K.M.M.), the National Science Foundation (grant no. DGE-1745038 to P.B.) and National Cancer Institute of NIH (grant no. F31CA228240 to D.A.A.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. [DOI] [PubMed] [Google Scholar]
- Alexeyev MF, Fayzulin R, Shokolenko IN, and Pastukh V. (2010). A retro-lentiviral system for doxycycline-inducible gene expression and gene knockdown in cells with limited proliferative capacity. Mol. Biol. Rep 37, 1987–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DA III, Grajales-Reyes GE, Satpathy AT, Vasquez Hueichucura CE, Murphy TL, and Murphy KM (2017). Revisiting the specificity of the MHC class II transactivator CIITA in classical murine dendritic cells in vivo. Eur. J. Immunol 47, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajana S, Roach K, Turner S, Paul J, and Kovats S. (2012). IRF4 promotes cutaneous dendritic cell migration to lymph nodes during homeostasis and inflammation. J. Immunol 189, 3368–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajana S, Turner S, Paul J, Ainsua-Enrich E, and Kovats S. (2016). IRF4 and IRF8 Act in CD11c+ Cells To Regulate Terminal Differentiation of Lung Tissue Dendritic Cells. J. Immunol 196, 1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovolenta C, Driggers PH, Marks MS, Medin JA, Politis AD, Vogel SN, Levy DE, Sakaguchi K, Appella E, Coligan JE (1994). Molecular interactions between interferon consensus sequence binding protein and members of the interferon regulatory factor family. Proc. Natl Acad. Sci. U S A 91, 5046–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahler S, Zinselmeyer BH, Raju S, Nitschke M, Suleiman H, Saunders BT, Johnson MW, Bohner AMC, Viehmann SF, Theisen DJ, et al. (2017). Opposing Roles of Dendritic Cell Subsets in Experimental GN. J. Am. Soc. Nephrol 29, 138–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briseno CG, Haldar M, Kretzer NM, Wu X, Theisen DJ, KC W, Durai V, Grajales-Reyes GE, Iwata A, Bagadia P, et al. (2016). Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep. 15, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotta S, Dakic A, D’Amico A, Pang SH, Greig KT, Nutt SL, and Wu L. (2010). The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity 32, 628–641. [DOI] [PubMed] [Google Scholar]
- Chen HM, Zhang P, Voso MT, Hohaus S, Gonzalez DA, Glass CK, Zhang DE, and Tenen DG (1995). Neutrophils and monocytes express high levels of PU.1 (Spi-1) but not Spi-B. Blood 85, 2918–2928. [PubMed] [Google Scholar]
- Chopin M, Lun AT, Zhan Y, Schreuder J, Coughlan H, D’Amico A, Mielke LA, Almeida FF, Kueh AJ, Dickins RA, et al. (2019). Transcription Factor PU.1 Promotes Conventional Dendritic Cell Identity and Function via Induction of Transcriptional Regulator DC-SCRIPT. Immunity 50, 77–90. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, et al. (2012). A validated regulatory network for th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M. and Bigley V. (2018). Human dendritic cell subsets: an update. Immunol. 154, 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K, Tamoutounour S, Vu Manh TP, Fossum E, Luche H, Ardouin L, Guilliams M, Azukizawa H, Bogen B, Malissen B, et al. (2011). Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8alpha+ type. J. Immunol 187, 4411–4415. [DOI] [PubMed] [Google Scholar]
- Driggers PH, Ennist DL, Gleason SL, Mak WH, Marks MS, Levi BZ, Flanagan JR, Appella E, and Ozato K. (1990). An interferon gamma-regulated protein that binds the interferon-inducible enhancer element of major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. U.S.A 87, 3743–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai V, Bagadia P, Briseno CG, Theisen DJ, Iwata A, Davidson JT, Gargaro M, Fremont DH, Murphy TL, and Murphy KM (2018). Altered compensatory cytokine signaling underlies the discrepancy between Flt3(−/−) and Flt3l(−/−) mice. J. Exp. Med 215, 1417–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai V, Bagadia P, Granja JM, Satpathy AT, Kulkarni DH, Davidson JT, Wu R, Patel SJ, Iwata A, Liu TT, et al. (2019). Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol 20, 1161–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durai V. and Murphy KM (2016). Functions of Murine Dendritic Cells. Immunity 45, 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbeis CF, Singh H, and Storb U. (1995). Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 9, 1377–1387. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, et al. (2009). The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med 206, 3115–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander LB, Khan AA, Ciofani M, Spooner C, Rutz S, et al. (2012). A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1-IRF Complexes. Science 338, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, KC W, Kretzer NM, Briseno CG, Durai V, Bagadia P, et al. (2015). Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat. Immunol 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, et al. (2011). IRF8 mutations and human dendritic-cell immunodeficiency. N. Engl. J. Med 365, 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, Painter MW, and Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Hess JL, Small JA, and Clements JE (1989). Sequences in the visna virus long terminal repeat that control transcriptional activity and respond to viral trans-activation: involvement of AP-1 sites in basal activity and trans-activation. J. Virol 63, 3001–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, and Feuerer M. (2013). Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol 14, 821–830. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Mizutani T, and Taniguchi T. (2004). Negative regulation of IFN-alpha/beta signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc. Natl. Acad. Sci. U.S.A 101, 2416–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humblin E, Thibaudin M, Chalmin F, Derangere V, Limagne E, Richard C, Flavell RA, Chevrier S, Ladoire S, Berger H, et al. (2017). IRF8-dependent molecular complexes control the Th9 transcriptional program. Nat. Commun 8, 2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa E, Hida S, Omatsu Y, Shimoyama S, Takahara K, Miyagawa S, Inaba K, and Taki S. (2004). Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc. Natl. Acad. Sci U.S.A 101, 3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Durai V, Tussiwand R, Briseno CG, Wu X, Grajales-Reyes GE, Egawa T, Murphy TL, and Murphy KM (2017). Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol 18, 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Levi BZ, Tamura T, and Ozato K. (2005). Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J. Interferon Cytokine Res 25, 770–779. [DOI] [PubMed] [Google Scholar]
- Klemsz MJ, McKercher SR, Celada A, Van Beveren C, and Maki RA (1990). The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell 61, 113–124. [DOI] [PubMed] [Google Scholar]
- Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, Hayden MS, Akira S, Sher A, and Ghosh S. (2013). Recognition of Profilin by Toll-like Receptor 12 Is Critical for Host Resistance to Toxoplasma gondii. Immunity 38, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzer NM, Theisen DJ, Tussiwand R, Briseno CG, Grajales-Reyes GE, Wu X, Durai V, Albring J, Bagadia P, Murphy TL, et al. (2016). RAB43 facilitates cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells. J. Exp. Med 213, 2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, Sievert E, Corse E, Chong A, and Sciammas R. (2017). The IRF4 Gene Regulatory Module Functions as a Read-Write Integrator to Dynamically Coordinate T Helper Cell Fate. Immunity 47, 481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwata T, Gongora C, Kanno Y, Sakaguchi K, Tamura T, Kanno T, Basrur V, Martinez R, Appella E, Golub T, et al. (2002). Gamma interferon triggers interaction between ICSBP (IRF-8) and TEL, recruiting the histone deacetylase HDAC3 to the interferon-responsive element. Mol. Cell. Biol 22, 7439–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Tiniakou I, Perez OA, Kirkling ME, Yap GS, Hock H, and Reizis B. (2018). Transcription factor Etv6 regulates functional differentiation of cross-presenting classical dendritic cells. J. Exp. Med 215, 2265–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Zhou YJ, Ma W, Zhang W, Aljoufi A, Luh T, Lucero K, Liang D, Thomsen M, Bhagat G, et al. (2017). Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat. Immunol 18, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Spolski R, Liao W, Wang L, Murphy TL, Murphy KM, and Leonard WJ (2012). BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Chauvistre H, Costa IG, Gusmao EG, Mitzka S, Hanzelmann S, Baying B, Klisch T, Moriggl R, Hennuy B, et al. (2015). Epigenetic program and transcription factor circuitry of dendritic cell development. Nucleic Acids Res. 43, 9680–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley RC, Gill JG, Murphy TL, Langer EM, Cai M, Mashayekhi M, Wang W, Niwa N, Nerbonne JM, Kyba M, et al. (2008). Mesp1 coordinately regulates cardiovascular fate restriction and epithelial-mesenchymal transition in differentiating ESCs. Cell Stem Cell 3, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Medina KL, Lancki DW, and Singh H. (2003). IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 17, 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Turetsky A, Trinh L, and Lu R. (2006). IFN regulatory factor 4 and 8 promote Ig light chain kappa locus activation in pre-B cell development. J. Immunol 177, 7898–7904. [DOI] [PubMed] [Google Scholar]
- Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, et al. (2011). CD8a+ Dendritic Cells Are the Critical Source of Interleukin-12 that Controls Acute Infection by Toxoplasma gondii Tachyzoites. Immunity 35, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKercher SR, Torbett BE, Anderson KL, Henkel GW, Vestal DJ, Baribault H, Klemsz M, Feeney AJ, Wu GE, Paige CJ, et al. (1996). Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO Journal 15, 5647–5658. [PMC free article] [PubMed] [Google Scholar]
- Meraro D, Hashmueli S, Koren B, Azriel A, Oumard A, Kirchhoff S, Hauser H, Nagulapalli S, Atchison ML, and Levi BZ (1999). Protein-protein and DNA-protein interactions affect the activity of lymphoid-specific IFN regulatory factors. J. Immunol 163, 6468–6478. [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. (2012). Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol 13, 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, and Kitamura T. (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, Brehm MA, Swain SL, Welsh RM, and Berg LJ (2014). Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J. Immunol 192, 5881–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai K, Maienschein-Cline M, Simonetti G, Chen J, Rosenthal R, Brink R, Chong AS, Klein U, Dinner AR, Singh H, et al. (2013). Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38, 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, Gudjonsson S, Hakansson U, Reizis B, Kotarsky K, et al. (2013). IRF4 Transcription-Factor-Dependent CD103(+)CD11b(+) Dendritic Cells Drive Mucosal T Helper 17 Cell Differentiation. Immunity 38, 958–969. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz M, Kibardin A, Sturge CR, Pifer R, Li H, Burstein E, Ozato K, Larin S, and Yarovinsky F. (2013). Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J. Immunol 191, 4818–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, KC W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. (2013). Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol 14, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, KC W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, and Murphy KM (2012). Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med 209, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC III, Belardelli F, and Gabriele L. (2002). ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J. Exp. Med 196, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, et al. (2013). IRF4 Transcription Factor-Dependent CD11b(+) Dendritic Cells in Human and Mouse Control Mucosal IL-17 Cytokine Responses. Immunity 38, 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, Malleret B, Zhang S, Larbi A, Zolezzi F, et al. (2015). Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol 16, 718–728. [DOI] [PubMed] [Google Scholar]
- Scott EW, Simon MC, Anastasi J, and Singh H. (1994). Requirement of Transcription Factor Pu.1 in the Development of Multiple Hematopoietic Lineages. Science 265, 1573–1577. [DOI] [PubMed] [Google Scholar]
- Shih DS, Carruth LM, Anderson M, and Clements JE (1992). Involvement of FOS and JUN in the activation of visna virus gene expression in macrophages through an AP-1 site in the viral LTR. Virology 190, 84–91. [DOI] [PubMed] [Google Scholar]
- Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, et al. (2016). IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, and Ozato K. (2005). IFN regulatory factor-4 and −8 govern dendritic cell subset development and their functional diversity. J. Immunol 174, 2573–2581. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, and Tanaka N. (2001). IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol 19, 623–655. [DOI] [PubMed] [Google Scholar]
- Theisen DJ, Davidson JT, Briseno CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, et al. (2018). WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen DJ, Ferris ST, Briseno CG, Kretzer N, Iwata A, Murphy KM, and Murphy TL (2019). Batf3-Dependent Genes Control Tumor Rejection Induced by Dendritic Cells Independently of Cross-Presentation. Cancer Immunol. Res 7, 29–39. [DOI] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, et al. (2015). Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42, 916–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussiwand R, Lee WL, Murphy TL, Mashayekhi M, Wumesh KC, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM, et al. (2012). Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander LB, Khan AA, Hackney JA, Agrawal S, Lesch J, Zhou M, Lee WP, Park S, Xu M, DeVoss J, et al. (2014). Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol 15, 161–167. [DOI] [PubMed] [Google Scholar]
- Veals SA, Santa MT, and Levy DE (1993). Two domains of ISGF3 gamma that mediate protein-DNA and protein-protein interactions during transcription factor assembly contribute to DNA-binding specificity. Mol. Cell. Biol 13, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M, Takami M, Ichino M, Nakazawa M, Matsuyama T, et al. (2011). Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS One 6, e25812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Genes preferentially induced by retroviral reconstitution of Irf4 or Irf8 with or without co-expression of BATF3, related to Figure 3
Table S2 Genes specific for DC and macrophage, related to Figure 4
Table S3 Genes differentially expressed in splenic cDC2 from WT and Batf−/− Batf3−/− mice, related to Figure 5
Data Availability Statement
Microarrays are available on the Gene Expression Omnibus (GEO) database with the accession no. GSE140451 and GSE140452. Following data sets were downloaded and reanalyzed: microarrays for splenic cDC (GSE110789) (Durai et al., 2018), ChIP-seq data sets for IRF4, IRF8, BATF3, H3K4me1, H3K27ac (GSE66899) (Grajales-Reyes et al., 2015), for PU.1 (GSE57563) (Lin et al., 2015), Immunological Genome Project (ImmGen) data sets for ATAC-seq (GSE100738), and microarrays (GSE15907 and GSE37448) (Heng et al., 2008).