

Abstract
Extracellular magnesium ion ([Mg2+]) is a well-known voltage-dependent blocker of NMDA receptors, which plays a critical role in the regulation of neuronal plasticity, learning, and memory. It is generally, believed that NMDA receptor activation involves in Mg2+ being removed into extracellular compartment from the channel pore. On the other hand, Mg2+ is one of the most abundant intracellular cations, and involved in numerous cellular functions. However, we do not know if extracellular magnesium ions can influx into neurons to affect intracellular signaling pathways. In our current study, we found that extracellular [Mg2+] elevation enhanced CREB activation by NMDA receptor signaling in both mixed sex rat cultured neurons and brain slices. Moreover, we found that extracellular [Mg2+] led to CREB activation by NMDA application, albeit in a delayed manner, even in the absence of extracellular calcium, suggesting a potential independent role of magnesium in CREB activation. Consistent with this, we found that NMDA application leads to an NMDAR-dependent increase in intracellular free [Mg2+] in cultured neurons in the absence of extracellular calcium. Chelating this magnesium influx or inhibiting P38 mitogen-activated protein kinase (p38 MAPK) blocked the delayed pCREB by NMDA. Finally, we found that NMDAR signaling in the absence of extracellular calcium activates p38 MAPK. Our studies thus indicate that magnesium influx, dependent on NMDA receptor opening, can transduce a signaling pathway to activate CREB in neurons.
Keywords: neurons, magnesium influx, second messenger, NMDA receptor, CREB, p38 MAPK
Introduction
The presence of high concentrations of Mg2+ in the mammalian cells is essential for the regulation of a broad number of cellular functions and the activity of many enzymes (Romani, 2007). Accumulating evidence suggests Mg2+ has neuroprotective effects in the central nervous system; the decline of brain free [Mg2+] is related to the cognitive impairment in aging, trauma, ischemia/stroke, and neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (Turner and R, 2006; Billard, 2011; Vink and Nechifor, 2011). On the other hand, magnesium supplement has been implicated in memory boosting (Slutsky et al., 2010). On a molecular level, magnesium ion was identified as an NMDA receptor open channel blocker and neuronal depolarization can relieve the Mg2+ block (Mayer et al., 1984; Nowak et al., 1984; Mayer and Westbrook, 1987); hence, Mg2+ is considered a voltage dependent NMDAR open channel blocker. This gives rise to the importance of NMDAR as a co-incidence detector critical for associative memory as its activation needs both the binding of its ligand neurotransmitter, glutamate (released from presynaptic neurons), and postsynaptic neuron membrane depolarization. Even though magnesium is well known as an NMDAR open channel blocker, many studies have demonstrated that substantial magnesium can pass through the NMDA receptor channel pore. In 1993, Brocard et al (Brocard et al., 1993) reported intracellular free [Mg2+] was increased in cortical cultures following NMDAR stimulation. Most of the [Mg2+] increase is Ca2+-trigged intracellular Mg2+ release; interestingly, they found extracellular [Mg2+] also contributed to a delayed intracellular [Mg2+] increase, which can be further amplified by extracellular sodium removal. Further studies indicated that [Mg2+] can influx through the NMDA receptor by both electrophysiological and imaging techniques, and both sodium and calcium ions negatively regulate the permeability of NMDA receptors to magnesium ions (Stout et al., 1996; Antonov and Johnson, 1999; Qian et al., 2002; Qian and Johnson, 2006). However, there are no reports on whether magnesium influx by NMDAR receptor activation can elicit intracellular signaling.
The transcription factor CREB regulates the transcription of thousands of genes and it plays important roles in numerous cellular processes in central nervous system, such as neuronal development, survival, synaptic plasticity and learning and memory (Lonze and Ginty, 2002). Serine 133 is the key regulatory site for CREB activation. Phosphorylation of CREB at Ser-133 (pCREB) triggers the recruitment of the coactivator CBP (CREB binding protein); this CBP interaction can bring CREB to DNA transcriptional machinery promoting gene transcription and protein synthesis(West et al., 2001). NMDA receptor mediated Ca2+ influx is critical in the regulation of CREB activity in neurons (Deisseroth et al., 2003). Interestingly, mild stimulation of NMDA receptors (20 μM NMDA) results in a temporal CREB activation profile starkly different from that of strong stimulation of NMDA receptors (100 μM NMDA). Mild stimulation of NMDA receptors leads to the quick onset of pCREB which then decreases to a persistent value well above baseline pCREB. On the other hand, strong stimulation of NMDA receptors gives a quick onset of CREB activation which then decreases dramatically to a value below baseline pCREB value, due to the inhibition of pCREB at later time points by extrasynaptic NMDA receptor signaling pathway (Sala et al., 2000; Hardingham et al., 2002; Siddoway B, 2011). This dominant pCREB inhibition by extrasynaptic NMDAR signaling has been termed by Hardingham et al (Hardingham et al., 2002) as pCREB shut-off which is relevant to glutamate toxicity in ischemia stroke during which glutamate transporter malfunctions or functions in reverse, leading to excess glutamate in the extracellular space. Manipulation leading to blockade of pCREB shut-off is thus desirable.
Ca2+ influx through NMDARs initiates multiple kinase pathways, including MAPK and CaM-dependent kinases (CaMK), which lead to CREB phosphorylation at Ser-133, CREB activation and CREB targeted gene expression (Deisseroth et al., 2003). MAPK family includes Erk1/2, JNK and p38MAPK (Park et al., 2005; Guindi et al., 2018) and they signal to Msk, which translocate to nucleus to phosphorylate CREB at Ser133. On the other hand, MAPK including p38K and other kinases such as PKA require magnesium as co-factor for their enzymatic activity (Waas et al., 2004; Roskoski, 2015), providing prime targets for regulation by magnesium influx.
In our present work, we have discovered that extracellular magnesium ion can exert a positive role in pCREB phosphorylation induced by NMDAR activation in neurons. The effect of extracellular magnesium increased the duration of pCREB in response to continued NMDA application. We have observed magnesium influx in calcium free solutions and our data indicate that chelating the magnesium influx blocked the positive effect of magnesium on CREB phosphorylation. Finally, we identified p38 MAPK transduces the signaling effect of magnesium CREB phosphorylation.
Methods
Neuronal cultures and acute brain slices
Primary rat cortical neurons were prepared from cortices of mixed sex E18 Sprague Dawley rat embryos. Cells were plated on poly-L-lysine coated wells or glass cover slips in 12-well culture plates (500K cells per plate) in Neurobasal medium supplemented with 2% B27 and 1% GlutaMAX (Siddoway et al., 2014a; Siddoway et al., 2014b; Gao et al., 2018). 13–18 days in vitro (DIV) neurons was used in this study. The experimental protocols for acute mouse brain slices were approved by the Institutional Animal Care and Use Committee of University of Rochester. The transverse cortical slices (300 μm) were prepared from male and female P14 C57B6 mice as described previously (Hu et al., 2006; Hou et al., 2013). In brief, p14 rats were anesthetized with i.p. injection of ketamine/xylazine (90:10 mg/kg) before being euthanized by decapitation with guillotine. The brain was removed quickly and placed in chilled ACSF (concentration in mM: NaCl 125, KCl 2.5, MgCl2 1, CaCl2 2, NaHCO3 25, NaH2PO4 1.25, and dextrose 25). The transverse section of the cortex was then quickly sliced with a vibratome (Leica). Slices were bubbled with oxygen in ACSF (with 1 or 3 mM Mg2+) for 1 hr before NMDA incubation.
Chemicals and antibodies.
Anti CREB pS133 (1:1000; Millipore); anti-CREB (1:500; Santa Cruz Biotechnology); anti P38 MAPK pThr180/182 (1:1000; Cell signal); anti P38 MAPK (1:1000; Cell signal); anti MAP2 (1:500; Millipore); anti NeuN (1:500; Millipore). Chemicals from Sigma: NMDA, DAPV, spermine, PD0325901; Chemicals from Tocris: MK801, SB202190, SB203580, TTX, TEA, KN93, STO-609, LY292004, KT5720, SB415286, PD98059, SP600125, Wortmannin, Dantrolene, Thapsigargin, Ruthenium Red, Naltriben; Imipramine was from Abcam; Cyclopiazonic acid was from MP Biomedicals. Magnesium Green™ AM and Fura-2 AM were from Millipore.
NMDAR stimulation in Western blotting studies.
Normal ACSF (mM: 125 NaCl; 2.5 KCl; 1 MgCl2; 2 CaCl2; 33 D-Glucose; 25 HEPES; pH7.3) or modified ACSF were made fresh, 10 μM Glycine was added before experiment. ACSF was conditioned in incubator for 2–3 hours before use. Neurons were washed briefly with ACSF 3 times, then incubated in the same ACSF for 1 hour before the time-course of NMDA or other compound. Experiments in conditioned medium: 10 mM EGTA was applied for 1 hour to chelate free-Ca2+, 10 mM EDTA was used to Chelate free-Ca2+/Mg2+. [Mg2+] concentration in medium is about 0.814 mM (http://www.thermofisher.com/us/en/home/technical-resources/media-formulation.251.html), which was adjusted to 3 or 9 mM for 1 hour before NMDA application. Compounds were applied in Ca2+ free ACSF for 1 hour before NMDA treatment.
The 12-well plates were put on ice immediately after treatment, with the culture medium aspirated quickly, before 200 ul 1xloading buffer (contains protease and phosphatase inhibitor) was added to each well for about 10 minutes on ice. The cell lysates were then harvested and heated for 10 minutes at 95 C. For the NMDA time course experiment, one 12-well plate of neurons were used each time and repeated at least 4 times in separate independent experiments.
Image J was used for the quantification of Western Blot results. We inverted and thresholded the target bands before measuring the integrated density of each bands. Normalized value of pCREB to CREB was obtained to get relative pCREB value for each sample. If necessary, it will then be normalized to the value of control, with control finally set as 1. We used SPSS16.0 for quantification, One-way Anova (LSD) method for statistical data analysis.
Mg2+ and Ca2+ imaging.
For the Mg2+ imaging measurement, primary neurons grown on coverslips were loaded with 4 μM Magnesium Green AM for 20 min. Cells were then perfused with Ca2+ free ACSF, followed by the stimulation of 20 μM NMDA. Cells were excited at 488 nm and emission was monitored at 505 nm. Peak responses were recorded and normalized to the baseline. For the Ca2+ imaging measurement, 2 μM Fura-2 AM was used as an intracellular Ca2+ indicator, loaded for 20 min, and followed by the stimulation of 20 μM NMDA in normal ACSF. Cells were alternately excited at 340 and 380 nm, and emission was monitored at 505 nm. Imaging was performed using an inverted epifluorescence Nikon microscope with a 40 oil immersion objective (NA 1.3). Images were captured every second with an exposure of 10 ms and 4×4 binning using a digital camera (Cooke Sensicam QE) driven by TILL Photonics software.
Immunostaining.
2-week-old neurons grown on coverslips were incubated in ACSF containing different ions for 1 to 2 hours, then were fixed in ice cold 4% PFA for 20 minutes. Primary antibodies (anti-MAP2 and anti-NeuN) were diluted in 0.5% Triton X-100/5% goat serum in PBS, and incubated overnight at 4°C. Fluorophore conjugated secondary antibodies were incubated for 1 hour at room temperature. Images were captured with a Carl Zeiss confocal system.
Results
Extracellular Magnesium ions positively regulates NMDAR-mediated pCREB signaling
We investigated the extracellular Mg2+ effects on pCREB by NMDA in cultured neurons. We first replicated the different temporal effects of mild (20μM NMDA) and strong (100μM NMDA) stimulation of NMDA receptors on pCREB in literature with normal extracellular magnesium concentration (~ 1mM) (Sala et al., 2000; Hardingham et al., 2002; Karpova et al., 2013) (Fig. 1ab). Interestingly, we found that elevation of the extracellular Mg2+ concentration significantly enhanced pCREB phosphorylation by NMDA (in neurons in either normal ACSF (Fig. 1a) or conditioned medium (Fig. 1b)) for almost all time points. In general, the higher extracellular magnesium have significantly increased effect over lower extracellular magnesium condition. The notable observation is that magnesium effect is more pronounced at later time points for mild NMDAR stimulation while the it is more robust at early time points for strong NMDAR stimulation. The enhancement effects on pCREB by NMDA is [Mg2+] specific, as the substitution of extracellular Mg2+ with Ba2+, another divalent cation, does not have the effect (Fig. S1–1), even though that Ba2+ incubation increased basal pCREB, which is opposite to that by Mg2+ (Fig. 1ab, Fig. 2a). We next found that magnesium also enhances pCREB by NMDA in brain slices (Fig. 1c), indicating that extracellular magnesium also potentiate pCREB signaling in neurons organized in brain slices.
Fig. 1. Extracellular Mg2+ enhances NMDA trigged CREB phosphorylation.

a-b. Time-course of 20 or 100 μM NMDA induced CREB phosphorylation in ACSF (a) or conditioned medium (b) with 1, 3 or 9 mM Mg2+; Mg2+ elevation enhanced NMDA-induced CREB phosphorylation at Ser133 (pCREB). Magnesium ion concentration in medium is around 0.8 mM. Adding 2.2 and 8.2 mM extra magnesium in conditioned medium led to approximately 3 mM and 9 mM external magnesium concentrations. Neurons was incubated with extra Mg2+ containing ACSF or condition medium for one hour before the NMDA application. c. Mg2+ enhances NMDA-induced CREB phosphorylation in mouse brain slices. Acute mouse brain slices was preconditioned with ACSF containing 3 mM Mg2+ for 1 hour. Brain slices were then incubated with 20 μM NMDA for 10 minutes. (*, P<0.05; **, P<0.01, N=3). Error bar in this figure and all other figures indicate standard error of the mean (SEM). Con: indicating control. In supplemental figure legends, C indicates control for space reasons.
Fig. 2. Extracellular Mg2+ is necessary for NMDA caused delayed CREB phosphorylation in neurons with Ca2+ chelated in conditioned medium.
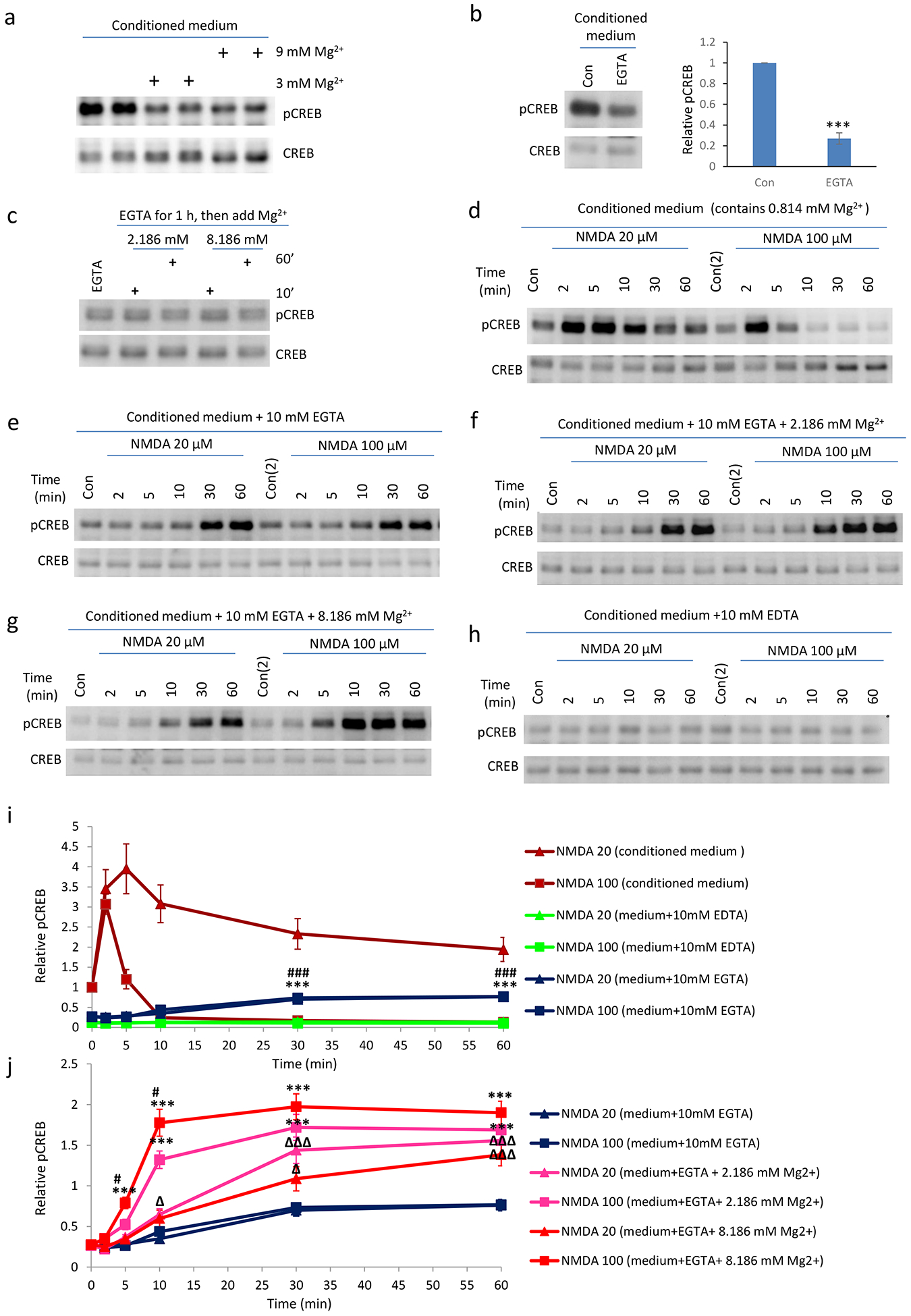
a. Increasing Mg2+ in conditioned medium decreases basal pCREB (1 hour time point). b. pCREB levels after the chelation of Ca2+ alone (with 10 mM EGTA). Neurons were treated for 1 hour before cell harvesting. c. Increasing Mg2+ in conditioned medium did not affect basal pCREB levels when calcium is chelated by EGTA in conditioned medium. 10 mM EGTA was added to conditioned medium for 1 hour before extra MgCl2 was included for 10 or 60 minutes. d-h. Mg2+ dependent CREB phosphorylation in response to NMDA application in neurons. d. NMDA treatment led to increased pCREB in neurons in conditioned medium. e: Delayed CREB phosphorylation was detected after the removal of external Ca2+ with EGTA. (f-g) Increase of Mg2+ concentrations in conditioned medium (with EGTA) enhanced NMDA effects on CREB phosphorylation. h: Further chelating magnesium in conditioned medium blocked the delayed pCREB by NMDA as in (e). i, j. Quantifications of d-h. (**, P<0.01; **, P<0.001)
Extracellular Magnesium ions negatively regulates basal pCREB
Basal pCREB is mediated by calcium influx from NMDA receptors (Hou et al., 2014), while it is believed that extracellular magnesium inhibits NMDA receptor function via acting as an open channel blocker of NMDAR and inhibiting membrane excitability (Zhu et al., 2000). Consistent with this, we found that including more magnesium (Fig. 1a–1b, 2a) or chelating extracellular calcium (Fig. 2b) in conditioned medium decreased basal pCREB levels. On the other hand, increasing magnesium concentration in conditioned medium with EGTA does not have an effect on basal pCREB (Fig. 2c). This is because that pCREB is at its minimum at this state as EGTA has chelated all calcium, thus no calcium mediated glutamate release in the culture can occur. Our study thus indicates that extracellular magnesium ions have opposing roles on basal pCREB and stimulus generated pCREB by NMDA.
Extracellular magnesium ions positively regulates NMDAR-mediated pCREB signaling in the absence of extracellular calcium ions
In order to study whether extracellular magnesium effect on pCREB required calcium, we performed experiment in which extracellular calcium is either chelated or not include in ACSF. To our surprise, we discovered that, when extracellular calcium is chelated by EGTA, similar stimulation of NMDA receptors still led to a sustained pCREB, albeit in a delayed manner (Fig. 2e). Unlike the pattern of NMDAR mediated CREB phosphorylation in normal ACSF or conditioned medium (Fig. 1ab, 2d and 3d), this CREB phosphorylation gradually increased after NMDA application, and remained relatively stable after 30 minutes. Interestingly, for strong NMDAR stimulation, there is no pCREB shut-off (Fig. 2e). Our dose-dependent studies indicate that increase of extracellular Mg2+ in general potentiates pCREB by NMDA (Fig 2f–g, i–j), suggesting that the delayed pCREB by NMDA is Mg2+ dependent. On the other hand, chelating both extracellular calcium and magnesium (with 10 mM EDTA) blocked the NMDA effect on delayed pCREB (Fig. 2h) when compared with when only extracellular calcium was chelated, indicating the critical role of extracellular magnesium in this delayed pCREB induction by NMDA.
We observed similar phenomena when experiments were conducted with neurons incubated in artificial cerebral spinal fluid (ACSF). First, basal pCREB is decreased in calcium free ACSF, but increased when magnesium is removed from normal ACSF (Fig. 3a). Increasing magnesium concentration in ACSF with normal calcium (2 mM) decreased basal pCREB (Fig.3b), but did not have effect when experiments were conducted in calcium free ACSF (Fig. 3c). These data are consistent with our conclusions from experiments conducted in conditioned medium (Fig. 2a–c). pCREB induction by NMDA treatment of neurons incubated in ACSF exhibited a similar temporal profile (Fig. 3d) as that in conditioned medium, but differed in detailed kinetics. Strong NMDA stimulation led to an increase of pCREB which decreased much slower than that in conditioned medium (Fig. 3d). Nevertheless, we still observed a sustained pCREB by NMDA, but with delayed onset, when calcium was not included in ACSF (Fig. 3e). Similarly, increased extracellular magnesium concentration in general potentiated the pCREB by NMDA (Fig. 3f–g, k–l). Further removing magnesium from ACSF blocked this pCREB by NMDA (Fig. 3h), indicating the critical role of extracellular magnesium in pCREB induction by NMDA in the absence of extracellular calcium. In order to eliminate the possible effect of trace calcium on pCREB in our calcium-free ACSF, we performed an NMDA/pCREB experiment in calcium-free ACSF supplemented with added EGTA. Our data indicate that NMDA treatment still elicited a sustained, but delayed, pCREB response (Fig. 3i). On the other hand, the increase of pCREB by NMDA in normal ACSF is attenuated in neurons incubated with Mg-free ACSF (Fig. 3j), consistent with a positive role of magnesium on pCREB by NMDA. Neurons incubated in various ACSF including calcium-free ACSF appear to be healthy as pCREB is relatively stable (Fig. S3–1) and we did not observe differences in morphology when compared with controls (Fig. S3–2).
Fig. 3. Extracellular Mg2+ is responsible for the delayed CREB phosphorylation induced by NMDA in neurons incubated with Ca2+-free ACSF.
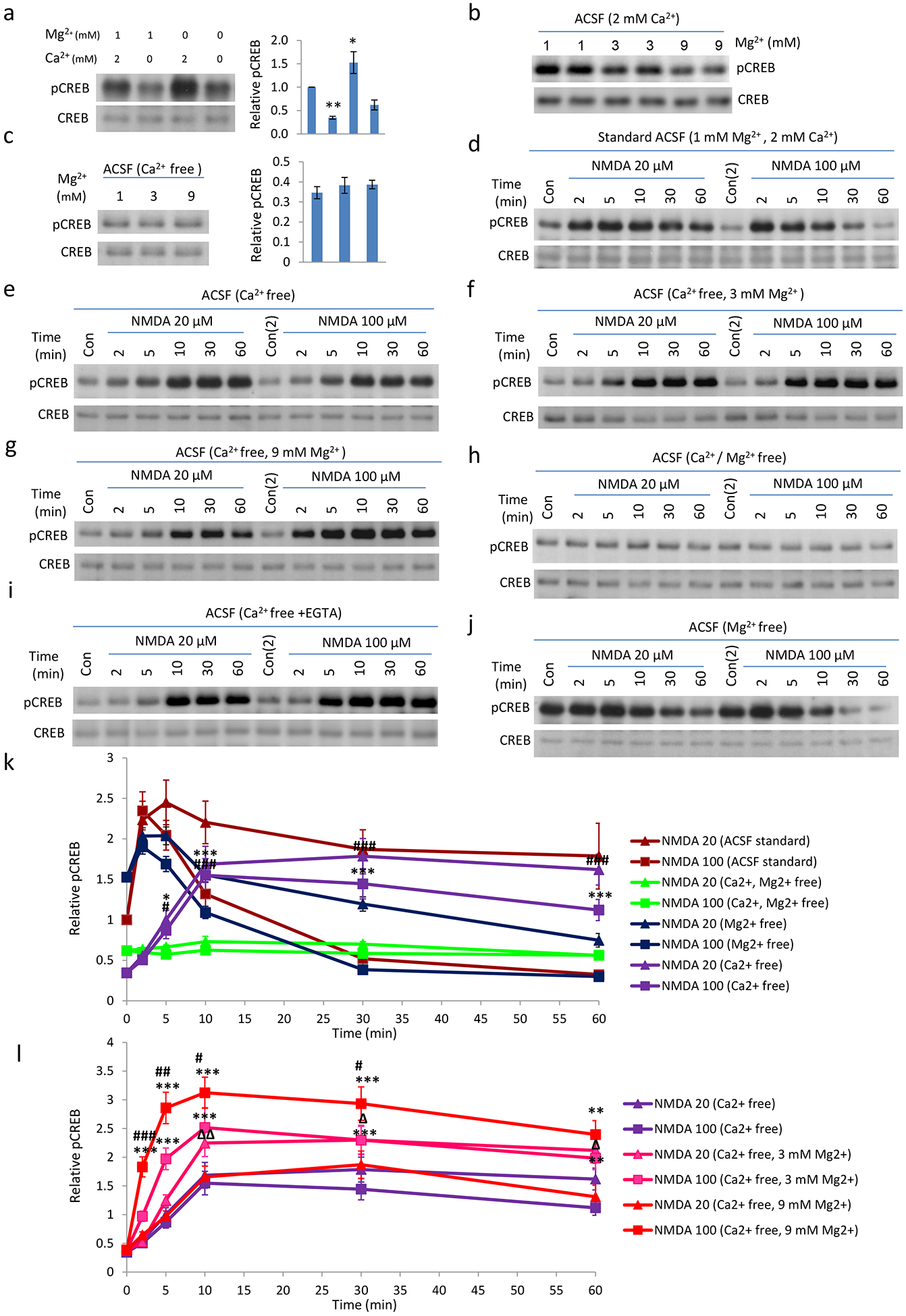
a. Basal pCREB level in Standard, Ca2+ free, Mg2+ free, and Ca2+ /Mg2+ free ACSF. Neurons were briefly washed with different ACSF (preconditioned in incubator for >1 hour) for 3 times, and then incubated in the same ACSF for 1 hour. Components of normal ACSF(mM: 125 NaCl; 2.5 KCl; 1 MgCl2; 2 CaCl2; 33 D-Glucose; 25 HEPES; pH7.3); 10 μM glycine was added freshly before experiments. b: Increasing magnesium concentration in ACSF decreases basal pCREB. c. Basal pCREB in Ca2+ free ACSF with different concentration of Mg2+. d-j. Time course of 20 or 100 μM NMDA in the regulation of CREB phosphorylation in neurons in different ACSF. Neurons were pre-incubated with different ACSF for 1 hour, before the pCREB time-course by NMDA was determined. Delayed CREB phosphorylation can be detected in Ca2+ free ACSF (e); Strong NMDAR stimulation (100 μM) cannot shut-off pCREB (e-g). Removal of Ca2+/Mg2+ abolished NMDA induced CREB phosphorylation (h); i: potential residual calcium in calcium free ACSF did not produce the delay pCREB by NMDA as delayed pCREB by NMDA Is still produced in calcium free ACSF with further inclusion of 1–2 mM EGTA. j: Basal pCREB level was high in Mg2+ free ACSF. NMDA receptor activation only very limitedly increased pCREB level, before CREB phosphorylation was shut-off, even with the low dose NMDA stimulation. k and l. Quantifications of d-h, j. (*, P<0.05; **, P<0.01)
NMDAR-dependent magnesium ion influx is necessary for delayed pCREB by NMDA application
Incubation of neurons with D-APV or MK801, competitive and non-competitive antagonists of NMDARs respectively, blocked the effect of extracellular magnesium on pCREB by either mild or strong NMDA stimulation at 10 minutes time point (Fig 4a), indicating the causal role of NMDAR in promoting pCREB. On the other hand, incubation of neurons with inhibitors to the Na+/Mg2+ exchanger (imipramine) (Romani, 2011), or TRPM7 (spermine) did not affect the Mg2+ effect on pCREB by NMDA (Fig. 4a). We next performed an imaging experiment to directly determine whether magnesium influx occurs. Primary neurons were loaded with 4 μM of Magnesium Green AM for 20 min. Cells were then perfused with Ca2+ free ACSF solution (supplemented with EGTA) followed by a 20 μM NMDA perfusion. We observed a gradual increase in the fluorescence signal (Fig. 4b, movie 1) which was blocked by either pre-incubation of neurons with MK801 for 2 min (Fig. 4c, e) or by removing the extracellular Mg2+ (Fig. 4d, e). We also performed calcium-imaging experiments. We loaded neurons in normal or calcium free ACSF with 2 μM intracellular Ca2+ indicator, Fura-2 AM, for 20 min followed by perfusion of neurons with 20 μM NMDA. We did not observe a calcium signal in neurons incubated in calcium free ACSF (with 2 mM EGTA) (Fig. 4f), but observed robust Ca2+ responses in neurons incubated in normal ACSF (Fig. 4g). In order to determine whether the magnesium influx induced by NMDA we observed is necessary for the delayed pCREB, we incubated neurons with EDTA-AM, a membrane permeable version of the EDTA which can be cleaved inside cells to yield membrane non-permeable EDTA, for one hour before NMDA/pCREB experiments were performed. Our data indicate that this manipulation blocked the pCREB by NMDA in calcium-free ACSF (Fig. 4h). Consistent with a potential role of NMDAR as a channel for magnesium influx, we found that the incubation of neurons with TRPM7 activator Naltriben did not induce CREB phosphorylation in Ca2+ free ACSF (Fig. S4–1A). In addition, we found that inhibiting intracellular calcium store, mitochondrial calcium store, ryanodine receptors, sodium channels and potassium channels did not have an effect on pCREB by NMDA (Fig. S4–1B).
Fig. 4. NMDA receptor dependent Mg2+ influx and p38 MAPK activation in delayed pCREB by NMDA in primary neurons.
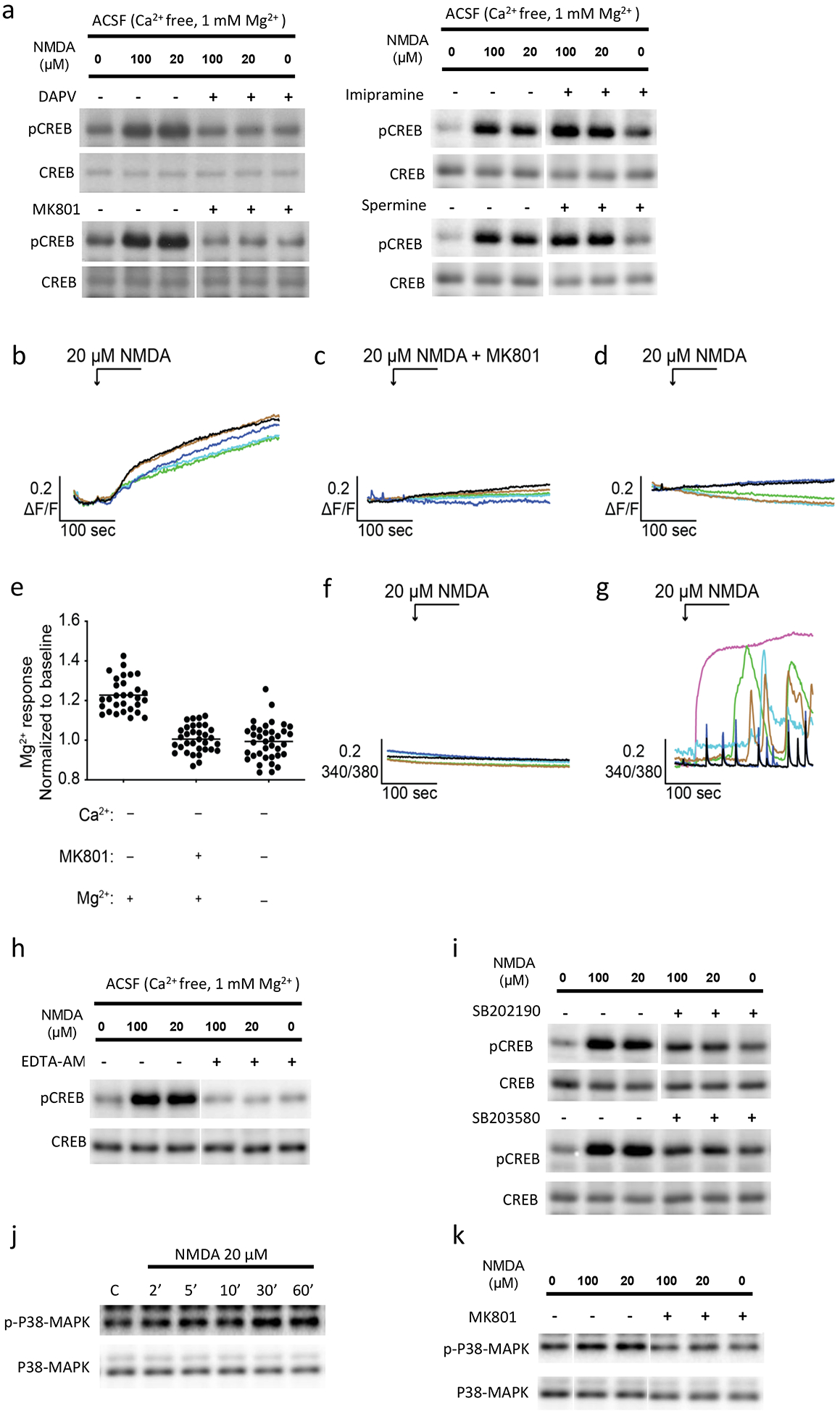
a: D-APV and MK801, but not imipramine and spermine, blocked the delayed pCREB by NMDA at 10 minute time point. D-APV, competitive NMDA receptor antagonist, 50 μM; MK801, NMDA receptor open channel blocker, 10 μM; impramine, Na+-Mg2+ exchanger inhibitor, 100 μM; or spermine, TRPM2 and TRPM7 channel blocker, 10 μM, was included in calcium free ACSF incubation with neurons for 1 hour before NMDA application. b: Primary neurons were loaded with 4 μM of Magnesium Green AM in normal ACSF for 20 min. Cells were then perfused with Ca2+ free ACSF, followed by the stimulation of 20 μM NMDA. This resulted in a gradual increase in the concentration of the intracellular Mg2+. This increase was blocked by either preincubation with 50 μM MK801 for 2 min (c, e) or by removing the extracellular Mg2+ (Ca2+ free ACSF) (d, e). Further, cells were loaded with 2 μM intracellular Ca2+ indicator, Fura-2 AM, for 20 min, followed by the stimulation of 20 μM NMDA in ACSF. Intracellular Ca2+ signals were not changed after NMDA stimulation in Ca2+ free ACSF (with 2 mM EGTA)(f). Robust Ca2+ responses were observed in the presence of extracellular Ca2+ (normal ACSF) (g). (*, P<0.05). h: EDTA-AM (200 μM) incubation with neurons abolished the effect of Mg2+ on delayed pCREB by NMDA. i. p38 MAPK inhibitor SB202190 or SB203580 blocked the effect of delayed pCREB by NMDA in calcium free ACSF. j. Time-course of p38 MAPK phosphorylation in neurons in Ca2+ free ACSF. k. Pre-treatment of MK801 (10 μM) blocked NMDA induced p38 MAPK phosphorylation in Ca2+ free ACSF.
P38 MAPK mediates the effect of extracellular magnesium ions on delayed pCREB by NMDA
What is the link between NMDA receptor dependent magnesium influx and CREB phosphorylation at Ser133? It is known that PP1 is a major CREB phosphatase in neurons (Bito et al., 1996; Yang et al., 2015), however we did not observe change in PP1 phosphorylation at Threonine 320, a marker for PP1 activity (Hou et al., 2013), in response to NMDA in calcium free ACSF (data not shown). We next turned to kinases that could phosphorylate CREB at Ser133 (Lonze and Ginty, 2002). We treated neurons with various kinase inhibitors and our results indicate that the inhibition of p38 MAPK, but not other kinases, efficiently attenuated Mg2+ dependent CREB phosphorylation by NMDA (Fig. 4i; Fig. S4–1C). Our result is consistent with report that magnesium is important for p38 MAPK activity (Easom et al., 1998; Kim et al., 2008; Billard, 2011). In further supporting the role of p38 kinase in pCREB by NMDA in calcium-free ACSF, we found that NMDA incubation with neurons in calcium free ACSF increased the levels of phosphorylated p38 MAPK at Thr180/Tyr182 (Fig. 4j), a marker for p38 MAPK activation (Rouse et al., 1994). We further found that the increase of phosphorylated p38 was blocked by MK801, an NMDA receptor open channel blocker (Fig. 4k). Synaptic NMDA receptor signaling promotes pCREB while extrasynaptic receptor signaling inhibits pCREB (Hardingham et al., 2002). Interestingly we did not observe significant effect of extracellular magnesium on pCREB induced by synaptic NMDA receptor activation (via incubating neurons with bicuculline, an GABAA receptor antagonist) (data not shown). This suggests that extracellular magnesium functions to counteract the extrasynaptic NMDA receptor’s pCREB shut-off function (Hardingham et al., 2002).
Discussion
Intracellular free [Mg2+] at resting state is at the millimolar level, whereas intracellular free Ca2+ is around 100 nM. In response to stimulus, while intracellular Ca2+ increases can be tens of thousands fold, intracellular free [Mg2+] fold change is minimal (Clapham, 2007). This made calcium a prominent second messenger in the cells. But nevertheless, in 2011, Li et al (Li et al., 2011) reported a role for [Mg2+] as an intracellular second messenger coupling cell-surface T-cell antigen receptor (TCR) to intracellular phospholipase Cγ1 (PLCγ1) in human T cells. Three fundamental features have been put forward for a molecule to qualify as a second messenger (Sutherland, 1972; Takaya et al., 2000; Li et al., 2011). First, its level must increase rapidly in response to a stimulus, typically in the form of the engagement of cell-surface receptor(s); second, it needs to alter the rate of one or more cellular processes; third, it exerts cell-type specific activity as different cells harbor different components of enzymes. Our results indicated Mg2+ conforms to all the three criteria for being a second messenger for CREB phosphorylation in neurons in response to NMDA receptor activation. Calcium is known to induce strong pCREB signaling pathways, but by using [Ca2+] free ACSF and calcium chelator in conditioned medium, we isolated Mg2+ influx as well as Mg2+ influx-trigged p38 MAPK activation signaling that was responsible for the delayed CREB activation. We showed that intracellular [Mg2+] increases rapidly within minutes after NMDAR stimulation, which is sufficient to activate p38 MAPK signaling pathways to phosphorylate CREB in a delayed manner. We also showed that extracellular [Mg2+] elevation further enhanced Ca2+-induced CREB activations in physiological conditions, revealing an unknown role of extracellular Mg2+ in CREB signaling.
P38 MAPK is a kinase whose activity requires magnesium binding and is a natural effector of magnesium influx. Our inhibitor (of p38MAPK) studies confirmed the necessary role of p38MAPK in transducing magnesium influx to pCREB. Moreover, we found that p38 can be activated by the magnesium influx; this probably suggests that p38MAPK is not maximally bound by magnesium inside resting neurons. Future studies are needed to determine p38 MAPK downstream signaling for pCREB.
Our Mg imaging/MK801 experiment indicates that magnesium ion influx is dependent on NMDA receptor activation. Moreover, our EDTA-AM/NMDA/pCREB experiment established the causality of magnesium influx in intracellular signaling pathways for CREB phosphorylation. What mediates the magnesium influx? Incubation with neurons an agonist to well-known magnesium transporter TRMP7 (Naltriben; extended figure 4.1) not being able to support pCREB suggest that TRMP7 is not likely the channel for the magnesium influx induced by NMDA receptor activation. Even though our use of existing magnesium transporter inhibitors did not block magnesium effect on pCREB by NMDA, because of the possibility of unknown magnesium transporters and magnesium exchangers present in the neurons, we could not rule out the contribution of magnesium transports and exchangers to the pCREB by NMDA. But the contributing magnesium transporters and exchangers in pCREB are still be subject to NMDA receptor regulation based on MK801 blocking studies. It would be interesting in future studies to identify the magnesium transporters. Moreover, we cannot rule out the possibility that our observed magnesium upregulation in neurons by imaging method is of an intracellular origin such as from mitochondria as in (Yamanaka et al., 2018).
Another route of magnesium influx, not necessarily mutually exclusive with possibilities discussed above is the passing of magnesium ion via NMDA receptor channel pore. Extracellular [Mg2+] is well known to be an open channel blocker for NMDA receptors. Membrane depolarization is generally believed to relieve this block via removing magnesium ion to extracellular space. However, magnesium ions indeed can pass through NMDA receptor channel pores via electrophysiological recordings (Stout et al., 1996), though the magnesium influx could be attenuated by competition from monovalent ions in calcium free solutions and additional from calcium ions in normal ACSF. The competition of calcium and sodium ions on magnesium ion passing through NMDA receptor channels suggests that the amount of magnesium ion flowing through NMDA receptor is low in response to NMDA receptor activation. However, this could explain the slow rise of magnesium influx and the delayed effect of magnesium on pCREB by NMDA in the absence of calcium in extracellular solution; longer time is needed for sufficient magnesium accumulation inside neurons to maximally activate p38 MAPK for eventual. Future work is needed to ascertain whether magnesium ion can pass through NMDA receptor channel pore to induce the effect on pCREB by NMDA.
We observed that 8 mM extra magnesium ions in conditioned medium completely reverses the normal pCREB shut-off induced by 100 μM NMDA. This is consistent with that 100 μM NMDA is the strong stimulus for NMDA receptor activation. This strong NMDA receptor stimulation is relevant to pathological conditions, such as glutamate toxicity during ischemia when glutamate transporter’s malfunction leads to excess extracellular glutamate. Even at this condition, however, 3 mM extracellular magnesium ions in conditioned medium also shows robust counteracting effect, especially at early time points. On the other hand, it is known that 20 μM NMDA/ 3 minutes is the minimum stimulus strength needed for induce long-term depression (LTD) (Lee et al., 1998), a physiological process. Mild NMDA receptor stimulation by 20 μM NMDA in our study thus represents a physiological stimulation. Under this physiological stimulation condition, 3 mM extracellular magnesium concentration also led to a significant and robust enhancement of pCREB by NMDA in normal ACSF or in conditioned medium (Fig. 1). Moreover, this increase of pCREB mainly occur over later time points that would indicate enhanced CREB-mediated gene expression. Because CREB-mediated gene expression critical for late phase LTP and memory formation (Barco et al., 2002), our data on the extracellular magnesium enhancement effect on pCREB by NMDA could thus provide another potential mechanism for reported memory boosting by dietary magnesium supplement (Slutsky et al., 2010).
There are studies with similar conclusions on magnesium effect on pCREB, but in different cell types, using different manipulations and with different signaling pathways. For example, Liao et al reported that elevating extracellular magnesium concentration directly promoted neuroprogenitor cell (NPC) differentiation into neurons, instead of astrocytes (Liao et al., 2017), probably via ERK1/2 activation and CREB phosphorylation at Ser133. Moreover, Yamanaka et al reported in 2018 that GABA application to DIV 7 hippocampal neurons led to magnesium release from mitochondria to activate mTOR as well as CREB signaling for neuronal maturation (Yamanaka et al., 2018). In these two studies, the elevation of magnesium level is around two folds, close to our studies of using 3 mM extracellular magnesium concentrations. On the other hand, in studies of using magnesium as a diet supplement, sustained increase of magnesium ion in cerebral spinal fluid (CSF) only needs to be 10% higher than normal to achieve measureable beneficial cognition outcome. Future studies are needed to determine whether the magnesium effects on CREB activation (our studies here and (Liao et al., 2017; Yamanaka et al., 2018)), NPC preferential differentiation to neurons (Liao et al., 2017) and/or neuronal maturation (Yamanaka et al., 2018) can be observed in a magnesium dietary type of study as in (Slutsky et al., 2010).
In summary, we have discovered that NMDA receptor dependent magnesium influx in neurons acts as a second messenger inducing signaling pathway phosphorylating CREB having potential relevance to both physiological and pathological conditions.
Supplementary Material
Fig. S1–1 Ba2+ does not mimic Mg2+ effects in the regulation of NMDAR mediate CREB signaling. A. Neurons were incubated with normal ACSF or ACSF plus 8 mM BaCl2 for 1 hour, before pCREB/NMDA experiments were performed. Contrary to Mg2+ effects, Ba2+ treatment increased basal pCREB level, and enhanced the shut-off effects of NMDA. B. Similar experiments as in A, except that neurons were incubated in conditioned medium.
Fig. S3–1 pCREB level is relatively stable when incubated in ACSF conditions including calcium-free ACSF. A-E. Basal pCREB of neurons in various modified ACSF at different time points. Neurons were washed with different pre-conditioned ACSF for 3 times, then incubated with the same ACSF for 1 hour, before being harvested at different time points. F. Neuronal response to NMDA treatments in neurons in normal medium, but experienced prior Ca2+ free ACSF incubation. Neurons were incubated with Ca2+ free ACSF for 2 hours, and then the ACSF was changed to conditioned medium for 30 minute before pCREB/NMDA experiment was performed.
Fig. S3–2 Normal morphology for neurons after incubation for 2 hours in different ACSFs including calcium free ACSF Neurons were incubated with different modified ACSF for 1 or 2 hours, before double labeling (with NeuN and MAP2 antibody) were performed. (Scale bar= 50 μM)
Fig. S4–1 Testing of signal pathways, channels and kinases that may underlie NMDA induced CREB phosphorylation in Ca2+ free ACSF in neurons. A. Blocking intracellular calcium sources as well as sodium and potassium channels does not have an effect on pCREB by NMDA in neurons incubated in calcium free ACSF. Cyclopiazonic acid, reversible inhibitor of sarcoplasmic reticulum Ca2+-ATPase, 20 μM; Thapsigargin, Ca2+-ATPase inhibitor, 10 μM; Ruthenium Red, mitochondria Ca2+ uptake and release blocker, 10 μM; Dantrolene, Ryanodine receptor inhibitor, 20 μM; TTX, Na+ channel blocker, 2 μM; TEA, K+ channel blocker, 10 mM. B. Time-course of the effect of TRMP7 activator naltriben on pCREB by NMDA. C. The effect of various kinase inhibitors on pCREB by NMDA. KN93, CaMK II inhibitor, 10 μM; PD0325901, MEK inhibitor, 10 μM; KT5720, 5 μM, PKA inhibitor; PD98059, MAPKK/MEK inhibitor, 20 μM; Wortmannin, PI3K inhibitor, 0.5 μM; LY294002, PI3K inhibitor, 20 μM; STO-609, CaMKK inhibitor, 10 μM; SB415286, GSK3β inhibitor, 20 μM; SP600125, JNK inhibitor, 20 μM; Compounds were applied in Ca2+ free ACSF for 1 hour before NMDA application. 10 minutes of NMDA treatment time point was used in these studies. These signaling pathways did not play a role in the regulation of pCREB by NMDA.
Movie 1. NMDA receptor activation induced Mg2+ increase in neurons in Ca2+ free ACSF (Totally 15 minutes). 20 μM NMDA is applied to neurons incubated in modified ACSF (3 mM Mg and 0 Ca).
Highlights:
Calcium is the well-known trigger for CREB phosphorylation and activation in neurons; however, we found that another divalent ion, extracellular magnesium, could also positively regulate CREB phosphorylation and activation, in an NMDA receptor dependent manner. Extracellular magnesium can flux into neurons to potentiate p38 MAPK pathway leading to increased CREB phosphorylation. This novel magnesium-mediated CREB signaling pathway has a slower onset and can be activated by both mild and strong NMDA receptor activation, indicating a potential relevance to both physiological and pathological role in our brain.
Acknowledgements:
This work is supported by NIH (R01 MH109719), NSF (IOS-1457336) to HX, NIH (R01 DE014756) to D. Y.. We would like to thank Dr. Jon Johnson (U. Pittsburgh) for helpful discussions regarding this work.
Reference:
- Antonov SM, Johnson JW (1999) Permeant ion regulation of N-methyl-D-aspartate receptor channel block by Mg(2+). Proc Natl Acad Sci U S A 96:14571–14576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER (2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108:689–703. [DOI] [PubMed] [Google Scholar]
- Billard JM (2011) Brain free magnesium homeostasis as a target for reducing cognitive aging. In: Magnesium in the Central Nervous System (Vink R, Nechifor M, eds). Adelaide (AU). [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. [DOI] [PubMed] [Google Scholar]
- Brocard JB, Rajdev S, Reynolds IJ (1993) Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron 11:751–757. [DOI] [PubMed] [Google Scholar]
- Clapham DE (2007) Calcium signaling. Cell 131:1047–1058. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13:354–365. [DOI] [PubMed] [Google Scholar]
- Easom RA, Tarpley JL, Filler NR, Bhatt H (1998) Dephosphorylation and deactivation of Ca2+/calmodulin-dependent protein kinase II in betaTC3-cells is mediated by Mg2+- and okadaic-acid-sensitive protein phosphatases. Biochem J 329 (Pt 2):283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Hu XD, Yang H, Xia H (2018) Distinct Roles of Protein Phosphatase 1 Bound on Neurabin and Spinophilin and Its Regulation in AMPA Receptor Trafficking and LTD Induction. Mol Neurobiol 55:7179–7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindi C, Cloutier A, Gaudreau S, Zerif E, McDonald PP, Tatsiy O, Asselin C, Dupuis G, Gris D, Amrani AA (2018) Role of the p38 MAPK/C/EBPbeta Pathway in the Regulation of Phenotype and IL-10 and IL-12 Production by Tolerogenic Bone Marrow-Derived Dendritic Cells. Cells 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- Hou H, Sun L, Siddoway BA, Petralia RS, Yang H, Gu H, Nairn AC, Xia H (2013) Synaptic NMDA receptor stimulation activates PP1 by inhibiting its phosphorylation by Cdk5. J Cell Biol 203:521–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou H, Chavez AE, Wang CC, Yang H, Gu H, Siddoway BA, Hall BJ, Castillo PE, Xia H (2014) The Rac1 inhibitor NSC23766 suppresses CREB signaling by targeting NMDA receptor function. J Neurosci 34:14006–14012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XD, Huang Q, Roadcap DW, Shenolikar SS, Xia H (2006) Actin-associated neurabin-protein phosphatase-1 complex regulates hippocampal plasticity. J Neurochem 98:1841–1851. [DOI] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Bera S, Bar J, Reddy PP, Behnisch T, Rankovic V, Spilker C, Bethge P, Sahin J, Kaushik R, Zuschratter W, Kahne T, Naumann M, Gundelfinger ED, Kreutz MR (2013) Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 152:1119–1133. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Lee SJ, Kim JS, Kang HS (2008) High extracellular [Mg2+]-induced increase in intracellular [Mg2+] and decrease in intracellular [Na+] are associated with activation of p38 MAP kinase and ERK2 in guinea-pig heart. Exp Physiol 93:1223–1232. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21:1151–1162. [DOI] [PubMed] [Google Scholar]
- Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, Cohen JI, Uzel G, Su HC, Lenardo MJ (2011) Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475:471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Jiang M, Li M, Jin C, Xiao S, Fan S, Fang W, Zheng Y, Liu J (2017) Magnesium Elevation Promotes Neuronal Differentiation While Suppressing Glial Differentiation of Primary Cultured Adult Mouse Neural Progenitor Cells through ERK/CREB Activation. Front Neurosci 11:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL (1987) Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol 394:501–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB (1984) Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309:261–263. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307:462–465. [DOI] [PubMed] [Google Scholar]
- Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, Hoffmann A, Montminy M, Karin M (2005) Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-kappaB as key regulators. Immunity 23:319–329. [DOI] [PubMed] [Google Scholar]
- Qian A, Johnson JW (2006) Permeant ion effects on external Mg2+ block of NR1/2D NMDA receptors. J Neurosci 26:10899–10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian A, Antonov SM, Johnson JW (2002) Modulation by permeant ions of Mg(2+) inhibition of NMDA-activated whole-cell currents in rat cortical neurons. J Physiol 538:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani AM (2007) Magnesium homeostasis in mammalian cells. Front Biosci 12:308–331. [DOI] [PubMed] [Google Scholar]
- Romani AM (2011) Cellular magnesium homeostasis. Arch Biochem Biophys 512:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr. (2015) A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res 100:1–23. [DOI] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR (1994) A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78:1027–1037. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M (2000) Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci 20:3529–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B, Hou H, Yang H, Petralia R, Xia H (2014a) Synaptic activity bidirectionally regulates a novel sequence-specific S-Q phosphoproteome in neurons. J Neurochem 128:841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B, Hou H, Yang J, Sun L, Yang H, Wang GY, Xia H (2014b) Potassium channel Kv2.1 is regulated through protein phosphatase-1 in response to increases in synaptic activity. Neurosci Lett 583:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway BHH, and Xia H (2011) Glutamatergic Synapses: Molecular Organisation. eLS DOI: 10.1002/9780470015902.a0000235.pub2. [DOI] [Google Scholar]
- Slutsky I, Abumaria N, Wu LJ, Huang C, Zhang L, Li B, Zhao X, Govindarajan A, Zhao MG, Zhuo M, Tonegawa S, Liu G (2010) Enhancement of learning and memory by elevating brain magnesium. Neuron 65:165–177. [DOI] [PubMed] [Google Scholar]
- Stout AK, Li-Smerin Y, Johnson JW, Reynolds IJ (1996) Mechanisms of glutamate-stimulated Mg2+ influx and subsequent Mg2+ efflux in rat forebrain neurones in culture. J Physiol 492 (Pt 3):641–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland EW (1972) Studies on the mechanism of hormone action. Science 177:401–408. [DOI] [PubMed] [Google Scholar]
- Takaya J, Higashino H, Kobayashi Y (2000) Can magnesium act as a second messenger? Current data on translocation induced by various biologically active substances. Magnes Res 13:139–146. [PubMed] [Google Scholar]
- Turner RJ, R V (2006) Magnesium in the Central Nervous System. In: New perspectives in magnesium research:338–355. [Google Scholar]
- Vink R, Nechifor M (2011) Magnesium in the Central Nervous System. Adelaide, Australia: Univ of Adelaide Press:338–355. [PubMed] [Google Scholar]
- Waas WF, Rainey MA, Szafranska AE, Cox K, Dalby KN (2004) A kinetic approach towards understanding substrate interactions and the catalytic mechanism of the serine/threonine protein kinase ERK2: identifying a potential regulatory role for divalent magnesium. Biochim Biophys Acta 1697:81–87. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka R, Shindo Y, Hotta K, Suzuki K, Oka K (2018) GABA-Induced Intracellular Mg(2+) Mobilization Integrates and Coordinates Cellular Information Processing for the Maturation of Neural Networks. Curr Biol 28:3984–3991 e3985. [DOI] [PubMed] [Google Scholar]
- Yang H, Hou H, Pahng A, Gu H, Nairn AC, Tang YP, Colombo PJ, Xia H (2015) Protein Phosphatase-1 Inhibitor-2 Is a Novel Memory Suppressor. J Neurosci 35:15082–15087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R (2000) Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 3:1098–1106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1–1 Ba2+ does not mimic Mg2+ effects in the regulation of NMDAR mediate CREB signaling. A. Neurons were incubated with normal ACSF or ACSF plus 8 mM BaCl2 for 1 hour, before pCREB/NMDA experiments were performed. Contrary to Mg2+ effects, Ba2+ treatment increased basal pCREB level, and enhanced the shut-off effects of NMDA. B. Similar experiments as in A, except that neurons were incubated in conditioned medium.
Fig. S3–1 pCREB level is relatively stable when incubated in ACSF conditions including calcium-free ACSF. A-E. Basal pCREB of neurons in various modified ACSF at different time points. Neurons were washed with different pre-conditioned ACSF for 3 times, then incubated with the same ACSF for 1 hour, before being harvested at different time points. F. Neuronal response to NMDA treatments in neurons in normal medium, but experienced prior Ca2+ free ACSF incubation. Neurons were incubated with Ca2+ free ACSF for 2 hours, and then the ACSF was changed to conditioned medium for 30 minute before pCREB/NMDA experiment was performed.
Fig. S3–2 Normal morphology for neurons after incubation for 2 hours in different ACSFs including calcium free ACSF Neurons were incubated with different modified ACSF for 1 or 2 hours, before double labeling (with NeuN and MAP2 antibody) were performed. (Scale bar= 50 μM)
Fig. S4–1 Testing of signal pathways, channels and kinases that may underlie NMDA induced CREB phosphorylation in Ca2+ free ACSF in neurons. A. Blocking intracellular calcium sources as well as sodium and potassium channels does not have an effect on pCREB by NMDA in neurons incubated in calcium free ACSF. Cyclopiazonic acid, reversible inhibitor of sarcoplasmic reticulum Ca2+-ATPase, 20 μM; Thapsigargin, Ca2+-ATPase inhibitor, 10 μM; Ruthenium Red, mitochondria Ca2+ uptake and release blocker, 10 μM; Dantrolene, Ryanodine receptor inhibitor, 20 μM; TTX, Na+ channel blocker, 2 μM; TEA, K+ channel blocker, 10 mM. B. Time-course of the effect of TRMP7 activator naltriben on pCREB by NMDA. C. The effect of various kinase inhibitors on pCREB by NMDA. KN93, CaMK II inhibitor, 10 μM; PD0325901, MEK inhibitor, 10 μM; KT5720, 5 μM, PKA inhibitor; PD98059, MAPKK/MEK inhibitor, 20 μM; Wortmannin, PI3K inhibitor, 0.5 μM; LY294002, PI3K inhibitor, 20 μM; STO-609, CaMKK inhibitor, 10 μM; SB415286, GSK3β inhibitor, 20 μM; SP600125, JNK inhibitor, 20 μM; Compounds were applied in Ca2+ free ACSF for 1 hour before NMDA application. 10 minutes of NMDA treatment time point was used in these studies. These signaling pathways did not play a role in the regulation of pCREB by NMDA.
Movie 1. NMDA receptor activation induced Mg2+ increase in neurons in Ca2+ free ACSF (Totally 15 minutes). 20 μM NMDA is applied to neurons incubated in modified ACSF (3 mM Mg and 0 Ca).