

Supplemental Digital Content is available in the text.
Keywords: acetylglucosamine, action potential, electrophysiology, hyperglycemia, phosphorylation
Abstract
Rationale:
Diabetic hyperglycemia is associated with cardiac dysfunction and increased arrhythmia risk, and CaMKII (calcium/calmodulin-dependent protein kinase II) function has been implicated. CaMKII activity is promoted by both oxidation and O-linked β-N-acetylglucosamine (O-GlcNAc) of known CaMKII sites.
Objective:
To investigate which posttranslational modifications occur in human diabetic hearts and how they alter electrophysiological and Ca2+ handling properties in hyperglycemia.
Methods and Results:
We assessed echocardiography, electrophysiology, Ca2+-handling, and protein expression in site-specific CaMKII mutant mice (O-GlcNAc-resistant S280A and oxidation-resistant MM281/2VV knock-ins, and global and cardiac-specific knockouts), in myocytes subjected to acute hyperglycemia and Ang II (angiotensin II) and mice after streptozotocin injections (to induce diabetes). Human patients with diabetes exhibit elevated CaMKII O-GlcNAcylation but not oxidation. In mice, acute hyperglycemia increased spontaneous diastolic Ca2+ sparks and waves and arrhythmogenic action potential changes (prolongation, alternans, and delayed afterdepolarizations), all of which required CaMKII-S280 O-GlcNAcylation. Ang II effects were dependent on NOX2 (NADPH oxidase 2)-mediated CaMKII MM281/2 oxidation. Diabetes led to much greater Ca2+ leak, RyR2 S2814 phosphorylation, electrophysiological remodeling, and increased susceptibility to in vivo arrhythmias, requiring CaMKII activation, predominantly via S280 O-GlcNAcylation and less via MM281/2 oxidation. These effects were present in myocytes at normal glucose but were exacerbated with the in vivo high circulating glucose. PLB (phospholamban) O-GlcNAcylation was increased and coincided with reduced PLB S16 phosphorylation in diabetes. Dantrolene, which reverses CaMKII-dependent proarrhythmic RyR-mediated Ca2+ leak, also prevented hyperglycemia-induced APD prolongation and delayed afterdepolarizations.
Conclusions:
We found that CaMKII-S280 O-GlcNAcylation is required for increased arrhythmia susceptibility in diabetic hyperglycemia, which can be worsened by an additional Ang II-NOX2-CaMKII MM281/2 oxidation pathway. CaMKII-dependent RyR2 S2814 phosphorylation markedly increases proarrhythmic Ca2+ leak and PLB O-GlcNAcylation may limit sarcoplasmic reticulum Ca2+ reuptake, leading to impaired excitation-contraction coupling and arrhythmogenesis in diabetic hyperglycemia.
Meet the First Author, see p 5
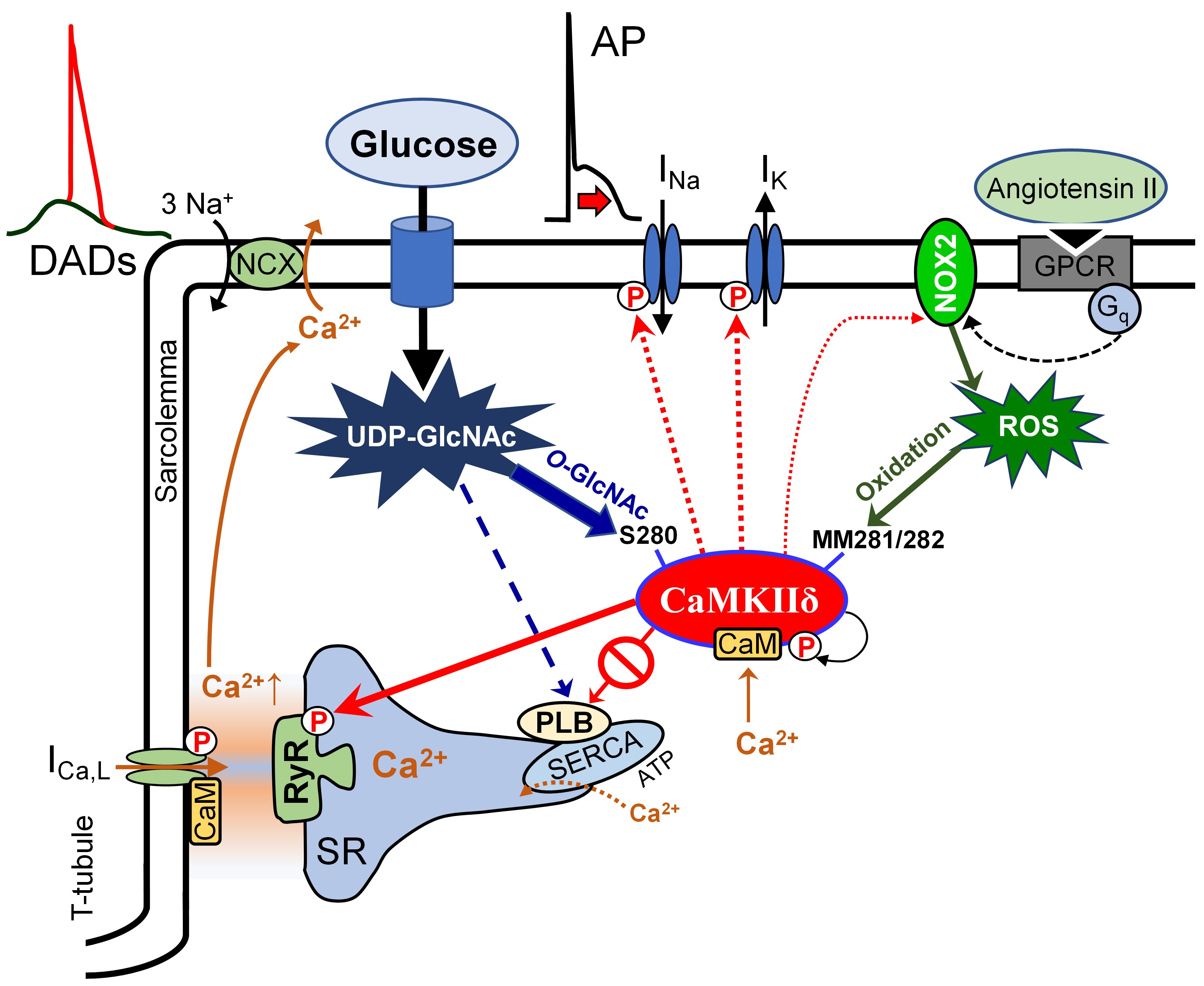
Cardiovascular complications including cardiomyopathy and arrhythmias are common sequelae of diabetes and the leading causes of mortality representing a major burden on human health.1,2 As the incidences of diabetes, prediabetes, and metabolic syndrome steadily increase worldwide while effective therapies for diabetic cardiomyopathy remain lacking, there is a pressing need for a better understanding of the exact molecular mechanisms by which diabetes promotes cardiac disease, to uncover novel therapeutic targets in precision medicine. Diabetic hyperglycemia and oxidative stress are major contributors to detrimental cardiac remodeling3 in which impaired intracellular Ca2+ handling and enhanced activity of CaMKII (calcium/calmodulin-dependent protein kinase II) have been implicated.4 CaMKII delicately senses and translates the periodic intracellular Ca2+ concentration rises in cardiomyocytes during excitation-contraction coupling to kinase activity.5 Besides the Ca2+/calmodulin-dependent activation of CaMKII that may lead to autophosphorylation (pT287),6 additional posttranslational modifications (PTMs) by oxidation7 and O-linked β-N-acetylglucosamine (O-GlcNAc)4,8 can also be enhanced in diabetes. Oxidation of MM281/282 pair9 and O-GlcNAcylation of S2804 have been identified in the regulatory hub region of CaMKII, and both promote autonomous, pathologically sustained kinase activity (molecular memory) leading to cardiac remodeling, heart failure (HF), and arrhythmias.10
Preventing the consequences of hyperglycemia and oxidative stress, including those by PTMs, is the focus of current research and clinical therapies in diabetes. Glycemic control is the basis of diabetes treatment and reduces cardiovascular complications and mortality.1 Postprandial hyperglycemia and glucose-variability may further increase cardiovascular risk,11 which may point to the memory function of CaMKII. O-GlcNAcylation of proteins, added by OGT (O-GlcNAc transferase) and removed by OGA (O-GlcNAcase),12 is generally enhanced in the diabetic heart; however, reduction of either OGT13 or OGA14 expression leads to deleterious effects on the diseased heart, suggesting a complex regulation of differential downstream targets including CaMKII,4 PLB (phospholamban),15 STIM1 (stromal interaction molecule 1),16 and HDAC4 (histone deacetylase 4)8 among >4000 potentially O-GlcNAcylated proteins.17 In line with this, intense glucose-lowering treatment may paradoxically increase the risk of HF,18 which again raises the need for a detailed understanding of the exact molecular mechanisms in this signaling network. Antioxidant therapies have also been proposed based on a large body of preclinical evidence,19 but to date, no clinical trial has shown clear benefit of antioxidant administration in patients with diabetes. Yet, the reduction of Ang (angiotensin)-induced oxidative stress and better glycemic control can contribute to the benefit of neprilysin-Ang inhibition (sacubitril/valsartan) therapy to treat HF in patients with diabetes in the PARADIGM-HF trial (prospective comparison of ARNI with ACEI to determine impact on global mortality and morbidity in heart failure trial).20 This evidence and unresolved questions prompted us to investigate the contribution of PTMs in CaMKII to the diabetic cardiac phenotype. A mechanistic understanding of this nuanced regulation may have important therapeutic implications by pointing to specific downstream targets that may explain ambiguous clinical results of glucose-lowering and antioxidant therapies in diabetes. Using PTM-resistant CaMKII mutant animals (MM281/2VV, S280A), we mechanistically investigated the contribution of each of these neighboring PTM sites in CaMKII to disease phenotype during acute and chronic diabetic hyperglycemia.
Here, we identified CaMKII S280 O-GlcNAcylation as the predominant regulator of increased arrhythmia risk in diabetic hearts. CaMKII MM281/2 oxidation was independent of hyperglycemia, was mediated by an Ang II—NOX2 (NADPH oxidase 2) pathway and played a secondary, yet synergistic role to promote autonomous kinase activity. The upregulated CaMKII-induced sarcoplasmic reticulum (SR) Ca2+ leak drove the arrhythmogenic electrophysiological remodeling in diabetic hyperglycemia.
Methods
Data Availability
The authors declare that all data and methods supporting the findings of this study are available in the Data Supplement or from the corresponding authors on reasonable request. Please see expanded methods and the Major Resources Table in the Data Supplement.
Human Heart Samples
Human right atrial appendage and left ventricular tissue samples were collected from patients undergoing on-pump coronary artery bypass graft surgery. Our informed consent practice, which required both verbal and written consent by each patient, conformed to the principles outlined in the Declaration of Helsinki. All human tissue work was performed as outlined in an Ethics Agreement approved by the Human and Disability Ethics Committee of New Zealand (LRS/12/01/001). For each of the 12 patients in the diabetic and nondiabetic cohorts, right atrial appendage samples were removed under normothermic conditions before cross clamping for cardiopulmonary bypass. For a subset of 6 patients in each cohort, a small wedge (1.5×1.5×10 mm) was surgically excised from the epicardium of the left ventricular anterior wall. Immediately after removal, tissue samples were placed in a sealed vial containing modified, low Ca2+ (0.5 mmol/L) Krebs-Henseleit buffer containing (in mmol/L): NaCl 118.5, KCl 4.5, NaH2PO4 0.3, MgCl2 1, NaHCO3 25, and glucose 11. Within 5 to 10 minutes after removal, all tissues were flash-frozen and stored at −80 °C.
Mouse Models and Animal Procedures
Several types of adult (10–12-week-old) C57BL/6J mice were used, including wild-type (WT, Jackson Laboratory, Stock No. 000664), NOX2 knockout (Jackson Laboratory, Stock No. 002365), CaMKIIδ cardiac-specific and global knockouts,21,22 CaMKIIδ-S280A-KI (that ablates the key O-GlcNAcylation site) created for this study,23 but also used in 2 parallel studies24,25 and mutated Met281Val and Met282Val (MMVV)-KI (that ablates key oxidation sites).7
Diabetes was induced in 6- to 8-week-old male mice by intraperitoneal injections with low-dose streptozotocin (50 mg/kg body weight in 40 mmol/L sodium citrate, pH=4.0) for 5 consecutive days and littermates received sodium citrate as vehicle control. Blood glucose levels were measured in fresh blood samples collected from the middle tail vein using OneTouch UltraMini blood glucose monitoring system and test strips (LifeScan; measurement range: 20–600 mg/dL). Only mice exhibiting >300 mg/dL blood glucose levels following streptozotocin injections (≈70% success rate) were included in the study.
Enzymatic isolation of left ventricular cardiomyocytes was performed as previously described.25 All animal handling and laboratory procedures were in accordance with the approved protocols (No. 19721 and No. 21064) of the Institutional Animal Care and Use Committee at University of California, Davis conforming to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (eighth edition, 2011).
Calcium Imaging
Intracellular Ca2+ transients and diastolic Ca2+ events (sparks and waves) were measured using confocal microscopy in freshly isolated ventricular cardiomyocytes loaded with Fluo-4 AM. Paired experiments were performed at room temperature in Tyrode’s solution containing normal glucose (100 mg/dL) then high glucose (540 mg/dL).
Cellular Electrophysiology
Recordings were performed in isolated ventricular cardiomyocytes using whole-cell patch-clamp with physiological solutions at 37 °C (for ionic composition see Data Supplement).26 Action potentials (APs) were evoked with short suprathreshold depolarizing pulses at 1 to 10 Hz pacing frequencies in current-clamp experiments. Arrhythmogenic diastolic activities were assessed during a 3-minute period following 1-minute tachypacing at 10 Hz.
In Vivo ECG and Pharmacological Stress Test
In vivo experiments were performed in anesthetized (isoflurane, 1%–2%) diabetic (streptozotocin-treated) and control (vehicle-treated) mice. For pharmacological stress test, mice were injected with isoproterenol (IP, 2 mg/kg) and caffeine (IP, 120 mg/kg). Premature ventricular complexes were counted during the next 60-minute period.
Murine Echocardiography
Transthoracic echocardiography was performed in anesthetized (isoflurane, 1.5%) animals before and after 4-week streptozotocin (or vehicle) treatment. Left ventricular B-mode, M-mode, and Doppler images were acquired using a Vevo 2100 echocardiography system (FUJIFILM VisualSonics) equipped with a 40 MHz transducer. Body temperature was carefully monitored, and anesthesia was adjusted to achieve 400 to 500 beats/min heart rate in each animal.
Fluorescence Recovery After Photobleaching
Fluorescence recovery after photobleaching experiments were performed in intact rabbit ventricular cardiomyocytes expressing GFP (green fluorescent protein)-tagged WT or S280A CaMKIIδC using an Olympus FluoView-1000 confocal microscope as previously described.27 Isolated myocytes were plated on laminin-coated coverslips, cultured, and used for experiments within 18 to 22 hours of adenoviral infection. Myocytes were electrically field stimulated at 0.5 Hz for 5 minutes at room temperature before initiating fluorescence recovery after photobleaching by bleaching a circular region with a diameter of 7.3 μm to 30% to 40% of the initial fluorescence value. Images were subsequently collected every 2.71 seconds to measure fluorescence recovery.
Statistical Analysis
Data are presented as mean±SEM. Normality of the data was assessed by Shapiro-Wilk test. Statistical significance of differences for normally distributed data was determined using 2-tailed Student t test (paired on unpaired) or ANOVA, when applicable. If the data were not normally distributed, we used nonparametric tests. Categorical outcomes were evaluated using Fisher exact test. In cellular experiments, we performed hierarchical statistical analyses (nested tests) to account for intersubject variability and nonindependent sampling (as multiple cells may come from one animal). GraphPad Prism 9 was used for data analysis. P<0.05 was considered statistically significant.
Results
CaMKII O-GlcNAcylation Is Enhanced in the Diabetic Human Heart
Figure 1A illustrates 5 known PTM sites in the highly conserved regulatory region of CaMKII, immediately adjacent to the CaM (calmodulin) binding site. These include oxidation (MM281/282) and O-GlcNAcylation (S280), autophosphorylation (pT287), and S-nitrosylation (C290), each of which promotes autonomous kinase activity after Ca2+/CaM dissociation (memory).
Figure 1.
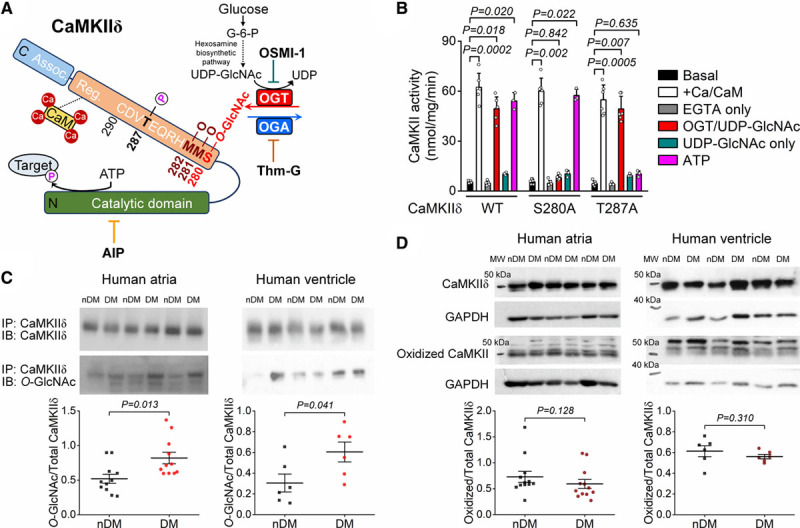
CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II) O-GlcNAcylation is increased in human diabetic hearts. A, Schematic of CaMKIIδ structure and sequence of the regulatory hub domain and the O-GlcNAcylation pathway. B, CaMKII activity measured as 32P incorporation in HEK293 cell lysates expressing wild type (WT), S280A, and T287A CaMKIIδ (n, basal=6, Ca/CaM [calmodulin]=6, EGTA=6, OGT [O-GlcNAc transferase]/UDP-GlcNAc=6, UDP-GlcNAc=3, ATP=3 in all 3 CaMKIIδ forms). Kruskal-Wallis 1-way ANOVA, followed by Dunn multiple comparisons test. C, Western blot data showing enhanced CaMKII O-GlcNAcylation in human atrial and ventricular samples from diabetic (DM) vs nondiabetic (nDM) patients (n, atrial-DM=11, atrial-nDM=11, ventricular-DM=6, ventricular-nDM=6). Mann-Whitney test. D, CaMKII oxidation was not increased in atrial and ventricular samples from patients with diabetes (n, atrial-DM=12, atrial-nDM=12, ventricular-DM=6, ventricular-nDM=6). Mann-Whitney test. MW indicates molecular weight; OGA, O-GlcNAcase; and OSMI-1, αR-[[(1,2-dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide.
To directly test whether CaMKIIδ S280 O-GlcNAcylation activates CaMKII autonomous activity, Figure 1B shows measurements of CaMKII-dependent incorporation of 32P from 32P-ATP into an artificial substrate, syntide-2 (as previously described28 and in the Data Supplement). Lysates of HEK293 cells expressing WT, S280A, and T287A CaMKIIδ were used. Maximal kinase activation was achieved by treating the CaMKII samples for 10 minutes with Ca2+ (200 µmol/L) and CaM (10 µmol/L; white bars), showing similar activation of all three CaMKIIδ forms by Ca2+-CaM. That activation is acutely reversed when Ca2+ is chelated with EGTA (10 mmol/L; gray bars). Robust autonomous kinase activity in WT CaMKIIδ was achieved by inclusion of either ATP (100 µmol/L) or a combination of UDP-GlcNAc (100 µmol/L) and recombinant human OGT (0.5 µg) during the Ca2+-CaM exposure and Ca2+-quench with EGTA (pink and red bars, respectively). In CaMKII-S280A, only ATP-induced autophosphorylation induced autonomic activity, whereas in T287A only O-GlcNAcylation-induced autonomous CaMKII activation. Notably, the direct substrate for O-GlcNAcylation by itself (UDP-GlcNAc) was insufficient to activate autonomy in any case (teal bars) without inclusion of the endogenous enzyme (OGT) that mediates O-GlcNAcylation (red bars). These data demonstrate that S280 and T287 are independently required for O-GlcNAcylation-dependent and autophosphorylation-dependent CaMKIIδ autonomy, respectively.
In human atrial and ventricular samples from patients with type 2 diabetes, we measured increased O-GlcNAcylation, but not oxidation, of CaMKII versus nondiabetic human samples (Figure 1C and 1D). Patients in the diabetic cohort exhibited higher fasting blood glucose and hemoglobin A1c values were obese and slightly hypertensive but did not exhibit chronic kidney disease, atrial fibrillation, or myocardial infarction, compared with age- and sex-matched nondiabetic patients (Table I in the Data Supplement). Importantly, the cardiac ejection fraction was preserved in both cohorts (ejection fraction >50%), although diabetic hearts exhibited diastolic dysfunction (60% increase in E/e′). Thus, cardiac CaMKII O-GlcNAcylation is elevated and might contribute to cardiac dysfunction in human diabetes.
To study the functional consequences of CaMKII O-GlcNAcylation in the heart, we used a knock-in mouse model in which a previously identified serine residue4 was mutated to alanine (S280A-KI) in CaMKIIδ conferring resistance to O-GlcNAcylation (Figure IA in the Data Supplement). CaMKII-S280A-KI mice exhibited unaltered baseline CaMKIIδ expression, and its functional readouts including CaMKII autophosphorylation (pT287) and phospholamban T17 phosphorylation (PLB pT17, a characteristic CaMKII target) at 12-week of age (Figure IB and IC in the Data Supplement). Likewise, the S280A-KI mice exhibited unaltered baseline cardiac contractile function on echocardiography, overall morphological parameters, and blood glucose levels (Table II in the Data Supplement). Thus, the new O-GlcNAc-resistant mutant mice exhibit a normal baseline phenotype.
High-Glucose Induced Diastolic SR Ca2+ Leak and CaMKII Mobility Require CaMKII S280 O-GlcNAcylation
CaMKIIδ phosphorylation of the cardiac RyR2 (ryanodine receptor 2) is a major contributor to pathological intracellular Ca2+ mishandling. We tested whether diabetic hyperglycemia might alter intracellular Ca2+ transients and SR Ca2+ release events (Ca2+ sparks and waves) in intact ventricular cardiomyocytes. We exposed freshly isolated cardiomyocytes to levels of high-glucose (540 mg/dL; 30 mmol/L) corresponding to what is observed in severe diabetes versus osmotically matched (glucose substituted with equimolar mannitol) low-glucose (100 mg/dL; 5.5 mmol/L) conditions. Acute high-glucose treatment (6 minutes) did not alter the pacing-induced Ca2+ transient amplitude or kinetics, but slightly reduced SR Ca2+ content in WT, cardiac-specific CaMKIIδ-knockout (cKO), or S280A-KI mice (Figure 2A and 2B). However, high glucose-induced a major increase in Ca2+ spark frequency, whether normalized to SR Ca2+ load or not (Figure 2C and 2D), but this effect was completely prevented in both the CaMKIIδ cKO and S280A-KI mice. Likewise, there were more arrhythmogenic Ca2+ waves induced by high-glucose in WT than cKO or S280A-KI mice (Figure II in the Data Supplement). We conclude that the glucose-induced increase in diastolic SR Ca2+ leak requires O-GlcNAcylation of CaMKIIδ at S280, which may contribute to arrhythmogenesis.
Figure 2.
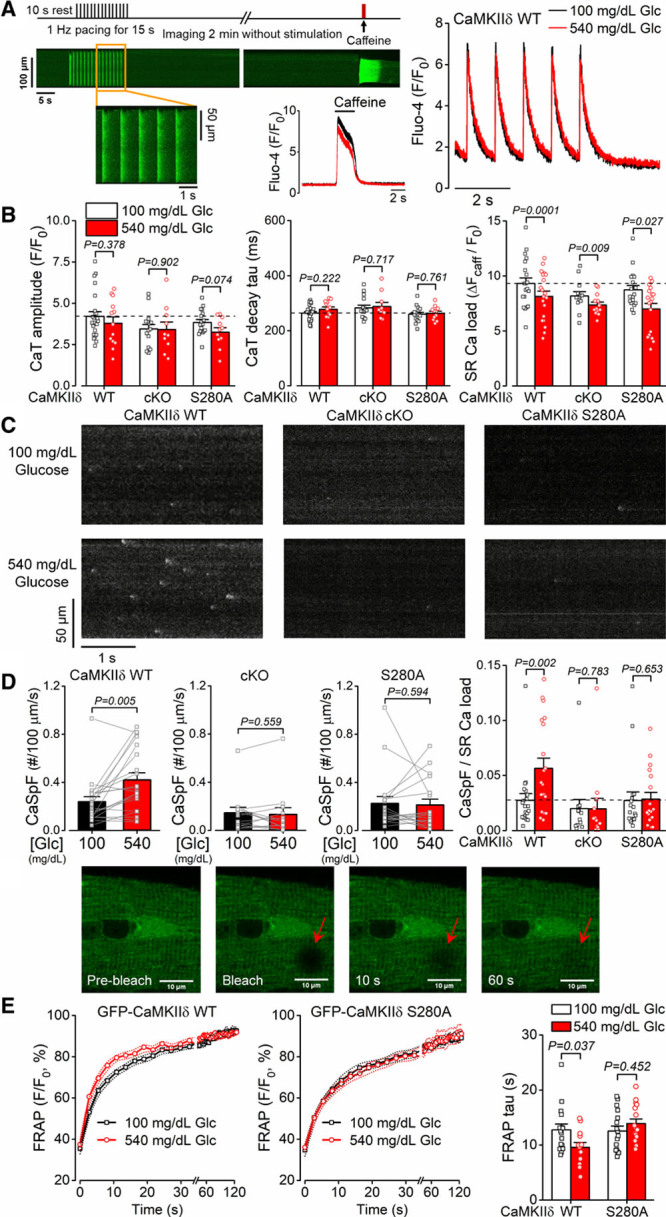
Glucose-induced diastolic sarcoplasmic reticulum (SR) Ca2+ leak is CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation dependent. A, Experimental protocol and representative intracellular Ca2+ signals (quantified as changes in Fluo-4 fluorescence). B, High-glucose treatment (540 mg/dL, 6 min) did not change the amplitude and decay of intracellular Ca2+ transient (CaT) but reduced SR Ca2+ load similarly in CaMKIIδ wild type (WT), cardiac-specific knockout (cKO), and O-GlcNAc-resistant CaMKIIδ-S280A knock-in (n[CaT]=total number of cells/animals, WT/normal glucose=24/8, WT/high-glucose=13/8, cKO/normal glucose=15/7, cKO/high-glucose=10/7, S280A/normal glucose=17/6, S280A/high-glucose=11/6; n[SR load]= total number of cells/animals, WT=20/11, cKO=13/9, S280A=18/9). Nested t test. C, Representative diastolic Ca2+ sparks were increased by high-glucose treatment in WT which was prevented in cKO and S280A. D, Increased Ca2+ spark frequency normalized to SR load indicates sensitized, leaky ryanodine receptors (n=total number of cells/animals, WT=20/11, cKO=13/9, S280A=18/9). Nested t test. E, Enhanced fluorescence recovery after photobleaching (FRAP) by high-glucose treatment in cardiomyocytes expressing GFP (green fluorescent protein)-tagged WT-CaMKIIδ (n=16 cells from 6 animals in normal glucose and n=13 cells from 6 animals in high-glucose) but not in GFP-CaMKIIδ-S280A (n=16 cells from 6 animals in both normal and high-glucose) indicates increased activation-dependent mobility of CaMKIIδ. Nested t test.
It was recently shown that CaMKIIδ activation promotes its subcellular mobility, which may facilitate phosphorylation of downstream targets outside the dyadic cleft to regulate contractile function, electrophysiology, and gene transcription.27 Figure 2E shows fluorescence recovery after photobleaching experiments in cardiomyocytes expressing GFP-tagged WT or S280A CaMKIIδ. High-glucose treatment increased the fluorescence recovery after photobleaching kinetics for WT but not for CaMKIIδ-S280A (Figure 2E), indicating that this increased CaMKIIδ mobility is also dependent upon S280 O-GlcNAcylation.
High-Glucose Induces Arrhythmogenic APs by CaMKII S280 O-GlcNAcylation
Enhanced CaMKIIδ activity is associated with cardiac arrhythmias. We tested whether acute diabetic hyperglycemia alters ventricular APD and arrhythmogenic activities. High-glucose treatment prolonged APD at lower pacing rates (1–3 Hz) and induced significant APD alternans in subsequent beats at high pacing rates (9–10 Hz; Figure 3A). Both of these arrhythmogenic AP changes were prevented in CaMKIIδ-cKO and S280A but not in the oxidation-resistant CaMKIIδ-MMVV (Figure 3B through 3D). Similar arrhythmogenic AP responses were observed in males and females (Figure III in the Data Supplement). Acutely blocking O-GlcNAcylation using the specific OGT inhibitor OSMI-1 (50 μmol/L) or chelating intracellular Ca2+ using EGTA in the pipette (10 mmol/L) both prevented the arrhythmogenic AP alterations by high-glucose (Figure 3E and 3F). Conversely, blocking O-GlcNAcase using the specific OGA inhibitor Thiamet-G (Thm-G, 100 nmol/L) induced these same APD changes even at normal glucose concentration and further enhanced the hyperglycemia effects. These results indicate that CaMKII O-GlcNAcylation but not oxidation mediate the acute hyperglycemic arrhythmogenic effects on APD.
Figure 3.
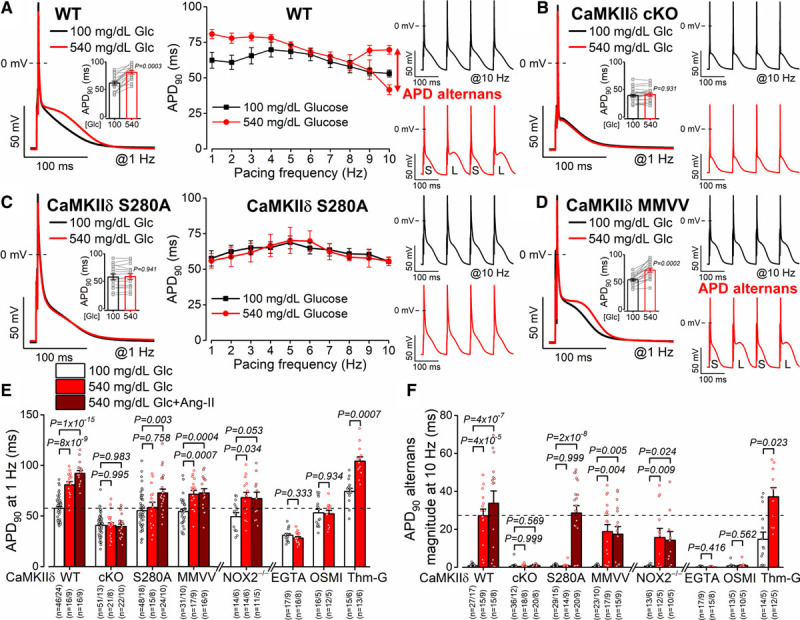
Glucose-induced arrhythmogenic action potentials are CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation dependent. A, Action potential duration (APD) prolongation and alternans (S, short; L, long) were increased by acute high-glucose treatment in wild-type (WT) myocytes (n=16 cells from 9 animals). B, Arrhythmogenic APD responses were prevented by CaMKIIδ-cardiac-specific knockout (cKO; n=21 cells from 8 animals). C and D, CaMKIIδ-S280A knock-in (n=15 ells from 8 animals) but not mutated Met281Val and Met282Val (MMVV; n=17 cells from 9 animals) was resistant to glucose-induced acute APD changes. Nested t test. E and F, Glucose-induced APD changes were prevented by CaMKIIδ-cKO, S280A, intracellular Ca2+ buffering (EGTA) or the O-GlcNAc transferase inhibitor OSMI-1 (αR-[[(1,2-dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide) but enhanced by the O-GlcNAcase inhibitor Thiamet-G (Thm-G) in WT. Ang II (angiotensin II) further enhanced arrhythmogenic APD changes, which was prevented by CaMKIIδ-cKO, MMVV, or in NOX2−/− (NADPH oxidase 2; n=total number of cells/animals is reported in the figure). Nested 1-way ANOVA, followed by Dunnett multiple comparisons test was used to compare 3 groups. Nested t test was used to compare 2 groups.
Ang II promotes both CaMKII activation and reactive oxygen species (ROS) production and could contribute to the diabetes disease phenotype. When we added Ang II (100 nmol/L) on top of high glucose it exacerbated the high-glucose effects on APD in WT myocytes (Figure 3E and 3F) but not in MMVV and NOX2-KO mice (NOX2−/−). Notably, both the high-glucose and Ang II effects on APD were completely absent in CaMKIIδ-cKO myocytes. Taken together, these data indicate that NOX2-mediated CaMKIIδ oxidation at MM281/2 as a separate and additive effect of Ang II to that caused by high glucose alone.
Diastolic arrhythmogenic activities such as delayed afterdepolarizations (DADs) and spontaneous APs (sAPs) were also enhanced by acute hyperglycemia and Ang II (Figure 4A through 4D and Figure IV in the Data Supplement). Indeed, the changes closely paralleled the glucose and Ang II effects on APD with respect to the 5 different mouse lines studied. That is, high glucose induction of DADs and sAPs required CaMKIIδ-S280 and O-GlcNAcylation, but not NOX2 or MM281/2 on CaMKIIδ, whereas Ang II required NOX2 and CaMKIIδ-MM281/2. CaMKIIδ was essential to both high glucose and Ang II effects, and those were only additive in the WT mice. As for APD effects above, these suggest 2 distinct signaling pathways that activate CaMKIIδ at neighboring regulatory sites. Dantrolene, which shifts back to normal the pathological and leaky RyR2 conformation that is induced by CaMKII phosphorylation,29 prevented hyperglycemia-induced arrhythmogenic AP changes and diastolic DADs (Figure V in the Data Supplement).
Figure 4.
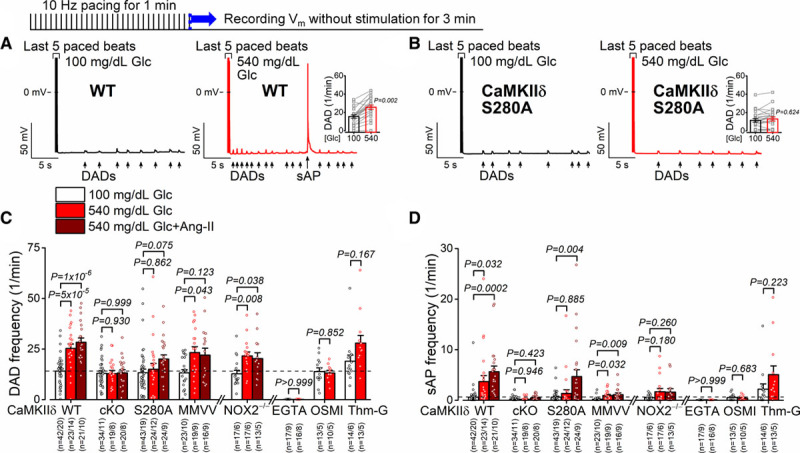
Glucose-induced delayed afterdepolarizations are CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation dependent. A and B, Delayed afterdepolarizations (DADs) and spontaneous action potentials (sAPs) were increased by high-glucose treatment in control (n=21 cells from 13 animals), the increases were prevented in CaMKIIδ-S280A (n=23 cells from 12 animals). Nested t test. C and D, Glucose-induced spontaneous diastolic activities were prevented by EGTA, OSMI-1 (αR-[[(1,2-dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide), and in CaMKIIδ cardiac-specific knockout (cKO) and S280A. Additional Ang II (angiotensin II) treatment further enhanced the arrhythmogenic activities, which were prevented in NOX2−/− (NADPH oxidase 2) and CaMKIIδ-cKO and mutated Met281Val and Met282Val (MMVV; n=total number of cells/animals is reported in the figure). Nested 1-way ANOVA, followed by Dunnett multiple comparisons test was used to compare 3 groups. Nested t test was used to compare 2 groups (EGTA, OSMI, Thm-G). WT indicates wild type.
Diabetes Increases Arrhythmia Susceptibility by CaMKII Activation
Next, we tested the contribution of these CaMKIIδ PTM sites to the disease phenotype in chronic diabetes. On repeated low-dose streptozotocin injections, mice developed severe hyperglycemia and were subject to morphometric and functional evaluation following 4 weeks of diabetes (Figure 5A). This time point was chosen based on literature data showing that 4 weeks following streptozotocin injections mice already exhibit cardiac remodeling8 without chronic kidney disease.30 This allowed us to focus primarily on the hyperglycemia effects on the heart rather than the secondary renal complications. Similar levels of hyperglycemia were seen in WT, S280A, and MMVV diabetic animals (Figure 5B), and similar weight losses with unaltered heart weight to body weight ratios were observed in all 3 mouse lines (Figure 5C). The ejection fraction and ventricular wall thickness in echocardiography were unchanged following streptozotocin in all experimental groups, indicating preserved systolic function at this early diabetic stage (Figure 5B and 5D). However, streptozotocin induced some changes in diastolic function (enlarged left atria, 21% decrease in E/A, 26% increase in E/e′) in WT, which were attenuated in CaMKIIδ-S280A and slightly also in MMVV (Figure 5E). Streptozotocin-treated WT mice also exhibited higher incidence of premature ventricular complexes on in vivo ECG recordings following caffeine+isoproterenol stress test, whereas premature ventricular complex incidence was decreased in streptozotocin-treated CaMKIIδ-KO (Figure 5F), indicating a CaMKIIδ-dependent marked increase in arrhythmia susceptibility in diabetes. In line with this, RyR2 S2814 phosphorylation in diabetic WT hearts was found to be 1.93× higher than in nondiabetic WT hearts, while phosphorylation of RyR S2814 was absent in CaMKIIδ-KO (Figure 5G). Moreover, there was a 26% increase in PLB O-GlcNAcylation in streptozotocin-treated WT and 6% increase in streptozotocin-treated CaMKIIδ-KO (Figure 5G), which was reported to occur at Ser16.15 There was a tendency for increased PLB expression (by 26%) in diabetic hearts, whereas PLB phosphorylation on S16 site was decreased by 22% and phosphorylation on T17 site was unchanged in diabetes (Figure VI in the Data Supplement).
Figure 5.
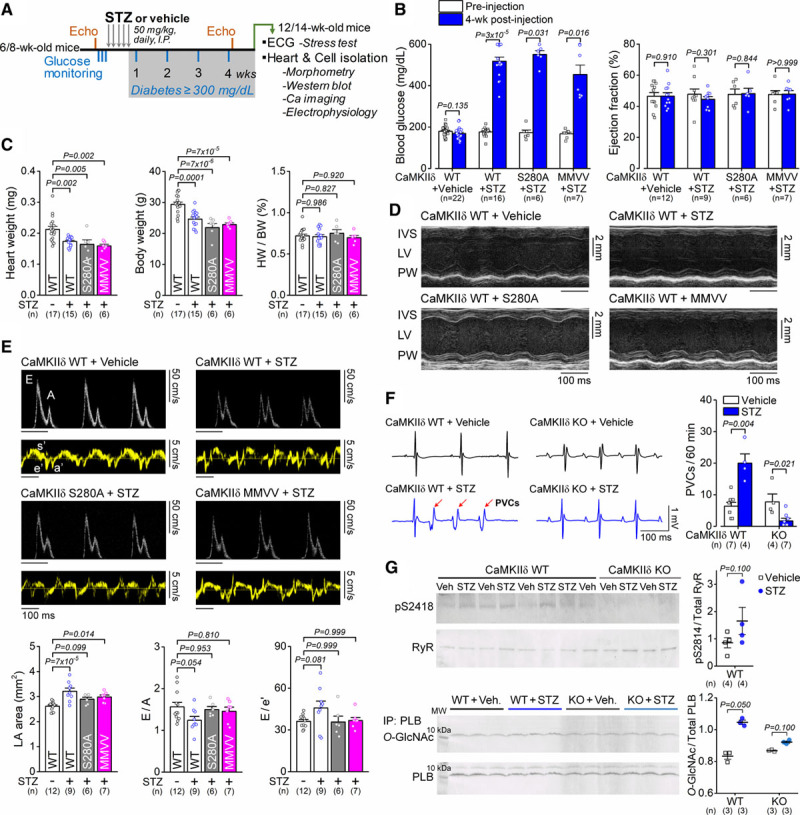
Diabetes-induced cardiac remodeling and arrhythmias are dependent on CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II) activation. A, Study protocol of streptozotocin (STZ)-induced diabetes and assessment of cardiac function. B, Blood glucose levels were highly elevated, whereas cardiac ejection fraction was preserved in STZ. Wilcoxon matched-pairs signed-rank test. C, Morphological parameters in either vehicle-treated or STZ-treated CaMKIIδ-wild type (WT), S280A, and mutated Met281Val and Met282Val (MMVV) mice. One-way ANOVA, followed by Dunnett multiple comparisons test. D, Preserved systolic heart function in STZ. E, Diastolic heart function (enlarged left atria, reduced E/A, increased E/e′) in 4-week diabetes. One-way ANOVA, followed by Dunnett multiple comparisons test. F, Incidence of premature ventricular complexes (PVCs) following isoproterenol+caffeine stress test was increased in diabetic WT animals, whereas CaMKIIδ-cardiac-specific knockout (cKO) was protective. Mann-Whitney test. G, Increased RyR (ryanodine receptor) S2814 phosphorylation and PLB (phospholamban) O-GlcNAcylation in STZ. Two technical replicates (blots) were performed for each protein sample. Mann-Whitney test. The number of biological replicates (n) is shown. HW/BW indicates heart weight to body weight ratio; IVS, interventricular septum; LA, left atrial; and LV, left ventricular.
Diabetes Promotes Diastolic SR Ca2+ Leak via CaMKII S280 O-GlcNAcylation
We assessed myocyte Ca2+ handling in ventricular cardiomyocytes freshly isolated from diabetic hearts. Streptozotocin induced a slight reduction in intracellular Ca2+ transient amplitude and rate of [Ca2+]i decay in all 3 genotypes (Figure 6A and 6B). SR Ca2+ load was preserved in normal glucose and slightly reduced in high-glucose conditions (Figure 6B), independent of CaMKIIδ PTMs. However, diabetes increased Ca2+ sparks and waves in WT in normal glucose and even further in high-glucose (as occur in vivo in streptozotocin-treated diabetic animals, Figure 6C and 6D and Figure IIC in the Data Supplement). Focusing on the Ca2+ spark frequency normalized to SR Ca2+ load (Figure 6D, right) streptozotocin induced an elevation of baseline Ca2+ sparks in WT and CaMKIIδ-MMVV-KI but not in S280-KI mice. Moreover, restoring glucose concentration to the levels seen in vivo in the streptozotocin mice (540 mg/dL) further increased Ca2+ sparks in all but the S280A myocytes. This has 2 important implications: (1) in the in vivo situation the WT diabetic mice have nearly 4× the rate of arrhythmogenic SR Ca2+ leak as in normal WT mice and (2) that the reversal of high glucose during cell isolation and incubation at normal 100 mg/dL glucose only partially reverses the chronic activation (ie, there is both an acutely reversible and a chronic component). Notably, these effects were completely prevented in CaMKIIδ-S280A and slightly attenuated in CaMKIIδ-MMVV.
Figure 6.
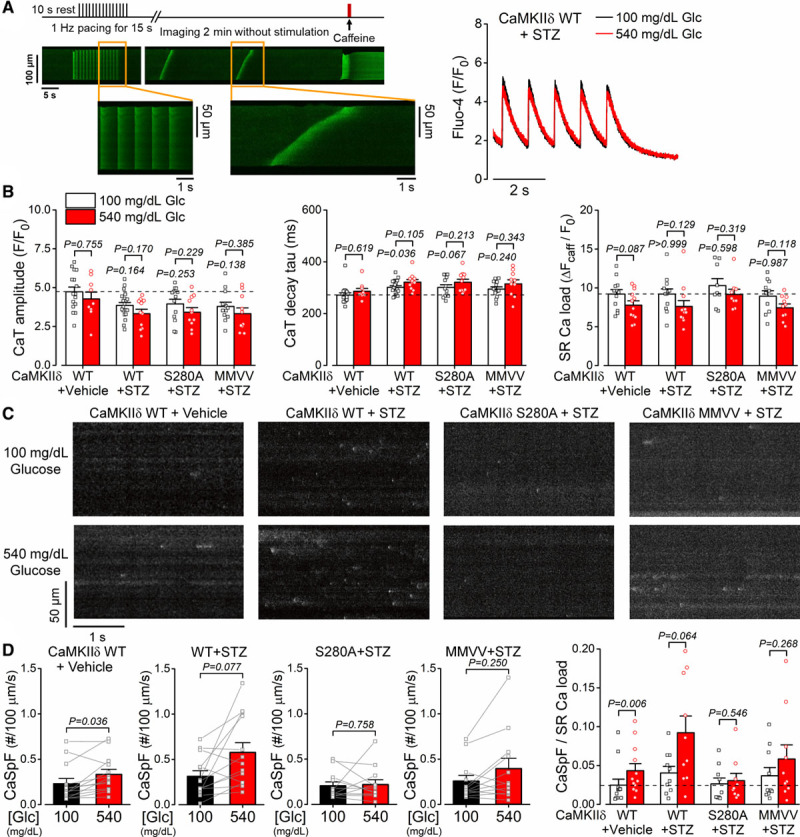
Diabetes-induced diastolic sarcoplasmic reticulum (SR) Ca2+ leak is CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation-dependent. A, Experimental protocol and representative intracellular Ca2+ signals (as changes in Fluo-4 fluorescence) showing spontaneous Ca2+ waves in wild type (WT) following 4-week of streptozotocin (STZ)-induced diabetes. B, Diabetes slightly reduced the amplitude of the intracellular Ca2+ transient and prolonged the Ca2+ transient decay without significantly altering SR Ca2+ load (n[CaT]=total number of cells/animals, WT+vehicle/normal glucose=16/7, WT+vehicle/high-glucose=9/7, WT+STZ/normal glucose=20/6, WT+STZ/high-glucose=11/6, S280A+STZ/normal glucose=13/5, S280A+STZ/high-glucose=11/5, mutated Met281Val and Met282Val [MMVV]+STZ/normal glucose=13/5, MMVV+STZ/high-glucose=10/5; n[SR load]=total number of cells/animals, WT+Vehicle=12/6, WT+STZ=12/7, S280A+STZ=9/5, MMVV+STZ=10/5). Acute effect of high-glucose within the same genotype/treatment was tested using nested t test. Effect of STZ-induced diabetes (in normal glucose) vs WT+Vehicle was tested using nested 1-way ANOVA, followed by Dunnett multiple comparisons test. C, Ca2+ sparks were increased in diabetic WT and further increased by acute high-glucose treatment. D, The increase in Ca2+ spark rate was prevented in CaMKIIδ-S280A and was attenuated in the CaMKIIδ-MMVV (n=total number of cells/animals, WT+Vehicle=12/6, WT+STZ=10/6, S280A+STZ=9/5, MMVV+STZ=10/5). Nested t test. CaSpF indicates Ca2+ spark frequency.
Diabetes Induces Arrhythmogenic APDs Predominantly by CaMKII S280 O-GlcNAcylation
Finally, in Figures 7 and 8, we subjected WT and diabetic myocytes to similar electrophysiological analysis as was done for acute hyperglycemia in Figures 3 and 4, informed by the in vivo proarrhythmic tendencies observed at the whole animal level in Figure 5F. Diabetes induction by streptozotocin led to pronounced AP remodeling even when measured at 100 mg/dL glucose, with APD prolonged versus vehicle controls and alternans already at physiological pacing rates (8 Hz) versus negligible alternans in vehicle controls even at 10 Hz (Figure 7A, 7E, and 7F). Raising glucose in streptozotocin myocytes to 540 mg/dL further increased APD and induced APD alternans already at 6 Hz. These arrhythmogenic AP changes were prevented by acute cell pretreatment with a selective CaMKII inhibitor peptide AIP (autocamtide-2-related inhibitory peptide; 1 μmol/L; Figure 7B). Importantly, high glucose also failed to increase APD or alternans in diabetic CaMKIIδ-S280A myocytes (Figure 7C), but in MMVV myocytes the effects were nearly as large as in WT+streptozotocin (Figure 7D). Conversely, Ang II did not induce additional APD prolongation or alternans in MMVV myocytes, although it did in the other 3 myocyte groups studied (Figure 7E and 7F). The conclusions here parallel the above for SR Ca2+ leak, in that CaMKIIδ-S280 appears essential for the high-glucose induced effects on APD and alternans, whereas the Ang II effect requires CaMKIIδ-MM281/2. Furthermore, the 4-week streptozotocin treatment alone produces effects that are incompletely reversed by simply lowering extracellular glucose from the high in vivo levels to those in the standard perfusate during cell isolation and incubation (leftmost 2 white bars in Figure 7E and 7F). However, even the acute restoration of the diabetic extracellular glucose level can further promote these APD effects and may more closely resemble the in vivo situation in these diabetic mice.
Figure 7.
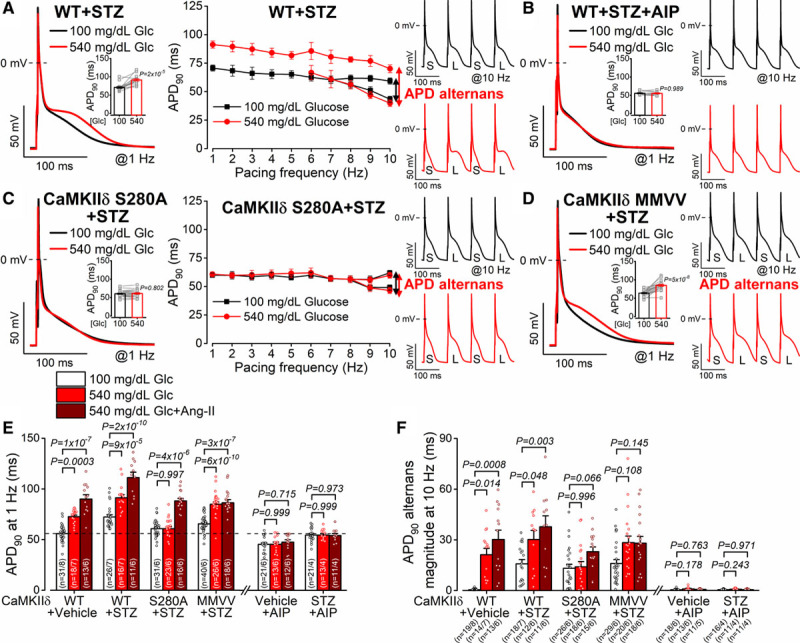
Diabetes-induced arrhythmogenic action potential remodeling is dependent predominantly on CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation. A, Action potential duration (APD) prolongation and alternans in streptozotocin (STZ)-treated wild-type (WT) murine ventricular myocytes (n=16 cells from 7 animals). B, Arrhythmogenic action potential (AP) remodeling in STZ was abolished by AIP (autocamtide-2-related inhibitory peptide; n=11 cells from 4 animals). C, APD prolongation was prevented in STZ-treated CaMKIIδ-S280A (n=22 cells from 6 animals). D, APD prolongation and alternans in STZ-treated CaMKIIδ-mutated Met281Val and Met282Val (MMVV; n=17 cells from 6 animals). Nested t test. E and F, AP remodeling was CaMKII-dependent and involved both hyperglycemia-dependent S280 O-GlcNAcylation (predominant mechanism) and Ang II-dependent 281/2MM oxidation (n=total number of cells/animals is reported in the figure). Nested 1 way ANOVA, followed by Dunnett multiple comparisons test. CaSpF indicates Ca2+ spark frequency.
Figure 8.
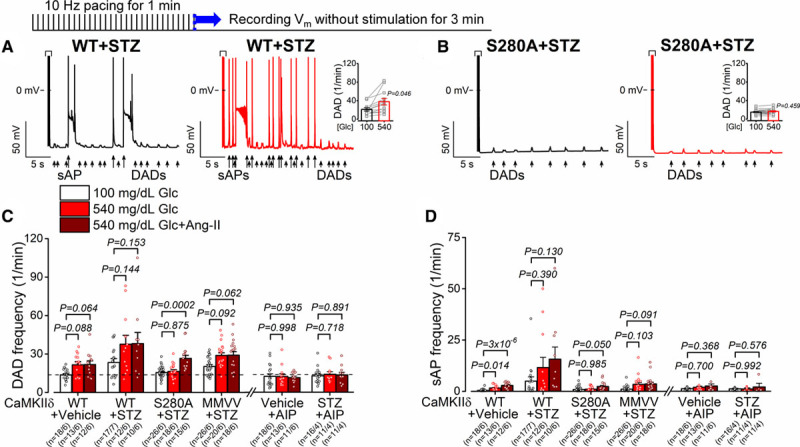
Diabetes-induced delayed afterdepolarizations are dependent on CaMKIIδ (Ca2+/calmodulin-dependent protein kinase II)-S280 O-GlcNAcylation. A and B, delayed afterdepolarizations (DADs) and spontaneous action potentials (sAPs) were increased by high-glucose in streptozotocin (STZ)-treated wild type (WT; n=12 cells from 6 animals), but it was prevented in STZ-treated CaMKIIδ-S280A (n=18 cells from 6 animals). Nested t test. C and D, Diabetic hyperglycemia largely enhanced spontaneous diastolic activities, which were prevented by AIP (autocamtide-2-related inhibitory peptide), in CaMKIIδ-cKO and S280A but not in mutated Met281Val and Met282Val (MMVV). Additional Ang II (angiotensin II) treatment further enhanced the arrhythmogenic activities, which were prevented in CaMKIIδ-cKO and MMVV but not in S280A (n=total number of cells/animals is reported in the figure). Nested 1-way ANOVA, followed by Dunnett multiple comparisons test.
Spontaneous diastolic DADs and sAPS were also measured in vehicle and streptozotocin-treated mice (Figure 8A through 8D and Figure IVC in the Data Supplement) and the effects qualitatively parallel and extend those for control animals (in Figure 4A through 4D) and for APD (Figure 7A through 7F). That is, streptozotocin promoted DADs and sAPs substantially, even at baseline glucose levels, in WT and CaMKIIδ-MMVV mice and this was exacerbated at the diabetic glucose level, especially in WT mice. However, those effects were completely prevented in CaMKIIδ-S280A mice. An interesting note here is that Ang II recruited a further increase in DADs (versus high glucose) only in the S280A mice (which had the lowest value in high glucose). But that may reflect a sort of ceiling effect for DADs in the WT cells, because Ang II still promoted sAPs in all but the MMVV mice (Figure 8C and 8D).
Discussion
CaMKII upregulation has been linked to various heart diseases including cardiac hypertrophy, HF, and arrhythmias.31 Altered intracellular Ca2+ signaling can be a cause but also a consequence of pathological CaMKII activation that leads to impairment of fundamental cellular processes such as excitation-contraction coupling, transcriptional regulation, cell mechanics, and energetics.10,31,32 PTMs of CaMKII may lead to sustained kinase activation when the intracellular Ca2+ transient has not yet been changed, thus promoting disease. CaMKII upregulation has been shown in diabetic cardiomyopathy, and CaMKII inhibition provided substantial benefit in various diabetic animal models.10 Increased O-GlcNAcylation of CaMKII has previously been identified in diabetes4,8; however, its contribution to disease remained elusive. Here, we show that a site-specific mutation of CaMKII that prevents O-GlcNAcylation (S280A knock-in) largely attenuated the cellular arrhythmogenic consequences of diabetic hyperglycemia without affecting the baseline phenotype.
Increased production of ROS, previously shown in diabetes,33 may further enhance CaMKII activation by oxidation of 2 neighboring methionines (281/2) in the regulatory domain of CaMKIIδ.9 Interestingly, it was previously shown that the increased ROS production and upregulation of p47phox and p67phox (NOX2 associated proteins) were attenuated following CaMKII inhibition in diabetic cardiomyocytes.34 However, we found that acute hyperglycemia led to CaMKII activation and its cellular consequences (RyR leak-mediated arrhythmogenic AP changes) exclusively via O-GlcNAcylation of S280 and not via MM281/2 oxidation. Even in chronic diabetes, CaMKIIδ-S280A was found to be largely protective, whereas CaMKIIδ-MMVV had only a minor role. These data suggest that CaMKII O-GlcNAcylation can be an early step in hyperglycemia-induced signaling, which then triggers kinase activation and may lead to further detrimental cellular remodeling, including increased ROS production.24 Moreover, a stimulated renin-Ang-aldosterone system, characteristic of type 2 diabetes and metabolic syndrome,35 can induce NOX-2-dependent ROS production and CaMKII oxidation.4,36 In contrast with our data here, Mesubi et al37 recently reported a predominant role for CaMKII oxidation in atrial fibrillation susceptibility in diabetes induced by one-time high-dose streptozotocin or repeated low-dose streptozotocin+high-fat diet. Therefore, the contribution of O-GlcNAcylation and oxidation of CaMKII may differ in different diabetes types, cardiac regions, and stages of disease. Overall, our results highlight the importance of tight blood glucose control in patients with diabetes and suggest that even significant temporary rises in blood glucose levels (impaired glucose tolerance) may lead to significant CaMKII activation by O-GlcNAcylation.
A dynamic cross-talk between O-GlcNAcylation and phosphorylation at the same or neighboring serine/threonine residues may occur in many nucleocytoplasmic proteins.38 In acute hyperglycemia, both S280 O-GlcNAcylation and T287 autophosphorylation of CaMKIIδ were previously found to be enhanced,4 suggesting a synergy between the 2 PTMs, creating molecular memory and prolonged active open state of the kinase. On the contrary, O-GlcNAcylation of PLB at S16 was shown to inhibit the phosphorylation of the same site by PKA (protein kinase A) and inhibit SERCA2 (SR Ca2+ ATPase 2), where only one of these modifications is possible at a given serine.15 This mechanism may also affect the CaMKII-dependent phosphorylation of the neighboring T17 in PLB because the Stokes radius of an O-GlcNAc moiety is ≈5× larger than a phosphate. In line with this, PLB O-GlcNAcylation was increased, pS16 was decreased, and pT17 levels were unchanged in diabetes (Figure 5G and Figure VI in the Data Supplement), and the intracellular Ca2+ transient decay was prolonged (Figure 6) indicating slowed SERCA and higher inhibition by PLB. These results agree with literature data reporting increased PLB expression and reduced pPLB levels in diabetes.39 Similarly, O-GlcNAcylation of HDAC4 at S642 was shown to prevent CaMKII-dependent phosphorylation at S632, which can be a protective mechanism against remodeling and systolic HF.8 However, the exact spatiotemporal details of these signaling networks (eg, which events may occur first) is yet to be determined. According to the current dogma, CaMKII gets activated predominantly in the dyadic cleft where local intracellular [Ca2+] may rise to 50 µmol/L during systolic SR Ca2+ release. From here, CaMKII may translocate to phosphorylate cytoplasmic and nuclear targets. As for Ca2+-dependent CaMKII activation,27 we found CaMKII-S280 O-GlcNAcylation in acute hyperglycemia further enhances CaMKIIδ mobility in the cytosol.
Remodeling in several ion channels, including voltage-gated Na+, Ca2+, and K+ channels have been shown in diabetes resembling the one that occurs via CaMKII signaling.10,31 We have recently shown that K+ channel downregulation in diabetes was markedly attenuated in CaMKII-S280 mice.25 Diabetes is also frequently associated with HF, which is known to induce complex ion channel remodeling26,40–42 and intracellular Ca2+ mishandling32,43,44 leading to impaired AP repolarization and SR Ca2+ leak (via RyR pS2814), and subsequent increase in the susceptibility to triggered activities and arrhythmias. This is in line with a recently demonstrated 2-hit arrhythmia model in diabetic hyperglycemia.45 CaMKII inhibition has been shown to prevent these arrhythmogenic events in various preclinical models of diabetes and HF.10,31 However, global CaMKII inhibition may have some detrimental effects in specific conditions relevant to diabetes, such as acidosis.46 Preventing just the O-GlcNAcylation of CaMKII and repressing its pathological autonomous activation may serve as a precision medicine approach to prevent arrhythmias in diabetic hyperglycemia.
Diastolic dysfunction is an early manifestation of diabetic cardiomyopathy and frequently reported in patients.47 Here, we show that CaMKII -S280 O-GlcNAcylation may contribute to diastolic dysfunction in diabetes. This effect could be mediated by the increased diastolic SR Ca2+ leak and also the phosphorylation of titin which increases diastolic stiffness in HF.48 However, assessing the contribution of CaMKII to diastolic dysfunction requires further investigation in additional animal models. Another limitation of our study is that we did not broadly assess possible phosphorylation differences attributed to CaMKII-S280 O-GlcNAcylation among the many CaMKII substrates in the diabetic hearts. HF with preserved ejection fraction is a major clinical challenge in diabetes, with very limited treatment options currently available.49 Empagliflozin, a SGLT2 (sodium-glucose cotransporter 2) inhibitor was demonstrated to significantly reduce cardiovascular mortality and hospitalization for HF in patients with diabetes in the EMPA-REG OUTCOME (empagliflozin cardiovascular event trial in type 2 diabetes mellitus patients).50 Importantly, empagliflozin was shown to reduce CaMKII activation and its cellular consequences in failing murine and human ventricular myocytes.51 Along those lines, the related drug exenatide induced of CASK (Ca/CaM dependent serine protein kinase) that inhibit CaMKII and could partially ameliorate hyperglycemic effects that we described here.52 Moreover, empagliflozin blunted postprandial glucose excursions and reduced glucose-variability in patients with diabetes, which may attenuate O-GlcNAc-dependent CaMKII activation.53 In line with this, dapagliflozin, another SGLT2 inhibitor was shown to reduce O-GlcNAcylated protein levels in a type 2 diabetic murine model.54
Enhanced O-GlcNAcylation was found in HF also in nondiabetic patients4,55 and animals13 as a result of altered cardiac metabolism. CaMKII O-GlcNAcylation was also enhanced in human failing hearts and even higher in HF patients with diabetes.4 Therefore, it may represent a therapeutic target in a broader range of heart diseases. Moreover, CaMKII O-GlcNAcylation is not restricted to the heart, but it has also been shown in brain samples of patients with diabetes and may occur in other organs as well.4 Impaired O-GlcNAcylation has been implicated in the pathogenesis of Alzheimer disease,56 neurodegeneration,57 and cancer.57 The potential involvement of CaMKII O-GlcNAcylation in those diseases is yet to be tested. Indeed, the ability of hyperglycemia to promote autonomous activation of the widespread and multifunctional CaMKII (already implicated in pathological heart disease) suggest value in further exploration of this pathway in other diseases.
Acknowledgments
We thank Mark E. Anderson (Johns Hopkins University) for kindly providing CaMKIIδ-mutated Met281Val and Met282Val (MMVV) mice. We thank Sonya Baidar, Johanna M. Borst, Logan R.J. Bailey, Austen J. Lucena, Nima Habibi, Sally Chesnut, Navya Chauhan, Juliana Mira Hernandez, Michael Nguyen, Irina Karashchuk, Natalie N. Pinna, and Maura Ferrero for help in cell isolation and laboratory tasks. We thank the surgeons and operating theatre staff of Dunedin Hospital, including Richard Bunton, Philip Davis, Krishna Bhagwat, Gareth Crouch, Marilyn Noye, Sean Coffey, and Michael J.A. Williams, as well as the New Zealand patients who generously donated tissue for this study. We thank Rachel Wallace for her help with human tissue Western blots.
Sources of Funding
This work was supported by grants from the National Institutes of Health (NIH) R01-HL030077 (D.M. Bers), P01-HL141084 (D.M. Bers), R01-HL142282 (D.M. Bers and J. Bossuyt), R01-HL111600 (C.M. Ripplinger), F32-HL144017 (C.Y. Ko), the Heart Research Council of New Zealand (HRC) 15/331 (J.R. Erickson), and the Royal Marsden Fund Project UOO1707 (J.R. Erickson).
Disclosures
None.
Supplemental Materials
Expanded Materials and Methods
Data Supplement Tables I–III
Data Supplement Figures I–VI
References 58,59
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- AIP
- autocamtide-2-related inhibitory peptide
- Ang II
- angiotensin II
- AP
- action potential
- CaM
- calmodulin
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- CASK
- Ca/CaM dependent serine protein kinase
- cKO
- cardiac-specific knockout
- DAD
- delayed afterdepolarization
- GFP
- green fluorescent protein
- HDAC4
- histone deacetylase 4
- HF
- heart failure
- MMVV
- CaMKII with mutated Met281Val and Met282Val
- NOX2
- NADPH oxidase 2
- OGA
- O-GlcNAcase
- O-GlcNAc
- O-linked β-N-acetylglucosamine
- OGT
- O-GlcNAc transferase
- OSMI-1
- αR-[[(1,2-dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-thienylmethyl)-benzeneacetamide
- PKA
- protein kinase A
- PLB
- phospholamban
- PTM
- posttranslational modification
- ROS
- reactive oxygen species
- RyR
- ryanodine receptor
- sAP
- spontaneous AP
- SERCA
- sarcoplasmic reticulum Ca2+ ATPase
- SGLT2
- sodium-glucose cotransporter2
- SR
- sarcoplasmic reticulum
- Thm-G
- Thiamet-G
- WT
- wild type
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.318402.
For Sources of Funding and Disclosures, see page 112.
Novelty and Significance
What Is Known?
In diabetes, excessive intracellular O-GlcNAcylation and reactive oxygen species have been reported.
-
Both O-GlcNAcylation and reactive oxygen species induce autonomous CaMKII (Ca2+/calmodulin-dependent protein kinase II)
activation.
Chronic CaMKII activation plays detrimental roles in the pathophysiology of impaired excitation-contraction coupling, cardiac remodeling, arrhythmias, and heart failure.
What New Information Does This Article Contribute?
We show that O-GlcNAcylation of CaMKII at serine 280 is the predominant mechanism for autonomous CaMKII activation in diabetic hyperglycemia. Oxidation of CaMKII at 2 neighboring methionine residues (281/2) is independent of hyperglycemia but is mediated by an angiotensin II—NOX2 (NADPH oxidase 2) pathway, and it plays a secondary, yet synergistic role to promote autonomous kinase activity.
CaMKII-dependent phosphorylation of the main sarcoplasmic Ca2+ release channel (RyR2 [ryanodine receptor 2]) at serine 2814 markedly increases proarrhythmic Ca2+ leak and drives proarrhythmic electrophysiological changes in diabetic hyperglycemia.
We found an increased O-GlcNAcylation of phospholamban that increases its inhibition of sarcoplasmic reticulum Ca2+ ATPase (SERCA) thus limiting SR Ca2+ reuptake and further impairing excitation-contraction coupling in diabetic hyperglycemia.
Clinically, diabetes is associated with increased risk of arrhythmias and heart failure. Excessive O-GlcNAcylation and reactive oxygen species production and upregulated CaMKII have been implicated in the pathophysiology of diabetic cardiomyopathy. However, the molecular details of this signaling were unclear. Here, we identified CaMKII S280 O-GlcNAcylation and subsequent SR Ca2+ leak as the predominant regulators of increased arrhythmia risk in diabetic hearts. This finding provides mechanistic insights into disease pathophysiology and may lead to better-targeted therapies in diabetes.
References
- 1.Cosentino F, Grant PJ, Aboyans V, Bailey CJ, Ceriello A, Delgado V, Federici M, Filippatos G, Grobbee DE, Hansen TB, et al. 2019 ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41:255–323. doi: 10.1093/eurheartj/ehz486 [DOI] [PubMed] [Google Scholar]
- 2.Jouven X, Lemaître RN, Rea TD, Sotoodehnia N, Empana JP, Siscovick DS. Diabetes, glucose level, and risk of sudden cardiac death. Eur Heart J. 2005;26:2142–2147. doi: 10.1093/eurheartj/ehi376 [DOI] [PubMed] [Google Scholar]
- 3.Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–638. doi: 10.1161/CIRCRESAHA.117.311586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. doi: 10.1038/nature12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 6.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–1274. doi: 10.1172/JCI65268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kronlage M, Dewenter M, Grosso J, Fleming T, Oehl U, Lehmann LH, Falcão-Pires I, Leite-Moreira AF, Volk N, Gröne HJ, et al. O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation. 2019;140:580–594. doi: 10.1161/CIRCULATIONAHA.117.031942 [DOI] [PubMed] [Google Scholar]
- 9.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. 2019;127:246–259. doi: 10.1016/j.yjmcc.2019.01.001 [DOI] [PubMed] [Google Scholar]
- 11.Ceriello A. Postprandial hyperglycemia and diabetes complications: is it time to treat? Diabetes. 2005;54:1–7. doi: 10.2337/diabetes.54.1.1 [DOI] [PubMed] [Google Scholar]
- 12.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815 [DOI] [PubMed] [Google Scholar]
- 13.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A. 2010;107:17797–17802. doi: 10.1073/pnas.1001907107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, Jones SP. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ Res. 2009;104:41–49. doi: 10.1161/CIRCRESAHA.108.189431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E, Suzuki K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20:1217–1226. doi: 10.1093/glycob/cwq071 [DOI] [PubMed] [Google Scholar]
- 16.Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U, Krebs-Haupenthal J, et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018;24:62–72. doi: 10.1038/nm.4452 [DOI] [PubMed] [Google Scholar]
- 17.Ma J, Hart GW. O-GlcNAc profiling: from proteins to proteomes. Clin Proteomics. 2014;11:8. doi: 10.1186/1559-0275-11-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boussageon R, Bejan-Angoulvant T, Saadatian-Elahi M, Lafont S, Bergeonneau C, Kassaï B, Erpeldinger S, Wright JM, Gueyffier F, Cornu C. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: meta-analysis of randomised controlled trials. BMJ. 2011;343:d4169. doi: 10.1136/bmj.d4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:375–415. doi: 10.1016/j.pharmthera.2014.01.003 [DOI] [PubMed] [Google Scholar]
- 20.Seferovic JP, Claggett B, Seidelmann SB, et al. Effect of sacubitril/valsartan versus enalapril on glycaemic control in patients with heart failure and diabetes: a post-hoc analysis from the PARADIGM-HF trial. Lancet Diabetes Endocrinol. 2017;5:333–340. doi: 10.1016/S2213-8587(17)30087-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegyi B, Borst JM, Lucena AJ, Bailey LRJ, Bossuyt J, Bers DM. Diabetic hyperglycemia regulates potassium channels and arrhythmias in the heart via autonomous CaMKII activation by O-linked glycosylation. Biophys J. 2019;116:98a. doi: 10.1016/j.bpj.2018.11.566 Abstract [Google Scholar]
- 24.Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, Heller Brown J, Molkentin JD, Bossuyt J, Bers DM. Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ Res. 2020;126:e80–e96. doi: 10.1161/CIRCRESAHA.119.316288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegyi B, Borst JM, Bailey LRJ, Shen EY, Lucena AJ, Navedo MF, Bossuyt J, Bers DM. Hyperglycemia regulates cardiac K+ channels via O-GlcNAc-CaMKII and NOX2-ROS-PKC pathways. Basic Res Cardiol. 2020;115:71. doi: 10.1007/s00395-020-00834-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegyi B, Bossuyt J, Griffiths LG, Shimkunas R, Coulibaly Z, Jian Z, Grimsrud KN, Sondergaard CS, Ginsburg KS, Chiamvimonvat N, et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc Natl Acad Sci U S A. 2018;115:E3036–E3044. doi: 10.1073/pnas.1718211115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wood BM, Simon M, Galice S, Alim CC, Ferrero M, Pinna NN, Bers DM, Bossuyt J. Cardiac CaMKII activation promotes rapid translocation to its extra-dyadic targets. J Mol Cell Cardiol. 2018;125:18–28. doi: 10.1016/j.yjmcc.2018.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erickson JR, Patel R, Ferguson A, Bossuyt J, Bers DM. Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ Res. 2011;109:729–738. doi: 10.1161/CIRCRESAHA.111.247148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, Chen-Izu Y, Cornea RL, Wehrens XHT, Bers DM. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72. doi: 10.1016/j.yjmcc.2016.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breyer MD, Böttinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K; AMDCC. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648 [DOI] [PubMed] [Google Scholar]
- 31.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455 [DOI] [PubMed] [Google Scholar]
- 33.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishio S, Teshima Y, Takahashi N, Thuc LC, Saito S, Fukui A, Kume O, Fukunaga N, Hara M, Nakagawa M, et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. 2012;52:1103–1111. doi: 10.1016/j.yjmcc.2012.02.006 [DOI] [PubMed] [Google Scholar]
- 35.Lim HS, MacFadyen RJ, Lip GY. Diabetes mellitus, the renin-angiotensin-aldosterone system, and the heart. Arch Intern Med. 2004;164:1737–1748. doi: 10.1001/archinte.164.16.1737 [DOI] [PubMed] [Google Scholar]
- 36.He BJ, Joiner ML, Singh MV, Luczak ED, Swaminathan PD, Koval OM, Kutschke W, Allamargot C, Yang J, Guan X, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat Med. 2011;17:1610–1618. doi: 10.1038/nm.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mesubi OO, Rokita AG, Abrol N, Wu Y, Chen B, Wang Q, Granger JM, Tucker-Bartley A, Luczak ED, Murphy KR, et al. Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. J Clin Invest. 2021;131:95747. doi: 10.1172/JCI95747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dillmann WH. Diabetic cardiomyopathy. Circ Res. 2019;124:1160–1162. doi: 10.1161/CIRCRESAHA.118.314665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6 [DOI] [PubMed] [Google Scholar]
- 41.Hegyi B, Bossuyt J, Ginsburg KS, Mendoza LM, Talken L, Ferrier WT, Pogwizd SM, Izu LT, Chen-Izu Y, Bers DM. Altered repolarization reserve in failing rabbit ventricular myocytes: calcium and β-Adrenergic effects on delayed- and inward-rectifier potassium currents. Circ Arrhythm Electrophysiol. 2018;11:e005852. doi: 10.1161/CIRCEP.117.005852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegyi B, Morotti S, Liu C, Ginsburg KS, Bossuyt J, Belardinelli L, Izu LT, Chen-Izu Y, Bányász T, Grandi E, et al. Enhanced depolarization drive in failing rabbit ventricular myocytes: calcium-dependent and β-adrenergic effects on late sodium, L-type calcium, and sodium-calcium exchange currents. Circ Arrhythm Electrophysiol. 2019;12:e007061. doi: 10.1161/CIRCEP.118.007061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89 [DOI] [PubMed] [Google Scholar]
- 45.Hegyi B, Ko CY, Bossuyt J, Bers DM. Two-hit mechanism of cardiac arrhythmias in diabetic hyperglycemia: reduced repolarization reserve, neurohormonal stimulation and heart failure exacerbate susceptibility. Cardiovasc Res. 2021:cvab006. doi: 10.1093/cvr/cvab006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neef S, Sag CM, Daut M, Bäumer H, Grefe C, El-Armouche A, DeSantiago J, Pereira L, Bers DM, Backs J, et al. While systolic cardiomyocyte function is preserved, diastolic myocyte function and recovery from acidosis are impaired in CaMKIIδ-KO mice. J Mol Cell Cardiol. 2013;59:107–116. doi: 10.1016/j.yjmcc.2013.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fontes-Carvalho R, Ladeiras-Lopes R, Bettencourt P, Leite-Moreira A, Azevedo A. Diastolic dysfunction in the diabetic continuum: association with insulin resistance, metabolic syndrome and type 2 diabetes. Cardiovasc Diabetol. 2015;14:4. doi: 10.1186/s12933-014-0168-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664–674. doi: 10.1161/CIRCRESAHA.111.300105 [DOI] [PubMed] [Google Scholar]
- 49.McHugh K, DeVore AD, Wu J, Matsouaka RA, Fonarow GC, Heidenreich PA, Yancy CW, Green JB, Altman N, Hernandez AF. Heart failure with preserved ejection fraction and diabetes: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:602–611. doi: 10.1016/j.jacc.2018.11.033 [DOI] [PubMed] [Google Scholar]
- 50.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. ; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720 [DOI] [PubMed] [Google Scholar]
- 51.Mustroph J, Wagemann O, Lücht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018;5:642–648. doi: 10.1002/ehf2.12336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mustroph J, Sag CM, Bähr F, Schmidtmann AL, Gupta SN, Dietz A, Islam MMT, Lücht C, Beuthner BE, Pabel S, et al. Loss of CASK accelerates heart failure development. Circ Res. 2021;128:1139–1155. doi: 10.1161/CIRCRESAHA.120.318170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishimura R, Tanaka Y, Koiwai K, Inoue K, Hach T, Salsali A, Lund SS, Broedl UC. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol. 2015;14:11. doi: 10.1186/s12933-014-0169-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, Dollet L, Le May C, Toumaniantz G, Manrique A, et al. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. 2017;66:1030–1040. doi: 10.2337/db16-0733 [DOI] [PubMed] [Google Scholar]
- 55.Lunde IG, Aronsen JM, Kvaløy H, Qvigstad E, Sjaastad I, Tønnessen T, Christensen G, Grønning-Wang LM, Carlson CR. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics. 2012;44:162–172. doi: 10.1152/physiolgenomics.00016.2011 [DOI] [PubMed] [Google Scholar]
- 56.Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014;289:34472–34481. doi: 10.1074/jbc.R114.601351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797 [DOI] [PubMed] [Google Scholar]
- 58.Hegyi B, Bányász T, Izu LT, Belardinelli L, Bers DM, Chen-Izu Y. β-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J Mol Cell Cardiol. 2018;123:168–179. doi: 10.1016/j.yjmcc.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res. 2011;109:1354–1362. doi: 10.1161/CIRCRESAHA.111.248401 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data and methods supporting the findings of this study are available in the Data Supplement or from the corresponding authors on reasonable request. Please see expanded methods and the Major Resources Table in the Data Supplement.