

Abstract
Tirzepatide (LY3298176), a dual GIP and GLP-1 receptor (GLP-1R) agonist, delivered superior glycemic control and weight loss compared with GLP-1R agonism in patients with type 2 diabetes. However, the mechanism by which tirzepatide improves efficacy and how GIP receptor (GIPR) agonism contributes is not fully understood. Here, we show that tirzepatide is an effective insulin sensitizer, improving insulin sensitivity in obese mice to a greater extent than GLP-1R agonism. To determine whether GIPR agonism contributes, we compared the effect of tirzepatide in obese WT and Glp-1r–null mice. In the absence of GLP-1R–induced weight loss, tirzepatide improved insulin sensitivity by enhancing glucose disposal in white adipose tissue (WAT). In support of this, a long-acting GIPR agonist (LAGIPRA) was found to enhance insulin sensitivity by augmenting glucose disposal in WAT. Interestingly, the effect of tirzepatide and LAGIPRA on insulin sensitivity was associated with reduced branched-chain amino acids (BCAAs) and ketoacids in the circulation. Insulin sensitization was associated with upregulation of genes associated with the catabolism of glucose, lipid, and BCAAs in brown adipose tissue. Together, our studies show that tirzepatide improved insulin sensitivity in a weight-dependent and -independent manner. These results highlight how GIPR agonism contributes to the therapeutic profile of dual-receptor agonism, offering mechanistic insights into the clinical efficacy of tirzepatide.
Keywords: Metabolism, Therapeutics
Keywords: Diabetes
Introduction
Type 2 diabetes mellitus (T2DM) is a global pandemic, placing significant strain on healthcare systems world wide (1). Driven primarily by excess body weight, systemic insulin resistance (IR) is a key driver of pancreatic β cell dysfunction and the subsequent progression to T2DM (2, 3). Although the etiology remains to be confirmed, current evidence implicates several factors (e.g., genetic predisposition, hyperlipidemia/lipotoxicity, impaired branched-chain amino acid (BCAA) catabolism, inflammation, endoplasmic reticulum stress, and mitochondrial dysfunction) in the development of whole-body IR (4). To date, caloric restriction–mediated weight loss, bariatric surgery, or pharmacotherapy with thiazolidinediones (TZDs, small-molecule agonists of the nuclear hormone receptor PPARγ) are the only effective approaches that specifically treat IR for improving glucose control (5). However, in the long term, caloric restriction is ineffective due to counterregulatory responses and compliance issues, bariatric surgery is an expensive procedure carrying significant risk, and the TZD drug class has fallen out of favor due to an unacceptable side effect profile (e.g., weight gain, edema, fracture, ref. 6). Therefore, new therapeutic agents that are capable of targeting multiple organs/signaling pathways to enhance insulin sensitivity may offer an attractive mechanism-based approach for treating T2DM (7).
Over the past decade, glucagon-like peptide-1 receptor agonists (GLP-1RAs) have emerged as popular medicines for the treatment of T2DM due to their safety and efficacy (8). The glycemic benefits associated with GLP-1RAs are due to enhanced β cell function, delayed gastric emptying, and weight loss–driven improvements in insulin sensitivity (9). However, although the GLP-1RA class has improved the standard of care, many patients still do not reach their glycemic targets (10), and thus, there remains a need to identify agents capable of complementing the actions of GLP-1RAs. As key sites of energy storage and oxidation, white and brown adipose tissue (WAT and BAT) are at the forefront of the link between obesity-induced IR and the subsequent development of T2DM (11, 12). Thus, one strategy to augment the therapeutic profile of GLP-1RAs is to complement their actions with agents that target WAT and BAT to improve their capacities for metabolic substrate disposal, thus lowering plasma levels of lipids, BCAAs, and other metabolites that may contribute to systemic IR (13, 14). Glucose-dependent insulinotropic polypeptide (GIP) is a hormone released from the gut following the consumption of food (15). Similar to GLP-1, the primary physiological role of GIP is to enhance glucose-dependent insulin secretion (16, 17). However, the metabolic effects of GIP extend beyond the pancreas and include several metabolically relevant organs (18). In contrast to the glucagon-like peptide 1 receptor (GLP-1R), the GIP receptor (GIPR) is expressed in WAT (19) and BAT (20). Via its effects in these tissues, GIP has been described primarily as a regulator of glucose and lipid metabolism (20–22).
Overexpression of GIP or administration of long-acting GIPR agonists (LAGIPRAs) enhances whole-body insulin sensitivity in models of obesity-induced IR (23, 24). Mechanistically, the benefits associated with chronic GIPR engagement are linked to improved adipose tissue health, characterized by enhanced adipocyte insulin signaling, reduced proinflammatory immune cell infiltration, recruitment of metabolic pathways associated with lipid storage and oxidation, and the release of insulin-sensitizing adipokines (23–26). Together, the ability of GIP to target adipose tissue and improve metabolic homeostasis highlights its attractiveness for partnering with GLP-1 (18, 22). As a therapeutic modality, next-generation antidiabetic agents engineered to simultaneously target the GIP and GLP-1 receptors (GLP-1Rs) are being pursued (27–29). The most advanced multireceptor agonist is tirzepatide (LY3298176), an imbalanced and biased GIP and GLP-1R agonist (28, 30). In a phase 2b trial of patients with T2DM, tirzepatide delivered superior weight loss and improved glycemic control when compared with a selective GLP-1RA (dulaglutide, 1.5 mg; ref. 31). Importantly, improved glucose homeostasis was associated with enhanced insulin sensitivity, and post hoc analysis of this study suggested that the majority of the insulin sensitization occurred independent of weight loss (32). Therefore, we investigated the mechanistic basis of how tirzepatide may improve insulin sensitivity and whether GIPR agonism directly contributes to this benefit.
Results
To determine if tirzepatide improves insulin sensitivity, we conducted a series of chronic studies in high-fat diet–fed obese IR mice. Treatment with tirzepatide reduced body weight, food intake, tissue weights, and plasma leptin (Figure 1, A–C). Fourteen days of treatment with tirzepatide reduced circulating triglyceride levels and free fatty acids (FFAs) and lowered hepatic lipid content (Figure 1, D and E). In line with improved glucose homeostasis, tirzepatide reduced fed (Figure 1F) and fasted (Figure 1G) blood glucose and plasma insulin, while increasing circulating levels of the insulin sensitizers adiponectin and IGFBP2 (refs. 33, 34 and Figure 1H). Furthermore, in accordance with a state of heightened insulin sensitivity, tirzepatide improved insulin tolerance (Figure 1I). Importantly, chronic treatment with tirzepatide reduced body weight and food intake and enhanced markers of insulin sensitivity when dosed at room temperature (24°C, Figure 1) and thermal neutral (27°C) conditions (Supplemental Figure 1, A–E; supplemental material available online with this article; https://doi.org/10.1172/JCI146353DS1). To directly investigate if tirzepatide improves systemic insulin sensitivity, we performed hyperinsulinemic-euglycemic clamp studies in obese IR mice (Figure 2). Tirzepatide enhanced systemic insulin sensitivity, such that a 5.2-fold increase in the glucose infusion rate (GIR) was required to maintain euglycemia when compared with vehicle-treated animals (Figure 2A). There was no effect of tirzepatide on endogenous glucose production (EGP, Figure 2B), suggesting that enhanced whole-body glucose disposal was driven by improved peripheral insulin sensitivity. To elucidate which tissues accounted for the enhanced insulin sensitization, we measured 2-[1-14C] deoxy-D-glucose uptake. Tirzepatide increased insulin stimulated glucose disposal in skeletal muscle (Figure 2, C–E) and WAT (Figure 2, F and G). These experiments indicate that tirzepatide is a highly effective insulin sensitizer in obese IR mice, with enhanced insulin-stimulated glucose disposal in skeletal muscle and adipose tissue, appearing to account for the glycemic benefits.
Figure 1. Chronic treatment with tirzepatide enhanced insulin tolerance in obese mice.

Obese insulin-resistant mice were dosed once daily for 14 days with vehicle or tirzepatide (TZP, 10 nmol/kg; n = 5–8 per group). (A) Daily body weight and food intake. (B) Tissue weights, (C) plasma, (D) leptin triglycerides and free fatty acids (FFA), and (E) liver triglyceride after 14 days of treatment. (F) Fed and (G) fasted blood glucose and plasma insulin. (H) Fed adiponectin and insulin-like growth factor binding protein 2 (IGFBP2). (I) Insulin tolerance test. Data are presented as mean ± SEM. *P < 0.05 compared with vehicle. Statistical analyses performed included (A) 2-way ANOVA, (B–H) Student’s unpaired t test, and (I) 1-way ANOVA followed by Dunnett’s multiple comparisons test, where appropriate.
Figure 2. Chronic treatment with tirzepatide ameliorates insulin resistance in mice.

Obese insulin-resistant mice were dosed once daily for 14 days with vehicle or tirzepatide (TZP, 10 nmol/kg; n = 13–15 per group). Following 14 days of treatment, insulin sensitivity was assessed via a hyperinsulinemic-euglycemic clamp. (A) Average glucose infusion rates (GIR) throughout and GIR during the final 30 minutes of the clamp. (B) Endogenous glucose production (EGP). Insulin-stimulated glucose disposal in (C) white gastrocnemius, (D) red gastrocnemius, (E) soleus skeletal muscle, (F) epididymal (eWAT), and (G) inguinal (iWAT) white adipose tissue. Data are presented as mean ± SEM. *P < 0.05 compared with vehicle. Statistical analyses performed included Kruskal-Wallis test followed by Bonferroni multiple comparisons test, where appropriate.
One of the most effective approaches to achieving clinically meaningful improvements in glycemic control is weight loss (35). Therefore, to determine if the effect of tirzepatide on systemic insulin sensitivity is driven in a weight-dependent and/or -independent manner, we compared the insulin-sensitizing action of tirzepatide to a selective GLP-1RA (semaglutide), a mechanism that has been shown to ameliorate IR entirely due to weight loss (36). For these studies, we matched weight loss in obese IR mice by dosing tirzepatide (3 nmol/kg) at a 3-fold lower dose than semaglutide (10 nmol/kg). To further control for weight-driven improvements in insulin sensitivity, we pair fed (PF) a group of animals the same daily food intake as animals dosed with tirzepatide. Chronic administration of either agent reduced body weight and food intake in obese IR mice (Figure 3A). Following 14 days of treatment, there was no difference in weight loss in animals treated with tirzepatide versus those treated with semaglutide. Tirzepatide reduced fasting glucose levels lower than those seen in semaglutide-treated or PF animals (Supplemental Figure 1G). Tirzepatide, semaglutide, and PF reduced fasting insulin when compared with that in vehicle-treated animals (Supplemental Figure 1H). To compare the insulin-sensitizing effects of the 2 treatments, we conducted hyperinsulinemic-euglycemic clamp experiments following the 14 days of dosing. Strikingly, despite eliciting equivalent weight loss, administration of tirzepatide increased whole-body insulin sensitivity more than that achieved in the semaglutide treatment and PF groups (Figure 3B). Tirzepatide increased the GIR by 4.7-fold, while the semaglutide and PF groups increased the GIR by 3.5-fold and 3-fold, respectively, when compared with the vehicle-treated group. There was a 1.4-fold and 1.5-fold increase in the GIR in animals administered tirzepatide when compared with treatment with semaglutide or PF, respectively (Figure 3C). Importantly, comparing the insulin-sensitizing action of tirzepatide to that of the weight-matched groups (semaglutide and PF) indicated that the weight-independent effect of tirzepatide accounts for 30% of the total improvement in insulin sensitivity in obese IR mice. No effect on EGP was observed for any of the groups (Figure 3D). Tirzepatide augmented insulin-stimulated glucose disposal in skeletal muscle significantly more than semaglutide (Figure 3, E–G), while inducing insulin stimulated uptake more than PF in epididymal WAT and BAT (P = 0.09; Figure 3, H–J). These data further support the notion that tirzepatide is indeed a highly effective insulin sensitizer capable of improving IR in both a weight-dependent and -independent manner. Such improvements in whole-body insulin sensitivity associated with the tirzepatide treatment shed light on the improvement in glycemic control observed clinically with tirzepatide when compared with GLP-1R agonism (31).
Figure 3. Tirzepatide enhances insulin sensitivity in a weight-dependent and -independent manner.

Obese insulin-resistant mice dosed once daily with vehicle, semaglutide, or tirzepatide for 14 days (n = 14–15 per group). (A) Daily body weight and food intake. Following 14 days of treatment, insulin sensitivity was assessed via a hyperinsulinemic-euglycemic clamp. (B) Average glucose infusion rates (GIR) throughout and during the final 30 minutes of the clamp. (C) Average GIR fold change when comparing TZP to that of weight-matched groups during the final 30 minutes of the clamp. (D) Endogenous glucose production (EGP). Insulin-stimulated glucose disposal in (E–G) skeletal muscle, (H and I) white adipose tissue (epididymal and inguinal adipose tissue [eWAT and iWAT]), and (J) brown adipose tissue (interscapular brown adipose tissue [iBAT]). Data are presented as mean ± SEM. *P < 0.05 compared with vehicle, #P < 0.05 compared with semaglutide, and §P < 0.05 compared with pair fed. Statistical analyses performed included 1-way ANOVA or Kruskal-Wallis test followed by Bonferroni multiple comparisons test, where appropriate.
To determine if GIPRA pharmacology accounts for the weight-independent insulin sensitization effect of tirzepatide, we utilized obese IR germline GLP-1R–null mice (Glp-1r–/– mice, ref. 28). This approach enabled determining whether tirzepatide improves insulin sensitivity by engaging only the GIPR, importantly, in the absence of weight loss. Glp-1r–/– mice are reported to be protected from diet-induced obesity (37). Thus, to maximize weight gain, circumvent protection from obesity, and ensure a state of IR, we exposed Glp-1r–/– animals to a 60% HFD and minimized thermal stress by housing animals in a thermal neutral (27°C) environment (38). Following 12 weeks of HFD, Glp-1r–/– mice had become obese (body weight >50g), gaining equivalent body weight and fat mass to WT animals (Figure 4, A and B). To investigate whether engagement of the GIPR was sufficient for tirzepatide to improve insulin sensitivity, Glp-1r–/– mice were dosed daily for 2 weeks with vehicle or tirzepatide. In accordance with previous studies using GIPR agonists in obese IR mice (24, 28), chronic treatment with tirzepatide had no effect on body weight, food intake, and fasted insulin, while there was a decrease in fasting glucose in Glp-1r–/– mice (Figure 4, C and D). Following 14 days of treatment, hyperinsulinemic-euglycemic clamp studies were performed and showed that, even without weight loss in the Glp-1r–/– mice, tirzepatide induced a 1.7-fold increase in the GIR compared with vehicle-treated animals (Figure 4E and Supplemental Figure 2A). Importantly, this was a similar magnitude to that observed for the weight-independent effect of tirzepatide on insulin sensitivity in WT mice (Figure 3C). There was no effect of tirzepatide on EGP in Glp-1r–/– mice (Figure 4F). Furthermore, in line with an adipose tissue–specific action of GIPRA, Glp-1r–/– mice treated with tirzepatide showed enhanced insulin-stimulated glucose uptake only in WAT (Figure 4J). There was no effect of tirzepatide on insulin-stimulated glucose uptake in skeletal muscle and subcutaneous WAT (Figure 4, G–I and K). In total, these data support the hypothesis that GIPR agonism accounts for the weight-independent insulin-sensitizing action of tirzepatide. Our findings in mice reported here provide translational insight into how GIPR agonism may contribute to the robust glycemic profile of tirzepatide treatment in patients with T2DM (31).
Figure 4. GIPR agonism contributes to the weight-independent insulin sensitization action of tirzepatide.

WT (C57BL/6J) and germline, whole-body, glucagon-like peptide 1 receptor–KO mice (Glp-1r–/– mice) were individually housed in a temperature-controlled (27˚C) environment with a 12-hour light/12-hour dark cycle and fed a high-fat (60% of calories from fat) diet for 12 weeks. (A) Weekly body weight and food intake. (B) Fat and lean mass after 12 weeks of high-fat feeding. Obese insulin-resistant Glp-1r–/– mice, dosed once daily with either vehicle (n = 12–15) or tirzepatide (TZP, 10 nmol/kg) for 14 days. (C) Daily body weight and food intake. (D) Fasting blood glucose and plasma insulin following 14 days of treatment. Following 14 days of treatment, insulin sensitivity was assessed via a hyperinsulinemic-euglycemic clamp. (E) Average glucose infusion rate (GIR) and that during the final 30 minutes of the clamp. (F) Endogenous glucose production (EGP). Insulin-stimulated glucose disposal in (G) soleus, (H) red, and (I) white gastrocnemius skeletal muscle as well as (J) epididymal white adipose tissue (eWAT) and (K) inguinal white adipose tissue (iWAT). Data are presented as mean ± SEM. *P < 0.05 compared with vehicle. Statistical analyses performed included Student’s unpaired t test, 2-way ANOVA, where appropriate.
To complement studies conducted in Glp-1r–/– mice with tirzepatide, we further investigated whether chronic engagement of the GIPR improves insulin sensitivity. Here, we developed an acylated (C-20) long-acting (Figure 5A), potent, and selective GIPR agonist (Table 1). Following a single s.c. dose of LAGIPRA (200 nmol/kg), the mean apparent clearance and mean half-life were 8.76 mL/h/kg and 6.29 hours, respectively (Table 1). In vivo, pharmacokinetic (PK) analysis in combination with glucose tolerance assays indicated that LAGIPRA had an exposure sufficient for a once-a-day dosing paradigm in mice (Table 1 and Figure 5B). Further, LAGIPRA showed selectivity for mouse GIPR, as it reduced hyperglycemia in intraperitoneal glucose tolerance test experiments in Glp-1r–/– mice (Figure 5, B and C) but had no effect in Gipr-deficient (Gipr–/–) animals (Figure 5D). Dose-response studies in WT animals indicated that maximum efficacy for glucose lowering with LAGIPRA occurred at 300 nmol/kg (Figure 5B). Therefore, we used this dose (300 nmol/kg/d) for all subsequent studies. Chronic treatment of obese mice with LAGIPRA (300 nmol/kg/d) had no effect on daily body weight, food intake, energy expenditure, or substrate utilization rates (Supplemental Figure 2, A–H). Next, we determined if LAGIPRA enhanced insulin sensitivity in obese IR mice. Chronic treatment of obese IR mice with LAGIPRA had a small (4% reduction) effect on body weight and transient reduction (during the first 24–48 hours) in food intake (Figure 6A). Following 14 days of treatment, LAGIPRA decreased fasting glucose and insulin (Figure 6B). To determine specifically if chronic engagement of the GIPR improves insulin sensitivity, we again conducted hyperinsulinemic-euglycemic clamp studies. Importantly, and similar to tirzepatide administration in Glp-1r–/– mice, chronic treatment with LAGIPRA improved whole-body insulin sensitivity, with a 1.7-fold increase in the GIR (Figure 6C). There was no effect of LAGIPRA on EGP (Figure 6D). While there was no effect of LAGIPRA treatment on skeletal muscle insulin sensitivity (Figure 6, E–G), LAGIPRA enhanced insulin sensitivity in both visceral and s.c. WAT, as demonstrated by increased glucose uptake (Figure 6, H and I). Thus, in line with the effect of tirzepatide in WT and Glp-1r–/– mice, chronic GIPR agonism improves systemic insulin sensitivity in obese IR mice, an effect that may be mediated via actions in adipose tissue.
Figure 5. Development and characterization of a long-acting GIPR agonist.

(A) Structure schematic of a long-acting glucose-dependent insulinotropic polypeptide receptor agonist (LAGIPRA). Intraperitoneal glucose tolerance tests in (B) WT and (C) germline whole-body glucagon-like peptide 1 receptor–KO mice (Glp-1r–/– mice) and (D) glucose-insulinotropic polypeptide receptor–null mice (Gipr–/– mice) dosed s.c. with vehicle (LAGIPRA, 1000 nmol/kg, n = 5) or long-acting glucagon-like peptide 1 receptor agonist (LAGLP-1RA; semaglutide [30 nmol/kg], n = 5) 16 hours prior to assay. Data are presented as mean ± SEM. *P < 0.05 compared with vehicle. Statistical analyses performed included a 1-way ANOVA, followed by Dunnett’s multiple comparisons test, where appropriate.
Table 1. In vitro potency and pharmacokinetic analysis.
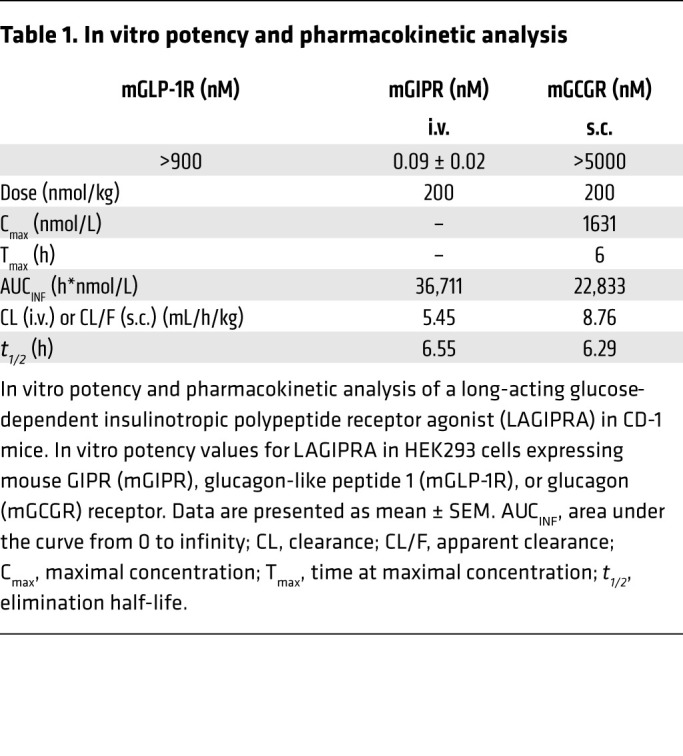
Figure 6. LAGIPRA improves insulin sensitivity in obese insulin-resistant mice.

High-fat diet–fed obese insulin-resistant mice dosed daily with either vehicle (n = 8–14) or a long-acting glucose-dependent insulinotropic polypeptide receptor agonist (LAGIPRA, n = 8–14) for 14 days. (A) Daily body weight and food intake. (B) Fasting blood glucose and plasma insulin following 14 days of treatment. Hyperinsulinemic-euglycemic clamp following 14 days of treatment. (C) Average glucose infusion rates throughout and during the final 30 minutes of clamp (GIR). (D) Endogenous glucose production (EGP). Insulin-stimulated glucose disposal in (E) soleus, (F) red, and (G) white gastrocnemius skeletal muscle and (H) epididymal white adipose tissue (eWAT) and (I) inguinal white adipose tissue (iWAT). Data are presented as mean ± SEM. *P < 0.05 compared with vehicle. Statistical analyses performed included Student’s unpaired t test, 2-way ANOVA, or Kruskal-Wallis test, where appropriate.
To elucidate the potential mechanism(s) by which tirzepatide (and GIPR agonism) improved insulin sensitivity, we conducted RNA-Seq analysis in several tissues. Interestingly, there was no effect of tirzepatide or LAGIPRA on metabolic genes in skeletal muscle or WAT (data not shown). Unexpectedly, the most marked differences were found in BAT, where both tirzepatide and LAGIPRA treatment robustly induced the expression of genes associated with the catabolism of glucose, lipid, and BCAAs (Figure 7). BAT is a metabolically active organ that has a robust capacity to lower circulating metabolites, including glucose, lipids, and BCAAs (39–41). Importantly, this metabolic action is tightly linked to its ability to improve metabolic homeostasis in preclinical models and humans with obesity and/or T2DM (41–43). Here, a total of 3671 and 1908 genes were found to be differentially regulated by treatment with tirzepatide or LAGIPRA, respectively, in BAT (Figure 7A). Overall, 1926 and 1296 genes were upregulated and 1745 and 612 genes were downregulated by tirzepatide and LAGIPRA, respectively. Consistent with GIPR engagement, 703 upregulated and 198 downregulated genes were shared between tirzepatide and LAGIPRA treatment. Accordingly, gene set enrichment analysis of differentially expressed genes indicated that tirzepatide and LAGIPRA effected similar molecular targets, including pathways associated with cellular and mitochondrial metabolism. Specifically, tirzepatide and/or LAGIPRA treatment induced the mRNA expression of metabolic transcriptional regulators (ChREBP, SREBP-1, RXRβ, PPARγ, and KLF-15; Figure 7B and Supplemental Figure 3) and recruited genes associated with the uptake, storage, breakdown, and oxidation of glucose and lipids in BAT (GLUT1, GLUT4, ATGL, DGAT1, and DGAT2; Supplemental Figure 3). Interestingly, tirzepatide and LAGIPRA treatment induced the expression of BCAT2 and BCKDH, enzymes that catalyze the first and second steps of BCAA catabolism, respectively (Figure 7B). Furthermore, LAGIPRA treatment, in particular, induced expression of genes associated with downstream steps in BCAA catabolism (e.g., HIBCH, HIBADH, ACAD8, ACADm, IVD, MCEE, and MCCC2; Figure 7B). Consistent with increased TCA cycle flux and electron transport, tirzepatide and/or LAGIPRA treatment stimulated the recruitment of genes associated with mitochondrial substrate transport (e.g., CPT1A, MPC2, MCAT, SLC25A39, SLC25A11, SLC25A22, and SLC25A29), fatty acid oxidation (PDK2, ACAD8, ACAD9, ACAD10, ACADL, ACADVL, and ACADM), electron transport activity (COX5A, COX5B, COX7A1, COX8b, COX17, COX6B1, COX10, and COX11), and thermogenic capacity (UCP1; Supplemental Figure 3). There was no effect of PF on several of these genes, suggesting that GIPR agonism mediates a direct and weight-independent induction of metabolic pathways associated with the oxidation of glucose, lipids, and BCAAs in BAT.
Figure 7. Tirzepatide and GIPR agonism induced BCAA catabolic gene expression in BAT in obese IR mice.

High-fat diet–fed obese insulin-resistant mice (C57BL/6J) were dosed once daily with vehicle (saline, n = 6), tirzepatide (TZP, n = 6), a long-acting glucose-dependent insulinotropic polypeptide receptor agonist (LAGIPRA, n = 6), or saline (pair fed, n = 6). Following 14 days of treatment, tissue samples were collected for metabolic and molecular analyses. (A) Venn diagram of differentially (up- and downregulated gene expression) expressed genes (FDR < 0.05). (B) Heatmap of RNA-Seq expression Z-scores computed for genes associated with the branched-chain amino acid (BCAA) pathway in brown adipose tissue (BAT). *P < 0.05 compared with vehicle and #P < 0.05 compared with pair fed. Statistical analyses was performed using 1-way ANOVA, followed by FDR correction, where appropriate.
Of particular interest, we found that TZP modulated BCAA metabolism in BAT. To determine if the induction of BCAA catabolic genes in BAT resulted in a functional outcome, we investigated the effect of tirzepatide and LAGIPRA on tissue and circulating levels of BCAA. There was an increase in BCAAs (valine, leucine, and isoleucine) in BAT of mice treated with either tirzepatide or LAGIPRA (Table 2). Furthermore, indicative of tissue level catabolism of BCAAs, there was an increase in the BCAA catalytic product glutamate (a product of the first step of BCAA catabolism) in BAT of mice treated with tirzepatide (Table 2). In line with tissue changes affecting circulating BCAA, tirzepatide treatment decreased plasma levels of BCAAs (valine, leucine, and isoleucine) and BCAA catalytic products glutamate and branched-chain α-ketoacids (BCKAs; α-ketoisocaproate and α-keto-β-methylvalerate; Table 2). Interestingly, while there was no effect of LAGIPRA treatment on BCAA levels, but consistent with the robust effect of LAGIPRA on BCAA catabolic genes, LAGIPRA decreased circulating levels of BCKAs α-ketoisocaproate and α-keto-β-methylvalerate (Table 2). The translational relevance of these preclinical studies is exemplified by post hoc analysis of clinical data, suggesting that the weight-independent insulin-sensitizing action of tirzepatide is strongly associated with reduced circulating BCAA/BCKA in patients with T2DM (44). Taken together, these data indicate that tirzepatide (and GIPR agonism) may improve systemic insulin sensitivity by activating metabolic pathways associated with the oxidation of glucose, lipids, and BCAAs in adipose tissue.
Table 2. Metabolite analysis.

Discussion
Multireceptor pharmacology is at the forefront of next-generation therapies for the treatment of metabolic diseases (27, 45). One of these investigational agents is tirzepatide, a dual GIP and GLP-1R agonist, which has shown enhanced glycemic control and weight loss in patients with T2DM when compared with GLP-1R agonist monotherapy therapy (31). Here, we took a comprehensive approach, combining measurements at the whole-body, tissue-specific, and molecular levels in a mouse model of obesity-induced IR to investigate how tirzepatide improves insulin sensitivity beyond what occurs by weight loss alone and, importantly, whether the GIPR contributes to the efficacy. The major findings of the current report are that tirzepatide improves insulin sensitivity in a weight-dependent and -independent manner in obese IR mice. Using Glp-1r–/– mice, we showed that GIPR agonism accounts for the weight-independent insulin sensitization. Further, mechanistic studies indicate that tirzepatide-mediated improvements in insulin sensitivity are associated with the induction of glucose, FFA, and BCAA oxidation in BAT.
Obesity-induced IR is a key component underlying the development and progression of T2DM (46). Currently, there are limited efficacious, safe, and durable medications that target this disfunction in T2DM (47). One of the most effective therapies for the treatment of T2DM is weight loss (48, 49). In line with this, GLP-1R agonists drive weight loss and improve insulin sensitivity (36). However, a caveat of improving insulin sensitivity principally through weight loss is that once body weight is regained, metabolic benefits may be compromised (50, 51). Thus, improving insulin action in both a weight-dependent and -independent manner could provide greater treatment efficacy and durability. A key finding of the current study is that tirzepatide ameliorated peripheral IR in a model of DIO. Importantly, the insulin sensitization was underscored by enhanced glucose disposal in skeletal muscle and in both visceral and s.c. WAT. Consequently, a key question arising from this study was whether a component of the observed insulin sensitization occurred independent of weight loss. To address this, we matched the body weight of animals treated with tirzepatide to that of a PF group and with animals dosed with a selective GLP-1R monoagonist (semaglutide), a mechanism that improves insulin action primarily due to weight loss (36). Remarkably, despite losing equivalent body weight, animals treated with tirzepatide had a greater improvement in systemic insulin sensitization when compared with the weight-matched groups. Moreover, this weight-independent insulin benefit was coupled with augmented glucose disposal in skeletal muscle and in both WAT and BAT. Collectively, these results suggest that tirzepatide delivers both weight-dependent and -independent insulin sensitization in obese IR mice. The clinical implications of these findings are that the weight-independent insulin sensitization identified here could offer greater treatment durability, in contrast to agents that provide only weight-driven benefits. In support of this hypothesis, HOMA-IR analysis has suggested that a significant proportion of tirzepatide-induced insulin sensitization occurs in the absence of weight loss in subjects with T2DM (52). Together with studies showing that GIP can improve insulin sensitivity (23, 24), this prompted the hypothesis that engagement of the GIPR may drive the weight-independent insulin sensitization action of tirzepatide.
Two therapeutic modalities have been proposed to target the GIPR for treating metabolic diseases (22, 53). Originally, the discovery that Gipr–/– mice are protected from obesity (54), in addition to studies showing that GIP enhances lipid storage in WAT (55, 56), led to the idea that GIP was obesogenic and consequently prompted several approaches to block GIP action (57). Indeed, antagonizing antibodies targeting the GIPR have been shown to protect mice from DIO (58, 59). To date, however, these agents have not been shown to drive weight loss in already obese animals; further, GIPR antagonism offers minimal glycemic benefit (53, 60). In addition, as GIP is the primary incretin in humans (17, 61), blockade of GIP action may be detrimental to glycemic control, while chronic GIPR agonism either has no effect on body weight or drives weight loss and improves insulin action in mice (23, 24, 28, 29, 62, 63). Here, to determine if GIPR agonism accounts for the weight-independent insulin sensitization, we treated obese IR Glp-1r–/– mice chronically with tirzepatide, and to complement this strategy, we dosed obese IR WT animals with a LAGIPRA. Consistent with data from other reports (23, 28, 29, 62), neither LAGIPRA nor tirzepatide (in GLP-1R–KO animals) reduced body weight, supporting the hypothesis that GLP-1R agonism is required to unlock the synergistic action GIPR agonism on weight loss (18). Importantly, this provides an experimental paradigm to investigate the weight- and GLP-1R–independent effect of tirzepatide (GIPR agonism) on systemic insulin sensitivity. Using hyperinsulinemic-euglycemic clamp studies, we found that both LAGIPRA and tirzepatide improved insulin sensitivity in obese mice in the absence of weight loss. Notably, the magnitude of enhanced insulin action matched the weight-independent insulin sensitization occurring when comparing tirzepatide to semaglutide. Further, in line with the GIPR being expressed in adipose tissue (20), we found that for both LAGIPRA and tirzepatide (in Glp-1r–/– mice) enhanced insulin sensitization was underlined by augmented glucose disposal in WAT. Thus, although the specific contribution of WAT to total postprandial glucose clearance is not comparable to that of skeletal muscle (64, 65), the cumulative findings indicate that treatment with tirzepatide results in weight-independent insulin sensitivity in obese IR mice and that engagement of the GIPR accounts for this benefit. These data agree with previous findings showing that GIPR agonism augments insulin sensitivity in obese mice (24). Mechanistically, GIP has been shown to improve insulin sensitivity due to the recruitment of metabolic pathways associated with the storage and oxidation of glucose and FFAs in adipose tissue (23, 24).
Although the cause of obesity-associated IR is intensely debated (4, 66, 67), recently, a strong association between elevated BCAAs/BCKAs, obesity, systemic IR, and T2DM has consistently been reported (66, 68). This has led to the suggestion that dysregulation of BCAA metabolism may play a causal role in developing IR (66). Indeed, dysregulation of BCAA catabolism in adipose tissue plays a role in the rise of circulating BCAAs due to reductions in the expression of enzymes in BCAA catabolism (69–71). Consequently, in models of obesity-induced IR, induction of BCAA catabolism either systemically or specifically in adipose tissue, reduces circulating BCAAs and alleviates IR (39, 67–69). In accordance, we showed that tirzepatide-mediated (and GIPR agonism–mediated) improvements in insulin sensitivity are coupled with the induction of enzymes responsible for the catabolism of BCAA in BAT. Specifically, both tirzepatide and LAGIPRA were found to increase expression of genes linked with glucose and lipid metabolism. The most robust effect occurred on the expression of BCAT2 and BCKDH, enzymes that catalyze the first and second (and rate limiting) steps in BCAA catabolism, respectively, as well as several downstream members of the BCAA catabolic pathway in BAT. Importantly, these molecular changes were found to be associated with increased tissue levels of BCAA and reduced circulating levels of BCAA and their product BCKA. Our studies are supported by data showing the GIPR is expressed in BAT and that GIP can act directly on brown adipocytes to induce metabolic gene expression (20). In addition, the potential clinical relevance of our findings is highlighted by post hoc analyses showing that tirzepatide reduces circulating BCAA in patients with T2DM and that this effect is strongly associated with biomarkers indicative of enhanced insulin sensitivity (44). These results lead to the intriguing hypothesis that one way in which tirzepatide may enhance insulin sensitivity in patients is through the catabolism of glucose, lipids, and BCAAs in BAT. In support of this, activated BAT is reported to oxidize circulating glucose, lipids, and BCAAs in BAT in humans (39). Therefore, while BAT has classically been considered to have a limited effect on human metabolism (72, 73), studies show that adult humans have functional BAT (74), which when activated, is capable of positively affecting systemic metabolism (43, 75, 76). This clearly highlights the pharmacological potential of this organ in humans (77) and is in line with recent clinical findings indicating that the presence of BAT in humans is associated with cardiometabolic health (78). Thus, while it is likely to make a smaller contribution in humans (compared with mice), our data suggest that BAT may aid weight-dependent mechanisms in the lowering of circulating lipids and BCAAs, enhancing the improvement of insulin sensitivity by tirzepatide in patients with T2DM (44, 52). We note that mechanistic studies in human subjects are further required to fully test this hypothesis.
Summary.
In summary, here we demonstrate that tirzepatide and LAGIPRA improve insulin sensitivity in obese IR mice. Importantly, we found that tirzepatide enhances insulin action to a greater extent than GLP-1RA treatment and that this additional efficacy occurs independent of changes in body weight. Using two complementary approaches, we demonstrate that this weight-independent insulin sensitization is accounted for by engagement of the GIPR. Mechanistically, tirzepatide appears to mediate its insulin-sensitizing action in a weight-dependent and -independent (via GIPR agonism) manner by the induction of metabolic pathways linked to the oxidation of glucose, lipids, and BCAAs. Together, this reduces excess nutrient delivery to metabolically relevant organs, subsequently enhancing systemic insulin sensitivity. The weight-independent insulin sensitization identified here could offer greater treatment durability, in contrast to agents that provide only weight-driven effects. Further, these preclinical data demonstrate how GIPR agonism contributes to enhanced glycemic control of tirzepatide, a dual GIP and GLP-1R agonist.
Methods
Peptide synthesis and in vitro characterization.
LAGIPRA, GLP-1R agonist semaglutide, and tirzepatide were synthesized at Eli Lilly and Company. cAMP assays were conducted in HEK293 cells expressing the mouse GLP-1R, GIPR, or glucagon receptor (GcgR). Using homogeneous time-resolved fluorescence methods to assess cAMP accumulation (CisBio cAMP Dynamic 2 HTRF Assay Kit, 62AM4PEJ), assays were conducted to determine the potency of LAGIPRA performed in the presence of casein (instead of serum albumin) as a nonspecific blocker, which does not interact with the fatty acid moiety of LAGIPRA. Intracellular cAMP levels were determined by extrapolation using a standard curve. Dose-response curves of compounds were plotted as the percentage of stimulation normalized to minimum (buffer only) and maximum (maximum concentration of each control ligand) values and analyzed using a 4-parameter nonlinear regression fit with a variable slope (Genedata Screener 13). EC50 is the concentration of compound causing half-maximal simulation in a dose-response curve.
Animals.
All animals were individually housed in a temperature-controlled (23°C ± 0.9°C to 27°C ± 0.9°C) facility with a 12-hour-light/12-hour-dark cycle. WT, Glp-1r–/–, and Gipr–/– mice on a C57BL/6 genetic background (28, 79) were maintained at Taconic Contract Breeding.
High-fat diet study in WT and Glp-1r–/– mice.
WT and Glp-1r–/– mice housed in a thermal neutral environment (27°C) were fed a 60% high-fat diet (D12492; Research diets) for a minimum of 12 weeks. Body composition was assessed before and after high-fat feeding using quantitative nuclear magnetic resonance. Body weight and food intake were recorded weekly.
Chronic dosing studies in obese insulin-resistant WT animals.
Male mice were maintained on a high-fat diet (consisting of 60% fat, 20% carbohydrate, and 20% protein caloric content D12492; Research Diets) and had free access to food and water before randomization by weight. Animals received s.c. injections of vehicle (n = 6–16), semaglutide (10 nmol/kg, n = 16), or tirzepatide (3 and 10 nmol/kg, n = 6–16) or LAGIPRA (300 nmol/kg) once a day for 14 days. Mice were sacrificed, plasma was collected in EDTA-coated tubes, and tissues were collected and snap frozen in liquid nitrogen.
Chronic treatment with tirzepatide in obese insulin-resistant Glp-1r–/– mice.
Male Glp-1r–/– mice were fed a high-fat diet (60% of calories from fat, D12492; Research diets) for a minimum of 16 weeks. Following this period animals were weighed and body composition was determined using quantitative nuclear magnetic resonance. Mice were then randomly assigned to treatment groups in which they received a daily injection of vehicle (n = 16) or tirzepatide (10 nmol/kg, n = 16) for 14 days. Body weight and food intake were recorded daily. Fourteen days after the start of each experiment, insulin sensitivity was determined via insulin clamp.
In vivo metabolic analysis.
Metabolic rate and substrate utilization rates were determined in obese mice treated with vehicle (n = 6) or LAGIPRA (300 nmol/kg, n = 6) using an open respirometer system (LabMaster System; TSE Systems). Briefly, oxygen consumption (VO2, mL/kg/h) and carbon dioxide production (VCO2, mL/kg/h) were measured throughout the final 7 days of a 14-day dosing period. VO2 (mL/kg/h) and VCO2 (mL/kg/h) were used to calculate energy expenditure and respiratory exchange ratio (RER = VCO2/VO2). Animals had ad libitum access to water and high-fat diet when in metabolic cages. All measurements were made at approximately 24°C to 25°C.
Insulin clamp.
Catheters were placed in the left carotid artery and the right jugular vein 7–8 days prior to euglycemic clamp. Animals were fasted overnight and, after 2 hours of study box acclimation, a bolus/continuous infusion of 3-3H-glucose (PerkinElmer; 6 μCi bolus; basal period, 0.05 μCi/min; clamp period, 0.125 μCi/min) was initiated and maintained throughout the test period. Blood glucose was measured from arterial blood via a glucometer every 5 minutes during the clamp period. An i.v. infusion of 3 mU/kg/min Humulin R (Eli Lilly and Company) and 22.5% glucose was started and periodically adjusted to maintain blood glucose concentration at 115–125 mg/dL. Somatostatin (5 μg/kg/min, Bachem) was administered i.v. to inhibit endogenous insulin secretion, and washed donor mouse erythrocytes were infused to maintain blood volume. Blood was collected at the end of the basal and clamp periods to determine EGP (80). A bolus dose of 2-[1-14C] Deoxy-D-glucose (PerkinElmer, 5 μCi) was administered i.v. to measure tissue glucose uptake under steady-state glucose concentrations. Animals were subsequently sacrificed by intraarterial administration of pentobarbital. Tissues were rapidly excised and frozen in liquid N2 to estimate tissue glucose uptake (81). Additional arterial blood samples were obtained to monitor hematocrit or were stored frozen as plasma for subsequent assays.
Metabolomic analysis.
BCAAs, BCKAs, and glutamate were measured using a mass spectrometry–based targeted metabolomics approach. Briefly, 2 overnight extraction protocols were used for the detection of the amino acids (A, acetonitrile-methanol solvent mixture, 50%:50%, v/v) and the ketoacids (B, methanol-water mixture, 80%:20%, v/v), respectively. Aliquots of 25 μL plasma were used for each extraction protocol. Amino acids were also measured in BAT. Tissue was snap frozen in liquid nitrogen and pulverized. A tissue aliquot of about 50 mg was weighted, and the extraction solution A was added to achieve a final concentration of 50 mg/mL. Stable labeled compounds were added to the extraction solutions for internal standard quantitation (Supplemental Table 1). Data were acquired using a Shidmadzu Nexera X2 UHPLC system coupled to a Sciex 6500 triple quadrupole mass spectrometer equipped with an electrospray source operating in positive and negative ion mode. BCAAs and glutamate were chromatographically separated using a Waters XBridge BEH Amide column (150 mm × 2.1 mm × 3.5 μm). BCKAs were separated on a Waters XSelect HSS T3 C18 column (150 mm × 2.1 mm × 3.5 μm). The columns were maintained at 40°C. Elution solvents were water with 0.1% formic acid with 10 mM ammonium formate and acetonitrile with 0.1% formic acid. The mobile phases eluted following a linear gradient for both the HILIC and reversed-phase method, leading to a total of 29 minutes of analysis time per run, including reequilibration (gradient not shown). Data were acquired in multiple-reaction monitoring mode. Details on the MRM conditions used for the analytes of interest are reported in Supplemental Table 1. All positive ions detected were protonated species ([M+H]+). All negative ions were detected as deprotonated species ([M−H]−).
Pharmacokinetic analysis.
The PK of LAGIPRA was evaluated in male CD-1 mice following a single i.v. or s.c. dose of 200 nmol/kg. Blood samples were collected over 168 hours, and the resulting individual concentrations from 2 animals/group/time point were used to calculate the reported PK parameters. The mean PK parameters are shown in Figure 4 (Table 2).
Analysis of metabolites and circulating factors.
Blood samples were collected on ice before storage of plasma at −80°C. Plasma triglycerides and FFAs were measured using a Hitachi 912 Clinical Chemistry Analyzer (Roche Diagnostics). Insulin (Crystal Chem Inc.), Adiponectin (BioVendor Inc.), IGFBP2 (R&D Systems), and leptin (Crystal Chem Inc.) were measured by ELISA. All samples for a given hormone were assayed in duplicate within a single assay.
Insulin tolerance test.
Briefly, on the morning of the procedure, animals were fasted for 4 hours. After which, animals received an intraperitoneal injection of insulin (0.5 U/kg, Humilin R, Eli Lilly and Company). Blood glucose was determined by tail clip at 0, 15, 30, 60, and 120 minutes after injection.
Glucose tolerance test.
Briefly, animals were fasted overnight (16 hours). After which, animals received an intraperitoneal injection of glucose (2 g/kg 50% dextrose). Blood glucose was determined by tail clip at 0, 15, 30, 60, and 120 minutes after injection.
Body composition analysis.
Body composition of mice was determined using Quantitative Nuclear Magnetic Resonance analysis (ECHO MRI, 3-1 Composition Analyzer; Echo Medical Systems).
RNA isolation and RNA-Seq analysis.
Total RNA was isolated from tissues using TRIzol reagent (Invitrogen) and the RNeasy lipid mini kit (Qiagen). RNA quality was confirmed via RNA integrity number (RIN) using an Agilent RNA ScreenTape Assay with the Agilent 4200 TapeStation (G2991AA). Only samples with RIN values ≥7.9 were chosen for RNA-Seq analysis. Samples were sequenced on an Illumina NovaSeq using paired 150 bp reads. Raw FastQ files were quality trimmed using cutadapt (cutadapt1.9.1) and aligned using GSNAP (v2013-11-27, command line parameters -B 5 -A sam -N 1 -t 8 -s splicesites --quality-protocol=sanger --gunzip --sam-multiple-primaries --maxsearch=1000 -- npaths=100) to build 38.p3 of the mouse genome. Read counts were quantified using a custom Perl script and summarized at the gene level (NCBI m38.p3 annotation). To map reads to genes and obtain gene level expression measures, RNA-Seq data were subjected to a “rollup” pipeline developed at Eli Lilly and Company. The following rules were applied for the rollup: (a) exon reads of multiple assays from the same libraries were summed; (b) exons were excluded if more than 80% of samples had less than 10 counts; (c) robust gene level signals across exons of a gene were determined by a robust linear model and were output for each library and each gene; and (d) mean signal of log2-transformed gene levels across all samples were median normalized. Read counts were then quantile normalized using a custom R script. Principal component analysis and outlier analysis were applied to the rollup output normalized data to check any potential pattern or outliers among the samples. As no pattern or outlier was detected, the 1-way ANOVA using R software was applied to the normalized data to test the pairwise comparisons among the treatments. For each comparison, the fold changes and P values as well as the FDR (82) were used to adjust for the multiple testing across the genes were reported. Raw and processed data have been deposited within the Gene Expression Omnibus repository (GSE173522).
Statistics.
Data are presented as mean ± SEM. Statistical analyses performed included Student’s unpaired 2-tailed t test, 1-way ANOVA or 2-way ANOVA, or Kruskal-Wallis test followed by Holm-Bonferroni or Dunnett’s multiple comparisons test, where appropriate. Differences were considered significant at P < 0.05.
Study approval.
All experiments were performed in accordance with protocols approved by the Eli Lilly and Co. IACUC.
Author contributions
AKH, JLF, SW, and DLK conducted insulin clamps. MEC conducted insulin clamps and contributed to data interpretation. XR and MD conducted LAGIPRA in vitro characterization and PK analysis. BAD, EPSCF, LD, OC, CCC, DAB, LSOF, RC, amd AML performed glucose and insulin tolerance tests and in vivo metabolic analysis, conducted ELISA analyses, and performed RNA isolation. VP and JAW performed metabolomic analysis and contributed data interpretation. KALC and YL were responsible for RNA-Seq analyses. KWS, MPC, REG, JSM, TC, and JAF contributed to data interpretation and writing of the manuscript. WCR was responsible for study conception, data interpretation, and contributed to writing of the manuscript. RJS was responsible for study conception, experimental designs, data analyses and interpretation, and drafting and writing of the manuscript.
Supplementary Material
Version 1. 05/18/2021
In-Press Preview
Version 2. 06/15/2021
Electronic publication
Footnotes
Conflict of interest: All authors are current or past employees of Eli Lilly and Company.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(12):e146353.https://doi.org/10.1172/JCI146353.
Contributor Information
Ricardo J. Samms, Email: samms_ricardo_j@lilly.com.
Michael E. Christe, Email: christe_michael_e@lilly.com.
Valentina Pirro, Email: pirro_valentina@lilly.com.
Brian A. Droz, Email: droz_brian_a@lilly.com.
Adrienne K. Holland, Email: holland_adrienne@lilly.com.
Jessica L. Friedrich, Email: friedrich_jessica_l@lilly.com.
Samantha Wojnicki, Email: wojnicki_samantha@lilly.com.
Debra L. Konkol, Email: konkol_debra_l@lilly.com.
Richard Cosgrove, Email: cosgrove_richard@lilly.com.
Ellen P.S. Conceição Furber, Email: furber_ellen_c@lilly.com.
Xiaoping Ruan, Email: ruan_xiaoping@lilly.com.
Annie M. Long, Email: annie.long@lilly.com.
Mridula Dogra, Email: dogra_mridula@lilly.com.
Jill A. Willency, Email: WILLENCY_JILL_AMANDA@LILLY.COM.
Yanzhu Lin, Email: lin_yanzhu@lilly.com.
Liyun Ding, Email: ding_liyun@lilly.com.
Christine C. Cheng, Email: cheng_christine@lilly.com.
Over Cabrera, Email: over.cabrera@lilly.com.
Daniel A. Briere, Email: briere_daniel_a@lilly.com.
Jorge Alsina-Fernandez, Email: alsina-fernandez_jorge@lilly.com.
Ruth E. Gimeno, Email: gimeno_ruth@lilly.com.
Julie S. Moyers, Email: moyers_julie@lilly.com.
Tamer Coskun, Email: coskun_tamer@lilly.com.
Matthew P. Coghlan, Email: coghlan_matthew@lilly.com.
Kyle W. Sloop, Email: sloop_kyle_w@lilly.com.
William C. Roell, Email: roell_william_c@lilly.com.
References
- 1.Seuring T, et al. The economic costs of type 2 diabetes: a global systematic review. PharmacoEconomics. 2015;33(8):811–831. doi: 10.1007/s40273-015-0268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee S, et al. Type 2 diabetes. Lancet. 2017;389(10085):2239–2251. doi: 10.1016/S0140-6736(17)30058-2. [DOI] [PubMed] [Google Scholar]
- 3.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88(4):787–835. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152(4):673–684. doi: 10.1016/j.cell.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 5.DeFronzo RA, et al. Pioglitazone: The forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab Vasc Dis Res. 2019;16(2):133–143. doi: 10.1177/1479164118825376. [DOI] [PubMed] [Google Scholar]
- 6.Lebovitz HE. Thiazolidinediones: the forgotten diabetes medications. Curr Diab Rep. 2019;19(12):151. doi: 10.1007/s11892-019-1270-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller TD, et al. Glucagon-like peptide 1 (GLP-1) Mol Metab. 2019;30:72–130. doi: 10.1016/j.molmet.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbel JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013;17(6):819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Sikirica MV, et al. Reasons for discontinuation of GLP1 receptor agonists: data from a real-world cross-sectional survey of physicians and their patients with type 2 diabetes. Diabetes Metab Syndr Obes. 2017;10:403–412. doi: 10.2147/DMSO.S141235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crewe C, et al. The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest. 2017;127(1):74–82. doi: 10.1172/JCI88883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carobbio S, et al. Adipose tissue function and expandability as determinants of lipotoxicity and the metabolic syndrome. Adv Exp Med Biol. 2017;960:161–196. doi: 10.1007/978-3-319-48382-5_7. [DOI] [PubMed] [Google Scholar]
- 13.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45(9):1201–1210. doi: 10.1007/s00125-002-0873-y. [DOI] [PubMed] [Google Scholar]
- 14.Virtue S, Vidul-Puig A. It’s not how fat you are, it’s what you do with it that counts. PLoS Biol. 2008;6(9):e237. doi: 10.1371/journal.pbio.0060237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nauck MA, Meier JJ. Incretin hormones: Their role in health and disease. Diabetes Obes Metab. 2018;20(Suppl 1):5–21. doi: 10.1111/dom.13129. [DOI] [PubMed] [Google Scholar]
- 16.Dupre J, et al. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37(5):826–828. doi: 10.1210/jcem-37-5-826. [DOI] [PubMed] [Google Scholar]
- 17.Gasbjerg LS, et al. Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes. 2019;68(5):906–917. doi: 10.2337/db18-1123. [DOI] [PubMed] [Google Scholar]
- 18.Samms RJ, et al. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab. 2020;31(6):410–421. doi: 10.1016/j.tem.2020.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Rudovich N, et al. GIP receptor mRNA expression in different fat tissue depots in postmenopausal non-diabetic women. Regul Pept. 2007;142(3):138–145. doi: 10.1016/j.regpep.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Beaudry JL, et al. Physiological roles of the GIP receptor in murine brown adipose tissue. Mol Metab. 2019;28:14–25. doi: 10.1016/j.molmet.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yip RG, Wolfe MM. GIP biology and fat metabolism. Life Sci. 2000;66(2):91–103. doi: 10.1016/s0024-3205(99)00314-8. [DOI] [PubMed] [Google Scholar]
- 22.Finan B, et al. Reappraisal of GIP pharmacology for metabolic diseases. Trends Mol Med. 2016;22(5):359–376. doi: 10.1016/j.molmed.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Kim SJ, et al. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS One. 2012;7(7):e40156. doi: 10.1371/journal.pone.0040156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varol C, et al. Long-acting glucose-dependent insulinotropic polypeptide ameliorates obesity-induced adipose tissue inflammation. J Immunol. 2014;193(8):4002–4009. doi: 10.4049/jimmunol.1401149. [DOI] [PubMed] [Google Scholar]
- 25.Mohammad S, et al. A naturally occurring GIP receptor variant undergoes enhanced agonist-induced desensitization, which impairs GIP control of adipose insulin sensitivity. Mol Cell Biol. 2014;34(19):3618–3629. doi: 10.1128/MCB.00256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad S, et al. Gastric inhibitory peptide controls adipose insulin sensitivity via activation of cAMP-response element-binding protein and p110β isoform of phosphatidylinositol 3-kinase. J Biol Chem. 2011;286(50):43062–43070. doi: 10.1074/jbc.M111.289009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gimeno RE, et al. Leveraging the gut to treat metabolic disease. Cell Metab. 2020;31(4):679–698. doi: 10.1016/j.cmet.2020.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coskun T, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab. 2018;18:3–14. doi: 10.1016/j.molmet.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finan B, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5(209):209ra151. doi: 10.1126/scitranslmed.3007218. [DOI] [PubMed] [Google Scholar]
- 30.Willard FS, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. 2020;5(17):e140532. doi: 10.1172/jci.insight.140532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frias JP, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–2193. doi: 10.1016/S0140-6736(18)32260-8. [DOI] [PubMed] [Google Scholar]
- 32.Thomas MK, et al. 980-P: Tirzepatide, a dual GIP and GLP-1 receptor agonist, improves markers of beta-cell function and insulin sensitivity in type 2 diabetes patients. Diabetes. 2019;68(Suppl 1):980 [Google Scholar]
- 33.Hedbacker K, et al. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab. 2010;11(1):11–22. doi: 10.1016/j.cmet.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab. 2013;2(3):133–141. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delahanty LM, et al. Effects of weight loss, weight cycling, and weight loss maintenance on diabetes incidence and change in cardiometabolic traits in the Diabetes Prevention Program. Diabetes Care. 2014;37(10):2738–2745. doi: 10.2337/dc14-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fonseca VA, et al. Reductions in insulin resistance are mediated primarily via weight loss in subjects with type 2 diabetes on semaglutide. J Clin Endocrinol Metab. 2019;104(9):4078–4086. doi: 10.1210/jc.2018-02685. [DOI] [PubMed] [Google Scholar]
- 37.Scrocchi LA, Drucker DJ. Effects of aging and a high fat diet on body weight and glucose tolerance in glucagon-like peptide-1 receptor –/– mice. Endocrinology. 1998;139(7):3127–3132. doi: 10.1210/endo.139.7.6092. [DOI] [PubMed] [Google Scholar]
- 38.Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol. 2011;214(Pt 2):242–253. doi: 10.1242/jeb.050989. [DOI] [PubMed] [Google Scholar]
- 39.Yoneshiro T. et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature. 2019;572(7771):614–619. doi: 10.1038/s41586-019-1503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartelt A, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17(2):200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 41.Chondronikola M, et al. Brown adipose tissue activation is linked to distinct systemic effects on lipid metabolism in humans. Cell Metab. 2016;23(6):1200–1206. doi: 10.1016/j.cmet.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwen KA, et al. Cold-induced brown adipose tissue activity alters plasma fatty acids and improves glucose metabolism in men. J Clin Endocrinol Metab. 2017;102(11):4226–4234. doi: 10.1210/jc.2017-01250. [DOI] [PubMed] [Google Scholar]
- 43.Chondronikola M, et al. Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes. 2014;63(12):4089–4099. doi: 10.2337/db14-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valentina Pirro JAW, et al. Effects of tirzepatide, a novel dual GIP and GLP-1 receptor agonist, on metabolic profile in patients with type 2 diabetes. Paper presented at: 56th Annual Meeting of the European Association for the Study of Diabetes; September 21–25, 2020. Virtual. https://www.easd.org/virtualmeeting/home.html#!resources/effects-of-tirzepatide-a-novel-dual-gip-and-glp-1-receptor-agonist-on-metabolic-profile-in-patients-with-type-2-diabetes-8f8ff293-dc81-460d-8ec8-a6e6734658ec Accessed May 6, 2021.
- 45.Sloop KW, et al. Beyond glucagon-like peptide-1: is G-protein coupled receptor polypharmacology the path forward to treating metabolic diseases? ACS Pharmacol Transl Sci. 2018;1(1):3–11. doi: 10.1021/acsptsci.8b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahn SE, et al. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 47.American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care. 2020;43(Supplement 1):S98. doi: 10.2337/dc20-S009. [DOI] [PubMed] [Google Scholar]
- 48.Wilding JP. The importance of weight management in type 2 diabetes mellitus. Int J Clin Pract. 2014;68(6):682–691. doi: 10.1111/ijcp.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.American Diabetes Association. 8. Obesity management for the treatment of type 2 diabetes: Standards of Medical Care in Diabetes — 2020. Diabetes Care. 2020;43(Suppl 1):S89–S97. doi: 10.2337/dc20-S008. [DOI] [PubMed] [Google Scholar]
- 50.Beavers KM, et al. Effects of weight regain following intentional weight loss on glucoregulatory function in overweight and obese adults with pre-diabetes. Obes Res Clin Pract. 2015;9(3):266–273. doi: 10.1016/j.orcp.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beavers DP, et al. Cardiometabolic risk after weight loss and subsequent weight regain in overweight and obese postmenopausal women. J Gerontol A Biol Sci Med Sci. 2013;68(6):691–698. doi: 10.1093/gerona/gls236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas MK, et al. Dual GIP and GLP-1 receptor agonist tirzepatide improves beta-cell function and insulin sensitivity in type 2 diabetes. J Clin Endocrinol Metab. 2021;106(2):388–396. doi: 10.1210/clinem/dgaa863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Killion EA, et al. Glucose-dependent insulinotropic polypeptide receptor therapies for the treatment of obesity, do agonists = antagonists? Endocr Rev. 2020;41(1):bnz002. doi: 10.1210/endrev/bnz002. [DOI] [PubMed] [Google Scholar]
- 54.Miyawaki K, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8(7):738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 55.Beck B, Max JP. Gastric inhibitory polypeptide enhancement of the insulin effect on fatty acid incorporation into adipose tissue in the rat. Regul Pept. 1983;7(1):3–8. doi: 10.1016/0167-0115(83)90276-8. [DOI] [PubMed] [Google Scholar]
- 56.Eckel RH, et al. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes. 1979;28(12):1141–1142. doi: 10.2337/diab.28.12.1141. [DOI] [PubMed] [Google Scholar]
- 57.Gasbjerg LS, et al. Glucose-dependent insulinotropic polypeptide (GIP) receptor antagonists as anti-diabetic agents. Peptides. 2018;100:173–181. doi: 10.1016/j.peptides.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 58.Killion EA, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. 2018;10(472):eaat3392. doi: 10.1126/scitranslmed.aat3392. [DOI] [PubMed] [Google Scholar]
- 59.Kaneko K, et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J Clin Invest. 2019;129(9):3786–3791. doi: 10.1172/JCI126107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Svendsen B, et al. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol Metab. 2020;32:44–55. doi: 10.1016/j.molmet.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nauck MA, Meier JJ. GIP and GLP-1: stepsiblings rather than monozygotic twins within the incretin family. Diabetes. 2019;68(5):897–900. doi: 10.2337/dbi19-0005. [DOI] [PubMed] [Google Scholar]
- 62.Mroz PA, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. 2019;20:51–62. doi: 10.1016/j.molmet.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campbell JE. Targeting the GIPR for obesity: to agonize or antagonize? Potential mechanisms. Mol Metab. 2020;46:101139. doi: 10.1016/j.molmet.2020.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frayn KN, et al. Metabolic characteristics of human adipose tissue in vivo. Clin Sci (Lond) 1989;76(5):509–516. doi: 10.1042/cs0760509. [DOI] [PubMed] [Google Scholar]
- 65.DeFronzo RA, et al. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76(1):149–155. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White PJ, Newgard CB. Branched-chain amino acids in disease. Science. 2019;363(6427):582–583. doi: 10.1126/science.aav0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. doi: 10.1152/physrev.00063.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15(5):606–614. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lackey DE, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab. 2013;304(11):1175–1187. doi: 10.1152/ajpendo.00630.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herman MA, et al. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285(15):11348–11356. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.She P, et al. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab. 2007;293(6):E1552–1563. doi: 10.1152/ajpendo.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cannon B, Nedergaard J. Metabolic consequences of the presence or absence of the thermogenic capacity of brown adipose tissue in mice (and probably in humans) Int J Obes (Lond) 2010;34(Suppl 1):S7–S16. doi: 10.1038/ijo.2010.177. [DOI] [PubMed] [Google Scholar]
- 73.Nedergaard J, et al. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293(2):E444–E452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 74.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360(15):1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Mara AE, et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest. 2020;130(5):2209–2219. doi: 10.1172/JCI131126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cypess AM, et al. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 2015;21(1):33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen KY, et al. Opportunities and challenges in the therapeutic activation of human energy expenditure and thermogenesis to manage obesity. J Biol Chem. 2020;295(7):1926–1942. doi: 10.1074/jbc.REV119.007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becher T, et al. Brown adipose tissue is associated with cardiometabolic health. Nat Med. 2021;27(1):58–65. doi: 10.1038/s41591-020-1126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Briere DA, et al. Mechanisms to elevate endogenous GLP-1 beyond injectable GLP-1 analogs and metabolic surgery. Diabetes. 2018;67(2):309–320. doi: 10.2337/db17-0607. [DOI] [PubMed] [Google Scholar]
- 80.Steele R, et al. Measurement of size turnover rate of body glucose pool by the isotope dilution method. Am J Physiol. 1956;187(1):15–24. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- 81.Kraegen EW, et al. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol. 1985;248(3 Pt 1):E353–E62. doi: 10.1152/ajpendo.1985.248.3.E353. [DOI] [PubMed] [Google Scholar]
- 82.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Royal Statist Soc. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.